

Abstract
We report the preparation of lithium‐salt‐free KDA (potassium diisopropylamide; 0.6 m in hexane) complexed with TMEDA (N,N,N′,N′‐tetramethylethylenediamine) and its use for the flow‐metalation of (hetero)arenes between −78 °C and 25 °C with reaction times between 0.2 s and 24 s and a combined flow rate of 10 mL min−1 using a commercial flow setup. The resulting potassium organometallics react instantaneously with various electrophiles, such as ketones, aldehydes, alkyl and allylic halides, disulfides, Weinreb amides, and Me3SiCl, affording functionalized (hetero)arenes in high yields. This flow procedure is successfully extended to the lateral metalation of methyl‐substituted arenes and heteroaromatics, resulting in the formation of various benzylic potassium organometallics. A metalation scale‐up was possible without further optimization.
Keywords: arenes, flow chemistry, heteroarenes, lateral metalation, potassium
Potassium in flow: Functionalized(hetero)arenes were metalated using a commercial continuous‐flow setup, leading to the corresponding potassium organometallics. Trapping with various electrophiles gave polyfunctionalized (hetero)aromatics in good yields. Further, the flow procedure was extended to the lateral metalation of methyl‐substituted (hetero)arenes.

Of all the alkali metals, lithium has by far received the most attention in organic synthesis.1 However, the use of sodium and potassium organometallic intermediates has been explored since more than a century2 and presents several specific advantages such as enhanced reactivity, low prices, and moderate toxicity of these alkali organometallics as well as opportunities for new metalation selectivities.3 Recently, we have reported that the use of continuous flow techniques4 considerably facilitates the use of sodium bases such as NaDA (sodium diisopropylamide) for the selective sodiation of aromatics and heterocycles.5 Herein, we wish to report a new metalation procedure making it possible to perform arene and heteroarene metalations as well as lateral metalations using potassium diisopropylamide (KDA) and N,N,N′,N′‐tetramethylethylenediamine (TMEDA) in continuous flow in a hexane/tetrahydrofuran (THF) mixture.
Whereas KDA was usually prepared by the Schlosser method by mixing LDA (lithium diisopropylamide) with tBuOK,6 we have envisioned preparing this base in the absence of any lithium salts, using a modified procedure decribed by Collum for the preparation of NaDA.7 Thus, small slices of oil‐free solid potassium suspended in hexane were mixed with diisopropylamine. The resulting suspension was cooled to 0 °C and isoprene was added dropwise. After 30 min of stirring at 0 °C, the suspension was warmed to 25 °C, leading after 6 h reaction time to a dark solution (Table 1, entries 1–6). The resulting KDA/TMEDA solution was titrated with a standardized solution of 0.40 m n‐butanol in hexane. In most cases, an excess of potassium (ca. 3 equiv) was used and the KDA/TMEDA yield was calculated based on diisopropylamine (1.0 equiv). We have varied the equivalents of TMEDA and isoprene (entries 1–4) and found that 1.0 equiv of TMEDA and 0.5 equiv of isoprene resulted in the best yield after 6 h reaction time (entry 4).6a Stirring for longer times did not improve the yield. Such KDA/TMEDA solutions were stable for at least one week at 25 °C. Similar yields were obtained using cyclohexane instead of hexane (entry 5). A quantitative yield was reached by setting potassium as the limiting reagent (1.0 equiv) and adding an excess of diisopropylamine (DIPA, 3.0 equiv), TMEDA (3.0 equiv), and isoprene (1.5 equiv; entry 6). Attempts to extend this preparation to 2,2,6,6‐tetramethylpiperidine (TMPH) or Cy2NH led to significantly lower yields (entries 7 and 8). For subsequent experiments performed in continuous flow, we have used the KDA/TMEDA preparation conditions described in entry 4.
Table 1.
Optimization of the preparation of potassium amide bases using solid potassium, secondary amides, TMEDA and isoprene in hexane.

