

Abstract
The human 2‐oxoglutarate (2OG)‐dependent oxygenase aspartate/asparagine‐β‐hydroxylase (AspH) is a potential medicinal chemistry target for anticancer therapy. AspH is present on the cell surface of invasive cancer cells and accepts epidermal growth factor‐like domain (EGFD) substrates with a noncanonical (i. e., Cys 1–2, 3–4, 5–6) disulfide pattern. We report a concise synthesis of C‐3‐substituted derivatives of pyridine‐2,4‐dicarboxylic acid (2,4‐PDCA) as 2OG competitors for use in SAR studies on AspH inhibition. AspH inhibition was assayed by using a mass spectrometry‐based assay with a stable thioether analogue of a natural EGFD AspH substrate. Certain C‐3‐substituted 2,4‐PDCA derivatives were potent AspH inhibitors, manifesting selectivity over some, but not all, other tested human 2OG oxygenases. The results raise questions about the use of pyridine‐carboxylate‐related 2OG analogues as selective functional probes for specific 2OG oxygenases, and should aid in the development of AspH inhibitors suitable for in vivo use.
Keywords: demethylases; hydroxylase inhibitors; aspartate/asparagine-beta-hydroxylase; oxygenases; pyridine-2,4-dicarboxylic acid
Sights on a new target: A concise synthesis of C‐3‐substituted pyridine‐2,4‐dicarboxylic acid (2,4‐PDCA) derivatives is reported. The derivatives are potent inhibitors of the human 2‐oxoglutarate‐dependent oxygenase aspartate/asparagine‐β‐hydroxylase (AspH), which is a potential medicinal chemistry target for cancer therapy. This work constitutes a step towards the development of AspH inhibitors suitable for in vivo use.
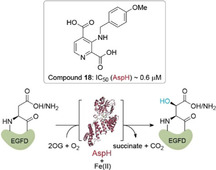
Introduction
Following the pioneering identification of the procollagen prolyl‐residue hydroxylases (CPHs) as FeII and 2‐oxoglutarate (2OG)‐dependent oxygenases,1 related enzymes, which play important roles in human biology, have emerged; some of these are validated medicinal chemistry targets.2 Human 2OG oxygenases have roles in lipid metabolism,3 processing proteins destined for secretion,1, 2 histone/chromatin modifications,4 DNA/RNA damage repair,5 and hypoxia sensing.6
Inhibition of the CPHs was pursued for the treatment of fibrotic diseases, but was suspended due to toxicity issues.7 The CPH inhibition work pioneered the use of 2OG analogues/competitors such as pyridine‐2,4‐dicarboxylic acid (2,4‐PDCA or 2,4‐lutidinic acid, 1; Figure 1)8 and N‐oxalylglycine (NOG, 2, Figure 1).9 This approach has been successfully applied to the development of the 2OG‐dependent hypoxia‐inducible transcription factor‐α (HIF‐α) prolyl‐residue hydroxylases (PHDs) inhibitors,10 some of which are approved for use in the treatment of anemia in patients suffering from chronic kidney disease (e. g., roxadustat 3,11 Figure 1). With the notable exception of mildronate (meldonium),12 an inhibitor of γ‐butyrobetaine dioxygenase (BBOX),13 most 2OG oxygenase inhibitors are active‐site FeII ligands/2OG competitors.10 Examples include pyridine‐carboxylate derivative QC‐6352 (4, Figure 1), which inhibits the Jmjc lysine‐specific demethylase 4 (KDM4, JMJD2) enzymes and which shows anti‐proliferative effects in cancer models.14
Figure 1.
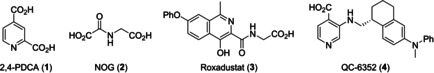
Structures of the broad‐spectrum 2OG oxygenase inhibitors 2,4‐PDCA (1) and NOG (2) and the PHD selective inhibitor roxadustat (3)11a and the KDM4‐selective inhibitor QC‐6352 (4).14
The human 2OG oxygenase aspartate/asparagine‐β‐hydroxylase (AspH, BAH, HAAH) catalyses the hydroxylation of conserved Asp and Asn residues in epidermal growth factor‐like domains (EGFDs),15 which include the extracellular domains of the notch receptor and its ligands.16 AspH levels are upregulated in multiple cancers (e. g., hepatocellular carcinoma17 and pancreatic cancer18). AspH is reported to be translocalised from the endoplasmic reticulum (ER) to the cell surface membrane resulting in enhanced tumour invasiveness and a diminished patient survival rate.17b, 19 AspH is also strongly hypoxically regulated20 and may have a role in hypoxia sensing.21 AspH is thus an interesting potential target from a cancer medicinal chemistry perspective.22
Only a few structure‐activity relationship (SAR) studies directed at identifying AspH inhibitors have been reported. Initial studies identified several nonselective small‐molecule AspH inhibitors in cell‐based experiments25 and l‐ascorbate based AspH inhibitors have been used in cellular and animal experiments.22a, 26 Studies focusing on the development of nitrogen‐containing heteroaromatic scaffolds for AspH inhibition have not been reported despite the prominence of these scaffolds in approved active pharmaceutical ingredients (APIs), including anticancer drugs and 2OG oxygenase inhibitors.27 The discrepancy between the potential of a AspH as a medicinal chemistry target and the limited effort of developing efficient small‐molecule AspH inhibitors for clinical use likely relates to the lack of efficient assays for isolated AspH.
Recently, we reported crystal structures of recombinant human AspH using truncated constructs.23 Together with biochemical studies, these reveal that AspH accepts EGFDs with a noncanonical (Cys 1–2, 3–4, 5–6; Figure 2C) disulfide pattern, rather than the well‐characterised canonical (Cys 1–3, 2–4, 5–6; Figure 2B) disulfide pattern.23 These findings prompted us to develop a semi‐automated high‐throughput assay using solid phase extraction (SPE) coupled to mass spectrometry (MS) and stable disulfide analogues (e. g., hFX‐CP101‐119; Figure 2D) to monitor AspH activity.24
Figure 2.
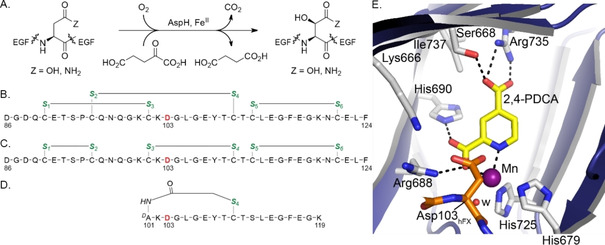
AspH catalyses the hydroxylation of the Asp/Asn‐residues of specific EGFD disulfide isomers. A) Scheme for the AspH‐catalysed hydroxylation of Asp/Asn‐residues in EGFDs. B) The canonical (Cys 1–3, 2–4, 5–6) EGFD1 disulfide pattern of human coagulation factor X (hFX amino acids 86–124); the AspH hydroxylation site (Asp103hFX) is in red, cystine thiols in green. C) The noncanonical (Cys 1–2, 3–4, 5–6) hFX EGFD1 disulfide isomer. D) A cyclic thioether peptide (hFX‐CP101‐119) mimicking the central noncanonical (Cys 3–4) hFX EGFD1 disulfide fold containing the AspH hydroxylation site Asp103hFX (red).23 E) Close‐up of the AspH active site in the AspH:2,4‐PDCA crystal structure (PDB ID: 5JTC).24 The key residues engaged in 2,4‐PDCA (Ser668, Arg688, His690, and Arg735) and metal binding (His679, His725) are shown. A pocket formed by AspH residues Ser668, Ile737, His690, Arg688, and Lys666 adjacent to the C‐3 position of 2,4‐PDCA is sufficiently large to accommodate substituents at the C‐3 position of 2,4‐PDCA. Colours: violet/grey: His6‐AspH315‐758; yellow: carbon‐backbone of 2,4‐PDCA; orange: carbon‐backbone of a synthetic hFX EGFD1‐derived peptide; purple: Mn; red: oxygen; blue: nitrogen. w: water.
Here, we report SAR studies on nitrogen‐containing heteroaromatic small‐molecule AspH inhibitors. Novel C‐3‐substituted derivatives of 2,4‐PDCA (1), which is a potent, but non‐selective AspH inhibitor competing with 2OG for 2OG binding in the AspH active site (Figure 2E),24, 25 were synthesised and their inhibitory properties investigated using SPE‐MS inhibition assays against AspH and other human 2OG oxygenases. The results reveal the potential for potent and selective AspH inhibition, and raise questions regarding the selectivity of PDCA‐type and related 2OG oxygenase inhibitors that have been reported in the literature.
Results and Discussion
Inhibition of AspH by isomers and derivatives of 2,4‐PDCA
The mechanism by which 2,4‐PDCA (1) inhibits AspH involves competition with 2OG for binding at the AspH active site.24 2,4‐PDCA (1) coordinates to the active site FeII through bidentate chelation with its nitrogen atom and an oxygen atom of the C‐2 carboxylate as evidenced by crystallography (PDB ID: 5JTC; Figure 2E).24 Isomers of 2,4‐PDCA lacking the C‐2 carboxylate do not inhibit AspH.25a For SAR studies, we therefore fixed the C‐2 2,4‐PDCA carboxylate group whilst varying the position of the other carboxylate. Half‐maximum inhibitory concentrations (IC50) of three 2,4‐PDCA regioisomers (5‐7; Figure 3) were determined using SPE‐MS high‐throughput in vitro AspH inhibition assays conducted at 2OG, substrate, and FeII concentrations close to their K m values.21
Figure 3.
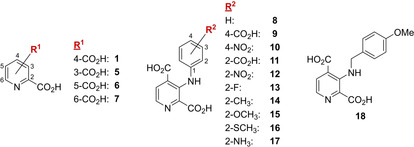
Structures of 2,x‐PDCAs (x=3‐6; 1 and 5–7) and C‐3‐substituted 2,4‐PDCA derivatives (8–18) initially screened for AspH inhibition.
Efficient inhibition of AspH by 2,3‐PDCA (quinolinic acid, 5), a natural product of the kynurenine pathway of tryptophan metabolism,28 was observed (IC50 ∼0.5 μM, Table 1), whereas 2,5‐PDCA (6) did not inhibit AspH (Table 1). However, the potency of 2,3‐PDCA (5) was lower than that of 2,4‐PDCA (1) by an order of magnitude (Table 1). Apparently, inverse behaviour with inhibitor potency has been observed in inhibition studies with KDM4E, which is inhibited by 2,5‐PDCA (6), but not by 2,3‐PDCA (5).29 This observation indicates that different PDCA regioisomers might selectively inhibit different subclasses of the human 2OG oxygenases. This proposal is further supported by the findings that 2,5‐PDCA (6) inhibits the CPHs with greater potency than 2,4‐PDCA (1), whereas 2,5‐PDCA (6) is significantly less potent than 2,4‐PDCA (1) in inhibiting the PHDs.8, 30
Table 1.
The Inhibition of human AspH by regioisomers and C‐3 derivatives of 2,4‐PDCA.
|
|
Compound |
IC50 [μM][a] |
|---|---|---|
|
1 |
2,4‐PDCA (1) |
0.03 ±0.01 |
|
2 |
2,3‐PDCA (5) |
0.51 ±0.05 |
|
3 |
2,5‐PDCA (6) |
inactive |
|
4 |
2,6‐PDCA (7) |
5.48 ±0.75 |
|
5 |
8 |
38.8 ±7.0 |
|
6 |
9 |
inactive |
|
7 |
10 |
inactive |
|
8 |
11 |
10.4 ±1.8 |
|
9 |
12 |
16.1 ±4.0 |
|
10 |
13 |
22.1 ±1.5 |
|
11 |
14 |
12.9 ±1.5 |
|
12 |
15 |
13.8 ±0.2 |
|
13 |
16 |
16.1 ±2.5 |
|
14 |
17 |
4.67 ±0.29 |
|
15 |
18 |
0.58 ±0.06 |
[a] Mean average of two independent runs (n=2; mean ± standard deviation, SD). AspH inhibition assays were performed as described in the Experimental Section using 50 nM His6‐AspH315‐758 and 1.0 μM hFX‐CP101‐119 (Figure 2D) as a substrate.
Moderate inhibition of AspH activity by 2,6‐PDCA (7) was observed (IC50 ∼5.5 μM, Table 1). Previous studies have indicated that 2,6‐PDCA (7) can inhibit 2OG oxygenases by (potential) active‐site FeII binding as well as by chelating FeII in solution, thus lowering available iron concentrations.8, 29
Although 2,4‐PDCA (1) is more potent than its regioisomers 5–7, it lacks selectivity for AspH over other human 2OG oxygenases, in particular the Jmjc KDMs.10 SAR studies were therefore initiated to explore the selectivity of 2,4‐PDCA derivatives. Based on the crystal structure of 2,4‐PDCA (1) in complex with AspH (PDB ID: 5JTC),24 we proposed that introduction of substituents at the C‐3 position of 2,4‐PDCA might increase inhibitor selectivity (Figure 2E). As we had previously synthesised C‐3‐substituted 2,4‐PDCA derivatives 8–18 (Figure 3) as KDM4E (JMJD2E) inhibitors,31 their potential inhibition of AspH was investigated (Table 1). The inhibition of AspH by 2,4‐PDCA bearing an aniline substituent at C‐3 (8) was >1000 fold less than the parent 2,4‐PDCA 1 (IC50∼38.8 μM, Table 1). Introducing electron‐deficient substituents at the aniline para position of compound 8 resulted in complete loss of activity (9, 10), while electron‐deficient substituents at the aniline ortho position manifested moderate potency (11–13). Electron‐donating substituents at the aniline ortho position (14–17) did not improve potency relative to 8; only in the case of pyridine 17 was a notable improvement observed (IC50∼4.7 μM, Table 1).
The most pronounced effect on inhibitor potency was observed when the aniline substituent at the C‐3 position of 2,4‐PDCA was changed to 4‐methoxybenzylamine: Compound 18 inhibited AspH efficiently, its IC50 value was only 20 fold above that of 2,4‐PDCA 1 (IC50∼0.6 μM, Table 1). The significantly higher potency of 18 compared to 2,4‐PDCA derivatives 8–17 may relate to the increased rotational freedom enabled by the additional methylene‐unit in 18.
Notably, 2,4‐PDCA (1) is a potent KDM4E inhibitor (IC50=0.44 μM), however, the 2,4‐PDCA derivative 18 is only a weak reported KDM4E inhibitor (IC50=41 μM),31 indicating that substituents at the C‐3 position of 2,4‐PDCA might enable selective AspH inhibition.
An improved synthesis of C‐3 aminoalkyl‐substituted 2,4‐PDCA derivatives
Having identified pyridine 18 as a potent and selective (with respect to KDM4E) AspH inhibitor, we aimed to synthesise other C‐3 aminoalkyl‐substituted 2,4‐PDCA derivatives to generate further SAR data and to investigate the selectivity of the inhibitor series with respect to other human 2OG oxygenases. However, the reported synthesis of C‐3‐substituted 2,4‐PDCA derivatives employs harsh reaction conditions and non‐selective reactions as manifested by low overall yields (Scheme 1A).31 Moreover, the HCl adduct of pyridine 18 slowly degraded over time, even when stored at −20 °C.
Scheme 1.
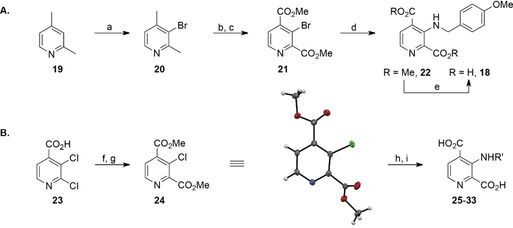
A) The reported synthesis of C‐3 aminoalkyl‐substituted 2,4‐PDCA derivative 18 31 compared with B) the new route to C‐3 aminoalkyl‐substituted 2,4‐PDCA derivatives. a) Br2, 20 % oleum, 165 °C, 35 %; b) KMnO4, NaOH, H2O, reflux; c) H2SO4, MeOH, reflux, 45 % (2 steps); d) amine (1.2 equiv), Pd2(dba)3 (2 mol%), Xantphos (6 mol%), Cs2CO3, toluene, 110 °C, 69 %; e) NaOH, MeOH/H2O, then: HCl, 96 %; f) SOCl2, MeOH, reflux, 89 %; g) CO (1.5 atm), Cl2Pd‐rac‐BINAP (1 mol%), iPr2NEt, MeOH, 100 °C (sand bath temperature, sealed flask), 90 %; h) alkylamine (H2NR′, 1.5 equiv), Pd(OAc)2 (4 mol%), Xantphos (6 mol%), pyridine, toluene, 190 °C (sand bath temperature, sealed flask), 32–71 %; i) LiOH, MeOH/H2O, 0 °C to RT, then: Dowex® 50XW8, 61–88 %. Crystal structure colour code: grey: carbons; white: hydrogens; blue: nitrogen; red: oxygens; green: chlorine.
An optimised route for synthesis of C‐3 aminoalkyl‐substituted 2,4‐PDCA derivatives was developed which shortens the reported synthesis by one step and overcomes the described shortcomings (Scheme 1B). Thus, commercial 2,3‐dichloroisonicotinic acid (23) was converted into its methyl ester, which was then submitted to regioselective Pd‐catalysed carbonylation32 yielding dimethyl ester 24 (90 %, over two steps).
1‐ and 2‐dimensional NMR analysis of the crude reaction product 24 indicated full substrate conversion with exclusive formation of the desired regioisomer in the carbonylation reaction as confirmed by single‐crystal X‐ray diffraction analysis of purified 24.
Buchwald‐Hartwig amination33 of dimethyl ester 24 with alkylamines required optimisation due to the reduced reactivity of its C−Cl bond compared to the C−Br bond of dimethyl ester 21 (Scheme 1). Undesired reaction pathways (ester saponification and amide bond formation) occurring instead of Buchwald‐Hartwig amination under the reported conditions31 were circumvented by replacing the inorganic base Cs2CO3 with pyridine. Conversion was further improved by substituting the palladium precatalyst Pd2(dba)3 with Pd(OAc)2, retaining Xantphos34 as ligand. Both linear and α‐branched alkylamines were successfully applied in the amination reactions whereas α,α‐disubstituted alkylamines did not react, presumably due to steric hindrance. The use of alkylamines containing heteroaromatic moieties (e. g., thiophene, furan) was unsuccessful. Crystallographic analysis confirmed the anticipated regioselectivity of the Buchwald‐Hartwig reaction (Figure S1 and Table S1 in the Supporting Information).
The intermediate dimethyl esters were subjected to lithium hydroxide‐mediated saponification to afford the desired C‐3 aminoalkyl‐substituted 2,4‐PDCA derivatives 18 and 25–33 (Figure 4) in good yields; excess base was removed by acidic ion exchange chromatography to yield the salt‐free inhibitors which were stable as solids and in aqueous solution (see the Experimental Section).
Figure 4.
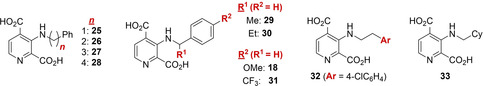
Structures of C‐3 aminoalkyl‐substituted 2,4‐PDCA derivatives 18 and 25–33 (pyridines 29 and 30 were prepared as racemic mixtures).
SAR studies on the inhibition of AspH by C‐3 aminoalkyl‐substituted 2,4‐PDCA derivatives
The SAR studies reveal that increasing the length of the C‐3 aminoalkyl chain from methylene to butylene has a detrimental effect on potency (25–28, Figure 5 and Table 2). Introducing a substituent α to the amino group of the alkyl chain increases potency; the effect appears to be more pronounced for shorter alkyl groups (methyl, 29, IC50∼0.3 μM versus ethyl, 30, IC50∼1.2 μM; Table 2). Note that for pyridines 29 and 30 racemic mixtures were tested. Potency was reduced with a saturated C‐3 aminoalkyl chain (33) compared to a C‐3 aminoalkyl side chain bearing an aromatic residue (25). This might relate to increased steric bulk or lack of π‐π stacking effects. Overall para‐substituted aromatic moieties in the C‐3 aminoalkyl side chain (18, 31, 32) increase potency compared to non‐substituted aromatics (25, 26). This effect is more pronounced for electron donating substituents (18) compared to electron withdrawing substituents (31).
Figure 5.
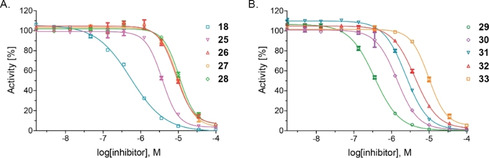
Representative dose‐response curves used to determine IC50 values for C‐3 aminoalkyl‐substituted 2,4‐PDCA derivatives 18 and 25–33. Two dose‐response curves each composed of technical duplicates were independently determined by using SPE‐MS AspH inhibition assays and manifest high Z’‐factors36 and signal‐to‐noise ratios (Figure S2). Assays were performed as described in the Experimental Section by using 50 nM His6‐AspH315‐758, 1 μM hFX‐CP101‐119 (Figure 2D), 100 μM l‐ascorbic acid (LAA), 3 μM 2OG, and 2 μM FeII in 50 mM HEPES buffer (pH 7.5, 20 °C). Data are shown as the mean average of two technical duplicates (n=2; mean ± SD).
Table 2.
The inhibition of human AspH by C‐3 aminoalkyl‐substituted 2,4‐PDCA derivatives.
|
|
Compound |
IC50 [μM][a] |
|---|---|---|
|
1 |
18 |
0.58 ±0.06 |
|
2 |
25 |
3.95 ±0.33 |
|
3 |
26 |
7.66 ±1.45 |
|
4 |
27 |
10.91 ±1.83 |
|
5 |
28 |
10.76 ±0.28 |
|
6 |
29 |
0.28 ±0.11 |
|
7 |
30 |
1.22 ±0.26 |
|
8 |
31 |
1.86 ±1.06 |
|
9 |
32 |
3.67 ±0.74 |
|
10 |
33 |
13.13 ±4.84 |
[a] Mean average of two independent runs (n=2; mean ± SD). AspH inhibition assays were performed as described in the Experimental Section by using 50 nM His6‐AspH315‐758 and 1.0 μM hFX‐CP101‐119 (Figure 2D) as a substrate.
Selectivity studies
To investigate the selectivity of the C‐3 functionalised 2,4‐PDCA derivatives for AspH, their inhibitory activities against the isolated human 2OG oxygenases PHD2, which is a validated medicinal chemistry target,35 factor inhibiting HIF‐α (FIH), and the Jmjc histone demethylase KDM4E were determined. The inhibition assays all employed SPE‐MS to help ensure comparability with the AspH results and minimise possible discrepancies due to different assay methods (Table 3).
Table 3.
The inhibition of human AspH, PHD2, FIH, and KDM4E by selected C‐3 functionalised 2,4‐PDCA derivatives.
|
Inhibitor |
IC50 AspH [μM][a,b] |
IC50 PHD2 [μM][a,c] |
IC50 FIH [μM][a,d] |
IC50 KDM4E [μM][a,e] |
|---|---|---|---|---|
|
1 |
0.03 ±0.01 |
5.29±3.35 |
5.03±2.06 |
0.18±0.0139 |
|
17 |
4.67 ±0.29 |
2.60 ±1.20 |
6.59 ±0.05 |
0.43 ±0.05 |
|
18 |
0.58 ±0.06 |
>50 |
43.0 ±3.9 |
2.45 ±0.59 |
|
25 |
3.95 ±0.33 |
>50 |
>50 |
2.36±0.13 |
|
26 |
7.66 ±1.45 |
>50 |
>50 |
5.71±0.04 |
|
29 |
0.28 ±0.11 |
>50 |
14.9 ±1.5 |
0.99 ±0.31 |
|
30 |
1.22 ±0.26 |
>50 |
46.1 ±3.2 |
3.69 ±0.85 |
|
31 |
1.86 ±1.06 |
>50 |
>50 |
3.57 ±0.38 |
|
32 |
3.67 ±0.74 |
>50 |
26.1 ±3.9 |
4.14±0.28 |
[a] Mean of two independent runs (n=2; mean ± SD). [b] Using 50 nM His6‐AspH315‐758 and 1.0 μM hFX‐CP101‐119 (Figure 2D). [c] Using 150 nM PHD2 and 5.0 μM C‐terminal oxygen‐dependent degradation domain fragment (HIF‐1α CODD, amino acids 558–574). [d] Using 150 nM FIH and 5.0 μM C‐terminal transactivation domain fragment (HIF‐1α CAD, amino acids 788–822). [e] Using 150 nM KDM4E and 10.0 μM of a variant of histone 3 (H31‐15K9me3, amino acids 1–15; Experimental Section)40. Enzyme inhibition assays were performed as described in the Experimental Section.
Most of the C‐3 aminoalkyl‐substituted 2,4‐PDCA derivatives investigated did not inhibit PHD2, with the exception of 17 which bears an aniline substituent and is a rather weak AspH inhibitor (Table 3). These results are consistent with studies showing that the parent 2,4‐PDCA (1) is a relatively weak inhibitor of PHD2.30
FIH was substantially inhibited by C‐3 aminoalkyl‐substituted 2,4‐PDCA derivatives 17 and 29 (IC50∼6.6 and 14.9 μM, respectively; Table 3), whereas all the other investigated 2,4‐PDCA derivatives displayed only moderate potency (Table 3). Thus, most inhibitors displayed >40 fold selectivity for AspH with the exceptions of 17 (non‐selective) and 32 (ca. seven‐fold selective for AspH; Table 3). Analysis of a crystal structure of FIH complexed with FeII and 2,4‐PDCA37 suggests that 2,4‐PDCA derivatives 17 and 29 likely inhibit FIH by replacing 2OG in the active site with the C‐3 substituent facing into a hydrophobic region as observed for the (partially) selective FIH inhibitor N‐oxalyl‐D‐phenylalanine (NOFD).38
The inhibition of KDM4E activity by the C‐3 aminoalkyl‐substituted 2,4‐PDCA derivatives might have been considered likely to be relatively weak, as the 2,4‐PDCA C‐3 derivative 18 has been previously described to be a significantly less potent KDM4E inhibitor (IC50∼41 μM) compared to the C‐3 aniline‐substituted 2,4‐PDCAs 8–17.31 However, C‐3 aminoalkyl‐substituted 2,4‐PDCA derivatives 18, 25, 26, and 29–32 inhibited KDM4E efficiently (Table 3) with IC50 values as low as ∼1.0 μM for 29. The results may in part reflect improved SPE‐MS based KDM4E inhibition assays39, 40, 41 compared to the previously used FDH‐coupled KDM4E inhibition assays requiring higher enzyme and cofactor/(co‐)substrate concentrations.31 Selectivities for AspH versus KDM4E did not exceed 4 : 1 and, considering that KDM4E assay concentrations were approximately three times higher than those of AspH, the selectivity for AspH may even be lower. The results also indicate that structurally similar KDM4 inhibitors lacking the C‐2 carboxylate moiety, which display high potency against KDM4E,14, 41a could inhibit AspH activity substantially. This observation might, at least in part, account for the observed antiproliferative effects of some of these compounds observed in cell‐based experiments.14 Considering the prominence of the isonicotinic acid scaffold for the development of small‐molecule KDM4 inhibitors,42 concerns on the selectivity of these compounds arise.
Conclusions
The results described herein demonstrate that it is possible to efficiently modulate the potent AspH inhibitor 2,4‐PDCA (1, Figure 1) using a novel four‐step synthetic sequence (Scheme 1), which includes two palladium‐catalysed reactions, to identify compounds of approximately equal potency. Out of the eight most potent identified AspH inhibitors, 29 is the most efficient AspH inhibitor (IC50∼0.3 μM); C‐3 aminoalkyl‐substituted 2,4‐PDCA derivative 26 (IC50∼7.7 μM) is the least efficient (Table 3).
Although further work is required, the results reveal that it should be possible to develop small‐molecule AspH inhibitors of suitable potency and selectivity for in vivo studies aimed at validating AspH as a medicinal chemistry target to develop novel cancer therapeutics and at exploring the function of EGFD hydroxylation in greater detail. Considering that AspH is translocated to the cell membrane of invasive cancer cells,19 the dicarboxylic acid motif of the 2,4‐PDCA derivatives synthesised might be beneficial to minimise the cell‐wall permeability of the inhibitors and thus reducing the possibility of undesired off‐target effects through inhibiting other 2OG oxygenases, such as the Jmjc KDMs, in cells.
In terms of the selectivity of the C‐3‐substituted 2,4‐PDCA derivatives, the results presented here demonstrate a substantial overlap between AspH inhibition with that of KDM4E (Table 3). By implication, this will likely extend to at least the other Jmjc KDM4 enzymes (i. e., human KDM4A‐D). Crystallographic analysis of the active site structures of the two types of 2OG oxygenases suggest that it should be possible to develop selective inhibitors for AspH or the KDM4 class of 2OG oxygenases.23, 43 Now that a reliable assay for isolated AspH has been established24 and Jmjc KDMs including KDM4E are actively being pursued as medicinal chemistry targets with several pyridine‐based and related small‐molecule inhibitors for cancer treatment,44 AspH should be included in Jmjc KDM selectivity counter‐screens in order to develop improved inhibitors and safe medicines.
It should also be noted that 2,4‐PDCA (1)24 and 2,3‐PDCA (quinolinic acid, 5) are potent AspH inhibitors. The latter observation raises the possibility of natural inhibition of AspH by small‐molecules such as quinolinic acid. A prodrug form of 2,4‐PDCA might be useful in probing AspH function in vivo bearing in mind possible “off‐target” effects through the inhibition of other 2OG oxygenases including the Jmjc KDMs (Table 3). In this regard, it is notable that dimethyl N‐oxalylglycine (DMOG), a prodrug form of NOG (2, Figure 1), has been used to mimic the cellular hypoxic response by inhibiting the PHDs, though it inhibits other 2OG oxygenases and other enzymes.20 Like 2,3‐PDCA, NOG is a natural product, at least in plants.45 NOG is a less potent inhibitor of AspH and the Jmjc KDMs than 2,4‐PDCA;24 conversely, 2,4‐PDCA inhibits the PHDs significantly less efficient than AspH (Table 3). Thus, providing care is taken to consider “off‐target” effects, incompletely selective small‐molecule inhibitors can be of use in biological functional assignment work.
Experimental Section
General information: All reagents were from commercial sources (Sigma‐Aldrich, Inc.; Fluorochem Ltd; Tokyo Chemical Industries; Alfa Aesar) and used as received unless otherwise stated. 2,4‐PDCA (1) and its regioisomers (5–7) were from Sigma‐Aldrich. The synthesis and characterisation of 2,4‐PDCA derivatives 8–17 has been described elsewhere.31 Anhydrous solvents were from Sigma‐Aldrich, Inc. and kept under an atmosphere of nitrogen. Solvents, liquids, and solutions were transferred using nitrogen‐flushed stainless steel needles and syringes. All reactions were carried out under an atmosphere of nitrogen unless stated otherwise. Milli‐Q ultrapure (MQ‐grade) water was used for buffers; LCMS grade solvents (Merck) were used for solid phase extraction (SPE)‐MS.
Purifications were performed using an automated Biotage Isolera One purification machine (wavelength monitored: 254 and 280 nm) equipped with pre‐packed Biotage® SNAP KP‐Sil or Biotage® SNAP Ultra flash chromatography cartridges. The cartridge size and solvent gradients (in column volumes, CV) used, are specified in the individual experimental procedures. HPLC grade solvents (ethyl acetate and cyclohexane; Sigma‐Aldrich Inc.) were used for purifications, reaction work‐ups, and extractions.
Thin‐layer chromatography (TLC) was carried out using Merck silica gel 60 F254 TLC plates and visualised under UV light. Melting points (MP) were determined using a Stuart SMP‐40 automated melting point apparatus. Infrared (IR) spectroscopy was performed using a Bruker Tensor‐27 Fourier transform infrared (FTIR) spectrometer. High‐resolution mass spectrometry (HRMS) was performed using electro‐spray ionisation (ESI) mass spectrometry (MS) in the positive or negative ionisation modes employing a Thermo Scientific Exactive mass spectrometer (ThermoFisher Scientific); data are presented as a mass‐to‐charge ratio (m/z) in Daltons.
Single crystal X‐ray diffraction data were collected using an Oxford Diffraction SuperNova diffractometer (Rigaku). Structures were solved using SUPERFLIP software46 before refinement with the CRYSTALS software suite.47 Crystallographic data can be obtained from the Cambridge Crystallographic Data Centre (CCDC 1988141–42).
Nuclear magnetic resonance (NMR) spectroscopy was performed using a Bruker AVANCE AVIIIHD 600 machine equipped with a 5 mm BB‐F/1H Prodigy N2 cryoprobe. Chemical shifts for protons are reported in parts per million (ppm) downfield from tetramethylsilane and are referenced to residual protium in the NMR solvent (CDCl3: δ=7.28 ppm; D2O: δ=4.79 ppm). For 13C NMR, chemical shifts are reported in the scale relative to the NMR solvent (i. e., CDCl3: δ=77.00 ppm). For 19F NMR, chemical shifts are reported in the scale relative to CFCl3; coupling constants are accurate to 0.1 Hz. The number of C atoms in brackets indicates overlapping signals in 13C NMR; chemical shift numbers in brackets indicate close signals that can be differentiated taking into account second respectively third decimal numbers.
General synthetic procedures
General Procedure A: To dimethyl 3‐chloropyridine‐2,4‐dicarboxylate 24 (1.0 equiv), palladium acetate (0.04 equiv), and xantphos34 (0.06 equiv) in a capped 5 or 20 mL microwave reaction vial were added anhydrous toluene (0.25 M), pyridine (2.5 equiv), then N‐alkylamine (1.5 equiv), at ambient temperature. N2 gas was bubbled through the reaction mixture for 15 min, which was then placed into a preheated sand bath (190 °C) and stirred for 22–24 h. The reaction mixture was then cooled to ambient temperature, evaporated under reduced pressure, and purified by column chromatography to afford the desired purified dimethyl C‐3‐aminoalkyl 2,4‐pyridinedicarboxylic acid ester.
General Procedure B: To a solution of the dimethyl 3‐aminoalkylpyridine‐2,4‐dicarboxylate (1.0 equiv) in methanol (0.2 M, HPLC grade) was added an aqueous solution of lithium hydroxide (0.4 M, 2.8 equiv) under an ambient atmosphere at 0 °C. The reaction mixture was allowed to slowly warm to ambient temperature overnight (14–18 h); the methanol was then removed under reduced pressure. The solution was extracted three times with CH2Cl2 (the organic extracts were discarded); the aqueous phase was acidified (pH∼7.7) by adding Dowex® 50XW8 (H+‐form, mesh 200–400), filtered, and lyophilised to afford the solid C‐3‐aminoalkyl‐substituted pyridine‐2,4‐dicarboxylic acid. The crude product was sufficiently purified as judged by 1H and 13C NMR and used without further purification in the biological assays.
Synthetic procedures and compound characterisations
Methyl 2,3‐dichloroisonicotinate (23): To a solution of 2,3‐dichloroisonicotinic acid (12.3 g, 63.8 mmol, 1.0 equiv) in anhydrous methanol (125 mL) was added dropwise thionyl chloride (7.0 mL, 95.6 mmol, 1.5 equiv), at ambient temperature under a nitrogen atmosphere. The reaction mixture was stirred under reflux for 2 h, then cooled to ambient temperature and concentrated. The residue was dissolved in ethyl acetate, washed twice with saturated aqueous NaHCO3 solution, then once with brine; The organic solution was dried over anhydrous Na2SO4, filtered, evaporated, and purified by column chromatography (100 g KP‐Sil; 80 mL/min; 100 % cyclohexane (2 column volumes, CV) followed by a linear gradient (7 CV): 0→40 % ethyl acetate in cyclohexane) to afford 11.6 g (89 %) of purified methyl ester 23. White solid, m.p.: 33–35 °C; 1H NMR (600 MHz, 300 K, CDCl3): δ=8.36 (d, J=4.9 Hz, 1H), 7.51 (d, J=4.8 Hz, 1H), 3.97 ppm (s, 3H); 13C NMR (150 MHz, 300 K, CDCl3): δ=164.0, 151.2, 146.9, 140.4, 128.5, 122.7, 53.2 ppm; IR (film): =3002, 2956, 1738, 1576, 1532, 1447, 1432, 1352, 1294, 1278, 1202, 1158, 1110, 1047, 966 cm−1; HRMS (ESI): m/z calcd for C7H6O2NCl2 [M+H]+: 205.9770, found: 205.9772.
Dimethyl 3‐chloropyridine‐2,4‐dicarboxylate (24): To a solution of methyl 2,3‐dichloroisonicotinate (23) (2.06 g, 10.0 mmol, 1.0 equiv) and dichloro[2,2’‐bis(diphenylphosphino)‐1,1’‐binaphthyl]palladium(II) [(rac‐BINAP)PdCl2] (80 mg, 0.1 mmol, 0.01 equiv) in anhydrous methanol (50 mL) in a 250 mL J‐Young Schlenk tube was added N,N‐diisopropylethylamine (2.61 mL, 15.0 mmol, 1.5 equiv) under an ambient temperature. Carbon monoxide gas (synthesis grade) was bubbled through the solution for 10 minutes. CAUTION: Carbon monoxide is a highly toxic and flammable gas; it should be handled in a well‐vented fume cupboard taking appropriate safety measures. The Schlenk tube was then sealed under CO‐pressure (∼1.5–2.0 atm) and placed in a sand bath, which was heated under stirring behind a safety shield at 100 °C for 12 h. The reaction mixture was cooled to ambient temperature, then concentrated and purified by column chromatography (50 g KP‐Sil; 60 mL/min; 100 % cyclohexane (3 CV) followed by a linear gradient (10 CV): 0→30 % ethyl acetate in cyclohexane) to afford 2.16 g (94 %) the pure dimethyl ester 24. Single‐crystals suitable for X‐ray diffraction analysis were obtained from a concentrated solution of analytically pure 24 in cyclohexane/CH2Cl2 by slow solvent evaporation at ambient temperature and atmosphere. CCDC 1988141 contains the complete supplementary crystallographic data file; selected crystallographic data are shown in Table S1. White solid, m.p.: 60–62 °C; 1H NMR (600 MHz, 300 K, CDCl3): δ=8.64 (d, J=4.8 Hz, 1H), 7.70 (d, J=4.8 Hz, 1H), 4.04 (s, 3H), 4.00 ppm (s, 3H); 13C NMR (150 MHz, 300 K, CDCl3): δ=164.7, 164.2, 150.4, 147.3, 139.7, 127.9, 125.5, 53.2, 53.1(8) ppm; IR (film): =3005, 2957, 1737, 1579, 1543, 1453, 1433, 1389, 1316, 1269, 1199, 1172, 1154, 1115, 1047, 987, 953 cm−1; HRMS (ESI): m/z calcd for C9H9O4NCl [M+H]+: 230.0215, found: 230.0215.
Dimethyl 3‐((4‐methoxybenzyl)amino)pyridine‐2,4‐dicarboxylate (22): According to General Procedure A, pyridine 22 (471 mg, 71 %) was obtained from dimethyl 3‐chloropyridine‐2,4‐dicarboxylate 24 (459 mg, 2.0 mmol) after column chromatography (25 g Ultra; 45 mL/min; 100 % cyclohexane (3 CV) followed by a linear gradient (10 CV): 0→30 % ethyl acetate in cyclohexane). The analytical data of pyridine 22 correspond to those reported.31 Yellow solid, m.p.: 72–74 °C; 1H NMR (600 MHz, 300 K, CDCl3): δ=8.33 (t, J=4.8 Hz, 1H), 8.04 (d, J=4.7 Hz, 1H), 7.63 (d, J=4.7 Hz, 1H), 7.22−7.20 (m, 2H), 6.88−6.86 (m, 2H), 4.19 (d, J=4.7 Hz, 2H), 3.96 (s, 3H), 3.89 (s, 3H), 3.80 ppm (s, 3H); 13C NMR (150 MHz, 300 K, CDCl3): δ=167.6, 167.3, 159.2, 145.4, 135.9, 133.4, 129.9, 129.1, 127.5, 122.7, 114.1, 55.2, 52.8, 52.6, 50.4 ppm; IR (film): =3328, 2998, 2952, 2838, 1722, 1696, 1612, 1584, 1567, 1512, 1438, 1294, 1247, 1192, 1161, 1127, 1106, 1078, 1033, 1000 cm−1; HRMS (ESI): m/z calcd for C17H19O5N2 [M+H]+: 331.1289, found: 331.1285.
3‐((4‐Methoxybenzyl)amino)pyridine‐2,4‐dicarboxylic acid (18): Pyridine 18 (133 mg, 88 %) was obtained from dimethyl pyridine‐2,4‐dicarboxylate 22 (165 mg, 0.5 mmol) according to General Procedure B. Yellow solid, m.p.: >250 °C (decomposition); 1H NMR (600 MHz, 300 K, D2O): δ=7.93 (d, J=5.0 Hz, 1H), 7.43 (d, J=5.0 Hz, 1H), 7.30 (d, J=8.5 Hz, 2H), 6.96 (d, J=8.6 Hz, 2H), 4.35 (s, 2H), 3.82 ppm (s, 3H); 13C NMR (150 MHz, 300 K, D2O): δ=174.6, 172.7, 158.0, 142.0, 141.2, 136.9, 136.4, 131.9, 129.4, 124.4, 114.1, 55.3, 49.4 ppm; IR (film): =3262, 2960, 2926, 1609, 1512, 1465, 1388, 1301, 1246, 1177, 1107, 1030 cm−1; HRMS (ESI): m/z calcd for C15H13O5N2 [M−H]−: 301.0830, found: 301.0826.
Dimethyl 3‐(benzylamino)pyridine‐2,4‐dicarboxylate (34): According to General Procedure A, pyridine 34 (191 mg, 32 %) was obtained from dimethyl 3‐chloropyridine‐2,4‐dicarboxylate 24 (459 mg, 2.0 mmol) after column chromatography (25 g Ultra; 50 mL/min; 100 % cyclohexane (3 CV) followed by a linear gradient (10 CV): 0→20 % ethyl acetate in cyclohexane). Single‐crystals suitable for X‐ray diffraction analysis were obtained from a concentrated solution of analytically pure 34 in cyclohexane/CH2Cl2 by slow solvent evaporation at ambient temperature and atmosphere. CCDC 1988142 contains the complete supplementary crystallographic data file; the molecular structure is shown in Figure S1 and selected crystallographic data in Table S1. Yellow solid, m.p.: 71–72 °C; 1H NMR (600 MHz, 300 K, CDCl3): δ=8.45 (brt, J=4.2 Hz, 1H), 8.06 (d, J=4.6 Hz, 1H), 7.64 (d, J=4.7 Hz, 1H), 7.37–7.35 (m, 2H), 7.32−7.29 (m, 3H), 4.29 (d, J=4.9 Hz, 2H), 3.97 (s, 3H), 3.89 ppm (s, 3H); 13C NMR (150 MHz, 300 K, CDCl3): δ=167.7, 167.3, 145.5, 137.9, 136.1, 133.5, 128.8, 127.8, 127.7, 127.6, 122.9, 52.8, 52.6, 50.9 ppm; IR (film): =3327, 3030, 2952, 1721, 1697, 1585, 1568, 1498, 1437, 1344, 1293, 1241, 1192, 1162, 1127, 1108, 1000 cm−1; HRMS (ESI): m/z calcd for C16H17O4N2 [M+H]+: 301.1183, found: 301.1184.
3‐(Benzylamino)pyridine‐2,4‐dicarboxylic acid (25): Pyridine 25 (86 mg, 63 %) was obtained from dimethyl pyridine‐2,4‐dicarboxylate 34 (150 mg, 0.5 mmol) according to General Procedure B. Yellow solid, m.p.: >240 °C (decomposition); 1H NMR (600 MHz, 300 K, D2O): δ=7.95 (d, J=5.5 Hz, 1H), 7.67 (d, J=5.5 Hz, 1H), 7.43–7.39 (m, 4H), 7.37–7.35 (m, 1H), 4.58 ppm (s, 2H); 13C NMR (150 MHz, 300 K, D2O): δ=172.9, 166.3, 144.0, 141.1, 138.3, 130.9 (br), 129.0 (br), 128.8, 127.7, 127.6(5), 126.5, 48.5 ppm; IR (film): =3182, 3031, 2980, 1618, 1526, 1498, 1453, 1386, 1291, 1244, 1207, 1104 cm−1; HRMS (ESI): m/z calcd for C14H13O4N2 [M+H]+: 273.0870, found: 273.0869.
Dimethyl 3‐(phenethylamino)pyridine‐2,4‐dicarboxylate (35): According to General Procedure A, pyridine 35 (414 mg, 66 %) was obtained from dimethyl 3‐chloropyridine‐2,4‐dicarboxylate 24 (459 mg, 2.0 mmol) after column chromatography (25 g Ultra; 50 mL/min; 100 % cyclohexane (3 CV) followed by a linear gradient (10 CV): 0→20 % ethyl acetate in cyclohexane). Clear yellow oil; 1H NMR (600 MHz, 300 K, CDCl3): δ=8.07 (brs, 1H), 8.02 (d, J=4.7 Hz, 1H), 7.60 (d, J=4.7 Hz, 1H), 7.32–7.30 (m, 2H), 7.25–7.21 (m, 3H), 3.98 (s, 3H), 3.91 (s, 3H), 3.34–3.31 (m, 2H), 2.95 ppm (t, J=7.2 Hz, 2H); 13C NMR (150 MHz, 300 K, CDCl3): δ=167.6, 167.4, 145.7, 138.2, 136.0, 133.7, 128.7, 128.6, 127.4, 126.6, 122.9, 52.8, 52.6, 48.2, 36.6 ppm; IR (film): =3326, 3028, 2951, 1725, 1698, 1570, 1499, 1438, 1349, 1293, 1243, 1193, 1160, 1126, 1108, 1089, 996 cm−1; HRMS (ESI): m/z calcd for C17H19O4N2 [M+H]+: 315.1339, found: 315.1336.
3‐(Phenethylamino)pyridine‐2,4‐dicarboxylic acid (26): Pyridine 26 (101 mg, 71 %) was obtained from dimethyl pyridine‐2,4‐dicarboxylate 35 (157 mg, 0.5 mmol) according to General Procedure B. Yellow solid, m.p.: >220 °C (decomposition); 1H NMR (600 MHz, 300 K, D2O): δ=7.85 (d, J=5.5 Hz, 1H), 7.63 (d, J=5.5 Hz, 1H), 7.37–7.34 (m, 2H), 7.31–7.30 (m, 2H), 7.28–7.26 (m, 1H), 3.66 (t, J=6.5 Hz, 2H), 2.97 ppm (t, J=6.5 Hz, 2H); 13C NMR (150 MHz, 300 K, D2O): δ=173.1, 165.6, 144.8, 141.0, 139.0, 129.0, 128.9(8), 128.7, 127.4 (br), 126.6, 126.4 (br), 45.9 (br), 35.6 (br) ppm; IR (film): =3194, 3028, 2924, 1617, 1531, 1497, 1455, 1387, 1293, 1244, 1210, 1107 cm−1; HRMS (ESI): m/z calcd for C15H13O4N2 [M−H]−: 285.0881, found: 285.0879.
Dimethyl 3‐((3‐phenylpropyl)amino)pyridine‐2,4‐dicarboxylate (36): According to General Procedure A, pyridine 36 (379 mg, 58 %) was obtained from dimethyl 3‐chloropyridine‐2,4‐dicarboxylate 24 (459 mg, 2.0 mmol) after column chromatography (25 g Ultra; 50 mL/min; 100 % cyclohexane (3 CV) followed by a linear gradient (10 CV): 0→20 % ethyl acetate in cyclohexane). Clear yellow oil; 1H NMR (600 MHz, 300 K, CDCl3): δ=8.11 (brt, J=4.7 Hz, 1H), 8.02 (d, J=4.7 Hz, 1H), 7.60 (d, J=4.6 Hz, 1H), 7.30–7.27 (m, 2H), 7.22–7.19 (m, 1H), 7.17–7.15 (m, 2H), 3.97 (s, 3H), 3.89 (s, 3H), 3.06–3.03 (m, 2H), 2.72 (t, J=7.6 Hz, 2H), 2.02–1.96 ppm (m, 2H); 13C NMR (150 MHz, 300 K, CDCl3): δ=167.7, 167.5, 145.9, 141.0, 135.7, 133.3, 128.4, 128.3(7), 127.4, 126.1, 122.6, 52.8, 52.5, 46.2, 33.1, 32.0 ppm; IR (film): =3329, 3027, 2950, 2865, 1727, 1696, 1586, 1571, 1499, 1438, 1294, 1243, 1195, 1157, 1126, 1108, 997 cm−1; HRMS (ESI): m/z calcd for C18H21O4N2 [M+H]+: 329.1496, found: 329.1493.
3‐((3‐Phenylpropyl)amino)pyridine‐2,4‐dicarboxylic acid (27): Pyridine 27 (91 mg, 61 %) was obtained from dimethyl pyridine‐2,4‐dicarboxylate 36 (164 mg, 0.5 mmol) according to General Procedure B. Yellow solid, m.p.: >220 °C (decomposition); 1H NMR (600 MHz, 300 K, D2O): δ=7.84 (d, J=5.5 Hz, 1H), 7.61 (d, J=5.5 Hz, 1H), 7.28–7.25 (m, 2H), 7.22–7.21 (m, 2H), 7.19–7.17 (m, 1H), 3.32 (t, J=6.3 Hz, 2H), 2.73 (t, J=7.3 Hz, 2H), 1.99 ppm (app. quint., J=6.8 Hz, 2H); 13C NMR (150 MHz, 300 K, D2O): δ=173.1, 166.0, 144.6, 141.7, 140.3, 128.7 (br), 128.5 (2 C), 127.2 (br), 126.4, 125.8, 44.2, 32.6, 30.8 ppm; IR (film): =3194, 2980, 2935, 2864, 1618, 1603, 1531, 1497, 1467, 1454, 1385, 1291, 1244, 1207, 1175, 1104, 1030 cm−1; HRMS (ESI): m/z calcd for C16H15O4N2 [M−H]−: 299.1037, found: 299.1034.
Dimethyl 3‐((3‐phenylbutyl)amino)pyridine‐2,4‐dicarboxylate (37): According to General Procedure A, pyridine 37 (398 mg, 58 %) was obtained from dimethyl 3‐chloropyridine‐2,4‐dicarboxylate 24 (459 mg, 2.0 mmol) after column chromatography (25 g Ultra; 50 mL/min; 100 % cyclohexane (3 CV) followed by a linear gradient (10 CV): 0→15 % ethyl acetate in cyclohexane). Clear yellow oil; 1H NMR (600 MHz, 300 K, CDCl3): δ=8.06 (brs, 1H), 8.01 (d, J=4.7 Hz, 1H), 7.60 (d, J=4.6 Hz, 1H), 7.30–7.28 (m, 2H), 7.21–7.17 (m, 3H), 3.99 (s, 3H), 3.93 (s, 3H), 3.06–3.04 (m, 2H), 2.66 (t, J=7.2 Hz, 2H), 1.77–1.67 ppm (m, 4H); 13C NMR (150 MHz, 300 K, CDCl3): δ=167.8, 167.5, 145.9, 141.8, 135.6, 133.2, 128.3 (2 C), 127.4, 125.9, 122.5, 52.8, 52.5, 46.7, 35.4, 29.9, 28.5 ppm; IR (film): =3330, 3027, 2948, 2859, 1727, 1696, 1585, 1571, 1504, 1438, 1294, 1243, 1193, 1153, 1126, 1109, 999 cm−1; HRMS (ESI): m/z calcd for C19H23O4N2 [M+H]+: 343.1652, found: 343.1656.
3‐((4‐Phenylbutyl)amino)pyridine‐2,4‐dicarboxylic acid (28): Pyridine 28 (99 mg, 63 %) was obtained from dimethyl pyridine‐2,4‐dicarboxylate 37 (171 mg, 0.5 mmol) according to General Procedure B. Yellow solid, m.p.: >210 °C (decomposition); 1H NMR (600 MHz, 300 K, D2O): δ=7.88 (d, J=5.5 Hz, 1H), 7.61 (d, J=5.5 Hz, 1H), 7.32–7.30 (m, 2H), 7.26–7.25 (m, 2H), 7.21–7.19 (m, 1H), 3.33 (t, J=6.5 Hz, 2H), 2.65 (t, J=7.2 Hz, 2H), 1.77–1.72 (m, 2H), 1.68–1.64 ppm (m, 2H); 13C NMR (150 MHz, 300 K, D2O): δ=173.1, 166.1, 144.3 (br), 142.9, 140.8, 129.5 (br), 128.5, 128.4(9), 128.1 (br), 126.3, 125.7, 44.2, 34.3, 28.3, 27.5 ppm; IR (film): =3182, 2980, 2931, 2858, 1619, 1532, 1453, 1384, 1290, 1244, 1205, 1105 cm−1; HRMS (ESI): m/z calcd for C17H17O4N2 [M−H]−: 313.1194, found: 313.1191.
rac ‐Dimethyl 3‐((1‐phenylethyl)amino)pyridine‐2,4‐dicarboxylate (38): According to General Procedure A, pyridine 38 (304 mg, 48 %) was obtained from dimethyl 3‐chloropyridine‐2,4‐dicarboxylate 24 (459 mg, 2.0 mmol) after column chromatography (25 g Ultra; 50 mL/min; 100 % cyclohexane (3 CV) followed by a linear gradient (10 CV): 0→30 % ethyl acetate in cyclohexane). Yellow solid, m.p.: 95–96 °C; 1H NMR (600 MHz, 300 K, CDCl3): δ=8.67 (brd, J=6.6 Hz, 1H), 7.99 (d, J=4.7 Hz, 1H), 7.50 (d, J=4.6 Hz, 1H), 7.27–7.24 (m, 2H), 7.21–7.19 (m, 1H), 7.13 (d, J=7.4 Hz, 2H), 4.74 (app. quint., J=6.7 Hz, 1H), 3.94 (s, 3H), 3.78 (s, 3H), 1.60 ppm (d, J=6.7 Hz, 3H); 13C NMR (150 MHz, 300 K, CDCl3): δ=167.8, 167.2, 144.7, 143.0, 136.3, 134.1, 128.6, 127.5, 127.3, 126.2, 124.3, 55.1, 52.8, 52.5, 25.6 ppm; IR (film): =3322, 3029, 2952, 1721, 1697, 1589, 1568, 1509, 1437, 1375, 1292, 1243, 1193, 1166, 1126, 999 cm−1; HRMS (ESI): m/z calcd for C17H19O4N2 [M+H]+: 315.1339, found: 315.1341.
rac ‐3‐((1‐Phenylethyl)amino)pyridine‐2,4‐dicarboxylic acid (29): Pyridine 29 (103 mg, 72 %) was obtained from dimethyl pyridine‐2,4‐dicarboxylate 38 (157 mg, 0.5 mmol) according to General Procedure B. Yellow solid, m.p.: >270 °C (decomposition); 1H NMR (600 MHz, 300 K, D2O): δ=7.92 (d, J=5.4 Hz, 1H), 7.55 (d, J=5.4 Hz, 1H), 7.36–7.31 (m, 4H), 7.29–7.26 (m, 1H), 4.97 (q, J=6.7 Hz, 1H), 1.56 ppm (d, J=6.7 Hz, 3H); 13C NMR (150 MHz, 300 K, D2O): δ=172.7, 167.2 (br), 144.1, 143.2, 141.7, 133.5 (br), 130.9 (br), 128.7, 127.3, 126.3, 126.2, 54.6, 23.8 ppm; IR (film): =3182, 3030, 2927, 1612, 1519, 1452, 1374, 1288, 1243, 1211, 1125, 1102, 1019 cm−1; HRMS (ESI): m/z calcd for C15H13O4N2 [M−H]−: 285.0881, found: 285.0877.
rac ‐Dimethyl 3‐((1‐phenylpropyl)amino)pyridine‐2,4‐dicarboxylate (39): According to General Procedure A, pyridine 39 (268 mg, 41 %) was obtained from dimethyl 3‐chloropyridine‐2,4‐dicarboxylate 24 (459 mg, 2.0 mmol) after column chromatography (25 g Ultra; 50 mL/min; 100 % cyclohexane (3 CV) followed by a linear gradient (12 CV): 0→20 % ethyl acetate in cyclohexane). Yellow solid, m.p.: 83–85 °C; 1H NMR (600 MHz, 300 K, CDCl3): δ=8.77 (brd, J=6.7 Hz, 1H), 7.97 (d, J=4.6 Hz, 1H), 7.49 (d, J=4.6 Hz, 1H), 7.26–7.24 (m, 2H), 7.21–7.19 (m, 1H), 7.09 (d, J=7.7 Hz, 2H), 4.52 (q, J=6.7 Hz, 1H), 3.94 (s, 3H), 3.77 (s, 3H), 2.00–1.93 (m, 1H), 1.90–1.83 (m, 1H), 1.00 ppm (t, J=7.4 Hz, 3H); 13C NMR (150 MHz, 300 K, CDCl3): δ=167.8, 167.2, 145.1, 141.4, 136.1, 133.9, 128.4, 127.5, 127.2, 126.9, 124.2, 61.1, 52.7, 52.4, 32.6, 10.6 ppm; IR (film): =3307, 3002, 2948, 2874, 1713, 1678, 1585, 1503, 1455, 1431, 1370, 1285, 1239, 1190, 1165, 1121, 1103, 1043, 1000, 962 cm−1; HRMS (ESI): m/z calcd for C18H21O4N2 [M+H]+: 329.1496, found: 329.1495.
rac ‐3‐((1‐Phenylpropyl)amino)pyridine‐2,4‐dicarboxylic acid (30): Pyridine 30 (66 mg, 88 %) was obtained from dimethyl pyridine‐2,4‐dicarboxylate 39 (82 mg, 0.25 mmol) according to General Procedure B. Yellow solid, m.p.: >240 °C (decomposition); 1H NMR (600 MHz, 300 K, D2O): δ=7.90 (d, J=5.5 Hz, 1H), 7.59 (d, J=5.6 Hz, 1H), 7.37–7.35 (m, 2H), 7.31–7.27 (m, 3H), 4.83 (t, J=6.7 Hz, 1H), 1.96–1.86 (m, 2H), 1.56 ppm (t, J=7.4 Hz, 3H); 13C NMR (150 MHz, 300 K, D2O): δ=172.3, 165.8, 144.4, 142.5, 142.4, 130.3, 128.6, 128.5(7), 127.3, 127.0, 126.7, 60.1, 31.3, 9.9 ppm; IR (film): =3196, 3065, 2965, 2933, 1621, 1596, 1523, 1494, 1455, 1382, 1286, 1242, 1208, 1103 cm−1; HRMS (ESI): m/z calcd for C16H15O4N2 [M−H]−: 299.1037, found: 299.1033.
Dimethyl 3‐((4‐(trifluoromethyl)benzyl)amino)pyridine‐2,4‐dicarboxylate (40): According to General Procedure A, pyridine 40 (307 mg, 42 %) was obtained from dimethyl 3‐chloropyridine‐2,4‐dicarboxylate 24 (459 mg, 2.0 mmol) after column chromatography (25 g Ultra; 50 mL/min; 100 % cyclohexane (3 CV) followed by a linear gradient (10 CV): 0→25 % ethyl acetate in cyclohexane). Clear yellow oil; 1H NMR (600 MHz, 300 K, CDCl3): δ=8.51 (brt, J=4.7 Hz, 1H), 8.10 (d, J=4.7 Hz, 1H), 7.67 (d, J=4.6 Hz, 1H), 7.62 (d, J=8.2 Hz, 2H), 7.43 (d, J=8.0 Hz, 2H), 4.37 (d, J=5.3 Hz, 2H), 3.97 (s, 3H), 3.88 ppm (s, 3H); 19F NMR (565 MHz, 300 K, CDCl3): δ=−62.6 ppm (s, 3F); 13C NMR (150 MHz, 300 K, CDCl3): δ=167.7, 167.3, 145.4, 142.0, 136.7, 133.8, 130.1 (q, J=32.4 Hz), 127.9, 127.7, 125.8 (q, J=3.6 Hz), 124.0 (q, J=272.1 Hz), 123.1, 52.9, 52.7, 50.4 ppm; IR (film): =3325, 2955, 1723, 1698, 1620, 1587, 1570, 1505, 1439, 1418, 1325, 1293, 1242, 1194, 1162, 1122, 1067, 1018, 957 cm−1; HRMS (ESI): m/z calcd for C17H16O4N2F3 [M+H]+: 369.1057, found: 369.1055.
3‐((4‐(Trifluoromethyl)benzyl)amino)pyridine‐2,4‐dicarboxylic acid (31): Pyridine 31 (121 mg, 71 %) was obtained from dimethyl pyridine‐2,4‐dicarboxylate 40 (184 mg, 0.5 mmol) according to General Procedure B. Yellow solid, m.p.: >220 °C (decomposition); 1H NMR (600 MHz, 300 K, D2O): δ=7.94 (dd, J=5.5, 0.5 Hz, 1H), 7.69 (d, J=8.2 Hz, 2H), 7.62 (d, J=5.5 Hz, 1H), 7.52 (d, J=8.1 Hz, 2H), 4.66 ppm (s, 2H); 19F NMR (565 MHz, 300 K, D2O): δ=−62.2 ppm (s, 3F); 13C NMR (150 MHz, 300 K, D2O): δ=173.0, 166.9 (br), 143.7, 142.9, 140.7, 132.1 (br), 129.7 (br), 128.8 (q, J=32.0 Hz), 127.9, 126.2, 125.5 (q, J=3.7 Hz), 124.2 (q, J=271.4 Hz), 48.0 ppm; IR (film): =3264, 2926, 2852, 1601, 1532, 1450, 1386, 1325, 1294, 1164, 1114, 1067, 1018 cm−1; HRMS (ESI): m/z calcd for C15H10O4N2F3 [M−H]−: 339.0598, found: 339.0595.
Dimethyl 3‐((4‐chlorophenethyl)amino)pyridine‐2,4‐dicarboxylate (41): According to General Procedure A, pyridine 41 (400 mg, 57 %) was obtained from dimethyl 3‐chloropyridine‐2,4‐dicarboxylate 24 (459 mg, 2.0 mmol) after column chromatography (25 g Ultra; 50 mL/min; 100 % cyclohexane (3 CV) followed by a linear gradient (10 CV): 0→20 % ethyl acetate in cyclohexane). Clear yellow oil; 1H NMR (600 MHz, 300 K, CDCl3): δ=8.06 (brt, J=4.7 Hz, 1H), 8.04 (d, J=4.7 Hz, 1H), 7.61 (d, J=4.7 Hz, 1H), 7.29–7.27 (m, 2H), 7.16–7.15 (m, 2H), 3.98 (s, 3H), 3.92 (s, 3H), 3.32–3.29 (m, 2H), 2.92 ppm (t, J=7.1 Hz, 2H); 13C NMR (150 MHz, 300 K, CDCl3): δ=167.6, 167.3, 145.6, 136.7, 136.1, 133.7, 132.5, 130.0, 128.7, 127.4, 122.9, 52.8, 52.6, 48.0, 35.9 ppm; IR (film): =3325, 2995, 2951, 2875, 1725, 1698, 1585, 1570, 1493, 1438, 1349, 1293, 1243, 1195, 1159, 1126, 1109, 1091, 1015, 996 cm−1; HRMS (ESI): m/z calcd for C17H18O4N2Cl [M+H]+: 349.0950, found: 349.0949.
3‐((4‐Chlorophenethyl)amino)pyridine‐2,4‐dicarboxylic acid (32): Pyridine 32 (111 mg, 69 %) was obtained from dimethyl pyridine‐2,4‐dicarboxylate 41 (174 mg, 0.5 mmol) according to General Procedure B. Yellow solid, m.p.: >230 °C (decomposition); 1H NMR (600 MHz, 300 K, D2O): δ=7.86 (d, J=5.5 Hz, 1H), 7.62 (d, J=5.5 Hz, 1H), 7.33–7.31 (m, 2H), 7.25–7.24 (m, 2H), 3.64 (t, J=6.5 Hz, 2H), 2.94 ppm (t, J=6.5 Hz, 2H); 13C NMR (150 MHz, 300 K, D2O): δ=173.1, 165.7, 144.7, 141.1, 137.6, 131.6, 130.5, 129.4 (br), 128.4, 127.7 (br), 126.3 (br), 45.8 (br), 35.1 (br) ppm; IR (film): =3251, 2934, 2854, 1602, 1533, 1493, 1449, 1387, 1325, 1298, 1245, 1164, 1111, 1067, 1017 cm−1; HRMS (ESI): m/z calcd for C15H12O4N2Cl [M−H]−: 319.0491, found: 319.0488.
Dimethyl 3‐((cyclohexylmethyl)amino)pyridine‐2,4‐dicarboxylate (42): According to General Procedure A, pyridine 42 (262 mg, 43 %) was obtained from dimethyl 3‐chloropyridine‐2,4‐dicarboxylate 24 (459 mg, 2.0 mmol) after column chromatography (25 g Ultra; 50 mL/min; 100 % cyclohexane (3 CV) followed by a linear gradient (15 CV): 0→16 % ethyl acetate in cyclohexane). Yellow solid, m.p.: 45–47 °C; 1H NMR (600 MHz, 300 K, CDCl3): δ=8.00 (d, J=4.6 Hz, 1H), 7.60 (d, J=4.6 Hz, 1H), 4.00 (s, 3H), 3.94 (s, 3H), 2.88 (d, J=6.5 Hz, 2H), 1.80–1.74 (m, 4H), 1.71–1.69 (m, 1H), 1.62–1.56 (m, 1H), 1.31–1.25 (m, 2H), 1.22–1.14 (m, 1H), 1.03–0.97 ppm (m, 2H), the NH proton was not detected; 13C NMR (150 MHz, 300 K, CDCl3): δ=167.8, 167.6, 146.2, 135.4, 133.0, 127.5, 122.5, 53.6, 52.8, 52.5, 38.6, 30.8, 26.3, 25.8 ppm; IR (film): =3323, 2998, 2924, 2852, 1721, 1697, 1587, 1505, 1437, 1385, 1340, 1291, 1237, 1190, 1161, 1143, 1102, 1072, 997 cm−1; HRMS (ESI): m/z calcd for C16H23O4N2 [M+H]+: 307.1652, found: 307.1653.
3‐((Cyclohexylmethyl)amino)pyridine‐2,4‐dicarboxylic acid (33): Pyridine 33 (53 mg, 76 %) was obtained from dimethyl pyridine‐2,4‐dicarboxylate 42 (76 mg, 0.25 mmol) according to General Procedure B. Yellow solid, m.p.: >230 °C (decomposition); 1H NMR (600 MHz, 300 K, D2O): δ=7.90 (d, J=5.5 Hz, 1H), 7.66 (d, J=5.4 Hz, 1H), 3.19 (d, J=6.5 Hz, 2H), 1.76–1.71 (m, 4H), 1.66–1.59 (m, 2H), 1.30–1.16 (m, 3H), 1.06–1.00 ppm (m, 2H); 13C NMR (150 MHz, 300 K, D2O): δ=173.2, 166.2, 144.7 (br), 140.7, 129.1 (br), 127.8 (br), 126.4, 51.2, 37.8, 30.1, 26.0, 25.4 ppm; IR (film): =3253, 2924, 2851, 1617, 1533, 1450, 1388, 1292, 1245, 1207, 1109 cm−1; HRMS (ESI): m/z calcd for C14H19O4N2 [M+H]+: 279.1339, found: 279.1340.
Biochemical procedures
Recombinant AspH production and purification: N‐Terminally His6‐tagged human AspH315‐758 (His6‐AspH315‐758) was produced in Escherichia coli BL21 (DE3) cells using a pET‐28a(+) vector and purified by NiII‐affinity chromatography (HisTrap HP column, GE Healthcare; 1 mL/min flow rate) and size‐exclusion chromatography (HiLoad 26/60 Superdex 75 pg 300 mL column; 1 mL/min) using an ÄKTA Pure machine (GE Healthcare), as previously reported.21, 23 AspH was >95 % pure as analysed by SDS‐PAGE and ESI‐MS analysis and had the anticipated mass as reported (m/z calcd for His6‐AspH315–758: 54519 Da, found: 54519 Da).23 Purified AspH was stored in 50 mM HEPES buffer (pH 7.5, 150 mM NaCl) at a concentration of 125 μM at −78 °C; fresh aliquots were used for all AspH inhibition assays.
AspH substrate synthesis: A synthetic thioether linked cyclic peptide, hFX‐CP101‐119 (Figure 2D),23 was used as AspH substrate. hFX‐CP101‐119 was designed based on 19 EGFD1 amino acid residues of the sequence of human coagulation factor X (hFX amino acids 101–119), which is a reported cellular AspH substrate.48 hFX‐CP101−119 was prepared with a C‐terminal amide; it was synthesised by an intramolecular thioetherification cyclisation reaction from the corresponding linear peptide (d‐Ala replacing Cys101hFX and Ser replacing Cys112hFX) which was obtained by microwave‐assisted solid phase peptide synthesis using the Fmoc‐protection strategy.21
AspH inhibition assays: AspH inhibition assays were performed at 2OG and FeII concentrations close to the relevant K m values as previously described.24 Co‐substrate/cofactor stock solutions (l‐ascorbic acid, LAA: 50 mM in MQ‐grade water; 2‐oxoglutarate, 2OG: 10 mM in MQ‐grade water; ammonium iron(II) sulfate hexahydrate, FAS, (NH4)2Fe(SO4)2 ⋅ 6H2O: 400 mM in 20 mM HCl diluted to 1 mM in MQ‐grade water) were freshly prepared from commercial solids (Sigma Aldrich) on the day the assays were performed.
Solutions of the 2,4‐PDCA derivatives (100 % DMSO) were dry dispensed across 384‐well polypropylene assay plates (Greiner) in a 3‐fold and 11‐point dilution series (100 μM top concentration) using an ECHO 550 acoustic dispenser (Labcyte). DMSO and 2,4‐PDCA (1) were used as negative and positive controls. The DMSO concentration was kept constant at 0.5 % (v/v) throughout all experiments (using the DMSO backfill option of the acoustic dispenser). Each reaction was performed in technical duplicates in adjacent wells of the assay plates; additionally, assays were performed in two independent duplicates on different days using different DMSO inhibitor solutions.
The Enzyme Mixture (25 μL/well), containing 0.1 μM His6‐AspH315‐758 in 50 mM HEPES buffer (pH 7.5), was dispensed across the inhibitor‐containing 384‐well assay plates with a multidrop dispenser (ThermoFischer Scientific) at 20 °C under ambient atmosphere. The plates were subsequently centrifuged (1000 rpm using a Heraeus Megafuge 16 centrifuge equipped with a M‐20 rotor, 15 s) and incubated for 15 min. The Substrate Mixture (25 μL/well), containing 2.0 μM hFX‐CP101‐119, 200 μM LAA, 6.0 μM 2OG, and 4.0 μM FAS in 50 mM HEPES buffer (pH 7.5), was added using the multidrop dispenser. Note: The multidrop dispenser ensured proper mixing of the enzyme and the substrate mixtures which was essential for assay reproducibility. The plates were centrifuged (1000 rpm using a Heraeus Megafuge 16 centrifuge equipped with a M‐20 rotor, 15 s) and after incubating for 7 min, the enzyme reaction was stopped by addition of 10 % (v/v) aqueous formic acid (5 μL/well). The plates were centrifuged (1000 rpm using a Heraeus Megafuge 16 centrifuge equipped with a M‐20 rotor, 60 s) and analysed by MS.
MS‐analyses were performed using a RapidFire RF 365 high‐throughput sampling robot (Agilent) attached to an iFunnel Agilent 6550 accurate mass quadrupole time‐of‐flight (Q‐TOF) mass spectrometer operated in the positive ionisation mode. Assay samples were aspirated under vacuum for 0.4 s and loaded onto a C4 solid phase extraction (SPE) cartridge. After loading, the C4 SPE cartridge was washed with 0.1 % (v/v) aqueous formic acid to remove non‐volatile buffer salts (5 s, 1.5 mL/min). The peptide was eluted from the SPE cartridge with 0.1 % (v/v) aqueous formic acid in 85/15 (v/v) acetonitrile/water into the mass spectrometer (5 s, 1.25 mL/min) and the SPE cartridge re‐equilibrated with 0.1 % (v/v) aqueous formic acid (1 s, 1.25 mL/min). The mass spectrometer parameters were: capillary voltage (4000 V), nozzle voltage (1000 V), fragmentor voltage (365 V), gas temperature (280 °C), gas flow (13 L/min), sheath gas temperature (350 °C), sheath gas flow (12 L/min). The m/z +2 charge states of the cyclic peptide (substrate) and the hydroxylated cyclic peptide (product) were used to extract ion chromatogram data, peak areas were integrated using RapidFire Integrator software (Agilent). The data were exported into Microsoft Excel and used to calculate the % conversion of the hydroxylation reaction using the equation: % conversion=100 x (integral product cyclic peptide) / (integral substrate cyclic peptide+integral product cyclic peptide). Normalised dose‐response curves (2,4‐PDCA and DMSO controls) were obtained from the raw data by non‐linear regression (GraphPad Prism 5) and used to determine IC50 values. The standard deviation (SD) of two independent IC50 determinations (n=2) was calculated using GraphPad Prism 5. Z’‐factors and signal‐to‐noise (S/N) ratios were calculated according to the cited literature using Microsoft Excel (Figure S2).36
PHD2, FIH, and KDM4E inhibition assays: The in vitro PHD2,49 FIH,49 and KDM4E39, 40 inhibition assays were performed as described in the cited literature using recombinant human enzymes (PHD2181‐426,49 FIH,49 and KDM4E39, 40 were prepared according to established procedures). Synthetic peptide substrates were used: HIF‐α CODD, amino acids 558–574 for PHD2;49 HIF‐α CAD, amino acids 788–822 for FIH;49 H31‐15K9me3, histone 3 (H3) amino acids 1–15 with Lys9 of H3 bearing three methyl groups at the N ϵ position, Lys4 of H3 was substituted by an Ala and Lys14 of H3 by an Ile residue,40 for KDM4E. All peptides were prepared as C‐terminal amides by GL Biochem (Shanghai) Ltd.), monitoring peptide hydroxylation in the case of PHD2 and FIH (+16 Da mass shift) or peptide demethylation in the case of KDM4E (‐14 Da mass shift) by SPE‐MS.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the Wellcome Trust (106244/Z/14/Z), Cancer Research UK (C8717/A18245), and the Biotechnology and Biological Sciences Research Council (BB/J003018/1 and BB/R000344/1) for funding. L.B. thanks the Deutsche Forschungsgemeinschaft for a fellowship (BR 5486/2‐1).
L. Brewitz, A. Tumber, A. Thalhammer, E. Salah, K. E. Christensen, C. J. Schofield, ChemMedChem 2020, 15, 1139.
References
- 1.
- 1a. Veis A., Brownell A. G., Kefalides N., CRC Crit. Rev. Biochem. 1975, 2, 417–453; [DOI] [PubMed] [Google Scholar]
- 1b. Myllyharju J., Matrix Biol. 2003, 22, 15–24; [DOI] [PubMed] [Google Scholar]
- 1c. Gorres K. L., Raines R. T., Crit. Rev. Biochem. Mol. Biol. 2010, 45, 106–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. 2-Oxoglutarate-Dependent Oxygenases (Eds.: C. J. Schofield, R. P. Hausinger), The Royal Society of Chemistry, Cambridge, 2015.
- 3. Vaz F. M., Wanders R. J. A., Biochem. J. 2002, 361, 417–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Black J. C., Van Rechem C., Whetstine J. R., Mol. Cell 2012, 48, 491–507; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Greer E. L., Shi Y., Nat. Rev. Genet. 2012, 13, 343–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Rasmussen K. D., Helin K., Genes Dev. 2016, 30, 733–750; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Shen L., Song C.-X., He C., Zhang Y., Annu. Rev. Biochem. 2014, 83, 585–614; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c. Jia G., Fu Y., Zhao X., Dai Q., Zheng G., Yang Y., Yi C., Lindahl T., Pan T., Yang Y.-G., He C., Nat. Chem. Biol. 2011, 7, 885–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Kaelin W. G., Ratcliffe P. J., Mol. Cell 2008, 30, 393–402; [DOI] [PubMed] [Google Scholar]
- 6b. Schofield C. J., Ratcliffe P. J., Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Vasta J. D., Raines R. T., J. Med. Chem. 2018, 61, 10403–10411; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Kivirikko K. I., Myllyharju J., Matrix Biol. 1998, 16, 357–368; [DOI] [PubMed] [Google Scholar]
- 7c. Fuller G. C., J. Med. Chem. 1981, 24, 651–658. [DOI] [PubMed] [Google Scholar]
- 8. Majamaa K., Hanauske-Abel H. M., Günzler V., Kivirikko K. I., Eur. J. Biochem. 1984, 138, 239–245. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Cunliffe C. J., Franklin T. J., Hales N. J., Hill G. B., J. Med. Chem. 1992, 35, 2652–2658; [DOI] [PubMed] [Google Scholar]
- 9b. Baader E., Tschank G., Baringhaus K. H., Burghard H., Günzler V., Biochem. J. 1994, 300, 525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rose N. R., McDonough M. A., King O. N. F., Kawamura A., Schofield C. J., Chem. Soc. Rev. 2011, 40, 4364–4397. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Akizawa T., Iwasaki M., Otsuka T., Reusch M., Misumi T., Adv. Ther. 2019, 36, 1438–1454; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Dhillon S., Drugs 2019, 79, 563–572. [DOI] [PubMed] [Google Scholar]
- 12. Leung I. K. H., Krojer T. J., Kochan G. T., Henry L., von Delft F., Claridge T. D. W., Oppermann U., McDonough M. A., Schofield C. J., Chem. Biol. 2010, 17, 1316–1324. [DOI] [PubMed] [Google Scholar]
- 13. Simkhovich B. Z., Shutenko Z. V., Meirēna D. V., Khagi K. B., Mežapuķe R. J., Molodchina T. N., Kalvlņš I. J., Lukevics E., Biochem. Pharmacol. 1988, 37, 195–202. [DOI] [PubMed] [Google Scholar]
- 14. Chen Y. K., Bonaldi T., Cuomo A., Del Rosario J. R., Hosfield D. J., Kanouni T., Kao S.-c., Lai C., Lobo N. A., Matuszkiewicz J., McGeehan A., O'Connell S. M., Shi L., Stafford J. A., Stansfield R. K., Veal J. M., Weiss M. S., Yuen N. Y., Wallace M. B., ACS Med. Chem. Lett. 2017, 8, 869–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Stenflo J., Holme E., Lindstedt S., Chandramouli N., Huang L. H., Tam J. P., Merrifield R. B., Proc. Natl. Acad. Sci. USA 1989, 86, 444–447; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Korioth F., Gieffers C., Frey J., Gene 1994, 150, 395–399. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Kopan R., Ilagan M. X. G., Cell 2009, 137, 216–233; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b. Chillakuri C. R., Sheppard D., Lea S. M., Handford P. A., Semin. Cell Dev. Biol. 2012, 23, 421–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. Lavaissiere L., Jia S., Nishiyama M., de la Monte S., Stern A. M., Wands J. R., Friedman P. A., J. Clin. Invest. 1996, 98, 1313–1323; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17b. Wang K., Liu J., Yan Z.-L., Li J., Shi L.-H., Cong W.-M., Xia Y., Zou Q.-F., Xi T., Shen F., Wang H.-Y., Wu M.-C., Hepatology 2010, 52, 164–173. [DOI] [PubMed] [Google Scholar]
- 18. Guofang H., Boran X., Yanghui B., Chuanlin W., Beibei R., Bei S., Xuewei B., Bosn. J. Basic Med. Sci. 2018, 18, 297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.
- 19a. Ince N., de la Monte S. M., Wands J. R., Cancer Res. 2000, 60, 1261–1266; [PubMed] [Google Scholar]
- 19b. Zou Q., Hou Y., Wang H., Wang K., Xing X., Xia Y., Wan X., Li J., Jiao B., Liu J., Huang A., Wu D., Xiang H., Pawlik T. M., Wang H., Lau W. Y., Wang Y., Shen F., EBioMedicine 2018, 31, 287–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.
- 20a. Elvidge G. P., Glenny L., Appelhoff R. J., Ratcliffe P. J., Ragoussis J., Gleadle J. M., J. Biol. Chem. 2006, 281, 15215–15226; [DOI] [PubMed] [Google Scholar]
- 20b. Benita Y., Kikuchi H., Smith A. D., Zhang M. Q., Chung D. C., Xavier R. J., Nucleic Acids Res. 2009, 37, 4587–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brewitz L., Tumber A., Schofield C. J., J. Biol. Chem. 2020, 10.1074/jbc.RA1119.012202.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.
- 22a. Aihara A., Huang C.-K., Olsen M. J., Lin Q., Chung W., Tang Q., Dong X., Wands J. R., Hepatology 2014, 60, 1302–1313; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22b. Chang W. H., Forde D., Lai A. G., Cancer Commun. 2019, 39, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pfeffer I., Brewitz L., Krojer T., Jensen S. A., Kochan G. T., Kershaw N. J., Hewitson K. S., McNeill L. A., Kramer H., Münzel M., Hopkinson R. J., Oppermann U., Handford P. A., McDonough M. A., Schofield C. J., Nat. Commun. 2019, 10, 4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brewitz L., Tumber A., Pfeffer I., McDonough M. A., Schofield C. J., Sci. Rep. 2020, 10, 8650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.
- 25a. Derian C. K., VanDusen W., Przysiecki C. T., Walsh P. N., Berkner K. L., Kaufman R. J., Friedman P. A., J. Biol. Chem. 1989, 264, 6615–6618; [PubMed] [Google Scholar]
- 25b. Gronke R. S., Welsch D. J., VanDusen W. J., Garsky V. M., Sardana M. K., Stern A. M., Friedman P. A., J. Biol. Chem. 1990, 265, 8558–8565. [PubMed] [Google Scholar]
- 26.
- 26a. Dong X., Lin Q., Aihara A., Li Y., Huang C.-K., Chung W., Tang Q., Chen X., Carlson R., Nadolny C., Gabriel G., Olsen M., Wands J. R., Oncotarget 2014, 6, 1231–1248; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26b. Huang C.-K., Iwagami Y., Aihara A., Chung W., de la Monte S., Thomas J.-M., Olsen M., Carlson R., Yu T., Dong X., Wands J., PLoS One 2016, 11, e0150336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vitaku E., Smith D. T., Njardarson J. T., J. Med. Chem. 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- 28. Badawy A. A.-B., Int. J. Tryptophan Res. 2017, 10, 1178646917691938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rose N. R., Ng S. S., Mecinović J., Liénard B. M. R., Bello S. H., Sun Z., McDonough M. A., Oppermann U., Schofield C. J., J. Med. Chem. 2008, 51, 7053–7056. [DOI] [PubMed] [Google Scholar]
- 30. Hirsilä M., Koivunen P., Günzler V., Kivirikko K. I., Myllyharju J., J. Biol. Chem. 2003, 278, 30772–30780. [DOI] [PubMed] [Google Scholar]
- 31. Thalhammer A., Mecinović J., Loenarz C., Tumber A., Rose N. R., Heightman T. D., Schofield C. J., Org. Biomol. Chem. 2011, 9, 127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Albaneze-Walker J., Bazaral C., Leavey T., Dormer P. G., Murry J. A., Org. Lett. 2004, 6, 2097–2100. [DOI] [PubMed] [Google Scholar]
- 33.
- 33a. Guram A. S., Rennels R. A., Buchwald S. L., Angew. Chem. Int. Ed. 1995, 34, 1348–1350; [Google Scholar]; Angew. Chem. 1995, 107, 1456–1459; [Google Scholar]
- 33b. Louie J., Hartwig J. F., Tetrahedron Lett. 1995, 36, 3609–3612. [Google Scholar]
- 34. Kamer P. C. J., van Leeuwen P. W. N. M., Reek J. N. H., Acc. Chem. Res. 2001, 34, 895–904. [DOI] [PubMed] [Google Scholar]
- 35. Rabinowitz M. H., J. Med. Chem. 2013, 56, 9369–9402. [DOI] [PubMed] [Google Scholar]
- 36. Zhang J.-H., Chung T. D. Y., Oldenburg K. R., J. Biomol. Screening 1999, 4, 67–73. [DOI] [PubMed] [Google Scholar]
- 37. Conejo-Garcia A., McDonough M. A., Loenarz C., McNeill L. A., Hewitson K. S., Ge W., Liénard B. M., Schofield C. J., Clifton I. J., Bioorg. Med. Chem. Lett. 2010, 20, 6125–6128. [DOI] [PubMed] [Google Scholar]
- 38. McDonough M. A., McNeill L. A., Tilliet M., Papamicaël C. A., Chen Q.-Y., Banerji B., Hewitson K. S., Schofield C. J., J. Am. Chem. Soc. 2005, 127, 7680–7681. [DOI] [PubMed] [Google Scholar]
- 39. Bonnici J., Tumber A., Kawamura A., Schofield C. J., Phil. Trans. R. Soc. B 2018, 373, 20170071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hutchinson S. E., Leveridge M. V., Heathcote M. L., Francis P., Williams L., Gee M., Munoz-Muriedas J., Leavens B., Shillings A., Jones E., Homes P., Baddeley S., Chung C.-w., Bridges A., Argyrou A., J. Biomol. Screening 2012, 17, 39–48. [DOI] [PubMed] [Google Scholar]
- 41.
- 41a. Westaway S. M., Preston A. G. S., Barker M. D., Brown F., Brown J. A., Campbell M., Chung C.-w., Diallo H., Douault C., Drewes G., Eagle R., Gordon L., Haslam C., Hayhow T. G., Humphreys P. G., Joberty G., Katso R., Kruidenier L., Leveridge M., Liddle J., Mosley J., Muelbaier M., Randle R., Rioja I., Rueger A., Seal G. A., Sheppard R. J., Singh O., Taylor J., Thomas P., Thomson D., Wilson D. M., Lee K., Prinjha R. K., J. Med. Chem. 2016, 59, 1357–1369; [DOI] [PubMed] [Google Scholar]
- 41b. Westaway S. M., Preston A. G. S., Barker M. D., Brown F., Brown J. A., Campbell M., Chung C.-w., Drewes G., Eagle R., Garton N., Gordon L., Haslam C., Hayhow T. G., Humphreys P. G., Joberty G., Katso R., Kruidenier L., Leveridge M., Pemberton M., Rioja I., Seal G. A., Shipley T., Singh O., Suckling C. J., Taylor J., Thomas P., Wilson D. M., Lee K., Prinjha R. K., J. Med. Chem. 2016, 59, 1370–1387. [DOI] [PubMed] [Google Scholar]
- 42. Lin H., Li Q., Li Q., Zhu J., Gu K., Jiang X., Hu Q., Feng F., Qu W., Chen Y., Sun H., J. Enzyme Inhib. Med. Chem. 2018, 33, 777–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hillringhaus L., Yue W. W., Rose N. R., Ng S. S., Gileadi C., Loenarz C., Bello S. H., Bray J. E., Schofield C. J., Oppermann U., J. Biol. Chem. 2011, 286, 41616–41625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.
- 44a. Kaniskan H. Ü., Martini M. L., Jin J., Chem. Rev. 2018, 118, 989–1068; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44b. Thinnes C. C., England K. S., Kawamura A., Chowdhury R., Schofield C. J., Hopkinson R. J., Biochim. Biophys. Acta Gene Regul. Mech. 2014, 1839, 1416–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Al-Qahtani K., Jabeen B., Sekirnik R., Riaz N., Claridge T. D. W., Schofield C. J., McCullagh J. S. O., Phytochemistry 2015, 117, 456–461. [DOI] [PubMed] [Google Scholar]
- 46. Palatinus L., Chapuis G., J. Appl. Crystallogr. 2007, 40, 786–790. [Google Scholar]
- 47.
- 47a. Parois P., Cooper R. I., Thompson A. L., Chem. Cent. J. 2015, 9, 30; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47b. Cooper R. I., Thompson A. L., Watkin D. J., J. Appl. Crystallogr. 2010, 43, 1100–1107. [Google Scholar]
- 48. McMullen B. A., Fujikawa K., Kisiel W., Sasagawa T., Howald W. N., Kwa E. Y., Weinstein B., Biochemistry 1983, 22, 2875–2884. [DOI] [PubMed] [Google Scholar]
- 49. Holt-Martyn J. P., Chowdhury R., Tumber A., Yeh T.-L., Abboud M. I., Lippl K., Lohans C. T., Langley G. W., W. Figg Jr , McDonough M. A., Pugh C. W., Ratcliffe P. J., Schofield C. J., ChemMedChem 2020, 15, 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary