

Abstract
Objective
The antiepileptic drug candidate, padsevonil, is the first in a novel class of drugs designed to interact with both presynaptic and postsynaptic therapeutic targets: synaptic vesicle 2 proteins and γ‐aminobutyric acid type A receptors (GABAARs), respectively. Functional aspects of padsevonil at the postsynaptic target, GABAARs, were characterized in experiments reported here.
Methods
The effect of padsevonil on GABA‐mediated Cl− currents was determined by patch clamp on recombinant human GABAARs (α1β2γ2) stably expressed in a CHO‐K1 cell line and on native GABAARs in cultured rat primary cortical neurons. Padsevonil selectivity for GABAAR subtypes was evaluated using a two‐electrode voltage clamp on recombinant human GABAARs (α1‐5/β2/γ2) in Xenopus oocytes.
Results
In recombinant GABAARs, padsevonil did not evoke Cl− currents in the absence of the agonist GABA. However, when co‐administered with GABA at effective concentration (EC)20, padsevonil potentiated GABA responses by 167% (EC50 138 nmol/L) and demonstrated a relative efficacy of 41% compared with zolpidem, a reference benzodiazepine site agonist. Similarly, padsevonil demonstrated GABA‐potentiating activity at native GABAARs (EC50 208 nmol/L) in cultured rat cortical neurons. Padsevonil also potentiated GABA (EC20) responses in GABAARs expressed in oocytes, with higher potency at α1‐ and α5‐containing receptors (EC50 295 and 281 nmol/L) than at α2‐ and α3‐containing receptors (EC50 1737 and 2089 nmol/L). Compared with chlordiazepoxide—a nonselective, full GABAAR agonist—the relative efficacy of padsevonil was 60% for α1β2γ2, 26% for α2β2γ2, 56% for α3β2γ2, and 41% for α5β2γ2; no activity was observed at benzodiazepine‐insensitive α4β2γ2 receptors.
Significance
Results of functional investigations on recombinant and native neuronal GABAARs show that padsevonil acts as a positive allosteric modulator of these receptors, with a partial agonist profile at the benzodiazepine site. These properties may confer better tolerability and lower potential for tolerance development compared with classic benzodiazepines currently used in the clinic.
Keywords: benzodiazepine, drug‐resistant epilepsy, patch clamp, SV2 proteins
Key Points.
Padsevonil, an antiepileptic drug candidate, was rationally designed to interact with presynaptic (SV2) and postsynaptic (GABAA receptor) targets
Padsevonil binds with low‐to‐moderate affinity to recombinant and native GABAA receptors
Padsevonil acts as a partial agonist at the benzodiazepine site of GABAA receptors
1. INTRODUCTION
Padsevonil, a novel antiepileptic drug (AED) candidate, is the first in a class of drugs with both presynaptic and postsynaptic mechanism of action. The rationale for the design and development of such a drug was the observation that in nonclinical models, co‐administration of levetiracetam (LEV), a synaptic vesicle 2A (SV2A) ligand together with agents acting on γ‐aminobutyric acid type A (GABAA) receptors (GABAARs) led to enhanced protection against seizures, without any detrimental effect on safety. 1 Consequently, it was hypothesized that a single molecular entity acting simultaneously on these presynaptic and postsynaptic targets may translate into improved clinical efficacy in the treatment of patients with epilepsy, particularly those with drug‐resistant epilepsy.
The selective interaction of Padsevonil with its molecular targets, as intended in the rational drug discovery program, has been confirmed in both in vitro and in vivo studies. 2 Specifically, Padsevonil was designed to bind to the presynaptic SV2 proteins with high affinity and to postsynaptic GABAARs, with low‐to‐moderate affinity. In radioligand displacement studies, Padsevonil has been shown to bind to SV2A with nanomolar affinity (pKi 8.5) that is approximately 100‐ and 2000‐fold greater than that of brivaracetam (BRV) and LEV, respectively. 2 Another major differentiating factor between Padsevonil and the SV2A selective ligands LEV and BRV is that Padsevonil also binds with high affinity to the B and C isoforms of SV2 proteins (pKi 7.9 and 8.5, respectively). 2 Padsevonil also demonstrates different binding properties to SV2A in that it displays significantly slower dissociation kinetics, translating into longer target occupancy. 2 Furthermore, unlike LEV and BRV, the interaction between Padsevonil and SV2A is not affected by UCB1244283, a positive allosteric modulator of SV2A, suggesting a different binding site for Padsevonil in the SV2A protein. 2 , 3 , 4 At recombinant GABAARs, Padsevonil displayed low‐to‐moderate affinity (pKi 6.4) for the benzodiazepine (BZD) binding site. This binding profile has been confirmed in receptor occupancy studies in vivo (mice), where Padsevonil exhibited SV2A occupancy at low doses (median effective dose [ED50] 0.2 mg/kg), and BZD site occupancy at higher doses (ED50 36 mg/kg). 2 Overall, these observations support the unique target profile of Padsevonil that should translate into presynaptic modulation of neurotransmitter release and the postsynaptic potentiation of GABAergic inhibitory transmission.
The objective of the experiments reported here was to fully characterize the functional effects of Padsevonil on different GABAAR subunits in recombinant and native neuronal systems using electrophysiologic techniques.
2. MATERIAL AND METHODS
2.1. Animals
All experiments were conducted in compliance with guidelines issued by the ethics committee for animal experimentation according to Belgian law. All efforts were made to minimize animal pain and discomfort.
For patch‐clamp recordings in rat primary cortical neurons, pregnant female Wistar rats were obtained from Charles River (Lyon, France) and embryonic animals were surgically removed at embryonic days E17‐E18. Animals were housed in a holding room under a 12‐hour light–dark cycle with lights on at 06:00 hours. Temperature was maintained at 20‐24°C, relative humidity at 40%‐70%, and the rate of air replacement was at least 15 times per hour. Animals had on‐demand access to standard dry pellet food and tap water.
2.2. Compounds
Padsevonil ((4R)‐4‐(2‐chloro‐2,2‐difluoroethyl)‐1‐{[2‐(methoxymethyl)‐6‐(trifluoromethyl)imidazo[2,1‐b][1,3,4]thiadiazol‐5‐yl]methyl}pyrrolidin‐2‐one) was synthesized at UCB Pharma. Zolpidem was obtained from Tocris (Ref 0655; B5A/91713), GABA from Sigma (Ref A2129; B045K00721), chlordiazepoxide from Sequoia Research (Cat. No. SRP02170c), and Ro15‐4513 from Tocris (Cat. No. 1997). All stock solutions were prepared at a maximum of 10 mmol/L in dimethyl sulfoxide (DMSO, 100%) and stored at −20°C. When required, the compounds were diluted into the extracellular medium yielding a final DMSO concentration of 0.1%‐0.5%.
2.3. Patch‐clamp recordings on recombinant human GABAA receptors
GABAAR Cl− currents were recorded using automated patch clamp from recombinant human GABAARs consisting of the following subunits: α1 (accession number NM000806), β2 (NM021911), and γ2 (NM198904) stably expressed in a CHO‐K1 cell line. Full details have been described previously. 5 Briefly, CHO cells expressing these subunits were cultured in Ham's F‐12 medium with glutamine (GIBCO, Carlsbad, CA, USA) containing 400 μg/mL G418 (GIBCO), 250 μg/mL Zeocin (Invitrogen), 10% fetal bovine serum, and 1% penicillin/streptomycin (Lonza). Cells were harvested using Accumax treatment (Sigma) and allowed to recover for 90 min at room temperature before recording started.
For patch‐clamp recording, the extracellular solution contained 135 mmol/L NaCl, 4.7 mmol/L KCl, 2.5 mmol/L CaCl2, 1.2 mmol/L MgCl2, 11 mmol/L glucose, and 10 mmol/L HEPES (adjusted to pH 7.4 with NaOH 5 N), and intracellular solution 100 mmol/L KCl, 40 mmol/L KF, 2 mmol/L MgCl2, 11 mmol/L EGTA, 10 mmol/L glucose, and 10 mmol/L HEPES (adjusted to pH 7.2 with KOH 1 N).
Currents were recorded using the automated patch‐clamp platform PatchXpress 7000A (Molecular Devices), with the holding potential set to −60 mV. Current traces were recorded at room temperature by patch‐clamp amplifier (Multiclamp 700A Computer‐Controlled Patch Clamp Dual Headstage Amplifier, Molecular Devices) at a sampling rate of 2 KHz. Data were analyzed using DataXpress 2 software (version 2.0.4.2; Molecular Devices). To measure the effects of an allosteric modulator binding to the BZD site of GABAARs, a low concentration of agonist is needed to potentiate the drug response. For these experiments, 5 µmol/L GABA was used, a concentration that mediates 20% of GABAAR activation (EC20). To quantify the potentiation of GABAAR Cl− currents by Padsevonil relative to zolpidem, the experimental protocol described by Ghisdal et al was followed. 6 Briefly, potentiation of currents following application of 5 μmol/L GABA and 10 or 30 μmol/L Padsevonil is determined and compared with the maximum potentiation elicited by application of 5 μmol/L GABA and 1 μmol/L zolpidem in the same cell after washout.
2.4. Two‐electrode voltage clamp recordings on recombinant human GABAA receptors in Xenopus oocytes
Different subunits of the human GABAAR, including α1 (NM000806), α2 (NM000807), α3 (NM000808), α4 (NM000809), α5 (NM000810), β2 (NM021911), and γ2 (BC059389) were cloned into pcDNA3.1(+) (Invitrogen). GABAAR subunit mRNA for oocyte injections were prepared by RD‐Biotech (Besançon, France). Xenopus leavis oocytes (stage V–VI), dissected and defolliculated, were purchased from EcoCyte Bioscience (Castrop‐Rauxel, Germany). Microinjections of mRNA for the GABAAR subunits (α1‐5:β2: γ2; ratio 1:1:5.5) were performed with the automated system Roboocyte (Multi Channel Systems MCS, Reutlingen, Germany) using a total volume ~50 nL mRNA dissolved in RNAse‐free water (Ambion). Injected oocytes were kept at 17°C for 3‐6 days in Barth's solution containing 88 mmol/L NaCl, 2.4 mmol/L NaHCO3, 1 mmol/L KCl, 0.33 mmol/L Ca(NO3)2, 0.41 mmol/L CaCl2, 0.82 mmol/L MgSO4, 5 mmol/L Tris/HCl, supplemented with penicillin (100 UI/mL)/streptomycin (100 μg/mL) (Whittaker Cambrex) (adjusted to pH 7.4 with NaOH 1 N).
Two‐electrode voltage clamp (TEVC) recordings were performed with the automated Roboocyte system (Multi Channel Systems MCS) using standard recording protocols. Oocytes were impaled and voltage clamped at a holding potential of −60 mV. After impalement, the oocytes were rinsed with normal frog ringer buffer for 60 s, and allowed to stabilize for a further 60 s. Normal frog ringer solution contained 115 mmol/L NaCl, 2.5 mmol/L KCl, 1.8 mmol/L CaCl2, 10 mmol/L HEPES (adjusted to pH 7.2 with NaOH 5 N). For each oocyte, GABA was applied twice for 20 s with a 6 min interval to evoke inward Cl− currents and was used as the control GABA current. The concentration–response curve was started by a 6‐min pre‐incubation of Padsevonil (1 nmol/L to 30 μmol/L) followed by co‐application of GABA and Padsevonil for 20 s.
Drugs were applied via a liquid dispenser (Gilson GX271) coupled to a peristaltic pump (Gilson MINIPULS 3). The perfusion rate of oocytes in the 96‐well plates was set at approximately 3 mL/min. All solutions were prepared freshly before each experiment. During measurements, oocytes were perfused with normal frog ringer solution.
GABA‐evoked Cl‐ currents were analyzed by the Roboocyte Software version 2.2 (Multi Channel Systems MCS). The magnitude of the potentiation of the currents by the drug was calculated based on the ratio I (GABA + drug)/IGABA where I (GABA + drug) is the current response evoked by co‐application of drug and GABA and IGABA represents the current amplitude evoked by GABA alone.
Dose–response curves were analyzed using GraphPad Prism software and EC50 values calculated by nonlinear regression analysis using a sigmoidal dose–response equation, where Y = Bottom + (Top‐Bottom)/(1 + 10^((LogEC50‐X)). Data were normalized to maximum potentiation values of the drug.
2.5. Patch‐clamp recordings of native GABAA receptor currents in rat primary cortical neurons
Rat cortical neurons were prepared as described previously. 6 Briefly, neocortex was removed from Wistar embryonic rats (E17‐E18) of either sex, dissociated in 0.25% trypsin, and triturated in complete neurobasal‐based medium composed of neurobasal medium, 2% B27 serum‐free supplement, 0.5 mmol/L glutamax (all from Invitrogen), 10% horse serum, and 1% penicillin/streptomycin (Lonza, Basel, Switzerland). Coverslips coated with poly‐d‐lysine and laminin (Sigma) were seeded, and neurons (40 000‐50 000 cells/mL) grown in complete neurobasal‐based medium. Cultures were maintained for 15 days at 37°C in a 5% CO2/95% air incubator.
Voltage clamp recordings of GABAAR currents were performed on primary cortical neurons (days in‐vitro, 10‐14) in tight‐seal whole‐cell patch‐clamp configuration. Patch pipettes were pulled from 1.5 mm (inside diameter) borosilicate glass capillaries (Harvard Apparatus, MA, USA) and had a resistance of 4‐5 MΩ when filled with intracellular solution composed of 140 mmol/L KCl, 10 mmol/L EGTA, 2 mmol/L MgCl2, 2 mmol/L Na2‐ATP, and 10 mmol/L HEPES (adjusted to pH 7.2 with KOH). Currents were recorded at room temperature. Neuronal preparations were continuously perfused at a rate of ~1 mL/min with extracellular solution composed of 140 mmol/L NaCl, 5.4 mmol/L KCl, 1 mmol/L MgCl2, 1.3 mmol/L CaCl2, 12.5 mmol/L HEPES, 10 mmol/L glucose, 0.001 mmol/L tetrodotoxin, and 0.01 mmol/L CGP 35 348, a GABAB receptor antagonist (adjusted to pH 7.3 with NaOH). The junction potential between the intracellular and extracellular solutions was measured at 4 mV and compensated for by on‐line subtraction. Series resistances were compensated by 70% on the amplifier (Axopatch 200B, Molecular Devices). After seal formation, the pipette capacitive current was cancelled, and following breakthrough, the whole‐cell capacitive current was cancelled. Currents were filtered at 100 kHz and digitized at 5 kHz using a Digidata 1322A converter and the acquisition program pClamp (both Molecular Devices). According to the Cl− equilibrium potential measured at −1 mV, the holding membrane potential was set at −70 mV for recording GABA‐activated Cl− currents. Control of drug application, data acquisition, and data analysis was achieved using pClamp software (Molecular Devices).
Padsevonil was diluted in the extracellular solution and applied by a rapid microperfusion system (VC83, ALA Scientific Instruments). GABA was applied at 2 µmol/L (concentration previously determined as EC10) for 10 s to evoke the basal inward GABAAR Cl‐ currents, followed by a series of GABA applications together with Padsevonil at increasing concentrations (0, 0.1, 1, 10, and 30 µmol/L). Each GABA application in the protocol was followed by a 4‐min washout period to allow recovery from receptor desensitization. The intensity of GABA‐evoked currents was measured at the peak.
3. RESULTS
3.1. Padsevonil is a positive allosteric modulator of GABAA receptors with a partial agonist profile
The functional activity of Padsevonil on recombinant GABAARs was determined using patch‐clamp recordings from the human α1β2γ2 GABAAR stably expressed in a CHO cell line, for which the pharmacological profile has been previously established. 6 The effects of Padsevonil (0.01‐30 µmol/L) on the receptor was first evaluated in the presence of the agonist, GABA, at EC20 (a concentration that produces 20% of the maximum response) set to 100%. Padsevonil potentiated the agonist response by 167%, with an EC50 of 138 nmol/L (Figure 1A), but at concentrations up to 30 µmol/L, it did not evoke Cl− currents in the absence of GABA, confirming that it modulates GABAAR activity through an allosteric site on the receptor (Figure 1B). The effects of Padsevonil on the maximal activation of the receptor were also evaluated using GABA at a concentration of 30 µmol/L (EC90), confirming that Padsevonil does not overstimulate the receptor at high GABA concentrations (data not shown).
Figure 1.
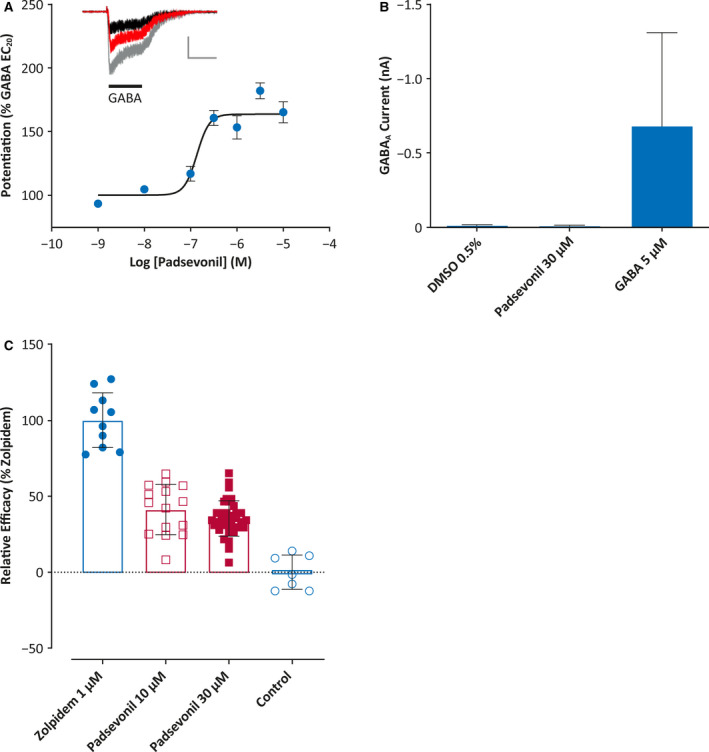
Functional properties of padsevonil on recombinant γ‐aminobutyric acid type A (GABAA) receptors. A, Patch‐clamp recordings of human recombinant GABAA receptor (α1β2γ12) currents in CHO cells. Padsevonil (1 nmol/L–10 µmol/L) was tested in the presence of GABA at EC20 (5 µmol/L) (n = 7‐15 cells; ± SEM). Inset: Representative GABAA currents recorded from the same cell under control conditions (black), with padsevonil 10 µmol/L (red) and with 1 µmol/L zolpidem (gray). Horizontal bar represents 5 s and vertical bar 0.5 nA. B, Effects of padsevonil and GABA on GABAA receptor currents (n = 4‐7 cells; ± standard error of the mean [SEM]). C, Relative efficacy of padsevonil compared with zolpidem in potentiating GABA‐mediated (EC20) Cl− currents in human α1β2γ12 GABAA receptors (n = 7‐35 cells; ± SEM). Error bars are ± SEM
To evaluate the relative efficacy of Padsevonil binding to the BZD site of the GABAAR, the effects of Padsevonil and zolpidem were tested sequentially on the same cell. Zolpidem, a full agonist at the BZD site, potentiates GABA (EC20) with a maximal efficacy at 1 µmol/L, defined as the maximal response (100%). At concentrations of 10 and 30 µmol/L, Padsevonil stimulated the GABA EC20 response to 41% and 35% of that of zolpidem, respectively, indicating that Padsevonil elicits a partial agonist response on human α1β2γ2 GABAARs in comparison with zolpidem (Figure 1C).
3.2. Padsevonil is a nonselective modulator of benzodiazepine‐sensitive GABAA receptors
The selectivity of Padsevonil for specific GABAAR subtypes was evaluated in three sets of experiments using TEVC. In the first, activity of Padsevonil at different recombinant GABAARs containing BZD‐sensitive subunits was measured. In a transient expression system in Xenopus oocytes, mRNA encoding α1, α2, α3, and α5 was expressed in the presence of β2/γ2 subunits. Dose–response values of Padsevonil (1‐30 µmol/L) on the different receptor subtypes are shown in Figure 2. Padsevonil potentiated GABA responses (EC20) at receptors containing α1 and α5 subunits with similar potency (EC50 295 and 281 nmol/L; Table 1), whereas potentiation of receptors with α2 and α3 subunits was noted at higher concentrations (EC50 1737 and 2089 nmol/L) (Figure 2A, Table 1).
Figure 2.

Padsevonil selectivity for γ‐aminobutyric acid type A (GABAA) receptor subtypes as determined by two‐electrode voltage clamp recordings (TEVC) on human recombinant GABAA receptors expressed in Xenopus oocytes. A, Dose–response of padsevonil (1 nmol/L–30 µmol/L). Data are shown as mean ± SEM (n = 6‐26 cells). B, Relative efficacy of padsevonil (10 µM) compared with chlordiazepoxide (CDP, 10 µmol/L) on GABAA receptor subtypes. Data are shown as mean ± SEM. SEM, standard error of mean
Table 1.
Characteristics of the interaction of Padsevonil with human recombinant and native rat GABAA receptors
| Function EC50(nmol/L) | Function relative efficacy (%) | |
|---|---|---|
| Human recombinant receptors | ||
| α1β2γ2 | 138 ± 29 (n = 22) a 295 ± 38 (n = 26) b | 41 ± 4 (n = 15) a 60 ± 6 (n = 16) b |
| α2β2γ2 | 1737 ± 258 (n = 12) b | 26 ± 4 (n = 13) b |
| α3β2γ2 | 2089 ± 227 (n = 17) b | 56 ± 6 (n = 18) b |
| α4β2γ2 | No potentiation | Not evaluated |
| α5β2γ2 | 281 ± 73 (n = 6) b | 41 ± 10 (n = 10) b |
| Primary rat cultured neurons | ||
| Primary cortical neurons | 208 ± 99 (n = 8) a | Not evaluated |
Values are mean ± standard error of the mean.
Potentiation of GABA EC20 measured by patch clamp. Relative efficacy of Padsevonil (10 µmol/L) is normalized toward zolpidem (1 µmol/L).
Potentiation of GABA EC20 measured by two‐electrode voltage clamp. Relative efficacy of Padsevonil (10 µmol/L) is normalized toward chlordiazepoxide (10 µmol/L).
In the second set of experiments, the relative efficacy of Padsevonil across different receptor combinations was evaluated using chlordiazepoxide as a comparator. Chlordiazepoxide is a nonselective full agonist of different GABAAR subtypes; its relatively low affinity and water solubility enables easy washout before subsequent incubations with other drugs being tested. 7 The efficacy of Padsevonil was evaluated using GABA at EC20 and the comparison with chlordiazepoxide (10 µmol/L) showed a relative efficacy of 60 ± 25% for α1, 26 ± 13% for α2, 56 ± 27% for α3, and 41 ± 29% for α5 (Figure 2B). These results suggest that Padsevonil acts as a nonselective partial agonist at the BZD site of GABAARs.
In the third set, the activity of Padsevonil on GABAARs containing the α4 subunit was evaluated. These receptors are reported to be expressed at synaptic and extrasynaptic locations and are insensitive to BZDs. Results confirmed that GABAARs comprising α4β2γ2 subunits expressed in Xenopus oocytes are potentiated by the reference compound Ro 15‐4513 (1 µmol/L) 8 but are insensitive to zolpidem, a BZD site ligand (Figure 3). Padsevonil (10 and 30 µmol/L) did not potentiate the GABA (EC20) response on α4β2γ2 receptors, confirming the lack of interaction with BZD‐insensitive GABAAR subtypes.
Figure 3.

Effects of padsevonil, zolpidem, and the reference compound Ro 15‐4513 on potentiation of the γ‐aminobutyric acid (GABA; control) response on α4β2γ2 receptors expressed in Xenopus oocytes. Currents were recorded using two‐electrode voltage clamp in the presence of GABA at EC20. Error bars are mean ± SEM. SEM, standard error of mean
3.3. Padsevonil modulates the activity of native GABAA receptors in cultured neurons
The activity of Padsevonil on native GABAARs expressed in rat primary neuronal cultures was evaluated using manual patch‐clamp recordings to measure whole‐cell GABAAR currents. In the presence of GABA at EC20, Padsevonil produced a concentration‐dependent potentiation of activity at neuronal GABAARs (Figure 4A); pEC50 was calculated to be 6.68. These results also confirm the previous observation that Padsevonil acts via an allosteric site on native receptors.
Figure 4.

Effects of padsevonil on native γ‐aminobutyric acid type A (GABAA) receptors in cultured neurons. A, Dose–response of padsevonil on native GABAA receptor currents in rat primary cortical neurons (days in vitro [DIV] 10‐14). Data have been normalized to the basal evoked current with GABA at EC10 (2 µmol/L) (n = 8 neurons; mean ± SEM). Inset: Representative traces of GABAA receptor currents evoked under control conditions (in black), with padsevonil 10 µmol/L (in red) and with zolpidem 1 µmol/L (in gray) in the same cortical neuron (DIV 11). Horizontal bar represents 5 s and vertical bar 1 nA. B, quantitative polymerase chain reaction analysis of GABAA receptor subunits in rat primary cortical neurons (average of three different cultures; DIV 10‐14). Data have been normalized to the maximal levels (mean ± standard deviation)
To characterize the diversity of GABAARs expressed in cultured neurons (10‐14 days in vitro), quantitative polymerase chain reaction (qPCR) analysis was performed (Figure 4B). With focus on the BZD‐sensitive subtypes, results showed that most subunits were expressed except for the α3 and δ subunits.
4. DISCUSSION
Padsevonil is an AED candidate rationally designed to bind with high affinity to SV2 proteins and with low‐to‐moderate affinity to the BZD binding site of the GABAAR. 2 The focus of the studies reported here was on the functional aspects of the interaction between Padsevonil and the BZD site of the GABAAR to gain further insight into the molecular mechanism of action of Padsevonil.
Patch‐clamp experiments on recombinant GABAARs revealed that Padsevonil had no intrinsic activity at these receptors—in the absence of the endogenous agonist GABA, Padsevonil did not elicit Cl‐ currents. However, when applied in the presence of GABA, Padsevonil potentiated the agonist response by 167%. These results indicate that similar to many BZDs, Padsevonil acts as a positive allosteric modulator of the GABAAR. Such modulators bind to the GABAAR at a site distinct from the agonist (GABA) binding site, in a pocket at the interface between the extracellular domains of α and γ subunits. 9 Drug binding leads to a conformational change in the receptor that increases the apparent affinity for channel gating by GABA, which can bind simultaneously to two sites at the interfaces between α and β subunits. 10 , 11 Different BZDs show varying levels of potency or activity at the GABAAR, acting as full, partial, or inverse agonists or antagonists at the BZD site. 12 Padsevonil was designed specifically to function as a partial agonist, given that in animal models, partial agonists were reported to be associated with fewer sedative adverse effects and lack of development of tolerance and dependence with long‐term administration compared with classic BZDs such as diazepam, which act as full agonists. 13 Strategies to reduce BZD limitations have included development of GABAAR subtype–selective agents or agents with low relative efficacy to limit receptor activation. 14 In experiments reported here, the relative efficacy of Padsevonil was compared with that of zolpidem, a reference compound that acts as a full agonist at the BZD site with a maximal response defined as 100%. Results showed that the relative efficacy of Padsevonil was 41%, confirming that it acts as a partial agonist.
As part of the ligand‐gated ion channel superfamily, GABAARs are organized as pentameric membrane‐spanning proteins surrounding a central pore that forms the ion channel. 15 So far, 19 mammalian genes encoding GABAAR subunits, forming eight subunit classes, have been cloned. 15 The large number of subunits assembling in a variety of configurations gives rise to specific GABAAR subtypes, with different cellular and subcellular distribution, and importantly, different pharmacological profiles. 11 , 12 Notably, not all pentameric configurations are sensitive to BZDs. Receptors containing an α/γ subunit interface, with α1, 2, 3, or 5 and predominantly γ2 subunits, form a high affinity binding site for BZDs. 9 , 16 In contrast, receptor subtypes containing α4 or α6 subunits are described as BZD‐insensitive because of a substitution mutation where a conserved histidine residue, which confers BZD sensitivity, is replaced by an arginine residue. 17 , 18 , 19 Given these differences in sensitivity, Padsevonil activity at BZD‐sensitive and BZD‐insensitive GABAAR subtypes was determined. Results showed that Padsevonil binds to all BZD‐sensitive GABAAR subtypes; however, it displays approximately sixfold higher potency for receptors with α1 or α5 subunits than for those with α2 or α3 subunits. Similar to results described above, the relative efficacy of Padsevonil at these GABAAR subtypes was lower than that of chlordiazepoxide, a reference compound that acts as a nonselective full agonist of different GABAAR subtypes, confirming that Padsevonil displays a partial agonist profile. Results further showed that Padsevonil does not activate GABAARs subtypes containing the α4 subunit. The BZD‐insensitive receptor subtypes containing α4 or α6 subunits are predominantly extrasynaptic and are thought to mediate tonic inhibitory currents, distinguishable from phasic synaptic transmission mediated by postsynaptic GABAAR subtypes. 12 , 16 Tonic inhibition is a consequence of persistent activation of GABAARs dispersed over the neuronal surface by low levels of ambient GABA, and is important for regulating the overall excitability of neurons; phasic inhibition on the other hand is spatially and temporally discrete, allowing the rapid, point‐to‐point synaptic communication used to control the flow of specific signals. 11 , 16 , 20 Phasic inhibition also plays a central role in the generation and maintenance of neuronal network rhythmicity, and consequently, synchronization of inhibitory/excitatory networks, especially under conditions of hypersynchronicity observed during seizure activity. 16 , 21 Results of experiments reported here suggest that Padsevonil may preferentially modulate the phasic inhibition mediated by postsynaptic GABAARs. Finally, experiments performed on rat primary cortical neurons indicated that Padsevonil has a similar potency for native and recombinant GABAARs, with native receptors mainly represented by the α1‐, α2‐, and α5‐containing receptor subtypes in this neuronal system.
The properties of Padsevonil described above may confer advantages over classic BZDs used in the clinic. Specifically, the low‐to‐moderate affinity binding combined with the partial agonist profile could limit the level of occupancy and activation of GABAARs in the brain. Although sufficient to contribute to antiseizure activity, the risk of developing side effects and tolerance could be reduced. Nonclinical studies have shown that Padsevonil is highly active in models of drug‐resistant epilepsy 22 at doses that lead to >95% SV2A occupancy and <20% occupancy on GABAARs. 2 The potential of Padsevonil to induce tolerance was evaluated in the validated pentylenetetrazol (PTZ)–induced clonic seizure threshold test, where the ability of AEDs to increase the seizure threshold is evaluated after acute and repeated dosing. 23 With both acute and repeated (twice daily for 4 consecutive days) dosing, Padsevonil increased the threshold for PTZ‐induced seizures to the same extent, indicating that tolerance was not developed. 22 In contrast, diazepam, a full agonist at the BZD site, showed a reduced ability to increase the PTZ threshold after repeated dosing. 22 Clinical evidence also supports the observation that partial agonists may be associated with a lower risk of tolerance. Clobazam, a partial agonist at the BZD site, has been used for the treatment of patients with Lennox–Gastaut syndrome for many years. 24 , 25 Furthermore, in a long‐term extension trial, sustained seizure control was observed at stable dosages over a 3‐year period, indicating lack of tolerance development. 26 , 27 Abecarnil, another partial agonist, has shown efficacy in the treatment of patients with photosensitive epilepsy without development of tolerance. 28
In conclusion, Padsevonil is a novel AED candidate with a unique mechanism of action, interacting with both presynaptic and postsynaptic targets. Via its dual impact on neuronal activity, dampening excitation, and enhancing inhibition, it could potentially curtail neuronal hyperactivity during seizures more rapidly and strongly than currently available AEDs. It has shown robust efficacy in patients with highly treatment‐resistant focal epilepsy in a Phase IIa proof‐of‐concept trial. 29 Experiments reported here have shed further light on the functional effects of Padsevonil on GABAARs. It is notable, however, that the activity of Padsevonil on GABAARs during epileptogenesis or under conditions of chronic epilepsy remains to be evaluated. GABAARs undergo major plasticity during epileptogenesis, with changes involving modulation of the expression of receptor subunits and functional alterations. 30 , 31 Furthermore, it has been shown that paradoxically, in brain tissue obtained from patients with drug‐resistant epilepsy, GABA can cause depolarization resulting in excitatory signals, 32 an observation attributed to disruption of Cl− homeostasis. 33 The impact of Padsevonil on such excitatory currents needs further evaluation. Additional experiments based on relevant ex or in vivo models, for example, brain slices from animals with chronic epilepsy, are also required to provide a better understanding of Padsevonil activity on GABAARs that have gone plasticity changes due to pathophysiologic conditions.
CONFLICT OF INTERESTS
All authors were employees of UCB Pharma at the time of research. MW has now retired, RMK is currently employed by OncoArendi Therapeutics SA, Warsaw, Poland, and PG by Pi life sciences consultancy, Ghent, Belgium. The authors confirm having read the Journal's position on ethical publication and affirm that this report is consistent with Journal guidelines.
ACKNOWLEDGMENTS
The authors thank Anne Vandendriessche and Nadine Noel for their contribution to the automated patch‐clamp recordings. The authors also thank Fabien Debailleul, PhD, UCB Pharma, Brussels, Belgium for overseeing the development of the manuscript, and Azita Tofighy for providing writing support, funded by UCB Pharma.
Niespodziany I, Ghisdal P, Mullier B, et al. Functional characterization of the antiepileptic drug candidate, padsevonil, on GABAA receptors. Epilepsia. 2020;61:914–923. 10.1111/epi.16497
Funding information
Studies reported here were funded by UCB Pharma.
DATA AVAILABILITY STATEMENT
Data from nonclinical studies are outside the scope of UCB’s data sharing policy.
REFERENCES
- 1. Kaminski RM, Matagne A, Patsalos PN, Klitgaard H. Benefit of combination therapy in epilepsy: a review of the preclinical evidence with levetiracetam. Epilepsia. 2009;50:387–97. [DOI] [PubMed] [Google Scholar]
- 2. Wood M, Daniels V, Provins L, Wolff C, Kaminski RM, Gillard M. Pharmacological profile of the antiepileptic drug candidate padsevonil – interactions with synaptic vesicle 2 proteins and GABAA receptors. J Pharmacol Exp Ther. 2020;372:1–10. [DOI] [PubMed] [Google Scholar]
- 3. Daniels V, Wood M, Leclercq K, Kaminski RM, Gillard M. Modulation of the conformational state of the SV2A protein by an allosteric mechanism as evidenced by ligand binding assays. Br J Pharmacol. 2013;169:1091–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wood MD, Gillard M. Evidence for a differential interaction of brivaracetam and levetiracetam with the synaptic vesicle 2A protein. Epilepsia. 2017;58:255–62. [DOI] [PubMed] [Google Scholar]
- 5. Niespodziany I, André VM, Leclère N, Hanon E, Ghisdal P, Wolff C. Brivaracetam differentially affects voltage‐gated sodium currents without impairing sustained repetitive firing in neurons. CNS Neurosci Ther. 2015;21:241–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ghisdal P, Noel N, Pacico N, Martini M, Foerch P, Hanon E, et al. Determining the relative efficacy of positive allosteric modulators of the GABAA receptor: design of a screening approach. J Biomol Screen. 2014;19:462–7. [DOI] [PubMed] [Google Scholar]
- 7. Atack JR, Wafford KA, Tye SJ, Cook SM, Sohal B, Pike A, et al. TPA023 [7‐(1,1‐dimethylethyl)‐6‐(2‐ethyl‐2H‐1,2,4‐triazol‐3‐ylmethoxy)‐3‐(2‐fluorophenyl )‐1,2,4‐triazolo[4,3‐b]pyridazine], an agonist selective for alpha2‐ and alpha3‐containing GABAA receptors, is a nonsedating anxiolytic in rodents and primates. J Pharmacol Exp Ther. 2006;316:410–22. [DOI] [PubMed] [Google Scholar]
- 8. Whittemore ER, Yang W, Drewe JA, Woodward RM. Pharmacology of the human gamma‐aminobutyric acidA receptor alpha 4 subunit expressed in Xenopus laevis oocytes. Mol Pharmacol. 1996;50:1364–75. [PubMed] [Google Scholar]
- 9. Sigel E, Ernst M. The benzodiazepine binding sites of GABAA receptors. Trends Pharmacol Sci. 2018;39:659–71. [DOI] [PubMed] [Google Scholar]
- 10. Sigel E, Steinmann ME. Structure, function, and modulation of GABA(A) receptors. J Biol Chem. 2012;287:40224–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chua HC, Chebib M. GABAA Receptors and the diversity in their structure and pharmacology. Adv Pharmacol. 2017;79:1–34. [DOI] [PubMed] [Google Scholar]
- 12. Olsen RW. GABAA receptor: Positive and negative allosteric modulators. Neuropharmacology. 2018;136:10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rundfeldt C, Löscher W. The pharmacology of imepitoin: the first partial benzodiazepine receptor agonist developed for the treatment of epilepsy. CNS Drugs. 2014;28:29–43. [DOI] [PubMed] [Google Scholar]
- 14. Rudolph U, Knoflach F. Beyond classical benzodiazepines: novel therapeutic potential of GABA receptor subtypes. Nat Rev Drug Disc. 2011;10:685–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Olsen RW, Sieghart W. International Union of Pharmacology. LXX. Subtypes of gamma‐aminobutyric acid(A) receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol Rev. 2008;60:243–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat Rev Neurosci. 2005;6:215–29. [DOI] [PubMed] [Google Scholar]
- 17. Wisden W, Herb A, Wieland H, Keinänen K, Lüddens H, Seeburg PH. Cloning, pharmacological characteristics and expression pattern of the rat GABAA receptor alpha 4 subunit. FEBS Lett. 1991;289:227–30. [DOI] [PubMed] [Google Scholar]
- 18. Wieland HA, Luddens H, Seeburg PH. A single histidine in GABAA receptors is essential for benzodiazepine agonist binding. J Biol Chem. 1992;267:1426–9. [PubMed] [Google Scholar]
- 19. Wafford KA, Thompson SA, Thomas D, Sikela J, Wilcox AS, Whiting PJ. Functional characterization of human γ‐aminobutyric acid A receptors containing the α4 subunit. Mol Pharmacol. 1996;50:670–8. [PubMed] [Google Scholar]
- 20. Mody I, Pearce RA. Diversity of inhibitory neurotransmission through GABA(A) receptors. Trends Neurosci. 2004;27:569–75. [DOI] [PubMed] [Google Scholar]
- 21. Jiruska P, de Curtis M, Jefferys JGR, Schevon CA, Schiff SJ, Schindler K. Synchronization and desynchronization in epilepsy: controversies and hypotheses. J Physiol. 2013;591:787–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Leclercq K, Matagne A, Provins L, Klitgaard H, Kaminski RM. Pharmacological profile of the antiepileptic drug candidate padsevonil – characterization in rodent seizure and epilepsy models. J Pharmacol Exp Ther. 2020;372:11–20. [DOI] [PubMed] [Google Scholar]
- 23. Rundfeldt C, Wlaź P, Hönack D, Löscher W. Anticonvulsant tolerance and withdrawal characteristics of benzodiazepine receptor ligands in different seizure models in mice. Comparison of diazepam, bretazenil and abecarnil. J Pharmacol Exp Ther. 1995;275:693–702. [PubMed] [Google Scholar]
- 24. Faulkner MA. Comprehensive overview: efficacy, tolerability, and cost‐effectiveness of clobazam in Lennox‐Gastaut syndrome. Ther Clin Risk Manag. 2015;11:905–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gauthier AC, Mattson RH. Clobazam: a safe, efficacious, and newly rediscovered therapeutic for epilepsy. CNS Neurosci Ther. 2015;21:543–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Conry JA, Ng YT, Kernitsky L, Mitchell WG, Veidemanis R, Drummond R, et al. OV‐1004 Study Investigators. Stable dosages of clobazam for Lennox‐Gastaut syndrome are associated with sustained drop‐seizure and total‐seizure improvements over 3 years. Epilepsia. 2014;55:558–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gidal BE, Wechsler RT, Sankar R, Montouris GD, White HS, Cloyd JC, et al. Deconstructing tolerance with clobazam: Post hoc analyses from an open‐label extension study. Neurology. 2016;87:1806–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kasteleijn‐Nolst Trenité DG, Groenwold R, Schmidt B, Löscher W. Single dose efficacy evaluation of two partial benzodiazepine receptor agonists in photosensitive epilepsy patients: A placebo‐controlled pilot study. Epilepsy Res. 2016;122:30–6. [DOI] [PubMed] [Google Scholar]
- 29. Muglia P, Toledo M, Steinhoff BJ, et al.Efficacy and tolerability of adjunctive padsevonil in adults with drug‐resistant focal onset seizures: a randomized, double‐blind, placebo‐controlled, proof‐of‐concept trial. American Epilepsy Society (AES), 2017. Annual Meeting Abstract Database. AESnet.org.
- 30. González MI, Grabenstatter HL, Cea‐Del Rio CA, Cruz Del Angel Y, Carlsen J, Laoprasert RP, et al. Seizure‐related regulation of GABAA receptors in spontaneously epileptic rats. Neurobiol Dis. 2015;77:246–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mazzuferi M, Palma E, Martinello K, Maiolino F, Roseti C, Fucile S, et al. Enhancement of GABA(A)‐current run‐down in the hippocampus occurs at the first spontaneous seizure in a model of temporal lobe epilepsy. Proc Natl Acad Sci USA. 2010;107:3180–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cohen I, Navarro V, Clemenceau S, Baulac M, Miles R. On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science. 2002;298:1418–21. [DOI] [PubMed] [Google Scholar]
- 33. Cohen I, Navarro V, Le Duigou C, Miles R. Mesial temporal lobe epilepsy: a pathological replay of developmental mechanisms? Biol Cell. 2003;95:329–33. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data from nonclinical studies are outside the scope of UCB’s data sharing policy.