|
Entry |
R2NH 1.0 equiv |
TMEDA X equiv |
Isoprene X equiv |
t [h] |
Molarity (K base) |
Yield [%] |
|---|---|---|---|---|---|---|
|
1 |
DIPA |
2.7 |
0.5 |
6 |
0.33 |
33 |
|
2 |
DIPA |
1.0 |
1.0 |
6 |
0.40 |
40 |
|
3 |
DIPA |
2.7 |
1.0 |
6 |
0.50 |
50 |
|
4 |
DIPA |
1.0 |
0.5 |
6 |
0.56 |
56 |
|
5 |
DIPA |
1.0 |
0.5 |
18 |
0.57 (0.49) |
57 (49)[a] |
|
6 |
DIPA |
3.0 |
1.5 |
18 |
0.33 |
99[b] |
|
7 |
TMPH |
1.0 |
0.5 |
6 |
0.20 |
20 |
|
8 |
HNCy2 |
1.0 |
0.5 |
6 |
0.28 |
28 |
[a] Yield of KDA/TMEDA in cyclohexane. [b] Potassium was used as the limiting reagent, DIPA was used in excess (3.0 equiv).
In preliminary experiments, we have optimized the reaction conditions for performing metalations with KDA/TMEDA in hexane and in continuous flow using benzofuran (1 a) in THF as the substrate and adamantanone (2 a) as the quenching reagent. We have varied the temperature, the flow rate, and the reactor size (reactor volume) and have found that it was best to perform the metalation at −78 °C using 1.5 equiv of KDA/TMEDA, a 4 mL tube reactor, and a combined flow rate of 10 mL min−1, leading to a reaction time of 24 s for the metalation.8 The resulting potassium organometallic 3 a was then quenched with adamantanone (2 a, 1.5 equiv) at −40 °C for 10 min, leading after workup to the tertiary alcohol 4 aa in 95 % isolated yield (Scheme 1).
Scheme 1.

Metalation of benzofuran (1 a) with KDA/TMEDA and subsequent trapping with adamantanone (2 a) in continuous flow. [a] Isolated yield of analytically pure product. [b] Cyclohexane was used as solvent.
These potassium organometallics display a high reactivity and the metalation of benzothiazole under optimum conditions9 (flow rate: 10 mL min−1; reaction time: 0.18 s; reactor volume: 0.03 mL; reaction temperature: −78 °C) furnished the potassium intermediate 3 b, which was trapped with various electrophiles such as ketones (adamantanone (2 a) and norcamphor (2 b)), leading to the tertiary alcohols 4 ba and 4 bb in 74–77 % yield (Table 2, entries 1 and 2).
Table 2.
Metalation of benzothiazole (1 b) using KDA/TMEDA in continuous flow and subsequent batch quenching with various electrophiles of type 2 leading to functionalized benzothiazole derivatives of type 4.

|
Entry |
Electrophile of type 2 |
Product of type 4 [a] |
|
Entry |
Electrophile of type 2 |
Product of type 4 [a] |
|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
|
|
1 |
2a |
4ba: 77%, (74%)[b] |
|
4 |
2d |
4bd: 91% |
|
|
|
|
|
|
|
|
|
2 |
2b |
4bb: 77% |
|
5 |
2e |
4be: 93% |
|
|
pivaldehyde |
|
|
|
Bu2S2 |
|
|
3 |
2c |
4bc: 75% |
|
6 |
2f |
4bf: 92%, (47%)[b] |










[a] Yield of analytically pure isolated product. [b] Barbier‐type reaction using a premixed solution of benzothiazole (1.00 equiv) and electrophile (1.50 equiv), instant quenching with NH4Cl.
Using Barbier‐type conditions,10 that is, metalation of a mixture of 1 b (1.00 equiv) with 2 a (1.50 equiv) with KDA/TMEDA (1.50 equiv) under the same flow conditions, led to the alcohol 4 ba in 74 % yield (entry 1). Quenching of 3 b with pivaldehyde (2 c) afforded the alcohol 5 bc in 75 % yield. Weinreb amides were excellent acylation reagents for potassium organometallics and the trapping of 3 b with 2 d and 2 e gave the corresponding ketones in 91–93 % yield (entries 4 and 5). Thiolation of 3 b with Bu2S2 (2 f) led to the thioether 4 bf in 92 % yield. The corresponding Barbier reaction proceeded in this case with only 47 % yield (entry 6).
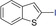
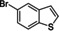
We have extended the reaction scope to various heterocyclic and aromatic substrates. For example, benzothiophene derivatives 1 c and 1 d were metalated with KDA/TMEDA and quenched with iodine (2 g) or the aromatic aldehyde 2 h as well as the disulfide 2 i, leading to the expected products (4 cg, 4 dh, and 4 di) in 63–98 % yield (Table 3, entries 1–3). Complete regioselectivity of the metalation of 3‐octylthiophene (1 e) was observed and addition to dicyclopropyl ketone (2 j) gave the tertiary alcohol 4 ej in 65 % yield (entry 4). Similarly, 2‐phenylthiophene 1 f was metalated with KDA/TMEDA and trapped with 2 a, affording 4 fa in 80 % yield (entry 5). 2‐Methoxypyrazine (1 g) was regioselectively metalated at position 3 with KDA/TMEDA (−78 °C, 0.18 s using a combined flow rate of 10 mL min−1). Addition of ketone 2 a gave the desired alcohol 4 ga in 81 % yield (entry 6).
Table 3.
Metalation of (hetero)arenes of type 1 using KDA/TMEDA in continuous flow and subsequent batch quench with various electrophiles of type 2 leading to functionalized (hetero)arenes of type 4.
|
Entry |
Substrate of type 1; T [°C], t [s], Flow rate [mL min−1] |
Electrophile of type 2 [a] |
Product of type 4 [b] |
|---|---|---|---|
|
|
|
I2 |
|
|
1 |
1c: −78, 24, 10 |
2g |
4cg: 63%[c] |
|
|
|
|
|
|
2 |
1d: −78, 0.18, 10 |
2h |
4dh: 98% |
|
|
|
|
|
|
3 |
1d: −78, 0.18, 10 |
2i |
4di: 93% |
|
|
|
|
|
|
4 |
1e: −78, 0.18, 10 |
2j |
4ej: 65% |
|
|
|
|
|
|
5 |
1f: −78, 0.18, 10 |
2a |
4fa: 80% |
|
|
|
|
|
|
6 |
1g: −78, 0.18, 10 |
2a |
4ga: 81% |
|
|
|
|
|
|
7 |
1h: −78, 24, 10 |
2a |
4ha: 42% |
|
|
|
|
|
|
8 |
1i: −78, 24, 10 |
2k |
4ik: 82% |
|
|
|
Bu2S2 |
|
|
9 |
1i: −78, 24, 10 |
2f |
4if: 73% |
|
|
|
|
|
|
10 |
1j: −78, 0.18, 10 |
2h |
4jh: 71% |























[a] 1.50 equiv of electrophile was used. [b] Yield of analytically pure isolated product. [c] KDA/TMEDA was prepared in cyclohexane. [d] Barbier‐type reaction using a premixed solution of 1,3‐dimethoxybenzene (1 i, 28 mg, 0.20 mmol, 1.00 equiv) and adamantanone (2 a, 45 mg, 0.30 mmol, 1.50 equiv), instant quenching with NH4Cl.
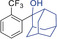
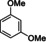
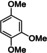
Extension to various aromatic substrates was possible. Electron‐poor trifluoromethylbenzene (1 h) was metalated in ortho‐position with KDA/TMEDA (−78 °C, 24 s reaction time, 10 mL min−1 combined flow rate), providing after addition of 2 a the alcohol 4 ha in 42 % yield (entry 7). Electron‐rich substrates such as 1,3‐dimethoxybenzene (1 i) and 1,2,4‐trimethoxybenzene (1 j) were metalated with KDA/TMEDA and gave after batch quenching with aldehydes 2 k and 2 h and Bu2S2 (2 f) the corresponding adducts 4 ik, 4 if, and 4 jh in 71–82 % yield (entries 8–10).
Remarkably, aromatic nitriles were tolerated in such metalations and 3‐methoxybenzonitrile (1 k) was deprotonated at the 2‐position by KDA/TMEDA (−78 °C, reaction time: 0.18 s). The resulting arylpotassium derivative 3 k reacted with various electrophiles (ketone 2 j, pivaldehyde (2 c), and TMS‐Cl (2 l)), leading to the expected products 4 kj, 4 kc, and 4 kl in 62–88 % yield. Batch metalation of 1 k with KDA/TMEDA followed by Me3SiCl quenching afforded the product 4 kl in 78 % yield. A Wurtz‐type coupling11 using primary alkyl iodides such as dodecyl iodide (2 m) led to the alkylated 3‐methoxybenzonitrile 4 km in 53 % yield. (Scheme 2).
Scheme 2.

Metalation of 3‐methoxybenzonitrile (1 k) with KDA/TMEDA in continuous flow and subsequent trapping with various electrophiles. [a] Yield of analytically pure isolated product. [b] Yield of analytically pure isolated product obtained under batch conditions.
Then, we turned our attention to substrates able to undergo lateral metalation. Thus, thioanisole (5 a) was previously lithiated with BuLi and DABCO or HMPA leading to PhSCH2Li (6 a).12 However, LDA did not achieve a lithiation, neither under batch nor under flow conditions.13 On the other hand, KDA/TMEDA successfully deprotonated 5 a under batch as well as flow conditions (Scheme 3) affording PhSCH2K (7 a), which was quenched with ketones 2 a and 2 j and alkyl iodide 2 m resulting in the desired products 8 aa, 8 aj, and 8 am in 62–99 % yield.
Scheme 3.

Metalation of thioanisole (5 a) using various lithium and potassium bases under batch and flow conditions. [a] Yield of analytically pure isolated product obtained in continuous flow. [b] Yield of analytically pure isolated product obtained under batch conditions.
Whereas lateral alkali‐metalations of arenes were well described under batch conditions,14 the corresponding reactions in flow are rare.15 The use of KDA/TMEDA was quite advantageous for the metalation of methyl‐substituted arenes (Table 4). Preliminary results show that a 0.2 m solution of toluene (9 a) led to unsatisfactory results; however, the injection of neat toluene (9 a) considerably improved the flow metalation with KDA/TMEDA. Interestingly, this metalation was performed at 25 °C (in contrast to previously described metalations of arenes and heteroarenes). In this case, the reaction time was increased to 24 s at a flow rate of 10 mL min−1. Under these convenient conditions, the subsequent batch‐trapping with ketone 2 a gave 11 aa in 69 % yield. Similarly, p‐xylene (9 b) provided the monopotassium derivative 10 b, which after quenching with dodecyl iodide (2 m) or Weinreb amide 2 e afforded the products 11 bm and 11 be in 95–96 % yield. Mesitylene (9 c) was metalated neat and after quenching with ketone 2 n and dodecyl iodide (2 m) gave the arenes 11 cn and 11 cm in 89–92 % yield. In the case of the Wurtz‐type coupling with 2 m, the reaction was scaled up tenfold to a 3 mmol scale,16 providing 11 cm in 93 % yield. For 1‐methylnaphthalene (9 d), a 0.2 m solution in THF was used and standard KDA/TMEDA metalation led after trapping with ketone 2 a to the corresponding naphthylmethyl alcohol 11 da in 92 % yield. Functionalized substrates such as 2‐fluorotoluene (9 e) were metalated at the benzylic position, affording the potassium organometallic 10 e, which after quenching with ketone 2 j led to the tertiary alcohol 11 ej in 66 % yield. N,N‐diisopropyl‐2‐methylbenzamide (9 f) led upon reaction with KDA/TMEDA at −40 °C (reaction time: 24 s) solely to the lateral metalated species 10 f, completely avoiding ortho metalation.1d, 17 Trapping with various electrophiles such as ketone 2 j, alkyl iodide 2 m, and Weinreb amide 2 o gave the expected products 11 fj, 11 fm, and 11 fo in 75–93 % yield. Further, ketones were tolerated. For example, lateral metalation of ketone 9 g using KDA/TMEDA proceeded smoothly at −40 °C within 0.18 s using a flow rate of 10 mL min−1. Batch trapping with ketone 2 j and cinnamyl bromide (2 p) in the presence of 10 % CuCN⋅2LiCl resulted in the tertiary alcohol 11 gj and the allylated ketone 11 gp in 72–90 % yield. We further extended the substrate scope to methyl‐substituted heterocycles such as 2‐chloro‐3‐methylpyridine (9 h). Metalation of 9 h at the meta‐methyl substituent using KDA/TMEDA led to the corresponding organopotassium species 10 h, which after batch quenching with various carbonyl electrophiles (2 a, 2 q, 2 n, and 2 r) gave the corresponding alcohols 11 ha, 11 hq, 11 hn, and 11 hr in 75–97 % yield.
Table 4.
Lateral metalation of methyl‐substituted (hetero)arenes using KDA/TMEDA in continuous flow leading to organopotassium species of type 10. Subsequent batch trapping with various electrophiles afforded functionalized methyl‐substituted (hetero)arenes of type 11.
|
|

Yields of analytically pure isolated products. [a] Substrate (neat), E‐X (0.30 mmol, 1.00 equiv), KDA/TMEDA (1.10 equiv), 25 °C, 24 s, 10 mL min−1. [b] Wurtz‐type coupling with the corresponding iodide. [c] From the corresponding Weinreb amide. [d] Scale‐up to 2.0 mmol using the optimized flow conditions. [e] 25 °C, 24 s, 10 mL min−1 [f] −40 °C, 24 s, 10 mL min−1. [g] 40 °C, 0.18 s, 10 mL min−1. [h] 10 mol% CuCN⋅2 LiCl. [i] −78 °C, 0.18, 10 mL min−1.
Trapping 10 h with alkyl iodide 2 m and cinnamyl bromide 2 p (in the presence of 10 % CuCN⋅2 LiCl) led to the corresponding products 11 hm and 11 hs in 66–77 % yield. Pyrazine 9 i was metalated in continuous flow with KDA/TMEDA. We have found that after metalation at the methyl substituent, the heterobenzylic potassium organometallic 10 i was obtained. Batch trapping with dibutyl disulfide (2 f) and dodecyl iodide (2 m) gave the functionalized pyrazines 11 if and 11 im in 79–95 % isolated yield.
In summary, we have reported a preparation of the potassium base KDA/TMEDA in the absence of lithium salts and have demonstrated its utility for the metalation of (hetero)arenes containing sensitive functional groups using a flow setup. The resulting potassium organometallics react upon batch quenching instantly with various electrophiles, affording functionalized (hetero)arenes in high yields. This flow procedure was successfully extended to the lateral metalation of methyl‐substituted arenes and heteroaromatics, resulting in benzylic potassium organometallics, which were trapped with a range of electrophiles. Scaling up was possible without further optimization. Further investigations of flow metalations using KDA/TMEDA are currently under way in our laboratories.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
N.W. thanks the German Academic Scholarship Foundation for a fellowship. We thank the DFG and LMU for financial support. We further thank BASF (Ludwigshafen) and Albemarle (Frankfurt) for the generous gift of chemicals and Uniqsis for technical support.
J. H. Harenberg, N. Weidmann, P. Knochel, Angew. Chem. Int. Ed. 2020, 59, 12321.
In memory of Rolf Huisgen
References
- 1.
- 1a. Clayden J., Organolithiums: Selectivity for Synthesis (Eds.: J. E. Baldwin, R. M. Williams), Pergamon, Oxford, 2002; [Google Scholar]
- 1b. Rathman T., Schwindeman J. A., Org. Process Res. Dev. 2014, 18, 1192; [Google Scholar]
- 1c. Wu G., Huang M., Chem. Rev. 2006, 106, 2596; [DOI] [PubMed] [Google Scholar]
- 1d. Snieckus V., Chem. Rev. 1990, 90, 879; [Google Scholar]
- 1e. Whisler M. C., MacNeil S., Snieckus V., Beak P., Angew. Chem. Int. Ed. 2004, 43, 2206; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 2256. [Google Scholar]
- 2.
- 2a. Seyferth D., Organometallics 2006, 25, 2; [Google Scholar]
- 2b. Seyferth D., Organometallics 2009, 28, 2; [Google Scholar]
- 2c. Buckton G. B., Proc. R. Soc. London 1859, 9, 685; [Google Scholar]
- 2d. Buckton G. B., Liebigs Ann. Chem. 1859, 112, 220; [Google Scholar]
- 2e. Carothers W. H., Coffman D. D., J. Am. Chem. Soc. 1930, 52, 1254; [Google Scholar]
- 2f. Wanklyn J. A., Liebigs Ann. Chem. 1858, 108, 67. [Google Scholar]
- 3.
- 3a. Ma Y., Woltornist R. A., Algera R. F., Collum D. B., J. Org. Chem. 2019, 84, 9051; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Algera R. F., Ma Y., Collum D. B., J. Am. Chem. Soc. 2017, 139, 11544; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3c. Mulvey R. E., Robertson S. D., Angew. Chem. Int. Ed. 2013, 52, 11470; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 11682; [Google Scholar]
- 3d. Schlosser M., Organometallics in Synthesis, Wiley, Hoboken, 2013; [Google Scholar]
- 3e. Schlosser M., Hartmann J., Stähle M., Kramer J., Walde A., Mordini A., Chimia 1986, 40, 306. [Google Scholar]
- 4.
- 4a. Kim H., Nagaki A., Yoshida J.-i., Nat. Commun. 2011, 2, 264; [DOI] [PubMed] [Google Scholar]
- 4b. Battilocchio C., Feist F., Hafner A., Simon M., Tran D. N., Allwood D. M., Blakemore D. C., Ley S. V., Nat. Chem. 2016, 8, 360; [DOI] [PubMed] [Google Scholar]
- 4c. Roesner S., Buchwald S. L., Angew. Chem. Int. Ed. 2016, 55, 10463; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 10619; [Google Scholar]
- 4d. Teci M., Tilley M., McGuire M. A., Organ M. G., Org. Process Res. Dev. 2016, 20, 1967; [Google Scholar]
- 4e. Gutmann B., Kappe C. O., J. Flow Chem. 2017, 7, 65; [Google Scholar]
- 4f. Britton J., Jamison T. F., Nat. Protoc. 2017, 12, 2423; [DOI] [PubMed] [Google Scholar]
- 4g. Price G. A., Bogdan A. R., Aguirre A. L., Iwai T., Djuric S. W., Organ M. G., Catal. Sci. Technol. 2016, 6, 4733; For recent reviews about flow chemistry see: [Google Scholar]
- 4h. Plutschack M. B., Pieber B., Gilmore K., Seeberger P. H., Chem. Rev. 2017, 117, 11796; [DOI] [PubMed] [Google Scholar]
- 4i. Colella M., Nagaki A., Luisi R., Chem. Eur. J. 2020, 26, 19. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Weidmann N., Ketels M., Knochel P., Angew. Chem. Int. Ed. 2018, 57, 10748; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10908. [Google Scholar]
- 6.
- 6a.The crystal structure of KDA complexed with 1.0 equiv of TMEDA was reported: Clegg W., Kleditzsch S., Mulvey R. E., O'Shaughnessy P., J. Organomet. Chem. 1998, 558, 193; [Google Scholar]
- 6b. Lochmann L., Trekoval J., J. Organomet. Chem. 1979, 179, 123; [Google Scholar]
- 6c. Lochmann L., Pospišil J., Lim D., Tetrahedron Lett. 1966, 7, 257; [Google Scholar]
- 6d. Mordini A., Peruzzi D., Russo F., Valacchi M., Reginato G., Brandi A., Tetrahedron 2005, 61, 3349; [Google Scholar]
- 6e. Lochmann L., Janata M., Cent. Eur. J. Chem. 2014, 12, 537; [Google Scholar]
- 6f.for preparation of KTMP using Me3SiCH2K see: Conway B., Kennedy A. R., Mulvey R. E., Robertson S. D., Alvarez J. G., Angew. Chem. Int. Ed. 2010, 49, 3182; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 3250. [Google Scholar]
- 7. Ma Y., Algera R. F., Collum D. B., J. Org. Chem. 2016, 81, 11312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Commercially available equipment from Uniqsis was used. For a detailed optimization, see pp. 8–9 in the Supporting Information.
- 9.Optimization studies of the flow conditions were conducted separately for each substrate.
- 10.
- 10a. Barbier P., Compt. Rend. 1899, 128, 110; [Google Scholar]
- 10b. Barbier P., C. R. Acad. Sci. Paris 1899, 128, 110; [Google Scholar]
- 10c. Blomberg C., Hartog F. A., Synthesis 1977, 18. [Google Scholar]
- 11.
- 11a. Wurtz A., Ann. Chim. Phys. 1955, 44, 275; [Google Scholar]
- 11b. Wurtz A., Ann. Chim. Phys. 1855, 96, 364. [Google Scholar]
- 12.
- 12a. Corey E. J., Seebach D., J. Org. Chem. 1966, 31, 4097; [Google Scholar]
- 12b. Semmelhack M. F., Herndon J. W., Organometallics 1983, 2, 363; [Google Scholar]
- 12c.The use of TMEDA and 2.2 equiv of nBuLi leads to dimetalation of thioanisole; Cabiddu S., Floris C., Melis S., Tetrahedron Lett. 1986, 27, 4625. [Google Scholar]
- 13.For detailed screening conditions, see pp. 6–7 in the Supporting Information.
- 14.
- 14a. Fleming P., O′Shea D. F., J. Am. Chem. Soc. 2011, 133, 1698; [DOI] [PubMed] [Google Scholar]
- 14b. Manvar A., Fleming P., O'Shea D. F., J. Org. Chem. 2015, 80, 8727; [DOI] [PubMed] [Google Scholar]
- 14c. Gualtieri F., Mordini A., Pecchi S., Scapecchi S., Synlett 1996, 5, 447; [Google Scholar]
- 14d. Miah M. A. J., Sibi M. P., Chattopadhyay S., Familoni O. B., Snieckus V., Eur. J. Org. Chem. 2018, 440; [Google Scholar]
- 14e. Fässler J., McCubbin J. A., Roglans A., Kimachi T., Hollett J. W., Kunz R. W., Tinkl M., Zhang Y., Wang R., Campbell M., Snieckus V., J. Org. Chem. 2015, 80, 3368; [DOI] [PubMed] [Google Scholar]
- 14f. MacNeil S. L., Familoni O. B., Snieckus V., J. Org. Chem. 2001, 66, 3662; [DOI] [PubMed] [Google Scholar]
- 14g. Zhai D.-D., Zhang X.-Y., Liu Y.-F., Zheng L., Guan B.-T., Angew. Chem. Int. Ed. 2018, 57, 1650; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 1666. [Google Scholar]
- 15.
- 15a. Venturoni F., Nikzad N., Ley S. V., Baxendale I. R., Org. Biomol. Chem. 2010, 8, 1798; [DOI] [PubMed] [Google Scholar]
- 15b. Wong J. Y. F., Tobin J. M., Vilela F., Barker G., Chem. Eur. J. 2019, 25, 12439; [DOI] [PubMed] [Google Scholar]
- 15c. Lee H.-J., Kim H., Kim D.-P., Chem. Eur. J. 2019, 25, 11641. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Teci M., Tilley M., McGuire M. A., Organ M. G., Chem. Eur. J. 2016, 22, 17407; [DOI] [PubMed] [Google Scholar]
- 16b. Hafner A., Filipponi P., Piccioni L., Meisenbach M., Schenkel B., Venturoni F., Sedelmeier J., Org. Process Res. Dev. 2016, 20, 1833. [Google Scholar]
- 17.
- 17a. Balloch L., Kennedy A. R., Mulvey R. E., Rantanen T., Robertson S. D., Snieckus V., Organometallics 2011, 30, 145; [Google Scholar]
- 17b. Singh K. J., Hoepker A. C., Collum D. B., J. Am. Chem. Soc. 2008, 130, 18008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary