

Abstract
Observations of actin dynamics in living cells using fluorescence microscopy have been foundational in the exploration of the mechanisms underlying cell migration. We used CRISPR/Cas9 gene editing to generate neutrophil‐like HL‐60 cell lines expressing GFP‐β‐actin from the endogenous locus (ACTB). In light of many previous reports outlining functional deficiencies of labeled actin, we anticipated that HL‐60 cells would only tolerate a monoallelic edit, as biallelic edited cells would produce no normal β‐actin. Surprisingly, we recovered viable monoallelic GFP‐β‐actin cells as well as biallelic edited GFP‐β‐actin cells, in which one copy of the ACTB gene is silenced and the other contains the GFP tag. Furthermore, the edited cells migrate with similar speeds and persistence as unmodified cells in a variety of motility assays, and have nearly normal cell shapes. These results might partially be explained by our observation that GFP‐β‐actin incorporates into the F‐actin network in biallelic edited cells at similar efficiencies as normal β‐actin in unedited cells. Additionally, the edited cells significantly upregulate γ‐actin, perhaps helping to compensate for the loss of normal β‐actin. Interestingly, biallelic edited cells have only modest changes in global gene expression relative to the monoallelic line, as measured by RNA sequencing. While monoallelic edited cells downregulate expression of the tagged allele and are thus only weakly fluorescent, biallelic edited cells are quite bright and well‐suited for live cell microscopy. The nondisruptive phenotype and direct interpretability of this fluorescent tagging approach make it a promising tool for studying actin dynamics in these rapidly migrating and highly phagocytic cells.
Keywords: cell motility, CRISPR/Cas9, persistence, β‐actin
1. INTRODUCTION
Fluorescence microscopy of actin dynamics in living cells has been a fundamental tool for developing our understanding of the mechanisms of cell motility (Kreis, Geiger, & Schlessinger, 1982; Wang, 1985). Several methods for imaging in vivo actin dynamics have been employed over the past several decades, including microinjection and electroporation of covalently labeled purified actin (Kreis et al., 1982; Taylor & Wang, 1978; Wang, 1985) or phalloidin (Tsugiyama, Okimura, Mizuno, & Iwadate, 2013; Yam et al., 2007), expression of GFP‐labeled actin from an ectopic locus (Choidas et al., 1998; Westphal et al., 1997), expression of tagged actin‐binding proteins or peptides such as LifeAct, Utrophin, and F‐tractin (Belin, Goins, & Mullins, 2014), and use of membrane‐permeable small molecule fluorescent probes such as SiR‐actin (Lukinavičius et al., 2014). Each of these methods has its drawbacks; labeled actin subunits have been shown to have defects in polymerization and in interactions with actin‐binding proteins (Aizawa, Sameshima, & Yahara, 1997; Amann & Pollard, 2001; Chen, Nag, & Pollard, 2012; Hammer, Wang, Saeed, & Pedrosa, 2019; Kuhn & Pollard, 2005; Murugesan et al., 2016; Nagasaki et al., 2017; Westphal et al., 1997; Yi, Wu, Crites, & Hammer, 2012), while probes that bind to F‐actin may alter its dynamic behavior in vivo (Dancker, Löw, Hasselbach, & Wieland, 1975).
With the emergence of new CRISPR/Cas9 genome‐editing tools, it has become possible to routinely fluorescently tag genes at their endogenous loci. Using CRISPR/Cas9 constructs and methodologies designed for systematic gene tagging in human‐induced pluripotent stem cells (hiPSCs) (Roberts et al., 2017), we sought to create a GFP‐β‐actin HL‐60 cell line to study the roles of actin dynamics in these highly motile and phagocytic cells. This leukemia‐derived cell line can be differentiated into a neutrophil‐like phenotype that exhibits rapid motility and chemotaxis, and has been shown to rescue neutropenic mice in a fungal infection model (Millius & Weiner, 2009; Spellberg et al., 2005; Tsai et al., 2019).
Various labeled actin constructs have been shown many times in the literature to be defective in incorporating into F‐actin networks as compared to unlabeled actin. For example, purified actin chemically labeled with Oregon Green or Rhodamine at Cys‐374 has been shown to act as a kinetically inactive tracer (i.e., it incorporates into filaments with very low efficiency such that it has no effect on filament growth kinetics) (Amann & Pollard, 2001; Kuhn & Pollard, 2005). Deficiencies of labeled actin have also been measured in vivo. In MDCK cells ectopically expressing GFP‐β‐actin, the ratio of filamentous to monomeric GFP‐actin is much lower than that of normal β‐actin (Robbins et al., 1999). Furthermore, a systematic characterization of actin imaging methodologies applied to S2, XTC, B16‐F10, and U2‐OS cells revealed that, while GFP‐actin incorporates into F‐actin structures at the cell periphery, it does not incorporate into radial fibers, transverse arcs, or stress fibers as seen by phalloidin staining (Belin et al., 2014). Labeled actin monomers are also defective in their association with actin‐binding proteins. When purified mixtures of unlabeled actin and GFP‐actin are copolymerized in vitro, filaments containing more than 30% GFP‐actin are unable to undergo myosin‐based filament gliding (Aizawa et al., 1997; Westphal et al., 1997). Furthermore, formins, which have recently been identified as having important roles in cell polarity and motility (Kage et al., 2017; Shi et al., 2009), are unable to incorporate labeled actin into the F‐actin network (Chen et al., 2012).
As human cells carry only a single gene for β‐actin (Pollard, 2001), the major isoform expressed in non‐muscle cells (Vandekerckhove & Weber, 1978a, 1978b; Vedula & Kashina, 2018), we expected that edited HL‐60 lines would tolerate only one tagged allele, requiring one normal allele. To our great surprise, we were able to generate both a monoallelic GFP‐β‐actin HL‐60 line and a biallelic edited line in which the wild type allele is silenced and only the GFP‐β‐actin allele is expressed. Monoallelic lines downregulate expression of the GFP‐β‐actin allele, so their overall fluorescence intensity is rather dim. Biallelic edited cells, however, have no choice but to express GFP‐tagged β‐actin. They are therefore intensely fluorescent and, in our hands, easy to image by live cell microscopy. Despite expressing no normal β‐actin, the biallelic GFP‐β‐actin cells migrate with similar speeds and persistence as unedited (KW) and monoallelic edited cells. In addition, both monoallelic and biallelic edited lines have KW‐like cell shapes. Consistent with these results, yet in stark contrast with previously published work on labeled actin, we find HL‐60 cells incorporate GFP‐β‐actin into the F‐actin network at very similar efficiencies as unlabeled β‐actin. Perhaps to compensate for loss of normal β‐actin, both the monoallelic and biallelic lines upregulate expression of γ‐actin.
Given the direct interpretability and nondisruptive phenotype of endogenous GFP‐β‐actin expression in HL‐60 cells, we anticipate this tagging method will prove to be a highly useful tool for monitoring actin dynamics in the context of the extraordinarily rapid migration of these cells, and also during other actin‐dependent cell behaviors such as phagocytosis.
2. RESULTS AND DISCUSSION
2.1. Confirmation of biallelic and monoallelic edited cells
We transfected undifferentiated HL‐60 cells with both a GFP donor plasmid and a ribonucleoprotein (RNP) complex containing purified Cas9 protein and a guide RNA targeting the N‐terminus of the gene encoding β‐actin (ACTB), in order to induce homology‐directed‐repair‐based incorporation of the GFP sequence into the ACTB gene (Figure 1a, adapted from Roberts et al., 2017). Upon RNP transfection, it was very clear by both fluorescence microscopy and FACS analysis that there were two viable populations of GFP‐positive cells: one dim and one bright, with the bright population having ~5 times higher intensity than the dim population (Figure 1b). We confirmed by both fixed cell microscopy (Figure 1c) and live cell microscopy (Figure 1d; Supporting Video S1) that the GFP tag had the expected subcellular localization for actin in differentiated cells. HL‐60 cells are largely diploid, with a modal chromosome number of 45–46, an average genome copy number of 1.9, and no reported copy number variations for the ACTB gene (Gallagher et al., 1979; Jacobson et al., 2019; Liang et al., 1999; Shiau, Gu, Chen, Lin, & Jou, 2011; Tate et al., 2019). We thus hypothesized that these two populations consisted of a dim monoallelic (AG+) edited population and a bright biallelic (AG++) edited population. We confirmed by Western blot for N‐terminal β‐actin that the AG++ cells carry a biallelic edit, and thus produce no untagged β‐actin (Figure 2a; Supporting Figure S1). We also confirmed by Western blot that the edited cells produce a GFP‐positive band around 70 kDa, the expected molecular weight for GFP‐β‐actin (Figure 2a; Figure S1). A biallelic edit can produce two possible genotypes consistent with our results (Figure 1a); either (a) the cells contain a biallelic GFP insertion, or (b) the cells contain one tagged allele and one silenced allele that was damaged during nonhomologous end joining, as previously reported for β‐actin tagging in hiPSCs (Roberts et al., 2017). In the latter case, cells overexpress their remaining functional allele, leading to a brighter GFP‐β‐actin signal as compared to monoallelic edited cells. To further characterize our edited lines, we performed PCR genotyping of the ACTB gene; we determined that AG++ cells contain an insertion/silencing biallelic edit (Figure S2). The AG+ cells are too dim to image actin dynamics by live cell microscopy; in their differentiated neutrophil‐like state, the AG+ edited cells are >10‐fold less fluorescent than AG++ cells as imaged by FACS (data not shown) and as probed by Western blot for GFP (Figure 2a; Figure S1). The AG++ cells, however, are bright enough for live cell imaging. We designed these cell lines in order to study the rapid migration of differentiated HL‐60 cells, which is the subject of the remainder of this work, but we also briefly explored imaging of cell division in undifferentiated cells. This work revealed that GFP‐β‐actin localizes to the cytokinetic furrow only at late stages of division, likely due to the fact that formins do not incorporate labeled actin (Chen et al., 2012; Figure S3).
Figure 1.
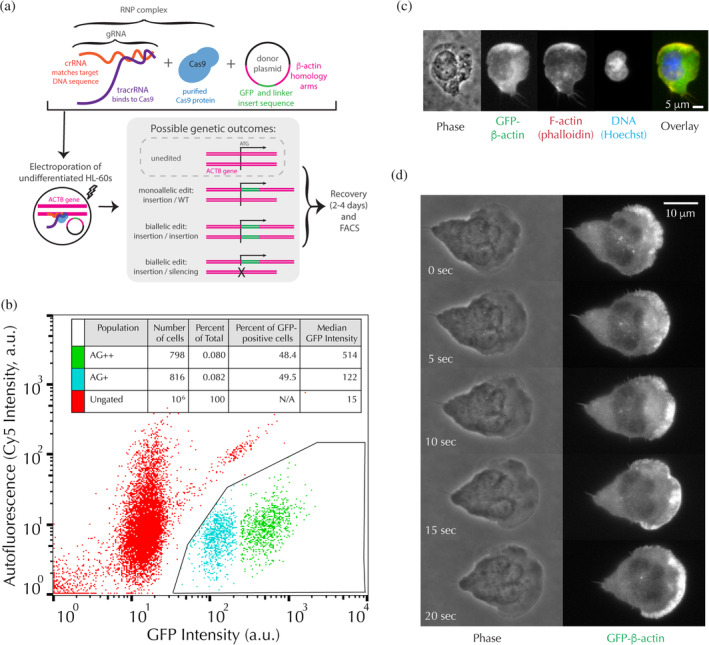
Biallelic and monoallelic edited cells are viable and exhibit the expected localization of actin. (a) Schematic of the RNP‐transfection protocol used to endogenously tag β‐actin (adapted from Roberts et al., 2017). (b) FACS results from the transfection showing two GFP‐positive populations of cells, henceforth referred to as AG+ and AG++. Fluorescence intensity through a Cy5 filter, used as a measure of autofluorescence, is plotted as a function of GFP fluorescence intensity. Inset: Quantification of relative populations by FACS. (c) Example fluorescence image of a fixed, differentiated AG++ HL‐60 cell. Scale bar: 5 μm. (d) Example time‐lapse montage of Video S1, showing an AG++ cell migrating under agarose imaged simultaneously with far‐red transmitted light for phase contrast and epi‐illumination for GFP excitation. Scale bar: 10 μm
Figure 2.
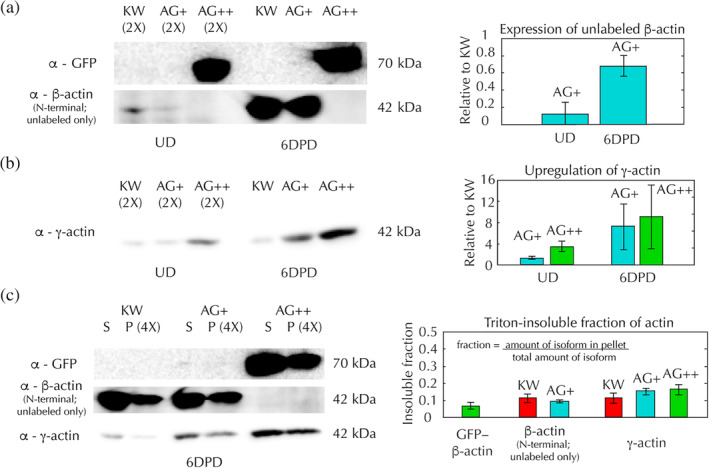
Biallelic and monoallelic edited cells preferentially upregulate their normal actin isoforms. (a–c) Western blots for β‐actin, γ‐actin, and GFP‐β‐actin expression in KW (red), AG+ (blue), and AG++ (green) HL‐60 cells, along with quantification of band ratios averaged over at least three blots. Error bars in the right panels indicate the standard error. Western blots were performed on whole protein extracts harvested from undifferentiated (UD) and 6 days post‐differentiation (6DPD) HL‐60 cells and on Triton‐extracted protein samples harvested from 6DPD HL‐60 cells. All Western band examples shown were from the same blot, which was stripped and re‐stained for each antibody in series. (a) (Left) Example of a Western blot stained for either GFP (top) or β‐actin (bottom) in UD or 6DPD HL‐60 cell whole protein extracts. Note that the N‐terminal β‐actin antibody, which must be used in order to exclude γ‐actin, does not bind to GFP‐β‐actin due to the N‐terminal GFP labeling. β‐Actin bands are overexposed to show that β‐actin is completely absent from AG++ cells. (Right) Ratio of unlabeled β‐actin bands in UD and 6DPD AG+ cells, relative to UD and 6DPD KW cells. (b) (Left) Western blot results for γ‐actin on whole protein extracts of UD and 6DPD HL‐60 cells. (Right) Ratio of γ‐actin bands in UD and 6DPD AG+ and AG++ cells, relative to UD and 6DPD KW cells. (c) (Left) Western blot results from Triton‐insoluble (P, pellet) and Triton‐soluble (S, supernatant) protein extracts from 6DPD HL‐60 cells. (Right) Fraction of GFP‐β‐actin, normal β‐actin, or γ‐actin in the Triton‐insoluble pellet
Using Western blot analysis we found that AG+ cells produce ~90% less normal β‐actin than unedited (KW) cells in their undifferentiated state, and ~30% less normal β‐actin than KW cells at 6 days post‐differentiation (6DPD) (Figure 2a; Figure S1). Both AG+ and AG++ edited lines may be compensating with γ‐actin, as they upregulate γ‐actin 1.3‐fold and 3.3‐fold in undifferentiated cells and 7.1‐fold and 9‐fold in differentiated cells at 6DPD, relative to KW (Figure 2b; Figure S1). We also performed Western blots on Triton‐extracted protein samples, in which the Triton‐soluble (G‐actin, supernatant) and Triton‐insoluble (F‐actin, pellet) populations are separated. We found that the ratio of Triton‐insoluble γ‐actin relative to the total amount of γ‐actin is similar to that of β‐actin in KW cells (Figure 2c; Figure S1), meaning that γ‐actin is able to incorporate into the F‐actin network at similar efficiencies as β‐actin in KW cells. Surprisingly, GFP‐β‐actin is also able to incorporate into the F‐actin network in AG++ cells at similar efficiencies as β‐actin in KW cells (Figure 2c; Figure S1), contrary to previously published defects in polymerization of labeled actin subunits. This is perhaps permitted by the particular flexible linker inserted between the GFP tag and β‐actin in our edited lines. Interestingly, all three HL‐60 lines retained only ~10% of their unlabeled β‐actin and γ‐actin in the Triton‐insoluble cytoskeleton, which is similar to previous measurements in HL‐60s and primary human neutrophils (of ~30%) (Watts, 1995), but quite low as compared to other cell types such as activated platelets (60–80%) (Rosenberg, Stracher, & Lucas, 1981) and bovine arterial endothelial cells (40–80%) (McGrath, Osborn, Tardy, Dewey, & Hartwig, 2000).
2.2. Motility of biallelic and monoallelic edited cells
We evaluated motility of the edited cell lines using three distinct assays: a 1D microfluidic assay, in which cells are constrained to migrate along straight channels (Figure 3a; Videos S2–S4), a 2D under‐agarose assay, in which cells are sandwiched between a coverslip and an agarose pad overlay (Figure 3d; Videos S5–S7), and a 3D collagen migration assay (Figure 3g; Videos S8–S10). By performing multiple replicates for each assay (1D: two experiments, 2 days, one field of view per experiment, and 33, 38, and 37 cell tracks for KW, AG+, and AG++ cells, respectively; 2D: two experiments, 2 days, six fields of view per experiment, and 199, 240, and 284 cell tracks for KW, AG+, and AG++ cells, respectively; 3D: three experiments, 3 days, two fields of view per experiment, and 166, 184, and 230 cell tracks for KW, AG+, and AG++ cells, respectively—see Methods section and Supporting Tables S1–S3), we were able to use ANOVA to determine relative contributions of cell type, cell‐to‐cell, coverslip‐to‐coverslip, and day‐to‐day variability to the total variance observed in our measurements.
Figure 3.
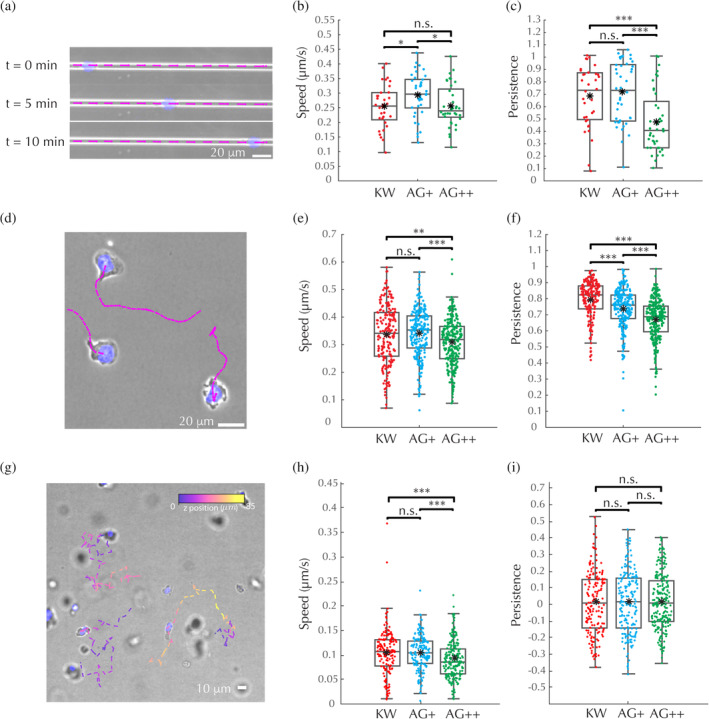
Biallelic and monoallelic edited cells have nearly normal motility. (a–c) 1D microfluidic channel motility assays. (a) Schematic of experiment and velocity measurement: Velocity measurement windows (pink), depicted as line segments, overlaid onto the microscopy image (blue, nucleus; gray, phase contrast) for three representative time points from a single experiment on KW cells. Scale bar: 20 μm. (b) Average persistent‐state (persistence>0.9, see Materials and Methods section) speed for each track. (c) Average persistence score assigned by Bayesian inference algorithm for each track. (d–f) 2D under‐agarose motility assays. (d) Schematic of experiment and velocity measurement: Velocity measurement windows (pink), depicted as line segments, overlaid onto the microscopy image (blue, nucleus; gray, phase contrast) of the final frame of a single experiment on KW cells. Scale bar: 20 μm. (e) Average persistent‐state (persistence >0.6) speed for each track. (f) Average persistence score assigned by Bayesian inference algorithm for each track. (g–i) 3D collagen migration assays. (g) Schematic of experiment and velocity measurement: Velocity measurement windows, colored by z‐height, depicted as line segments, overlaid onto 2D maximum intensity projections (blue, nucleus; gray, phase contrast) of a single frame of a single experiment on KW cells. Scale bar: 10 μm. (h) Average persistent‐state (persistence >0) speed for each track. (i) Average persistence score assigned by Bayesian inference algorithm for each track. (a,b,d,e,h,i) Central line, median; black dot, mean; boxes, 25th and 75th percentiles; whiskers, furthest data point that is not an outlier; outliers, any point that is more than 1.5 times the interquartile‐range past the 25th and 75th percentiles; significance values calculated using a non‐parametric rank‐sum test: *p < .05, **p < .01, ***p < .001, and n.s. indicates not significant
In 1D, 2D, and 3D, AG+ and AG++ cells behaved similarly to KW (Figure 3a–i; Tables S1–S3). Although AG++ cells move slightly slower than KW under agarose and in collagen, and AG+ cells move slightly faster than KW in 1D channels, the differences in mean speed are within 15% and are much smaller than the cell‐to‐cell or coverslip‐to‐coverslip variability, as confirmed by ANOVA (Figure 3b,e,h; Tables S4, S6, and S8), suggesting that these differences lack biological significance.
In addition to measuring speed, we employed a Bayesian inference algorithm (Metzner et al., 2015), based on a model of a heterogeneous random walk, to assign a persistence measurement to each time point for each cell track (Figures S4–S6). We were able to detect a measurable reduction in persistence in both AG+ and AG++ cells in 2D under agarose, and in AG++ cells in 1D channels. Still, in no case was the variability due to genotype larger than cell‐to‐cell variability, and only in the case of persistence in the 2D under‐agarose assay was the variability due to genotype larger than coverslip‐to‐coverslip variability (Figure 3c,f,i; Tables S5, S7, and S9).
Overall, while we did observe statistically significant changes in speed and persistence in the edited lines, the effect size is small compared to normal variability observed in these assays, such that the cells are viable for the study of actin dynamics in the context of motility.
2.3. Cell shape distributions of biallelic and monoallelic edited cells
To determine whether edited cells have altered cell shape, we performed principal components analysis on a pooled data set consisting of aligned contours from all three cell lines (Figure 4a). Contours were extracted by segmenting images of fixed, phalloidin‐stained cells plated under agarose. Our analyses focused on the first six principal component axes, which explain 88% of the variance. We found that the edited cell lines had only minor differences in shape compared to KW, with each genotype's mean being within half of a standard deviation from the overall mean shape across all six principal component axes (Figure 4b). Both edited lines were slightly smaller, and the AG++ cells were slightly less polarized, than KW cells (Figure 4c).
Figure 4.
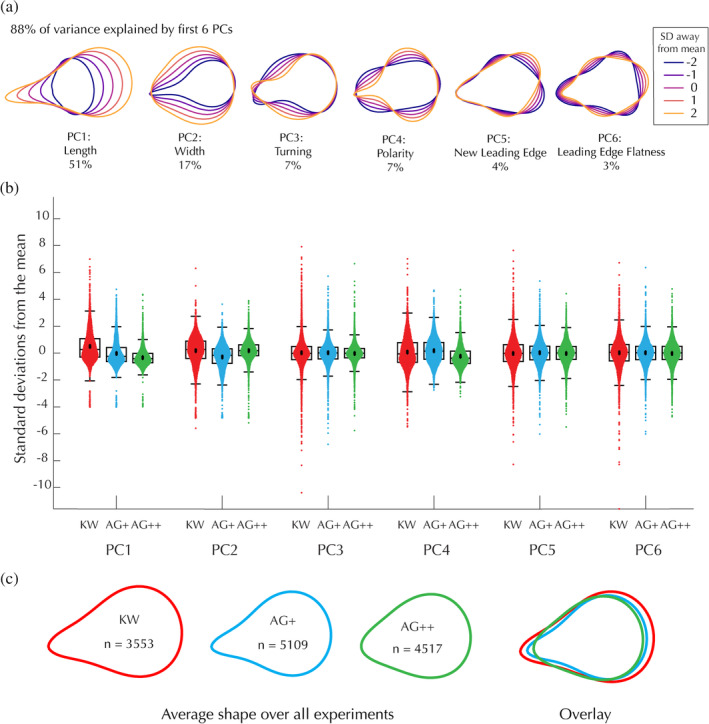
Biallelic and monoallelic edited cells have nearly normal shape. (a) Principal component axes of the shape space plotted at different standard deviations away from the average shape. Percent of variance explained by each axis is written below each principal component. (b) Distribution of each cell line along each principal component axis in the shape space. Each point represents an individual cell. Box plots were generated as in Figure 2. (c) Average shape for each cell line. Cell shapes were measured in three different experiments (coverslips) performed on three different days. (Left) Average cell shapes for each line including data from all three experiments, where n represents the number of cells analyzed. (Right) The same average cell shapes plotted on top of each other
2.4. Changes in gene expression due to the CRISPR gene editing
HL‐60s undergo a wide variety of known changes in gene expression upon differentiation (Lee et al., 2002; Rincón, Rocha‐Gregg, & Collins, 2018; Santos‐Beneit & Mollinedo, 2000; Tamayo et al., 1999). We used RNA sequencing (RNA‐seq) to explore whether the genome editing process (including the transfection and selection) or GFP‐labeling of β‐actin induces additional changes in gene expression. Upon performing differential expression analysis on all pairwise comparisons between undifferentiated and differentiated KW, AG+, and AG++ cells, we found that the largest gene expression changes occurred between undifferentiated and differentiated cells, as expected, with each cell line having about ten thousand genes differentially expressed more than 2‐fold in their differentiated state as compared to their undifferentiated state (Table S10). The next‐largest gene expression changes we observed were between the CRISPR‐edited lines and the KW line. AG++ and AG+ cells differentially express approximately a thousand genes more than 2‐fold relative to KW, including approximately fifty actin‐related genes (Figure 5a,b; Tables S10–S14). Interestingly, the smallest gene expression differences we observed were between the AG+ and AG++ lines. There are barely any genes, actin‐related or not, that are differentially expressed more than 2‐fold between AG+ and AG++ cells (Figure 5c; Tables S10, S15, and S16). We believe this striking similarity is unlikely to be due to a bottlenecking event, as the AG+ and AG++ lines were made from a heterogeneous transfected population, not clonal isolates.
Figure 5.
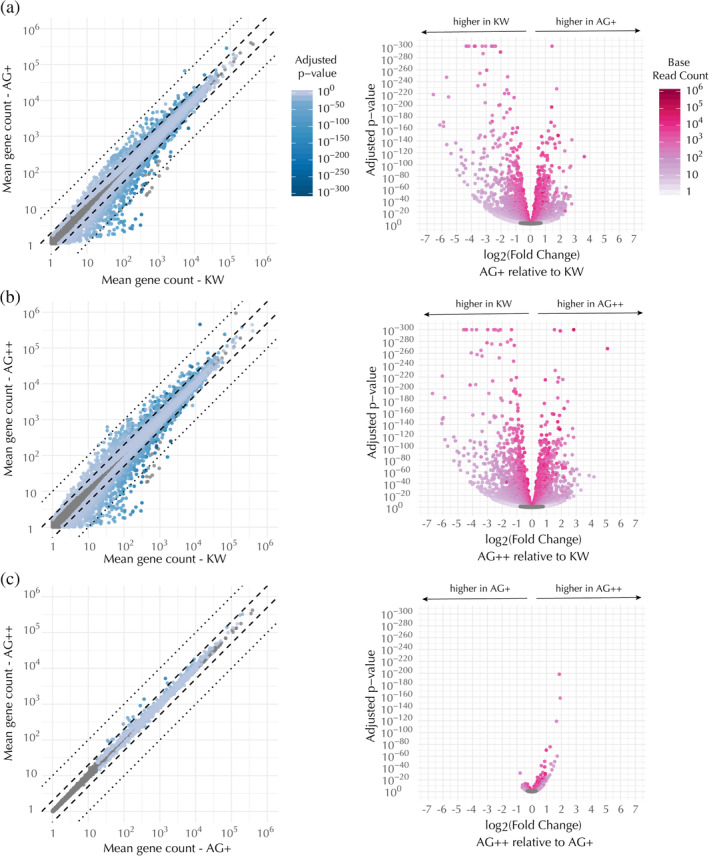
Transcriptional profiles of the edited lines are similar to each other but distinct from the KW line. Pairwise comparisons of differential expression for each mapped gene, as follows: (a) AG+ versus KW, (b) AG++ versus KW, and (c) AG++ versus AG+. Differential expression levels listed by gene name can be found in Tables S11, S13, and S15, respectively. (Left) Scatter plots of mean gene count in the compared cell types, colored by the adjusted p‐value for detecting a difference between the means. (Right) Volcano plots of the same data showing the adjusted p‐value for detecting a difference between the means as a function of the fold change in expression in the first sample (y‐axis in the corresponding scatter plot) as compared to the second sample (x‐axis in the corresponding scatter plot). In both the scatter and volcano plots, each dot represents a single gene. Means were calculated over RNA‐seq runs from seven biological replicates. Genes that were not significantly differentially expressed (adjusted p‐value >.05) are shown as gray dots
Given that AG++ cells express no normal β‐actin, we reasoned that they might compensate for deficient β‐actin activity by differentially regulating other actin‐related genes as compared to AG+ cells. In addition, we hypothesized that GFP‐labeling of actin might affect its known activity as a transcriptional regulator (Miralles & Visa, 2006). If the major gene expression changes observed in the edited lines as compared to KW were due to the GFP‐labeling of β‐actin, then we would have expected these changes to be more extreme in the AG++ cells. However, the similarity between the two edited lines implies that the substantial gene expression changes observed in the edited lines compared to KW are most likely due to the genome editing process—including concomitant selection—rather than the GFP‐labeling of β‐actin.
In addition to measuring global gene expression changes, we wanted to determine whether the increase in β‐actin and γ‐actin expression that we observed at the protein level (Figure 2a,b) was also reflected at the transcriptional level. Indeed, we found that total β‐actin was upregulated about 45% in AG+ cells (Tables S11 and S12) and 2‐fold in AG++ cells (Tables 13 and S14) relative to KW, although we did not explore how the GFP tag may interfere with RNA‐seq read alignment to the ACTB region of the genome. Total γ‐actin mRNA was upregulated about 3.6‐fold in AG+ cells (Tables S11 and S12) and 7‐fold in AG++ cells (Tables S13 and S14), which is consistent with, but much more drastic than, the change in γ‐actin protein concentration as measured by Western blot (Figure 2b).
2.5. Conclusion
We were very excited that we were able to generate viable AG++ GFP‐tagged β‐actin HL‐60 cells. Despite many previous reports of functional defects in labeled actin, we found that HL‐60 cells are able to efficiently incorporate GFP‐β‐actin into their F‐actin network, allowing AG++ cells expressing no normal β‐actin to migrate almost normally. While the Triton extraction assay reported nearly‐normal incorporation of GFP‐β‐actin overall, it is possible that certain actin structures, such as formin‐mediated filament bundles, do not incorporate labeled actin. For these types of structures, our edited lines may be compensating for loss of normal β‐actin by upregulating γ‐actin. γ‐Actin and β‐actin are structurally very similar, differing by only four amino acids at their N‐terminus. This structural similarity should in theory allow them to interact with similar actin‐binding proteins. Given that γ‐actin generally localizes to stress fibers (Belin et al., 2014; Otey, Kalnoski, Lessard, & Bulinski, 1986), which are largely formin‐mediated, it is most likely the case that formins can incorporate γ‐actin. Additionally, our RNA sequencing data suggests β‐actin and γ‐actin are expressed at comparable levels in HL‐60 cells, while most common cell lines produce much more β‐actin than γ‐actin. It is also interesting to note that our results show the edited lines have undergone a host of gene expression changes, many which are much larger than that of γ‐actin, which were not explored in this paper. Most importantly, the edited cells expressing only GFP‐tagged β‐actin have only minor disruptions to cell motility and cell shape, and thus are likely to be a useful tool to study either of these phenomena.
3. MATERIALS AND METHODS
3.1. HL‐60 cell culture and differentiation
HL‐60 cells were cultured and differentiated into a neutrophil‐like state as described previously (Millius & Weiner, 2009). Briefly, cells were cultured in RPMI‐full medium (see below) at 37°C and 5% CO2, in a standard tissue culture incubator. Cells were passaged every 2–3 days, maintaining a cell density between 0.1 and 1 × 105 cells·ml−1. Unedited, AG+, and AG++ cells could all be passaged using the same passaging frequency and dilution, indicating the edited lines have no severe defect in cell growth or division. To induce a neutrophil‐like phenotype, cells were differentiated in 1.57% DMSO in RPMI‐full medium and used for experiments at 6 days post‐differentiation. RPMI‐full medium: RPMI 1640 plus L‐glutamine and 25 mM HEPES (Gibco RPMI 1640 Medium, HEPES, Thermo Fisher Scientific, Cat. #22400089), supplemented with 10% heat‐inactivated fetal bovine serum (hiFBS) (Foundation Fetal Bovine Serum, Gemini Bio Products Cat. #900–108, heated in a water bath for 40 min at 56C), 100 U·ml−1 penicillin, 10 μg·ml−1 streptomycin, and 0.25 μg·ml−1 Amphotericin B (Gibco Antibiotic‐Antimycotic, Thermo Fisher Scientific, Cat. #15240).
3.2. Cas9 RNP transfection
HL‐60 cells were transfected using a previously published protocol (Roberts et al., 2017), adapted for HL‐60/suspension cells (Gundry et al., 2016).
3.2.1. RNP complex
All handling of the RNP reagents was performed in a Class II A2 biological safety cabinet to avoid contamination of the cell lines. In addition, all RNAs and proteins were kept on ice when not being directly handled. CRISPR RNA (crRNA) (Dharmacon Custom Edit‐R CRISPR‐Cas9 Synthetic crRNA: GCCGTTGTCGACGACGAGCG, sequence from the Allen Institute for Cell Science Cell Catalog), and trans‐activating crRNA (tracrRNA) (Edit‐R CRISPR‐Cas9 Synthetic tracrRNA, Dharmacon Cat. #U‐002005) were resuspended at 100 μM in sterile, RNAase‐free TE buffer (IDTE pH 7.5 1X TE Solution Integrated DNA Technologies Cat. #11‐01‐02‐02) according to the Dharmacon resuspension protocol. To make the duplex, the crRNA and tracrRNA were combined at 40 μM (2:5 dilution of each) in duplex buffer (Integrated DNA Technologies Nuclease‐Free Duplex Buffer Cat. #11‐01‐03‐01), then incubated at 95°C for 5 min, followed by incubation at room temperature for 2 hr. To make the RNP complex, 1 μg of purified Cas9 protein and 1 μg RNA duplex were coincubated at room temperature for 10–15 min before transfection. RNP complex was coelectroporated with a GFP donor plasmid (Addgene Cat. #87425), using the following protocol.
3.2.2. Electroporation
Cells were transfected using the Neon transfection system (Thermo Fisher) according to the manufacturers 10 μl transfection protocol. Briefly, 2 × 105 cells were concentrated at 500g for 5 min, washed in 1 ml sterile PBS (without magnesium and calcium), and concentrated again. The supernatant was removed, and the cells were resuspended in buffer R. The RNP mixture was added to the cells, along with 0.5 μg GFP donor plasmid. The cells were then transfected with a single 1,350 V, 35 ms pulse, and recovered in 500 μl pre‐warmed RPMI‐full (see HL‐60 Cell Culture and Differentiation). Cells were allowed to proliferate in normal culturing conditions for 10 days before FACS. RNP transfection efficiency was approximately 0.1–0.2% in our hands using the 10 μl kit (Figure 1b). Interestingly, within the GFP‐positive cells, there were equal numbers of AG+ and AG++ cells. This would not be expected if editing of the two alleles were random independent processes.
3.3. Fluorescence activated cell sorting (FACS)
Undifferentiated transfected cells were resuspended at 20 × 106 cells·ml−1 in a 5% solution of hiFBS in PBS (without magnesium and calcium). Cells were then sorted for GFP‐positive cells using fluorescence through a Cy5 filter as a control for autofluorescence, such that GFP‐positive cells lie below the diagonal shown in Figure 1b. Interestingly, the HL‐60s exhibited a large Cy5‐positive population, presumably because they uptake phenol red from their normal growth media. In addition to Cy5 and GFP fluorescence, cells were gated by their forward‐scatter to side‐scatter ratio, to eliminate dead cells, and their pulse width to forward‐scatter ratio, to eliminate clumps of cells (data not shown). We performed a second round of FACS to separate the GFP‐positive cells into the AG+ and AG++ populations (data not shown).
3.4. Western blots
For whole protein extraction, cells were washed with PBS, concentrated, resuspended in Laemmli SDS‐PAGE sample buffer, boiled, and sonicated. For Triton extraction, the same procedure was followed, except the concentrated samples were instead resuspended in Triton extraction buffer, spun at 250,000g for 5 min, and the Triton soluble and insoluble fractions were than separated before being resuspended in sample buffer, as described further in Robbins et al. (1999). The samples were run on 10% polyacrylamide gels, and then stained with Coomassie (Lämmli, 1970) to normalize the amount of protein in each sample. The samples were then run again on 10% acrylamide gels using equivalent amounts of protein, imaged with a reversible membrane protein stain (Novex Reversible Membrane Protein Stain Kit, Thermo Fisher Cat. #IB7710 or Ponceau), and analyzed by Western blot. A single blot was probed three separate times by striping the blot after each round of detection. Antibodies were used to detect ACTB (Abcam ab6276 1:1000 in 5% non‐fat dry milk or Blotto), GFP (Abcam ab6556 1:1000 in 5% non‐fat dry milk or Blotto), and γ‐actin (EMD Millipore MABT824 1:1000 in 5% non‐fat dry milk or Blotto). The secondary antibody was an HRP conjugate and detected using chemiluminescence (Sigma CPS1) and a digital gel documentation system (Azure c600 or Biorad Versadoc MP4000), using a series of exposure times. Three to four blots were performed for each measurement.
3.5. PCR genotyping
Genomic DNA was extracted from undifferentiated KW, AG+, and AG++ cells using the QIAGEN Blood and Cell Culture DNA Mini kit (QIAGEN Cat. #13323) and extraction protocol. PCR amplification of the ACTB gene was performed on genomic DNA using the primer pair 5′‐CGATGGGGTACTTCAGGGTG‐3′ and 5′‐CTGGGACTCAAGGCGCTAAC‐3′. PCR products were run alongside a DNA ladder (Invitrogen TrackIt 1 Kb Plus DNA Ladder, Thermo Fisher Cat. #10488085) on a 1% agarose gel with 0.1 μg·ml−1 ethidium bromide.
3.6. Fluorescence microscopy
All fluorescence microscopy was performed on a Nikon microscopy system with transmitted light using phase contrast and Nikon LWD 0.52 NA condenser and with epifluorescence imaging using a Lambda XL light source. Images were acquired either on an Andor Zyla 4.2 sCMOS camera (high speed imaging), or an Andor iXon EMCCD camera (all other imaging) using μ‐Manager software (Edelstein et al., 2014).
3.7. Live‐cell imaging of cell division
3.7.1. Overview
Undifferentiated AG++ cells were cell cycle synchronized using serum starvation, then imaged by GFP epifluorescence and phase contrast on poly‐L‐lysine‐coated coverslips.
3.7.2. Cell synchronization
Undifferentiated AG++ cells were split down to 1.5 × 105 cells·ml−1 and allowed to grow in 10% hiFBS in RPMI for 24 hr. Cells were then washed with PBS, resuspended in RPMI without serum, and allowed to grow for 22.5 hr, thereby arresting cells in the G0/G1 phase of the cell cycle. Cells were then released from the G0/G1 phase of the cell cycle by addition of 10% hiFBS. Cells were imaged 23 hr after re‐addition of serum.
3.7.3. Plating and imaging
μ‐90‐washed 25 mm diameter coverslips were incubated in 0.01% w/v poly‐L‐lysine in water (EMS Cat. #19320‐B, diluted 1:10 in Type 1 water) for 5 min, rinsed 3× with Type 1 water, then dried overnight at RT. Synchronized AG++ cells were spun down at 500g for 5 min, resuspended in 10% hiFBS in L15, and allowed to settle by gravity onto a poly‐L‐lysine‐coated coverslip at 37°C. Cells were then imaged at 30 s intervals using sequential phase contrast and epifluorescence illumination through a standard GFP filter set.
3.8. Fixed‐cell imaging of cell division
3.8.1. Overview
Undifferentiated AG++ cells were plated on fibronectin‐coated coverslips under an agarose pad. Cells were then fixed in PFA, stained with Hoechst and phalloidin, and imaged—all while still under‐agarose.
3.8.2. Plating and imaging
Undifferentiated, unsynchronized AG++ cells were plated, fixed, and stained exactly as in Methods: Preparation, imaging, and analysis of fixed‐cell samples section with only one exception—as undifferentiated cells are only weakly adherent to the coverslip, staining and imaging were performed without removing the agarose pad.
3.9. Microfluidic channel (1D) motility assays
3.9.1. Overview
At 6 days post‐differentiation, cells were stained with a cell‐permeable Hoechst nuclear dye and then loaded on a microfluidic device harboring straight migratory channels primed with fMLP. Cells were then imaged as they migrated towards the chemoattractant gradient with acquisitions taken at 3 s intervals for 60 min.
3.9.2. Microfluidic devices
Microfluidic devices were prepared as described previously (Boneschansker, Yan, Wong, Briscoe, & Irimia, 2014; Irimia, 2014), harboring straight migratory channels of 6 μm width and 3 μm height.
3.9.3. Microfluidic device priming
Priming of each device commenced by pipetting 10 μl of an fMLP‐based chemoattractant solution through the loading port. For this, fMLP was diluted in L‐15 media containing 20% hiFBS to a final concentration of 100 nM. The device was then placed in a vacuum chamber connected to house vacuum (27 inHg) for 10 min. Upon removal, the device was rested for an additional 10 min, until the chemoattractant filled entirely the array of migration channels connected orthogonally to the central loading channel. The device was washed twice by pipetting into the loading port 200 μl of 10 μg·ml−1 fibronectin diluted in L‐15 media containing 20% hiFBS. These washing steps removed the chemoattractant from the central loading channel and its passive diffusion from the migration channels into the central loading channel established a chemoattractant gradient.
3.9.4. Cell staining and loading
2 × 105 cells were spun down at 500g for 5 min and resuspended in 1 ml L‐15 media containing Hoechst 33324 at a final concentration of 1 μg·ml−1. The Hoechst‐stained cell suspension was incubated at 37°C for 15 min. Stained cells were spun down at 500g for 5 min and were resuspended in ~30 μl of L‐15. Out of this cell suspension, 5 μl were pipetted into the loading port of the device. The sample was then incubated on the microscope at 37°C, where the cells could be monitored.
3.9.5. Imaging
Cells were incubated and imaged at 37°C on an epifluoresence microscope (see Fluorescence Microscopy section of Materials and Methods) at ×20 magnification (20x 0.75NA Plan Apo Phase Contrast air objective, Nikon MRD30205) using sequential phase contrast and epifluorescence illumination through a standard DAPI filter set. For each experiment, a 60 min time‐lapse movie was acquired at 3 s intervals. Two such experiments, each on a different day, were performed for each cell line.
3.10. Under‐agarose (2D) motility assays
3.10.1. Overview
At 6 days post‐differentiation, cells were stained with a cell‐permeable Hoechst nuclear dye, plated on a fibronectin‐coated coverslip, and then overlaid with an agarose pad supplemented with hiFBS and fMLP to stimulate migration. Cells were then imaged at 3 s intervals for 10 min.
3.10.2. Coverslip preparation
Glass coverslips (25 mm diameter #1.5 Micro Coverglass, Electron Microscopy Sciences Cat. #72225‐01) were boiled in 0.1% Micro‐90 cleaning solution in deionized water, rinsed in deionized water, rinsed in ethanol, and dried in an oven at 150°C. On the day of each experiment, coverslips were incubated with 10 μg·ml−1 fibronectin in PBS (Fibronectin human plasma, Sigma Aldrich Cat. #F2006, reconstituted following the manufacturer's instructions) at room temperature for 1.5 hr. Following incubation, coverslips were washed once in RPMI‐full media, and then placed into L‐15 media at room temperature until plating cells.
3.10.3. Agarose pad
On the day of each experiment, a 20 mg·ml−1 (2X) solution of agarose (UltraPure Low Melting Point Agarose Powder, Invitrogen Cat. #16520100) in L‐15 media was dissolved by microwaving and stored at 37°C. Then a 2X solution of imaging media was prepared, containing 20% hiFBS (see HL‐60 Cell Culture and Differentiation) and 2 nM fMLP (N‐Formyl‐Met‐Leu‐Phe, Sigma Cat. #F3506, stock solutions prepared at 1 μM in DMSO) in L‐15. Immediately before pouring the gel, the 2X agarose solution was mixed 1:1 with 2X imaging media, for a final concentration of 1% agarose, 10% hiFBS, and 1 nM fMLP. To prepare the pad, 900 μl of agarose solution was pipetted into a 25 mm diameter mold and then allowed to gel at room temperature.
3.10.4. Cell staining and plating
2 × 105 cells were concentrated at 500g for 5 min and incubated in 1 μg·ml−1 Hoechst 33324 in L‐15 for ~15 min. The cells were then concentrated at 500g for 5 min, resuspended in ~20 μl supernatant, and placed dropwise onto the coverslip. The agarose pad was then immediately laid on top of the cells. The sample was then incubated on the microscope at 37°C, where the cells could be monitored. It is necessary to allow the sample to dry slightly to reduce the gap between the agarose pad and the coverslip. Once the cells began migrating, in about 45 min, 1 ml of mineral oil was placed on top of the pad to keep the sample from drying out further.
3.10.5. Imaging
Cells were incubated and imaged at 37°C on an epifluoresence microscope (see Fluorescence Microscopy section of Materials and Methods) at ×20 magnification (20x 0.75NA Plan Apo Phase Contrast air objective, Nikon MRD30205) using sequential phase contrast and epifluorescence illumination through a standard DAPI filter set. For each sample, a 10 min time‐lapse movie was acquired at 3 s intervals. Six such movies were captured sequentially for six different fields of view, giving a total of 1 hr of imaging per sample. Two such experiments, each on a different day, were performed for each cell line. For the high‐magnification supplemental videos, cells were imaged on an epifluoresence microscope at ×100 magnification (100x 1.45NA Plan Apo oil objective, Nikon MRD31905), with an intermediate magnification of ×1.5. Cells were observed simultaneously with far‐red transmitted light for phase contrast and epi‐illumination for GFP excitation. The GFP emission and far red light was passed through a beamsplitter (Cairn research OptoSplit III) to split the channels and simultaneously project a phase contrast and GFP image side‐by‐side on the camera.
3.11. 3D motility assays
3.11.1. Overview
At 6 days post‐differentiation, cells were stained with a cell‐permeable Hoechst nuclear dye and then embedded in a collagen matrix. Cells were then imaged as they moved through the collagen matrix with acquisitions taken at 66 s intervals for 66 min. Note that the cells move more slowly in 3D compared to 1D and 2D, so 3D assays were imaged at a lower frame rate to capture the equivalent track dynamics seen in 1D and 2D.
3.11.2. Cell staining and collagen matrix embedding
3 × 105 cells were concentrated at 300g for 5 min and incubated in 1 mg·ml−1 Hoechst 33324 in L‐15 with 10% hiFBS for 15 min. The cells were then concentrated at 300g for 5 min, and then resuspended in 20 μl of the supernatant. During incubation with Hoechst stain, a 180 μl collagen aliquot was prepared according to manufacturer recommendations and placed on ice. Briefly, 3.2 μl sterile water, 7.2 μl 10x PBS, 12.5 μl 0.1 M NaOH, 90 μl L‐15, and 18 μl hiFBS were combined with 50 μl 3 mg·ml−1 collagen (ThermoFisher, cat. # A1048301). The 20 μl cell suspension was added to the collagen mixture for a final concentration of 0.75 mg·ml−1 collagen, mixed well by pipetting, and then added to the channel of an Ibidi μ‐Slide I (IbiTreat, Cat. #80106). After 1 min incubation at room temperature, the slide was inverted to help prevent cell sedimentation, and incubated at 37C for gel formation. After 25 min, the μ‐Slide I media reservoirs were filled with 2 ml total L‐15 media containing 10% hiFBS and left to incubate between 30 min to 1.5 hr prior to imaging.
3.11.3. Imaging
Cells were incubated and imaged at 37°C on an epifluoresence microscope (see Fluorescence Microscopy section of Materials and Methods) at ×20 magnification (20x 0.75NA Plan Apo Phase Contrast air objective, Nikon MRD30205) using sequential phase contrast and epifluorescence illumination through a standard DAPI filter set. For each sample, a 66 min time‐lapse movie was acquired at 66 s intervals. A z‐stack was acquired over 85 μm with images every 5 μm. Two movies were captured for each collagen gel preparation, and three such experiments, each on a different day, were performed for each cell line.
3.12. Motility tracking and analysis
3.12.1. Overview
Cell tracks were extracted from time‐lapse microscopy images using custom MATLAB (1D and 2D) and Python (3D) code. To analyze the tracks, nonoverlapping nonadjacent velocities were calculated over 6 s (1D and 2D) and 66 s (3D) windows. Then, persistence scores for each time point were inferred using a previously published sequential Bayesian algorithm (Metzner et al., 2015).
3.12.2. Image analysis
Nuclear images for 1D and 2D assays were contrast‐enhanced and then segmented by thresholding. For 3D, nuclear images were segmented without contrast‐enhancement.
3.12.3. Tracking
For 1D and 2D assays, the centroids of each nuclear mask were tracked using the u‐track 2.0 (Jaqaman et al., 2008; Ng, Besser, Danuser, & Brugge, 2012) MATLAB software, with parameters chosen to allow random motion, directed motion, and instantaneous direction reversal (parameters.linearMotion = 2), but not track merging or splitting (gapCloseParam.mergeSplit = 0). For 1D assays, only the first cell to enter each channel was tracked. For 3D images, the centroids of each nuclei were tracked using custom code written in Python (available upon request). Briefly, cell trajectories were determined by first identifying the x and y positions from the centroid of segmented nuclei. Using a 30 pixel × 30 pixel region centered around each x,y position, z positions were then estimated using a weighted‐intensity average across the vertical image stacks. Cell tracks were then determined by first calculating all possible cell‐to‐cell displacements between consecutive time points, and then matching cells through minimization of the total displacement across cells. The cell density within the 3D gel was such that individual tracks could be easily identified with this approach. Cells that were found in the bottom or topmost z position for more than 20% of the tracked positions were ignored due to the possibility that their focus was outside the acquisition range.
3.12.4. Velocity calculations
From these tracks, nonoverlapping velocity calculations were made using a 6 s (2‐frame) discretization for 1D and 2D tracks and a 66 s discretization (1‐frame) for 3D tracks.
3.12.5. Persistence estimations
To estimate the persistence of each track at each time point, velocities were analyzed using a previously published Bayesian inference algorithm based on a persistent random walk (Metzner et al., 2015). Parameters were chosen empirically to best capture persistence changes in the tracks (grid size = 200, pMin = 10−5, persistence box kernel radius = 2, activity box kernel radius = 2). Persistences were allowed to range from −1.5 to 1.5, and activities were allowed to range from 0 to 0.5. For 3D tracks, we only considered cell trajectories containing 20 or more time points.
3.12.6. Statistical analysis
Histograms of cell speed presented as a Gaussian superimposed with a decaying exponential as a function of speed. We found that the removal of points in which the cell was not persistent also removed the decaying exponential, which is consistent with the decaying exponential being caused by blebbing events. Thus for all comparisons of cell speed, we limited our comparisons to time points where the cells were relatively persistent. To classify each velocity measurement as either persistent or not persistent, an empirical threshold of persistence (0.9 for 1D assays, 0.6 for 2D assays, and 0 for 3D assays) was chosen. A Wilcoxon rank‐sum test was run to determine p values for the difference between medians of these distributions. Furthermore, a nested ANOVA was then run to determine the relative effects of day‐to‐day variability, cell‐to‐cell variability, coverslip‐to‐coverslip variability, and genotype on persistence and persistent cell speed.
3.13. Preparation, imaging, and analysis of fixed‐cell samples
3.13.1. Overview
Cells were fixed using a previously published protocol (Millius & Weiner, 2009), adapted for under‐agarose assays. Fixed cell samples were stained with fluorescent phalloidin (actin) and Hoechst (nucleus) and imaged. From these images, cells were segmented using a custom MATLAB pipeline. Cell mask alignment and principal components analysis were done using CellTool, an open‐source cell shape analysis software written in python (Keren et al., 2008; Pincus & Theriot, 2007). A single shape space was created from the combined KW, AG+, and AG++ segmented images. A nested ANOVA was then run to determine the relative effects of day‐to‐day variability, cell‐to‐cell variability, coverslip‐to‐coverslip variability, and genotype on the locations of cell populations along each principal mode of this combined shape space.
3.13.2. Fixation
Cells were plated as described previously (see Materials and Methods: Under‐agarose motility assays section), with a few modifications. First, a much thinner agarose pad was used to increase permeability of the fixative (0.51 mm, 3 #1.5 coverslips). We confirmed by microscopy that cells were fixed within 4 s using this agarose thickness. Second, no mineral oil was placed on the sample. Instead, a stainless steel washer was placed over the agarose pad to keep the pad from floating off and/or shearing the cells during the fixing process. Finally, the Hoechst stain was not necessary before fixation. Once the cells began migrating, the sample was fixed by pipetting 100 μl of fixative (3.7% formaldehyde, 0.32 M sucrose, 0.14 M KCl, 1 mM MgCl2, 2 mM EGTA, 20 mM HEPES, pH 7.5) dropwise over the center of the agarose pad. The samples were incubated in fixative for 20 min at room temperature, and then placed in TBS solution at 4°C overnight to allow the agarose pad to gently float off of the coverslip.
3.13.3. Staining
Samples were permeabilized in stain buffer (330 mM Phalloidin Alexa‐Fluor‐647, 1 μg·ml−1 Hoechst 33342 in 0.2% TX‐100 in TBS) and protected from light for 20 min. Coverslips were then rinsed once in 0.2% TX‐100 in TBS, rinsed twice in TBS, and mounted on a slide with Pro‐Long Diamond anti‐fade medium.
3.13.4. Imaging
Fixed cell samples were imaged on an epifluoresence microscope (see Fluorescence Microscopy section of Materials and Methods) at ×40 magnification (0.95 NA Plan Apo Phase Contrast 40X air objective) with ×1.5 intermediate magnification, illuminating for phase contrast, GFP, Cy5, and DAPI. A 20 × 20 grid of images was acquired for each coverslip.
3.13.5. Image segmentation and principal components analysis
Cell masks were created using custom MATLAB scripts. Briefly, masks were extracted by segmenting the phalloidin images using an adaptive thresholding scheme and filtered using the nuclear signal (i.e., there should be only one nucleus per cell mask). The masks of all segmented cells from the various cell lines and experimental replicates were combined into a single image set. CellTool then extracted 100 x,y‐contour points from each mask, and aligned these contours to each other. This combined contour set was then used to create a single shape space, in which CellTool calculated the principal modes of variation within this shape space, and assigned each mask a position along each principal mode.
3.14. RNA sequencing
3.14.1. Sample preparation
Seven T25 flasks with 5 ml each of undifferentiated KW, AG+, and AG++ cells were allowed to grow 2–3 days, to approximately 100,000 cells·ml−1. Likewise, seven flasks with 5 ml each of differentiated KW, AG+, and AG++ cells were allowed to grow to 6‐days post‐differentiation, reaching approximately 100,000 cells·ml−1. At this point, total RNA was extracted using the RNEasy Mini kit (Qiagen Cat# 74104). RNA quality was assayed via bioanalyzer analysis performed by the Stanford Protein and Nucleic Acid (PAN) Facility.
3.14.2. Library preparation and RNA sequencing
RNA libraries were prepared by the Stanford Functional Genomics Facility using the KAPA stranded RNA‐seq kit with RiboErase (Kit code KK8483, Roche Cat. # 07962282001), for a fragment length of 200–300 bp. Sequencing was run on the Illumina NextSeq 500 System using the High‐Output Kit with 2 × 75 read length.
3.14.3. Analysis
Reads were initially checked for quality control using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). Read alignment to the human genome (Ensembl genome assembly GRCh38.p12) was performed using HISAT2 (https://ccb.jhu.edu/software/hisat2) (Kim, Langmead, & Salzberg, 2015; Zerbino et al., 2018). Read counting was performed using HTSeq (Anders, Pyl, & Huber, 2015). Differential gene expression analysis was performed using DESeq2 (Love, Huber, & Anders, 2014). Extraction of actin‐related genes was performed by isolating genes with the term “actin” included either in the gene name or in the gene's Gene Ontology Consortium (GO) molecular function terms (The Gene Ontology Consortium, 2000, 2017).
AUTHOR CONTRIBUTIONS
R.M.G. and G.S. performed the Cas9 RNP complex preparation and transfection. R.M.G. and M.F. performed the Western blots. A.H. performed the microfluidic channel experiments. N.M.B. performed the 3D migration experiments. G.S. performed the RNA extraction for RNA sequencing. R.M.G. and A.S. performed the PCR genotyping. R.M.G. performed all other experiments with contributions from Stanford facilities: FACS sorting and fluorescence measurements were performed by the Stanford Shared FACS Facility; RNA quality control via Bioanalyzer was run by the Stanford Protein and Nucleic Acid Facility; and RNA library prep, quality control, and RNA sequencing were run by the Stanford Functional Genomics Facility. N.B. and R.M.G. performed the 3D motility tracking and analysis. R.M.G. performed all analysis for FACS, Western blot, 1D motility, 2D motility, cell shape, and RNA sequencing experiments. R.M.G. and J.A.T. prepared the manuscript. All authors read and approved the final version of the manuscript.
Supporting information
Supplementary Figure 1 Full images of the example Western blot from Figure 2. Full Western blots are displayed at a single imaging exposure for the reversible membrane total protein stain, along with a series of imaging exposure times for the β‐actin, GFP, and γ‐actin stains. All images were taken from a single blot stripped and re‐stained in the order displayed from left to right.
Supplementary Figure 2 PCR genotyping of the edited cell lines. (A) Schematic of the PCR‐based genotyping strategy. (B) Top: Agarose gel results for the PCR products, displayed alongside the DNA ladder. Bottom left: Magnification of ~800 bp bands. Bottom right: Quantification of band locations. Pixel intensities were summed over the long axis of each band. Peak intensity positions, shown as dashed lines, mark the centers of each band. The AG++ band runs at a lower position on the gel, indicating a smaller PCR product, consistent with a deletion of about 10–11 bp.
Supplementary Figure 3 GFP‐β‐actin in the cytokinetic ring. (A‐B) Live‐ and fixed‐cell imaging of cytokinesis in undifferentiated AG++ cells. Scale bar: 5 μm. (A) GFP epifluorescence time lapse montage of cytokinesis in AG++ cells plated on poly‐L‐lysine‐coated coverslips. Frames are displayed in 60 sec intervals. (B) Four example AG++ cells fixed and stained to image cell shape (phase contrast, gray), DNA (Hoechst, blue), labeled actin (GFP‐β‐actin, green), and F‐actin (phalloidin, red).
Supplementary Figure 4 Example of Bayesian persistence fitting on a track from a 1D microfluidic channel motility assay. (A) Track of a single cell plotted over time (B) Fitted persistence value plotted over time. Dashed line – persistence threshold used to stratify velocity measurements. (C) Fitted activity values plotted over time. (D) Heatmap of fitted persistence and activity values.
Supplementary Figure 5 Example of Bayesian persistence fitting on a track from a 2D under‐agarose motility assay. (A) Track of a single cell plotted over time (B) Fitted persistence value plotted over time. Dashed line – persistence threshold used to stratify velocity measurements. (C) Fitted activity values plotted over time. (D) Heatmap of fitted persistence and activity values.
Supplementary Figure 6 Example of Bayesian persistence fitting on a track from a 3D collagen motility assay. (A) Track of a single cell plotted over time (B) Fitted persistence value plotted over time. Dashed line – persistence threshold used to stratify velocity measurements. (C) Fitted activity values plotted over time. (D) Heatmap of fitted persistence and activity values.
Supplementary Table 1 Summary of 1D motility assay results
Supplementary Table 2: Summary of 2D motility assay results
Supplementary Table 3: Summary of 3D motility assay results
Supplementary Table 4: ANOVA results for persistent speed distributions in 1D motility assays
Supplementary Table 5: ANOVA results for persistence distributions in 1D motility assays
Supplementary Table 6: ANOVA results for persistent speed distributions in 2D motility assays
Supplementary Table 7: ANOVA results for persistence distributions in 2D motility assays
Supplementary Table 8: ANOVA results for persistent speed distributions in 3D motility assays
Supplementary Table 9: ANOVA results for persistence distributions in 3D motility assays
Supplementary Table 10: Number of significantly differentially regulated genes
Supplementary Table 11 Differentially regulated genes between AG+ and KW at 6 days post‐differentiation (.xlsx)
Supplementary Table 12 Differentially regulated actin‐related genes between AG+ and KW at 6 days post‐differentiation (.xlsx)
Supplementary Table 13 Differentially regulated genes between AG++ and KW at 6 days post‐differentiation (.xlsx)
Supplementary Table 14 Differentially regulated actin‐related genes between AG++ and KW at 6 days post‐differentiation (.xlsx)
Supplementary Table 15 Differentially regulated genes between AG++ and AG+ at 6 days post‐differentiation (.xlsx)
Supplementary Table 16 Differentially regulated actin‐related genes between AG++ and AG+ at 6 days post‐differentiation (.xlsx)
Video S1 Live cell phase‐contrast and epifluorescence imaging of AG++ cell migration under agarose
Video S2 Example 1D motility assay for KW cells
Video S3 Example 1D motility assay for AG+ cells
Video S4 Example 1D motility assay for AG++ cells
Video S5 Example 2D motility assay for KW cells
Video S6 Example 2D motility assay for AG+ cells
Video S7 Example 2D motility assay for AG++ cells
Video S8 Example 3D motility assay for KW cells
Video S9 Example 3D motility assay for AG+ cells
Video S10 Example 3D motility assay for AG++ cells
ACKNOWLEDGEMENTS
We thank Ruwanthi Gunawardane for sharing detailed protocols for the RNP‐transfection of hiPSCs, as well as Michael Gundry, Lorenzo Brunetti, Margaret Goodell, and Daisuke Nakada for sharing detailed protocols for the RNP‐transfection of HL‐60 cells. We also thank Amanda Haupt and Brock Roberts for their insight and protocols for genotyping the edited lines. We are grateful to Felix Ellett and Daniel Irimia for graciously sharing their microfluidic devices. Finally, we thank James Russell and Effie Bastounis for helpful advice on RNA sequencing analysis. This study was supported by Howard Hughes Medical Institute, Stanford University, Gerald J. Lieberman Fellowship to R.M.G., NSF Graduate Research Fellowship to R.M.G., Stanford Bio‐X Bowes Fellowship to A.H., Fellowships from the Alexander S. Onassis Public Benefit Foundation and the Foundation for Education and European Culture to A.H., and an NIH S10 Shared Instrument Grant S10RR027431‐01 to the Stanford Shared FACS Facility.
Garner RM, Skariah G, Hadjitheodorou A, et al. Neutrophil‐like HL‐60 cells expressing only GFP‐tagged β‐actin exhibit nearly normal motility. Cytoskeleton. 2020;77:181–196. 10.1002/cm.21603
Funding information Alexander S. Onassis Public Benefit Foundation; Foundation for Education and European Culture; Gerald J. Lieberman Fellowship; Howard Hughes Medical Institute; National Institutes of Health, Grant/Award Number: S10RR027431‐01; National Science Foundation Graduate Research Fellowship; Stanford Bio‐X Bowes Fellowship
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Aizawa, H. , Sameshima, M. , & Yahara, I. (1997). A green fluorescent protein‐actin fusion protein dominantly inhibits cytokinesis, cell spreading, and locomotion in dictyostelium. Cell Structure and Function, 22(3), 335–345. 10.1247/csf.22.335 [DOI] [PubMed] [Google Scholar]
- Amann, K. J. , & Pollard, T. D. (2001). Direct real‐time observation of actin filament branching mediated by Arp2/3 complex using total internal reflection fluorescence microscopy. Proceedings of the National Academy of Sciences, 98(26), 15009–15013. 10.1073/pnas.211556398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders, S. , Pyl, P. T. , & Huber, W. (2015). HTSeq‐A python framework to work with high‐throughput sequencing data. Bioinformatics, 31(2), 166–169. 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belin, B. J. , Goins, L. M. , & Mullins, R. D. (2014). Comparative analysis of tools for live cell imaging of actin network architecture. BioArchitecture, 4(6), 189–202. https://doi.org/10.1080/19490992.2014.1047714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boneschansker, L. , Yan, J. , Wong, E. , Briscoe, D. M. , & Irimia, D. (2014). Microfluidic platform for the quantitative analysis of leukocyte migration signatures. Nature Communications, 5, 1–12. 10.1038/ncomms5787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Q. , Nag, S. , & Pollard, T. D. (2012). Formins filter modified actin subunits during processive elongation. Journal of Structural Biology, 177(1), 32–39. 10.1016/j.jsb.2011.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choidas, A. , Jungbluth, A. , Sechi, A. , Murphy, J. , Ullrich, A. , & Marriott, G. (1998). The suitability and application of a GFP‐actin fusion protein for long‐term imaging of the organization and dynamics of the cytoskeleton in mammalian cells. European Journal of Cell Biology, 77(2), 81–90. 10.1016/S0171-9335(98)80075-7 [DOI] [PubMed] [Google Scholar]
- Dancker, P. , Löw, I. , Hasselbach, W. , & Wieland, T. (1975). Interaction of actin with phalloidin: Polymerization and stabilization of F‐actin. BBA ‐ Protein Structure, 400(2), 407–414. 10.1016/0005-2795(75)90196-8 [DOI] [PubMed] [Google Scholar]
- Edelstein, A. D. , Tsuchida, M. A. , Amodaj, N. , Pinkard, H. , Vale, R. D. , & Stuurman, N. (2014). Advanced methods of microscope control using μManager software. Journal of Biological Methods, 1(2), 10 10.14440/jbm.2014.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher, R. , Collins, S. , Trujillo, J. , McCredie, K. , Ahearn, M. , Tsai, S. , … Gallo, R. (1979). Characterization of the continuous, differentiating myeloid cell line (HL‐60) from a patient with acute promyelocytic leukemia. Blood, 54(3), 713–733. [PubMed] [Google Scholar]
- Gundry, M. C. , Brunetti, L. , Lin, A. , Mayle, A. E. , Kitano, A. , Wagner, D. , … Nakada, D. (2016). Highly efficient genome editing of murine and highly efficient genome editing of murine and human hematopoietic progenitor cells by CRISPR / Cas9. Cell Reports, 17(5), 1453–1461. 10.1016/j.celrep.2016.09.092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer, J. A. , Wang, J. C. , Saeed, M. , & Pedrosa, A. T. (2019). Origin, organization, dynamics, and function of actin and actomyosin networks at the T cell immunological synapse. Annual Review of Immunology, 37(1), 201–224. 10.1146/annurev-immunol-042718-041341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irimia, D. (2014). Cell migration in confined environments. Methods in Cell Biology, 121, 141–153. 10.1016/B978-0-12-800281-0.00010-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson, E. C. , Grand, R. S. , Perry, J. K. , Vickers, M. H. , Olins, A. L. , Olins, D. E. , & O'Sullivan, J. M. (2019). Hi‐C detects novel structural variants in HL‐60 and HL‐60/S4 cell lines. Genomics, 112, 151–162. 10.1016/j.ygeno.2019.05.009 [DOI] [PubMed] [Google Scholar]
- Jaqaman, K. , Loerke, D. , Mettlen, M. , Kuwata, H. , Grinstein, S. , Schmid, S. L. , & Danuser, G. (2008). Robust single‐particle tracking in live‐cell time‐lapse sequences. Nature Methods, 5(8), 695–702. 10.1038/NMETH.1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kage, F. , Winterhoff, M. , Dimchev, V. , Mueller, J. , Thalheim, T. , Freise, A. , … Rottner, K. (2017). FMNL formins boost lamellipodial force generation. Nature Communications, 8, 14832 10.1038/ncomms14832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren, K. , Pincus, Z. , Allen, G. M. , Barnhart, E. L. , Marriott, G. , Mogilner, A. , & Theriot, J. A. (2008). Mechanism of shape determination in motile cells. Nature, 453(7194), 475–480. 10.1038/nature06952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D. , Langmead, B. , & Salzberg, S. L. (2015). HISAT: A fast spliced aligner with low memory requirements. Nature Methods, 12(4), 357–360. 10.1038/nmeth.3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreis, T. E. , Geiger, B. , & Schlessinger, J. (1982). Mobility of microinjected rhodamine actin within living chicken gizzard cells determined by fluorescence photobleaching recovery. Cell, 29(3), 835–845. 10.1016/0092-8674(82)90445-7 [DOI] [PubMed] [Google Scholar]
- Kuhn, J. R. , & Pollard, T. D. (2005). Real‐time measurements of actin filament polymerization by total internal reflection fluorescence microscopy. Biophysical Journal, 88(2), 1387–1402. 10.1529/biophysj.104.047399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lämmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227(5259), 680–685. 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
- Lee, K.‐H. , Chang, M.‐Y. , Ahn, J.‐I. , Yu, D.‐H. , Jung, S.‐S. , Choi, J.‐H. , … Ahn, M.‐J. (2002). Differential gene expression in retinoic acid‐induced differentiation of acute promyelocytic leukemia cells, NB4 and HL‐60 cells. Biochemical and Biophysical Research Communications, 296(5), 1125–1133. 10.1016/S0006-291X(02)02043-0 [DOI] [PubMed] [Google Scholar]
- Liang, J. C. , Ning, Y. , Wang, R. Y. , Padilla‐Nash, H. M. , Schröck, E. , Soenksen, D. , … Ried, T. (1999). Spectral karyotypic study of the HL‐60 cell line: Detection of complex rearrangements involving chromosomes 5, 7, and 16 and delineation of critical region of deletion on 5q31.1. Cancer Genetics and Cytogenetics, 113(2), 105–109. 10.1016/S0165-4608(99)00030-8 [DOI] [PubMed] [Google Scholar]
- Love, M. I. , Huber, W. , & Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biology, 15(12), 1–21. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukinavičius, G. , Reymond, L. , D'Este, E. , Masharina, A. , Göttfert, F. , Ta, H. , … Johnsson, K. (2014). Fluorogenic probes for live‐cell imaging of the cytoskeleton. Nature Methods, 11(7), 731–733. 10.1038/nmeth.2972 [DOI] [PubMed] [Google Scholar]
- McGrath, J. L. , Osborn, E. A. , Tardy, Y. S. , Dewey, C. F. , & Hartwig, J. H. (2000). Regulation of the actin cycle in vivo by actin filament severing. Proceedings of the National Academy of Sciences, 97(12), 6532–6537. 10.1073/pnas.100023397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzner, C. , Mark, C. , Steinwachs, J. , Lautscham, L. , Stadler, F. , & Fabry, B. (2015). Superstatistical analysis and modelling of heterogeneous random walks. Nature Communications, 6(May), 1–8. 10.1038/ncomms8516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millius, A. , & Weiner, O. D. (2009). Chemotaxis in neutrophil‐like HL‐60 cells In Chemotaxis (Vol. 571, pp. 167–177). Totowa, NJ: Humana Press; 10.1007/978-1-60761-198-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miralles, F. , & Visa, N. (2006). Actin in transcription and transcription regulation. Current Opinion in Cell Biology, 18(3), 261–266. 10.1016/j.ceb.2006.04.009 [DOI] [PubMed] [Google Scholar]
- Murugesan, S. , Hong, J. , Yi, J. , Li, D. , Beach, J. R. , Shao, L. , … Hammer, J. A. (2016). Formin‐generated actomyosin arcs propel T cell receptor microcluster movement at the immune synapse. Journal of Cell Biology, 215(3), 383–399. 10.1083/jcb.201603080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasaki, A. , Kijima, T. S. , Yumoto, T. , Imaizumi, M. , Yamagishi, A. , Kim, H. , … Uyeda, Q. P. T. (2017). The position of the GFP tag on actin affects the filament formation in mammalian cells. Cell Structure and Function, 42(2), 131–140. 10.1247/csf.17016 [DOI] [PubMed] [Google Scholar]
- Ng, M. R. , Besser, A. , Danuser, G. , & Brugge, J. S. (2012). Substrate stiffness regulates cadherin‐dependent collective migration through myosin‐II contractility. Journal of Cell Biology, 199(3), 545–563. 10.1083/jcb.201207148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otey, C. A. , Kalnoski, M. H. , Lessard, J. L. , & Bulinski, J. C. (1986). Immunulocalization of the gamma isoform of nonmuscle actin in cultured cells. Journal of Cell Biology, 102(5), 1726–1737. 10.1083/jcb.102.5.1726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pincus, Z. , & Theriot, J. A. (2007). Comparison of quantitative methods for cell‐shape analysis. Journal of Microscopy, 227(2), 140–156. 10.1111/j.1365-2818.2007.01799.x [DOI] [PubMed] [Google Scholar]
- Pollard, T. D. (2001). Genomics, the cytoskeleton and motility. Nature, 409(6822), 842–843. 10.1038/35057029 [DOI] [PubMed] [Google Scholar]
- Rincón, E. , Rocha‐Gregg, B. L. , & Collins, S. R. (2018). A map of gene expression in neutrophil‐like cell lines. BMC Genomics, 19(1), 1–17. 10.1186/s12864-018-4957-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins, J. R. , Barth, A. I. , Marquis, H. , De Hostos, E. L. , Nelson, W. J. , & Theriot, J. A. (1999). Listeria monocytogenes exploits normal host cell processes to spread from cell to cell. Journal of Cell Biology, 146(6), 1333–1349. 10.1083/jcb.146.6.1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, B. , Haupt, A. , Tucker, A. , Grancharova, T. , Arakaki, J. , Fuqua, M. A. , … Gunawardane, R. N. (2017). Systematic gene tagging using CRISPR/Cas9 in human stem cells to illuminate cell organization. Molecular Biology of the Cell, 28(21), 2854–2874. 10.1091/mbc.E17-03-0209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg, S. , Stracher, A. , & Lucas, R. C. (1981). Isolation and characterization of actin and actin‐binding protein from human platelets. Journal of Cell Biology, 91(1), 201–211. 10.1016/0014-5793(81)80352-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos‐Beneit, A. M. , & Mollinedo, F. (2000). Expression of genes involved in initiation, regulation, and execution of apoptosis in human neutrophils and during neutrophil differentiation of HL‐60 cells. Journal of Leukocyte Biology, 67(5), 712–724. 10.1002/jlb.67.5.712 [DOI] [PubMed] [Google Scholar]
- Shi, Y. , Zhang, J. , Mullin, M. , Dong, B. , Alberts, A. S. , & Siminovitch, K. A. (2009). The mDial formin is required for neutrophil polarization, migration, and activation of the LARG/RhoA/ROCK signaling axis during chemotaxis. The Journal of Immunology, 182(6), 3837–3845. 10.4049/jimmunol.0803838 [DOI] [PubMed] [Google Scholar]
- Shiau, C. K. , Gu, D. L. , Chen, C. F. , Lin, C. H. , & Jou, Y. S. (2011). IGRhCellID: Integrated genomic resources of human cell lines for identification. Nucleic Acids Research, 39(SUPPL. 1), 520–524. 10.1093/nar/gkq1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellberg, B. J. , Collins, M. , French, S. W. , Edwards, J. E. , Fu, Y. , & Ibrahim, A. S. (2005). A phagocytic cell line markedly improves survival of infected neutropenic mice. Journal of Leukocyte Biology, 78(2), 338–344. 10.1189/jlb.0205072 [DOI] [PubMed] [Google Scholar]
- Tamayo, P. , Slonim, D. , Mesirov, J. , Zhu, Q. , Kitareewan, S. , Dmitrovsky, E. , … Golub, T. R. (1999). Interpreting patterns of gene expression with self‐organizing maps: Methods and application to hematopoietic differentiation. Proceedings of the National Academy of Sciences, 96(March), 2907–2912. 10.1073/pnas.96.6.2907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate, J. G. , Bamford, S. , Jubb, H. C. , Sondka, Z. , Beare, D. M. , Bindal, N. , … Forbes, S. A. (2019). COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Research, 47(D1), D941–D947. 10.1093/nar/gky1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, D. L. , & Wang, Y. L. (1978). Molecular cytochemistry: Incorporation of fluorescently labeled actin into living cells. Proceedings of the National Academy of Sciences, 75(2), 857–861. 10.1073/pnas.75.2.857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Gene Ontology Consortium . (2000). Gene ontology: Tool for the unification of biology. Nature Genetics, 25(1), 25–29. 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Gene Ontology Consortium . (2017). Expansion of the gene ontology knowledgebase and resources: The gene ontology consortium. Nucleic Acids Research, 45(D1), D331–D338. 10.1093/nar/gkw1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, T. Y. C. , Collins, S. R. , Chan, C. K. , Hadjitheodorou, A. , Lam, P. Y. , Lou, S. S. , … Theriot, J. A. (2019). Efficient front‐rear coupling in neutrophil chemotaxis by dynamic myosin II localization. Developmental Cell, 49(2), 189–205.e6. 10.1016/j.devcel.2019.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsugiyama, H. , Okimura, C. , Mizuno, T. , & Iwadate, Y. (2013). Electroporation of adherent cells with low sample volumes on a microscope stage. Journal of Experimental Biology, 216(19), 3591–3598. 10.1242/jeb.089870 [DOI] [PubMed] [Google Scholar]
- Vandekerckhove, J. , & Weber, K. (1978a). At least six different actins are expressed in a higher mammal: An analysis based on the amino acid sequence of the amino‐terminal tryptic peptide. Journal of Molecular Biology, 126(4), 783–802. 10.1016/0022-2836(78)90020-7 [DOI] [PubMed] [Google Scholar]
- Vandekerckhove, J. , & Weber, K. (1978b). Mammalian cytoplasmic actins are the products of at least two genes and differ in primary structure in at least 25 identified positions from skeletal muscle actins. Proceedings of the National Academy of Sciences, 75(3), 1106–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedula, P. , & Kashina, A. (2018). The makings of the “actin code”: regulation of actin's biological function at the amino acid and nucleotide level. Journal of Cell Science, 131(9), jcs215509 10.1242/jcs.215509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. L. (1985). Exchange of actin subunits at the leading edge of living fibroblasts: Possible role of treadmilling. Journal of Cell Biology, 101(2), 597–602. 10.1083/jcb.101.2.597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts, R. G. (1995). Role of gelsolin in the formation and organization of triton‐soluble F‐actin during myeloid differentiation of hl‐60 cells. Blood, 85(8), 2212–2221. [PubMed] [Google Scholar]
- Westphal, M. , Jungbluth, A. , Heidecker, M. , Mühlbauer, B. , Heizer, C. , Schwartz, J. M. , … Gerisch, G. (1997). Microfilament dynamics during cell movement and chemotaxis monitored using a GFP‐actin fusion protein. Current Biology, 7(3), 176–183. 10.1016/S0960-9822(97)70088-5 [DOI] [PubMed] [Google Scholar]
- Yam, P. T. , Wilson, C. A. , Ji, L. , Hebert, B. , Barnhart, E. L. , Dye, N. A. , … Theriot, J. A. (2007). Actin‐myosin network reorganization breaks symmetry at the cell rear to spontaneously initiate polarized cell motility. Journal of Cell Biology, 178(7), 1207–1221. 10.1083/jcb.200706012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, J. , Wu, X. S. , Crites, T. , & Hammer, J. A. (2012). Actin retrograde flow and actomyosin II arc contraction drive receptor cluster dynamics at the immunological synapse in Jurkat T cells. Molecular Biology of the Cell, 23(5), 834–852. 10.1091/mbc.e11-08-0731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino, D. R. , Achuthan, P. , Akanni, W. , Amode, M. R. , Barrell, D. , Bhai, J. , … Flicek, P. (2018). Ensembl 2018. Nucleic Acids Research, 46(D1), D754–D761. 10.1093/nar/gkx1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 Full images of the example Western blot from Figure 2. Full Western blots are displayed at a single imaging exposure for the reversible membrane total protein stain, along with a series of imaging exposure times for the β‐actin, GFP, and γ‐actin stains. All images were taken from a single blot stripped and re‐stained in the order displayed from left to right.
Supplementary Figure 2 PCR genotyping of the edited cell lines. (A) Schematic of the PCR‐based genotyping strategy. (B) Top: Agarose gel results for the PCR products, displayed alongside the DNA ladder. Bottom left: Magnification of ~800 bp bands. Bottom right: Quantification of band locations. Pixel intensities were summed over the long axis of each band. Peak intensity positions, shown as dashed lines, mark the centers of each band. The AG++ band runs at a lower position on the gel, indicating a smaller PCR product, consistent with a deletion of about 10–11 bp.
Supplementary Figure 3 GFP‐β‐actin in the cytokinetic ring. (A‐B) Live‐ and fixed‐cell imaging of cytokinesis in undifferentiated AG++ cells. Scale bar: 5 μm. (A) GFP epifluorescence time lapse montage of cytokinesis in AG++ cells plated on poly‐L‐lysine‐coated coverslips. Frames are displayed in 60 sec intervals. (B) Four example AG++ cells fixed and stained to image cell shape (phase contrast, gray), DNA (Hoechst, blue), labeled actin (GFP‐β‐actin, green), and F‐actin (phalloidin, red).
Supplementary Figure 4 Example of Bayesian persistence fitting on a track from a 1D microfluidic channel motility assay. (A) Track of a single cell plotted over time (B) Fitted persistence value plotted over time. Dashed line – persistence threshold used to stratify velocity measurements. (C) Fitted activity values plotted over time. (D) Heatmap of fitted persistence and activity values.
Supplementary Figure 5 Example of Bayesian persistence fitting on a track from a 2D under‐agarose motility assay. (A) Track of a single cell plotted over time (B) Fitted persistence value plotted over time. Dashed line – persistence threshold used to stratify velocity measurements. (C) Fitted activity values plotted over time. (D) Heatmap of fitted persistence and activity values.
Supplementary Figure 6 Example of Bayesian persistence fitting on a track from a 3D collagen motility assay. (A) Track of a single cell plotted over time (B) Fitted persistence value plotted over time. Dashed line – persistence threshold used to stratify velocity measurements. (C) Fitted activity values plotted over time. (D) Heatmap of fitted persistence and activity values.
Supplementary Table 1 Summary of 1D motility assay results
Supplementary Table 2: Summary of 2D motility assay results
Supplementary Table 3: Summary of 3D motility assay results
Supplementary Table 4: ANOVA results for persistent speed distributions in 1D motility assays
Supplementary Table 5: ANOVA results for persistence distributions in 1D motility assays
Supplementary Table 6: ANOVA results for persistent speed distributions in 2D motility assays
Supplementary Table 7: ANOVA results for persistence distributions in 2D motility assays
Supplementary Table 8: ANOVA results for persistent speed distributions in 3D motility assays
Supplementary Table 9: ANOVA results for persistence distributions in 3D motility assays
Supplementary Table 10: Number of significantly differentially regulated genes
Supplementary Table 11 Differentially regulated genes between AG+ and KW at 6 days post‐differentiation (.xlsx)
Supplementary Table 12 Differentially regulated actin‐related genes between AG+ and KW at 6 days post‐differentiation (.xlsx)
Supplementary Table 13 Differentially regulated genes between AG++ and KW at 6 days post‐differentiation (.xlsx)
Supplementary Table 14 Differentially regulated actin‐related genes between AG++ and KW at 6 days post‐differentiation (.xlsx)
Supplementary Table 15 Differentially regulated genes between AG++ and AG+ at 6 days post‐differentiation (.xlsx)
Supplementary Table 16 Differentially regulated actin‐related genes between AG++ and AG+ at 6 days post‐differentiation (.xlsx)
Video S1 Live cell phase‐contrast and epifluorescence imaging of AG++ cell migration under agarose
Video S2 Example 1D motility assay for KW cells
Video S3 Example 1D motility assay for AG+ cells
Video S4 Example 1D motility assay for AG++ cells
Video S5 Example 2D motility assay for KW cells
Video S6 Example 2D motility assay for AG+ cells
Video S7 Example 2D motility assay for AG++ cells
Video S8 Example 3D motility assay for KW cells
Video S9 Example 3D motility assay for AG+ cells
Video S10 Example 3D motility assay for AG++ cells
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.