

Abstract

The leishmaniases, caused by Leishmania species of protozoan parasites, are neglected tropical diseases with millions of cases worldwide. Current therapeutic approaches are limited by toxicity, resistance, and cost. N-Myristoyltransferase (NMT), an enzyme ubiquitous and essential in all eukaryotes, has been validated via genetic and pharmacological methods as a promising anti-leishmanial target. Here we describe a comprehensive structure–activity relationship (SAR) study of a thienopyrimidine series previously identified in a high-throughput screen against Leishmania NMT, across 68 compounds in enzyme- and cell-based assay formats. Using a chemical tagging target engagement biomarker assay, we identify the first inhibitor in this series with on-target NMT activity in leishmania parasites. Furthermore, crystal structure analyses of 12 derivatives in complex with Leishmania major NMT revealed key factors important for future structure-guided optimization delivering IMP-105 (43), a compound with modest activity against Leishmania donovani intracellular amastigotes and excellent selectivity (>660-fold) for Leishmania NMT over human NMTs.
Introduction
Leishmaniases are diseases caused by protozoan parasites of the genus Leishmania, which are transmitted to humans by female sandflies of the genera Phlebotomus and Lutzomyia. The clinical symptoms depend on the Leishmania species involved and range from skin ulcers with permanent scars (cutaneous leishmaniasis, CL) to swelling of the spleen and liver (visceral leishmaniasis, VL). Leishmaniasis is distributed worldwide: in 2015, 87 countries were considered endemic for CL and 75 for VL, with 12–15 million people infected and up to 30 000 deaths per year. Three hundred and fifty million people are at risk of infection, and 1 million new cases occur each year.1,2 Therapeutic approaches currently rely on four main drugs: pentavalent antimonials (sodium stibogluconate, SSG), miltefosine, amphotericin B, and paromomycin. However, these drugs are limited by toxicity, cost, and problematic treatment regimens, while drug resistance to SSG has emerged in India.3 It is therefore important to develop new drugs and therapeutic strategies for leishmaniases.
N-Myristoyltransferase (NMT) catalyzes the co-translational N-terminal myristoylation of a wide variety of proteins in all eukaryotes. Recent studies identified NMT as a potential target in drug discovery against diseases caused by fungi4,5 and several protozoan parasites, including Plasmodium falciparum,6−9Plasmodium vivax,10 and Trypanosoma brucei.11−13 Human (host) NMT has also been explored in viral infections, including the common cold, poliovirus, foot-and-mouth disease virus,14 enterovirus,15 and HIV,16 as well as in cancer.17,18 In Leishmania, more than 30 high-confidence NMT substrates have been identified, including a number of proteins known to be critical for parasite function, suggesting that NMT inhibition will have pleiotropic effects on the parasite.19−24 The recent discovery and characterization of DDD100097 have provided pharmacological validation of NMT as a drug target in Leishmania;(25) however, significant challenges remain in translating its potent enzymatic activity into in vivo activity while retaining selectivity over human (Hs) NMTs. Despite DDD100097’s potency against the Leishmania major (Lm) NMT and modest activity in intracellular amastigotes (EC50 2.4 μM), it only has a 10-fold selectivity over HsNMTs.
We have previously reported peptidomimetic inhibitors of LmNMT with sub-micromolar potency but minimal selectivity over the two human NMTs 1 and 2,26 and a series of small-molecule inhibitors targeting Leishmania donovani (Ld) NMT from an initial benzo[b]thiophene scaffold, with optimized analogues showing minimal host cell toxicity.27 However, neither series inhibited intramacrophage L. donovani amastigotes, the clinically relevant life stage of the parasite, implying problems with stability or poor cellular uptake. In a previous publication, we reported a high-throughput screen (HTS) of Plasmodium falciparum (Pf) NMT and LdNMT against a 150 000 compound diversity set28 using a scintillation proximity assay,29 identifying four series with good to excellent selectivity against LdNMT over other NMTs, based on aminoacylpyrrolidines, piperidinylindoles, thienopyrimidines, and biphenyl derivatives. Subsequently, we reported the binding mode of all four series, elucidated by cocrystallization with LmNMT,30 and these findings were used to improve the potency of the aminoacylpyrrolidines and piperidinylindoles in a structure-guided approach, leading to a 40-fold increased potency and good selectivity over HsNMTs.31 However, due to poor cellular uptake, these inhibitors also lacked cell-based activity.32
Here we report a comprehensive structure–activity relationship (SAR) study of the thienopyrimidine series of potent Leishmania NMT inhibitors, improving their selectivity over human NMTs and identifying the first cell-active on-target LdNMT inhibitors within this series.
Results and Discussion
A more synthetically accessible analogue of the initial thienopyrimidine HTS-hit, PF-00349412, resulted in a recently published high-resolution crystal structure of derivative 1 (also known as IMP-083) in complex with LmNMT30 (Figure 1). Interestingly, the structure indicates that two molecules occupy the peptide-binding groove. The ligand proximal to myristoyl-CoA is proposed to be of higher affinity due to its better fit to the electron density maps and its lower mean atomic temperature (B) factor, while the distal ligand is sometimes absent (Figures 1C and S2A). The basic center in the N-methylpiperidine ring in 1 is predicted to be 92% protonated at pH 7.4, and in the crystal structure, this group in the proximal ligand is protonated. It forms a hydrogen bond with a water molecule, which in turn may interact with LmNMT Tyr80, Tyr92, and Asn167, and ion-pairing with the carboxylate of Leu421 is also observed. Aromatic stacking interactions between LmNMT Tyr217 and the thienopyrimidine system also contribute to the overall recognition (Figure 1D). The N4 atom of the pyrimidine ring additionally forms a direct hydrogen bond with Tyr345. The third structural feature of the inhibitor, the nitrile group, is surrounded by LmNMT Val81, Ala204, and Gly205. The corresponding moiety in the distal ligand may form a hydrogen bond with Asn376. Although the major interactions between LmNMT and compound 1 are known, explaining the 17-fold selectivity (IC50 (HsNMT1)/IC50 (LdNMT)) of the inhibitor (1) for Leishmania NMTs over the human enzyme is not straightforward since all amino acid side chains involved in significant interactions are conserved in human NMT. We hypothesized that conformational preferences in LdNMT and LmNMT Tyr217 compared with that of the equivalent HsNMT1 residue might cause the observed potency differences.9,30
Figure 1.

Binding pocket of compound 1 in LmNMT. (A) Ribbon rendering of LmNMT color ramped from amino (blue) to carboxy (red) terminus. The atoms of the myristoyl-CoA and 1 ligands are shown in space-filling representations and colored by atom types C (gray), O (red), N (blue), P (magenta), and S (yellow). (B) Structure of 1 (also known as IMP-083). (C) Cylinder rendering of the ligands emphasizing the proximal and distal binding sites for a pair of molecules of 1, which pack against one another. (D) Binding pocket for 1 showing the Cα and side chains of 1-interacting residues together with the C-terminal residue Leu421 whose α-carboxylate makes an ion-pairing interaction with the tertiary amino group of the proximal ligand. Water molecules in the neighborhood of 1 are shown as red spheres, and polar protein–ligand interactions are denoted by dashed lines. PDB ID: 4cgo.
Synthesis
In this SAR study, variation in the ligand core and substitutions at the C2, C4, C6, and C7 positions were all of interest (Figure 2B). The syntheses of lead ligand 1 and analogues were designed to allow variation at each of these positions, exploiting the ability to selectively displace the 4-chloro substituent of 2,4-dichloropyrimidines to thieno[3,2-d]pyrimidines and other fused heterocyclic cores of interest.33 After synthesis of the heterocyclic core according to literature methods, the C4 substituent was selectively incorporated by 4-chloro displacement using stoichiometric quantities of the appropriate amine and controlled temperatures. Displacement at the 2-chloro position to generate C2 analogues, such as 2, was then achieved using an excess of a second amine and higher reaction temperatures, often under microwave irradiation (Scheme 1).34
Figure 2.

(A) Correlation of LmNMT-based and LdNMT-based pIC50 values obtained with the CPM assay. The Pearson coefficient (ρ) and P-value are shown. (B) Schematic representation of the SAR results. The four structural segments of the inhibitor are highlighted. All symbols are annotated in the lowest box.
Scheme 1. General Scheme for the Synthesis of 1 and Related Analogues.

Reagents and conditions: (i) amine (1.0 equiv), N,N-diisopropylethylamine (DIPEA, 1.2 equiv), EtOAc, room temperature (rt), 18 h; (ii) amine (4–10 equiv), EtOAc, microwave, 120–200 °C, 30–90 min; (iii) 1-chloroethyl chloroformate (1.0 equiv), DCE, reflux, 1 h; (iv) MeOH, reflux, 1 h; and (v) aldehyde (1.0 equiv), NaBH(OAc)3, DCE, rt 3–18 h.
However, it was not possible to use a one-step 2-chloro displacement to access compound 1 or later analogues such as 3, 10–13, or 15–16, either due to the presence of two possible nucleophilic centers in the amine reagent, e.g., N-methylpiperidin-4-amine for analogue 10, or limited commercial availability. Attempts to synthesize des-methyl derivatives, for example, using 4-amino-N-methylpiperidine, failed due to a competing demethylation/displacement reaction that led to an inseparable mix of products.35 Instead, the additional basic center was protected using a more sterically hindering N-benzyl group during the displacement reaction and then deprotected in a two-step process, generally using 1-chloroethyl chloroformate and methanol.36 The resulting secondary amine could be derivatized using reductive amination to give the desired products. This methodology was successfully applied to generate compound 1 and also analogues with quinazoline cores (3) or molecules containing linear- (11, 15), pyrrolidine- (26, 52, 53), or piperidine-based amines at the C2 position (12, 13). Detailed procedures for the different cores and scaffolds are given in the Supporting Information.
Activity
A fluorogenic assay was used to analyze the potency of all inhibitors against Ld, Lm NMTs, and HsNMT1.37 The assay quantifies the amount of coenzyme A liberated during the acyl transfer reaction using the fluorogenic thiol-reactive dye 7-diethylamino-3-(4-maleimido-phenyl)-4-methylcoumarin (CPM). Initial analysis of a selection of 11 compounds against both Leishmania NMTs revealed a strong correlation in activity (Figure 2A), as expected from the sequence alignment of the enzymes (Figure S1), with an identity of 98%. Due to the varied availability of the LdNMT and LmNMT enzymes, enzymatic activity data for ligands against both LdNMT and LmNMT enzymes was considered during the SAR analysis. Cocrystal structures were generated solely with LmNMT thanks to the capacity to solve structures rapidly by soaking ligands into crystals of this protein. The SAR findings are summarized schematically in Figure 2B.
In our previous structural analysis,30 it was proposed that conformational variations between human and Leishmania NMTs in LmNMT Tyr217 might be responsible for selectivity differences.9 To assess the impact of the π interaction, we initially investigated the importance of the inhibitor core by exchanging the thienopyrimidine (1, 2) with a quinazoline (3, 4), a pyridopyrimidine (5), a purine (6), or a monocyclic pyrimidine (7) (Table 1). Thienopyrimidines (1, 2), quinazolines (3, 4), and pyridopyrimidine (5) exhibit comparable potency and selectivity profiles in this series, supported by an overlay of the crystal structures of compounds 1 and 3 in complex with LmNMT (Figure 3A), which reveals an identical binding mode for both compounds. In contrast, inhibitory potency is entirely lost in the case of purine (6) and pyrimidine (7) derivatives, which may be due to reduced lipophilicity compared with those of 2 and 4. We therefore focused our subsequent SAR study on C2, C4, and C6/C7 substituents on thienopyrimidine and quinazoline cores to obtain small-molecule inhibitors with enhanced potency.
Table 1. Effect of Changing Core Structures on Enzyme- and Cell-Based Activitiesb.
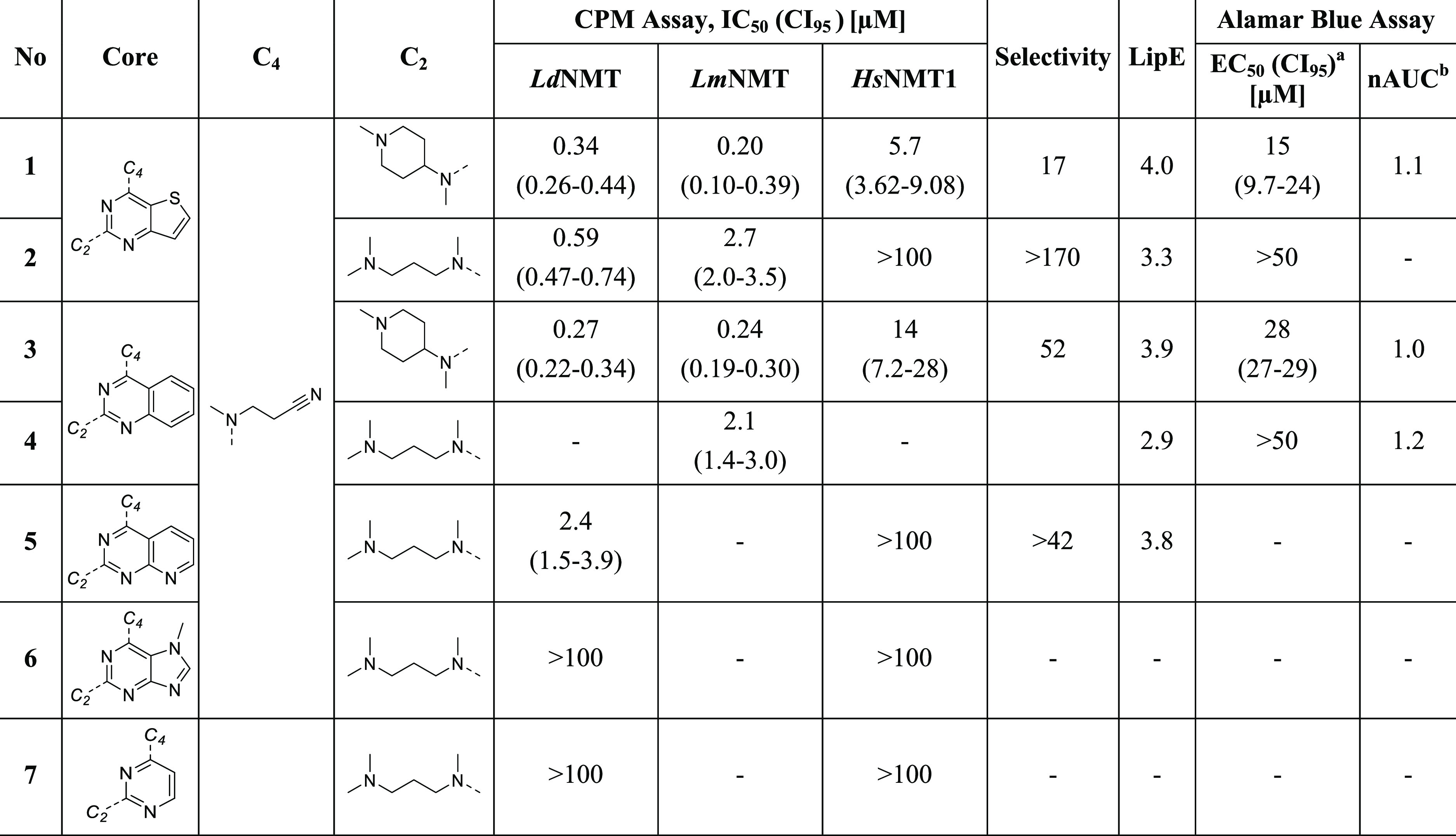
Selectivity = IC50 (HsNMT1)/IC50 (LdNMT). LipE = pIC50 – clog P; where values for LdNMT and LmNMT were obtained, the average LipE is reported. EC50 for intracellular L. donovani amastigotes.
Toxicity against bone-marrow-derived macrophages; nAUC, normalized area under the curve. The dose–response curves are shown in Figures S6–S10.
Figure 3.

Comparison of the modes of binding of selected inhibitors with varied cores or C2 substitution. MyrCoA is not shown in the binding site, for clarity. Stereo renderings can be found in Figure S3. (A) 1 and 3 emphasizing the highly similar binding modes of the proximal ligand pair, which differ only in the core substitution of the thienopyrimidine for a quinazoline. Protein carbons: light green or green; ligand carbons: gray, dark gray; and waters: crimson or red. PDB ID 4cgo, 6qda. (B) 1, 10, and 12 exploring N-substitutions on the piperidine ring. As before, the proximal ligands are closely superimposable. The same is true of the distal ligands, with the exception of the piperidine ring, which is significantly displaced in the ethyl-substituted 12. Protein carbons: light green, green, lawn green; ligand carbons: gray, dark gray, light gray; and waters: crimson, red, violet red. PDB ID 4cgo, 6qdb, 6qdc. (C) 1 and 2 probing the significance of linear versus cyclic amine extensions from C2 of the core. The proximal ligand’s binding mode is altered in 2, with tilting of the thienopyrimidine core, dual conformations of the C2 amine extension, and repositioning of the C4 aminopropionitrile. No distal ligand was observed in the LmNMT complex with 2. Protein carbons: light green or green; ligand carbons: gray, dark gray; and waters: crimson or red. PDB ID 4cgo, 6qd9. (D) Chemical structures.
Effect of C2 Substitution on LdNMT and LmNMT Enzyme Activities
We first looked at the effect of variation in the C2 unit (Table 2). Exchanging the piperidine for a tetrahydropyran (8) or hydroxypropyl substituent (9) resulted in complete loss of activity. Considering the striking differences in basicity and hydrogen-bond interaction energies,38 these findings support the importance of the basic center, consistent with the binding mode of compound 1 and trends in previously reported ligand series.11,25,30 The potency deviation between a tertiary (1–4) or secondary amine as the basic center (10–11, 23) is negligible, presumably due to calculated pK differences of less than one unit (compound 2 pKb = 4.6; compound 11 pKb = 3.8). However, further extending the N-methyl substituent or inserting an aromatic system (12–16, 20) gradually decreases the inhibitory potency, presumably because the steric demand of the moiety is limited by the size of the pocket formed by LmNMT Tyr80, Tyr92, Asn167, Thr203, and Leu421, and myristoyl-CoA (MyrCoA). While the crystal structures of 1, 10, and 12 in complex with LmNMT (Figure 3B) show that these compounds are well accommodated and show similar binding modes, the basic centers in the C2 substituents of the distal ligands deviate from 10 to 1 by 0.8 Å and from 1 to 12 by 1.5 Å due to steric interactions with Asp396. However, these small differences are likely unimportant since the proximal ligand appears to drive inhibition. Between the overlaid proximal ligands, there is even greater similarity; for example, the basic centers deviate by only 0.2 Å. Enhancing the flexibility of the basic center by applying acyclic amine-based C2 substituents (2, 4) has no effect on the inhibitory activity against LdNMT. Considering sequence alignment (Figure S1), the same result was expected for LmNMT; however, 2 and 4 exhibited a 10-fold decreased activity against the L. major enzyme. The corresponding crystal structure of 2 with LmNMT shows a small displacement of the core and dual conformations of the C2 derivatives in the linear amine side chain compared with 1, while no electron density was observed for the distal ligand, suggesting it is absent (Figure 3C). There is also a significant difference observed in the position of the aminopropionitrile group: instead of occupying a groove composed of LmNMT Val81 and Gly205 as in 1, the moiety is vertically aligned in compound 2. It is possible therefore that the orientation of the C2 substituent in 2 and 4 varies between the two Leishmania enzymes.
Table 2. Effect of Changing C2 Substitution Using Thienopyrimidine and Quinazoline Cores (Compounds 8–30)a,b.

95% CI = 95% confidence interval. Selectivity = IC50 (HsNMT)/IC50 (LdNMT). LipE = pIC50 – clog P; where values for LdNMT and LmNMT were obtained, the average LipE is reported. EC50 for intracellular L. donovani amastigotes.
Toxicity against bone-marrow-derived macrophages; nAUC, normalized area under the curve. The dose–response curves are shown in Figures S6–S10.
Significantly changing the geometry of the C2 substituent and thereby decreasing the distance between the basic center and the core resulted in decisively reduced inhibitory potency and selectivity (Figure 2B). This occurs if piperazine- (17–18), pyrrolidine-amine- (19, 25–26, 53–55), or inverted piperidine-amine (20–22)-based moieties are substituted on either thienopyrimidine or quinazoline cores, suggesting that the basic center and the bicyclic core must adopt a specific conformation to permit optimal interactions in the peptide-binding pocket of LmNMT. The crystal structure of compound 52, which is discussed further below, supports this rationalization (Figure 4D). The geometry of the basic center and bicyclic core seems to be optimal in compound 1. Further increasing degrees of freedom (2, 4) appears to be counterproductive and results in a decreased activity in LmNMT as discussed above. Increasing the rigidity and bulkiness of the piperidine ring using a bicyclic system (27) diminishes the inhibitory activity against both Ld and LmNMT.
Figure 4.

Comparison of the modes of binding of selected inhibitors with varied C4 or C6 substitution. For clarity, MyrCoA is not shown in the binding site. Stereo renderings can be found in Figure S4. (A) 1 and 43 demonstrate similar binding interactions despite the rigidification of the aminopropionitrile. The distal ligand does not appear, and 43 demonstrates dual conformations of the C2 substituent. Protein carbons: light green, green; ligand carbons: gray, dark gray; and waters: crimson, red, violet red. PDB ID 4cgo, 6qdd. (B) Chemical structures of relevant ligands. (C, D) tert-Butyl C6 substitution is accommodated in the proximal ligands for 51 and 52. However, the distal ligand conformation is inverted in 51 and dual C2 conformations are observed in 52, which is present at partial occupancy PDB ID 4cgo, 6qde, 6qdf. (E) 2 and 56 have analogous binding modes with an additional hydrogen bond in 56 mediated by the bromine substituent. PDB ID 4cgo, 6qdg.
Finally, we explored whether variation at the first amine in the C2 substituent was tolerated. Extending the alkyl chain with a hydroxyethyl (28) or hydroxypropyl (29–30) group decreases inhibitory potency, presumably due to the increased steric demand.
Effect of C4 Substitution on LdNMT and LmNMT Enzyme Activities
Substitution at C4 was shown to be crucial as compound 31, a derivative of 2 lacking the aminopropionitrile in position C4, was no longer active. We explored the effect of differing substituents at this position using linear moieties of similar size that, like the aminopropionitrile, were also potential hydrogen-bond acceptors. The results indicate that alkyl ethers (32–33), amino alcohols (34–35, 39), and amino amides (36–38) are not tolerated but increasing the rigidity of the alkyl ethers with a pyrrolidine motif (40–42) restores the inhibitory activity. However, applying the same strategy to the aminopropionitriles (43–45) failed to improve their potency.
It had been noted that the aminopropionitrile side chains had different geometries in the proximal and distal ligands (Figure S5). Therefore, only one enantiomer was made for compounds 40–45, in which the geometry was proposed to match the pro-stereochemistry observed in the proximal ligand in previous structures such as Figure 3A. Theoretically, the proximal ligands should still be able to bind, but the C4 side chain would clash in the distal ligand and be unable to bind (Table 3).
Table 3. Effect of Changing C4 and C2 Substitutions on Enzyme- and Cell-Based Activities (Compounds 31–49)a.

95% CI = 95% confidence interval. Selectivity = IC50 (HsNMT)/IC50 (LdNMT). LipE = pIC50 – clog P; where values for LdNMT and LmNMT were obtained, the average LipE is reported. nAUC, normalized area under the curve. The dose–response curves are shown in Figures S6–S10.
This was supported by the crystal structures of 1 and 43 in complex with LmNMT, which indicated a similar binding mode for both proximal ligands, with little deviation between the cores and C4 substituents, and no distal ligand present. However, the structure of 43 does show two possible conformations of the C2 substituent (Figure 4A). This result also helps confirm that the proximal ligand is of primary importance for activity.
To test the impact of the nitrile group, analogues with a simple amino ethyl moiety at C4 were synthesized (46–49). Interestingly, this simple group is sufficient to deliver small-molecule inhibitors with moderate activity, although at the cost of a slight drop in selectivity over HsNMT1 (46 compared with 1 or 43). This finding supports the hypothesis that the main role of the substituent in position C4 is to facilitate ideal positioning of the major interaction sites—the aromatic core and the basic center in the C2 moiety—mainly via hydrophobic interactions.
Effect of Substitution at C6 on LdNMT and LmNMT Enzyme Activities
Finally, we probed the effect of substitution at C6 in combination with a range of C2 substituents. Modifications here are generally tolerated and do not prevent binding to Leishmania NMTs. Introducing a bulky tert-butyl group (50–54) results in a 10-fold increased inhibitory potency (50) and a similar selectivity profile when compared with derivatives without a C6 substituent. Compound 50 is the most potent inhibitor in this SAR study, with a 7-fold improved IC50 value against LdNMT of 46 nM (95% CI: 35–60 nM) and a similar selectivity (IC50(HsNMT1)/IC50(LdNMT) = 12-fold) to that of compound 1. The crystal structures of 51 and 52 in complex with LmNMT reveal that the tert-butyl group has only a modest impact on the binding mode of the proximal ligand (Figure 4C,D). However, the distal ligand exhibits a flipped orientation in 51, most likely due to the steric demand of the C6 moiety that alters the conformation of LmNMT His219 (Figure 4C). In the structure with 52, there is only partial ligand occupancy; moreover, the pyrrolidine moiety at the C2 position in the proximal ligand displays two alternate conformations. As a result, key interactions of the basic center with the Leu421 carboxylate and the water molecule bound to LmNMT Tyr80, Tyr92, and Asn167 are weakened and make a smaller contribution to binding (Figure 4D). Considering the potency difference between 51 and 52, this observation corroborates our hypothesis that decreased activities of 18–22, 25–26, and 52–54 are caused by conformational deviations in the C2 moiety.
Substitution at C6/C7 on the quinazoline core was also explored. Inhibitors with bromo (55–57), methoxy (58–59), or aromatic substituents (60–62) in position C6 exhibit comparable activity and selectivity in comparison to compounds lacking a C6 moiety. The crystal structure of compound 56 in complex with LmNMT indicates similar binding modes of the bromo (56) and the corresponding thienopyrimidine derivative (2). An additional interaction is potentially formed between the bromide and a water molecule bound to His219 and the backbone amide of Asp396 (Figure 4E). In contrast to substitution at C6, moving the methoxy group from C6 (59) to C7 (63) significantly decreases the inhibitory potency.
Although the SAR discussed so far (compounds 1–63) revealed some derivatives with significantly improved nanomolar potency with respect to initial compound 1 (e.g., 43 and 50), none of them exhibit lipophilic efficiency (LipE; LipE = pIC50 – clog P) values above 4 (Figure 5A). Previous work on inhibitors for NMT in Trypanosoma brucei(39) and P. vivax(8) established that interactions around PvNMT Ser319 (LmNMT Ser330) are important for higher affinity compounds. In this thienopyrimidine series, the proximal ligand does not extend into this pocket, which is occupied by the distal ligand. To try and grow into this pocket, and to further improve the molecules’ drug-like properties, a fragment merging approach with DDD8564611,39 a T. brucei NMT inhibitor was trialed. Crystallography performed on DDD85646 in LmNMT had shown that the sulfonamide group projected the pyrazole into the desired pocket, forming a hydrogen bond to LmNMT Ser330 (Table 4).11
Figure 5.

(A) Plot of pIC50 against clog P. Diagonal lines represent areas of corresponding lipophilic efficiency (LipE) values. (B) Plot of LipE against selectivity (selectivity = IC50 (HsNMT)/IC50 (LdNMT)). (C) Schematic representation of fragment merging strategy between 3 and DDD85646(11) to generate 64 and related analogues. (D) Crystal structure of 1 and 64 in complex with LmNMT. 64 principally occupies the site of the proximal ligand in 1 extending through the sulfonamide group, which forms hydrogen bonds with the LmNMT backbone amide. PDB ID 4cgo, 6qdh.
Table 4. Effect of Changing C2 and C6/7 Substitutions and Fragment Merging on Enzyme- and Cell-Based Activities (Compounds 50–67).
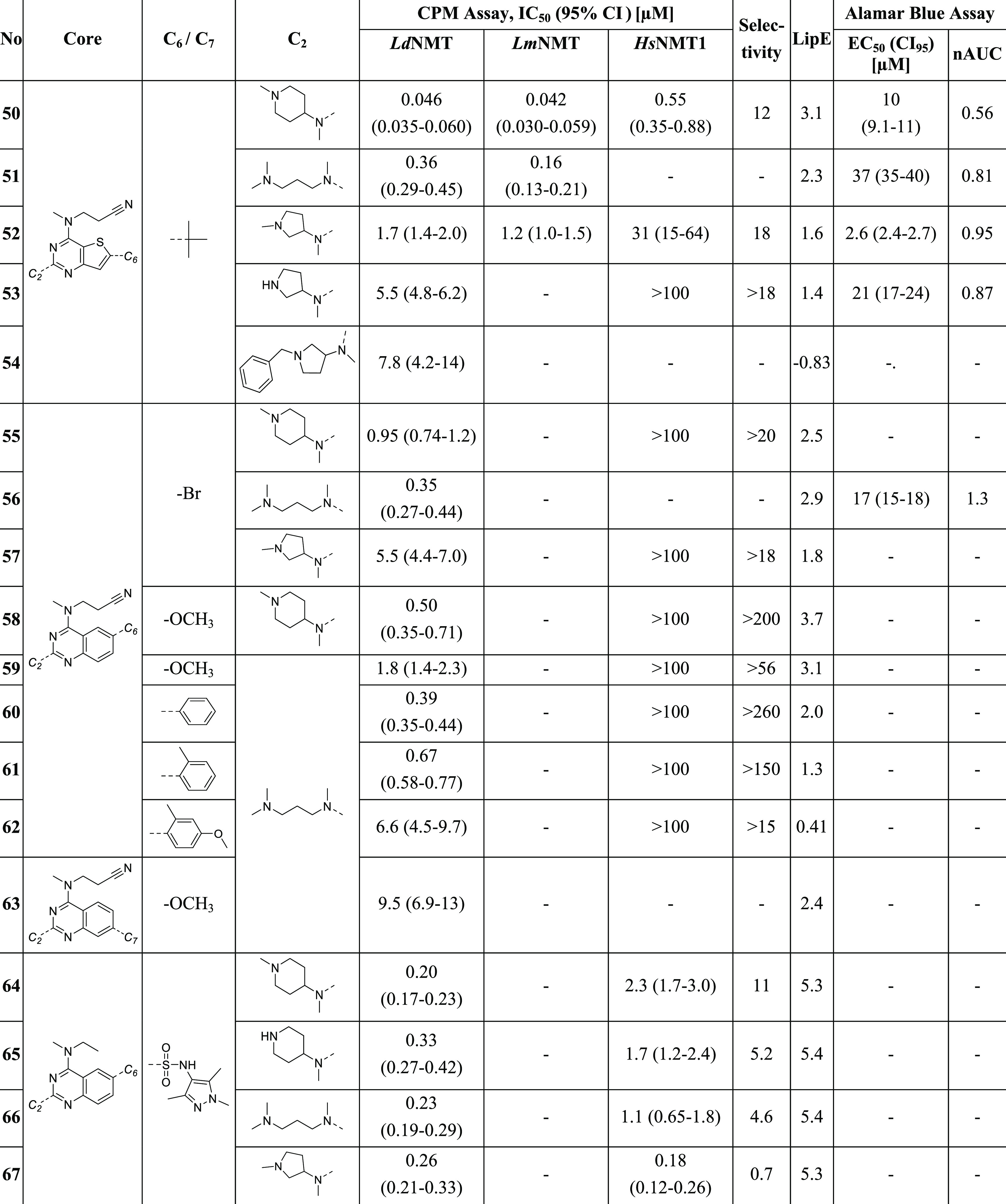
95% CI = 95% confidence interval. Selectivity = IC50 (HsNMT)/IC50 (LdNMT). LipE = pIC50 – log P; where values for LdNMT and LmNMT were obtained, the average LipE is reported. nAUC, normalized area under the curve. The dose–response curves are shown in Figures S6–S10.
Merged ligands were therefore proposed, in which a sulfonamide and pyrazole group would grow off the C6 position (Figure 5C), The strongly acidic reaction conditions required for chlorosulfonylation of the quinazoline core were not compatible with the aminopropionitrile side chain; hence, this modification was attempted on the more accessible N-ethyl derivative, given that analogues 46–49 still retained some activity. These efforts resulted in the pyrazole-based sulfonamide derivatives 64–67.
These compounds have lower clog P values and combine the major interactions of the distal and the proximal ligands. The crystal structure of 64 in complex with LmNMT (Figure 5D) demonstrates the success of this approach. The quinazoline core and C2 substituent of 64 exhibit the same conformation in the peptide-binding pocket as the proximal ligand of 1, while the pyrazole moiety of 64 coincides with the aromatic system of the distal ligand of 1. Additionally, the sulfonamide group of 64 mediates a hydrogen bond with the backbone amides of Gly395 and Asn396. Analysis of the potency of all sulfonamide derivatives (64–67) revealed decisively improved IC50 values in comparison to the corresponding thienopyrimidine derivatives, resulting in LipE values above 5 (Figure 5A).
However, a significant drawback of this new subseries is their lower selectivity in relation to HsNMT1. For instance, inhibitors 65 and 66, respectively, exhibit a decreased and marginal selectivity of only 5.2- and 4.6-folds (Figure 5B). It is proposed that exchanging the amino ethyl C4 substituent with aminopropionitrile could result in sulfonamides with improved selectivity over HsNMT1 and better early lead-compound properties. However, these molecules have proven synthetically challenging, and the corresponding derivatives will be part of future studies.
Cell-Based Assays
A selection of 22 compounds was further studied in an Alamar blue-based cellular assay, which had been previously validated against known anti-leishmanial drugs amphotericin B and miltefosine,32 to quantify their cytotoxicity against macrophages and their activity against intracellular amastigotes of L. donovani. Due to the steep slope of the toxicity profile and the correspondingly low accuracy of the CC50 values, the normalized area under the curve (nAUC) was used as the toxicity parameter (Figure 6A). Strikingly, all compounds with an EC50 below 50 μM that show a toxic effect against macrophages (nAUC ≪ 1) share the tert-butyl group as C6 substituent (50–53). This increased effect on macrophages may be due to the larger clog P and clog D values of compounds 50–53, potentially improving membrane permeability or increasing compound promiscuity. Comparing pIC50 values against Leishmania NMT with intracellular amastigote pEC50 values indicates a good correlation for all compounds apart from 26, 52, and 53 (Figure 6B), which show greater potency in cells than anticipated. Interestingly, these three inhibitors are the only derivatives tested that share a pyrrolidine-based C2 moiety (Figure 4D).
Figure 6.
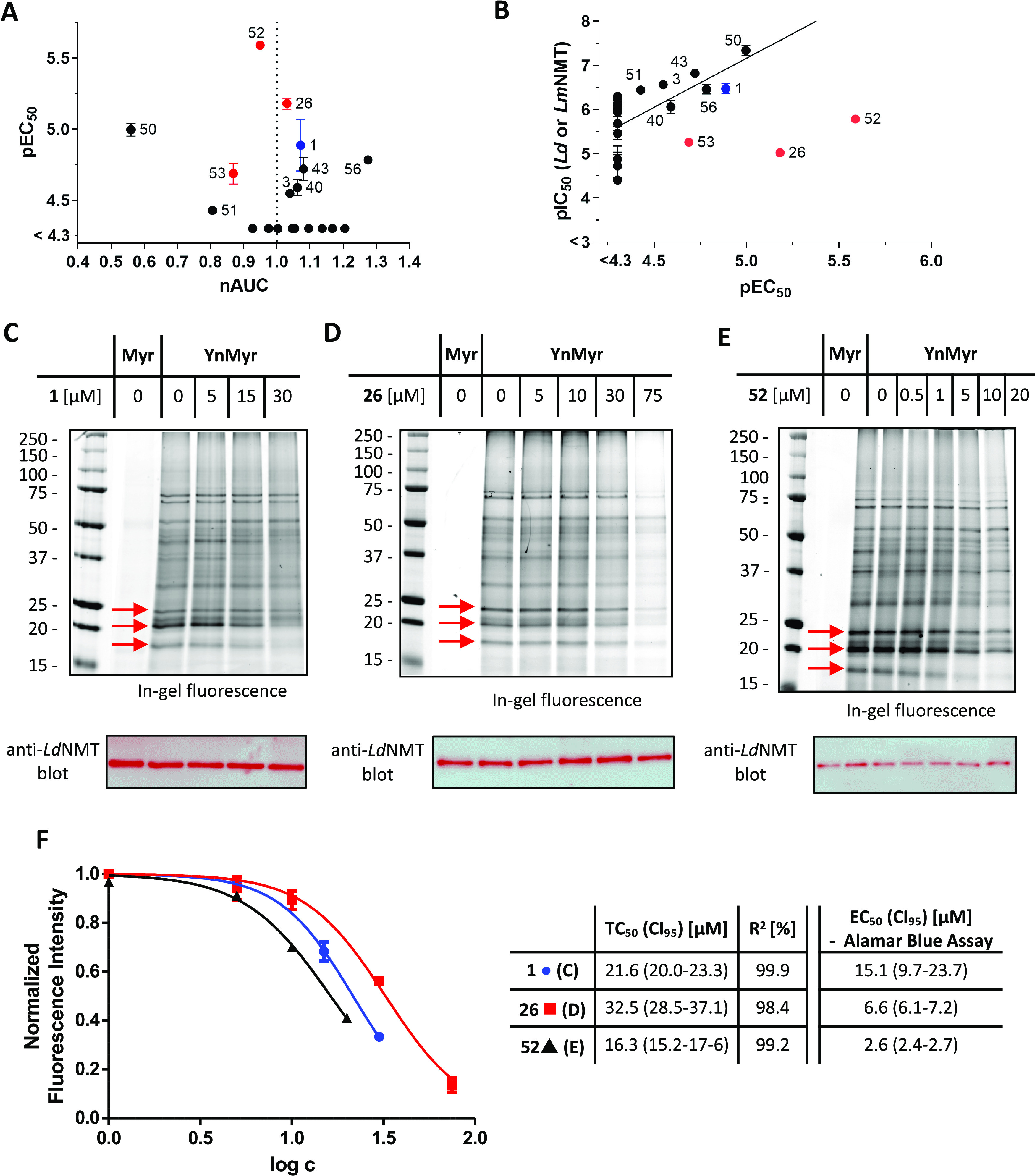
(A) Plot of anti-L. donovani amastigote activity (pEC50) against macrophage toxicity (normalized area under the curve, nAUC). Thienopyrimidine 1 is highlighted in blue. (B) Plot of Leishmania NMT pIC50 (either Ld or Lm) against cell-based pEC50. A significant correlation (ρ = 0.75, P = 0.0006) is evident if 26, 52, and 53 (highlighted in red) are excluded. Compound 1 is highlighted in blue. (C–E) In-gel fluorescence and anti-LdNMT Western blot results for the cell-based tagging assay. (F) Fluorescence intensities of several individual bands between 25 and 15 kDa were quantified in (C–E) and normalized to the corresponding intensities of the control samples (without inhibitor) and the anti-LdNMT blot results. Tagging IC50 values (TC50) were extracted by nonlinear regression with a sigmoidal dose–response model (constraints: bottom = 0; top = 1.0) to the mean normalized intensity.
Furthermore, 26 and 52 exhibit a 3- to 6-fold improved EC50 value with respect to compound 1 (Figure 6A,B). To test the hypothesis that this improved potency is due to an on-target effect, a cell-based tagging assay22 was performed with 1, 26, and 52 (Figure 6C–F). Briefly, L. donovani amastigotes were treated with varying concentrations of the corresponding compound and YnMyr, an alkyne analogue of myristic acid.40 Following cell lysis, labeling with a trifunctional capture reagent41 via copper-catalyzed azide–alkyne cycloaddition facilitated the visualization of myristoylated proteins by in-gel fluorescence. Western blot analysis using an antibody against LdNMT revealed no variations of the total protein amount or the NMT expression level over the investigated inhibitor concentration range (Figure 6C–E), and all three compounds (1, 26, and 52) significantly decreased myristoylation in a dose-dependent manner. To obtain the tagging IC50 value (TC50), a quantitative measure of target engagement, the fluorescence intensities of several individual bands between 25 and 15 kDa were quantified and normalized to the corresponding intensities of the control sample (no inhibitor) and the anti-LdNMT blot results (loading control). The TC50 and EC50 values of compound 1 are in good agreement, indicating that the anti-L. donovani activity is due to on-target NMT inhibition (Figure 6F). However, the TC50 values of compounds 26 and 52 are both 5- to 6-fold above the EC50 values obtained with the Alamar blue assay (Figure 6B). Although compounds 26 and 52 do not show toxic off-target effects against macrophages, these findings suggest that nonspecific effects may also contribute to the anti-L. donovani activity for these compounds.
Conclusions
NMT inhibitors with a fused pyrimidine core exhibit three sites involved in interactions with the enzyme: the aromatic system of the core, an amine-based C4 moiety, and the basic center in the C2 substituent. The central scaffold forms stacking interactions with Tyr217, with scaffold lipophilicity particularly being important. The reason for C4 preference is less evident; although it does not appear to form any important interactions with the protein, its contribution to affinity may be through the quality of its packing with the C2 substituent. In the case of the C2 substituent, our data show that a specific geometry is essential for the appropriate positioning of the basic center. Although linear C2 moieties are well tolerated, exchanging the piperidine with a pyrrolidine system results in decreased activity against NMT due to a flipped orientation in the binding pocket, leading to a different set of hydrogen bonds. Remarkably, the corresponding derivatives exhibit a significantly improved anti-Leishmanial activity. In fact, compounds 26 and 52 are the most potent derivatives in the cellular assay that additionally show no toxic effect against macrophages. However, results from the tagging assay suggest that although 26 and 52 cause a dose-dependent decrease in protein-myristoylation, off-target effects may also contribute to anti-leishmanial activity. This explanation is supported by the fact that the pyrrolidine-based derivatives (26, 52, and 53) are the only inhibitors that deviate from the correlation between enzyme- and cell-based potencies. A particularly promising new inhibitor, IMP-105 (43), exhibits enhanced selectivity for Leishmania NMT over human NMT1 of >660-fold, while both enzyme- and cell-based activities are maintained (Figure 7). Considering the importance of selectivity, IMP-105 (43) is a useful tool molecule to investigate the impact of NMT inhibition in Leishmania. However, as yet, these compounds do not have the optimal potency or pharmacokinetic properties to enable in vivo experiments to be undertaken to confirm whether these compounds can reduce parasite burden in vivo. Future studies to further improve the potency and properties of these compounds will focus on the combination of the improved selectivity features shown in compound 43 with the improved inhibitory activities and LipE values of the pyrazole-based sulfonamides displayed by compounds 64–67.
Figure 7.

Comparison between ligands 1 and 43.
Experimental Section
Chemistry General Methods
With the exception of those described in the following sections, all chemicals were purchased from Sigma-Aldrich Ltd. (Gillingham, U.K.), Apollo Scientific (Stockport, U.K.), Acros Organics (Geel, Belgium), Alfa Aesar (Heysham, U.K.), or TCI-UK (Oxford, U.K.) and were used without further purification.
Microwave-assisted experiments were undertaken on a Biotage Initiator using Biotage microwave vials (2–5 mL), which were sealed and heated to the targeted temperature using variable power to maintain the set temperature.
Flash column chromatography of compounds was undertaken either using an Isolera One (Biotage) or manually on silica gel (silicagel 60 [40–63 μm]) using glass columns and an appropriate solvent mixture.
High-performance liquid chromatography (HPLC) of compounds was undertaken using a Gilson semipreparative reverse-phase HPLC system equipped with a HICHROM C18 Column (250 mm × 21.2 mm), #306 pumps, and a Gilson UV/vis detector, detecting at 220 nm. For the latter, the mobile phase consisted of water + 0.1% formic acid (solvent A) and methanol + 0.1% formic acid (solvent B), with an elution method of 0–2 min 5% B, 2–30 min 5–98% B, 30–32 min 98% B, and 32–32.5 min 2% B at a flow rate of 12 mL/min.
For liquid chromatography-mass spectrometry (LC-MS), the compounds were purified and analyzed on an LC-MS system equipped with both an XBridge prep C18 5 μm, 19 mm × 100 mm OBD column, and an XBridge C18 5 μm, 4.6 mm × 100 mm column. Unless specified otherwise, all compounds were analyzed over a gradient of methanol in water (5–98% over 12 min and then 98% methanol for 3 min at a flow rate of 1.2 mL/min), both containing 0.1% formic acid. Alternative gradient elutions started from 20 or 50% methanol for the same time periods. Retention times (Rt) and mass peaks (MH+) were recorded, and purity was determined as >95% by integration of the diode array trace, unless otherwise stated.
1H and 13C NMR spectra were recorded at 400 and 101 MHz, respectively, on Bruker AV instruments at room temperature and referenced to residual solvent signals. Data are presented as follows: chemical shift in ppm, multiplicity (br = broad, app = apparent, s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet), coupling constants in Hz, and integration.
High-resolution mass spectra were obtained from the Mass Spectrometry Service of the Department of Chemistry, Imperial College London.
General Synthetic Methods
General Method for C4 Displacements (Method A)
To a solution of the appropriate 2,4-dichloro-fused pyrimidine intermediate (1.0 equiv) in EtOAc (10 mL) was added DIPEA (1.2 equiv) followed by the appropriate amine (1.0 equiv), and the reaction was stirred at room temperature overnight. The reaction mixture was washed with sodium hydroxide solution (1 M, 10 mL), and the aqueous layer was re-extracted with EtOAc (10 mL). The combined organic layers were dried (Na2SO4) and then concentrated under reduced pressure. Where necessary, the crude product was purified by flash column chromatography.
General Method for Displacement at C2 (Method B)
The appropriate 4-amino-2-chloro-fused pyrimidine (1.0 equiv) was suspended in EtOAc (1 mL) in a 2 mL Biotage microwave vial. An excess of the appropriate amine (4–10 equiv) was added, and the vial was sealed. The closed vial was heated in a Biotage Initiator microwave oven for 30–90 min to a fixed target temperature of between 120 and 180 °C depending on the molecule. The reaction mixture was partitioned between EtOAc (20 mL) and sodium hydroxide solution (1 M, 10 mL), and then the aqueous layer was re-extracted with EtOAc (10 mL). The combined organic layers were dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified by flash column chromatography or by reverse-phase HPLC. Fractions containing the desired product were combined and evaporated under reduced pressure or partially evaporated and lyophilized after HPLC purification.
General Method for Debenzylation (Method C)
A solution of the appropriate N-benzyl derivative in 1,2-dichloroethane (DCE, 5.0 mL) was cooled to 0 °C before the addition of 1-chloroethyl chloroformate (ACE-Cl, 1.0 equiv). The reaction mixture was heated under reflux for 1 h, cooled to room temperature, and then evaporated to dryness. The intermediate was redissolved in methanol (5.0 mL) and returned to reflux for a further 1 h. The reaction mixture was evaporated once again and partitioned between EtOAc (10 mL) and sodium hydroxide solution (1 M, 10 mL), and then the aqueous layer was re-extracted with EtOAc (10 mL). The combined organic layers were dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified by flash column chromatography or by reverse-phase HPLC. Fractions containing the desired product were combined and evaporated under reduced pressure or partially evaporated and lyophilized after HPLC purification.
General Method for Reductive Amination (Method D)
A solution of the appropriate amine in DCE was treated with a solution of the appropriate aldehyde (1–10 equiv) in DCE. The solution was stirred at room temperature for 15 min before being treated with solid sodium triacetoxyborohydride (4–10 mol equiv). The mixture was stirred for 3–18 h at room temperature until complete by thin-layer chromatography (TLC) and then partitioned between CH2Cl2 (20 mL) and saturated NaHCO3 solution (10 mL). The organic phase was dried over Na2SO4, concentrated under reduced pressure, and the crude product was purified by reverse-phase HPLC. Fractions containing the desired product were partially evaporated and lyophilized after HPLC purification.
Synthesis of Final Compounds
3-(Methyl(2-(methyl(1-methylpiperidin-4-yl)amino)thieno[3,2-d]pyrimidin-4-yl)amino)propanenitrile (1)
According to general method D for reductive amination, 3-(methyl(2-(methyl(piperidin-4-yl)amino)thieno[3,2-d]pyrimidin-4-yl)amino)propanenitrile (11, 0.033 g, 0.1 mmol) was dissolved in DCE (3 mL) and treated with paraformaldehyde (6.0 mg, 0.2 mmol). After stirring at room temperature for 10 min, solid sodium triacetoxyborohydride (85 mg, 0.4 mmol) was added, and the resulting suspension was stirred at room temperature overnight. After workup, the crude product was purified by reverse-phase HPLC (50–98% MeOH/water/0.1% formic acid), partially evaporated, and then lyophilized to give the title compound (1) as a formate salt (24.9 mg, 0.064 mmol, 64%). LC-MS Rt = 5.9 min; MH+ 345; 1H NMR (400 MHz, CD3OD) δ 8.38 (s, 2H), 7.94 (d, J = 5.5 Hz, 1H), 7.20 (d, J = 5.5 Hz, 1H), 4.99–4.95 (m, 1H), 4.08 (t, J = 6.6 Hz, 2H), 3.64–3.59 (m, 2H), 3.56 (s, 3H), 3.20 (dt, J = 12.8, 3.0 Hz, 2H), 3.07 (s, 3H), 2.93 (t, J = 6.6 Hz, 2H), 2.88 (s, 3H), 2.19 (dq, J = 12.9, 3.6 Hz, 2H), 2.01–1.95 (m, 2H). ESI HRMS, found 345.1875 (C17H25N6S, [M + H]+, requires 345.1861).
3-((2-((3-(Dimethylamino)propyl)(methyl)amino)thieno[3,2-d]pyrimidin-4-yl)(methyl)amino)propanenitrile (2)
According to general method B for C2 displacements, intermediate IM1 (50.0 mg, 0.20 mmol) was suspended in EtOAc (1 mL) in a microwave vial and treated with excess N,N,N′-trimethyl-1,3-propane-diamine (0.5 mL, 2 mmol). After workup, the crude product was purified by reverse-phase HPLC (20–98% MeOH/water/0.1% formic acid) to give the title compound (2) as the formate salt (12 mg, 0.032 mmol, 16%). LC-MS Rt = 3.8 min; MH+ 333; 1H NMR (400 MHz, CDCl3) δ 8.55 (s, 1H), 7.61 (d, J = 5.5 Hz, 1H), 7.14 (d, J = 5.5 Hz, 1H), 4.00 (t, J = 7.0 Hz, 2H), 3.70 (t, J = 7.0 Hz, 2H), 3.55 (s, 3H), 3.15 (s, 3H), 2.81 (t, J = 7.0 Hz, 2H), 2.65 (t, J = 7.0 Hz, 2H), 2.48 (s, 6H), 2.03–1.88 (m, 2H); 13C NMR (101 MHz, CDCl3), δ 186.3, 167.8, 163.9, 160.3, 157.9, 132.1, 124.2, 118.6, 56.6, 47.7, 47.3, 44.4, 38.5, 35.6, 24.8, 16.3; ESI HRMS, found 333.1861 (C16H25N6S [M + H]+, requires 333.1869).
3-(Methyl(2-(methyl(1-methylpiperidin-4-yl)amino)quinazolin-4-yl)amino)propanenitrile (3)
3-(Methyl(2-(methyl(piperidin-4-yl)amino)quinazolin-4-yl)amino)propanenitrile (23, 0.022 g, 0.068 mmol) was dissolved in DCE (1 mL) and treated with paraformaldehyde (15.0 mg, 0.5 mmol). After stirring at room temperature for 10 min, solid sodium triacetoxyborohydride (44 mg, 0.21 mmol) was added, and the resulting suspension was stirred at room temperature overnight. After workup, the crude product was purified by reverse-phase HPLC (50–98% MeOH/water/0.1% formic acid), partially evaporated, and then lyophilized to give the title compound (3) as a di-formate salt (2.9 mg, 0.007 mmol, 10%). LC-MS Rt = 5.98 min; MH+ 339; 1H NMR (400 MHz, CD3OD) δ 8.39 (s, 3H), 8.20 (dd, J = 8.4, 1.0 Hz, 1H), 7.78–7.74 (m, 1H), 7.69 (dd, J = 8.4, 1.1 Hz, 1H), 7.39 (dt, J = 8.3, 1.2 Hz, 1H), 5.06–4.98 (m, 1H), 4.19 (t, J = 6.8 Hz, 2H), 3.65 (s, 3H), 3.65–3.58 (m, 2H), 3.24–3.18 (m, 4H), 3.18 (s, 3H), 3.03 (t, J = 6.8 Hz, 2H), 2.85 (s, 3H), 2.31–2.19 (m, 2H), 2.05–2.00 (m, 2H). ESI HRMS, found 339.2299 (C19H27N6 [M + H]+ requires 339.2297).
3-((2-((3-(Dimethylamino)propyl)(methyl)amino)quinazolin-4-yl)(methyl)amino)propanenitrile (4)
According to general method B for C2 displacements, intermediate IM2 (0.04 g, 0.16 mmol) was suspended in EtOAc (1 mL) in a microwave vial and treated with excess N,N,N′-trimethyl-1,3-propane-diamine (200 μL, 1.6 mmol). The vial was sealed and irradiated for 30 min at 150 °C. After workup, the crude product was purified by reverse-phase HPLC (30–98% MeOH/water/0.1% formic acid), partially evaporated, and then lyophilized to give the title compound, 4, as a colorless di-formate salt (9.2 mg, 0.022 mmol, 14%). LC-MS Rt = 6.8 min; MH+ 327; 1H NMR (400 MHz, CD3OD) δ 8.52 (s, 2H), 8.14 (dd, J = 8.5, 1.4 Hz, 1H), 7.72 (ddd, J = 8.5, 7.0, 1.4 Hz, 1H), 7.60 (d, J = 8.5 Hz, 1H), 7.31 (ddd, J = 8.5, 7.0, 1.4 Hz, 1H), 4.15 (t, J = 6.7 Hz, 2H), 3.84 (t, J = 7.0 Hz, 2H), 3.61 (s, 3H), 3.30 (s, 3H), 3.16 (t, J = 7.0 Hz, 2H), 3.05 (t, J = 6.7 Hz, 2H), 2.88 (s, 6H), 2.18 (p, J = 7.0 Hz, 2H); ESI HRMS, found 327.2302 (C18H27N6 [M + H]+, requires 327.2297).
3-((2-((3-(Dimethylamino)propyl)(methyl)amino)pyrido[2,3-d]pyrimidin-4-yl)(methyl)amino)propanenitrile (5)
According to general method B for C2 displacements, intermediate IM3 (0.022 g, 0.09 mmol) was suspended in EtOAc (1 mL) in a 2 mL microwave vial and treated with excess N,N,N′-trimethyl-1,3-propane-diamine (50 μL, 0.40 mmol). The vial was sealed and irradiated for 30 min at 150 °C. After workup, the crude product was purified by reverse-phase HPLC (20–98% MeOH/water/0.1% formic acid), partially evaporated and then lyophilized to give the title compound, 5, as a pale yellow formate salt (21.1 mg, 0.056 mmol, 64%). LC-MS Rt = 1.7 min; MH+ 328; 1H NMR (400 MHz, CD3OD) δ 8.67 (dd, J = 4.5, 1.7 Hz, 1H), 8.53 (dd, J = 8.4, 1.7 Hz, 1H), 8.45 (s, 1H), 7.18 (dd, J = 8.4, 4.5 Hz, 1H), 4.11 (t, J = 6.7 Hz, 2H), 3.79 (m, 2H), 3.58 (s, 3H), 3.26 (s, 3H), 3.13–3.07 (m, 2H), 3.01 (t, J = 6.7 Hz, 2H), 2.88 (s, 6H), 2.19–2.11 (m, 2H); ESI HRMS, found 328.2263 (C17H26N7 [M + H]+ requires 328.225).
3-((2-((3-(Dimethylamino)propyl)(methyl)amino)-7-methyl-7H-purin-6-yl)(methyl)amino)propanenitrile (6)
According to general method B for C2 displacements, intermediate IM4 (0.021g, 0.08 mmol) was suspended in EtOAc (1 mL) in a 2 mL microwave vial and treated with excess N,N,N′-trimethyl-1,3-propane-diamine (200 μL, 0.8 mmol). The vial was sealed and irradiated for 30 min at 150 °C. After workup, the crude residue was purified by reverse-phase HPLC (50–98% MeOH/water/0.1% formic acid) to give the title compound as a colorless formate salt (13 mg, 0.035 mmol, 44%). LC-MS Rt = 1.6 min; MH+ 331; 1H NMR (400 MHz, CD3OD) δ 8.55 (s, 1H), 8.04 (s, 1H), 4.00 (s, 3H), 3.84 (t, J = 6.6 Hz, 2H), 3.69 (t, J = 6.6 Hz, 2H), 3.25 (s, 3H), 3.17 (s, 3H), 2.92 (t, J = 6.6 Hz, 2H), 2.84 (t, J = 6.6 Hz, 2H), 2.67 (s, 6H), 2.02 (p, J = 6.6 Hz, 2H). ESI HRMS, found 331.2365 (C16H27N8 [M + H]+, requires 331.2359).
3-((2-((3-(Dimethylamino)propyl)(methyl)amino)pyrimidin-4-yl)(methyl)amino)propanenitrile (7)
3-((2-Chloropyrimidin-4-yl)(methyl)amino)propanenitrile (IM22, 50 mg, 0.25 mmol) and N,N,N′-trimethyl-1,3-propane-diamine (149 μL, 1.02 mmol) were dissolved in n-BuOH (1.0 mL) and heated at 120 °C for 4 h. After cooling, the solution was diluted with EtOAc, washed with H2O and then brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by reverse-phase HPLC (50–98% MeOH/water/0.1% formic acid) to give the title compound 7 (67 mg, 0.20 mmol, 82%). LC-MS Rt = 1.04 min; MH+ 277.4; 1H NMR (400 MHz, CDCl3) δ 8.47 (s, 1H), 7.95 (d, J = 5.9 Hz, 1H), 5.81 (d, J = 6.0 Hz, 1H), 3.86 (t, J = 6.6 Hz, 2H), 3.66 (t, J = 6.9 Hz, 2H), 3.09 (s, 6H), 2.93–2.85 (m, 2H), 2.71 (t, J = 6.5 Hz, 2H), 2.65 (s, 6H), 2.07–1.99 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 167.8, 161.7, 161.1, 156.4, 92.4, 55.4, 46.5, 45.8, 42.8, 36.6, 35.2, 23.2, 16.2; ESI HRMS, found 277.2142 (C14H25N6 [M + H]+, requires 277.2141).
3-(Methyl(2-(methyl(tetrahydro-2H-pyran-4-yl)amino)thieno[3,2-d]pyrimidin-4-yl)amino)propanenitrile (8)
According to general method B for C2 displacements, intermediate IM1 (0.025 g, 0.1 mmol) was suspended in EtOAc (1 mL) in a 2 mL Biotage microwave vial and treated with excess N-methyltetrahydro-2H-pyran-4-amine (92 mg, 0.8 mmol). After workup, the crude product was purified by reverse-phase HPLC (50–98% MeOH/water/0.1% formic acid) to give the title compound (8), as a formate salt (5.0 mg, 0.013 mmol, 13%). 1H NMR (400 MHz, CD3OD) δ 8.31 (s, 2H), 7.98 (d, J = 5.6 Hz, 1H), 7.22 (d, J = 5.6 Hz, 1H), 4.13–4.04 (m, 6H), 3.59 (s, 3H), 3.07 (s, 3H), 2.94–2.91 (m, 1H), 2.92 (t, J = 6.7 Hz, 2H), 1.98–1.60 (m, 4H), ESI HRMS, found 332.1549 (C16H22N5OS [M + H]+, requires 332.1545).
3-((2-((3-Hydroxypropyl)(methyl)amino)thieno[3,2-d]pyrimidin-4-yl)(methyl)amino)propanenitrile (9)
According to general method B for C2 displacements, intermediate IM1 (0.025 g, 0.1 mmol) was suspended in EtOAc (1 mL) in a microwave vial and treated with excess 3-(methylamino)propan-1-ol (80 mg, 0.8 mmol). The vial was sealed and irradiated for 30 min at 120 °C. After workup, the crude residue was purified by reverse-phase HPLC (50–98% MeOH/water/0.1% formic acid) to give the title compound (9) as a formate salt (5.0 mg, 0.014 mmol, 14%). LC-MS Rt = 9.3 min; MH+ 306; 1H NMR (400 MHz, CD3OD) δ 8.58 (s, 1H), 7.83 (d, J = 5.5 Hz, 1H), 7.12 (d, J = 5.5 Hz, 1H), 4.05 (t, J = 6.5 Hz, 2H), 3.74 (t, J = 6.5 Hz, 2H), 3.61 (t, J = 6.5 Hz, 2H), 3.54 (s, 3H), 3.17 (s, 3H), 3.15–3.04 (m, 1H), 2.90 (t, J = 6.5 Hz, 2H), 1.94–1.78 (m, 2H); 13C NMR (101 MHz, CD3OD) δ 162.7, 161.9, 160.1, 157.9, 132.5, 122.5, 118.4, 59.1, 58.7, 46.0, 37.1, 34.6, 30.2 15.1; ESI HRMS, found 306.1402 (C14H20N5OS [M + H]+, requires 306.1389).
3-(Methyl(2-(methyl(piperidin-4-yl)amino)thieno[3,2-d]pyrimidin-4-yl)amino)propanenitrile (10)
According to general method C for debenzylation, a solution of 3-((2-((1-benzylpiperidin-4-yl)(methyl)amino)thieno[3,2-d]pyrimidin-4-yl)(methyl)amino)propanenitrile (14, 380 mg, 0.9 mmol) in DCE (7 mL) was reacted with 1-chloroethyl chloroformate (107 μL, 0.9 mmol). After workup, the crude product was purified by flash column chromatography (CH2Cl2/MeOH/aq. NH3; 95:5:0.5–90:10:1–80:20:1) to give the title compound (10) as an orange foam (150 mg, 0.45 mmol, 50%). LC-MS Rt = 6.0 min; MH+ 331; 1H NMR (400 MHz, CDCl3) δ 7.56 (d, J = 5.4 Hz, 1H), 7.11 (d, J = 5.4 Hz, 1H), 4.70 (m, 1H), 3.94 (t, J = 6.8 Hz, 2H), 3.49 (s, 3H), 3.20 (m, 2H), 3.05 (br, 1H), 3.01 (s, 3H), 2.80–2.73 (m, 4H), 1.80–1.68 (m, 4H); ESI HRMS, found 331.1718 (C16H23N6S [M + H]+, requires 331.1705).
3-(Methyl-(2-(methyl-(3-(methylamino)propyl)amino)thieno[3,2-d]pyrimidin-4-yl)amino)propanenitrile (11)
According to general method B for C2 displacements, intermediate IM1 (0.025 g, 0.1 mmol) was suspended in EtOAc (1 mL) in a microwave vial and treated with excess N,N′-dimethylpropane-1,3-diamine (80 mg, 0.8 mmol). The vial was sealed and irradiated for 30 min at 120 °C. After workup, the crude residue was purified by reverse-phase HPLC (30–98% MeOH/water/0.1% formic acid) to give the title compound (11) as a formate salt (1.6 mg, 0.044 mmol, 4%). LC-MS Rt = 6.1 min; MH+ 319; 1H NMR (400 MHz, CD3OD) δ 8.44 (s, 2H), 7.91 (d, J = 5.5 Hz, 1H), 7.20 (d, J = 5.5 Hz, 1H), 4.09 (t, J = 6.5 Hz, 2H), 3.74 (t, J = 6.5 Hz, 2H), 3.57 (s, 3H), 3.18 (s, 3H), 2.97 (t, J = 6.5 Hz, 2H), 2.91 (t, J = 6.5 Hz, 2H), 2.64 (s, 3H), 2.10–1.98 (m, 2H); ESI HRMS, found 319.1714 (C15H23N6S [M + H]+, requires 319.1705).
3-(Methyl(2-(methyl(1-ethylpiperidin-4-yl)amino)thieno[3,2-d]pyrimidin-4-yl)amino)propanenitrile (12)
According to general method D for reductive amination, 3-(methyl(2-(methyl(piperidin-4-yl)amino)thieno[3,2-d]pyrimidin-4-yl)amino)propanenitrile (10, 0.030 g, 0.1 mmol) was dissolved in DCE (3 mL), treated with acetaldehyde (10 mL, 0.2 mmol) and solid sodium triacetoxyborohydride (85 mg, 0.4 mmol), and then stirred at room temperature overnight. After workup, the crude residue was purified by reverse-phase HPLC (50–98% MeOH/water/0.1% formic acid), partially evaporated, and then lyophilized. The product was impure and so was repurified by reverse-phase HPLC (20–98% MeOH/water/0.1% formic acid) to give the title compound (12) as a di-formate salt (13.8 mg, 0.031 mmol, 31%). LC-MS Rt = 6.6 min; MH+ 359; 1H NMR (400 MHz, CD3OD) δ 8.43 (s, 2H), 7.89 (d, J = 5.5 Hz, 1H), 7.17 (d, J = 5.5 Hz, 1H), 5.01–4.93 (m, 1H), 4.08 (t, J = 7.0 Hz, 2H), 3.71–3.65 (m, 2H), 3.55 (s, 3H), 3.20 (q, J = 7.0 Hz, 2H), 3.14 (dt, J = 13.1, 2.6 Hz, 2H), 3.07 (s, 3H), 2.90 (t, J = 7.0 Hz, 2H), 2.21–2.10 (m, 2H), 2.04–1.97 (m, 2H), 1.37 (t, J = 7.0 Hz, 3H); ESI HRMS, found 359.2001 (C18H27N6S [M + H]+, requires 359.2018).
3-((2-((1-Isobutylpiperidin-4-yl)(methyl)amino)thieno[3,2-d]pyrimidin-4-yl)(methyl)amino)propanenitrile (13)
According to general method D for reductive amination, 3-(methyl(2-(methyl(piperidin-4-yl)amino)thieno[3,2-d]pyrimidin-4-yl)amino)propanenitrile (10, 0.030 g, 0.1 mmol) was dissolved in DCE (3 mL) and treated with isobutyraldehyde (7.2 mg, 0.1 mmol) and then sodium triacetoxyborohydride (85 mg, 0.4 mmol) before stirring at room temperature overnight. After workup, the crude product was purified by reverse-phase HPLC (50–98% MeOH/water/0.1% formic acid), partially evaporated, and then lyophilized to give the title compound (13) as a formate salt (22.0 mg, 0.051 mmol, 51%). LC-MS Rt = 7.8 min; MH+ 387; 1H NMR (CD3OD, δ, ppm) 8.41 (s, 2H), 7.91 (d, J = 5.5 Hz, 1H), 7.18 (d, J = 5.5 Hz, 1H), 4.96 (m, 1H), 4.08 (t, J = 7.0 Hz, 2H), 3.72–3.66 (m, 2H), 3.56 (s, 3H), 3.15 (dt, J = 13.0, 2.9 Hz, 2H), 3.07 (s, 3H), 2.98 (d, J = 7.0 Hz, 2H), 2.90 (t, J = 7.0 Hz, 2H), 2.29–2.13 (m, 3H), 2.00–1.95 (m, 2H), 1.10 (d, J = 7.0 Hz, 6H); ESI HRMS, found 387.2323 (C20H31N6S, [M + H]+, requires 387.2331).
3-((2-((1-Benzylpiperidin-4-yl)(methyl)amino)thieno[3,2-d]pyrimidin-4-yl)(methyl)amino)propanenitrile (14)
According to general method B for C2 displacements, intermediate IM1 (0.25 g, 1.0 mmol) was suspended in EtOAc (3 mL) in a microwave vial and treated with excess 1-benzyl-N-methylpiperidin-4-amine (830 mg, 4 mmol). The vial was sealed and irradiated for 90 min at 180 °C. After workup, the crude product was purified by column chromatography on silica by elution with CH2Cl2 and then CH2Cl2/MeOH/aq. NH3 solution (97:3:0.5). A second column (EtOAc, 100%) gave the title compound (14) as a colorless oil (380 mg, 0.090 mmol, 90%). LC-MS Rt = 8.8 min; MH+ 421; 1H NMR (400 MHz, CDCl3) δ 7.59 (d, J = 5.5 Hz, 1H), 7.37–7.27 (m, 5H), 7.14 (d, J = 5.5 Hz, 1H), 4.62 (t, J = 11.5 Hz, 1H), 3.98 (t, J = 6.8 Hz, 2H), 3.55 (m, 5H), 3.03 (m, 5H), 2.79 (t, J = 6.8 Hz, 2H), 2.20–2.10 (m, 2H), 1.94–1.84 (m, 2H), 1.671 (d, J = 11.5 Hz, 2H); ESI HRMS, found 421.2187 (C23H29N6S [M + H]+, requires 421.2174).
3-((2-((3-(Isobutyl(methyl)amino)propyl)(methyl)-amino)thieno[3,2-d]pyrimidin-4-yl)(methyl)-amino)propanenitrile (15)
According to general method D, 3-(methyl(2-(methyl(3-(methylamino)propyl)amino)thieno[3,2-d]pyrimidin-4-yl)amino)propanenitrile (11, 0.032 g, 0.1 mmol) was dissolved in DCE (3 mL) and treated with isobutyraldehyde (7.2 mg, 0.1 mmol). After stirring at room temperature for 10 min, solid sodium triacetoxyborohydride (85 mg, 0.4 mmol) was added, and the resulting suspension was stirred at room temperature overnight. After workup, the crude product was purified by reverse-phase HPLC (50–98% MeOH/water/0.1% formic acid), partially evaporated, and then lyophilized to give the title compound 15 as a formate salt (5.0 mg; 0.012 mol, 12%). LC-MS Rt = 7.9 min; MH+ 375; 1H NMR (400 MHz, CDCl3) δ 8.51 (s, 1H), 7.61 (d, J = 5.4 Hz, 1H), 7.13 (d, J = 5.4 Hz, 1H), 3.99 (t, J = 6.6 Hz, 2H), 3.68 (t, J = 6.6 Hz, 2H), 3.54 (s, 3H), 3.14 (s, 3H), 2.82–2.75 (m, 4H), 2.52–2.47 (m, 5H), 2.00–1.99 (m, 3H), 0.97 (d, J = 6.6 Hz, 6H); ESI HRMS, found 375.2345 (C19H31N6S [M + H]+, requires 375.2331).
3-((2-((3-(Benzyl(methyl)amino)propyl)(methyl)amino)thieno-[3,2-d]pyrimidin-4-yl)(methyl)amino)propanenitrile (16)
According to general method D, 3-(methyl(2-(methyl(3-(methylamino)propyl)amino)thieno[3,2-d]pyrimidin-4-yl)amino)propanenitrile (11, 0.032 g, 0.1 mmol) was dissolved in DCE (3 mL) and treated with benzaldehyde (10 mg, 0.1 mmol) and then sodium triacetoxyborohydride (85 mg, 0.4 mmol). The resulting suspension was stirred at room temperature overnight, and after workup, the crude product was purified by reverse-phase HPLC (50–98% MeOH/water/0.1% formic acid), partially evaporated, and then lyophilized to give the title compound (16) as a formate salt (7.0 mg, 0.015 mmol, 15%). LC-MS Rt = 8.4 min; MH+ 409; 1H NMR (400 MHz, CDCl3) δ 8.53 (s, 1H), 8.46 (s, 1H), 7.62 (d, J = 5.5 Hz, 1H), 7.36–7.31 (m, 5H), 7.09 (d, J = 5.5 Hz, 1H), 4.00 (s, 2H), 3.96 (t, J = 6.6 Hz, 2H), 3.68 (t, J = 6.6 Hz, 2H), 3.55 (s, 3H), 3.11 (s, 3H), 2.90–2.85 (m, 2H), 2.76 (t, J = 6.6 Hz, 2H), 2.53 (s, 3H), 2.11–2.02 (m, 2H); ESI HRMS found 409.2165, (C22H29N6S [M + H]+, requires 409.2174).
3-(Methyl(2-(piperazin-1-yl)thieno[3,2-d]pyrimidin-4-yl)amino)propanenitrile (17)
According to general method B for C2 displacements, intermediate IM1 (0.025 g, 0.1 mmol) was suspended in EtOAc (1 mL) in a microwave vial and treated with excess piperazine (70 mg, 0.8 mmol). The vial was sealed and irradiated for 30 min at 120 °C. After workup, the crude product was purified by reverse-phase HPLC (50–98% MeOH/water/0.1% formic acid) to give the title compound (17) as a formate salt (1.5 mg, 0.004 mmol, 4%). LC-MS Rt = 8.1 min; MH+ 303; 1H NMR (400 MHz, CD3OD) δ 8.42 (s, 1H), 7.91 (d, J = 5.5 Hz, 1H), 7.15 (d, J = 5.5 Hz, 1H), 4.08 (t, J = 6.5 Hz, 2H), 4.06–4.02 (m, 4H), 3.79–3.70 (m, 2H), 3.57 (s, 3H), 3.29–3.25 (m, 4H), 2.88 (t, J = 6.5 Hz, 2H); ESI HRMS, found 303.1394 (C14H19N6S [M + H]+, requires 303.1392).
3-(Methyl(2-(4-methylpiperazin-1-yl)thieno[3,2-d]pyrimidin-4-yl)amino)propanenitrile (18)
According to general method B for C2 displacements, intermediate IM1 (0.05 g, 0.2 mmol) was suspended in ethyl acetate (1 mL) in a microwave vial and treated with excess N-methylpiperazine (0.3 mL, 2 mmol). The vial was sealed and irradiated for 30 min at 120 °C. After workup, the crude product was purified by flash column chromatography using CH2Cl2 and then CH2Cl2/MeOH/aq. NH3 solution (95:5:0.5) to give the title compound (18) as a colorless solid (45 mg, 0.14 mmol, 72%). LC-MS Rt = 5.2 min; MH+ 317; 1H NMR (400 MHz, CDCl3) δ 7.65 (d, J = 5.7 Hz, 1H), 7.18 (d, J = 5.7 Hz, 1H), 4.01 (t, J = 6.6 Hz, 2H), 3.84 (dd, J = 6.1, 4.1 Hz, 4H), 3.57 (s, 3H), 2.81 (t, J = 6.6 Hz, 2H), 2.60–2.47 (m, 4H), 2.37 (s, 3H); ESI HRMS, found 317.1542 (C15H21N6S [M + H]+, requires 317.1548).
3-((2-((1-Benzylpyrrolidin-3-yl)(methyl)amino)thieno[3,2-d]pyrimidin-4-yl)(methyl)amino)propanenitrile (19)
According to general method B for C2 displacements, intermediate IM1 (0.10 g, 0.4 mmol) was suspended in EtOAc (1.5 mL) in a microwave vial and treated with excess 1-benzyl-N-methylpyrrolidin-3-amine (385 μL, 2.0 mmol). The vial was sealed and irradiated for 90 min at 180 °C. After workup, the crude product was purified by flash column chromatography using EtOAc (100%) and then CH2Cl2/MeOH/aq. NH3 (97:3:0.5) to give the title compound (19) as a colorless oil (95 mg, 0.23 mmol, 58%). LC-MS Rt = 8.5 min; MH+ 407; 1H NMR (400 MHz, CDCl3) δ 7.61 (d, J = 5.4 Hz, 1H), 7.43–7.25 (m, 5H), 7.17 (d, J = 5.4 Hz, 1H), 5.57–5.50 (m, 1H), 3.99 (t, J = 6.6 Hz, 2H), 3.73 (d, J = 12.8 Hz, 1H), 3.61 (d, J = 12.8 Hz, 1H), 3.56 (s, 3H,), 3.15 (s, 3H), 2.93–2.87 (m,1H), 2.79 (t, J = 6.6 Hz, 2H), 2.74–2.65 (m, 2H), 2.54–2.46 (m, 1H), 2.28–2.19 (m, 1H), 1.95–1.86 (m, 1H); ESI HRMS, found 407.2010 (C22H27N6S [M + H]+, requires 407.2018).
3-((2-(4-(Ethylamino)piperidin-1-yl)thieno[3,2-d]pyrimidin-4-yl)(methyl)amino)propanenitrile (20)
According to general method B for C2 displacements, intermediate IM1 (0.025 g, 0.1 mmol) was suspended in EtOAc (1.5 mL) in a microwave vial and treated with excess N-ethyl-1-methylpiperidin-4-amine (0.4 mmol). The vial was sealed and irradiated for 90 min at 180 °C. After workup, the crude product was purified by reverse-phase HPLC (50–98% MeOH/water) to give the title compound (20) as a di-formate salt (3.5 mg, 0.008 mmol, 8%). 1H NMR (400 MHz, CDCl3) δ 8.38 (br s, 2H), 7.89 (d, J = 5.5 Hz, 1H), 7.14 (d, J = 5.5 Hz), 4.88–4.84 (m, 2H), 4.07 (t, J = 6.7 Hz, 2H), 3.56 (s, 3H), 3.43–3.35 (m, 1H), 3.42–3.35 (m, 1H), 3.15–3.10 (q, J = 7.3 Hz, 2H), 3.02–2.94 (m, 2H), 2.88 (t, J = 6.4 Hz), 2.18–2.10 (m, 2H), 1.62–1.53 (m, 2H), 1.32 (t, J = 7.3 Hz, 3H). ESI HRMS, found 345.1886 (C17H25N6S [M + H]+, requires 345.1861).
3-((2-((3S,4S)-3-Fluoro-4-(methylamino)piperidin-1-yl)thieno[3,2-d]pyrimidin-4-yl)(methyl)amino)propanenitrile (21)
According to general method B for C2 displacements, intermediate IM1 (0.025 g, 0.1 mmol) was suspended in EtOAc (1.5 mL) in a microwave vial and treated with excess (3S,4S)-3-fluoro-N,1-dimethylpiperidin-4-amine (0.088 g, 0.4 mmol). The vial was sealed and irradiated for 90 min at 180 °C. After workup, the crude product was purified by reverse-phase HPLC (20–98% MeOH/water) to give the title compound (21) as a formate salt (7.0 mg, 0.018 mmol, 18%). 1H NMR (400 MHz, MeOD) δ 8.55 (br s, 1H), 7.91 (d, J = 5.5 Hz, 1H), 7.18 (d, J = 5.6 Hz, 1H), 5.08 (dtd, J = 12.4, 5.1, 2.0 Hz, 1H), 4.79 (ddd, J = 13.7, 4.7, 2.4 Hz, 1H), 4.59 (dtd, J = 50.0, 9.8, 5.3 Hz, 1H), 4.09 (t, J = 6.6 Hz, 2H), 3.58 (s, 3H), 3.11–2.95 (m, 2H), 2.91 (t, J = 6.6 Hz, 3H), 2.70 (s, 3H), 2.23 (ddt, J = 12.9, 5.0, 2.5 Hz, 1H), 1.58 (qd, J = 12.4, 4.5 Hz, 1H); ESI HRMS, found 349.1623 (C16H22N6SF [M + H]+, requires 349.1611).
3-((2-((3R,4S)-3-Fluoro-4-(methylamino)piperidin-1-yl)thieno[3,2-d]pyrimidin-4-yl)(methyl)amino)propanenitrile (22)
According to general method B for C2 displacements, intermediate IM1 (0.025 g, 0.1 mmol) was suspended in EtOAc (1.5 mL) in a microwave vial and treated with excess (3R,4S)-3-fluoro-N,1-dimethylpiperidin-4-amine (0.088 g, 0.4 mmol). The vial was sealed and irradiated for 90 min at 180 °C. After workup, the crude product was purified by reverse-phase HPLC (50–98% MeOH/water) to give the title compound (22) as a formate salt (6.4 mg, 0.016 mmol, 16%). 1H NMR (400 MHz, MeOD) δ 8.40 (s, 1H), 7.88 (d, J = 5.5 Hz, 1H), 7.14 (d, J = 5.6 Hz, 1H), 5.26 (ddt, J = 14.9, 12.0, 2.9 Hz, 1H), 5.14 (d, J = 49.7 Hz, 1H), 4.99–4.92* (m, 1H), 4.14–4.00 (m, 2H), 3.59–3.46 (m, 4H), 3.28–3.12 (m, 1H), 3.04–2.96 (m, 1H), 2.89 (td, J = 6.7, 1.8 Hz, 2H), 2.11–2.05 (m, 1H), 1.98–1.87 (m, 1H); ESI HRMS, found 349.1594 (C16H22N6SF [M + H]+, requires 349.1611). *Partially obscured by the H2O signal.
3-(Methyl(2-(methyl(piperidin-4-yl)amino)quinazolin-4-yl)amino)propanenitrile (23)
3-((2-Chloroquinazolin-4-yl)(methyl)amino)propanenitrile (IM2) (50 mg, 0.20 mmol, 1.0 equiv) was dissolved in n-BuOH (1.0 mL), and 1-benzyl-N-methylpiperidin-4-amine (163 mg, 0.80 mmol, 4.0 equiv) was added. The reaction was heated at 200 °C for 2 h. After cooling to room temperature, the reaction mixture was partitioned between EtOAc (20 mL) and sodium hydroxide solution (1 M, 10 mL), and then the aqueous layer was re-extracted with EtOAc (10 mL). The combined organic layers were dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified by flash column chromatography with CH2Cl2/MeOH (10:1) to give 3-((2-((1-benzylpyrrolidin-3-yl)(methyl)amino)quinazolin-4-yl)(methyl)amino)propanenitrile (88 mg). This intermediate was then debenzylated using 10% Pd/C (22 mg) and ammonium formate (126 mg) in EtOH (2.0 mL). After filtration through celite and concentration in vacuo, the residue was purified by reverse-phase HPLC (20–98% MeOH/water/0.1% formic acid), partially evaporated, and then lyophilized to give the title compound (23) as a formate salt (33 mg, 0.089 mmol, 44% over two steps). LC-MS Rt = 8.8 min. 1H NMR (400 MHz, CDCl3) δ 8.48 (s, 2H), 8.17 (dd, J = 8.4, 1.2 Hz, 1H), 7.78–7.61 (m, 2H), 7.35 (ddd, J = 8.4, 6.8, 1.5 Hz), 5.02*, 4.17 (t, J = 7.1 Hz, 2H), 3.57 (m, 1H), 3.55 (m, 1H), 3.26–3.18 (m, 2H), 3.18 (s, 3H), 2.16 (m, 2H), 2.04 (m, 1H), 2.01 (m, 1H). ESI HRMS, found 325.2166 (C18H25N6 [M + H]+, requires 325.2141). *Partially obscured by the H2O signal and identified by analogy with compound 3.
3-((2-((1-Ethylpiperidin-4-yl)(methyl)amino)quinazolin-4-yl)(methyl)amino)propanenitrile (24)
In a variation on general method D for reductive amination, 3-(methyl(2-(methyl(piperidin-4-yl)amino)quinazolin-4-yl)amino)propanenitrile (23, 0.009 g, 0.028 mmol) was dissolved in DCM (1 mL) and treated with isobutyraldehyde (5.0 μL, 0.14 mmol). After stirring at room temperature for 10 min, solid sodium triacetoxyborohydride (30 mg, 0.14 mmol) was added, and the resulting suspension was stirred at room temperature overnight. After workup, the crude product was purified by reverse-phase HPLC (5–98% MeOH/water/0.1% formic acid), partially evaporated, and then lyophilized to give the title compound (24) as a 2.5 × formate salt (2.0 mg, 0.004 mmol, 15%). 1H NMR (400 MHz, CDCl3) δ 8.35 (br s, 2H), 8.14 (d, J = 8.4 Hz, 1H), 7.71 (ddd, J = 8.3, 7.0, 1.0 Hz, 1H), 7.63 (dd, J = 8.4, 0.9 Hz, 1H), 7.31 (ddd, J = 8.3, 7.1, 1.3 Hz, 1H), 5.50 (tt, J = 12.1, 4.0 Hz, 1H), 4.14 (t, J = 6.9 Hz, 2H), 3.67 (m, 2H), 3.60 (s, 3H), 3.21–3.10 (m, 7H), 3.02 (t, J = 6.9 Hz, 2H), 2.19 (m, 2H), 2.04 (m, 2H), 1.37 (t, J = 7.2 Hz, 3H); ESI HRMS, found 353.2449 (C20H29N6 [M + H]+, requires 353.2454).
3-(Methyl(2-(methyl(pyrrolidin-3-yl)amino)quinazolin-4-yl)amino)propanenitrile (25)
3-((2-Chloroquinazolin-4-yl)(methyl)amino)propanenitrile (IM2) (50 mg, 0.20 mmol, 1.0 equiv) was dissolved in n-BuOH (1.0 mL), and 1-benzyl-N-methylpyrrolidin-3-amine (154 μL, 0.80 mmol, 4.0 equiv) was added. The reaction was heated at 120 °C for 2 h. After cooling to room temperature, the reaction mixture was partitioned between EtOAc (20 mL) and sodium hydroxide solution (1 M, 10 mL), and then the aqueous layer was re-extracted with EtOAc (10 mL). The combined organic layers were dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified by flash column chromatography with CH2Cl2/MeOH (10:1) to give 3-((2-((1-benzylpyrrolidin-3-yl)(methyl)amino)quinazolin-4-yl)(methyl)amino)propanenitrile (41 mg, 0.10 mmol). This intermediate (24 mg, 0.06 mmol) was then debenzylated using 10% Pd/C (7 mg) and ammonium formate (38 mg, 0.6 mmol) in EtOH (2.0 mL). After filtration through celite and concentration in vacuo, the residue was purified by reverse-phase HPLC (5–98% MeOH/water/0.1% formic acid), partially evaporated, and then lyophilized to give the title compound (25) as a formate salt (1.5 mg, 0.004 mmol, 67%). LC-MS Rt = 9.6 min. 1H NMR (400 MHz, CDCl3) δ 8.48 (s br, 1H), 8.04 (dd, J = 8.4, 0.9 Hz, 1H), 7.61 (m, 1H), 7.53 (dd, J = 8.4, 1.0 Hz, 1H), 7.19 (ddd, J = 8.3, 6.8, 1.4 Hz, 1H), 5.22 (m, 1H), 4.03 (t, J = 6.7 Hz, 2H), 3.71 (ddd, J = 11.8, 8.6, 3.3 Hz, 1H), 3.52–3.42 (m, 5H), 3.31–3.26 (m, 1H), 3.25 (s, 3H), 2.99 (t, J = 6.6 Hz, 2H), 2.45–2.34 (m, 1H), 2.31–2.20 (m, 1H); ESI HRMS, found 311.1985 (C17H23N6 [M + H]+, requires 311.1984).
3-(Methyl(2-(methyl(1-methylpyrrolidin-3-yl)amino)quinazolin-4-yl)amino)propanenitrile (26)
In a variation on general method D for reductive amination, 3-(methyl(2-(methyl(pyrrolidin-3-yl)amino)quinazolin-4-yl)amino)propanenitrile (25, 3.1 mg, 0.01 mmol, 1.0 equiv) was dissolved in DCM (1 mL) and treated with formaldehyde (10 equiv). After stirring at room temperature for 10 min, solid sodium triacetoxyborohydride (11 mg, 0.05 mmol, 5.0 equiv) was added, and the resulting suspension was stirred at room temperature overnight. After workup, the crude product was purified by reverse-phase HPLC (5–98% MeOH/water/0.1% formic acid), partially evaporated, and then lyophilized to give the title compound (26) as a yellow oil (2.4 mg, 0.007 mmol, 74%). LC-MS Rt = 2.7 min; MH+ 325; 1H NMR (400 MHz, CDCl3) δ 7.96 (dd, J = 8.5, 1.1 Hz, 1H), 7.53 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.46 (dd, J = 8.5, 1.1 Hz, 1H), 7.10 (ddd, J = 8.3, 6.6, 1.2 Hz, 1H), 5.68–5.60 (m, 1H), 3.46 (s, 3H), 3.14 (s, 3H), 2.97 (t, J = 6.8 Hz, 2H), 2.85–2.77 (m, 2H), 2.70–2.61 (m, 2H), 2.42 (s, 3H), 2.28–2.18 (m, 1H), 2.00–1.91 (m, 1H); ESI HRMS, found 325.2132 (C18H25N6 [M + H]+, requires 325.2141).
3-(Methyl(2-(methyl((1R,3R,5S)-8-methyl-8-azabicyclo[3.2.1]octan-3-yl)amino)quinazolin-4-yl)amino)propanenitrile (27)
According to general method B for C2 displacements, intermediate IM2 (0.03 g, 0.12 mmol) was suspended in EtOAc (1 mL) in a microwave vial and treated with excess (1R,3R,5S)-N,8-dimethyl-8-azabicyclo[3.2.1]octan-3-amine (0.115 g, 0.75 mmol). The vial was sealed and irradiated for 90 min at 150 °C. After workup, the crude residue was purified by reverse-phase HPLC (30–98% MeOH/water/0.1% formic acid), partially evaporated, and then lyophilized. The product was repurified using gradient elution (10–98% MeOH/water/0.1% formic acid), partially evaporated, and then lyophilized to give the title compound (27) as a colorless di-formate salt (2.9 mg, 0.006 mmol, 5%). LC-MS Rt = 7.1 min; MH+ 365; 1H NMR (400 MHz, CD3OD) δ 8.47 (s, 2H), 8.08 (dd, J = 8.5, 1.4 Hz, 1H), 7.65 (ddd, J = 8.5, 6.8, 1.4 Hz, 1H), 7.57 (dd, J = 8.5, 1.4 Hz, 1H), 7.24 (ddd, J = 8.5, 6.8, 1.4 Hz, 1H), 5.44 (m, 1H), 4.12 (t, J = 7.0 Hz, 2H), 4.07–4.00 (m, 2H), 3.56 (s, 3H), 3.14 (s, 3H), 3.04 (t, J = 7.0 Hz, 2H), 2.87 (s, 3H), 2.48 (m, 2H), 2.39–2.22 (m, 4H), 1.98 (m, 2H). ESI HRMS, found 365.246 (C21H29N6 [M + H]+, requires 365.2454).
3-((2-((2-Hydroxyethyl)(piperidin-4-yl)amino)quinazolin-4-yl)(methyl)amino)propanenitrile (28)
According to general method B for C2 displacements, intermediate IM2 (50 mg, 0.20 mmol) was suspended in n-BuOH (1 mL) in a microwave vial and treated with excess 2-((1-benzylpiperidin-4-yl)amino)ethanol (188 mg, 0.80 mmol). The vial was sealed and irradiated at 200 °C for 2 h. After workup, the crude residue was purified by flash column chromatography (CH2Cl2/MeOH; 95:5) to give the intermediate 3-((2-((1-benzylpiperidin-4-yl)(2-hydroxyethyl)amino)quinazolin-4-yl)(methyl)amino)propanenitrile (32 mg, 0.072 mmol, 36%). The intermediate was debenzylated with 10% Pd/C (10 mg) and ammonium formate (50 mg, 0.79 mmol) in EtOH (2.0 mL). After filtration through celite, the filtrate was concentrated in vacuo and then purified by reverse-phase HPLC (5–98% MeOH/water/0.1% formic acid) to give the title compound, 28, as a di-formate salt (2.0 mg, 0.004 mmol, 5%). LC-MS Rt = 6.2 min; MH+ 355; 1H NMR (400 MHz, CD3OD) δ 8.39 (s, 2H), 8.08 (dd, J = 8.3, 1.0 Hz, 1H), 7.64 (ddd, J = 8.5, 7.2, 1.3 Hz, 1H), 7.48 (dd, J = 8.3, 0.8 Hz, 1H), 7.24 (ddd, J = 8.3, 6.9, 1.3 Hz, 1H), 5.02–4.96* (m, 1H), 4.10 (t, J = 7.0 Hz, 2H), 3.82 (d, J = 5.6 Hz, 2H), 3.71 (d, J = 5.6 Hz, 2H), 3.59–3.52 (m, 5H), 3.25–3.18 (m, 2H), 3.00 (t, J = 7.0 Hz, 2H), 2.10–2.04 (m, 4H); ESI HRMS, found 355.2242 (C19H27N6O [M + H]+, requires 355.2246). *Partially obscured by the H2O signal.
3-((2-((3-Hydroxypropyl)(piperidin-4-yl)amino)quinazolin-4-yl)(methyl)amino)propanenitrile (29)
In a variation on general method B for C2 displacements, intermediate IM2 (50 mg, 0.20 mmol) was suspended in n-BuOH (1 mL) in a microwave vial and treated with excess 3-((1-benzylpiperidin-4-yl)amino)propan-1-ol (198 mg, 0.80 mmol). The vial was sealed and irradiated at 200 °C for 2 h. After workup, the crude residue was purified by flash column chromatography (CH2Cl2/MeOH; 95/5) to give the intermediate 3-((2-((1-benzylpiperidin-4-yl)(3-hydroxypropyl)amino)quinazolin-4-yl)(methyl)amino)propanenitrile (45 mg, 0.098 mmol, 49%). The intermediate was debenzylated with 10% Pd/C (11 mg) and ammonium formate (63 mg, 1.0 mmol) in EtOH (2.0 mL). After filtration through celite, the filtrate was concentrated in vacuo and then purified by reverse-phase HPLC (5–98% MeOH/water/0.1% formic acid) to give the title compound, 29, as a di-formate salt (12.0 mg, 0.026 mmol, 13%). LC-MS Rt = 7.0 min; MH+ 369; 1H NMR (400 MHz, CD3OD) δ 8.41 (s, 2H), 8.14 (dd, J = 8.4, 1.0 Hz, 1H), 7.69 (ddd, J = 8.3, 7.0, 1.2 Hz, 1H), 7.51 (d, J = 8.2 Hz, 1H), 7.30 (ddd, J = 8.2, 7.2, 1.1 Hz, 1H), 4.99–4.92* (m, 1H), 4.14 (t, J = 7.0 Hz, 2H), 3.73–3.69 (m, 4H), 3.60 (s, 3H), 3.60–3.53 (m, 2H), 3.25–3.17 (m, 2H), 3.00 (t, J = 6.8 Hz, 2H), 2.19–2.07 (m, 4H), 1.92 (qt, J = 6.2 Hz, 2H); ESI HRMS, found 369.2399 (C20H29N6O [M + H]+, requires 369.2403). *Partially obscured by the H2O signal.
3-((2-((3-Hydroxypropyl)(1-methylpiperidin-4-yl)amino)quinazolin-4-yl)(methyl)amino)propanenitrile (30)
In a variation of general method D for reductive amination, 3-((2-((3-hydroxypropyl)(piperidin-4-yl)amino)quinazolin-4-yl)(methyl)amino)propanenitrile (29, 9.0 mg, 0.024 mmol, 1.0 equiv) was dissolved in DCM (2.0 mL) and treated with formaldehyde (10 equiv). After stirring at room temperature for 10 min, solid sodium triacetoxyborohydride (16 mg, 0.072 mmol, 3.0 equiv) was added, and the resulting suspension was stirred at room temperature overnight. After workup, the crude product was purified by reverse-phase HPLC (5–98% MeOH/water/0.1% formic acid), partially evaporated, and then lyophilized to give the title compound, 30, as a formate salt (2.0 mg, 0.005 mmol, 19%). LC-MS Rt = 6.9 min; MH+ 383.5; 1H NMR (400 MHz, CD3OD) δ 8.49 (s, 1H), 8.05 (dd, J = 8.4, 1.0 Hz, 1H), 7.61 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.44 (dd, J = 8.4, 1.0 Hz, 1H), 7.20 (ddd, J = 8.4, 6.8, 1.3 Hz, 1H), 4.97–4.90* (m, 1H), 4.07 (t, J = 7.0 Hz, 2H), 3.68 (dt, J = 11.7, 6.3 Hz, 2H), 5.54–5.49 (m, 5H), 3.07 (dt, J = 12.8, 2.8 Hz, 2H), 2.99 (t, J = 7.0 Hz, 2H), 2.80 (s, 3H), 2.21–2.20 (m, 4H), 1.88 (qt, J = 6.4 Hz, 2H); ESI HRMS, found 383.2578 (C21H31N6O [M + H]+, requires 383.2559). *Partially obscured by the H2O signal.
N1,N1,N3-Trimethyl-N3-(thieno[3,2-d]pyrimidin-2-yl)propane-1,3-diamine (31)
2-Chlorothieno[3,2-d]pyrimidine (10 mg, 0.059 mmol) was dissolved in N,N-dimethylacetamide (DMA, 1.0 mL), and N,N,N′-trimethyl-1,3-propane-diamine (52 μL) was added and then sealed and heated at 120 °C for 4 h. The reaction mixture was partitioned between EtOAc (10 mL) and 1 M NaOH solution (5 mL), and then the aqueous layer was re-extracted with EtOAc (10 mL). The combined organic layers were dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified by flash column chromatography to give the product (31) as a colorless oil (9.0 mg, 0.036 mmol, 61%) with a purity of 90%. LC-MS Rt = 8.3 min; MH+ 251.3 1H NMR (400 MHz, CDCl3) δ 8.81 (s, 1H), 7.76 (d, J = 5.4 Hz, 1H), 7.20 (d, J = 5.4 Hz, 1H), 3.75 (t, J = 7.2 Hz, 2H), 3.23 (s, 3H), 2.35 (t, J = 7.4 Hz, 2H), 2.26 (s, 6H), 1.84 (m, 2H). ESI HRMS, found 251.1331 (C12H19N4S [M + H]+, requires 251.1330).
N2-(3-(Dimethylamino)propyl)-N4-(2-methoxyethyl)-N2,N4-dimethylthieno[3,2-d]pyrimidine-2,4-diamine (32)
According to general method B for C2 displacements, intermediate IM5 (50 mg, 0.19 mmol) was suspended in EtOAc (1 mL) in a microwave vial and treated with excess N,N,N′-trimethyl-1,3-propane-diamine (0.5 mL, 2.0 mmol) and then sealed and irradiated for 30 min at 120 °C. After workup, the crude product was purified by reverse-phase HPLC (20–98% MeOH/water/0.1% formic acid) to give the title compound (33) (5.3 mg, 0.016 mmol, 8%). LC-MS Rt = 7.6 min; MH+ 338; 1H NMR (400 MHz, CDCl3) δ 7.54 (d, J = 5.5 Hz, 1H), 7.14 (d, J = 5.5 Hz, 1H), 3.90 (t, J = 6.0 Hz, 2H), 3.75–3.58 (m, 4H), 3.44 (s, 3H), 3.36 (s, 3H), 3.15 (s, 3H), 2.56 (t, J = 6.0 Hz, 2H), 2.41 (s, 6H), 1.93 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 163.2, 160.3, 158.3, 131.3, 124.0, 104.8, 71.3, 59.2, 57.0, 50.8, 47.4, 44.8, 38.7, 35.6, 25.1; ESI HRMS, found 338.2014 (C16H28N5OS [M + H]+ requires 338.2015).
N-(2-Methoxyethyl)-N-methyl-2-(4-methylpiperazin-1-yl)thieno[3,2-d]pyrimidin-4-amine (33)
According to general method B for C2 displacements, intermediate IM5 (50 mg, 0.19 mmol) was suspended in EtOAc (1 mL) in a microwave vial and treated with excess N-methylpiperazine (0.3 mL, 2.0 mmol). The vial was sealed and irradiated for 30 min at 120 °C. After workup, the crude product was purified by reverse-phase HPLC (20–98% MeOH/water/0.1% formic acid) to give the title compound (33) as a formate salt (21 mg, 0.057 mmol, 30%). LC-MS Rt = 7.0 min; MH+ 322; 1H NMR (400 MHz, CDCl3) δ 8.49 (s, 1H), 7.57 (d, J = 5.5 Hz, 1H), 7.13 (d, J = 5.5 Hz, 1H), 3.90 (t, J = 5.8 Hz, 2H), 3.87–3.83 (m, 4H), 3.66 (t, J = 5.8 Hz, 2H), 3.45 (s, 3H), 3.36 (s, 3H), 2.57 (m, 4H), 2.39 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 167.9, 163.2, 160.2, 158.3, 131.4, 124.0, 71.1, 59.1, 54.8, 50.8, 45.8, 43.9, 38.7; ESI HRMS, found 322.1696 (C15H24N5OS [M + H]+ requires 322.1688).
2-((2-((3-(Dimethylamino)propyl)(methyl)amino)thieno[3,2-d]pyrimidin-4-yl)(methyl)amino)ethanol (34)
According to general method B for C2 displacements, intermediate IM6 (50 mg, 0.21 mmol) was suspended in EtOAc (1 mL) in a microwave vial and treated with excess N,N,N′-trimethyl-1,3-propane-diamine (0.5 mL, 2 mmol). The vial was sealed and irradiated for 30 min at 120 °C. After workup, the crude product was purified by reverse-phase HPLC (20–98% MeOH/water/0.1% formic acid) to give the title compound (34) as a formate salt (22 mg, 0.06 mmol, 28%). LC-MS Rt = 6.1 min; MH+ 324; 1H NMR (400 MHz, CDCl3) δ 8.54 (s, 1H), 7.64 (d, J = 5.5 Hz, 1H), 7.20 (d, J = 5.5 Hz, 1H), 4.47 (s, 1H), 3.98–3.84 (m, 4H), 3.78 (t, J = 6.9 Hz, 2H), 3.52 (s, 3H), 3.18 (s, 3H), 3.06–2.95 (m, 2H), 2.70 (s, 6H), 2.17–1.99 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 168.6, 162.5, 159.8, 159.0, 32.1, 123.7, 60.0, 55.4, 53.2, 47.0, 42.8, 38.1, 35.4, 23.3; ESI HRMS, found 324.1863 (C15H26N5OS [M + H]+ requires 324.1858).
3-((2-((3-(Dimethylamino)propyl)(methyl)amino)thieno[3,2-d]pyrimidin-4-yl)(methyl)amino)propan-1-ol (35)
2-((2-Chlorothieno[3,2-d]pyrimidin-4-yl)(methyl)amino)ethanol IM7 (23 mg, 0.088 mmol, 1.0 equiv) was dissolved in n-BuOH, and N,N,N′-trimethyl-1,3-propane-diamine (78 μL, 0.53 mmol, 6.0 equiv) was added. The reagents were heated at 150 °C under MW radiation for 1 h and then concentrated in vacuo. The residue was purified by reverse-phase HPLC (5–98% MeOH/water/0.1% formic acid) to give the title compound (35) as a formate salt (5.7 mg, 0.015 mmol, 17%). LC-MS Rt = 7.16 min; MH+ 338; 1H NMR (400 MHz, CD3OD) δ 8.57 (s, 1H), 7.89 (d, J = 5.5 Hz, 1H), 7.19 (d, J = 5.5 Hz, 1H), 3.90 (t, J = 7.3 Hz), 3.72 (t, J = 6.3 Hz, 2H), 3.67 (t, J = 6.1 Hz, 2H), 3.45 (s, 3H), 3.18 (s, 3H), 3.08 (t, J = 6.9 Hz, 2H), 2.85 (s, 6H), 2.09 (q, J = 6.6 Hz, 2H), 1.99–1.89 (m, 2H); ESI HRMS, found 338.2013 (C16H36N5OS, [M + H]+, requires 338.2015).
3-((2-((3-(Dimethylamino)propyl)(methyl)amino)thieno[3,2-d]pyrimidin-4-yl)(methyl)amino)propenamide (36)
3-((2-Chlorothieno[3,2-d]pyrimidin-4-yl)(methyl)amino)propenamide (IM8) (24 mg, 0.088 mmol, 1.0 equiv), was dissolved in EtOAc (1.0 mL) and N,N-dimethylformamide (DMF, 1.0 mL), and then N,N,N′-trimethyl-1,3-propane-diamine (78 μL, 0.53 mmol, 6.0 equiv) was added. The reagents were heated at 150 °C under MW radiation for 1 hr and then concentrated in vacuo. The residue was purified by reverse-phase HPLC (5–98% MeOH/water/0.1% formic acid) to give the title compound (36) as a formate salt (8.8 mg, 0.022 mmol, 25%). Purity 85%; LC-MS Rt = 5.4 min; MH+ 351; 1H NMR (CD3OD, δ, ppm) δ 8.07 (d, J = 5.5 Hz, 1H), 7.30 (d, J = 5.5 Hz, 1H), 4.16–4.09 (m, 2H), 3.77 (t, J = 6.8 Hz, 2H), 3.57 (s, 3H), 3.24 (s, 3H), 3.22–3.16 (m, 2H), 2.91 (s, 6H), 2.68 (s, 6H), 2.72–2.62 (m, 2H); ESI HRMS, found 351.1957 (C16H27N6OS, [M + H]+, requires 351.1967).
N-(2-((2-((3-(Dimethylamino)propyl)(methyl)amino)thieno[3,2-d]pyrimidin-4-yl)(methyl)amino)ethyl)-N-methylacetamide (37)
According to general method B for C2 displacements, intermediate IM9 (20 mg, 0.07 mmol) was suspended in EtOAc in a microwave vial and treated with excess N,N,N′-trimethyl-1,3-propane-diamine (100 μL, 0.8 mmol). The vial was sealed and irradiated for 30 min at 150 °C. After workup, the crude product was purified by reverse-phase HPLC (20–98% MeOH/water/0.1% formic acid) to give the title compound (37) as a formate salt as a mixture of rotamers (7.1 mg, 0.017 mmol, 23%). LC-MS Rt = 6.9 min; MH+ 379; 1H NMR (400 MHz, CD3OD) δ 8.58 (s, 1H), 7.87 (d, J = 5.4 Hz, 0.3H), 7.85 (d, J = 5.4 Hz, 0.7H), 7.22–7.13 (m, 1H), 4.04 (t, J = 6.0 Hz, 0.6H), 3.99 (t, J = 6.0 Hz, 1.4H), 3.78–3.65 (m, 4H), 3.50 (d, J = 2.0 Hz, 3H), 3.19 (s, 3H), 3.10 (s, 2H), 3.01 (m, 1H), 2.63 (m, 2H), 2.49 (s, 3H), 2.46 (s, 2H), 2.04 (s, 2H), 2.02 (s, 1H), 1.95 (m, 2H); ESI HRMS, found 379.2287 (C18H31N6OS [M + H]+ requires 379.228).
3-((2-((3-(Dimethylamino)propyl)(methyl)amino)thieno[3,2-d]pyrimidin-4-yl)(methyl)amino)-N,N-dimethylpropanamide (38)
3-((6-(tert-Butyl)-2-chlorothieno[3,2-d]pyrimidin-4-yl)(methyl)amino)propanenitrile IM10 (42 mg, 0.14 mmol) was suspended in DMA (1 mL) in a microwave vial, treated with excess N,N,N′-trimethyl-1,3-propane-diamine (80 μL, 0.55 mmol), and then heated for 24 h at 100 °C. After workup, the crude product was purified by reverse-phase HPLC (20–98% MeOH/water/0.1% formic acid) to give the title compound (38) as a formate salt (11.6 mg, 0.027 mmol, 19%). LC-MS Rt = 7.1 min; MH+ 379; 1H NMR (400 MHz, CD3OD) δ 8.58 (s, 1H), 7.85 (d, J = 5.5 Hz, 1H), 7.17 (d, J = 5.5 Hz, 1H), 4.13–4.01 (m, 2H), 3.70 (t, J = 7.0 Hz, 2H), 3.48 (s, 3H), 3.18 (s, 3H), 3.06 (s, 3H), 2.97 (s, 3H), 2.89–2.79 (m, 2H), 2.70 (t, J = 7.0 Hz, 2H), 2.53 (s, 6H), 1.97 (p, 2H); ESI HRMS, found 379.2282 (C18H31N6OS [M + H]+ requires 379.228).
(S)-(1-(2-((3-(Dimethylamino)propyl)(methyl)amino)thieno[3,2-d]pyrimidin-4-yl)pyrrolidin-2-yl)methanol (39)
According to general method B for C2 displacements, intermediate IM11 (27 mg, 0.1 mmol) was suspended in EtOAc (1 mL) in a microwave vial and treated with excess N,N,N′-trimethyl-1,3-propane-diamine (127 μL, 1.0 mmol). The vial was sealed and irradiated for 30 min at 120 °C. After workup, the crude product was purified by reverse-phase HPLC (30–98% MeOH/water/0.1% formic acid) to give the title compound (39) as a formate salt (12.1 mg, 0.031 mmol, 31%). LC-MS Rt = 7.0 min; MH+ 350; 1H NMR (CD3OD, δ, ppm) 8.45 (s, 2H), 7.93 (d, J = 5.4 Hz, 1H), 7.20 (d, J = 5.4 Hz, 1H), 4.63–4.56 (m, 1H), 4.09–4.01 (m, 1H), 3.97–3.89 (m, 1H), 3.89–3.84 (m, 1H), 3.74–3.64 (m, 2H), 3.69–3.84 (m, 1H), 3.19 (s, 3H), 3.10 (t, J = 7.1 Hz, 2H), 2.86 (s, 6H), 2.78–2.68 (m, 1H), 2.32–2.01 (m, 6H); ESI HRMS, found 350. 2017 (C17H28N5OS [M + H]+ requires 350.2015).
(S)-4-(2-(Methoxymethyl)pyrrolidin-1-yl)-N-methyl-N-(1-methylpiperidin-4-yl)thieno[3,2-d]pyrimidin-2-amine (40)
According to general method D for reductive amination, (S)-4-(2-(methoxymethyl)pyrrolidin-1-yl)-N-methyl-N-(piperidin-4-yl)thieno[3,2-d]pyrimidin-2-amine (41, 15 mg, 0.04 mmol) was dissolved in DCE (5 mL) and treated with paraformaldehyde (6.0 mg, 0.2 mmol). After stirring at room temperature for 10 min, solid sodium triacetoxyborohydride (85 mg, 0.4 mmol) was added, and the resulting suspension was stirred at room temperature overnight. After workup, the crude product was purified by reverse-phase HPLC (30–98% MeOH/water/0.1% formic acid), partially evaporated, and then lyophilized to give the title compound (40) as a colorless formate salt (6.8 mg, 0.016 mmol, 40%). LC-MS Rt = 8.3 min; MH+ 376; 1H NMR (400 MHz, CD3OD) δ 8.49 (s, 2H), 7.87 (d, J = 5.4 Hz, 1H), 7.15 (d, J = 5.4 Hz, 1H), 4.87–4.84* (m, 1H), 4.68–4.61 (m, 1H), 4.05–3.99 (m, 1H), 3.91–3.84 (m, 1H), 3.73 (dd, J = 9.0, 3.1 Hz, 1H), 3.59–3.51 (m, 2H), 3.43 (t, J = 8.4 Hz, 1H), 3.38 (s, 3H), 3.12–2.99 (m, 5H), 2.84 (s, 3H), 2.26–2.01 (m, 6H), 2.00–1.91 (m, 2H); ESI HRMS, found 376.2185 (C19H30N5OS [M + H]+ requires 376.2171). *Partially obscured by the H2O signal.
(S)-4-(2-(Methoxymethyl)pyrrolidin-1-yl)-N-methyl-N-(piperidin-4-yl)thieno[3,2-d]pyrimidin-2-amine (41)
According to general method B for C2 displacements, intermediate IM12 (0.10 g, 0.35 mmol) was suspended in EtOAc (1.5 mL) in a microwave vial and treated with excess 1-benzyl-N-methylpiperidin-4-amine (430 μL, 2.1 mmol). The vial was sealed and irradiated for 90 min at 180 °C. After workup, the crude product was purified by flash column chromatography (EtOAc, 100%) to give the intermediate, N-(1-benzylpiperidin-4-yl)-4-(2-(methoxymethyl)pyrrolidin-1-yl)-N-methylthieno[3,2-d]pyrimidin-2-amine, a colorless oil (140 mg, 0.31 mmol, 89%). 1H NMR (400 MHz, CDCl3) δ 7.56 (d, J = 5.4 Hz, 1H), 7.39–7.24 (m, 5H), 7.15 (d, J = 5.4 Hz, 1H), 4.71 (m, 1H), 4.67–4.62 (m, 1H), 4.00 (m, 1H), 3.84 (m, 1H), 3.77 (m, 1H), 3.56 (s, 2H), 3.38 (s, 3H), 3.35 (m, 1H), 3.08 (s, 3H), 3.02 (m, 2H), 2.25–2.08 (m, 5H), 1.89 (m, 3H), 1.71 (m, 2H).
The intermediate (140 mg, 0.31 mmol) was then debenzylated according to general method C. After workup, the crude product was purified by reverse-phase HPLC (50–98% MeOH/water/0.1% formic acid), partially evaporated and then lyophilized to give the title compound (41) as a di-formate salt (18.9 mg, 0.042 mmol, 13%). LC-MS Rt = 8.3 min; MH+ 362; 1H NMR (400 MHz, CD3OD) δ 8.42 (s, 1H), 7.96 (d, J = 5.5 Hz, 1H), 7.22 (d, J = 5.5 Hz, 1H), 4.91–4.87* (m, 1H), 4.69–4.64 (m, 1H), 4.05 (td, J = 9.1, 2.4 Hz, 1H), 3.90 (td, J = 9.4, 7.0 Hz, 1H), 3.73 (dd, J = 9.0, 3.1 Hz, 1H), 3.61–3.52 (m, 2H), 3.43 (dd, J = 9.0, 7.9 Hz, 1H), 3.38 (s, 3H), 3.22–3.11 (m, 2H), 3.09 (s, 3H), 2.29–2.13 (m, 1H), 2.16–1.95 (m, 7H) *Partially obscured by the H2O signal.
(S)-N1-(4-(2-(Methoxymethyl)pyrrolidin-1-yl)thieno[3,2-d]pyrimidin-2-yl)-N1,N3,N3-trimethylpropane-1,3-diamine (42)
According to general method B for C2 displacements, intermediate IM12 (15 mg, 0.053 mmol) was suspended in EtOAc (1 mL) in a microwave vial and treated with excess N,N,N′-trimethyl-1,3-propane-diamine (100 μL, 0.78 mmol). The vial was sealed and irradiated for 30 min at 150 °C. After workup, the crude product was purified by reverse-phase HPLC (30–98% MeOH/water/0.1% formic acid) to give the title compound (42) as a formate salt (3.3 mg, 0.008 mmol, 15%). LC-MS Rt = 8.4 min; MH+ 364; 1H NMR (400 MHz, CD3OD) δ 8.55 (s, 1H), 7.83 (d, J = 5.5 Hz, 1H), 7.14 (d, J = 5.5 Hz, 1H), 4.68–4.61 (m, 1H), 4.04–3.94 (m, 1H), 3.91–3.84 (m, 1H), 3.73 (dd, J = 9.2, 3.1 Hz, 1H), 3.71–3.63 (m, 2H), 3.48 (dd, J = 9.0, 7.8 Hz, 1H), 3.37 (s, 3H), 3.16 (s, 3H), 2.72 (t, J = 7.3 Hz, 2H), 2.55 (s, 6H), 2.23–2.01 (m, 4H), 2.01–1.91 (m, 2H); ESI HRMS, found 364.2182 (C18H30N5OS [M + H]+ requires 364.2171).
(S)-2-(1-(2-(Methyl(1-methylpiperidin-4-yl)amino)thieno[3,2-d]pyrimidin-4-yl)pyrrolidin-2-yl)acetonitrile (43)
According to general method D for reductive amination, (S)-2-(1-(2-(methyl(piperidin-4-yl)amino)thieno[3,2-d]pyrimidin-4-yl)pyrrolidin-2-yl)acetonitrile (44, 15 mg, 0.042 mmol) was reacted with paraformaldehyde (6.0 mg, 0.2 mmol) and sodium triacetoxyborohydride (85 mg, 0.4 mmol). After workup, the crude product was purified by reverse-phase HPLC (30–98% MeOH/water/0.1% formic acid), partially evaporated, and then lyophilized to give the title compound (43) as a formate salt (9.6 mg, 0.023 mmol, 55%). LC-MS Rt = 7.5 min; MH+ 371; 1H NMR (400 MHz, CD3OD) δ 8.42 (s, 2H), 7.90 (d, J = 5.5 Hz, 1H), 7.17 (d, J = 5.5 Hz, 1H), 5.03–4.93 (m, 1H), 4.78–4.72 (m, 1H), 4.15–4.09 (m, 1H), 4.00–3.93 (m, 1H), 3.65–3.57 (m, 2H), 3.25–3.15 (m, 2H), 3.13–3.08 (m, 1H), 3.07 (s, 3H), 2.92–2.84 (m, 1H), 2.88 (s, 3H), 2.35–2.11 (m, 5H), 2.04–1.93 (m, 3H); ESI HRMS, found 371.2020 (C19H27N6S [M + H]+ requires 371.2018).
(S)-2-(1-(2-(Methyl(piperidin-4-yl)amino)thieno[3,2-d]pyrimidin-4-yl)pyrrolidin-2-yl)acetonitrile (44)
According to general method B for C2 displacements, intermediate IM13 (0.10 g, 0.35 mmol) was suspended in EtOAc (1.5 mL) in a microwave vial and treated with excess 1-benzyl-N-methylpiperidin-4-amine (430 μL, 2.1 mmol). The vial was sealed and irradiated for 90 min at 180 °C. After workup, the crude product was purified by flash column chromatography (EtOAc; 100%) to give (S)-2-(1-(2-((1-benzylpiperidin-4-yl)(methyl)amino)thieno[3,2-d]pyrimidin-4-yl)pyrrolidin-2-yl)acetonitrile, as a colorless oil (126 mg, 0.28 mmol, 80%). 1H NMR (400 MHz, CDCl3) δ 7.60 (d, J = 5.4 Hz, 1H), 7.42–7.23 (m, 5H), 7.16 (d, J = 5.4 Hz, 1H), 4.68 (m, 3H), 4.09 (m, 1H), 3.99–3.88 (m, 1H), 3.57 (s, 2H), 3.11 (m, 1H), 3.08 (s, 3H), 3.04 (m, 2H), 2.81–2.69 (m, 1H), 2.33–2.08 (m, 4H), 1.99–1.84 (m, 2H), 1.70 (m, 2H).
The intermediate (126 mg, 0.28 mmol, 0.28 mmol) was debenzylated using 1-chloroethyl chloroformate according to general method C. After workup, the crude product was purified by flash column chromatography (CH2Cl2/MeOH/aq. NH3; 97:3:0.5–95:5:0.5–90:10:1) to give the title compound (44) as a colorless oil (25 mg, 0.070 mmol, 25%). LC-MS Rt = 7.6 min; MH+ 357; 1H NMR (400 MHz, CDCl3) δ 7.58 (d, J = 5.4 Hz, 1H), 7.14 (d, J = 5.4 Hz, 1H), 4.78–4.69 (m, 1H), 4.69–4.62 (m, 1H), 4.10–4.04 (m, 1H), 3.96–3.86 (m, 1H), 3.24–3.16 (m, 2H), 3.13–3.06 (m, 1H), 3.04 (s, 3H), 2.84–2.76 (m, 2H), 2.76–2.69 (m, 1H), 2.28–2.07 (m, 2H), 2.23–2.06 (m, 3H), 1.80–1.69 (m, 4H); ESI HRMS, found 357.1861 (C18H25N6S [M + H]+ requires 357.1861).
(S)-2-(1-(2-((3-(Dimethylamino)propyl)(methyl)amino)thieno[3,2-d]pyrimidin-4-yl)pyrrolidin-2-yl)acetonitrile (45)
According to general method B for C2 displacements, intermediate IM13 (15 mg, 0.042 mmol) was suspended in EtOAc (1 mL) in a microwave vial and treated with excess N,N,N′-trimethyl-1,3-propane-diamine (100 μL, 0.78 mmol). The vial was sealed and irradiated for 30 min at 150 °C. After workup, the crude product was purified by reverse-phase HPLC (20–98% MeOH/water/0.1% formic acid) to give the title compound (45) as a formate salt (6.3 mg, 0.016 mmol, 37%). LC-MS Rt = 7.8 min; MH+ 359; 1H NMR (400 MHz, CD3OD) δ 8.38 (s, 1H), 8.00 (d, J = 5.5 Hz, 1H), 7.31 (d, J = 5.5 Hz, 1H), 4.79–4.73* (m, 2H), 4.16–4.10 (m, 1H), 4.05–3.97 (m, 1H), 3.80–3.67 (m, 2H), 3.21 (s, 3H), 3.16–3.11 (m, 2H), 3.08–3.05 (m, 1H), 2.88 (s, 6H), 2.40–2.23 (m, 2H), 2.22–2.05 (m, 4H). ESI HRMS, found 359.2021, (C18H27N6S [M + H]+ requires 359.2018). * Partially obscured by the H2O signal.
N4-Ethyl-N2,N4-dimethyl-N2-(1-methylpiperidin-4-yl)thieno[3,2-d]pyrimidine-2,4-diamine (46)
According to general method D for reductive amination, N4-ethyl-N2,N4-dimethyl-N2-(piperidin-4-yl)thieno[3,2-d]pyrimidine-2,4-diamine (47, 18 mg, 0.045 mmol) was dissolved in DCE (5 mL) and treated with paraformaldehyde (6.0 mg, 0.2 mmol) and sodium triacetoxyborohydride (85 mg, 0.4 mmol). After workup, the crude product was purified by reverse-phase HPLC (20–98% MeOH/water/0.1% formic acid), partially evaporated, and then lyophilized to give the title compound (46) as a di-formate salt (14.0 mg, 0.034 mmol, 58%). LC-MS Rt = 7.8 min; MH+ 320; 1H NMR (400 MHz, CD3OD) δ 8.50 (br s, 2H), 7.88 (d, J = 5.5 Hz, 1H), 7.17 (d, J = 5.5 Hz, 1H), 4.93–4.90* (m, 1H), 3.85 (q, J = 7.0 Hz, 2H), 3.60–3.53 (m, 2H), 3.40 (s, 3H), 3.16–3.08 (m, 2H), 3.07 (s, 3H), 2.85 (s, 3H), 2.17 (dq, J = 13.3, 3.9 Hz, 2H), 2.00–1.93 (m, 2H), 1.29 (t, J = 7.0 Hz, 3H); ESI HRMS, found 320.1910, (C16H26N5S [M + H]+ requires 320.1909). *Partially obscured by the H2O signal.
N4-Ethyl-N2,N4-dimethyl-N2-(piperidin-4-yl)thieno[3,2-d]pyrimidine-2,4-diamine (47)
A solution of N2-(1-benzylpiperidin-4-yl)-N4-ethyl-N2,N4-dimethylthieno[3,2-d]pyrimidine-2,4-diamine (49, 31 mg, 0.078 mmol) was debenzylated using 1-chloroethyl chloroformate (20 μL) according to general method C. After workup, the crude product was purified by reverse-phase HPLC (20–98% MeOH/water/0.1% formic acid) to give the title compound (47) as a di-formate salt (22 mg, 0.055 mmol, 71%). LC-MS Rt = 7.9 min; MH+ 331; 1H NMR (400 MHz, CD3OD) δ 8.26 (s, 1H), 8.12 (d, J = 5.5 Hz, 1H), 7.37 (d, J = 5.5 Hz, 1H), 4.95–4.90* (m, 1H), 3.93 (q, J = 7.0 Hz, 2H), 3.60–3.54 (m, 2H), 3.50 (s, 3H), 3.27–3.18 (m, 2H), 3.14 (s, 3H), 2.15 (dq, J = 13.1, 4.2 Hz, 2H), 2.10–2.00 (m, 2H), 1.35 (t, J = 7.0 Hz, 3H); ESI HRMS, found 306.1750 (C15H24N5S [M + H]+ requires 306.1752). *Partially obscured by the H2O signal.
N2-(3-(Dimethylamino)propyl)-N4-ethyl-N2,N4-dimethylthieno[3,2-d]pyrimidine-2,4-diamine (48)
According to general method B for C2 displacements, intermediate IM14 (21 mg, 0.083 mmol) was suspended in EtOAc (1 mL) in a microwave vial and treated with excess N,N,N′-trimethyl-1,3-propane-diamine (117 μL, 0.8 mmol). The vial was sealed and irradiated for 30 min at 120 °C. After workup, the crude product was purified by reverse-phase HPLC (33–98% MeOH/water/0.1% formic acid) to give the title compound (48) (13 mg, 0.042 mmol, 51%). LC-MS Rt = 7.7 min; MH+ 308; 1H NMR (400 MHz, CD3OD) δ 7.75 (d, J = 5.5 Hz, 1H), 7.12 (d, J = 5.5 Hz, 1H), 3.80 (q, J = 7.1 Hz, 2H), 3.66 (t, J = 7.1 Hz, 2H), 3.34 (s, 3H), 3.13 (s, 3H), 2.43–2.36 (m, 2H), 2.28 (s, 6H), 1.90–1.81 (m, 2H), 1.26 (t, J = 7.1 Hz, 3H); ESI HRMS, found 308.1911 (C15H26N5S [M + H]+ requires 308.1909).
N2-(1-Benzylpiperidin-4-yl)-N4-ethyl-N2,N4-dimethylthieno[3,2-d]pyrimidine-2,4-diamine (49)
According to general method B for C2 displacements, intermediate IM14 (140 mg, 0.61 mmol) was suspended in EtOAc (1.5 mL) in a microwave vial and treated with excess 1-benzyl-N-methylpiperidin-4-amine (500 mg, 2.4 mmol). The vial was sealed and irradiated for 90 min at 180 °C. After workup, the crude product was purified by flash column chromatography (EtOAc; 100%) to give the title compound (49) as a colorless oil (87 mg, 0.22 mmol, 36%). LC-MS Rt = 9.0 min; MH+ 396; 1H NMR (400 MHz, CDCl3) δ 7.51 (d, J = 5.7 Hz, 1H), 7.36–7.22 (m, 5H), 7.12 (d, J = 5.7 Hz, 1H), 4.72–4.63 (m, 1H), 3.74 (q, J = 7.0 Hz, 2H), 3.52 (s, 2H), 3.30 (s, 3H), 3.06 (s, 2H), 3.02–2.96 (m, 2H), 2.13 (dt, J = 11.8, 2.5 Hz, 2H), 1.86 (dq, J = 12.1, 3.8 Hz, 2H), 1.72–1.76 (m, 2H), 1.24 (t, J = 7.0 Hz, 3H). ESI HRMS, found 396.2224 (C22H30N5S [M + H]+ requires 396.2223).
3-((6-(tert-Butyl)-2-(methyl(1-methylpiperidin-4-yl)amino)thieno[3,2-d]pyrimidin-4-yl)(methyl)amino)propanenitrile (50)
Intermediate IM15 (0.064 g, 0.21 mmol) was suspended in n-BuOH (1. mL) in a microwave vial and treated with excess 1-benzyl-N-methylpiperidin-4-amine (169 mg, 0.84 mmol). The vial was sealed and irradiated for 2 h at 200 °C. After workup, the crude product was purified by flash column chromatography (MeOH/CH2Cl2, 1:9) to give 3-((2-((1-benzylpiperidin-4-yl)(methyl)amino)-6-(tert-butyl)thieno[3,2-d]pyrimidin-4-yl)(methyl)amino)propanenitrile as a yellow oil (58 mg, 0.12 mmol, 58%). This was debenzylated using 10% Pd/C (13 mg) and ammonium formate (77 mg) in EtOH (2.0 mL). The crude product (0.024 g, 0.06 mmol) was then methylated using formaldehyde (6 mg, 0.29 mmol), sodium triacetoxyborohydride (27 mg, 0.125 mmol), and acetic acid (7.1 μL, 0.125 mmol) according to general method D to give the product (9.0 mg, 0.020 mmol, 10%) after purification by reverse-phase HPLC (30–98% MeOH/water/0.1% formic acid). LC-MS Rt = 8.3 min; MH+ 401; 1H NMR (CDCl3) δ 8.45 (s, 1.5H), 6.99 (s, 1H), 5.00–4.92 (m, 1H)*, 4.08 (t, J = 6.6 Hz, 2H), 3.64–3.58 (m, 2H), 3.55 (s, 3H), 3.23–3.16 (m, 2H), 3.07 (s, 3H), 2.91 (t, J = 6.7 Hz, 2H), 2.89 (s, 3H), 2.22–2.12 (m, 2H), 2.01–1.97 (m, 2H), 1.46 (s, 9H);. ESI HRMS, found 401.2491 (C21H33N6S [M + H]+ requires 401.2487). *Partially obscured by the water signal.
3-((6-(tert-Butyl)-2-((3-(dimethylamino)propyl)(methyl)amino)thieno[3,2-d]pyrimidin-4-yl)(methyl)amino)propanenitrile (51)
Intermediate IM15 (42 mg, 0.14 mmol) was suspended in DMA (1.0 mL) in a microwave vial and treated with excess N,N,N′-trimethyl-1,3-propane-diamine (80 μL, 0.55 mmol). The vial was sealed and heated for 24 h at 120 °C. The reaction mixture was partitioned between EtOAc (20 mL) and 1 M NaOH solution (10 mL), and then the aqueous layer was re-extracted with EtOAc (10 mL). The combined organic layers were dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified by reverse-phase HPLC (30–98% MeOH/water/0.1% formic acid) to give the title compound (51) (4.0 mg, 0.01 mmol, 7%). LC-MS Rt = 10.7 min (30–98% MeCN/water/0.1% formic acid); 1H NMR (CDCl3) δ 6.99 (s, 1H), 4.08 (t, J = 6.6 Hz, 2H), 3.72 (t, J = 6.3 Hz, 2H), 3.55 (s, 3H), 3.18 (s, 3H), 3.11 (t, J = 7.1 Hz, 2H), 2.91 (t, J = 6.6 Hz, 2H), 2.88 (s, 6H), 2.13–2.07 (m, 2H), 1.45 (s, 9H); ESI HRMS, found 389.2501 (C20H33N6S [M + H]+, requires 389.2487).
3-((6-(tert-Butyl)-2-(methyl(1-methylpyrrolidin-3-yl)amino)thieno[3,2-d]pyrimidin-4-yl)(methyl)amino)propanenitrile (52)
According to general method D for reductive amination, ((6-(tert-butyl)-2-(methyl(pyrrolidin-3-yl)amino)thieno[3,2-d]pyrimidin-4-yl)(methyl)amino)propanenitrile (53, 8 mg, 0.02 mmol) was methylated using paraformaldehyde (∼2 mg, 4.0 equiv) and sodium triacetoxyborohydride (17 mg, 4.0 equiv) in DCE (5.0 mL). After workup, the crude product was purified by reverse-phase HPLC (20–98% MeOH/water/0.1% formic acid), partially evaporated, and then lyophilized to give the title compound as a colorless formate salt (3.1 mg, 0.007 mmol, 36%). LC-MS Rt = 9.4 min; MH+ 387; 1H NMR (400 MHz, CD3OD) δ 8.54 (s, 3H), 7.01 (s, 1H), 5.13–5.05 (m, 1H), 4.05 (t, J = 6.6 Hz, 2H), 3.66–3.58 (m, 1H), 3.53 (s, 3H), 3.45–3.39 (m, 1H), 3.30–3.16 (m, 1H), 3.18 (s, 3H), 3.11–3.03 (m, 1H), 2.91–2.85 (m, 5H), 2.50–2.39 (m, 1H), 2.27–2.13 (m, 1H), 1.45 (s, 9H); ESI HRMS, found 387.2336 (C20H31N6S [M + H]+ requires 387.2331).
3-((6-(tert-Butyl)-2-(methyl(pyrrolidin-3-yl)amino)thieno[3,2-d]pyrimidin-4-yl)(methyl)amino)propanenitrile (53)
A solution of 3-((2-((1-benzylpyrrolidin-3-yl)(methyl)amino)-6-(tert-butyl)thieno[3,2-d]pyrimidin-4-yl)(methyl)amino)propanenitrile (54, 96 mg, 0.21 mmol) in EtOH (10 mL) was treated with an excess of ammonium formate (126 mg, 2 mmol) and a catalytic quantity of 10% Pd/C. The mixture was heated under reflux overnight, cooled to room temperature, filtered through celite, and evaporated under reduced pressure. The crude product was purified by reverse-phase HPLC (33–98% MeOH/water/0.1% formic acid), partially evaporated, and then lyophilized to give the title compound (53) as a colorless formate salt (9.7 mg, 0.023 mmol, 11%). LC-MS Rt = 9.6 min; MH+ 373; 1H NMR (400 MHz, CD3OD) δ 8.42 (s, 2H), 7.05 (s, 1H), 5.16–5.09 (m, 1H), 4.05 (t, J = 6.6 Hz, 2H), 3.72–3.76 (m, 1H), 3.53 (s, 3H), 3.49–3.38 (m, 2H), 3.30–3.23 (m, 1H), 3.18 (s, 3H), 2.88 (t, J = 6.6 Hz, 2H), 2.42–2.32 (m, 1H), 2.27–2.16 (m, 1H), 1.44 (s, 9H); ESI HRMS, found 373.2188 (C19H29N6S [M + H]+, requires 373.2174).
3-((2-((1-Benzylpyrrolidin-3-yl)(methyl)amino)-6-(tert-butyl)thieno[3,2-d]pyrimidin-4-yl)(methyl)amino)propanenitrile (54)
According to general method B for C2 displacements, intermediate IM15 (0.10 g, 0.32 mmol) was suspended in EtOAc (1.5 mL) in a microwave vial and treated with excess 1-benzyl-N-methylpyrrolidin-3-amine (250 μL, 1.3 mmol). The vial was sealed and irradiated for 90 min at 180 °C. After workup, the crude product was purified by flash column chromatography (EtOAc/hexane; 1:1 and then 1:0) to give the title compound (54) as a colorless oil (96 mg, 0.21 mmol, 65%). LC-MS Rt = 10.5 min; MH+ 463; 1H NMR (400 MHz, CDCl3) δ 7.38–7.22 (m, 5H), 6.94 (s, 1H), 5.55–5.46 (m, 1H), 3.95 (t, J = 6.6 Hz, 2H), 3.69 (d, J = 12.9 Hz, 1H), 3.57 (d, J = 12.9 Hz, 1H), 3.51 (s, 3H), 3.12 (s, 3H), 2.90–2.84 (m, 1H), 2.76 (t, J = 6.7 Hz, 2H), 2.70–2.56 (m, 2H), 2.48–2.36 (m, 1H), 2.26–2.16 (m, 1H), 1.98–1.82 (m, 1H), 1.42 (s, 9H); ESI HRMS found 463.2639 (C26H35N6S, [M + H]+, requires 463.2644).
3-((6-Bromo-2-(methyl(1-methylpiperidin-4-yl)amino)quinazolin-4-yl)(methyl)amino)propanenitrile (55)
3-((6-Bromo-2-chloroquinazolin-4-yl)(methyl)amino)propanenitrile IM16 (25.0 mg, 0.077 mmol) and N,1-dimethylpiperidin-4-amine (39 mg, 0.31 mmol) were dissolved in n-BuOH (2.0 mL) and heated at 150 °C for 3 h. After cooling, the solvent was concentrated in vacuo and the residue was purified by reverse-phase HPLC (50–98% MeOH/water/0.1% formic acid) to give the title compound 55 as a 50% formate salt (3.4 mg, 0.008 mmol, 10%). LC-MS Rt 6.73 min; 1H NMR (400 MHz, CDCl3) δ 8.50 (0.5H, s), 7.95 (1H, d, J = 2.2 Hz), 7.57 (1H, dd, J = 9.0, 2.2 Hz), 7.39 (1H, d, J = 9.0 Hz), 4.97 (1H, m), 3.94 (2H, t, J = 7.0 Hz), 3.49–3.44 (5H, m), 3.09 (3H, s), 2.86 (2H, t, J = 7.0 Hz), 2.85–2.78 (2H, m), 2.68 (s, 3H), 2.45–2.36 (m, 2H), 1.86–1.82 (2H, m); ESI HRMS, found 417.1402 (C19H26N679Br [M + H]+, requires 417.1402).
3-((6-Bromo-2-((3-(dimethylamino)propyl)(methyl)amino)quinazolin-4-yl)(methyl)amino)propanenitrile (56)
3-((6-Bromo-2-chloroquinazolin-4-yl)(methyl)amino)propanenitrile IM16 (50.0 mg, 0.15 mmol) and N1,N1,N3-trimethylpropane-1,3-diamine (90 μL, 0.61 mmol) were dissolved in EtOAc (1.5 mL) and heated at 180 °C in a microwave for 90 min. After cooling, the solution was washed with H2O, dried over MgSO4, and concentrated in vacuo to give a yellow oil, which was purified by reverse-phase HPLC (5–98% MeOH/water/0.1% formic acid) to give the title compound 56 as a formate salt (11 mg, 0.024 mmol, 16%). LC-MS Rt 8.29 min; 1H NMR (400 MHz, CDCl3) δ 8.53 (s, 1H), 7.93 (1H, d, J = 2.2 Hz), 7.55 (1H, dd, J = 9.0, 2.2 Hz), 7.35 (1H, d, J = 9.0 Hz), 3.93 (2H, t, J = 6.9 Hz), 3.73 (2H, t, J = 7.1 Hz), 3.44 (3H, s), 3.18 (3H, s), 2.87 (2H, t, J = 6.9 Hz), 2.74–2.70 (2H, m), 2.51 (6H, s), 2.02–1.95 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 168.6, 162.7, 158.2, 153.8, 135.7, 128.1, 127.8, 118.5, 112.4, 112.3, 56.1, 48.7, 47.0, 43.8, 41.8, 35.3, 24.3, 15.6; ESI HRMS, found 405.1391 (C18H26N679Br [M + H]+, requires 405.1402).
3-((6-Bromo-2-(methyl(1-methylpyrrolidin-3-yl)amino)quinazolin-4-yl)(methyl)amino)propanenitrile (57)
3-((6-Bromo-2-chloroquinazolin-4-yl)(methyl)amino)propanenitrile IM16 (25.0 mg, 0.077 mmol) and N1-dimethylpyrrolidin-3-amine (50 μL, 0.38 mmol) were dissolved in n-BuOH (2.0 mL) and heated at reflux for 3 h. After cooling, the solvent was concentrated in vacuo and the residue was dissolved in 5% MeOH and 95% H2O to give a suspension. After filtration, the residual solid was purified by reverse-phase HPLC (50–98% MeOH/water/0.1% formic acid) to give the title compound 57 as a di-formate salt (11 mg, 0.024 mmol, 32%). LC-MS Rt 6.71 min; 1H NMR (400 MHz, CDCl3) δ 8.41 (2H, s), 7.99 (1H, d, J = 2.2 Hz), 7.59 (1H, dd, J = 8.9, 2.2 Hz), 7.37 (1H, d, J = 9.0 Hz), 5.63 (1H, p, J = 8.6 Hz), 3.97 (2H, td, J = 6.6, 2.0 Hz), 3.61–3.52 (1H, m), 3.49 (3H, s), 3.47–3.29 (3H, m), 3.17 (3H, s), 2.87 (2H, t, J = 6.6 Hz), 2.84 (3H, s), 2.37–2.31 (2H, m); 13C NMR (101 MHz, CDCl3) δ 166.9, 162.4, 157.6, 153.0, 136.0, 128.0, 127.8, 118.5, 113.1, 112.7, 55.0, 54.7, 54.6, 48.8, 42.1, 41.1, 27.4, 15.7; ESI HRMS, found 403.1228 (C18H24N679Br [M + H]+, requires 403.1246).
3-((6-Methoxy-2-(methyl(1-methylpiperidin-4-yl)amino)quinazolin-4-yl)(methyl)amino)propanenitrile (58)
3-((2-Chloro-6-methoxyquinazolin-4-yl)(methyl)amino)propanenitrile IM17 (50.0 mg, 0.18 mmol) and N,1-dimethylpiperidin-4-amine (92 mg, 0.72 mmol) were dissolved in n-BuOH (2.0 mL) and heated at 120 °C for 3 h. After cooling, the reaction was concentrated in vacuo and the solid was dissolved in EtOAc and washed with H2O (5×). The solution was concentrated again, and the residue was purified by reverse-phase HPLC (50–98% MeOH/water/0.1% formic acid) to give the title compound 58 as a di-formate salt (2.0 mg, 0.004 mmol, 2%). LC-MS Rt 6.18 min; 1H NMR (400 MHz, MeOD) δ 8.36 (s, 2H), 7.57 (d, J = 9.1 Hz, 1H), 7.45 (d, J = 2.7 Hz, 1H), 7.39 (dd, J = 9.1, 2.7 Hz, 1H), 4.99–4.96 (1H, m)*, 4.09 (t, J = 6.8 Hz, 2H), 3.89 (s, 3H), 3.56 (s, 3H), 3.56–3.51 (2H, m), 3.13 (s, 3H), 3.13–3.03 (2H, m); 3.02 (t, J = 6.8 Hz, 3H), 2.82 (s, 3H), 2.19–2.09 (2H, m), 2.01–1.95 (2H, m); ESI HRMS, found 369.2393 (C20H29N6O [M + H]+, requires 369.2403). *Obscured by the H2O signal; the presence was confirmed by COSY coupling to multiplet at 2.19–2.09 Hz.
3-((2-((3-(Dimethylamino)propyl)(methyl)amino)-6-methoxyquinazolin-4-yl)(methyl)amino)propanenitrile (59)
3-((2-Chloro-6-methoxyquinazolin-4-yl)(methyl)amino)propanenitrile IM17 (50 mg, 0.18 mmol) and N1,N1,N3-trimethylpropane-1,3-diamine (106 μL, 0.72 mmol) were dissolved in EtOAc and heated at 180 °C in a microwave for 90 min. After cooling, the solution was washed with H2O and then brine, dried over MgSO4, and concentrated in vacuo to give a yellow oil, which was purified by reverse-phase HPLC (5–98% MeOH/water/0.1% formic acid) to give the title compound 59 (18 mg, 0.065 mmol, 36%). LC-MS Rt 7.22 min; 1H NMR (400 MHz, CDCl3) δ 7.47 (1H, d, J = 9.1 Hz), 7.24 (1H, dd, J = 9.1, 2.8 Hz), 7.16 (1H, d, J = 2.8 Hz), 3.89 (2H, t, J = 6.9 Hz), 3.85 (3H, s), 3.72 (2H, t, J = 7.1 Hz), 3.38 (3H, s), 3.19 (3H, s), 2.87 (2H, t, J = 6.9 Hz), 2.65 (2H, t, J = 7.7 Hz), 2.46 (6H, s), 2.01–1.90 (2H, m); 13C NMR (101 MHz, CDCl3) δ 163.7, 157.5, 153.3, 150.2, 127.6, 123.9, 118.7, 111.3, 105.4, 56.5, 55.9, 48.7, 47.1, 44.3, 41.3, 35.4, 24.7, 15.8; ESI HRMS, found 357.2386 (C19H29N6O [M + H]+, requires 357.2403).
3-((2-((3-(Dimethylamino)propyl)(methyl)amino)-6-phenylquinazolin-4-yl)(methyl)amino)propanenitrile (60)
3-((2-Chloro-6-phenylquinazolin-4-yl)(methyl)amino)propanenitrile IM18 (10 mg, 0.031 mmol) and N1,N1,N3-trimethylpropane-1,3-diamine (18 μL, 0.12 mmol) were dissolved in n-BuOH (0.5 mL) and heated at 120 °C for 4 h. After cooling, the solution was diluted with EtOAc, washed with H2O and then brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by reverse-phase HPLC (method C) to give the title compound 60 as a 0.5 formate salt (8 mg, 0.018 mmol, 61%). LC-MS Rt 8.87 min; 1H NMR (400 MHz, CDCl3) δ 8.55 (s, 0.5H), 8.01 (d, J = 2.0 Hz, 1H), 7.79 (dd, J = 8.7, 2.1 Hz, 1H), 7.61–7.54 (m, 3H), 7.45 (dd, J = 8.4, 7.0 Hz, 2H), 7.37–7.31 (m, 1H), 3.97 (t, J = 6.9 Hz, 2H), 3.78 (t, J = 7.0 Hz, 2H), 3.50 (s, 3H), 3.22 (s, 3H), 2.90 (t, J = 6.9 Hz, 2H), 2.75 (t, J = 7.8 Hz, 2H), 2.53 (s, 6H), 2.09–1.97 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 168.7, 163.9, 158.2, 154.3, 140.9, 133.2, 132.1, 129.1, 127.2, 126.9, 126.6, 123.7, 118.6, 111.4, 56.2, 48.7, 47.0, 43.9, 41.9, 35.4, 24.5, 15.7; ESI HRMS, found 403.2619 (C24H31N6 [M + H]+, requires 403.2610).
3-((2-((3-(Dimethylamino)propyl)(methyl)amino)-6-(o-tolyl)quinazolin-4-yl)(methyl)amino)propanenitrile (61)
3-((2-Chloro-6-(o-tolyl)quinazolin-4-yl)(methyl)amino)propanenitrile IM19 (30 mg, 0.089 mmol) and N1,N1,N3-trimethylpropane-1,3-diamine (52 μL, 0.36 mmol) were dissolved in n-BuOH (0.5 mL) and heated at 120 °C for 4 h. After cooling, the solution was diluted with EtOAc, washed with H2O and then brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by reverse-phase HPLC (50–98% MeOH/water/0.1% formic acid) to give the title compound 61 as a di-formate salt (6.2 mg, 0.012 mmol, 14%). LC-MS Rt 9.04 min (5–98% MeCN/water/0.1% formic acid); MH+ 417.4; 1H NMR (400 MHz, CDCl3) δ 8.40 (3H, br s), 7.85 (d, J = 8.6 Hz, 1H), 7.83 (d, J = 1.9 Hz, 1H), 7.63 (dd, J = 8.6, 1.8 Hz, 1H), 7.32–7.27 (m, 3H), 7.23–7.19 (m, 1H), 4.08 (t, J = 6.7 Hz, 2H), 3.87 (t, J = 7.1 Hz, 2H), 3.60 (s, 3H), 3.29 (s, 3H), 3.20–3.16 (m, 2H), 2.93 (t, J = 6.6 Hz, 2H), 2.80 (s, 6H), 2.27 (s, 3H), 2.20 (p, J = 7.6, 6.9 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 166.5, 162.8, 154.3, 140.3, 136.4, 135.7, 135.3, 130.7, 129.8, 127.9, 126.2, 126.0, 121.7, 117.9, 110.2, 54.7, 49.1, 47.1, 42.7, 42.3, 36.1, 23.0, 20.5, 15.5; ESI HRMS, found 417.2771 (C25H33N6 [M + H]+, requires 417.2767).
3-((2-((3-(Dimethylamino)propyl)(methyl)amino)-6-(4-methoxy-2-methylphenyl)quinazolin-4-yl)(methyl)amino)propanenitrile (62)
3-((2-Chloro-6-(4-methoxy-2-methylphenyl)quinazolin-4-yl)(methyl)amino)propanenitrile IM20 (22 mg, 0.060 mmol) and N1,N1,N3-trimethylpropane-1,3-diamine (36 μL, 0.24 mmol) were dissolved in n-BuOH (1 mL) and heated at 120 °C for 4 h. After cooling, the solution was diluted with EtOAc, washed with H2O and then brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by reverse-phase HPLC (50–98% MeOH/water/0.1% formic acid) to give the title compound 62 as a formate salt (5.5 mg, 0.011 mmol, 19%). LC-MS Rt 9.49 min; MH+ 447.4; 1H NMR (400 MHz, CDCl3) δ 8.35 (1H, s), 8.06 (m, 1H), 7.79 (d, J = 1.8 Hz, 1H), 7.61 (dd, J = 8.7, 1.9 Hz, 1H), 7.14 (d, J = 8.3 Hz, 1H), 6.85–6.81 (m, 2H), 4.07 (t, J = 6.6 Hz, 2H), 3.91 (t, J = 7.1 Hz, 2H), 3.85 (s, 3H), 3.59 (s, 3H), 3.29 (s, 3H), 3.24–3.19 (s, 2H), 2.92 (t, J = 6.8 Hz, 2H), 2.81 (s, 6H), 2.26 (m, 5H).
3-((2-((3-(Dimethylamino)propyl)(methyl)amino)-7-methoxyquinazolin-4-yl)(methyl)amino)propanenitrile (63)
3-((2-Chloro-7-methoxyquinazolin-4-yl)(methyl)amino)propanenitrile (IM21) and N1,N1,N3-trimethylpropane-1,3-diamine were dissolved in n-BuOH (1 mL) and heated at 120 °C for 4 h. After cooling, the solution was diluted with EtOAc, washed with H2O and then brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by reverse-phase HPLC (50–98% MeOH/water/0.1% formic acid) to give product 63 as a formate salt (17 mg, 0.042 mmol). LC-MS Rt 8.18 min; MH+ 357.3; 1H NMR (400 MHz, CDCl3) δ 8.49 (s, 1H), 7.85 (br s, 2H), 7.74 (d, J = 9.2 Hz, 1H), 6.99 (d, J = 2.6 Hz, 1H), 6.70 (dd, J = 9.2, 2.6 Hz, 1H), 3.95 (t, J = 6.8 Hz, 2H), 3.88 (s, 3H), 3.77 (t, J = 7.0 Hz, 2H), 3.45 (s, 3H), 3.20 (s, 3H), 2.99–2.92 (m, 2H), 2.86 (t, J = 6.8 Hz, 2H), 2.67 (s, 3H), 2.12–2.03 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 167.8, 163.4, 163.0, 157.3, 155.1, 127.1, 118.5, 112.8, 105.2, 104.2, 55.7, 55.3, 48.7, 46.8, 42.9, 41.9, 35.5, 23.4, 15.8; ESI HRMS, found 357.2397 (C19H29N6O [M + H]+, requires 357.2403).
4-(Ethyl(methyl)amino)-2-(methyl(1-methylpiperidin-4-yl)amino)-N-(1,3,5-trimethyl-1H-pyrazol-4-yl)quinazoline-6-sulfonamide (64)
According to general method D for reductive amination, 4-(ethyl(methyl)amino)-2-(methyl(piperidin-4-yl)amino)-N-(1,3,5-trimethyl-1H-pyrazol-4-yl)quinazoline-6-sulfonamide (67) was reacted with paraformaldehyde. After purification by reverse-phase HPLC, product 64 was obtained (1.2 mg, 0.002 mmol, 31%). LC-MS Rt 8.11 min (83%); MH+ 501.6; 1H NMR (400 MHz, CDCl3) δ 8.19 (br s, 3H), 7.97 (d, J = 1.9 Hz, 1H), 7.83 (dd, J = 8.9, 1.9 Hz, 1H), 7.49 (d, J = 9.0 Hz, 1H), 5.08–4.95 (m, 1H), 3.60 (s, 3H), 3.59–3.50 (m, 4H), 3.25–3.15 (m, 2H), 3.15 (s, 3H), 3.11 (s, 3H), 2.98 (s, 3H), 2.18–2.03 (m, 2H), 1.96 (s, 3H), 1.58 (s, 3H), 1.34–1.26 (m, 7H); ESI HRMS, found 501.2746 (C24H37N8O2S [M + H]+, requires 501.2760).
4-(Ethyl(methyl)amino)-2-(methyl(piperidin-4-yl)amino)-N-(1,3,5-trimethyl-1H-pyrazol-4-yl)quinazoline-6-sulfonamide (65)
2-Chloro-4-(ethyl(methyl)amino)-N-(1,3,5-trimethyl-1H-pyrazol-4-yl)quinazoline-6-sulfonamide (47 mg, 0.116 mmol, 1.0 equiv) and N-benzyl-N-methylpiperidin-4-amine (10 equiv) were combined in n-BuOH (1.0 mL) and stirred at 120 °C overnight. The reaction mixture was then concentrated in vacuo, and the crude residue was purified by flash column chromatography (CHCl3/MeOH/aq. NH3; 90:10:0.1) to give 2-((1-benzylpiperidin-4-yl)(methyl)amino)-4-(ethyl(methyl)amino)-N-(1,3,5-trimethyl-1H-pyrazol-4-yl)quinazoline-6-sulfonamide. This intermediate was debenzylated by dissolving it in 1 mL EtOH with ammonium formate (74 mg, 1.17 mmol, 10 equiv) and 10% Pd/C (13 mg) and then stirred at 80 °C overnight. The reaction mixture was then filtered through celite and concentrated in vacuo, before purification by reverse-phase HPLC to give product 65 as a light yellow oil (3.7 mg, 0.008 mmol, 7%). 1H NMR (400 MHz, CDCl3) δ 1H NMR (400 MHz, CDCl3) δ 8.25 (br, s, 3H), 7.99 (d, J = 1.9 Hz, 1H), 7.87 (dd, J = 8.9, 1.9 Hz, 1H), 7.55 (d, J = 8.8 Hz, 1H), 5.07–4.98 (m, 1H), 3.62 (s, 3H), 3.61–3.51 (m, 4H), 3.22–3.18 (m, 2H), 3.19 (s, 3H), 3.14 (s, 3H), 2.17–2.03 (m, 2H), 2.02–1.97 (m, 5H), 1.62 (s, 3H), 1.30 (t, J = 7.0 Hz, 3H).
2-((3-(Dimethylamino)propyl)(methyl)amino)-4-(ethyl(methyl)amino)-N-(1,3,5-trimethyl-1H-pyrazol-4-yl)quinazoline-6-sulfonamide (66)
2-Chloro-4-(ethyl(methyl)amino)quinazoline-6-sulfonyl chloride (50 mg, 0.156 mmol), 1,3,5-trimethyl-1H-pyrazol-4-amine (22 mg, 0.172 mmol), and triethylamine (43 μL, 0.312 mmol) were dissolved in DMF (1.0 mL). After purification by flash column chromatography, 2-chloro-4-(ethyl(methyl)amino)-N-(1,3,5-trimethyl-1H-pyrazol-4-yl)quinazoline-6-sulfonamide and N,N,N-trimethylpropane-1,3-diamine (58 μL) were combined in n-BuOH (1.0 mL) and stirred at 120 °C overnight. The reaction mixture was then concentrated in vacuo and purified by reverse-phase HPLC to give the title compound 66 (6 mg, 0.012 mmol, 12%). 1H NMR (400 MHz, CDCl3) δ 8.32 (br, s, 2H), 8.00 (d, J = 2.0 Hz, 1H), 7.84 (dd, J = 8.9, 2.0 Hz, 1H), 7.47 (d, J = 8.8 Hz, 1H), 3.78 (t, J = 6.4 Hz, 2H), 3.60 (s, 3H), 3.55 (q, J = 7.0 Hz, 2H), 3.22 (s, 3H), 3.18 (s, 3H), 3.10 (t, J = 7.0 Hz, 2H), 2.86 (s, 6H), 2.11 (qt, J = 6.5 Hz, 2H), 1.96 (s, 3H), 1.58 (s, 3H), 1.28 (t, J = 7.1 Hz, 3H); ESI HRMS, found 489.2739 (C23H37N8O2S [M + H]+, requires 489.2760).
4-(Ethyl(methyl)amino)-2-(methyl(1-methylpyrrolidin-3-yl)amino)-N-(1,3,5-trimethyl-1H-pyrazol-4-yl)quinazoline-6-sulfonamide (67)
2-Chloro-4-(ethyl(methyl)amino)quinazoline-6-sulfonyl chloride (50 mg, 0.156 mmol), 1,3,5-trimethyl-1H-pyrazol-4-amine (22 mg, 0.172 mmol), and triethylamine (43 μL, 0.312 mmol) were dissolved in DMF (1.0 mL). After purification by flash column chromatography, 2-chloro-4-(ethyl(methyl)amino)-N-(1,3,5-trimethyl-1H-pyrazol-4-yl)quinazoline-6-sulfonamide and N-dimethylpyrrolidin-3-amine (51 μL) were combined in n-BuOH (1.0 mL) and stirred at 120 °C overnight. The reaction mixture was then concentrated in vacuo and purified by reverse-phase HPLC to give the title compound 67 (5 mg, 0.01 mmol, 10%). 1H NMR (400 MHz, CDCl3) δ 8.30 (br, s, 2H), 8.02 (d, J = 2.0 Hz, 1H), 7.88 (dd, J = 8.9, 2.0 Hz, 1H), 7.53 (d, J = 8.8 Hz, 1H), 5.07* (m, 1H), 3.93–3.80 (m, 1H), 3.68–3.63 (m, 1H), 3.63 (s, 3H), 3.56 (q, J = 6.9 Hz, 2H), 3.46–3.40 (m, 1H), 3.28 (s, 3H), 3.28–2.23 (m, 1H), 3.18 (s, 3H), 3.03 (s, 3H), 2.60–2.51 (m, 1H), 2.35–2.25 (m, 1H), 1.99 (s, 3H), 1.61 (s, 3H), 1.29 (t, J = 7.0 Hz, 3H); ESI HRMS, found 487.2597 (C23H35N8O2S [M + H]+, requires 487.2604).*Obscured by the H2O signal.
Synthesis of Intermediate Compounds
3-((2-Chlorothieno[3,2-d]pyrimidin-4-yl)(methyl)amino)propanenitrile (IM1)
Using general method A, 2,4-dichlorothieno[3,2-d]pyrimidine (147 mg, 0.72 mmol) was treated with DIPEA (148 μL, 0.85 mmol) followed by 3-(methylamino)propanenitrile (67 μL, 0.71 mmol). After workup, the product, 3-((2-chlorothieno[3,2-d]pyrimidin-4-yl)(methyl)amino)propanenitrile (IM1), crystallized on evaporation of the solvent (173 mg, 0.68 mmol, 95%) without the need for further purification. Mp 156–158 °C; IR CN ν 2355 cm–1; MS (ESI) 253 (MH+); 1H NMR (400 MHz, CDCl3) δ 7.88 (d, J = 5.5 Hz, 1H), 7.45 (d, J = 5.5 Hz, 1H), 4.07 (t, J = 6.5 Hz, 2H), 3.73 (s, 3H), 2.90 (t, J = 6.5 Hz, 2H).
3-((2-Chloroquinazolin-4-yl)(methyl)amino)propanenitrile (IM2)
2,4-Dichloroquinazoline (0.7 g, 3.51 mmol) in DMF (3.0 mL) was treated with DIPEA (1.3 mL, 2.2 equiv) followed by 3-(methylamino)propanenitrile (720 μL, 2.2 equiv) and stirred at room temperature overnight. After the reaction was complete, H2O (50 mL) was added, and the resultant precipitate was collected by filtration and washed with water, giving the product (0.66 g, 2.68 mmol, 76%) without the need for further purification. 1H NMR (400 MHz, DMSO-d6) δ 8.25 (dd, J = 8.5, 1.3 Hz, 1H), 7.82 (ddd, J = 8.3, 6.9, 1.3 Hz, 1H), 7.69 (dd, J = 8.4, 1.3 Hz, 1H), 7.52 (ddd, J = 8.5, 7.0, 1.4 Hz, 1H), 4.03 (t, J = 6.8 Hz, 2H), 3.52 (s, 3H), 3.01 (t, J = 6.8 Hz, 2H).
3-((2-Chloropyrido[2,3-d]pyrimidin-4-yl)(methyl)amino)propanenitrile (IM3)
Using general method A, 2,4-dichloropyrido[2,3-d]pyrimidine (360 mg, 1.8 mmol) was treated with DIPEA (350 μL, 2.0 mmol) followed by 3-(methylamino)propanenitrile (170 μL, 1.8 mmol). After workup, the crude residue was purified by flash column chromatography (EtOAc/MeOH; 100:0 and then 90:10) to give the product, 3-((2-chloropyrido[2,3-d]pyrimidin-4-yl)(methyl)amino)propanenitrile (IM3), as a colorless solid (126 mg, 0.51 mmol, 28%). Mp 190–192 °C; 1H NMR (400 MHz, CDCl3), δ 9.06 (dd, J = 4.4, 1.8 Hz, 1H), 8.50 (dd, J = 8.5, 1.8 Hz, 1H), 7.42 (dd, J = 8.5, 4.4 Hz, 1H), 4.11 (t, J = 6.4 Hz, 2H), 3.72 (s, 3H), 2.99 (t, J = 6.4 Hz, 2H).
3-((2-Chloro-7-methyl-7H-purin-6-yl)(methyl)amino)propanenitrile (IM4)
A suspension of 2,6-dichloro-7-methyl-7H-purine (404 mg, 2.1 mmol, 1.0 equiv) in EtOAc/EtOH (10:1, 10 mL) was treated with DIPEA (1.2 equiv) followed by 3-(methylamino)propanenitrile (215 μL, 2.3 mmol, 1.2 equiv), and the reaction was heated under reflux for 4 h. The reaction mixture was cooled to room temperature and then washed with sodium carbonate solution (1 M, 10 mL), and the aqueous layer was re-extracted with EtOAc (10 mL). The combined organic layers were dried (Na2SO4) and concentrated under reduced pressure. The product, 3-((2-chloro-7-methyl-7H-purin-6-yl)(methyl)amino)propanenitrile (IM4), was obtained by flash column chromatography (EtOAc/MeOH; 95:5 and then 90:10) as a tan solid (300 mg, 1.2 mmol, 57%). 1H NMR (400 MHz, CDCl3), δ 8.05 (s, 1H), 4.08 (s, 3H), 3.86 (t, J = 6.5 Hz, 2H), 3.33 (s, 3H), 2.86 (t, J = 6.5 Hz, 2H).
2-Chloro-N-(2-methoxyethyl)-N-methylthieno[3,2-d]pyrimidin-4-amine (IM5)
Using general method A, 2,4-dichlorothieno[3,2-d]pyrimidine (146 mg, 0.71 mmol) was treated with DIPEA (148 μL, 0.85 mmol) followed by 2-methoxy-N-methylethanamine (63 mg, 0.71 mmol). After workup, the product, 2-chloro-N-(2-methoxyethyl)-N-methylthieno[3,2-d]pyrimidin-4-amine (IM5), crystallized on evaporation of the solvent (185 mg, 0.71 mmol, quant.) with no need for further purification. Mp 70–72 °C. MS (ESI) 258 (MH+); 1H NMR (400 MHz, CDCl3) δ 7.78 (d, J = 5.4 Hz, 1H), 7.35 (d, J = 5.4 Hz, 1H), 3.99 (t, J = 5.4 Hz, 2H), 3.71 (t, J = 5.4 Hz, 2H), 3.56 (s, 3H), 3.39 (s, 3H).
2-((2-Chlorothieno[3,2-d]pyrimidin-4-yl)(methyl)amino)ethanol (IM6)
Using general method A, 2,4-dichlorothieno[3,2-d]pyrimidine (146 mg, 0.71 mmol) was treated with DIPEA (148 μL, 0.85 mmol) followed by 2-(methylamino)ethanol (53 mg, 0.71 mmol). After workup, the product, 2-((2-chlorothieno[3,2-d]pyrimidin-4-yl)(methyl)amino)ethanol (IM6), crystallized on evaporation of the solvent as a colorless solid (158 mg, 0.65 mmol, 91%) with no need for further purification. Mp 182–183 °C. MS (ESI) 244 (MH+); 1H NMR (400 MHz, DMSO-d6) δ 8.26 (d, J = 5.8 Hz, 1H), 7.35 (d, J = 5.8 Hz, 1H), 4.94 (br. s, 1H), 3.82 (t, J = 5.8 Hz, 2H), 3.67 (t, J = 5.8 Hz, 2H), 3.42 (s, 3H).
3-((2-Chlorothieno[3,2-d]pyrimidin-4-yl)(methyl)amino)propan-1-ol (IM7)
In an adaptation of general method A, 2,4-dichlorothieno[3,2-d]pyrimidine (205 mg, 1.0 mmol) was treated with DIPEA (348 μL, 2.0 mmol) followed by 3-(methylamino)propanol (195 μL, 2.0 mmol). After workup, the residue was purified by flash column chromatography (CH2Cl2/MeOH; 20:1) to give the product, 2-((2-chlorothieno[3,2-d]pyrimidin-4-yl)(methyl)amino)ethanol (IM7), as a yellow solid (256 mg, 0.99 mmol, 99%). 1H NMR (400 MHz, CDCl3) δ 7.80 (d, J = 5.5 Hz, 1H), 7.35 (d, J = 5.5 Hz, 1H), 3.89 (t, J = 5.5 Hz, 2H), 3.60 (t, J = 5.5 Hz, 2H), 3.50 (s, 3H), 1.94–1.84 (m, 2H).
3-((2-Chlorothieno[3,2-d]pyrimidin-4-yl)(methyl)amino)propenamide (IM8)
2,4-Dichlorothieno[3,2-d]pyrimidine (50 mg, 0.24 mmol) was dissolved in chlorosulfuric acid (1.0 mL, excess) and stirred at room temperature for 16 h. The reaction was then cooled to 0 °C, quenched with H2O, and extracted with EtOAc. The aqueous layer was neutralized with NaOH, and the resulting precipitate was collected by filtration to give the product, IM8, as a white solid (26 mg, 0.096 mmol, 40%). LC-MS Rt = 10.5 min; MH+ 271/273; 1H NMR (400 MHz, DMSO-d6,) δ 8.29 (d, J = 5.5 Hz, 1H), 7.44 (s, 1H), 7.37 (d, J = 5.6 Hz, 1H), 6.94 (s, 1H), 3.94 (t, J = 7.1 Hz, 2H), 3.39 (s, 3H), 2.48 (d, J = 7.1 Hz, 2H).
N-(2-((2-Chlorothieno[3,2-d]pyrimidin-4-yl)(methyl)amino)ethyl)-N-methylacetamide (IM9)
Step 1: Using an adaptation of general method A, 2,4-dichlorothieno[3,2-d]pyrimidine (205 mg, 1.0 mmol) was treated with DIPEA (174 μL, 1.0 mmol) followed by N,N′-dimethylethylenediamine (1.0 mL, 9.3 mmol). After workup, the crude residue was purified by flash column chromatography (CH2Cl2/MeOH/aq. NH3; 95:5:0.5–97:3:0.5) to give the product, N-(2-chlorothieno[3,2-d]pyrimidin-4-yl)-N,N′-dimethylethylenediamine (230 mg, 0.90 mmol, 90%). 1H NMR (400 MHz, CDCl3), δ 7.78 (d, J = 5.5 Hz, 1H), 7.35 (d, J = 5.5 Hz, 1H), 3.96 (t, J = 6.5 Hz, 2H), 3.53 (s, 3H), 3.18 (s, 3H), 3.00 (t, J = 6.5 Hz, 2H), 2.55 (s, 3H).
Step 2: The product from Step 1 (230 mg, 0.9 mmol) was dissolved in CH2Cl2 (10 mL) and treated with Et3N (73 μL, 1.0 mmol) followed by acetyl chloride (72 μL, 1.0 mmol). The reaction was stirred at room temperature for 1 h and worked up by washing the reaction mixture with sodium hydroxide solution (1 M, 10 mL) followed by extraction of the aqueous layer with EtOAc (10 mL). The combined organic layers were dried (Na2SO4), concentrated under reduced pressure, and the crude residue was purified by flash column chromatography (EtOAc/MeOH; 95:5 and then 90:10) to give the product, N-(2-((2-chlorothieno[3,2-d]pyrimidin-4-yl)(methyl)amino)ethyl)-N-methylacetamide (IM9), as a low melting solid (70 mg, 0.23 mmol, 26%). 1H NMR as a 3:1 mixture of rotamers (400 MHz, CDCl3) δ 7.72 (d, J = 5.5 Hz, 0.3H), 7.69 (d, J = 5.5 Hz, 0.7H), 7.22 (d, J = 5.5 Hz, 0.3H), 7.19 (d, J = 5.5 Hz, 0.7H), 3.82 (t, J = 6.0 Hz, 1.4H), 3.75 (dd, J = 8.3, 6.0 Hz, 0.6H), 3.56 (t, J = 6.0 Hz, 1.4H), 3.51 (dd, J = 8.3, 6.0 Hz, 0.6H), 3.44 (s, 0.9H), 3.42 (s, 2.1H), 3.02 (s, 2.1H), 2.92 (s, 0.9H), 2.06 (s, 0.9H), 1.85 (s, 2.1H).
3-((2-Chlorothieno[3,2-d]pyrimidin-4-yl)(methyl)amino)-N,N-dimethylpropanamide (IM10)
Using general method A, 2,4-dichlorothieno[3,2-d]pyrimidine (205 mg, 1.0 mmol) was treated with DIPEA (174 μL, 1.0 mmol) followed by N,N-dimethyl-3-(methylamino)propanamide (170 mg, 1.5 mmol). After workup, the residue was purified by flash column chromatography (EtOAc/MeOH; 100:0 and then 95:5) to give the product, 3-((2-chlorothieno[3,2-d]pyrimidin-4-yl)(methyl)amino)-N,N-dimethylpropanamide (IM10), as a pale yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.78 (m, 1H), 7.41–7.32 (m, 1H), 4.10–4.03 (m, 2H), 3.57 (s, 3H), 3.11 (s, 3H), 2.98 (s, 3H), 2.83–2.76 (m, 2H).
(S)-(1-(2-Chlorothieno[3,2-d]pyrimidin-4-yl)pyrrolidin-2-yl)methanol (IM11)
Using general method A, 2,4-dichlorothieno[3,2-d]pyrimidine (205 mg, 1.0 mmol) was treated with DIPEA (209 μL, 1.2 mmol) followed by (S)-pyrrolidin-2-ylmethanol (100 μL, 1.0 mmol). After workup, the crude residue was purified by column chromatography (EtOAc/hexane; 50:50–100:0) to give the product, (S)-(1-(2-chlorothieno[3,2-d]pyrimidin-4-yl)pyrrolidin-2-yl)methanol (IM11), as a colorless solid (242 mg, 0.90 mmol, 90%). Mp 160–161 °C; 1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 5.5 Hz, 1H), 7.33 (d, J = 5.5 Hz, 1H), 4.59 (dq, J = 7.0, 3.5 Hz, 1H), 4.10 (dd, J = 7.0, 4.5 Hz, 2H), 3.87 (dd, J = 11.5, 3.5 Hz, 1H), 3.79 (dd, J = 11.5, 7.0 Hz, 1H), 2.27–2.08 (m, 3H), 1.93 (td, J = 8.1, 7.0, 3.5 Hz, 1H).
(S)-2-Chloro-4-(2-(methoxymethyl)pyrrolidin-1-yl)thieno[3,2-d]pyrimidine (IM12)
Using general method A, 2,4-dichlorothieno[3,2-d]pyrimidine (205 mg, 1.0 mmol) was treated with DIPEA (209 μL, 1.2 mmol) followed by (S)-2-(methoxymethyl)pyrrolidine (123 μL, 1.0 mmol). After workup, the product, (S)-2-chloro-4-(2-(methoxymethyl)pyrrolidin-1-yl)thieno[3,2-d]pyrimidine (IM12), crystallized on evaporation of the solvent as a colorless solid (250 mg, 0.88 mmol, 88%). Mp 110–112 °C; 1H NMR (400 MHz, CDCl3) δ 7.77 (d, J = 5.5 Hz, 1H), 7.35 (d, J = 5.5 Hz, 1H), 4.70 (td, J = 7.5, 3.0 Hz, 1H), 4.10–4.00 (m, 1H), 4.00–3.87 (m, 1H), 3.65 (dd, J = 9.5, 3.0 Hz, 1H), 3.63–3.53 (m, 1H), 3.40 (s, 3H), 2.32–2.13 (m, 2H), 2.13–2.01 (m, 2H).
(S)-2-(1-(2-Chlorothieno[3,2-d]pyrimidin-4-yl)pyrrolidin-2-yl)acetonitrile (IM13)
A solution of 2,4-dichlorothieno[3,2-d]pyrimidine (205 mg, 1.0 mmol, 1.0 equiv) in EtOAc (10 mL) was treated with DIPEA (1.2 equiv) followed by (S)-2-(pyrrolidin-2-yl)acetonitrile (125 mg, 1.1 mmol), and the reaction was heated under reflux for 4 h. The reaction mixture was cooled to room temperature and then washed with sodium hydroxide solution (1 M, 10 mL), and the aqueous layer was re-extracted with EtOAc (10 mL). The combined organic layers were dried (Na2SO4), concentrated under reduced pressure, and the residue was purified by flash column chromatography (DCM/hexane; 50:50–100:0 and then DCM/MeOH; 95:5) to give the product, (S)-(1-(2-chlorothieno[3,2-d]pyrimidin-4-yl)pyrrolidin-2-yl)methanol (IM13), as a yellow solid (115 mg, 0.41 mmol, 41%). 1H NMR (400 MHz, CDCl3) δ 7.82 (d, J = 5.5 Hz, 1H), 7.35 (d, J = 5.5 Hz, 1H), 4.68 (tt, J = 7.0, 3.5 Hz, 1H), 4.17 (ddd, J = 9.0, 7.5, 4.5 Hz, 1H), 4.07 (dt, J = 9.0, 7.5 Hz, 1H), 3.13 (dd, J = 16.8, 7.0 Hz, 1H), 2.93 (dd, J = 16.8, 3.5 Hz, 1H), 2.48–2.23 (m, 2H), 2.17 (tdd, J = 10.9, 6.3, 3.5 Hz, 2H).
2-Chloro-N-ethyl-N-methylthieno[3,2-d]pyrimidin-4-amine (IM14)
Using general method A, 2,4-dichlorothieno[3,2-d]pyrimidine (307 mg, 1.5 mmol) was treated with DIPEA (313 μL, 1.8 mmol) followed by N-methylethanamine (128 μL, 1.5 mmol). After workup, the product, 2-((2-chlorothieno[3,2-d]pyrimidin-4-yl)(methyl)amino)ethanol (IM14), crystallized on evaporation of the solvent as a pale yellow solid (305 mg, 1.3 mmol, 89%) with no need for further purification. Mp 105–107 °C; 1H NMR (400 MHz, CDCl3) δ 7.77 (d, J = 5.8 Hz, 1H), 7.37 (d, J = 5.8 Hz, 1H), 3.88 (q, J = 7.1 Hz, 2H), 3.44 (s, 3H), 1.34 (t, J = 7.1 Hz, 3H).
3-((6-(tert-Butyl)-2-chlorothieno[3,2-d]pyrimidin-4-yl)(methyl)amino)propanenitrile (IM15)
To a solution of 6-(tert-butyl)-2,4-dichlorothieno[3,2-d]pyrimidine (233 mg, 0.9 mmol, 1.0 equiv) in DMF (2.0 mL) was added DIPEA (310 μL, 1.8 mmol, 2.0 equiv) followed by 3-(methylamino)propanenitrile (167 μL, 1.8 mmol, 2.0 equiv), and the reaction was stirred at room temperature overnight. The reaction mixture was washed with sodium hydroxide solution (1 M, 10 mL), and the aqueous layer was re-extracted with EtOAc (10 mL). The combined organic layers were dried (Na2SO4) and then concentrated under reduced pressure. The crude product was purified by flash column chromatography by elution with ethyl acetate/hexane (2:1) giving the product (239 mg, 0.77 mmol, 86%).
3-((6-Bromo-2-chloroquinazolin-4-yl)(methyl)amino)propanenitrile (IM16)
To a suspension of 6-bromo-2,4-dichloroquinazoline (290 mg, 1.0 mmol) and 3-(methylamino)propanenitrile (195 μL, 2.1 mmol, 2.0 equiv) in DMF (3.0 mL) was added DIPEA (363 μL, 2.1 mmol, 2.0 equiv), and the solution was stirred at room temperature for 2 h. Water was added, and the solid formed was isolated by filtration, washed with water, and dried to give the product (156 mg, 0.48 mmol, 48%) as an orange powder. 1H NMR (400 MHz, CDCl3) δ 8.22 (d, J = 2.0 Hz, 1H), 7.82 (dd, J = 9.0, 2.0 Hz, 1H), 7.69 (d, J = 2.0 Hz, 1H), 4.04 (t, J = 6.4 Hz, 2H), 3.67 (s, 3H), 2.94 (t, J = 6.4 Hz, 3H).
3-((2-Chloro-6-methoxyquinazolin-4-yl)(methyl)amino)propanenitrile (IM17)
To a suspension of 2,4-dichloro-6-methoxyquinazoline (90 mg, 0.39 mmol, 1.0 equiv) and 3-(methylamino)propanenitrile (74 μL, 0.8 mmol, 2.0 equiv) in DMF (1.0 mL) was added DIPEA (137 μL, 0.8 mmol, 2.0 equiv), and the solution was stirred at room temperature for 2 h. Water was added, and the solid formed was isolated by filtration, washed with water, and dried to give the product (50 mg, 46%) as a yellow powder. 1H NMR (400 MHz, CDCl3) δ 7.80 (d, J = 9.2 Hz, 1H), 7.43 (dd, J = 9.2, 2.7 Hz, 1H), 7.36 (d, J = 2.7 Hz, 1H), 4.00 (t, J = 6.5 Hz, 2H), 3.91 (s, 3H), 3.61 (s, 3H), 2.94 (t, J = 6.5 Hz, 2H).
3-((2-Chloro-6-phenylquinazolin-4-yl)(methyl)amino)propanenitrile (IM18)
3-((6-Bromo-2-chloroquinazolin-4-yl)(methyl)amino)propanenitrile (25 mg, 0.08 mmol, 1.0 equiv) and phenyl boronic acid (9.4 mg, 0.08 mmol, 1.0 equiv) were dissolved in DMF (1.0 mL), and Pd(OAc)2 (1.0 mg, 0.004 mmol, 0.05 equiv) was added. A solution of Cs2CO3 (50 mg, 0.15 mmol) in H2O (0.3 mL) was added and stirred at room temperature for 2 h. The solution was diluted with EtOAc (15 mL) and then washed with H2O (5 × 10 mL) and then brine (10 mL) before drying over MgSO4. The compound was purified by flash column chromatography (EtOAc/hexane 5:95–30:70) to give the product as a white solid (10 mg, 43%). 1H NMR (400 MHz, CDCl3) δ 8.23 (d, J = 1.9 Hz, 1H), 8.00 (dd, J = 8.7, 1.9 Hz, 1H), 7.91 (d, J = 8.7 Hz, 1H), 7.65–7.57 (m, 2H), 7.55–7.46 (m, 2H), 7.46–7.38 (m, 1H), 4.08 (t, J = 6.5 Hz, 2H), 3.72 (s, 3H), 2.97 (t, J = 6.5 Hz, 2H).
3-((2-Chloro-6-phenylquinazolin-4-yl)(methyl)amino)propanenitrile (IM19)
3-((6-Bromo-2-chloroquinazolin-4-yl)(methyl)amino)propanenitrile (50 mg, 0.15 mmol, 1.0 equiv) and 2-methylphenyl boronic acid (21 mg, 0.15 mmol, 1.0 equiv) were dissolved in DMF (2.0 mL), and Pd(OAc)2 (2.0 mg, 0.008 mmol, 0.05 equiv) was added. A solution of Cs2CO3 (100 mg, 0.31 mmol) in H2O (0.6 mL) was added, and the solution was stirred at room temperature for 2 h. The solution was diluted with EtOAc (15 mL) and then washed with H2O (5 × 10 mL) and then brine (10 mL) before drying over MgSO4. The compound was purified by flash column chromatography (EtOAc/hexane 5:95–30:70) to give a colorless oil (34 mg, 0.10 mmol, 67%). 1H NMR (400 MHz, CDCl3) δ 8.00 (d, J = 1.8 Hz, 1H), 7.87 (d, J = 8.6 Hz, 1H), 7.74 (dd, J = 8.6, 1.8 Hz, 1H), 7.38–7.21 (m, 4H), 4.05 (t, J = 6.5 Hz, 2H), 3.65 (s, 3H), 2.96 (t, J = 6.5 Hz, 2H), 2.29 (s, 3H).
3-((2-Chloro-6-(4-methoxy-2-methylphenyl)quinazolin-4-yl)(methyl)amino)propanenitrile (IM20)
3-((6-Bromo-2-chloroquinazolin-4-yl)(methyl)amino)propanenitrile (50 mg, 0.15 mmol, 1.0 equiv) and (4-methoxy-2-methylphenyl)boronic acid (25 mg, 0.15 mmol, 1.0 equiv) were dissolved in DMF (2.0 mL), and Pd(OAc)2 (2.0 mg, 0.009 mmol, 0.06 equiv) was added. A solution of Cs2CO3 (100 mg, 0.31 mmol) in H2O (0.6 mL) was added, and the solution was stirred at room temperature for 2 h. The solution was diluted with EtOAc (15 mL) and then washed with H2O (5 × 10 mL) and then brine (10 mL) before drying over MgSO4. The compound was purified by flash column chromatography (EtOAc/hexane 5:95–25:75) to give a white solid (22 mg, 0.06 mmol, 40%). 1H NMR (400 MHz, CDCl3) δ 7.96 (dd, J = 1.8, 0.6 Hz, 1H), 7.84 (dd, J = 8.6, 0.6 Hz, 1H), 7.71 (dd, J = 8.6, 1.9 Hz, 1H), 7.18 (d, J = 8.3 Hz, 1H), 6.89–6.80 (m, 2H), 4.04 (t, J = 6.5 Hz, 2H), 3.85 (s, 3H), 3.64 (s, 3H), 2.95 (t, J = 6.5 Hz, 2H), 2.28 (s, 3H).
3-((2-Chloro-7-methoxyquinazolin-4-yl)(methyl)amino)propanenitrile (IM21)
To a suspension of 2,4-dichloro-7-methoxyquinazoline (30 mg, 0.13 mmol, 1.0 equiv) and 3-(methylamino)propanenitrile (25 μL, 0.26 mmol, 2.0 equiv) in DMF (0.5 mL) was added DIPEA (46 μL, 0.26 mmol, 2.0 equiv), and the solution was stirred at room temperature for 2 h. Water was added, and the solid formed was isolated by filtration, washed with water, and dried to give the product (10 mg, 0.036 mmol, 28%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.98 (d, J = 9.3 Hz, 1H), 7.18 (d, J = 2.7 Hz, 1H), 7.04 (dd, J = 9.4, 2.7 Hz, 1H), 4.02 (t, J = 6.5 Hz, 2H), 3.92 (s, 3H), 3.62 (s, 3H), 2.93 (t, J = 6.5 Hz, 2H).
3-((2-Chloropyrimidin-4-yl)(methyl)amino)propanenitrile (IM22)
3-(Methylamino)propanenitrile (571 μL, 6.7 mmol, 2.0 equiv) was added to a solution of 2,4-dichloropyrimidine (500 mg, 3.4 mmol, 1.0 equiv) and DIPEA (1.2 mL, 6.7 mmol, 2.0 equiv) in DMF (5.0 mL) and stirred at room temperature for 2 h. The solution was diluted with water and then extracted with EtOAc. The compound was purified by flash column chromatography (EtOAc/hexane 1:4) to give a solid (200 mg, 1.0 mmol 15%). 1H NMR (400 MHz, CDCl3) 1H NMR (400 MHz, chloroform-d) δ 8.13 (d, J = 6.1 Hz, 1H), 6.40 (d, J = 6.1 Hz, 1H), 3.91 (t, J = 6.4 Hz, 2H), 3.20 (s, 3H), 2.75 (t, J = 6.4 Hz, 2H).
Biochemical Methods
Protein Production and Crystallography
Protein production and crystallization of LmNMT-MyrCoA were as described in Brannigan et al. (2014).30 Ligand compounds (25 mM stocks in 50% dimethyl sulfoxide, DMSO) were used at a final concentration of 2.5 mM to soak LmNMT-MyrCoA crystals. X-ray diffraction data were collected on synchrotron beamlines at the Diamond Light Source (Didcot, U.K.) and processed using XDS42 and SCALA43 implemented within xia2.44 Data collection and refinement statistics are summarized in Table S3. For Rfree calculations, 5% of the data were excluded. Cycles of refinement using maximum-likelihood methods implemented in REFMAC45 were interspersed with model building and adjustment using COOT.46 The final refined protein structure models display good geometry with typically only a single amino acid residue (His347) lying outside of the preferred region of the Ramachandran plot.
CPM Assay
The enzyme-based inhibitory potency of all derivatives was determined for LdNMT, LmNMT, and HsNMT1 using the CPM assay format, as described previously.31,32 Data is summarized in Table S1, and the dose–response curves are shown in Figures S6–S8.
Alamar Blue-Based Assay
The assay was conducted as previously described and validated using amphotericin B and miltefosine as reference compounds. An Ethiopian strain of L. donovani (MHOM/ET/67/HU3, also known as LV9) was maintained in Rag-2–/– mice by serial passage. All experiments were approved by the University of York Animal Procedures and Ethics Committee and performed under the U.K. Home Office licence (“Immunology and Immunopathology of Leishmaniasis” Ref # PPL 60/4377). Isolation of amastigotes from infected spleens was performed by saponin lysis after homogenization, as previously described.37 Afterward, Ld amastigotes were plated at 4 × 105 parasites per well in 96-well plates followed by the addition of the inhibitor concentrations in RPMI medium supplemented with 20% heat-inactivated fetal calf serum (FCS), 100 μM adenine, 20 mM 2-[N-morpholino] ethanesulfonic acid (pH 5.5), 5 μM hemin, 3 μM biopterin, 1 μM biotin, penicillin (100 U/mL), and streptomycin (100 μg/mL). After 72 h at 26 °C, 1/5 volume Alamar blue (Trek Diagnostics) was added. Fluorescence was measured after another 6 h using a POLARstar Optima (BMG Labtech) plate reader (ex. 544 nm, em. 590 nm).
Macrophages (BMDM) were differentiated from the bone marrow of 6–8 week old female BALB/c mice, as described previously,37 and maintained at 37 °C and 5% CO2 in DMEM (GIBCO) supplemented with 4 mM l-glutamine (GIBCO) and 4% L929-cell-conditioned medium. BMDMs were seeded at 4.2 × 104 cells per well in 96-well plates and adhered for at least 4 h. Subsequently, inhibitor concentrations were added for 72 h, followed by the addition of a 1/10 volume Alamar blue (Trek Diagnostics). Fluorescence was measured after 6 h as described above. Data is summarized in Table S2, and the dose–response curves are shown in Figures S9 and S10.
Tagging Assay
Freshly isolated amastigotes (see above) were seeded at 7.5 × 107 parasites per mL and cultured in RPMI supplemented with 10% FCS and the corresponding inhibitor concentration. After 1 h at 37 °C, the parasites were collected by centrifugation (800g, 15 min, 4 °C) and resuspended in a fresh medium containing the inhibitor dilution and YnMyr or myristic acid (final concentration 50 μM). After 12 h, the parasites were pelleted and washed twice with cold phosphate-buffered saline (PBS). Subsequently, the cells were lysed on ice at 1 × 109 parasites per mL in lysis buffer (1% NP40, 1% sodium deoxycholate, 0.5% sodium dodecyl sulfate (SDS), 50 mM Tris (pH 7.4), 150 mM NaCl, EDTA-free protease inhibitor) by sonication (4 × 10 second burst, amplitude 45 with 1 min interval). All lysates were cleared by centrifugation (16 100g, 30 min, 4 °C), and protein concentrations were determined using the DC Protein Assay (Bio-Rad) following the manufacturer’s procedure.
Proteins were precipitated with chloroform/methanol (MeOH/CHCl3/H2O 4:1:3) and resuspended at 1 mg/mL in 1% SDS in PBS. Afterward, 1 μL AzTB41 (10 mM in DMSO, 0.1 mM final), 2 μL CuSO4 (50 mM in H2O, 1 mM final), 2 μL Tris(2 carboxyethyl)phosphine hydrochloride (TCEP) (50 mM in H2O, 1 mM final), and 1 μL Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA) (10 mM in DMSO, 0.1 mM final) were mixed. Six microliters of this click mixture was added to 100 μL of lysate, and the click reaction was shaken for 1 h at room temperature. Subsequently, the proteins were precipitated again by methanol/chloroform precipitation. The protein pellet was air-dried and redissolved in PBS containing 0.2% SDS and 0.1 mM dithiothreitol (DTT) using sonication to a final protein concentration of 1 mg/mL. All samples were further analyzed by SDS-PAGE, and fluorescence was scanned using a Typhoon imager (GE Healthcare; excitation laser: 532 nm, emission filter: LPG (575–700 nm); PMT: 750 V). For Western blot, the proteins were transferred from gels to poly(vinylidene fluoride) membranes (Immobilon-PSQ, Millipore) using a semidry system (Invitrogen). Following blocking (5% milk in TBS-T buffer), the membranes were incubated for 1 h at room temperature with the primary and overnight at 4 °C with the secondary HRP-conjugated antibody. Detection was performed using a Luminata Crescendo Western HRP substrate (Millipore) according to the manufacturer’s instructions with a Fujifilm LAS 3000 imager.
Acknowledgments
The authors would like to thank the Diamond Light Source for synchrotron beamtime (proposals mx1221 and 7864). This work was supported by The Wellcome Trust (grant 087792) and the Engineering and Physical Sciences Research Council (grants EP/F500416/1 and EP/K039946/1).
Glossary
Abbreviations
- CI
confidence interval
- CL
cutaneous leishmaniasis
- CPM
7-diethylamino-3-(4-maleimido-phenyl)-4-methylcoumarin
- DIPEA
diisopropylethylamine
- Ld
Leishmania donovani
- LipE
lipophilic ligand efficiency
- Lm
Leishmania major
- MyrCoA
myristoyl-CoA
- nAUC
normalized area under the curve
- NMT
N-myristoyltransferase
- SSG
sodium stibogluconate
- VL
visceral leishmaniasis
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00570.
Accession Codes
The coordinates and structure factor files have been deposited in the Protein Data Bank with accession codes 6qd9 (LmNMT-MyrCoA-2), 6qda (LmNMT-MyrCoA-3), 6qdb (LmNMT-MyrCoA-10), 6qdc (LmNMT-MyrCoA-12), 6qdd (LmNMT-MyrCoA-43), 6qde (LmNMT-MyrCoA-51), 6qdf (LmNMT-MyrCoA-52), 6qdg (LmNMT-MyrCoA-56), and 6qdh (LmNMT-MyrCoA-64).
Author Present Address
⊥ (D.P.) Adlai Nortye Biopharma, Hangzhou Economic & Technological Development Area, China.
Author Present Address
# (M.R.) Institute of Infection, Immunity & Inflammation, University of Glasgow, Glasgow, U.K.
Author Present Address
∇ (A.J.W.) Evotec, Hamburg, Germany.
The authors declare no competing financial interest.
Supplementary Material
References
- Torres-Guerrero E.; Quintanilla-Cedillo M. R.; Ruiz-Esmenjaud J.; Arenas R. Leishmaniasis: A Review. F1000Research 2017, 6, 750. 10.12688/f1000research.11120.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO, Leishmaniasis. https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (accessed 14 June, 2020).
- Hefnawy A.; Berg M.; Dujardin J. C.; De Muylder G. Exploiting Knowledge on Leishmania Drug Resistance to Support the Quest for New Drugs. Trends Parasitol. 2017, 33, 162–174. 10.1016/j.pt.2016.11.003. [DOI] [PubMed] [Google Scholar]
- Yamazaki K.; Kaneko Y.; Suwa K.; Ebara S.; Nakazawa K.; Yasuno K. Synthesis of Potent and Selective Inhibitors of Candida Albicans N-Myristoyltransferase Based on the Benzothiazole Structure. Bioorg. Med. Chem. 2005, 13, 2509–2522. 10.1016/j.bmc.2005.01.033. [DOI] [PubMed] [Google Scholar]
- Fang W.; Robinson D. A.; Raimi O. G.; Blair D. E.; Harrison J. R.; Lockhart D. E. A.; Torrie L. S.; Ruda G. F.; Wyatt P. G.; Gilbert I. H.; Van Aalten D. M. F. N -Myristoyltransferase Is a Cell Wall Target in Aspergillus Fumigatus. ACS Chem. Biol. 2015, 10, 1425–1434. 10.1021/cb5008647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowyer P. W.; Gunaratne R. S.; Grainger M.; Withers-Martinez C.; Wickramsinghe S. R.; Tate E. W.; Leatherbarrow R. J.; Brown K. A.; Holder A. A.; Smith D. F. Molecules Incorporating a Benzothiazole Core Scaffold Inhibit the N-Myristoyltransferase of Plasmodium Falciparum. Biochem. J. 2007, 408, 173–180. 10.1042/BJ20070692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z.; Brannigan J. A.; Moss D. K.; Brzozowski A. M.; Wilkinson A. J.; Holder A. A.; Tate E. W.; Leatherbarrow R. J. Design and Synthesis of Inhibitors of Plasmodium Falciparum N -Myristoyltransferase, a Promising Target for Antimalarial Drug Discovery. J. Med. Chem. 2012, 55, 8879–8890. 10.1021/jm301160h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rackham M. D.; Brannigan J. A.; Rangachari K.; Meister S.; Wilkinson A. J.; Holder A. A.; Leatherbarrow R. J.; Tate E. W. Design and Synthesis of High Affinity Inhibitors of Plasmodium Falciparum and Plasmodium Vivax N-Myristoyltransferases Directed by Ligand Efficiency Dependent Lipophilicity (LELP). J. Med. Chem. 2014, 57, 2773–2788. 10.1021/jm500066b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlott A. C.; Mayclin S.; Reers A. R.; Coburn-Flynn O.; Bell A. S.; Green J.; Knuepfer E.; Charter D.; Bonnert R.; Campo B.; Burrows J.; Lyons-Abbott S.; Staker B. L.; Chung C. W.; Myler P. J.; Fidock D. A.; Tate E. W.; Holder A. A. Structure-Guided Identification of Resistance Breaking Antimalarial N-Myristoyltransferase Inhibitors. Cell Chem. Biol. 2019, 26, 991–1000.e7. 10.1016/j.chembiol.2019.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves V.; Brannigan J. A.; Whalley D.; Ansell K. H.; Saxty B.; Holder A. A.; Wilkinson A. J.; Tate E. W.; Leatherbarrow R. J. Discovery of Plasmodium Vivax N -Myristoyltransferase Inhibitors: Screening, Synthesis, and Structural Characterization of Their Binding Mode. J. Med. Chem. 2012, 55, 3578–3582. 10.1021/jm300040p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frearson J. A.; Brand S.; McElroy S. P.; Cleghorn L. A. T.; Smid O.; Stojanovski L.; Price H. P.; Guther M. L. S.; Torrie L. S.; Robinson D. A.; Hallyburton I.; Mpamhanga C. P.; Brannigan J. A.; Wilkinson A. J.; Hodgkinson M.; Hui R.; Qiu W.; Raimi O. G.; Van Aalten D. M. F.; Brenk R.; Gilbert I. H.; Read K. D.; Fairlamb A. H.; Ferguson M. A. J.; Smith D. F.; Wyatt P. G. N-Myristoyltransferase Inhibitors as New Leads to Treat Sleeping Sickness. Nature 2010, 464, 728–732. 10.1038/nature08893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinks D.; Smith V.; Thompson S.; Robinson D. A.; Luksch T.; Smith A.; Torrie L. S.; McElroy S.; Stojanovski L.; Norval S.; Collie I. T.; Hallyburton I.; Rao B.; Brand S.; Brenk R.; Frearson J. A.; Read K. D.; Wyatt P. G.; Gilbert I. H. Development of Small-Molecule Trypanosoma Brucei N-Myristoyltransferase Inhibitors: Discovery and Optimisation of a Novel Binding Mode. ChemMedChem 2015, 10, 1821–1836. 10.1002/cmdc.201500301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright M. H.; Paape D.; Price H. P.; Smith D. F.; Tate E. W. Global Profiling and Inhibition of Protein Lipidation in Vector and Host Stages of the Sleeping Sickness Parasite Trypanosoma Brucei. ACS Infect. Dis. 2016, 2, 427–441. 10.1021/acsinfecdis.6b00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousnier A.; Bell A. S.; Swieboda D. P.; Morales-Sanfrutos J.; Pérez-Dorado I.; Brannigan J. A.; Newman J.; Ritzefeld M.; Hutton J. A.; Guedán A.; Asfor A. S.; Robinson S. W.; Hopkins-Navratilova I.; Wilkinson A. J.; Johnston S. L.; Leatherbarrow R. J.; Tuthill T. J.; Solari R.; Tate E. W. Fragment-Derived Inhibitors of Human N-Myristoyltransferase Block Capsid Assembly and Replication of the Common Cold Virus. Nat. Chem. 2018, 10, 599–606. 10.1038/s41557-018-0039-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbic Ramljak I.; Stanger J.; Real-Hohn A.; Dreier D.; Wimmer L.; Redlberger-Fritz M.; Fischl W.; Klingel K.; Mihovilovic M. D.; Blaas D.; Kowalski H. Cellular N-Myristoyltransferases Play a Crucial Picornavirus Genus-Specific Role in Viral Assembly, Virion Maturation, and Infectivity. PLoS Pathog. 2018, 14, e1007203 10.1371/journal.ppat.1007203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaton K. E.; Smith C. D. N-Myristoyltransferase Isozymes Exhibit Differential Specificity for Human Immunodeficiency Virus Type 1 Gag and Nef. J. Gen. Virol. 2008, 89, 288–296. 10.1099/vir.0.83412-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thinon E.; Morales-Sanfrutos J.; Mann D. J.; Tate E. W. N-Myristoyltransferase Inhibition Induces ER-Stress, Cell Cycle Arrest, and Apoptosis in Cancer Cells. ACS Chem. Biol. 2016, 11, 2165–2176. 10.1021/acschembio.6b00371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll S.; Freitas F. P.; Shah R.; Aldrovandi M.; da Silva M. C.; Ingold I.; Grocin A. G.; Xavier da Silva T. N.; Panzilius E.; Scheel C. H.; Mourão A.; Buday K.; Sato M.; Wanninger J.; Vignane T.; Mohana V.; Rehberg M.; Flatley A.; Schepers A.; Kurz A.; White D.; Sauer M.; Sattler M.; Tate E. W.; Schmitz W.; Schulze A.; O’Donnell V.; Proneth B.; Popowicz G. M.; Pratt D. A.; Angeli J. P. F.; Conrad M. FSP1 Is a Glutathione-Independent Ferroptosis Suppressor. Nature 2019, 575, 693–698. 10.1038/s41586-019-1707-0. [DOI] [PubMed] [Google Scholar]
- Sahin A.; Espiau B.; Tetaud E.; Cuvillier A.; Lartigue L.; Ambit A.; Robinson D. R.; Merlin G. The Leishmania ARL-1 and Golgi Traffic. PLoS One 2008, 3, e1620 10.1371/journal.pone.0001620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maclean L. M.; O’Toole P. J.; Stark M.; Marrison J.; Seelenmeyer C.; Nickel W.; Smith D. F. Trafficking and Release of Leishmania Metacyclic HASPB on Macrophage Invasion. Cell. Microbiol. 2012, 14, 740–761. 10.1111/j.1462-5822.2012.01756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills E.; Price H. P.; Johner A.; Emerson J. E.; Smith D. F. Kinetoplastid PPEF Phosphatases: Dual Acylated Proteins Expressed in the Endomembrane System of Leishmania. Mol. Biochem. Parasitol. 2007, 152, 22–34. 10.1016/j.molbiopara.2006.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright M. H.; Paape D.; Storck E. M.; Serwa R. A.; Smith D. F.; Tate E. W. Global Analysis of Protein N-Myristoylation and Exploration of N-Myristoyltransferase as a Drug Target in the Neglected Human Pathogen Leishmania Donovani. Chem. Biol. 2015, 22, 342–354. 10.1016/j.chembiol.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritzefeld M.; Wright M. H.; Tate E. W. New Developments in Probing and Targeting Protein Acylation in Malaria, Leishmaniasis and African Sleeping Sickness. Parasitology 2018, 145, 157–174. 10.1017/S0031182017000282. [DOI] [PubMed] [Google Scholar]
- Paape D.; Prendergast C. T.; Price H. P.; Doehl J. S. P.; Smith D. F. Genetic Validation of Leishmania Genes Essential for Amastigote Survival in Vivo Using N-Myristoyltransferase as a Model. Parasites Vectors 2020, 13, 132. 10.1186/s13071-020-3999-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corpas-Lopez V.; Moniz S.; Thomas M.; Wall R. J.; Torrie L. S.; Zander-Dinse D.; Tinti M.; Brand S.; Stojanovski L.; Manthri S.; Hallyburton I.; Zuccotto F.; Wyatt P. G.; De Rycker M.; Horn D.; Ferguson M. A. J.; Clos J.; Read K. D.; Fairlamb A. H.; Gilbert I. H.; Wyllie S. Pharmacological Validation of N-Myristoyltransferase as a Drug Target in Leishmania Donovani. ACS Infect. Dis. 2019, 5, 111–122. 10.1021/acsinfecdis.8b00226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olaleye T. O.; Brannigan J. A.; Roberts S. M.; Leatherbarrow R. J.; Wilkinson A. J.; Tate E. W. Peptidomimetic Inhibitors of N-Myristoyltransferase from Human Malaria and Leishmaniasis Parasites. Org. Biomol. Chem. 2014, 12, 8132–8137. 10.1039/C4OB01669F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rackham M. D.; Yu Z.; Brannigan J. A.; Heal W. P.; Paape D.; Barker K. V.; Wilkinson A. J.; Smith D. F.; Leatherbarrow R. J.; Tate E. W. Discovery of High Affinity Inhibitors of Leishmania Donovani N-Myristoyltransferase. Medchemcomm 2015, 6, 1761–1766. 10.1039/C5MD00241A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell A. S.; Bradley J.; Everett J. R.; Loesel J.; McLoughlin D.; Mills J.; Peakman M. C.; Sharp R. E.; Williams C.; Zhu H. Plate-Based Diversity Subset Screening Generation 2: An Improved Paradigm for High-Throughput Screening of Large Compound Files. Mol. Diversity 2016, 20, 789–803. 10.1007/s11030-016-9692-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell A. S.; Mills J. E.; Williams G. P.; Brannigan J. A.; Wilkinson A. J.; Parkinson T.; Leatherbarrow R. J.; Tate E. W.; Holder A. A.; Smith D. F. Selective Inhibitors of Protozoan Protein N-Myristoyltransferases as Starting Points for Tropical Disease Medicinal Chemistry Programs. PLoS Neglected Trop. Dis. 2012, 6, e1625 10.1371/journal.pntd.0001625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannigan J. A.; Roberts S. M.; Bell A. S.; Hutton J. A.; Hodgkinson M. R.; Tate E. W.; Leatherbarrow R. J.; Smith D. F.; Wilkinson A. J. Diverse Modes of Binding in Structures of Leishmania Major N-Myristoyltransferase with Selective Inhibitors. IUCrJ 2014, 1, 250–260. 10.1107/S2052252514013001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton J. A.; Goncalves V.; Brannigan J. A.; Paape D.; Wright M. H.; Waugh T. M.; Roberts S. M.; Bell A. S.; Wilkinson A. J.; Smith D. F.; Leatherbarrow R. J.; Tate E. W. Structure-Based Design of Potent and Selective Leishmania N-Myristoyltransferase Inhibitors. J. Med. Chem. 2014, 57, 8664–8670. 10.1021/jm5011397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paape D.; Bell A. S.; Heal W. P.; Hutton J. A.; Leatherbarrow R. J.; Tate E. W.; Smith D. F. Using a Non-Image-Based Medium-Throughput Assay for Screening Compounds Targeting N-Myristoylation in Intracellular Leishmania Amastigotes. PLoS Neglected Trop. Dis. 2014, 8, e3363 10.1371/journal.pntd.0003363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armarego W. L. F.Quinazolines. In Advances in Heterocyclic Chemistry; Katritzky A. R.; Boulton A. J., Eds.; Academic Press, 1979; Vol. 24, pp 1–62. [Google Scholar]
- Smits R. A.; Adami M.; Istyastono E. P.; Zuiderveld O. P.; Van Dam C. M. E.; De Kanter F. J. J.; Jongejan A.; Coruzzi G.; Leurs R.; De Esch I. J. P. Synthesis and QSAR of Quinazoline Sulfonamides As Highly Potent Human Histamine H4 Receptor Inverse Agonists. J. Med. Chem. 2010, 53, 2390–2400. 10.1021/jm901379s. [DOI] [PubMed] [Google Scholar]
- Yoshida K.; Taguchi M. Reaction of N-Substituted Cyclic Amines with 2,4-Dichloroquinazoline, 2,4- Dichloropyrimidine, and its 5-Methyl Derivative. J. Chem. Soc., Perkin Trans. 1 1992, 1, 919–922. 10.1039/p19920000919. [DOI] [Google Scholar]
- Olofson R. A.; Martz J. T.; Senet J. P.; Piteau M.; Malfroot T. A New Reagent for the Selective, High-Yield n-Dealkylation of Tertiary Amines: Improved Syntheses of Naltrexone and Nalbuphine. J. Org. Chem. 1984, 49, 2081–2082. 10.1021/jo00185a072. [DOI] [Google Scholar]
- Goncalves V.; Brannigan J. A.; Thinon E.; Olaleye T. O.; Serwa R.; Lanzarone S.; Wilkinson A. J.; Tate E. W.; Leatherbarrow R. J. A Fluorescence-Based Assay for N-Myristoyltransferase Activity. Anal. Biochem. 2012, 421, 342–344. 10.1016/j.ab.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziao N.; Laurence C.; Le Questel J. Y. Amino Nitrogen and Carbonyl Oxygen in Competitive Situations: Which Is the Best Hydrogen-Bond Acceptor Site?. CrystEngComm 2002, 4, 326–335. 10.1039/B202248F. [DOI] [Google Scholar]
- Brand S.; Cleghorn L. A. T.; McElroy S. P.; Robinson D. A.; Smith V. C.; Hallyburton I.; Harrison J. R.; Norcross N. R.; Spinks D.; Bayliss T.; Norval S.; Stojanovski L.; Torrie L. S.; Frearson J. A.; Brenk R.; Fairlamb A. H.; Ferguson M. A. J.; Read K. D.; Wyatt P. G.; Gilbert I. H. Discovery of a Novel Class of Orally Active Trypanocidal N-Myristoyltransferase Inhibitors. J. Med. Chem. 2012, 55, 140–152. 10.1021/jm201091t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broncel M.; Serwa R. A.; Ciepla P.; Krause E.; Dallman M. J.; Magee A. I.; Tate E. W. Multifunctional Reagents for Quantitative Proteome-Wide Analysis of Protein Modification in Human Cells and Dynamic Profiling of Protein Lipidation during Vertebrate Development. Angew. Chem., Int. Ed. 2015, 54, 5948–5951. 10.1002/anie.201500342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heal W. P.; Wright M. H.; Thinon E.; Tate E. W. Multifunctional Protein Labeling via Enzymatic N-Terminal Tagging and Elaboration by Click Chemistry. Nat. Protoc. 2012, 7, 105–117. 10.1038/nprot.2011.425. [DOI] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66 (2), 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. Scaling and Assessment of Data Quality. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2006, 62, 72–82. 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- Winter G. Xia2: An Expert System for Macromolecular Crystallography Data Reduction. J. Appl. Crystallogr. 2010, 43, 186–190. 10.1107/S0021889809045701. [DOI] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1997, 53, 240–255. 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and Development of COOT. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.