

Abstract

The repertoire of methods for the detection and chemotherapeutic treatment of prostate cancer (PCa) is currently limited. Prostate-specific membrane antigen (PSMA) is overexpressed in PCa tumors and can be exploited for both imaging and drug delivery. We developed and characterized four nanobodies that present tight and specific binding and internalization into PSMA+ cells and that accumulate specifically in PSMA+ tumors. We then conjugated one of these nanobodies to the cytotoxic drug doxorubicin, and we show that the conjugate internalizes specifically into PSMA+ cells, where the drug is released and induces cytotoxic activity. In vivo studies show that the extent of tumor growth inhibition is similar when mice are treated with commercial doxorubicin and with a 42-fold lower amount of the nanobody-conjugated doxorubicin, attesting to the efficacy of the conjugated drug. These data highlight nanobodies as promising agents for the imaging of PCa tumors and for the targeted delivery of chemotherapeutic drugs.
Introduction
Prostate cancer (PCa) is commonly detected by antibody-based assays that measure the serum concentration of the prostate-specific antigen (PSA),1−3 but these assays are prone to high error rates.4−6 In addition, although chemotherapies are often used to treat castration-resistant PCa,7 some potentially effective chemotherapies against PCa, such as doxorubicin (DOX), do not sufficiently accumulate within tumors and have a large distribution volume, resulting in low treatment efficacy and high nonspecific toxicity.8 Novel means for both the detection of PCa and the targeted delivery of cytotoxic agents are therefore urgently required.9 One promising target that can be employed to address both these issues is the prostate-specific membrane antigen (PSMA);10 a transmembrane protein that is overexpressed in PCa,11 possibly due to its folate hydrolase activity, which induces cell proliferation.11−13 PSMA is mostly expressed on the membranes of PCa cells, although it is also expressed on the neovasculature of many carcinomas, including PCa.11 Importantly, the overexpression of PSMA is associated with malignant, castration-resistant PCa, reduced androgen-receptor expression, and poor PCa prognosis;14−17 therefore, it can be used to detect PCa, identify the stage of the disease, and promote personalized, tumor-specific medicine.17,18 Notably, targeting PSMA can be especially important in the treatment of aggressive, androgen-independent PCa tumors, where its expression increases while that of PSA decreases,19 and where first-line treatments often fail making chemotherapeutic drugs a necessity.
PSMA has been extensively exploited as a target by multiple research groups, which presented promising compounds for PSMA-targeted diagnostics and inhibition, mostly in the field of nuclear medicine.20−25 Yet, to date, most proteins that were found to bind the extracellular region of PSMA with a sufficiently high affinity (nanomolar range) are monoclonal antibodies or antibody fragments,26−28 which have several caveats for both molecular imaging and cancer treatment purposes. For instance, the long serum half-life and broad biodistribution of antibodies often reduce the signal-to-noise ratio29 and maintain them in the circulation for long periods of time.30 These effects increase toxic side effects when the antibody is conjugated to a cytotoxic radioisotope or decrease specificity when the antibody is conjugated to a drug because the antibody–drug conjugate may internalize into nontumor cells. Moreover, the large size of antibodies often hinders their ability to penetrate into the core of the abnormal tumor tissue, thus dramatically reducing their drug-delivering efficiency.31 Antibody fragments may solve some of these caveats, but they often show weaker binding and low stability, and they may expose previously masked immunogenic epitopes.32 While some nonantibody PSMA binders and inhibitors have been described and show promising results,21−25,33 other engineered PSMA-binding peptides show low affinities, namely, at the high-nanomolar to micromolar range.34,35 An alternative approach, which combines the advantages of antibodies and smaller protein scaffolds to exploit the potential of PSMA as a target, could be found in the form of nanobodies (NBs).
NBs, also known as VHHs, are the single-chain variable domains of heavy-chain antibodies (HCAb).36,37 As the NB is the only fragment of the HCAb that mediates antigen binding, it can be expressed separately from the rest of the HCAb without reducing affinity,38 resulting in a minute (∼15 kDa), nonimmunogenic, highly target-specific protein, which is an excellent candidate for use as scaffold for in vivo imaging and targeted therapy applications.29,39 Indeed, in two separate pioneering studies, Evazalipour et al.40 and Zare et al.41 generated anti-PSMA NBs that successfully bound PSMA-expressing cells, both in vitro and in vivo. However, to generate a clinically applicable NB–drug conjugate that can specifically target PSMA, the structure of such compounds, their effects on PSMA activity and cell viability, and their potential as drug carriers should be determined empirically.
In the current study, we isolated and selected four NBs from the serum of a camel injected with a recombinant extracellular region of human PSMA. Then, we evaluated the structures, PSMA-binding epitopes, affinities, and specificities of these NBs and conjugated the most potent NB to DOX, thus generating a novel NB–drug conjugate that can specifically target PSMA-expressing cells. Our findings highlight the potential of using NB-based constructs for both the diagnosis and treatment of PCa.
Results
Isolation of Anti-PSMA NBs
RNA extracted from the lymphocytes of a PSMA-injected camel served as the basis for a NB phage-display library in the size of 107 variants. The phage-display panning process against PSMA yielded 47 bacterial colonies that express NB variants, wherein 23 unique NB sequences were identified. Of these, four NBs whose sequences repeated several times, and which showed the strongest binding to PSMA in ELISA, were chosen for purification (Figure S1A,B). The purified NBs (termed NB7, NB8, NB13, and NB37 (all sequences are presented in the Supporting Information) were of the expected size of ∼16 kDa (Figure S1C), and the yield was 4–18 mg/L culture.
NBs Bind to PSMA with a Pico- to Nanomolar Affinity
SPR revealed that the in vitro binding affinity of the four purified NBs to PSMA was in the pico- to nanomolar range but varied considerably between the NBs (Table 1, Figure 1A–D).
Table 1. Kinetic Binding Constants for the Interaction between PSMA and the NBs, As Measured by SPRa.
| NB | Kon [M–1 s–1] | Koff [s–1] | KD [nM] |
|---|---|---|---|
| NB7 | (7.1 × 105) ± (4.5 × 102) | (3.9 × 10–5) ± (2.8 × 10–7) | 0.055 |
| NB8 | (2.0 × 104) ± (4.2 × 101) | (1.2 × 10–4) ± (5.3 × 10–7) | 6.0 |
| NB13 | (3.6 × 104) ± (8.9 × 101) | (2.2 × 10–5) ± (9.9 × 10–7) | 0.60 |
| NB37 | (2.2 × 104) ± (5.3 × 101) | (7.5 × 10–5) ± (6.6 × 10–7) | 3.4 |
Values represent the mean ± SD.
Figure 1.

NBs bind to PSMA in vitro and to PSMA-expressing prostate cancer cells. The response units (RU), measured using SPR and a 1:1 Langmuir kinetic model, were used to calculate the affinity (KD) of immobilized NB7 (A), NB8 (B), NB13 (C), and NB37 (D) to PSMA. The PSMA concentrations were 25, 50, 100, 1600, or 3200 pM for the NB7 sensograms, and 2.94, 5.88, 11.75, 23.50, or 47.00 nM for the NB8, NB13, and NB37 sensograms. The bottom curve in each sensogram represents the lowest concentration, while the top curve represents the highest concentration. A FACS analysis (n = 3) was used to determine the binding of these NBs (0.1–1000 nM) to PC3-PIP (PSMA+) cells (E) and to PC3-flu (PSMA–) cells (F). For convenience, each fluorescence value was normalized to the fluorescence values at the highest and lowest concentrations of PC3-PIP cells.
FACS-based titration curves showed that all four NBs bind to PC3-PIP (PSMA+) prostate cancer cells in a dose-dependent manner (Figure 1E), but they do not bind to PC3-flu (PSMA–) cells (Figure 1F). Notably, the FACS binding curves did not reach a plateau, presumably because the NBs were internalized into the cells (see below); therefore, this data set was not used to calculate the KD values. An enzymatic activity assay revealed that the NBs do not compromise the enzymatic NAALDase activity of PSMA (Figure S2), suggesting that they bind to nonfunctional epitopes of the protein.
NB Structures and Their PSMA Binding Epitopes
The considerable variability (up to 100-fold) in the KD values of the four NBs could stem from their potentially different PSMA-binding epitopes. To test this possibility, the crystal structures of NB7, NB8, and NB37 were solved at a resolution of 1.5 Å (NB8, NB37) or 2.65 Å (NB7) (Figure 2A–C, Tables S1–S3); NB13 crystals could not be obtained under any of the attempted growth conditions. While the structures of NB8 and NB37 were very similar (in line with the high homology of their sequences, which differ in a single amino acid residue), the structure of NB7 was markedly different and included more β-sheets and fewer random regions. A SAXS analysis (Figure S3) indicated that the monomeric PSMA is stable in PBS and shows concentration-dependent intermolecular interactions, suggesting that PSMA monomers interact with each other in the solution, which corroborates with the ability of PSMA to form dimers.10,42 Scattering curves of the monomeric PSMA in the presence of increasing NB concentrations show that the Guinier region (S2 ≤ 0.006 Å–2) was linear, indicating little or no aggregation in any of the samples (Figure S4). However, adding low concentrations of NB13 to PSMA, namely, at PSMA:NB13 ratios between 1:0.5 and 1:3, increased the radius of gyration (Rg) of PSMA (Figure S5). The Rg of the PSMA–NB complexes shifted from the original Rg of PSMA (43 Å, Figure S5) to a higher Rg for NB7 and to a lower Rg for NB8, NB13, and NB37. The Rg of PSMA alone was comparable to the calculated Rg value based on the crystal structure of the PSMA monomer. The distribution of pairwise distances within the particle (Figure S6) is represented by P(r) (see Experimental Section). The Dmax of the PSMA P(r) was 115 Å (Figure S7), and the shape of the P(r) distribution indicates an elongated structure. The binding of PSMA to NB7 and to NB13 increased Dmax, while its binding to NB8 and to NB37 decreased it (Table S4).
Figure 2.

Structural analysis of NBs and their PSMA-binding epitopes. (A–C) Solved crystal structures of NB7 (A, red; 2.65 Å, PDB code 6XXN), NB8 (B, yellow; 1.5 Å, PDB code 6XXO), and NB37 (C, green; 1.5 Å, PDB code 6XXP). CDRs 1, 2, and 3 are labeled in magenta, orange, and cyan, respectively. (D–F) Structural reconstruction of protein complexes, based on their SAXS-resolved low-resolution structures (gray mesh), fitted with the crystal structure of PSMA (0.5 mg/mL), either alone (D; PDB code 1Z8L) or with 0.2 mg/mL NB7 (E, red; PDB code 6XXN) or NB37 (F, green; PDB code 6XXP). The PSMA monomers are labeled individually by Roman numerals; biological dimers are formed by I + II and III + IV, while nonbiological dimers are formed by I + III and II + IV. (G–H) Computational docking analyses of PSMA and NB7 (G) and of PSMA and NB37 (H). (G) PSMA monomers are colored in light blue and cyan, while NB7 is colored in red. Key interactions (according to Table S5) are shown as black dashed lines. (H) PSMA is colored in cyan, and NB37 is colored in green. Key interactions (Table S6) are shown as black dashed lines. .
Next, we calculated 20 reconstituted ab initio models from the data that were averaged using DAMMIN43 and DAMAVER.44 We used the crystal structures of PSMA, NB7, and NB37 and fit them to the reconstituted structures of a sample containing either PSMA alone (0.5 mg/mL) or PSMA (0.5 mg/mL) with NB7 or NB37 (0.2 mg/mL in each case) (Figure 2D–F). We assumed that the binding mechanism of NB8 is similar to that of NB37 due to their high sequence and structure homologies, and we did not generate a model of NB13 because we did not have its crystal structure.
The models suggested that PSMA forms a nonbiological dimer with interactions between the N-termini of both monomers (Figure 2D), similar to those observed in the tetrameric crystal structure of PSMA (PDB code 1Z8L). The low-resolution structure is asymmetrical such that one monomer appears to be smaller than the other; notably, however, this apparent asymmetry could have stemmed from the presence, in the solution, of both monomers and dimers such that the average size could reflect the combined size of both species. The models suggested that in the presence of NB7, another PSMA monomer is added to the dimer complex by forming a biological dimer with one of the PSMA monomers. According to this model, NB7 binds each monomer in the biological dimer with a different complementarity-determining region (CDR) (Figure 2E), leading to an increase in the complex size and in Rg. In the case of the PSMA–NB37 complex, NB37 appears to bind to PSMA at the N-terminus (Figure 2F), thus disrupting the nonbiological dimer, leading to a decrease in Rg.
Docking Analysis
SAXS results and molecular docking simulation of NB7 (PDB code 6XXN) with PSMA (PDB code 1Z8L) revealed that NB7 binds to PSMA close to the dimerization interface and simultaneously interacts with both monomers (Figure 2G, Table S5). NB7 interacts with one PSMA monomer mainly via CDR3 and CDR1, while CDR2 and several non-CDR residues interact with the second monomer in the homodimer (the main contributing interactions are presented in Figure 2G and are further detailed in Table S5). According to the docking simulation, NB37 (PDB code 6XXP) binds to an epitope close to the N-terminus of PSMA (Figure 2H, Table S6). The predicted interactions between NB37 and PSMA occur mainly via CDR2, and some occur via CDR3; the main contributing interactions are presented in Figure 2H and are detailed further in Table S6. In total, NB7 has more interactions than NB37, as the ligand contact surface area of the former is 969.34 Å2, as compared with 443.72 Å2 of the latter (Table S7).
NBs Accumulate in PSMA-Expressing Tumors in Vivo
Next, we aimed to determine whether the NBs bind specifically to PSMA-expressing PCa tumors in vivo and whether differences between their affinities correlate with their in vivo accumulation in tumors. To this end, we acquired whole-body near-infrared (NIR) optical images of nude mice inoculated with PC3-PIP and PC3-flu xenografts. We captured the images 3 and 6 h after injecting the labeled NB (early and middle time points, respectively) and again when the fluorescent signal could no longer be detected in vivo (late time point). In some mice, the signal was still detectable 56 h after the injection; we euthanized these mice due to ethical considerations and we denote the late time point in these cases as >56 h.
At the early imaging time point, the NBs were detected both in the kidneys and in the PC3-PIP tumors but not in the PC3-flu tumors. At the middle imaging time point, however, they were completely cleared from the kidneys and remained only in the PC3-PIP tumors (Figure 3, A−C). The duration until the fluorescent signal was no longer detected (late time point) depended on the affinity of the NB to PSMA such that NBs with higher affinities (lower KD) required longer durations for signal clearance (24 h for NB37, 32 h for NB8 and NB13, and >56 h for NB7). These low clearance rates suggest that all four NBs can potentially be used for in vivo applications, such as clinical imaging and tumor-specific drug delivery. In fact, even after the fluorescent signal was undetectable by whole-body imaging, it was still observed in the tumors ex vivo (Figure S8), where it increased in the PC3-PIP tumors, relative to the kidneys, over time (Figure 3D). The signal in other organs and in the PC3-flu tumors was much weaker throughout the experiment. For example, for NB8 after 3 h, the signal intensity was 171 000 (counts/cm2)/s in the PC3-PIP tumor and 94 700 (counts/cm2)/s in the kidneys, as compared with 21 300 (counts/cm2)/s in the PC3-flu tumor. This finding suggests that the NBs accumulated specifically in the PSMA+ tumors and were then cleared predominantly by the kidneys, as could be expected given the small size of NBs [16 kDa, while the renal cutoff is ∼60 kDa 45].
Figure 3.

In vivo whole-body NIR imaging of labeled NBs. PC3-flu (PSMA–) and PC3-PIP (PSMA+) PCa cells were co-injected as xenografts into the left and right upper flanks, respectively, of athymic nude mice. Nine days later, the mice were intravenously injected with fluorescently labeled NBs (from left to right: NB7, NB13, NB8, and NB37; the KD of each NB is shown in parentheses for convenience) and whole-body images were captured 3 h (A) and 6 h (B) postinjection and again when the signal was no longer detectable (C); 32 h for NB8 and NB13 and 24 h for NB37. Mice injected with NB7 still showed a fluorescent signal 56 h postinjection, at which point they were imaged and then euthanized. In each individual image, the left mouse was injected with tumor cells but not with NBs, the middle mouse was injected with NBs but not with tumor cells, and the right mouse was injected with both tumor cells and NBs. (D) Quantification of the AF680 fluorescent signals from the dissected organs at each time point for each NB. PC3-PIP tumors express PSMA, whereas PC3-flu tumors do not.
NBs Are Internalized into PSMA-Expressing Cells
For the NBs to be able to deliver chemotherapeutic agents into PSMA+ prostate tumor cells (a prerequisite for the efficacy of many existing drugs) they must be internalized specifically into PSMA+ cells. To test the internalization capability of the four NBs, we labeled them fluorescently and incubated them with either live PC3-PIP (PSMA+) or live PC3-flu (PSMA–) cells, together with a PE-anti-PSMA antibody and a Hoechst nuclear staining solution. Confocal imaging of the PC3-PIP cells revealed that the NBs colocalize with PSMA and appear both in the cell membranes and in clusters inside the cells (Figure 4A–D). Notably, the anti-PSMA antibody was not found inside the cells in the absence of a NB (Figure 4I), suggesting that the NB may prompt the internalization of PSMA while it is still bound to the anti-PSMA antibody. Imaging of the PC3-flu cells showed that neither the NBs nor the anti-PSMA antibodies bind to or internalize into the cells (Figure 4E–H). A long-term internalization assay revealed that NBs with higher affinities to PSMA (namely, NB7 and NB13) were internalized into the PSMA-expressing cells much faster than those with lower affinities (Figure S9). On the basis of the in vitro and in vivo affinities to PSMA and on the purification yields of each NB, we chose to generate a NB–drug conjugate using NB7.
Figure 4.
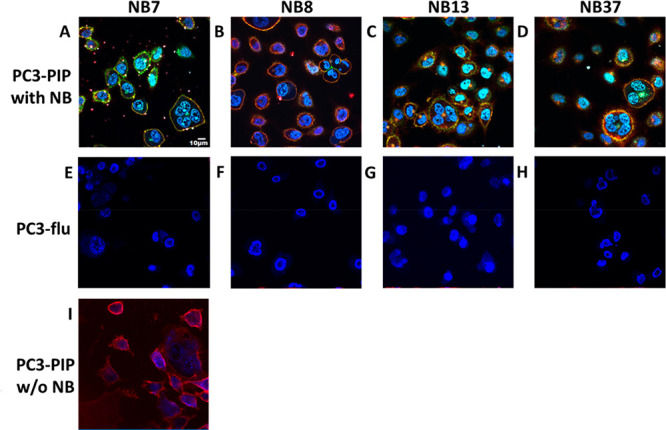
Confocal imaging of the internalization of NBs into prostate cancer cells. PC3-PIP (PSMA+) cells (A–D) and PC3-flu (PSMA–) cells (E–H) were incubated for 10 min with a Hoechst reagent (nuclei staining, blue), a PE-anti-PSMA antibody (red), and 100 nM of either NB7 (A, E), NB8 (B, F), NB13 (C, G), or NB37 (D, H), each labeled with Dylight 488 (green). Alternatively, PC3-PIP cells were incubated for 10 min with the PE-anti-PSMA antibody without any NB (I).
Conjugation of NB7cys to DOX
The clustered pattern of the NBs and PSMA inside the target cells suggests that their internalization is mediated by intracellular vesicles, as was previously shown for other PSMA binders.46−51 As both intracellular vesicles are typically acidic,52−55 we conjugated NB7 to DOX via the pH-sensitive linker N-(β-maleimidopropionic acid) hydrazide (BMPH) (Figures S10A and S11), which is hydrolyzed at pH < 6.0.55 We hypothesized that the acidic conditions in the vesicles would hydrolyze the covalent bond between the linker and DOX, thus releasing DOX from the conjugate and enabling it to diffuse outside the vesicles and into the cytosol, where it could penetrate the nucleus56 and, presumably, inhibit DNA transcription.
The conjugated protein, termed NB7cysDOX, was purified using size-exclusion chromatography (Figure S10B). The addition of DOX to NB7 slightly increased its size (namely, by ∼700 Da), but the hydrophobic nature of DOX reduced its elution rate, which allowed us to separate the conjugated from the nonconjugated proteins. The fluorescence of DOX (excitation 495 nm and emission 560 nm) led to the absorbance of only the conjugated protein at 488 nm, which is sufficiently close to 495 nm, and further distinguished between the conjugated and nonconjugated protein fractions. The NB7cysDOX fraction was further evaluated using mass spectrometry in acidic pH (pH = 4), in which DOX is cleaved from the linker and, thereby, from the NB. This analysis revealed that the mass of the conjugated protein is higher by 185 Da than that of NB7cys alone (namely, 16 141 Da, as compared with 16 326 Da, respectively; Figure S10C); this difference reflects the combined size of NB7cys and the BMPH linker, indicating that all NB molecules are conjugated to DOX and that DOX is released under acidic conditions. A FACS analysis of the binding of NB7cys and NB7cysDOX to the PSMA-expressing PC3-PIP cells (Figure S10D) revealed that the conjugation of DOX does not compromise the binding of NB7cys to these cells.
NB7cysDOX Is Cytotoxic to PSMA-Expressing Cells
To evaluate the ability of the NB7cysDOX conjugate to internalize specifically to PSMA-expressing cells and the successive detachment of DOX from the conjugate, we incubated PC3-PIP and PC3-flu cells for 15 min with Hoechst 33342 (nuclear staining) and with either 1.5 μg/mL DOX (which is fluorescent) or a molar equivalent of Dylight650-labeled NB7cys or NB7cysDOX (Figure 5). When incubated alone with the PCa cells, DOX (a small and hydrophobic molecule) diffused spontaneously into both PC3-PIP and PC3-flu cells, where it was found homogeneously scattered throughout the cytosol. Conversely, as expected from its PSMA-dependent internalization mechanism (see Figure 4A), NB7cys accumulated only in PC3-PIP cells, where it was found mostly in the cell membrane and had begun internalizing into the cytosol. The distribution of NB7cysDOX was very similar to that of NB7cys (namely, in defined regions on the membranes and cytosols of PC3-PIPcells but not of PC3-flu cells), but DOX was scattered in multiple regions within the cells, mostly separate from NB7cys (although small amounts of NB7cys were found within the DOX clusters). To test whether the DOX released from the internalized conjugate retains its cytotoxic activity, we incubated PC3-PIP cells for 24 h with 1.5 μg/mL DOX or with an equivalent molar amount of NB7cysDOX or NB7cys, counted the number of cells in each well, and compared it to that of untreated cells (Figure S12A). This assay revealed that the incubation with DOX or, to a greater extent, with NB7cysDOX significantly reduced the number of cells in the well. Next, in a different set of experiments, we incubated PC3-PIP cells for 24 h with either DOX, NB7cys, or NB7cysDOX (as described above) and then labeled the cells with propidium iodide (PI), a fluorescent marker of late apoptosis and necrosis. A FACS analysis (Figure S12B) revealed that whereas treating the cells with NB7cys did not change the PI signal, treating them with either DOX or, to a greater extent, NB7cysDOX considerably increased the signal. Treating PC3-PIP cells with TMRE (a reagent that labels active mitochondria57) revealed that DOX and NB7cysDOX similarly reduced the mitochondrial membrane potential (Figure S12C). Treating the cells with FCCP, which interrupts the mitochondrial membrane potential and served as a positive control, also significantly reduced the mitochondrial membrane potential of the PC3-PIP cells. In contrast, NB7cys alone did not change the TMRE signal, as compared with untreated cells. Taken together, these results indicate that NB7cysDOX is at least as cytotoxic to PSMA+ cells as DOX alone.
Figure 5.

Confocal imaging of the internalization of NB7cys, DOX, and the NB7cysDOX conjugate into PCa cells. PC3-PIP (PSMA+) and PC3-flu (PSMA–) cells were incubated with either DOX (green autofluorescence), NB7cys labeled with Dylight 650 (red), or NB7cysDOX labeled with Dylight 650 (red). Images were taken after 15 min of incubation. The separation between the red and green signals indicates the cleavage of DOX from the NB7cysDOX conjugate. Some of the DOX molecules were found separate from NB7cys (white arrows), while others colocalized with NB7cys (yellow arrows).
NB7cysDOX Inhibits Tumor Growth in Mice
To test the activity of NB7cysDOX in vivo, we created PC3-PIP tumor xenografts in athymic nude mice, and once the tumors reached ∼200 mm3, we intravenously treated them (twice a week for 3 weeks) with either saline (control), 2 mg/kg (2.86 μmol/kg) commercial DOX, which was previously shown to be effective in mice and is similar to that used in humans,58 or 1.4 mg/kg (0.087 μmol/kg) NB7cysDOX, which represents a molar dose of DOX that is 42-fold lower than that used for DOX alone. We measured the size of the tumor before each injection, but some mice had to be euthanized due to ethical considerations (namely, large tumor burden or physical deterioration) by 8 d following treatment initiation; in these mice, we estimated the tumor size in successive time points by extrapolation.59
The last time point at which tumors from all live mice were included in the analysis was 8 d following treatment initiation; at that time point, the average tumor size was significantly smaller in mice treated with NB7cysDOX than in those treated with saline (Figure 6A). An overtime analysis (Figure S13A) and rate-based growth slopes for treatment/control (T/C; Figure 6B) revealed that the tumor growth rate was, indeed, lower in mice treated with NB7cysDOX than in those treated with saline. Moreover, although the amount of DOX administered to NB7cysDOX-treated mice was significantly lower than that administered to DOX-treated mice (who received a 2 mg/kg dose of DOX), the tumor growth rate was similar in both groups, indicating the effectiveness of the NB7cysDOX conjugate relative to DOX administered alone. Notably, three mice were excluded from the DOX-treated group for the rate-based analysis, according to the procedure described in ref (59). In line with these findings, more mice reached a maximal tumor size (i.e., the size in which mice were euthanized due to ethical considerations) in the saline-treated group than in either the DOX-treated or the NB7cysDOX-treated groups, which were not significantly different from each other (Figure S13B).
Figure 6.

In vivo and in situ effects of NB7cysDOX on PC3-PIP (PSMA+) tumors. PC3-PIP xenografts in athymic nude mice were treated with either saline (control), 2 mg/kg commercial DOX, or 1.4 mg/kg NB7cysDOX. (A) Mean tumor volume after 8 d of treatment (before any mouse was excluded from the experiment due to ethical considerations). *p < 0.05 versus control (Student’s t test; n = 7 for controls and n = 8 for DOX and NB7cysDOX). (B) Slope of calculated logarithmic tumor growth in the treated versus control (T/C) groups. *p < 0.05 versus control (Student’s t test; n = 7 for controls, n = 4 for DOX, and n = 8 for NB7cysDOX; (C) Representative tissue section from a tumor, obtained 4 d after treatment termination, from one mouse treated with NB7cysDOX and labeled with PE-anti-PSMA (red) and FITC-anti-His (green) to identify PSMA and NB7, respectively. Blue staining indicates nuclei, and white arrows highlight the colocalization of PSMA and NB7. (D) H&E staining (top row) and TUNEL (green) and PI (red) staining (bottom row) of tissue sections from tumors obtained 4 d after treatment termination. White arrows indicate the colocalization of TUNEL and PI. Scale bars apply to all three images in the same row.
Next, we extracted tumors from the treated mice 4 d after the final dose of NB7cysDOX and labeled them with PE-anti-PSMA and FITC-anti-His. A histological analysis revealed that while PSMA was localized mostly to the membranes of the tumor cells, NB7cysDOX appeared either colocalized with PSMA or in the cytoplasm (Figure 6C), indicating that NB7cysDOX indeed reaches and remains within tumors for at least 4 days. Staining the tumors with H&E revealed that while tumors obtained from mice treated with saline were crowded and strongly labeled by H&E, tumors from mice treated with DOX or with NB7cysDOX were necrotic, fewer, and with large vacancies between cells (Figure 6D, top). A TUNEL assay revealed only a few apoptotic cells in tumors obtained from saline-treated mice, as compared with significant apoptosis in tumors obtained from DOX- or NB7cysDOX-treated mice (Figure 6D, bottom). These findings demonstrate that while the number of DOX molecules administered to NB7cysDOX-treated mice is less than 3% of that administered to the DOX-treated mice, the cytotoxic effect of the drug is similar in both groups.
Discussion and Conclusions
The chemotherapeutic treatment options for castration-resistant PCa are currently limited to taxanes, as most cytotoxic compounds exhibit high toxicity and severe side effects, even at doses that provide limited efficacy. In parallel, our ability to identify PCa tumors and determine their aggressiveness through imaging also remains a challenge. Several small-molecule inhibitors and ligands of PSMA have previously been shown to successfully detect, and potentially treat, PSMA-expressing tumors, particularity in the field of nuclear medicine,21,63−67 and some of these small molecules are clinically used to treat PSMA-expressing PCa lesions.66,68,69 Progress has also been made in the field of antibodies targeting PSMA, both for PCa detection and for radiotherapy purposes.70−72 In the current study, we report the development of PSMA-binding NBs that can detect PSMA+ PCa tumor with high specificity and deliver small-molecule cytotoxic drugs directly into the tumor cells. Our findings thus implicate NBs as a highly advantageous contribution to the rather limited repertoire of diagnostic and therapeutic agents for castration-resistant PCa.
The four NBs that we isolated demonstrated a wide range of affinities to PSMA, which presumably stems from their different PSMA-binding epitopes. Such a range of binding affinities, including in the picomolar range, has not been demonstrated in either of the two previous studies that developed anti-PSMA NBs.40,41 In addition, our NBs do not affect the enzymatic activity of PSMA, which we consider an advantage because our current understanding of the processes underlying PSMA overexpression in PCa is limited.
Of the four isolated NBs, NB7 showed the highest (picomolar) binding affinity to PSMA due to an avidity effect: it simultaneously interacts with two PSMA monomers via different CDRs such that when one CDR is detached from PSMA, the other CDRs remain bound. Consequently, the dissociation rate of NB7 is very low, resulting in a very low KD to soluble and cell-expressed PSMA. The different affinities of the four NBs enable us to designate them for different potential applications. For example, as the affinity of NB8 and NB37 to PSMA is relatively low, their clearance from the body is relatively fast and they are, therefore, more suitable for diagnostic purposes. In contrast, since the affinity of NB7 to PSMA is extremely high and its half-life is long and since it can effectively internalize into tumor cells, NB7 is more suitable for conjugation to cytotoxic drugs, such as DOX. Notably, however, these intended functions are not mutually exclusive, and each of the four NBs could serve for theranostic purposes in the future.
The NB7cysDOX conjugate accumulated in PSMA+ tumors and was almost completely internalized, within hours, only to PSMA+ cells; these characteristics would allow clinicians to minimize the dosage of the cytotoxic molecule. In addition, a small fraction (∼5%) of the conjugates remained bound to the membrane of tumor cells even after 16 h and can effectively deliver the drug to tumors even if they are not completely internalized. On the membrane, the conjugate is exposed to the slightly acidic tumor microenvironment, which could hydrolyze (although presumably at a lower rate) the bond between the linker and the drug. The release of the drug into the tumor microenvironment could be beneficial due to the “bystander effect”,60,61 i.e., by allowing the drug to penetrate to other (PSMA-negative or more distant) tumor-related cells while damaging as few healthy cells as possible. Importantly, the possible effect of a PSMA-targeting NB–drug conjugate could potentially be different in humans than in mice due to the expression of PSMA on the neovasculature of prostate carcinomas in humans, which was not observed in xenografts.62
To evaluate the efficacy of our NB–drug conjugate, we chose to use DOX, which is less potent than other available drugs and is therefore not the standard of care in PCa chemotherapy. While a low-potency drug better demonstrates the efficacy of targeted drug delivery as a proof-of-concept, clinical practice should preferably conjugate NB7cys to more potent drugs, such as docetaxel and cabazitaxel, which contain ketone groups that could covalently bind the BMPH linker. The efficacy of such conjugates should be tested directly in future studies.
We conclude that the four presented NBs are promising candidates for conjugation with cytotoxic small-molecule drugs due to their high affinity to the antigen and their ability to internalize into target cells without accumulating in other organs (as do conventional IgGs). Comprehensive pharmacokinetic, pharmacodynamic, and biodistribution studies are yet to be conducted, and the toxicity and side effect profiles of NB7cysDOX are yet to be determined. In addition, it is yet to be established whether the improved efficacy incurred by the targeted delivery of the drug is indeed clinically beneficial for PCa patients, and more clinically relevant methods (e.g., PET/CT) should be used to determine the efficacy of the NBs in clinical imaging applications. Notwithstanding, our findings add to accumulating evidence that strongly implicate NBs and NB–drug conjugates as promising alternatives for antibody-based therapeutics and imaging agents. Facilitating the development of efficient NBs for the detection of tumors and for the targeted delivery of drugs into specific cells will greatly enrich the toolbox for treating patients with various types of diseases.
Experimental Section
Animal Procedures
All animal experiments were approved by the Ethical Committee for Animal Experiments of Israel (authorization numbers 11-220-6 and 48-07-2012 for camel and mouse procedures, respectively). Extensive efforts were made to minimize the number and suffering of animals used in this study.
Generation and Purification of Anti-PSMA NBs
The protocol for NB generation was adapted from Pardon et al.73 and Vincke et al.74 Briefly, a camel (Camelus dromedarius) was immunized seven times, with 2 weeks between successive injections, with 1 mg of the purified extracellular domain of PSMA [residues 44–750;10 purchased from Caltech Protein Expression Center, CA]. The RNA from camel lymphocytes was then isolated and converted to DNA, and the DNA encoding for VHH was amplified and ligated to a pMECS vector. This DNA library was transformed to TG1 Escherichia coli competent cells, and the resulting library (107 clones) was subjected to selection using phage display through infection with an M13 helper phage. After two rounds of panning against PSMA, 47 bacterial colonies were individually evaluated for PSMA binding using ELISA and then sequenced (NIBN sequencing laboratory, Ben-Gurion University of the Negev, Israel). The DNA encoding for the four selected NBs (NB7, NB8, NB13, and NB37) and for NB7 with an added cysteine in the C-terminus (NB7cys) was transformed to WK6 E. coli. The bacteria were grown in TB medium (17 mM KH2PO4, 94 mM K2HPO4, 12 g/L peptone, 24 g/L yeast extract, 0.4% glycerol) at 37 °C and 250 rpm until they reached OD600 = 0.5. Then, 1 mM IPTG was added to the medium and the temperature was adjusted to 28 °C overnight, followed by a periplasmic extraction using 12 mL of TES buffer (500 mM sucrose, 200 mM Tris-HCl, 0.5 mM EDTA, pH 8) for 3 h and then using 24 mL of TES buffer (diluted 1:4) overnight. The NBs were further purified using affinity chromatography on Ni-NTA gravitational beads (Invitrogen, CA). The eluted fraction was subjected to FPLC purification using a Superdex 75 16/600 column (GE Healthcare, MA). The size and purity of the proteins were evaluated by using SDS–PAGE gel electrophoresis and mass spectrometry, confirming the expected size of ∼16 kDa and >95% purity.
Surface Plasmon Resonance Binding Assay
The affinity of each NB to PSMA was determined by using surface plasmon resonance (SPR) spectroscopy on a ProteOn XPR36 chip (Bio-Rad, CA). The chip was activated by using sulfo-NHS (0.1 M N-hydroxysuccinimide) and EDC [0.4 M 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide]. Each NB (0.2 μg) was immobilized in a 10 mM sodium acetate buffer, pH 5.0, at a flow rate of 30 μL/min. Bovine serum albumin (BSA) (3 μg) was immobilized on the chip as a negative control. Unbound esters were deactivated with 1 M ethanolamine HCl at pH 8.5. The soluble PSMA was then applied over the chip at concentrations of 2.94, 5.88, 11.75, 23.50, or 47.00 nM (for NBs 8, 13, and 37) or of 25, 50, 100, 1600, or 3200 pM (for NB7), at a flow rate of 25 μL/min. During this time, the association between the NBs and PSMA was measured. The dissociation was measured while flowing 50 μL/min PBST (namely, a phosphate buffered saline with 0.005% Tween). For each protein complex, a binding sensorgram was generated by subtracting the values of the PSMA response to BSA from those of the PSMA response to the NBs. The dissociation constant (KD) was determined from the Langmuir 1:1 kinetic model. The temperature throughout the binding measurements was set at 25 °C.
PSMA Activity Assay
The enzymatic NAALDase activity of PSMA was determined by using the assay protocol suggested by R&D systems for recombinant PSMA. Briefly, PSMA was diluted to 0.4 μg/mL and an Ac-Asp-Glu substrate (Sigma-Aldrich) was diluted to 40 μM in 50 mM HEPES, 0.1 M NaCl, pH 7.5. A working solution was generated by combining 125 μL of the PSMA and substrate solutions. For a negative control, the PSMA was deactivated by thermal denaturation. As a control for inhibition, 0.5 nM commercial PSMA inhibitor (PMPA, Tocris, Israel) was added to the solution containing the PSMA and the substrate. NB7, NB8, NB13, and NB37 (100 nM each) were added to this solution and incubated for 1 h at 37 °C and then for 5 min at 95 °C. Next, 250 μL of 15 mM phthaldialdehyde (Sigma-Aldrich) in 0.2 M NaOH and 0.1% β-mercaptoethanol were added to each sample. The samples were incubated at room temperature for 10 min, and their fluorescence was measured (excitation 330 nm, emission 450 nm). The fluorescence value of the untreated PSMA sample was set as 1, and all other samples were normalized accordingly.
Cell Binding Assay
PC3-PIP (PSMA-positive, PSMA+) cells and PC3-flu (PSMA-negative, PSMA–) cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), l-glutamine, penicillin, and streptomycin (Biological Industries, Israel). Once the cells reached 70% confluence, 105 cells were added to each well of 96-well U-shaped bottom plates (Greiner Bio-One, Austria), centrifuged at 150g for 5 min, and washed with PBSA (namely, PBS + 1 g/L BSA). NBs were added to the cells in concentrations of 0.1, 0.5, 2, 5, 10, 20, 50, 100, 500, or 1000 nM. The cells were incubated with the NBs for 2 h, followed by three PBSA washing steps. An anti-His antibody conjugated to fluorescein isothiocyanate (FITC) (Invitrogen) was then added at a dilution of 1:100, incubated with the cells for 1 h, and washed three times with PBSA. The cells were kept on ice throughout the experiment. The fluorescence of each sample was measured using an Accuri C6 flow cytometry analyzer (BD Biosciences, CA). Each experimental condition was repeated three times. To generate a titration curve, the value for each sample was determined using the equation
| 1 |
where Fsample is the mean fluorescence value, Flow is the fluorescence at the lowest concentration for PC3-PIP cells, and Fhigh is the fluorescence at the highest concentration for PC3-PIP cells. A binding curve was generated using GraphPad Prism 5.0.
Protein Crystallization, Data Collection, Structure Determination, and Refinement
NB7, NB8, and NB37 (5 mg/mL) were mixed at a 1:1 (v/v) ratio with a reservoir solution and crystallized, at room temperature, by the sitting-drop vapor diffusion method over a reservoir containing either 1.7 M ammonium sulfate and 6.57% 2-propanol (for NB7); 0.1 M trisodium citrate, pH 3.5, and 3 M NaCl (for NB8); or 0.1 M trisodium citrate, pH 3.5, and 25% polyethylene glycol 3350 (for NB37). The crystals were then harvested, cryoprotected, and flash-cooled in liquid nitrogen. X-ray diffraction (XRD) data were collected at beamline ID30B of the European Synchrotron Radiation Facility (ESRF, Grenoble, France). Data were collected at 100 K from one crystal of each NB that diffracted to a maximum resolution of 1.5 Å for NB8 and NB37 and of 2.65 Å for NB7. The NB7 crystal belongs to the space group P21, with unit cell dimensions of a = 53.563 Å, b = 171.716 Å, and c = 83.479 Å, and it contains eight copies of the protein in the asymmetric unit. The NB8 crystal belongs to the space group I222, with unit cell dimensions of a = 55.945 Å, b = 68.857 Å, and c = 75.647 Å, and it contains one copy of the protein in the asymmetric unit. The NB37 crystal belongs to the space group I222, with unit cell dimensions of a = 55.949 Å, b = 69.087 Å, and c = 75.869 Å, and it contains one copy of the protein in the asymmetric unit. X-ray data were merged and scaled using XDS75 and solved by molecular replacement using Phaser76 in CCP4.77 Protein Data Bank (PDB) code 5M7Q was used as a search model. Refinement included alternating cycles of manual rebuilding in COOT78 and automated refinement using Phenix.79 The coordinates and structure factors were submitted to the PDB under the accession codes 6XXN (NB7), 6XXO (NB8), and 6XXP (NB37).
Small-Angle X-ray Scattering, Analysis, and Three-Dimensional Structure Reconstruction
The small-angle X-ray scattering (SAXS) of monomeric PSMA was measured in PBS at a final concentration of 0.5–3 mg/mL. For PSMA–NB complex samples, the concentration of PSMA was 0.5 mg/mL and the concentrations of the NBs were 0.1–0.5 mg/mL. Measurements were performed in beamline BM29 at the ESRF. The X-ray wavelength was 1.5 Å, and the temperature was 4 °C. The detector was Pilatus 1 M, and the sample-to-detector distance was set at 2.86 m, with a scattering vector (q) range of 0.0025–0.5 Å–1. At a scattering angle of 2θ, the magnitude of the scattering vector (q) is defined as
| 2 |
The experimental SAXS data for all samples were linear in the low q, Guinier region. The radii of gyration (Rg) were derived from data in the qRg < 1 region by using the Guinier approximation:
| 3 |
We analyzed the small-angle region (0.012 < q < 0.08 Å–1) of the scattering profiles using the Guinier approximation embedded in the GNOM method.80 The scattering curve reflects structural characteristics in reciprocal space. Scattering profiles were translated into real space by Fourier transformation, resulting in the pairwise-distance distribution function P(r). This function reflects the distances between pairs of scattering points within the macromolecule, allowing the determination of the maximum dimension of the particle (Dmax). To obtain a reliable quantification of Dmax, we incorporated GNOM with in-house scripts.81 The Rg of monomeric PSMA extracted from SAXS data was compared to the calculated Rg from the crystal structure of monomeric PSMA (PDB code 3D7D) using CRYSOL.82 The overall three-dimensional ab initio models of PSMA and PSMA–NB complexes were restored from the experimental scattering data by using Dammin.44 Shape reconstruction was performed to represent the molecular shape as a closely packed sphere assembly within a search volume defined by Dmax, chosen with a χ2 < 1.3 for all models. For all samples, 20 low-resolution models were averaged using the program DAMAVER43 to yield an averaged model representing the general structural features of each reconstruction.
Computational Analysis of Binding Epitopes
The protein crystal structure of PSMA was selected for the docking procedure (PDB code 1Z8L).10 NB37 (PDB code 6XXP) and NB7 (PDB code 6XXN) were docked to a monomer form and to a homodimer form of the PSMA crystal structure by using Discovery Studio 4.5 (Biovia, Dassault Systems, San Diego, CA) with ZDOCK.83,84 The ZRANK method was then used to quickly and accurately rerank the docked protein complexes predicted by ZDOCK.85 For each docking simulation, the final top 2000 complexes of docking solution orientations were clustered into groups. Classification was based on the spatial proximities of the solution, using a maximal ligand interface RMSD cutoff of 6 Å from the cluster center and an interface cutoff of 9 Å, which defines the interface region between PSMA and the NB, to obtain better defined clusters. This process allowed us to select the most promising docking solutions for further analysis. The geometry of the selected docking solution was optimized by using an energy minimization protocol and the Biovia Smart Minimizer algorithm.86,87 For the selected minimized solution, the binding interface between two protein domains was identified and the interactions between the domains were calculated. The interface residues (namely, residues whose solvent-accessible surface area is different when the proteins are in a complex versus isolated) were identified, and the types of interaction (hydrogen bonds, electrostatic and hydrophobic interactions, etc.) were determined. Prior to docking all proteins, PSMA and the NBs were subjected to the Prepare Protein protocol, which corrects the enumeration of hydrogens by using either standard88 or predicted89 pKa values for Asp, Glu, Arg, Lys, His, Tyr, Cys, and the N-termini and C-termini of each chain, which are titratable. The outcomes of using this protocol are the preferred hydrogen representations and protonation states of chain termini and side chains.
In Vivo Optical Imaging
Tumor xenografts were generated in 6-week-old male athymic nude mice by using PC3-PIP and PC3-flu cells. Each mouse was simultaneously injected subcutaneously with 2 × 106 cells of each line, diluted 1:1 with Matrigel (Corning, USA); PC3-PIP cells were injected above the right upper flank, while PC3-flu cells were injected above the left one. Nine days after the inoculation, as the tumors reached a size of ∼200 mm3, these mice were injected intravenously with 1.5 nmole of either NB7, NB8, NB13, or NB37 (four mice per group) labeled with NHS-ester AlexaFluor680 (Invitrogen). In addition to these 16 mice, four tumor-bearing mice were not injected with any NB, while four other mice were injected with the labeled NBs (a different NB per mouse) but were not implanted with a xenograft. The mice were anesthetized with isoflurane at different time points (see below) and the distribution of the fluorescently labeled protein was measured in near-infrared (NIR) optical imaging using the IVIS Lumina system (PerkinElmer, USA). Exposure time was set at 1 s. The fluorescence signal was measured at the time of injection and 0.5, 1, 2, 3, 6, 10, 18, 24, 28, 32, 36, 48, and 56 h after injection. Images of the mice were acquired 3 and 6 h after injection and again when a signal was no longer detected (24–56 h after injection). At each time point, one mouse from each group of tumor-bearing mice that had been injected with a NB was euthanized for an ex vivo quantification of the fluorescent signal in its organs, using the Living Image software.
DOX Conjugation to NB7cys
The doxorubicin conjugate (presented in Figure S10) (1) was synthesized according to standard procedures (Figure S10A). N-(β-Maleimidopropionic acid) hydrazide trifluoroacetic acid salt (2, 39 mg, 0.13 mmol) was added to a solution of doxorubicin hydrochloride (DOX, 3, 29 mg, 0.05 mmol) in 10 mL of anhydrous methanol. Trifluoroacetic acid (3 μL) was added to the reaction mixture, which was then stirred at room temperature for 18 h in the dark. The reaction mixture was concentrated to a volume of 1 mL and added dropwise to acetonitrile (20 mL) while stirring. The resulting solution was allowed to stand at 4 °C for at least 24 h. The final product (1) was isolated by centrifugation, washed with fresh 1:10 methanol/acetonitrile solution, and dried under vacuum to yield 1, 25 mg, 71% yield. 1H NMR (DMSO-d6) δ = 10.46 (s, 1H), 7.94–7.92 (m, 2H), 7.67 (dd, J = 7.4 and 3.4 Hz, 1H), 6.87 (s, 2H), 5.78 (t, J = 4.9 Hz, 1H), 5.51 (s, 1H), 5.40 (d, J = 3.9 Hz, 1H), 5.26 (d, J = 2.0 Hz, 1H), 4.91 (t, J = 7.8 Hz, 1H), 4.40 (t, J = 4.4 Hz, 2H), 3.99 (s, 4H), 2.73 (d, J = 15.6 Hz, 1H), 2.34–2.24 (m, 2H), 2.15–2.10 (m, 2H), 1.88–1.81 (m, 2H), 1.71–1.66 (m, 2H), 1.14 (d, J = 6.8 Hz, 3H) ppm (Figure S11). MS (ESI) calculated for C34H37N4O13 [M + H]+: 709.23; observed, 709.14. By use of maleimide-based chemistry, NB7cys was then conjugated to 1 at a molar ratio of 1:20 (24 h at 4 °C). NB7cysDOX was separated from the unconjugated NB7cys by FPLC using Superdex 75 10/300 (GE Healthcare, MA). The conjugation of DOX to the protein was verified based on absorbance at 488 nm during the FPLC run and by mass spectrometry.
Confocal Imaging
The NBs were labeled at a 1:3 molar ratio with Dylight 488 NHS-ester (Thermo Scientific, IL). Phycoerythrin (PE)-anti PSMA antibody (BioLegend, CA) and Hoechst 33342 (Invitrogen) were incubated for 15 min with 3 × 104 PC3-PIP or PC3-flu cells, which were grown overnight in an 8-well μ-slide (ibidi GmbH, Germany) in the presence or absence of 100 nM labeled NB. NB7cys and NB7cysDOX were labeled at a 1:3 molar ratio with Dylight 650 NHS-ester. Hoechst 33342 and 1.5 μg/mL DOX (Teva, Israel) or an equivalent molar amount of labeled NB7cys or labeled NB7cysDOX were incubated with PC3-PIP and PC3-flu cells, grown as described above. The cells were imaged with an Olympus FV1000 confocal microscope (Olympus, Japan), with a long-working distance ×60/1.35 numerical aperture, oil-immersion objective.
Time-Dependent Quantification of NB Internalization
NB7, NB8, NB13, and NB37, each labeled with Dylight488, were individually incubated for 1 h (at 100 nM) in a 96-well plate. On each well, 1.5 × 104 PC3-PIP cells were seeded and grown overnight, and then the wells were imaged every 40 min for a total of 16 h, using the Operetta CLS high-content analysis system http://www.perkinelmer.com/Product/operetta-cls-system-hh16000000 (PerkinElmer). Each well was imaged as 24 fields, which were later combined to create an image of the entire well. Using the Operetta analysis software, the cells were qualitatively classified into two groups according to the distribution of NBs: (i) mostly on the cell membrane and (ii) mostly inside the cytoplasm. The number of cells in each group was quantified at each time point, and the ratio between the numbers of cells in each group was calculated.
Cell Quantification Assay
PC3-PIP cells (5 × 104) were seeded in 24-well plates. After the cells were attached to the plate, they were either left untreated or were treated with DOX (1.5 μg/mL) or an equivalent molar amount of NB7cys or NB7cysDOX. After 24 h of treatment, the number of cells in each well was counted using the Countess II automated cell counter (Invitrogen).
Cell Viability Assay
PC3-PIP cells were grown and treated as described in the Cell Quantification Assay section above. The cells were harvested, incubated with 0.5 μg of propidium iodide (PI; Biolegend), and their fluorescence intensity was measured in a BD C6 flow cytometer.
Mitochondrial Potential Assay
PC3-PIP cells (2 × 104) were seeded on 96-well plates. After the cells adhered to the plate, they were treated with either 1.5 μg/mL DOX or an equivalent molar amount of NB7cys or NB7cysDOX, or they were left untreated as a control. After 24 h, tetramethylrhodamine ethyl ester (TMRE; Abcam, U.K.) was added according to the protocol provided by the manufacturer. Fluorescence intensity was measured at an excitation wavelength of 549 nm and an emission wavelength of 575 nm. Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) served as a negative control, used according to the manufacturer’s protocol.
In Vivo Tumor Growth Inhibition
PC3-PIP xenografts were grown in athymic nude mice, as described in the In Vivo Optical Imaging section above. When the average tumor size reached 200 mm3, the mice were divided into three groups (controlled for average tumor size), each subjected to a different treatment: 150 μL of saline (n = 7), 2 mg/kg DOX (n = 8), or 1.4 mg/kg (∼40 μg) NB7cysDOX (n = 8). The treatment was administered to the tail vein twice a week for three consecutive weeks. At each sample point, the tumor volume was calculated (V = 0.5 × L × W × H), as previously described.90 A mouse was euthanized when tumor volume reached 1500 mm3 or when its physical condition deteriorated, according to the guidelines of the Committee for the Ethical Care and Use of Animals in Research at BGU. The estimated tumor volume prior to euthanasia and the rate-based T/C were determined as described previously.59
Histology
Four days following the final dose of each treatment in the in vivo tumor growth inhibition assay, the mice were euthanized and their xenografts were fixated in 4% formaldehyde and embedded in paraffin. Tumor sections (5 μm thickness) were subjected to hematoxylin and eosin (H&E) staining, TUNEL assay, and immunofluorescence (IF), as previously described.91,92 For IF, anti-PSMA conjugated to PE and anti-HIS conjugated to FITC were used to detect PSMA and NB7cysDOX, respectively. 4′,6-Diamidino-2-phenylindole (DAPI) was used for nuclei staining. H&E-stained sections were visualized using a panoramic MIDI II scanner (3DHISTECH Kft., Hungary). TUNEL, PI, and IF were visualized in a confocal microscope.
Statistical Analyses
Unless indicated otherwise, each experiment was performed in triplicate and the results indicate the mean ± SEM. Statistical significance was determined using Student’s t test.
Acknowledgments
We thank Prof. Serge Muyldermans and Dr. Cecile Vincke for providing the protocols for animal immunization and nanobody screening assay. We thank Dr. Alon Zilka for his technical assistance. SPR and microscopy experiments were performed at the NIBN Proteomics, Cytometry and Microscopy unit. We thank Prof. Varda Shoshan-Barmatz and Dr. Srinivas Pittala from Ben-Gurion University for the assistance with histology. The structural studies were performed on beamlines ID30-A3 and ID30-B at the European Synchrotron Radiation Facility (ESRF), Grenoble, France. We are grateful to the beamline scientists for providing assistance in using these beamlines. We thank the staff from the BM29-BioSAXS Beamline of the European Synchrotron Facility in Grenoble (ESRF). This work was supported by the European Research Council “Ideas Program” ERC-2013-StG (Grant 336041) and the Prostate Cancer Foundation (PCF) to N.P.
Glossary
Abbreviations Used
- PCa
prostate cancer
- PSA
prostate specific antigen
- DOX
doxorubicin
- PSMA
prostate specific membrane antigen
- NB
nanobody
- HCAb
heavy-chain antibodies
- NHS
N-hydroxysuccinimide
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- FBS
fetal bovine serum
- FITC
fluorescein isothiocyanate
- XRD
X-ray diffraction
- SAXS
small-angle X-ray scattering
- Rg
radii of gyration
- NIR
near-infrared
- PE
phycoerythrin
- H&E
hematoxylin and eosin
- DAPI
4′,6-diamidino-2-phenylindole
- PI
propidium iodide
- SPR
surface plasmon resonance
- CDR
complementarity determining region
- BMPH
N-(β-maleimidopropionic acid) hydrazide
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00418.
NB sequences; NB selection and purification; PSMA activity assay; SAXS analysis and the Rg of the monomeric PSMA; SAXS curves and the corresponding Guinier plots of PSMA with increasing concentrations of NBs; effect of NBs on the Rg of PSMA; results of the custom-made script analysis of the SAXS data; distance distribution function [P(r)] of PSMA and NBs at a molar ratio of 1:2; ex vivo optical imaging of prostate cancer xenografts; internalization of the NBs into PC3-PIP cells; conjugation of NB7cys to DOX; effect of NB7cysDOX on cell viability; in vivo tumor growth inhibition by NB7cysDOX; crystallographic statistics for NB7, NB8, and NB37; parameters for the SAXS analysis of PSMA with and without NBs; predicted interactions between NB7 and PSMA; predicted interactions between NB37 and PSMA; interface analysis for PSMA-NB7 and PSMA-NB37 (PDF)
Coordinates information for structure representation (PDB)
Coordinates information for structure representation (PDB)
Coordinates information for structure representation (PDB)
Molecular formula strings for the DOX-BMPH linker (compound 1) and compounds 2 and 3 (CSV)
Accession Codes
The coordinates and structure factors were submitted to the PDB under the accession codes 6XXN (NB7), 6XXO (NB8), and 6XXP (NB37). Authors will release the atomic coordinates and experimental data upon article publication.
Author Contributions
L.R. and N.P. designed the research. L.R., A.S., Y.Z., S.C., K.D., S.G., E.B.Z., and A.S. performed the research. L.R., A.S., S.C., L.L., B.A., E.A., and N.P. analyzed the data. L.R. and N.P. wrote the paper. All authors edited the manuscript and approved the final version.
The authors declare no competing financial interest.
This article was published ASAP on June 8, 2020. The Supporting Information file has been updated to include references. The corrected version was reposted on June 9, 2020.
Supplementary Material
References
- Schröder F. H.; Hugosson J.; Roobol M. J.; Tammela T. L. J.; Ciatto S.; Nelen V.; Kwiatkowski M.; Lujan M.; Lilja H.; Zappa M.; Denis L. J.; Recker F.; Berenguer A.; Määttänen L.; Bangma C. H.; Aus G.; Villers A.; Rebillard X.; van der Kwast T.; Blijenberg B. G.; Moss S. M.; de Koning H. J.; Auvinen A. Screening and Prostate-Cancer Mortality in a Randomized European Study. N. Engl. J. Med. 2009, 360 (13), 1320–1328. 10.1056/NEJMoa0810084. [DOI] [PubMed] [Google Scholar]
- Kibel a. S. Mortality Results from a Randomized Prostate-Cancer Screening Trial. Yearb. Urol. 2009, 2009, 145–147. 10.1016/S0084-4071(09)79273-5. [DOI] [Google Scholar]
- Catalona W. J.; Richie J. P.; Ahmann F. R.; Hudson M. A.; Scardino P. T.; Flanigan R. C.; Dekernion J. B.; Ratliff T. L.; Kavoussi L. R.; Dalkin B. L.; Waters W. B.; Macfarlane M. T.; Southwick P. C. Comparison of Digital Rectal Examination and Serum Prostate Specific Antigen in the Early Detection of Prostate Cancer: Results of a Multicenter Clinical Trial of 6,630 Men. J. Urol. 1994, 151 (5), 1283–1290. 10.1016/S0022-5347(17)35233-3. [DOI] [PubMed] [Google Scholar]
- Ilic D.; O’Connor D.; Green S.; Wilt T. J. Screening for Prostate Cancer: An Updated Cochrane Systematic Review. BJU Int. 2011, 107 (6), 882–891. 10.1111/j.1464-410X.2010.10032.x. [DOI] [PubMed] [Google Scholar]
- Thompson I.; Leach R. J.; Pollock B. H.; Naylor S. L. Prostate Cancer and Prostate-Specific Antigen: The More We Know, the Less We Understand. J. Natl. Cancer Inst. 2003, 95 (14), 1027–1028. 10.1093/jnci/95.14.1027. [DOI] [PubMed] [Google Scholar]
- Saini S. PSA and beyond: Alternative Prostate Cancer Biomarkers. Cell. Oncol. 2016, 39 (2), 97–106. 10.1007/s13402-016-0268-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidenreich A.; Bastian P. J.; Bellmunt J.; Bolla M.; Joniau S.; van der Kwast T.; Mason M.; Matveev V.; Wiegel T.; Zattoni F.; Mottet N. EAU Guidelines on Prostate Cancer. Part II: Treatment of Advanced, Relapsing, and Castration-Resistant Prostate Cancer. Eur. Urol. 2014, 65 (2), 467–479. 10.1016/j.eururo.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Harris K. A.; Harney E.; Small E. J. Liposomal Doxorubicin for the Treatment of Hormone-Refractory Prostate Cancer. Clin. Prostate Cancer 2002, 1 (1), 37–41. 10.3816/CGC.2002.n.005. [DOI] [PubMed] [Google Scholar]
- Zhao N.; C Woodle M.; Mixson A. J. Advances in Delivery Systems for Doxorubicin. J. Nanomed. Nanotechnol. 2018, 09 (05), 1000519. 10.4172/2157-7439.1000519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. I.; Bennett M. J.; Thomas L. M.; Bjorkman P. J. Crystal Structure of Prostate-Specific Membrane Antigen, a Tumor Marker and Peptidase. Proc. Natl. Acad. Sci. U. S. A. 2005, 102 (17), 5981–5986. 10.1073/pnas.0502101102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristau B. T.; O’Keefe D. S.; Bacich D. J. The Prostate-Specific Membrane Antigen: Lessons and Current Clinical Implications from 20 Years of Research. Urol. Oncol. 2014, 32 (3), 272–279. 10.1016/j.urolonc.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao V.; Bacich D. J. Prostate Specific Membrane Antigen (PSMA) Expression Gives Prostate Cancer Cells a Growth Advantage in a Physiologically Relevant Folate Environment in Vitro. Prostate 2006, 66 (8), 867–875. 10.1002/pros.20361. [DOI] [PubMed] [Google Scholar]
- Yao V.; Berkman C. E.; Choi J. K.; O’Keefe D. S.; Bacich D. J. Expression of Prostate-Specific Membrane Antigen (PSMA), Increases Cell Folate Uptake and Proliferation and Suggests a Novel Role for PSMA in the Uptake of the Non-Polyglutamated Folate, Folic Acid. Prostate 2010, 70 (3), 305–316. 10.1002/pros.21065. [DOI] [PubMed] [Google Scholar]
- Wright G. L.; Grob B. M.; Haley C.; Grossman K.; Newhall K.; Petrylak D.; Troyer J.; Konchuba A.; Schellhammer P. F.; Moriarty R. Upregulation of Prostate-Specific Membrane Antigen after Androgen- Deprivation Therapy. Urology 1996, 48 (2), 326–334. 10.1016/S0090-4295(96)00184-7. [DOI] [PubMed] [Google Scholar]
- Perner S.; Hofer M. D.; Kim R.; Shah R. B.; Li H.; Möller P.; Hautmann R. E.; Gschwend J. E.; Kuefer R.; Rubin M. A. Prostate-Specific Membrane Antigen Expression as a Predictor of Prostate Cancer Progression. Hum. Pathol. 2007, 38 (5), 696–701. 10.1016/j.humpath.2006.11.012. [DOI] [PubMed] [Google Scholar]
- von Eyben F. E.; Baumann G. S.; Baum R. P. PSMA Diagnostics and Treatments of Prostate Cancer Become Mature. Clin. Transl. Imaging 2018, 6 (2), 145–148. 10.1007/s40336-018-0270-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimadamore A.; Cheng M.; Santoni M.; Lopez-Beltran A.; Battelli N.; Massari F.; Galosi A. B.; Scarpelli M.; Montironi R. New Prostate Cancer Targets for Diagnosis, Imaging, and Therapy: Focus on Prostate-Specific Membrane Antigen. Front. Oncol. 2018, 8, 653. 10.3389/fonc.2018.00653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorff T. B.; Fanti S.; Farolfi A.; Reiter R. E.; Sadun T. Y.; Sartor O. The Evolving Role of Prostate-Specific Membrane Antigen-Based Diagnostics and Therapeutics in Prostate Cancer. Am. Soc. Clin. Oncol. Educ. B 2019, 39, 321–330. 10.1200/EDBK_239187. [DOI] [PubMed] [Google Scholar]
- Chang S. S. Overview of Prostate-Specific Membrane Antigen. Rev. Urol. 2004, 6 (Suppl 10), S13-8. [PMC free article] [PubMed] [Google Scholar]
- Lütje S.; Slavik R.; Fendler W.; Herrmann K.; Eiber M. PSMA Ligands in Prostate Cancer – Probe Optimization and Theranostic Applications. Methods 2017, 130, 42–50. 10.1016/j.ymeth.2017.06.026. [DOI] [PubMed] [Google Scholar]
- Benesová M.; Schäfer M.; Bauder-Wüst U.; Afshar-Oromieh A.; Kratochwil C.; Mier W.; Haberkorn U.; Kopka K.; Eder M. Preclinical Evaluation of a Tailor-Made DOTA-Conjugated PSMA Inhibitor with Optimized Linker Moiety for Imaging and Endoradiotherapy of Prostate Cancer. J. Nucl. Med. 2015, 56 (6), 914–920. 10.2967/jnumed.114.147413. [DOI] [PubMed] [Google Scholar]
- Rowe S. P.; Macura K. J.; Mena E.; Blackford A. L.; Nadal R.; Antonarakis E. S.; Eisenberger M.; Carducci M.; Fan H.; Dannals R. F.; Chen Y.; Mease R. C.; Szabo Z.; Pomper M. G.; Cho S. Y. PSMA-Based [18F]DCFPyL PET/CT Is Superior to Conventional Imaging for Lesion Detection in Patients with Metastatic Prostate Cancer. Mol. Imaging Biol. 2016, 18 (3), 411–419. 10.1007/s11307-016-0957-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatalic K. L. S.; Heskamp S.; Konijnenberg M.; Molkenboer-Kuenen J. D. M.; Franssen G. M.; Clahsen-van Groningen M. C.; Schottelius M.; Wester H. J.; van Weerden W. M.; Boerman O. C.; de Jong M. Towards Personalized Treatment of Prostate Cancer: PSMA I&T, a Promising Prostate-Specific Membrane Antigen-Targeted Theranostic Agent. Theranostics 2016, 6 (6), 849–861. 10.7150/thno.14744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillier S. M.; Maresca K. P.; Femia F. J.; Marquis J. C.; Foss C. A.; Nguyen N.; Zimmerman C. N.; Barrett J. A.; Eckelman W. C.; Pomper M. G.; Joyal J. L.; Babich J. W. Preclinical Evaluation of Novel Glutamate-Urea-Lysine Analogues That Target Prostate-Specific Membrane Antigen as Molecular Imaging Pharmaceuticals for Prostate Cancer. Cancer Res. 2009, 69 (17), 6932–6940. 10.1158/0008-5472.CAN-09-1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan X.; Liu F.; Kwon H.; Byun Y.; Minn I.; Cai X.; Zhang J.; Pomper M. G.; Yang Z.; Xi Z.; Yang X. (S)-3-(Carboxyformamido)-2-(3-(Carboxymethyl)Ureido)Propanoic Acid as a Novel PSMA Targeting Scaffold for Prostate Cancer Imaging. J. Med. Chem. 2020, 63 (7), 3563–3576. 10.1021/acs.jmedchem.9b02031. [DOI] [PubMed] [Google Scholar]
- Liu H.; Moy P.; Kim S.; Xia Y.; Rajasekaran a.; Navarro V.; Knudsen B.; Bander N. H. Monoclonal Antibodies to the Extracellular Domain of Prostate-Specific Membrane Antigen Also React with Tumor Vascular Endothelium. Cancer Res. 1997, 57 (17), 3629–3634. [PubMed] [Google Scholar]
- Chang S. S.; Reuter V. E.; Heston W. D. W.; Bander N. H.; Grauer L. S.; Gaudin P. B. Five Different Anti-Prostate-Specific Membrane Antigen (PSMA) Antibodies Confirm PSMA Expression in Tumor-Associated Neovasculature. Cancer Res. 1999, 59 (13), 3192–3198. [PubMed] [Google Scholar]
- Smith-Jones P. M.; Vallabahojosula S.; Goldsmith S. J.; Navarro V.; Hunter C. J.; Bastidas D.; Bander N. H. In Vitro Characterization of Radiolabeled Monoclonal Antibodies Specific for the Extracellular Domain of Prostate-Specific Membrane Antigen. Cancer Res. 2000, 60, 5237–5243. [PubMed] [Google Scholar]
- Vaneycken I.; D’huyvetter M.; Hernot S.; de Vos J.; Xavier C.; Devoogdt N.; Caveliers V.; Lahoutte T. Immuno-Imaging Using Nanobodies. Curr. Opin. Biotechnol. 2011, 22 (6), 877–881. 10.1016/j.copbio.2011.06.009. [DOI] [PubMed] [Google Scholar]
- Polakis P. Antibody Drug Conjugates for Cancer Therapy. Pharmacol. Rev. 2016, 68 (1), 3–19. 10.1124/pr.114.009373. [DOI] [PubMed] [Google Scholar]
- Xiang D.; Zheng C.; Zhou S. F.; Qiao S.; Tran P. H. L.; Pu C.; Li Y.; Kong L.; Kouzani A. Z.; Lin J.; Liu K.; Li L.; Shigdar S.; Duan W. Superior Performance of Aptamer in Tumor Penetration over Antibody: Implication of Aptamer-Based Theranostics in Solid Tumors. Theranostics 2015, 5 (10), 1083–1097. 10.7150/thno.11711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson A. L. Antibody Fragments: Hope and Hype. MAbs 2010, 2 (1), 77–83. 10.4161/mabs.2.1.10786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly T.; Dannoon S.; Hopkins M. R.; Murphy S.; Cahaya H.; Blecha J. E.; Jivan S.; Drake C. R.; Barinka C.; Jones E. F.; VanBrocklin H. F.; Berkman C. E. A High-Affinity [18F]-Labeled Phosphoramidate Peptidomimetic PSMA-Targeted Inhibitor for PET Imaging of Prostate Cancer. Nucl. Med. Biol. 2015, 42 (10), 780–787. 10.1016/j.nucmedbio.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitzmann S.; Mier W.; Schad A.; Kinscherf R.; Askoxylakis V.; Altmann A.; Kra S.; Eisenhut M.; Haberkorn U. A New Prostate Carcinoma Binding Peptide (DUP-1) for Tumor Imaging and Therapy A New Prostate Carcinoma Binding Peptide (DUP-1) for Tumor Imaging and Therapy. Clin. Cancer Res. 2005, 11, 139–146. [PubMed] [Google Scholar]
- Aggarwal S.; Singh P.; Topaloglu O.; Isaacs J. T.; Denmeade S. R. A Dimeric Peptide That Binds Selectively to Prostate-Specific Membrane Antigen and Inhibits Its Enzymatic Activity. Cancer Res. 2006, 66 (18), 9171–9177. 10.1158/0008-5472.CAN-06-1520. [DOI] [PubMed] [Google Scholar]
- Muyldermans S. Nanobodies: Natural Single-Domain Antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. 10.1146/annurev-biochem-063011-092449. [DOI] [PubMed] [Google Scholar]
- Yan J.; Li G.; Hu Y.; Ou W.; Wan Y. Construction of a Synthetic Phage-Displayed Nanobody Library with CDR3 Regions Randomized by Trinucleotide Cassettes for Diagnostic Applications. J. Transl. Med. 2014, 12, 343. 10.1186/s12967-014-0343-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muyldermans S.; Baral T. N.; Retamozzo V. C.; De Baetselier P.; De Genst E.; Kinne J.; Leonhardt H.; Magez S.; Nguyen V. K.; Revets H.; Rothbauer U.; Stijlemans B.; Tillib S.; Wernery U.; Wyns L.; Hassanzadeh-Ghassabeh G.; Saerens D. Camelid Immunoglobulins and Nanobody Technology. Vet. Immunol. Immunopathol. 2009, 128 (1–3), 178–183. 10.1016/j.vetimm.2008.10.299. [DOI] [PubMed] [Google Scholar]
- Massa S.; Xavier C.; De Vos J.; Caveliers V.; Lahoutte T.; Muyldermans S.; Devoogdt N. Site-Specific Labeling of Cysteine-Tagged Camelid Single-Domain Antibody-Fragments for Use in Molecular Imaging. Bioconjugate Chem. 2014, 25 (5), 979–988. 10.1021/bc500111t. [DOI] [PubMed] [Google Scholar]
- Evazalipour M.; D’Huyvetter M.; Tehrani B. S.; Abolhassani M.; Omidfar K.; Abdoli S.; Arezumand R.; Morovvati H.; Lahoutte T.; Muyldermans S.; Devoogdt N. Generation and Characterization of Nanobodies Targeting PSMA for Molecular Imaging of Prostate Cancer. Contrast Media Mol. Imaging 2014, 9 (3), 211–220. 10.1002/cmmi.1558. [DOI] [PubMed] [Google Scholar]
- Zare H.; Rajabibazl M.; Rasooli I.; Ebrahimizadeh W.; Bakherad H.; Ardakani L. S.; Gargari S. L. M. Production of Nanobodies against Prostate-Specific Membrane Antigen (PSMA) Recognizing LnCaP Cells. Int. J. Biol. Markers 2014, 29 (2), 169–179. 10.5301/jbm.5000063. [DOI] [PubMed] [Google Scholar]
- Schülke N.; Varlamova O. A.; Donovan G. P.; Ma D.; Gardner J. P.; Morrissey D. M.; Arrigale R. R.; Zhan C.; Chodera A. J.; Surowitz K. G.; Maddon P. J.; Heston W. D. W.; Olson W. C. The Homodimer of Prostate-Specific Membrane Antigen Is a Functional Target for Cancer Therapy. Proc. Natl. Acad. Sci. U. S. A. 2003, 100 (22), 12590–12595. 10.1073/pnas.1735443100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov V. V.; Svergun D. I. J. Appl. Crystallogr. 2003, 36, 860–864. 10.1107/S0021889803000268. [DOI] [Google Scholar]
- Svergun D. I. Restoring Low Resolution Structure of Biological Macromolecules from Solution Scattering Using Simulated Annealing. Biophys. J. 1999, 76 (6), 2879–2886. 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockel A.; Ohl B.; Gilge U.; Liewald A.; Heidland A. Elimination of Low Molecular Weight Proteins during Hemofiltration. Contrib. Nephrol. 1982, 32, 40–45. [DOI] [PubMed] [Google Scholar]
- Ghosh A.; Heston W. D. W. Tumor Target Prostate Specific Membrane Antigen (PSMA) and Its Regulation in Prostate Cancer. J. Cell. Biochem. 2004, 91, 528–539. 10.1002/jcb.10661. [DOI] [PubMed] [Google Scholar]
- Shen D.; Xie F.; Edwards W. B. Evaluation of Phage Display Discovered Peptides as Ligands for Prostate-Specific Membrane Antigen (PSMA). PLoS One 2013, 8 (7), e68339. 10.1371/journal.pone.0068339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Kopeckova P.; Buhler P.; Wolf P.; Pan H.; Bauer H.; Elsasser-Beile U.; Kopecek J. Biorecognition and Subcellular Trafficking of HPMA Copolymer - Anti-PSMA Antibody Conjugates by Prostate Cancer Cells. Mol. Pharmaceutics 2009, 6 (3), 959–970. 10.1021/mp8002682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anilkumar G.; Rajasekaran S. A.; Wang S.; Hankinson O.; Bander N. H.; Rajasekaran A. K. Prostate-Specific Membrane Antigen Association with Filamin A Modulates Its Internalization and NAALADase Activity 1. Cancer Res. 2003, 63, 2645–2648. [PubMed] [Google Scholar]
- Han D.; Wu J.; Han Y.; Wei M.; Han S.; Lin R.; Sun Z.; Yang F.; Jiao D.; Xie P.; Zhang L.; Yang A.-G.; Zhao A.; Wen W.; Qin W. A Novel Anti-PSMA Human ScFv Has the Potential to Be Used as a Diagnostic Tool in Prostate Cancer. Oncotarget 2016, 7 (37), 59471–59481. 10.18632/oncotarget.10697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G.; Vogt A.; Jiménez-franco L. D.; Rinscheid A.; Yousefzadeh-nowshahr E.; Solbach C.; Beer A. J.; Glatting G.; Kletting P. Modelling the Internalisation Process of Prostate Cancer Cells for PSMA- Speci Fi c Ligands. Nucl. Med. Biol. 2019, 72–73, 20–25. 10.1016/j.nucmedbio.2019.05.003. [DOI] [PubMed] [Google Scholar]
- Hu Y.-B.; Dammer E. B.; Ren R.-J.; Wang G. The Endosomal-Lysosomal System: From Acidification and Cargo Sorting to Neurodegeneration. Transl. Neurodegener. 2015, 4, 18. 10.1186/s40035-015-0041-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanamala M.; Wilson W. R.; Yang M.; Palmer B. D.; Wu Z. Mechanisms and Biomaterials in PH-Responsive Tumour Targeted Drug Delivery: A Review. Biomaterials 2016, 85, 152–167. 10.1016/j.biomaterials.2016.01.061. [DOI] [PubMed] [Google Scholar]
- Li H. J.; Du J. Z.; Liu J.; Du X. J.; Shen S.; Zhu Y. H.; Wang X.; Ye X.; Nie S.; Wang J. Smart Superstructures with Ultrahigh PH-Sensitivity for Targeting Acidic Tumor Microenvironment: Instantaneous Size Switching and Improved Tumor Penetration. ACS Nano 2016, 10 (7), 6753–6761. 10.1021/acsnano.6b02326. [DOI] [PubMed] [Google Scholar]
- Kolmel D. K.; Kool E. T. Oximes and Hydrazones in Bioconjugation: Mechanism and Catalysis. Chem. Rev. 2017, 117, 10358–10376. 10.1021/acs.chemrev.7b00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyomiya K.; Matsuo S.; Kurebe M. Mechanism of Specific Nuclear Transport of Adriamycin: The Mode of Nuclear Translocation of Adriamycin-Proteasome Complex 1. Cancer Res. 2001, 61, 2467–2471. [PubMed] [Google Scholar]
- Kuznetsov A. V.; Margreiter R.; Amberger A.; Saks V.; Grimm M. Changes in Mitochondrial Redox State, Membrane Potential and Calcium Precede Mitochondrial Dysfunction in Doxorubicin-Induced Cell Death. Biochim. Biophys. Acta, Mol. Cell Res. 2011, 1813 (6), 1144–1152. 10.1016/j.bbamcr.2011.03.002. [DOI] [PubMed] [Google Scholar]
- Aston W. J.; Hope D. E.; Nowak A. K.; Robinson B. W.; Lake R. A.; Lesterhuis W. J. A Systematic Investigation of the Maximum Tolerated Dose of Cytotoxic Chemotherapy with and without Supportive Care in Mice. BMC Cancer 2017, 17 (1), 1–10. 10.1186/s12885-017-3677-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hather G.; Liu R.; Bandi S.; Mettetal J.; Manfredi M.; Shyu W.-C.; Donelan J.; Chakravarty A. Growth Rate Analysis and Efficient Experimental Design for Tumor Xenograft Studies. Cancer Inf. 2014, 13s4, CIN.S13974. 10.4137/CIN.S13974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewhirst M. W.; Secomb T. W. Transport of Drugs from Blood Vessels to Tumour Tissue. Nat. Rev. Cancer 2017, 17 (12), 738–750. 10.1038/nrc.2017.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovtun Y. V.; Audette C. A.; Ye Y.; Xie H.; Ruberti M. F.; Phinney S. J.; Leece B. A.; Chittenden T.; Blättler W. A.; Goldmacher V. S. Antibody-Drug Conjugates Designed to Eradicate Tumors with Homogeneous and Heterogeneous Expression of the Target Antigen. Cancer Res. 2006, 66 (6), 3214–3221. 10.1158/0008-5472.CAN-05-3973. [DOI] [PubMed] [Google Scholar]
- Nguyen D. P.; Xiong P. L.; Liu H.; Pan S.; Leconet W.; Navarro V.; Guo M.; Moy J.; Kim S.; Ramirez-fort M. K.; Batra J. S.; Bander N. H. Induction of PSMA and Internalization of an Anti-PSMA MAb in the Vascular Compartment. Mol. Cancer Res. 2016, 14, 1045–1054. 10.1158/1541-7786.MCR-16-0193. [DOI] [PubMed] [Google Scholar]
- Hillier S. M.; Maresca K. P.; Lu G.; Merkin R. D.; Marquis J. C.; Zimmerman C. N.; Eckelman W. C.; Joyal J. L.; Babich J. W. 99mTc-Labeled Small-Molecule Inhibitors of Prostate-Specific Membrane Antigen for Molecular Imaging of Prostate Cancer. J. Nucl. Med. 2013, 54 (8), 1369–1376. 10.2967/jnumed.112.116624. [DOI] [PubMed] [Google Scholar]
- Zechmann C. M.; Afshar-Oromieh A.; Armor T.; Stubbs J. B.; Mier W.; Hadaschik B.; Joyal J.; Kopka K.; Debus J.; Babich J. W.; Haberkorn U. Radiation Dosimetry and First Therapy Results with a 124I/ 131I-Labeled Small Molecule (MIP-1095) Targeting PSMA for Prostate Cancer Therapy. Eur. J. Nucl. Med. Mol. Imaging 2014, 41 (7), 1280–1292. 10.1007/s00259-014-2713-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberkorn U.; Eder M.; Kopka K.; Babich J. W.; Eisenhut M.. Role of Radiolabelled Small Molecules Binding to PSMA in Diagnosis and Therapy of Prostate Cancer. In PET/CT in Prostate Cancer; Cook G., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp 51–58, 10.1007/978-3-319-57624-4_6. [DOI] [Google Scholar]
- Rowe S. P.; Gorin M. A.; Pomper M. G. Imaging of Prostate-Specific Membrane Antigen with Small-Molecule PET Radiotracers: From the Bench to Advanced Clinical Applications. Annu. Rev. Med. 2019, 70 (1), 461–477. 10.1146/annurev-med-062117-073027. [DOI] [PubMed] [Google Scholar]
- Szabo Z.; Mena E.; Rowe S. P.; Plyku D.; Nidal R.; Eisenberger M. A.; Antonarakis E. S.; Fan H.; Dannals R. F.; Chen Y.; Mease R. C.; Vranesic M.; Bhatnagar A.; Sgouros G.; Cho S. Y.; Pomper M. G. Initial Evaluation of [18F]DCFPyL for Prostate-Specific Membrane Antigen (PSMA)-Targeted PET Imaging of Prostate Cancer. Mol. Imaging Biol. 2015, 17 (4), 565–574. 10.1007/s11307-015-0850-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afshar-Oromieh A.; Holland-Letz T.; Giesel F. L.; Kratochwil C.; Mier W.; Haufe S.; Debus N.; Eder M.; Eisenhut M.; Schäfer M.; Neels O.; Hohenfellner M.; Kopka K.; Kauczor H. U.; Debus J.; Haberkorn U. Diagnostic Performance of 68Ga-PSMA-11 (HBED-CC) PET/CT in Patients with Recurrent Prostate Cancer: Evaluation in 1007 Patients. Eur. J. Nucl. Med. Mol. Imaging 2017, 44 (8), 1258–1268. 10.1007/s00259-017-3711-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietlein F.; Hohberg M.; Kobe C.; Zlatopolskiy B. D.; Krapf P.; Endepols H.; Täger P.; Hammes J.; Heidenreich A.; Neumaier B.; Drzezga A.; Dietlein M. An 18F-Labeled PSMA Ligand for PET/CT of Prostate Cancer: First-in-Humans Observational Study and Clinical Experience with 18F-JK-PSMA-7 during the First Year of Application. J. Nucl. Med. 2020, 61 (2), 202–209. 10.2967/jnumed.119.229542. [DOI] [PubMed] [Google Scholar]
- Tagawa S. T.; Milowsky M. I.; Morris M.; Vallabhajosula S.; Christos P.; Akhtar N. H.; Osborne J.; Goldsmith S. J.; Larson S.; Taskar N. P.; Scher H. I.; Bander N. H.; Nanus D. M. Phase II Study of Lutetium-177-Labeled Anti-Prostate-Specific Membrane Antigen Monoclonal Antibody J591 for Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2013, 19 (18), 5182–5191. 10.1158/1078-0432.CCR-13-0231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne J. R.; Green D. A.; Spratt D. E.; Lyashchenko S.; Fareedy S. B.; Robinson B. D.; Beattie B. J.; Jain M.; Lewis J. S.; Christos P.; Larson S. M.; Bander N. H.; Scherr D. S. A Prospective Pilot Study of 89Zr-J591/Prostate Specific Membrane Antigen Positron Emission Tomography in Men with Localized Prostate Cancer Undergoing Radical Prostatectomy. J. Urol. 2014, 191 (5), 1439–1445. 10.1016/j.juro.2013.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lütje S.; Rijpkema M.; Franssen G. M.; Fracasso G.; Helfrich W.; Eek A.; Oyen W. J.; Colombatti M.; Boerman O. C. Dual-Modality Image-Guided Surgery of Prostate Cancer with a Radiolabeled Fluorescent Anti-PSMA Monoclonal Antibody. J. Nucl. Med. 2014, 55 (6), 995–1001. 10.2967/jnumed.114.138180. [DOI] [PubMed] [Google Scholar]
- Pardon E.; Laeremans T.; Triest S.; Rasmussen S. G. F.; Wohlkönig A.; Ruf A.; Muyldermans S.; Hol W. G. J.; Kobilka B. K.; Steyaert J. A General Protocol for the Generation of Nanobodies for Structural Biology. Nat. Protoc. 2014, 9 (3), 674–693. 10.1038/nprot.2014.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincke C.; Gutierrez C.; Wernery U.; Devoogdt N.; Hassanzadeh-Ghassabeh G.; Muyldermans S. Generation of Single Domain Antibody Fragments Derived from Camelids and Generation of Manifold Constructs. Methods Mol. Biol. 2012, 907, 145–176. 10.1007/978-1-61779-974-7_8. [DOI] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr., Sect. D: Struct. Biol. 2010, 66, 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser Crystallographic Software. J. Appl. Crystallogr. 2007, 40 (Part 4), 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G. W.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. Overview of the CCP4 Suite and Current Developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67 (Part 4), 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60 (Part 12), 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkóczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L.-W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66 (Part 2), 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun D. I. Determination of the Regularization Parameter in Indirect-Transform Methods Using Perceptual Criteria. J. Appl. Crystallogr. 1992, 25 (4), 495–503. 10.1107/S0021889892001663. [DOI] [Google Scholar]
- Akabayov B.; Akabayov S. R.; Lee S.-J.; Tabor S.; Kulczyk A. W.; Richardson C. C. Conformational Dynamics of Bacteriophage T7 DNA Polymerase and Its Processivity Factor, Escherichia Coli Thioredoxin. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (34), 15033–15038. 10.1073/pnas.1010141107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun D.; Barberato C.; Koch M. H. J. CRYSOL—A Program to Evaluate X-Ray Solution Scattering of Biological Macromolecules from Atomic Coordinates. J. Appl. Crystallogr. 1995, 28, 768–773. 10.1107/S0021889895007047. [DOI] [Google Scholar]
- Chen R.; Weng Z. Docking Unbound Proteins Using Shape Complementarity, Desolvation, and Electrostatics. Proteins: Struct., Funct., Genet. 2002, 47 (3), 281–294. 10.1002/prot.10092. [DOI] [PubMed] [Google Scholar]
- Chen R.; Weng Z. A Novel Shape Complementarity Scoring Function for Protein-Protein Docking. Proteins: Struct., Funct., Genet. 2003, 51 (3), 397–408. 10.1002/prot.10334. [DOI] [PubMed] [Google Scholar]
- Pierce B.; Weng Z. ZRANK: Reranking Protein Docking Predictions with an Optimized Energy Function. Proteins: Struct., Funct., Genet. 2007, 67 (4), 1078–1086. 10.1002/prot.21373. [DOI] [PubMed] [Google Scholar]
- Fletcher R.; Reeves C. M. Function Minimization by Conjugate Gradients. Comput. J. 1964, 7 (2), 149–154. 10.1093/comjnl/7.2.149. [DOI] [Google Scholar]
- Fletcher R., Ed. Optimization, Symposium of the Institute of Mathematics and Its Applications, University of Keele, England; Academic Press, 1969. [Google Scholar]
- Creighton T.. PROTEINS: Structure and Molecular Properties, 2nd ed.; W.H. Freeman and Co.: New York, 2002. [Google Scholar]
- Bashford D.; Karplus M. Multiple-Site Titration Curves of Proteins: An Analysis of Exact and Approximate Methods for Their Calculation. J. Phys. Chem. 1991, 95 (23), 9556–9561. 10.1021/j100176a093. [DOI] [Google Scholar]
- Tomayko M. M.; Reynolds C. P. Determination of Subcutaneous Tumor Size in Athymic (Nude) Mice. Cancer Chemother. Pharmacol. 1989, 24 (3), 148–154. 10.1007/BF00300234. [DOI] [PubMed] [Google Scholar]
- Pittala S.; Krelin Y.; Shoshan-Barmatz V. Targeting Liver Cancer and Associated Pathologies in Mice with a Mitochondrial VDAC1-Based Peptide. Neoplasia (United States) 2018, 20 (6), 594–609. 10.1016/j.neo.2018.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A. H.; Jacobson K. A.; Rose J.; Zeller R. Hematoxylin and Eosin Staining of Tissue and Cell Sections. Cold Spring Harbor Protoc. 2008, 3 (5), 4986–4988. 10.1101/pdb.prot4986. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.