

Abstract
A method for the catalytic generation of functionalized aryl alkali metals is reported. These highly reactive intermediates are liberated from silyl‐protected aryl‐substituted diazenes by the action of Lewis basic alkali metal silanolates, resulting in desilylation and loss of N2. Catalytic quantities of these Lewis bases initiate the transfer of the aryl nucleophile from the diazene to carbonyl and carboxyl compounds with superb functional‐group tolerance. The aryl alkali metal can be decorated with electrophilic substituents such as methoxycarbonyl or cyano as well as halogen groups. The synthesis of a previously unknown cyclophane‐like [4]arene macrocycle from a 1,3‐bisdiazene combined with a 1,4‐dialdehyde underlines the potential of the approach.
Keywords: autocatalysis, chemoselectivity, Lewis bases, nucleophilic addition, silicon
Under cover of nitrogen: Silyl‐protected aryl‐substituted diazenes act as masked aryl nucleophiles. Upon initiation with Me3SiOM (M=Li, Na, and K), aryl transfer to carbonyl and carboxyl groups occurs at ambient temperature. This mild nucleophile generation is compatible with electron‐withdrawing and electron‐donating functional groups.
Synthetic chemistry without aryl nucleophiles based on lithium, magnesium (Grignard), and zinc is difficult to imagine.1 The usual methods for their preparation include reductive metalation and halogen–metal exchange of aryl halides, and the resulting polar organometallic reagents can be interconverted by transmetalation. These procedures are not always chemoselective, and the high reactivity of the nucleophiles is often detrimental to their functional‐group tolerance. In particular, Knochel and co‐workers have provided viable solutions to these problems, thereby turning polyfunctionalized zinc and Grignard reagents into everyday chemicals.2, 3, 4
An alternative to these reactive compounds are easy‐to‐handle and storable less polarized aryl pronucleophiles based on silicon, mainly the trimethylsilyl derivatives.5 Aside from the fact that these are typically accessed from one of the aforementioned reagents, their fluoride‐ or alkoxide‐promoted activation for aryl transfer to aldehydes is only applicable to electron‐deficient aryl groups attached to the silicon atom;6 even the parent Ph‐SiMe3 does not react6e, 7 (Scheme 1, Eq. 1). This gap was closed with the more electrophilic Ph‐Si(OMe)3 and Bu4N+F− (TBAF) as the Lewis basic activator (Scheme 1, Eq. 2).8 To overcome this limitation, we considered the related activation of N‐aryl‐N′‐silyldiazenes (Ar‐N=N‐SiR3), which can be readily synthesized in two steps from aryl hydrazines with no need for aryl halides.9 We envisioned that Lewis base activation of Ar‐N=N‐SiR3 could unleash a reactive aryl nucleophile equivalent by desilylation and denitrogenation. This conceptual framework was formulated by Bottaro forty years ago (Scheme 1, Eq. 3), yet with no demonstration of its synthetic value and using NaOMe as an overstoichiometric activator.10 This seminal contribution has been largely overlooked and, hence, has not witnessed any further development. The present work shows how this approach can be turned into a catalytic process with excellent functional‐group tolerance with respect to both the diazene and the carbonyl compound, including transformations of a difunctional building block (Scheme 1, Eq. 4).
Scheme 1.
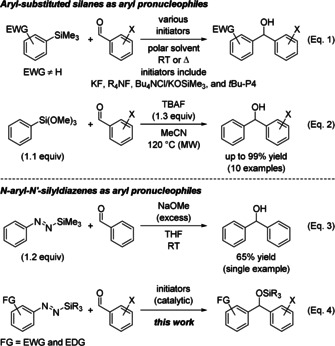
Silicon‐based aryl pronucleophiles in transition‐metal‐free 1,2‐addition to aldehydes (alcohols after hydrolysis). EWG=electron‐withdrawing group, EDG=electron‐donating group, FG=functional group, X=aryl substituent. MW=microwave irradiation, R=alkyl or aryl group, tBu‐P4=3‐tert‐butylimino‐1,1,1,5,5,5‐hexakis(dimethylamino)‐3‐{[tris(dimethylamino)phosphoranylidene]amino}‐1λ 5,3λ 5,5λ 5‐1,4‐triphosphazadiene (Schwesinger base), TBAF=tetrabutylammonium fluoride.
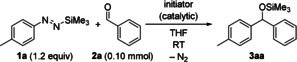
We began our investigation by testing various initiators in the reaction of 4‐tolyl‐substituted diazene 1 a and benzaldehyde (2 a) in THF (Table 1). Lithium salts such as the alkoxide tBuOLi [pK a (H2O)≈16.5] and the less basic silanolate Me3SiOLi11 [pK a (H2O)≈12.7] both initiated the reaction at room temperature, affording the silyl ether 3 aa in high yields within one hour (entries 1 and 2). With the same initiator loading of 10 mol %, improved reaction kinetics were achieved with the sodium and potassium salts of trimethylsilanol;11 full conversion was reached in less than five minutes, accompanied by vigorous evolution of N2 (entries 3 and 4). The same outcome was obtained with 5.0 mol % Me3SiONa but yields dwindled with 1.0 mol % Me3SiONa or Me3SiOK even at prolonged reactions times (entries 5–7). These results emphasize the influence of the alkali metal cation in this reaction. Polar co‐solvents such as N‐methylpyrrolidone (NMP) accelerated the already fast reactions. For completion, we included fluoride sources such as CsF and anhydrous TMAF into our screening. CsF did promote the aryl transfer but with a low reaction rate (entry 8), while essentially no conversion was seen with the poorly soluble ammonium fluoride (entry 9).
Table 1.
Selected examples from the optimization.
|
Entry |
Initiator |
mol % |
Time |
Yield [%][a] |
|---|---|---|---|---|
|
1 |
tBuOLi |
10 |
1 h |
91 |
|
2 |
Me3SiOLi |
10 |
1 h |
>95 |
|
3 |
Me3SiONa |
10 |
<5 min |
98 (70)[b] |
|
4 |
Me3SiOK |
10 |
<5 min |
90 |
|
5 |
Me3SiONa |
5 |
<5 min |
>95 |
|
6 |
Me3SiONa |
1 |
20 h |
6 |
|
7 |
Me3SiOK |
1 |
20 h |
58 |
|
8 |
CsF |
10 |
20 h |
60 |
|
9 |
TMAF |
10 |
20 h |
trace |
[a] Determined by calibrated GLC analysis with tetracosane as an internal standard. [b] Yield of isolated product on a 0.40 mmol scale after flash chromatography on silica gel in parentheses. TMAF=tetramethylammonium fluoride. 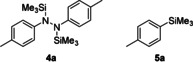
It is important to note that hydrazine 4 a was never detected by GLC analysis in those experiments. This would arise from the addition of the aryl nucleophile across the N=N double bond of the diazene followed by silylation.12 This result suggests that the aldehyde substrate outcompetes the diazene as the electrophile. The silylated arene 5 a, which formally arises from the silylation of the corresponding aryl anion, did usually form in trace amounts, likely because of the slight excess of the diazene reagent 1 a employed. In turn, compounds 4 a and 5 a were the major products of the alkoxide‐initiated degradation of 1 a in the absence of the aldehyde substrate (see the Supporting Information for details).
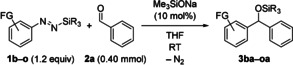
To demonstrate the scope of the new method, we continued with 10 mol % Me3SiONa in THF at room temperature as the standard procedure. Diazenes with silyl groups other than Me3Si were examined (1 b–d; Table 2, entries 1–3). It was only the Me2PhSi‐substituted derivative 1 d that behaved similarly to 1 a, affording the silyl ether 3 da in 86 % yield. Conversely, 1 b, which has a Et3Si group, and 1 c, which has a tBuMe2Si group, led to either low or no conversion of benzaldehyde. However, a little re‐optimization showed that Me3SiOK instead of Me3SiONa promotes aryl transfer from 1 b to benzaldehyde, and 3 ba was isolated in 76 % yield. The reaction of sterically more hindered 1 c required the addition of 18‐crown‐6, and the “more naked” silanolate and alkoxide intermediate enabled the formation of the silyl ether 3 ca in 68 % yield. We also prepared a wide range of Me3Si‐substituted diazenes with functionalized aryl groups (1 e–o; Table 2, entries 4–14). Without exception, these reacted in good yields under the standard setup. The successful reaction of electron‐rich 1 f to silyl ether 3 fa closes an important gap (cf. Scheme 1, top). Further notable examples include aryl transfers from diazenes 1 g, 1 h, and 1 n, which contain sensitive functional groups (CO2Me in 3 ga, CN in 3 ha, and NO2 in 3 na). Even the transfer of aryl nucleophiles containing a bromo or iodo group, as in 1 k and 1 l (competing halogen–metal exchange), or a fluorine substituent in the ortho position, as in 1 m (competing β‐elimination/aryne formation), proceeded in high yields. These examples highlight the chemoselectivity of the method and its orthogonality with classical carbonyl arylations. The productive combination of these diazenes and a broad range of aromatic, heteroaromatic, and cinnamic aldehydes (2 b–i) is further evidence of this (Figure 1). Enolizable aldehydes were not compatible but α‐branched 2‐ethylbutyraldehyde (2 j) yielded the silyl ether 3 aj in 70 % yield along with the silyl enol ether in 30 % yield. Other aliphatic aldehydes 2 k and 2 l reacted with high chemoselectivity to furnish 3 ak and 3 fl in good yields (Figure 1).
Table 2.
Scope I: Variation of the silyl and aryl groups of the diazene.
|
Entry |
Diazene |
SiR3 |
FG |
Silyl ether |
Yield [%] |
|---|---|---|---|---|---|
|
1[a] |
1 b |
SiEt3 |
4‐Me |
3 ba |
76 |
|
2[b] |
1 c |
SitBuMe2 |
4‐Me |
3 ca |
68 |
|
3 |
1 d |
SiMe2Ph |
4‐Me |
3 da |
86 |
|
4 |
1 e |
SiMe3 |
H |
3 ea |
72 |
|
5 |
1 f |
SiMe3 |
4‐OMe |
3 fa |
82 |
|
6[a] |
1 g |
SiMe3 |
4‐CO2Me |
3 ga |
80 |
|
7 |
1 h |
SiMe3 |
4‐CN |
3 ha |
72 |
|
8 |
1 i |
SiMe3 |
4‐F |
3 ia |
99 |
|
9 |
1 j |
SiMe3 |
4‐Cl |
3 ja |
81 |
|
10 |
1 k |
SiMe3 |
4‐Br |
3 ka |
87 |
|
11 |
1 l |
SiMe3 |
4‐I |
3 la |
83 |
|
12 |
1 m |
SiMe3 |
2‐F |
3 ma |
65 |
|
13[a] |
1 n |
SiMe3 |
3‐NO2 |
3 na |
67 |
|
14 |
1 o |
SiMe3 |
2,5‐Me2, 4‐F |
3 oa |
73 |
[a] Me3SiOK instead of Me3SiONa. [b] Me3SiOK/18‐crown‐6 (1.0:1.2 molar ratio) instead of Me3SiONa.
Figure 1.
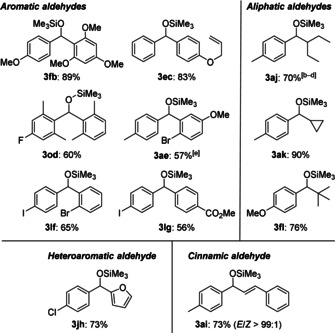
Scope II: Various diazene/aldehyde combinations.[a] [a] Unless noted otherwise, the reactions were performed on a 0.40 mmol scale with 10 mol % Me3SiONa in THF at room temperature. [b] Me3SiOK instead of Me3SiONa. [c] Formed along with the corresponding silyl enol ether in 30 % yield. [d] Yield determined by NMR spectroscopy using CH2Br2 as an internal standard. [e] The reaction was performed on a 1.8 mmol scale with 5.0 mol % Me3SiONa in THF at room temperature.
Less electrophilic ketones were also competent substrates but, as in the case of enolizable acetophenone, deprotonation was the predominant pathway to afford the corresponding silyl enol ether in 60 % yield (not shown, see the Supporting Information). Conversely, 6 a–d reacted in the planned way with Me3SiOK as the initiator (Scheme 2, top). No reaction or low conversion were observed with Me3SiOLi and Me3SiONa, presumably because of the low reactivity of the intermediate tertiary alkoxide, and hence its inability to maintain catalytic turnover. Unlike the 1,2‐selective aryl transfer to trans‐cinnamaldehyde (2 i→3 ai, Figure 1), the reaction of the aryl nucleophile with trans‐chalcone (6 c) led to the formation of both the 1,2‐adduct (7 ac, 65 %) and the 1,4‐adduct (6 %). Moreover, modification of the reaction setup with a higher loading of Me3SiOK (20 mol %) and slow addition of the diazene (3 equiv) to a solution of a methyl benzoates 8 and THF even enabled two‐fold aryl transfer to give the tertiary silyl ethers 9 in reasonable yields (Scheme 2, bottom). To the best of our knowledge, this is an unprecedented catalytic arylation of unactivated carboxylic acid derivatives with non‐stabilized carbanion equivalents.13 We note here that the occurrence of this nucleophilic addition is also diagnostic of the in situ formation of highly reactive aryl anions.14
Scheme 2.
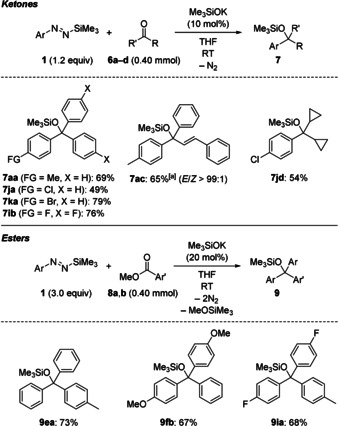
Scope III: Ketones and esters as electrophiles. [a] Formed along with the corresponding 1,4‐adduct in 6 % yield.
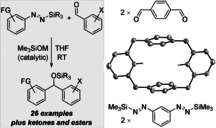
Our next plan was to explore whether the diazene platform would also enable reactions of aryl dinucleophiles.15 For this, we synthesized the 1,3‐bismetalated benzene equivalent 1 p from 1,3‐diaminobenzene in 26 % yield over three steps (see the Supporting Information for details and crystallographic characterization16). The bisdiazene 1 p is a storable deep‐blue crystalline solid with decent thermal stability (up to 130 °C). The reaction of 1 p and benzaldehyde (2 a, 1.4 equiv) in the presence of 20 mol % Me3SiOK afforded diol 10 pa in 65 % yield after deprotection with TBAF (Scheme 3, left).
Scheme 3.
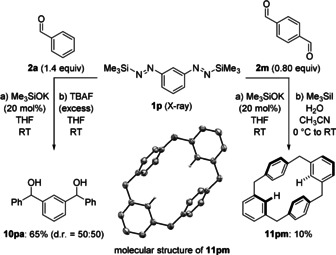
Scope IV: Equivalent of an aryl bisnucleophile in the reaction with an aldehyde (left) or a dialdehyde (right); molecular structure of a cyclophane‐like [4]arene macrocycle (middle, thermal ellipsoids are shown at the 50 % probability level and all hydrogen atoms except the two pointing toward the center are omitted for clarity).
The fact that 10 pa had been employed as a precursor of porphinoid macrocycles17 inspired us to make use of building block 1 p in the practical assembly of otherwise difficult‐to‐prepare macrocycles. The idea was to combine 1,3‐difunctional 1 p and terephthalaldehyde (2 m) with its 1,4‐substitution pattern, hoping that the alternate 1,3/1,4 motifs of the rings would result in cyclization rather than polymerization to poly(diarylcarbinols). The reaction of 1 p and 0.80 equiv 2 m initiated with 20 mol % Me3SiOK led to a complex product mixture of polymeric and oligomeric material, from which a tetrameric macrocyclic compound could be identified by HRMS analysis.18 Defunctionalization of the crude residue, that is removal of the silyl ethers by a reported procedure,19 considerably simplified the analysis and allowed the isolation of the unknown cyclophane‐like [4]arene macrocycle 11 pm in 10 % yield over two steps (Scheme 3, right).20 This seemingly low yield compares well with others from difficult macrocyclizations involving aromatic precursors lacking preorganization, especially in relatively high concentration (0.1 m).21 The molecular structure of 11 pm was confirmed by X‐ray diffraction16 (Scheme 3, middle) and shows a preference for a chair‐like conformation in the solid state, whereas a boat‐like conformation was computed to be more stable by approximately 0.5 kcal mol−1 (see the Supporting Information for details). Compound 11 pm is the simplest unfunctionalized member of an emerging class of hybrid macrocycles22 that currently comprises just two derivatives.23
Although an in‐depth mechanistic analysis is still pending, we propose the autocatalytic cycle outlined in Scheme 4. After initiation with either of the three trimethylsilanolate alkali salts, the aryl alkali metal will form as a result of desilylation and loss of N2. The in situ formed aryl nucleophile then adds to the carbonyl compound, forming an alkali metal alkoxide that will in turn engage in the desired degradation of the diazene reagent. This step then propagates the catalytic cycle.
Scheme 4.
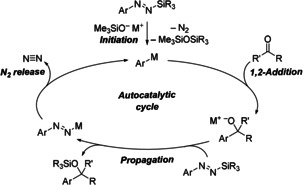
Proposed autocatalytic cycle. M=alkali metal, R and R′=alkyl or aryl, Ar=aryl.
To summarize, we have shown that N‐aryl‐N′‐silyldiazenes constitute a versatile platform from which various highly reactive and, at the same time, functionalized aryl nucleophiles can be released at ambient temperature. The reaction of a related bisdiazene illustrates the potential of the method to formally generate dinucleophiles. These reactive intermediates can be trapped in situ with functionalized carbonyl and carboxyl compounds. Conceptually, these arylation reactions are similar to a Barbier‐like setup,24 where the polar organometallics are generated in situ, thereby avoiding their delicate preparation and handling. However, our method makes use of Me3SiOM (with M=Li, Na, and K)11 species as initiators, whereas Barbier reactions typically rely on overstoichiometric amounts of reducing metals. Aside from its excellent functional‐group tolerance, this new method differs from established ones in that it is halide‐free, starting from aryl hydrazines rather than aryl halides.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
C.C. gratefully acknowledges the Alexander von Humboldt Foundation for a postdoctoral fellowship (2018–2019). M.O. is indebted to the Einstein Foundation Berlin for an endowed professorship.
C. Chauvier, L. Finck, E. Irran, M. Oestreich, Angew. Chem. Int. Ed. 2020, 59, 12337.
In memory of Rolf Huisgen and dedicated to Professor Reinhard Brückner on the occasion of his 65th birthday
Contributor Information
Dr. Clément Chauvier, http://www.organometallics.tu‐berlin.de.
Prof. Dr. Martin Oestreich, Email: martin.oestreich@tu-berlin.de.
References
- 1. Organometallics in Synthesis: Third Manual (Ed.: M. Schlosser), Wiley, Hoboken, 2013. [Google Scholar]
- 2.
- 2a. Benischke A. D., Ellwart M., Becker M. R., Knochel P., Synthesis 2016, 48, 1101–1107; [Google Scholar]
- 2b. Dagousset G., François C., León T., Blanc R., Sansiaume-Dagousset E., Knochel P., Synthesis 2014, 46, 3133–3171; [Google Scholar]
- 2c. Knochel P., Dohle W., Gommermann N., Kneisel F. F., Kopp F., Korn T., Sapountzis I., Vu V. A., Angew. Chem. Int. Ed. 2003, 42, 4302–4320; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 4438–4456. [Google Scholar]
- 3. Parham W. E., Bradsher C. K., Acc. Chem. Res. 1982, 15, 300–305. [Google Scholar]
- 4.Yoshida and co-workers introduced flow technology as an alternative to batch reactions:
- 4a. Nagaki A., Tetrahedron Lett. 2019, 60, 150923; [Google Scholar]
- 4b. Nagaki A., Yoshida J.-i., Top. Organomet. Chem. 2016, 57, 137–175. [Google Scholar]
- 5. Reich H. J. in Lewis Base Catalysis in Organic Synthesis (Eds.: E. Vedejs, S. E. Denmark), Wiley-VCH, Weinheim, 2016, pp. 233–280. [Google Scholar]
- 6.
- 6a. Ishikawa N., Isobe K.-i., Chem. Lett. 1972, 435–436; [Google Scholar]
- 6b. Effenberger F., Spiegler W., Angew. Chem. Int. Ed. Engl. 1981, 20, 265–266; [Google Scholar]; Angew. Chem. 1981, 93, 287–288; [Google Scholar]
- 6c. Effenberger F., Spiegler W., Chem. Ber. 1985, 118, 3872–3899; [Google Scholar]
- 6d. Effenberger F., Spiegler W., Chem. Ber. 1985, 118, 3900–3914; [Google Scholar]
- 6e. Ueno M., Hori C., Suzawa K., Ebisawa M., Kondo Y., Eur. J. Org. Chem. 2005, 1965–1968; [Google Scholar]
- 6f. Suzawa K., Ueno M., Wheatley A. E. H., Kondo Y., Chem. Commun. 2006, 4850–4852; [DOI] [PubMed] [Google Scholar]
- 6g. Das M., O‘Shea D. F., J. Org. Chem. 2014, 79, 5595–5607. [DOI] [PubMed] [Google Scholar]
- 7. Pilcher A. S., DeShong P., J. Org. Chem. 1996, 61, 6901–6905. [DOI] [PubMed] [Google Scholar]
- 8. Lerebours R., Wolf C., J. Am. Chem. Soc. 2006, 128, 13052–13053. [DOI] [PubMed] [Google Scholar]
- 9. Chauvier C., Finck L., Hecht S., Oestreich M., Organometallics 2019, 38, 4679–4686. [Google Scholar]
- 10. Bottaro J. C., J. Chem. Soc. Chem. Commun. 1978, 990. [Google Scholar]
- 11.For a review of trimethylsilanolate alkali salts, see: Bürglová K., Haláč J., Synthesis 2018, 50, 1199–1208. [Google Scholar]
- 12. Katritzky A. R., Wu J., Verin S. V., Synthesis 1995, 651–653. [Google Scholar]
- 13.For the addition of stabilized heteroaryl anions to lactones, see: Ricci A., Fiorenza M., Grifagni M. A., Bartolini G., Tetrahedron Lett. 1982, 23, 5079–5082. [Google Scholar]
- 14. Murphy J. A., Zhou S.-z., Thomson D. W., Schoenebeck F., Mahesh M., Park S. R., Tuttle T., Berlouis L. E. A., Angew. Chem. Int. Ed. 2007, 46, 5178–5183; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 5270–5275. [Google Scholar]
- 15.
- 15a. Fossatelli M., den Besten R., Verkruijsse H. D., Brandsma L., Recl. Trav. Chim. Pays-Bas 1994, 113, 527–528; [Google Scholar]
- 15b. Bickelhaupt F., Angew. Chem. Int. Ed. Engl. 1987, 26, 990–1005; [Google Scholar]; Angew. Chem. 1987, 99, 1020–1035. [Google Scholar]
- 16.CCDC 1971069 (1p) and 1971057 (11pm) contain the supplementary crystallographic data for this paper. These data can be obained free of charge from The Cambridge Crystallographic Data Centre.
- 17. Stępień M., Latos-Grażyński L., Chem. Eur. J. 2001, 7, 5113–5117. [DOI] [PubMed] [Google Scholar]
- 18.Larger macrocycles such as the [6]arene derivative were also observed by HRMS but could not be isolated nor characterized after defunctionalization.
- 19. Stoner E. J., Cothron D. A., Balmer M. K., Roden B. A., Tetrahedron 1995, 51, 11043–11062. [Google Scholar]
- 20.For the synthesis of parent [1,1,1,1]metacyclophane, see:
- 20a. McMurry J. E., Phelan J. C., Tetrahedron Lett. 1991, 32, 5655–5658; for the synthesis of parent [1,1,1,1]paracyclophane, see: [Google Scholar]
- 20b. Miyahara Y., Inazu T., Yoshino T., Tetrahedron Lett. 1983, 24, 5277–5280. [Google Scholar]
- 21.Examples are provided in the following review: Martí-Centelles V., Pandey M. D., Burguete M. I., Luis S. V., Chem. Rev. 2015, 115, 8736–8834. [DOI] [PubMed] [Google Scholar]
- 22.An example of an application of a hybrid macrocycle: Fowler D. A., Rathnayake A. S., Kennedy S., Kumari H., Beavers C. M., Teat S. J., Atwood J. L., J. Am. Chem. Soc. 2013, 135, 12184–12187. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Boinski T., Cieszkowski A., Rosa B., Szumna A., J. Org. Chem. 2015, 80, 3488–3495; for a brief discussion of so-called hybrid[n]arenes, see: [DOI] [PubMed] [Google Scholar]
- 23b. Wu J.-R., Yang Y.-W., Chem. Commun. 2019, 55, 1533–1534. [DOI] [PubMed] [Google Scholar]
- 24. Barbier P., C. R. Chim. 1899, 128, 110. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary