

Abstract
Aims
Chronic kidney disease (CKD) challenges diabetes management and is associated with increased cardiovascular morbidity and mortality. We examined whether clinical outcomes with insulin glargine 300 U/mL (Gla‐300) and insulin degludec 100 U/mL (IDeg‐100) are affected by renal function in a prespecified subgroup analysis from the BRIGHT trial.
Materials and methods
BRIGHT (NCT02738151) was a multicentre, open‐label, randomized, active‐controlled, two‐arm, parallel‐group, 24‐week study in insulin‐naïve uncontrolled type 2 diabetes (T2D). Participants were randomized 1:1 to evening Gla‐300 (n = 466) or IDeg‐100 (n = 463) and stratified based on baseline estimated glomerular filtration rate (eGFR) for this analysis.
Results
Heterogeneity of treatment effect across renal function subgroups was observed (P = .02), reflecting a greater mean glycated haemoglobin (HbA1c) reduction from baseline to week 24 with Gla‐300 versus IDeg‐100 in the eGFR <60 mL/min/1.73 m2 subgroup (least squares mean difference: −0.43% [95% confidence interval: −0.74% to −0.12%]), while there were no differences in hypoglycaemia incidence or rates over 24 weeks in that subgroup. HbA1c reductions were similar between treatments in the other eGFR subgroups. However, heterogeneity was observed for annualized rates of anytime (24 hours) or nocturnal (00:00‐05:59 hours) confirmed hypoglycaemia (≤70 mg/dL [≤3.9 mmol/L]) over 24 weeks showing less hypoglycaemia with Gla‐300 versus IDeg‐100 in the ≥90 mL/min/1.73 m2.
Conclusions
Kidney function seems to affect the glucose‐lowering effects of Gla‐300 versus IDeg‐100 in insulin‐naïve T2D. Greater HbA1c reductions with Gla‐300 without increase in hypoglycaemia risk, were observed in patients with eGFR <60 mL/min/1.73 m2.
Keywords: basal insulin, diabetes complications, insulin analogues, randomized trial, type 2 diabetes
1. INTRODUCTION
Chronic kidney disease (CKD) occurs in approximately 20%–40% of people with diabetes and is an independent risk factor for both cardiovascular disease (CVD) 1 and hypoglycaemia.2, 3 Hypoglycaemia is a key issue for people with diabetes and, along with CKD, is associated with increased morbidity and mortality, particularly in relation to CVD. 2 However, good glycaemic control may slow the progression of CKD in people with type 2 diabetes (T2D). 4
There is a need for well‐tolerated and effective glucose‐lowering treatments in people with T2D and CKD, which will allow people with T2D to reach glycaemic targets while minimizing hypoglycaemia. Comorbid CKD increases the difficulty of managing diabetes because some treatment options such as metformin, sulfonylureas, α‐glucosidase inhibitors and sodium glucose co‐transporter‐2 inhibitors (SGLT‐2i) are either contraindicated or not recommended as renal function deteriorates either because of the risk of hypoglycaemia, drug accumulation or lack of glycaemic efficacy. 2 Basal and/or rapid‐acting insulin is the therapeutic option that remains viable from early to most advanced stages of CKD, as it mimics physiological regulation of metabolism. However, because of the reduced renal insulin catabolism and impaired hypoglycaemia counter‐regulation in CKD, more careful titration of insulin is required in people with reduced renal function, to minimize the risk of hypoglycaemia. Therefore, it would be valuable to identify insulin options with a more desirable safety profile for people with T2D and CKD. 2
The second‐generation basal insulin analogues, insulin glargine 300 U/mL (Gla‐300) and insulin degludec 100 U/mL (IDeg‐100), have more stable and prolonged pharmacokinetic/pharmacodynamic profiles compared with first‐generation analogues such as insulin glargine 100 U/mL (Gla‐100).5, 6 Furthermore, when individual clinical doses are compared, Gla‐300 mimics the physiology of basal insulin on glucose and lipid metabolism, and suppresses glucagon with lower within‐day variability than Gla‐100.7, 8 The EDITION and BEGIN clinical trial programmes demonstrated comparable glycaemic control and reduced hypoglycaemia risk with Gla‐300 or IDeg‐100 compared with Gla‐100.9, 10, 11, 12 IDeg‐100 has been suggested to have comparable pharmacokinetics in people with and without renal impairment;13 US prescribing information for IDeg‐100 reports higher total and peak exposure in patients with mild to severe renal impairment versus those with normal kidney function, but concludes that any differences are not clinically relevant. 14 To our knowledge there are no clinical data currently available evaluating IDeg‐100 in people with T2D and renal impairment in comparison with either Gla‐100 or Gla‐300.
The BRIGHT trial, a head‐to‐head comparison of Gla‐300 and IDeg‐100 in insulin‐naïve people with T2D, demonstrated similar improvements in glycaemic control and hypoglycaemia over the entire study period with both insulins, but showed that Gla‐300 was associated with a lower incidence and rate of anytime (24 hours) confirmed hypoglycaemia (≤70 and <54 mg/dL) during the initial 12‐week active titration period. 15 Therefore, to examine whether clinical outcomes with Gla‐300 and IDeg‐100 are affected by renal function, a prespecified subgroup analysis of the primary endpoint was performed based on eGFR categories using data from the BRIGHT trial.
2. MATERIALS AND METHODS
2.1. Study design and participants
The study design and methods of the BRIGHT study have been reported previously. 15 In brief, BRIGHT (NCT02738151) was a multicentre, open‐label, randomized, active‐controlled, two‐arm, parallel‐group, 24‐week, non‐inferiority study in adult (≥18 years old) participants with uncontrolled T2D at screening, receiving oral antihyperglycaemic drugs with/without a glucagon‐like peptide‐1 (GLP‐1) receptor agonist at a stable dose for at least 3 months. Participants were randomized 1:1 to evening dosing with Gla‐300 (n = 466) or IDeg‐100 (n = 463).
Titration was performed at least weekly but no more than every 3 days, to a target fasting self‐monitored plasma glucose (SMPG) of 80–100 mg/dL (4.4–5.6 mmol/L) while avoiding hypoglycaemia (Table S1; see Supporting Information). Dose adjustments were based on median fasting SMPG values from the last three measurements, including the day of titration. Background therapies were unchanged unless safety concerns necessitated dose reduction or discontinuation.
All participants provided written informed consent and the study was conducted in accordance with the Declaration of Helsinki principles and the International Conference on Harmonization guidelines for Good Clinical Practice.
2.2. Outcomes
The primary endpoint of BRIGHT was changed in glycated haemoglobin (HbA1c) from baseline to week 24, results of which have been reported previously. 15 As per the statistical plan for BRIGHT, several predefined subgroup analyses of the primary endpoint were performed [age (<65/≥65 years), sex (male/female), baseline body mass index (<30/30–<35/≥35 kg/m2), screening HbA1c (<8/≥8%), diabetes duration (<10/≥10 years) and baseline eGFR (<60/60–<90/≥90 mL/min/1.73 m2)]. The present analysis focused on the outcomes according to renal function subgroups (eGFR ≥90 [normal], 60 to <90 [mild] and <60 mL/min/1.73m2 [moderate/severe]). In addition to the prespecified analysis of HbA1c change by renal function subgroups, secondary efficacy outcomes, including change in mean 24‐hour SMPG (from eight‐point profiles) and change in fasting SMPG over 24 weeks, and eight‐point SMPG profiles at baseline and week 24, were also analysed in a post‐hoc fashion according to renal function subgroups.
Safety endpoints included incidence and rates of confirmed [≤70 mg/dL (≤3.9 mmol/L) and <54 mg/dL (<3.0 mmol/L)] hypoglycaemia over 24 weeks. Hypoglycaemia incidence was also analysed during the active titration period (the first 12 weeks) and during the maintenance period (weeks 13–24). Confirmed hypoglycaemia included documented symptomatic or asymptomatic hypoglycaemia, as well as any severe events, which were defined as an event requiring assistance from another person to administer carbohydrate, glucagon or other resuscitative actions. Basal insulin dose was also assessed.
2.3. Data analysis and statistics
Safety endpoints were analysed in the safety population, defined as all randomized participants who received at least one dose of study insulin, according to the actual treatment received. All efficacy endpoints were assessed in the intention‐to‐treat population, defined as all randomized participants who received at least one dose of the study insulin, analysed according to the treatment group allocated by randomization. All continuous secondary efficacy endpoints were analysed by a mixed‐effect model with repeated measures (MMRM), using the missing at random framework, with fixed categorical effects of treatment, baseline eGFR categories, visit, treatment‐by‐visit interaction, baseline eGFR categories‐by‐treatment group interaction, baseline eGFR categories‐by‐visit interaction, baseline eGFR categories‐by‐treatment group‐by‐visit interaction, randomization strata of HbA1c at screening, randomization strata of sulfonylurea or glinide use at screening (yes; no), and the continuous fixed covariates of baseline efficacy parameter value and baseline efficacy parameter value‐by‐visit interaction.
Binary efficacy endpoints were assessed during the 24‐week on‐treatment period and before any rescue treatment, which were analysed using a logistic regression model and adjusted on randomization strata. For participants who discontinued study treatment prematurely, or for those who received rescue therapy during the 24‐week on‐treatment period, time windows were applied to retrieve assessments performed at premature end‐of‐treatment and pre‐rescue visits for the MMRM analyses. No multiplicity adjustments were made on secondary efficacy variables; only 95% confidence intervals (CIs) were reported.
For safety endpoints, the proportion of participants experiencing ≥1 hypoglycaemic event was analysed using logistic regression, including randomization strata as covariates. Hypoglycaemic event rates were analysed using an overdispersed Poisson regression model adjusted on randomization strata.
3. RESULTS
3.1. Participant disposition and baseline characteristics
BRIGHT included 466 participants in the Gla‐300 group and 463 in the IDeg‐100 group (Figure S1; see Supporting Information). All but one participant, who did not have a baseline eGFR measurement, were included in the analysis.
Baseline characteristics were comparable between the two treatment groups within each renal function subgroup (Table 1). However, some differences were observed between renal function subgroups. Those with impaired renal function tended to be older, with longer T2D duration and more commonly had a history of diabetic nephropathy than did those with normal renal function. Sulfonylurea treatment was comparable between the two treatment groups and was present in >60% of participants in the <60 mL/min/1.73 m2 subgroup.
TABLE 1.
Baseline characteristics according to renal function
| Baseline characteristic by baseline eGFR, mL/min/1.73 m2 | Gla‐300 | IDeg‐100 | ||||
|---|---|---|---|---|---|---|
| ≥90 (n = 246) | 60 to <90 (n = 172) | <60 (n = 47) | ≥90 (n = 221) | 60 to <90 (n = 193) | <60 (n = 49) | |
| Age, years | 56.9 ± 8.8 | 63.6 ± 8.4 | 68.8 ± 9.0 | 56.6 ± 9.7 | 62.9 ± 8.1 | 68.8 ± 7.7 |
| Age ≥65 years, n (%) | 51 (20.7) | 82 (47.7) | 34 (72.3) | 49 (22.2) | 82 (42.5) | 34 (69.4) |
| Sex (male), % | 51.2 | 57.0 | 48.9 | 52.9 | 60.6 | 36.7 |
| BMI, kg/m2 | 31.7 ± 4.2 | 31.8 ± 4.4 | 31.8 ± 4.9 | 31.4 ± 4.5 | 31.2 ± 4.4 | 31.1 ± 3.9 |
| Duration of T2D, years | 9.3 ± 5.7 | 11.6 ± 6.6 | 12.3 ± 5.5 | 9.5 ± 6.1 | 10.9 ± 5.5 | 15.4 ± 9.3 |
| Duration of T2D ≥10 years, n (%) | 107 (43.5) | 93 (54.1) | 31 (66.0) | 96 (43.4) | 107 (55.4) | 35 (71.4) |
| HbA1c, % | 8.82 ± 0.82 | 8.62 ± 0.83 | 8.58 ± 0.83 | 8.68 ± 0.82 | 8.51 ± 0.79 | 8.30 ± 0.67 |
| 24 h average 8‐point SMPG, mg/dL | 198.7 ± 47.4 | 186.3 ± 43.0 | 185.2 ± 47.7 | 193.3 ± 48.0 | 182.6 ± 43.0 | 186.5 ± 45.8 |
| eGFR, mL/min/1.73 m2 | 112.0 ± 18.7 | 76.5 ± 8.3 | 47.9 ± 9.5 | 111.6 ± 18.9 | 77.6 ± 8.4 | 49.0 ± 9.6 |
| Haematocrit, % | 44.0 ± 3.9 | 43.4 ± 3.6 | 41.5 ± 4.2 | 43.6 ± 3.7 | 43.6 ± 3.7 | 41.1 ± 4.2 |
| Haemoglobin, g/dL | 14.0 ± 1.4 | 13.9 ± 1.2 | 13.1 ± 1.5 | 13.9 ± 1.3 | 13.9 ± 1.3 | 12.9 ± 1.4 |
| History of diabetic nephropathy, n (%) | 7 (2.8) | 17 (9.9) | 15 (31.9) | 6 (2.7) | 15 (7.8) | 13 (26.5) |
| Microalbuminuriaa | 7 (100) | 11 (64.7) | 3 (20.0) | 6 (100) | 10 (66.7) | 3 (23.1) |
| Proteinuriaa | 0 | 3 (17.6) | 2 (13.3) | 0 | 2 (13.3) | 1 (7.7) |
| Impaired renal functiona | 0 | 3 (17.6) | 10 (66.7) | 0 | 3 (20.0) | 9 (69.2) |
| Antihyperglycaemic treatment, n (%) | ||||||
| Metformin | 236 (95.9) | 157 (91.3) | 34 (72.3) | 210 (95.0) | 175 (91.1) | 37 (75.5) |
| Sulfonylurea | 161 (65.4) | 103 (59.9) | 36 (76.6) | 140 (63.3) | 135 (70.3) | 34 (69.4) |
| Glinides | 5 (2.0) | 6 (3.5) | 1 (2.1) | 3 (1.4) | 4 (2.1) | 2 (4.1) |
| Thiazolidinediones | 8 (3.3) | 11 (6.4) | 2 (4.3) | 15 (6.8) | 7 (3.6) | 2 (4.1) |
| DPP‐4 inhibitors | 51 (20.7) | 54 (31.4) | 15 (31.9) | 44 (19.9) | 46 (24.0) | 16 (32.7) |
| SGLT‐2i | 31 (12.6) | 22 (12.8) | 9 (19.1) | 35 (15.8) | 25 (13.0) | 2 (4.1) |
| GLP‐1 receptor agonists | 27 (11.0) | 16 (9.3) | 3 (6.4) | 24 (10.9) | 33 (17.2) | 8 (16.3) |
| Alpha‐glucosidase inhibitors | 4 (1.6) | 2 (1.2) | 3 (6.4) | 1 (0.5) | 5 (2.6) | 1 (2.0) |
Data are mean ± SD unless otherwise stated.
Abbreviations: BMI, body mass index; DPP‐4, dipeptidyl peptidase‐4; eGFR, estimated glomerular filtration rate; Gla‐300, insulin glargine 300 U/mL; GLP‐1, glucagon‐like peptide‐1 (receptor agonist); IDeg‐100, insulin degludec 100 U/mL; SD, standard deviation; SGLT‐2i, sodium glucose co‐transporter‐2 inhibitors; SMPG, self‐monitored plasma glucose; T2D, type 2 diabetes mellitus.
Percentages based on participants with a history of diabetic nephropathy.
3.2. Glycated haemoglobin
A predefined subgroup analysis investigated the potential impact of age, sex, body mass index, screening HbA1c, diabetes duration and renal function on HbA1c reduction with Gla‐300 versus IDeg‐100 in the BRIGHT study (Figure S2; see Supporting Information). Heterogeneity of treatment effect was only observed across eGFR subgroups (P = .02), prompting the additional post‐hoc analyses by renal function that are the focus of this article.
Gla‐300 was associated with significantly greater mean HbA1c reductions from baseline to week 24 (8.58% to 6.94%) versus IDeg‐100 (8.30% to 7.28%) in the eGFR <60 mL/min/1.73 m2 subgroup (Figure 1: least squares mean difference −0.43% [95% CI: −0.74 to −0.12]). HbA1c reductions over 24 weeks were similar with either treatment in the other renal function subgroups. The HbA1c target (<7%) achievement did not differ between renal function subgroups at week 12 or week 24 (Table S3; see Supporting Information).
FIGURE 1.
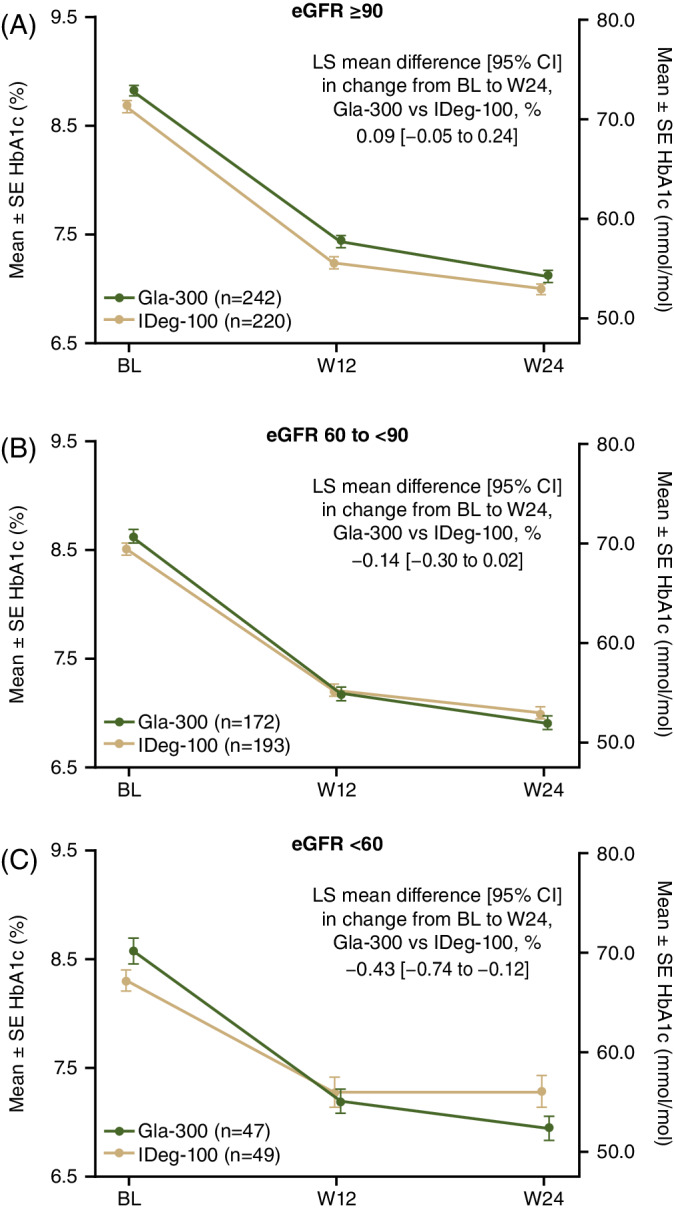
Mean ± SE HbA1c from baseline to W24 by renal function subgroup. A, ≥90 mL/min/1.73 m2. B, 60 to <90 mL/min/1.73 m2. C, <60 mL/min/1.73 m2. BL, baseline; CI, confidence interval; eGFR, estimated glomerular filtration rate; Gla‐300, insulin glargine 300 U/mL; HbA1c, glycated haemoglobin; IDeg‐100, insulin degludec 100 U/mL; LS, least squares; SE, standard error; W, week
3.3. Self‐monitored plasma glucose
Mean 24‐hour SMPG and mean fasting SMPG reductions (based on eight‐point profiles) showed a similar pattern to that seen for HbA1c in the eGFR <60 mL/min/1.73 m2 subgroup (Figure 2). Eight‐point SMPG profiles were similar for Gla‐300 and IDeg‐100 for all subgroups at baseline, but at week 24 there was a trend for lower values with Gla‐300 than IDeg‐100 at the post‐lunch and pre‐dinner time points (Figure S3; see Supporting Information).
FIGURE 2.
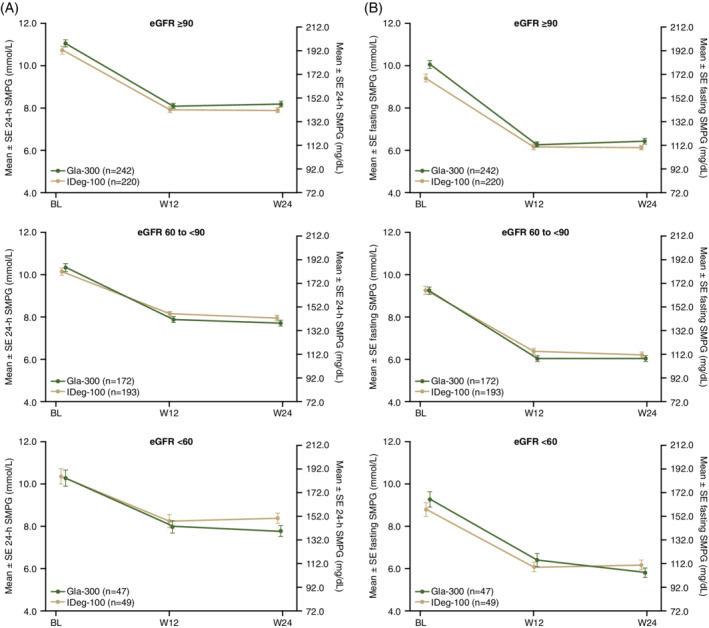
A, Mean ± SE 24‐h SMPG (from eight‐point profile), and B, mean ± SE fasting SMPG (pre‐breakfast) from baseline to W24 by renal function subgroup. eGFR, estimated glomerular filtration rate; Gla‐300, insulin glargine 300 U/mL; IDeg‐100, insulin degludec 100 U/mL; SE, standard error; SMPG, self‐monitored plasma glucose; W, week
3.4. Hypoglycaemia
Overall, incidence and annualized rates of hypoglycaemia increased with decreasing renal function (Figure 3). No heterogeneity of treatment effect was seen across eGFR subgroups for the incidence or annualized rates of anytime (24 hours) or nocturnal (00:00–05:59 hours) confirmed (<54 mg/dL [<3.0 mmol/L]) hypoglycaemia and for the incidence of anytime (24 hours) confirmed (≤70 mg/dL [≤3.9 mmol/L]) hypoglycaemia over 24 weeks. Hypoglycaemia incidence and rates were similar between treatments in the <60 mL/min/1.73 m2 subgroup, where the HbA1c difference was observed. However, there was significant heterogeneity of treatment effect across subgroups for the annualized rates of anytime (24 hours) and nocturnal (00:00–05:59 hours) confirmed (≤70 mg/dL [≤3.9 mmol/L]) hypoglycaemia showing less hypoglycaemia with Gla‐300 versus IDeg‐100 in the ≥90 mL/min/1.73 m2 subgroup (Figure 3).
FIGURE 3.
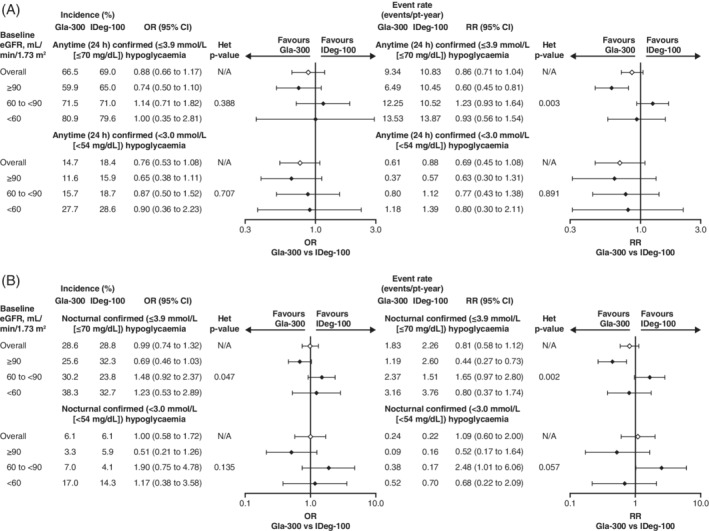
Incidence and rates of A, anytime (24 h) hypoglycaemic and B, nocturnal (00:00–05:59 h) hypoglycaemia with Gla‐300 versus IDeg‐100 over 24 weeks according to renal function subgroup. Rate ratios and CIs are based on an overdispersed Poisson regression model. Odds ratios and CIs are based on a logistic regression analysis. CI, confidence interval; eGFR, estimated glomerular filtration rate; Gla‐300, insulin glargine 300 U/mL; IDeg‐100, insulin degludec 100 U/mL; N/A, not applicable; OR, odds ratio; pt, patient; RR, rate ratio
Analysing hypoglycaemia by study period (0–12‐week active titration period or the 13–24‐week maintenance period) showed similar patterns in either time period to those observed over the full 24‐week period (Figure S4; see Supporting Information).
3.5. Insulin dose
In line with the overall BRIGHT population, within each renal function subgroup the mean starting dose of Gla‐300 was higher than IDeg‐100 (as per label instructions of 0.2 U/kg for Gla‐300 and 10 U for IDeg‐100) and it remained higher throughout the study. Daily doses of both insulins were highest in the subgroup with normal renal function and lowest in those with moderate/severe renal impairment (Figure 4, Table S4; see Supporting Information). However, interestingly, doses of Gla‐300 decreased from the eGFR ≥90 mL/min/1.73 m2 subgroup to the eGFR 60 to <90 mL/min/1.73 m2 subgroup, with no further decrease in the eGFR <60 mL/min/1.73 m2 subgroup. The opposite pattern was seen with IDeg‐100, with the difference in dose observed between the mild and moderate/severe renal function subgroups. Frequency of insulin dose change in each treatment by renal function subgroup is shown in Figure S5 (see Supporting Information). In all renal function subgroups, patients changing basal insulin dose >14 times were more likely to be on Gla‐300 than on IDeg‐100; this pattern was most pronounced in the <60 mL/min/1.73 m2 group.
FIGURE 4.
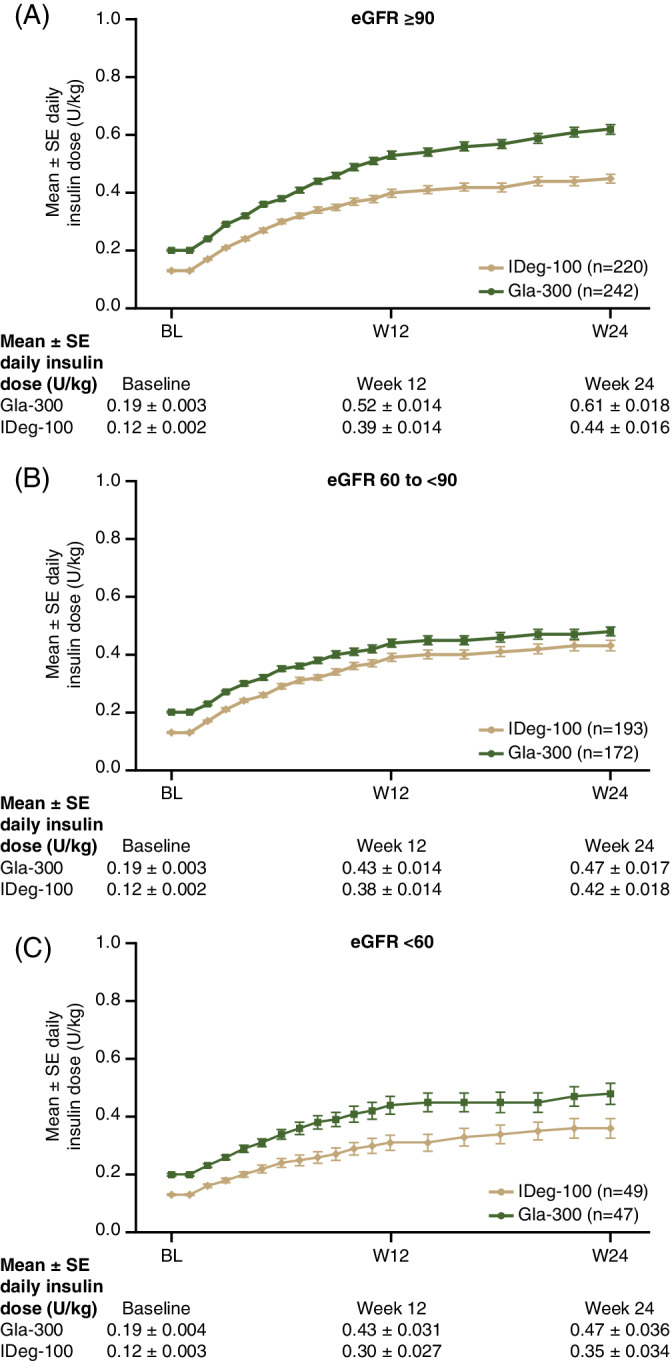
Mean daily insulin dose from baseline to W24 by renal function subgroup: A, eGFR ≥90 mL/min/1.73 m2, B, 60 to <90 mL/min/1.73 m2 and C, <60 mL/min/1.73 m2. eGFR, estimated glomerular filtration rate; Gla‐300, insulin glargine 300 U/mL; IDeg‐100, insulin degludec 100 U/mL; SE, standard error; W, week
4. DISCUSSION
This subanalysis of data from the BRIGHT study investigated the efficacy and safety of Gla‐300 and IDeg‐100 in people with T2D grouped by renal function. A prespecified subgroup of the primary endpoint demonstrated heterogeneity of treatment effect across baseline eGFR levels and greater HbA1c reduction with Gla‐300 compared with IDeg‐100 among those with eGFR <60 mL/min/1.73 m2, with no difference observed in those with eGFR ≥60 mL/min/1.73 m2. Based on this finding, post‐hoc analyses were performed looking at the same eGFR subgroups. Despite the greater reduction in HbA1c with Gla‐300 compared with IDeg‐100 among those with lower eGFR, there was no difference in hypoglycaemia over the 24 weeks, suggesting that the ability to achieve a lower glycaemic target with Gla‐300 does not compromise safety. Patients with diabetes and renal impairment have a higher risk of hypoglycaemia for multiple reasons, including lower insulin clearance, impaired hypoglycaemia counter‐regulation, older age and longer diabetes duration. 2 Therefore, an insulin treatment that decreases HbA1c without increasing the risk of hypoglycaemia is clinically important in this population.
The difference in HbA1c reduction between Gla‐300 and IDeg‐100 in the eGFR <60 mL/min/1.73 m2 group is secondary to the lower mean 24‐hour plasma glucose (nearly 11 mg/dL) with Gla‐300 at week 24, driven primarily by the post‐lunch and pre‐dinner plasma glucose from the eight‐point SMPG profile. The greatest difference at week 24 appears to be the post‐lunch plasma glucose and there is no clear explanation for this finding. A more detailed analysis of daily blood glucose fluctuations may have been possible using continuous glucose monitoring, but these data were not collected in BRIGHT. The observed HbA1c results are probably not because of differences in concomitant medications, as the use of sulfonylureas was similar. Among those with eGFR <60 mL/min/1.73 m2, more participants in the Gla‐300 group used SGLT‐2 inhibitors but this would have minimal impact on glycaemic control given the lower eGFR. A higher proportion of participants receiving IDeg‐100 used GLP‐1 receptor agonists versus Gla‐300, which could affect glycaemic control favourably. C‐peptide, a biomarker predictive of glycaemic control in basal insulin initiation, 16 was not measured in BRIGHT. However, in the present subanalysis, patients appeared to be older in the eGFR <60 mL/min/1.73 m2 group, whereas T2D duration appeared to be longer with IDeg‐100 (although there was high variability around this estimate, as measured by the standard deviations). The difference in glycaemic control observed, shown by HbA1c reduction, may have been related to insulin characteristics such as pharmacokinetics/pharmacodynamics or mechanism of action (subcutaneous precipitate formation for Gla‐300 vs. multi‐hexamer formation and albumin binding for IDeg‐100), 17 and/or to possible differences in renal handling of insulin catabolism at lower eGFR. Indirect support for this latter interpretation are the differential changes in dose of Gla‐300 and IDeg‐100 when moving across the subgroups from normal to slightly impaired, or to more impaired renal function. The fact that the dose of Gla‐300 did not decrease between the 60 to <90 and the <60 mL/min/1.73 m2 eGFR subgroups, unlike IDeg‐100, suggests differential handling of Gla‐300 and IDeg‐100 metabolism when renal function becomes impaired. Ad hoc studies are needed to confirm this hypothesis. Greater risk of hypoglycaemia has been observed with degludec versus Gla‐300 in a study with several limitations, which hypothesized that the cause could be the altered pharmacokinetics/pharmacodynamics of the acylated degludec, but not Gla‐300, in people with low circulating albumin.17, 18 In theory, this might be, at least in part, the case in participants in the present study with renal insufficiency, who generally tend to have lower than normal serum albumin, but in the BRIGHT study serum albumin was not measured.
It is possible that the lower risk of hypoglycaemia during the early weeks of treatment with Gla‐300 versus IDeg‐100 (previously reported in the overall population) enables some patients to titrate their basal insulin dose as intended to improve glycaemic control, without delayed titration. 15 Participants using Gla‐300 more often tended to change dose a greater number of times than those using IDeg‐100, a pattern particularly apparent in the subgroup of people with renal impairment. This more patient‐driven dynamic titration with Gla‐300 might suggest different titration needs of Gla‐300 versus IDeg‐100 in people with renal impairment given the lower bioavailability of Gla‐300 versus IDeg‐100,7, 8 but the interpretation remains hypothetical in the absence of an ad hoc study. Also of note, a lower annualized rate of hypoglycaemia was observed with Gla‐300 versus IDeg‐100 among those with eGFR ≥90 mL/min/1.73 m2 over the 24‐week study period, which is an interesting observation that needs to be confirmed in future analyses.
A strength of this analysis, derived from a randomized controlled head‐to‐head trial, is that it provides valuable information regarding Gla‐300 and IDeg‐100 in a patient population with renal impairment that is generally understudied.
This subgroup analysis of BRIGHT is limited as it was not a dedicated prospective trial in people with CKD, although the analysis of HbA1c change by renal function subgroup was pre‐planned. The number of patients in each subgroup was not controlled, and subsequently there were substantially fewer patients in the <60 mL/min/1.73 m2 group than the ≥90 mL/min/1.73 m2 group (96 vs. 467). The lack of randomization by renal subgroup also means that baseline characteristics that may have differed between subgroups were not controlled for in this. In addition, the lowest prespecified eGFR subgroup was <60 mL/min/1.73 m2, which indicates renal impairment, but it would be of further interest to analyse those with even lower eGFR levels of <45 or <30 mL/min/1.73 m2.
In conclusion, use of Gla‐300 versus IDeg‐100 in insulin‐naïve people with T2D and impaired renal function resulted in greater HbA1c reduction over the full study period of BRIGHT without between‐treatment differences in hypoglycaemia incidence or rates. The differences in outcomes observed in the present subanalysis reflect the importance of studies in special populations where the results are not necessarily similar to those seen in the general T2D population. Further investigation is required to confirm these results and to determine if Gla‐300 may allow more effective glycaemic management in this vulnerable population, and if so, to establish the mechanism by which these outcomes differ.
CONFLICT OF INTEREST
M.H. has served on an advisory panel for Eli Lilly, Novo Nordisk, Sanofi, AstraZeneca and Mundipharma; and has served as a consultant for Eli Lilly, Novo Nordisk, Sanofi, AstraZeneca and Mundipharma; and has received research support for AstraZeneca, Eli Lilly and Bristol‐Meyers Squibb; and has received honoraria or consulting fees from Amgen, AstraZeneca, Boehringer Ingelheim, Eli Lilly, Janssen, Johnson & Johnson, Novartis, Novo Nordisk, Pfizer, Medtronic and Sanofi. A.C. has served on an advisory panel for Abbott, AstraZeneca, Boehringer Ingelheim, Eli Lilly, HLS Therapeutics, Janssen, Merck, Medtronic, Novartis, Novo Nordisk and Sanofi; and has served as a co‐investigator in trials supported by Applied Therapeutics, Boehringer Ingelheim, Sanofi and Novo Nordisk; and has received honoraria for speaking from Abbott, AstraZeneca, Boehringer Ingelheim, Eli Lilly, Janssen, Medtronic, Merck, Novo Nordisk and Sanofi. D.M.‐W. has served as a consultant for Bayer; and has served on scientific advisory boards and received honoraria or consulting fees from Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, MSD, Novo Nordisk and Sanofi; and has served on a Speaker’s Bureau for Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, MSD, Novo Nordisk and Sanofi. J.K. has served on scientific advisory boards and received honoraria or consulting fees from Eli Lilly, Novo Nordisk, Sanofi, Napp, AstraZeneca and Boehringer Ingelheim; and has received grants/research support from Sanofi and AstraZeneca. J.W., Z.B. and F.L. are employees/shareholders of Sanofi. L.M.‐M. is an employee of IVIDATA Group, providing consultancy to Sanofi. J.R. has served on scientific advisory boards and received honoraria or consulting fees from Eli Lilly, Novo Nordisk, Sanofi, Janssen, Boehringer Ingelheim and Intarcia; and has received grants/research support from Merck, Pfizer, Sanofi, Novo Nordisk, Bristol‐Myers Squibb, Eli Lilly, GlaxoSmithKline, AstraZeneca, Janssen, Genentech, Boehringer Ingelheim, Intarcia and Lexicon. G.B. has received honoraria or consulting fees for Menarini, Sanofi; and has received research support/speaker’s bureau from Sanofi.
AUTHOR CONTRIBUTIONS
M.H., J.W., Z.B., F.L. and G.B.B. were involved in the concept and design of this analysis. L.M.‐M. performed statistical analyses. All authors were involved in the interpretation of the data, writing and reviewing drafts of the manuscript and approved the final version for submission.
Supporting information
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
ACKNOWLEDGEMENTS
The authors thank the study participants, trial staff and investigators for their participation. Editorial assistance was provided by Tamsin Brown, MSc, and Simon Rees, PhD, of Fishawack Communications Ltd, and was funded by Sanofi.
Haluzík M, Cheng A, Müller‐Wieland D, et al. Differential glycaemic control with basal insulin glargine 300 U/mL versus degludec 100 U/mL according to kidney function in type 2 diabetes: A subanalysis from the BRIGHT trial. Diabetes Obes Metab. 2020;22:1369–1377. 10.1111/dom.14043
Peer Review The peer review history for this article is available at https://publons.com/publon/10.1111/dom.14043.
Funding information Sanofi
REFERENCES
- 1. Papademetriou V, Lovato L, Doumas M, et al. Chronic kidney disease and intensive glycemic control increase cardiovascular risk in patients with type 2 diabetes. Kidney Int. 2015;87(3):649‐659. [DOI] [PubMed] [Google Scholar]
- 2. Alsahli M, Gerich JE. Hypoglycemia, chronic kidney disease, and diabetes mellitus. Mayo Clin Proc. 2014;89(11):1564‐1571. [DOI] [PubMed] [Google Scholar]
- 3. Rabkin R. Diabetic nephropathy. Clin Cornerstone. 2003;5(2):1‐11. [DOI] [PubMed] [Google Scholar]
- 4. MacIsaac RJ, Jerums G, Ekinci EI. Effects of glycaemic management on diabetic kidney disease. World J Diabetes. 2017;8(5):172‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Becker RH, Nowotny I, Teichert L, Bergmann K, Kapitza C. Low within‐ and between‐day variability in exposure to new insulin glargine 300 U/ml. Diabetes Obes Metab. 2015;17(3):261‐267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Heise T, Hermanski L, Nosek L, Feldman A, Rasmussen S, Haahr H. Insulin degludec: four times lower pharmacodynamic variability than insulin glargine under steady‐state conditions in type 1 diabetes. Diabetes Obes Metab. 2012;14(9):859‐864. [DOI] [PubMed] [Google Scholar]
- 7. Porcellati F, Lucidi P, Candeloro P, et al. Pharmacokinetics, pharmacodynamics, and modulation of hepatic glucose production with insulin glargine U300 and glargine U100 at steady state with individualized clinical doses in type 1 diabetes. Diabetes Care. 2019;42(1):85‐92. [DOI] [PubMed] [Google Scholar]
- 8. Lucidi P, Porcellati F, Cioli P, et al. Greater suppression of glucagon, lipolysis, and ketogenesis with insulin glargine U300 as compared with glargin U100 in type 1 diabetes mellitus. Diabetes Technol Ther. 2020;22(1):57–61. [DOI] [PubMed] [Google Scholar]
- 9. Riddle MC, Bolli GB, Ziemen M, et al. New insulin glargine 300 units/mL versus glargine 100 units/mL in people with type 2 diabetes using basal and mealtime insulin: glucose control and hypoglycemia in a 6‐month randomized controlled trial (EDITION 1). Diabetes Care. 2014;37(10):2755‐2762. [DOI] [PubMed] [Google Scholar]
- 10. Yki‐Jarvinen H, Bergenstal RM, Bolli GB, et al. Glycaemic control and hypoglycaemia with new insulin glargine 300 U/ml versus insulin glargine 100 U/ml in people with type 2 diabetes using basal insulin and oral antihyperglycaemic drugs: the EDITION 2 randomized 12‐month trial including 6‐month extension. Diabetes Obes Metab. 2015;17(12):1142‐1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bolli GB, Riddle MC, Bergenstal RM, et al. New insulin glargine 300 U/ml compared with glargine 100 U/ml in insulin‐naive people with type 2 diabetes on oral glucose‐lowering drugs: a randomized controlled trial (EDITION 3). Diabetes Obes Metab. 2015;17(4):386‐394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Roussel R, Ritzel R, Boelle‐Le Corfec E, Balkau B, Rosenstock J. Clinical perspectives from the BEGIN and EDITION programmes: trial‐level meta‐analyses outcomes with either degludec or glargine 300U/mL vs glargine 100U/mL in T2DM. Diabetes Metab. 2018;44(5):402‐409. [DOI] [PubMed] [Google Scholar]
- 13. Kiss I, Arold G, Roepstorff C, Bøttcher S, Kilm S, Haahr H. Insulin degludec: pharmachokinetics in patients with renal impairment. Clin Pharmacokinet. 2014;53:175‐183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nordisk Novo. TRESIBA® (insulin degludec) Prescribing Information. U.S Food and Drug Administration. www.accessdata.fda.gov/drugsatfda_docs/label/2019/203314s015s016lbl.pdf. 2019. Accessed November 29, 2019.
- 15. Rosenstock J, Cheng A, Ritzel R, et al. More similarities than differences testing insulin glargine 300 units/mL versus insulin Degludec 100 units/mL in insulin‐naive type 2 diabetes: the randomized head‐to‐head BRIGHT trial. Diabetes Care. 2018;41(10):2147‐2154. [DOI] [PubMed] [Google Scholar]
- 16. Landgraf W, Owens DR, Frier BM, Zhang M, Bolli GB. Fasting C‐peptide, a biomarker for hypoglycaemia risk in insulin‐naïve people with type 2 diabetes initiating basal insulin glargine 100 U/mL. Diabetes Obes Metab. 2020;22(3):315–323. [DOI] [PubMed] [Google Scholar]
- 17. Kawaguchi Y, Sawa J, Hamai C, Kumeda Y. Differential effect of hypoalbuminemia on hypoglycemia on type 2 diabetes patients treated with insulin glargine 300 U/mL and insulin degludec. Diabetes Ther. 2019;10(4):1535‐1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pieber TR, Bardtrum L, Isendahl J, Wagner L, Nishimura R. Commentary to "Differential Effect of Hypoalbuminemia on Hypoglycemia on Type 2 Diabetes Patients Treated with Insulin Glargine 300 U/ml and Insulin Degludec" by Kawaguchi et al. Diabetes Therapy 2019″. Diabetes Ther. 2020;11(2):561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5