

Abstract
Covalently linked photosensitizer–polyoxometalate (PS‐POM) dyads are promising molecular systems for light‐induced energy conversion processes, such as “solar” hydrogen generation. To date, very little is known of their fundamental photophysical properties which affect the catalytic reactivity and stability of the systems. PS‐POM dyads often feature short‐lived photoinduced charge‐separated states, and the lifetimes of these states are considered crucial for the function of PS‐POM dyads in molecular photocatalysis. Hence, strategies have been developed to extend the lifetimes of the photoinduced charge‐separated states, either by tuning the PS photophysics or by tuning the POM redox properties. Recently, some of us reported PS‐POM dyads based on cyclometalated IrIII complexes covalently linked to Anderson‐type polyoxometalate. Distinct hydrogen evolution reactivity (HER) of the dyads was observed, which was tuned by varying the central metal ion M of the POMM (M=Mn3+, Co3+, Fe3+). In this manuscript, the photoinduced electron‐transfer processes in the three Ir‐POMM dyads are investigated to rationalize the underlying reasons for the differences in HER activity observed. We report that upon excitation of the IrIII complex, ultrafast (sub‐ps) charge separation occurs, leading to different amounts of the charge‐separated states (Ir.+‐POMM .−) generated in the different dyads. However, in all dyads studied, the resulting Ir.+‐POMM .− species are short‐lived (sub‐ns) when compared to reference electron acceptors (e.g. porphyrins or fullerenes) reported in the literature. The reductive quenching of Ir.+‐POMM .− by a sacrificial donor, triethyl amine (1 m), to generate the intermediate Ir‐POMM .− is estimated to be very efficient (70–80 %) for all dyads studied. Based on this analyses, we conclude that the yield instead of the lifetime of the Ir.+‐POMM .− charge‐separated state determines the catalytic capacity of the dyads investigated. This new feature in the PS‐POM photophysics could lead to new design criteria for the development of novel PS‐POM dyads.
Keywords: catalytic mechanisms, hydrogen evolution, photochemistry, photophysics, polyoxometalates
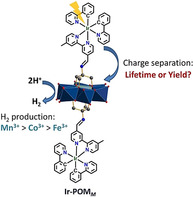
A mechanistic study on the distinct hydrogen evolution reactivity (HER) of three cyclometalated IrIII complex–polyoxometalate (Ir‐POMM) dyads. Their HER activity was tuned by modifying the central metal ion M of the POMM. The lifetime or the yield of Ir.+‐POMM .− determined the different HER activity of Ir‐POMM (see figure).
Introduction
Converting sunlight into chemical fuels is a key goal for green and sustainable energy technologies.1 The generation of “solar hydrogen” requires the combination of several fundamental steps such as light‐harvesting, charge‐separation, charge‐accumulation and catalysis (i.e. reduction of protons to H2).2 One strategy in artificial photosynthesis is to integrate all these fundamental processes into one supramolecular assembly, which at least contains a photoactive donor unit combined with an acceptor as catalytic unit for reduction reactions, respectively.2 As a result of this molecular design, photoexcitation of the supramolecular assembly leads to the formation of a charge‐separated state (CSS), in which the photoactive donor is oxidized, while the catalytic center is reduced. Ideally, the CSS is long lived and can thus react in a collision‐induced reaction with a sacrificial electron donor or other reactants to give the desired product.2 To‐date a number of typically organic electron acceptors such as porphyrin or fullerene have been combined in this fashion with photoactive transition metal complex photosensitizers as donors.2c, 3 More recently, molecular metal oxides, or polyoxometalates (POMs), have emerged as inorganic electron acceptors in the field of artificial photosynthesis.4 The beneficial properties of POMs as electron acceptors arise from their strong electron acceptor properties, multi‐electron redox capabilities and catalytic activity in their reduced states.4 The functionalization of POMs with visible light absorbing PS is critical, as POMs themselves typically only absorb light in the UV region. Thus, the design of covalently linked PS‐POM dyads has recently emerged as a new design concept in POMs energy conversion.5
However, due to the synthetic challenges involved,5j, 5k currently there are only a few pioneering studies available. In these initial studies, ReI, RuII and IrIII complexes, porphyrins as well as BODIPY dyes have been used as photosensitizers, leading to possible applications in light‐driven hydrogen evolution and photoelectrochemistry photovoltaics.5 In these studies, the main focus was on Keggin‐, Dawson‐ and Anderson‐derivatives as their covalent functionalization is well‐established.5 However, in the reported PS‐POM dyads either no charge‐separated states, that is, PS.+‐POM.−, or only relatively short‐lived PS.+‐POM.− were detected upon excitation of the photosensitizers.4, 5a, 5b, 5d, 5e The lifetimes of PS.+‐POM.− range from a few ps to the longest value reported up to now, which is about 500 ns.5c, 5g, 5h However, the lifetime of the photoinduced primary CSS, that is, PS.+‐POM.−, is generally considered key to the function of PS‐POM dyads in an artificial photosynthetic Scheme, as a long‐lived CSS allows for reductive quenching of the oxidized PS by a sacrificial donor and subsequent light‐driven or—depending on the specific reaction mechanism—dark processes. As POM‐based dyads do not excel in terms of long lifetime of the CSS, this might become one of the limiting factors for developing POM‐based artificial systems for solar fuels generation via multi‐electron processes. Compared to the more frequently utilized Keggin‐ and Dawson‐POMs, photoinduced dynamics in Anderson‐POMs based assemblies are scarcely investigated:5i Hasenknopf and co‐workers reported the linkage of ZnII porphyrins to Dawson‐ and Anderson‐POM and observed that photoinduced electron transfer takes place from the excited ZnII porphyrin to the Dawson‐ but not to the Anderson‐POM.5i
Some of us recently reported three PS‐POM dyads based on Anderson‐POM as electron acceptors, which were covalently linked to two cyclometalated IrIII complexes as photosensitizer and electron donor ((nBu4N)[MMo6O18{(OCH2)3CNCH(IrC33H26N4)}2] molecular structure see the inset in Figure 1).6 In these dyads, the central metal ion M of the POMM was varied from M=Mn3+ to Co3+ to Fe3+.6b It was found that all Ir‐POMM dyads displayed hydrogen evolution reactivity (HER) in the presence of triethyl amine as a sacrificial electron donor and acetic acid as the proton source.6b The HER activity decreased from Ir‐POM Mn (TON=80, measured over seven days) to Ir‐POM Co (TON=34) to Ir‐POM Fe (TON=20).6b Electrochemical data and DFT calculations indicated that the central M alters the redox properties of POMM.6b For M=Mn3+ the LUMO energy was the lowest.6b The modulation of the LUMO energy (3.95, 4.58 and 4.74 eV for POMMn, POMCo and POMFe, respectively)6b in turn leads to a variation of the driving force for electron transfer, and hence, distinct rate constants for charge separation (which would significantly affect the yield of Ir.+‐POMM .−) and charge recombination (the stability of Ir.+‐POMM .−) are expected.
Figure 1.
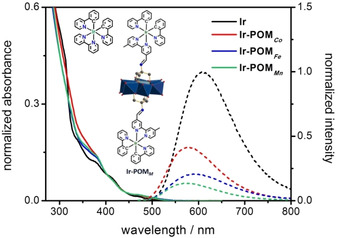
Normalized UV/Vis absorption spectra (isoabsorbing at 400 nm, OD400 nm=0.08) and normalized emission spectra (dashed line, divided by the emission maximum of Ir, that is, [Ir(bpy)(ppy)2]PF6) in aerated DMF. The extinction coefficients for all compounds were reported in ref. 6b. The metal oxo core of the Anderson‐anion is {MMo6O24} (M=Co3+, Fe3+, Mn3+). Color code: MoO6 octahedra, blue; metal cation M, cyan. POM and IrIII complex are covalently connected via a ‐C=N‐ (imine) bond.
In this work, we explore the charge separation and charge recombination underlying the different HER activity of the aforementioned Ir‐POMM dyads. In order to do so, we performed femtosecond and nanosecond time‐resolved spectroscopy and combine them with steady‐state and time‐resolved emission spectroscopy. The data presented indicates that the yield of the initial CSS is an important factor influencing the overall catalytic activity of Ir‐POMM. Thus, not only the lifetime of the CSS, Ir.+‐POMM .−, is to be considered as a photophysical metric to relate to the activity of such photocatalytically active dyads. Furthermore, on a more detailed notice, this work provides to the best of our knowledge the first example of a spectral characterization of a CSS in Anderson‐POM based PS‐POM dyads.
Results and Discussion
Steady‐state emission spectra of Ir‐POMM dyads are depicted in Figure 1. The emission spectra of each compound were recorded at an optical density of 0.08 upon excitation at 400 nm. Compared to Ir, the emission intensity from the IrIII unit is significantly decreased in Ir‐POM Mn and Ir‐POM Fe (by 86 and 79 %, respectively). For Ir‐POM Co the emission intensity is only reduced by 58 %. The partially quenched emission indicates that combining the IrIII complex with the POMM provides additional non‐radiative decay channels for the excited Ir‐POMM dyads.
Photoinduced dynamics
The non‐radiative decay pathways within the Ir‐POMM dyads were studied by fs transient absorption (TA) spectroscopy. Figure 2 shows the fs TA spectra of Ir, Ir‐POM Co and Ir‐POM Mn obtained in aerated DMF upon excitation at 400 nm. The data recorded for Ir‐POM Fe are depicted in the ESI (Figure S1) as all dyads show very similar spectral features but different kinetics. The fs TA spectra of Ir upon excitation of the singlet metal‐to‐ligand charge transfer (MLCT) states resemble the spectral‐temporal evolution as previously reported in literature:7 A broad excited‐state absorption (ESA) spanning from 350 to 750 nm corresponds to the absorption of 3MLCT states in the photosensitizer and is observed immediately upon photoexcitation.7b, 7c Subsequently, two bands at 375 and at around 500 nm increase on a sub‐10 ps timescale (Figure 2 a, b). Based on spectro‐electrochemical data, the ESA band at 375 nm (the orange dashed line in Figure 2 c, for more information see Figure S2 b) is assigned to the absorption of the reduced bpy ligand.7b, 7c, 8 Its increase at early times is attributed to interligand electron transfer (ILET) from an upper‐lying 3MLCTppy state to the lower‐lying 3MLCTbpy state, a processes which has been established in literature.7a, 7b
Figure 2.
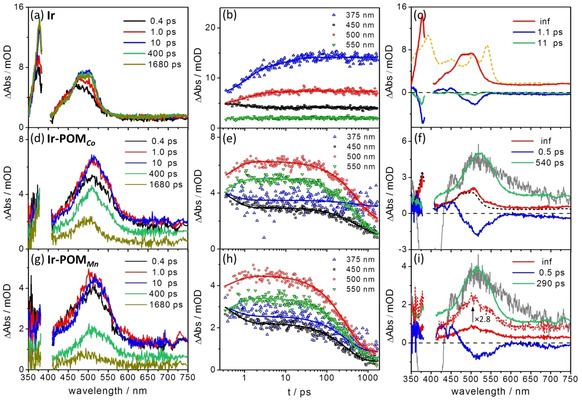
fs transient absorption spectra at selected delay times (left), selected kinetic traces with corresponding fit (middle) and decay‐associated spectra (DAS) resulted from the global fit of the fs TA data (right) obtained upon excitation at 400 nm in aerated DMF for (a–c) Ir, (d–f) Ir‐POM Co and (g–i) Ir‐POM Mn. The orange dashed line in (c) is the spectroelectrochemical UV/Vis absorption difference spectrum of the reduced bpy ligand. The black dashed line in (f) is the infinite component in Ir. The grey line shows the simulated absorption spectrum of Ir.+‐POMCo .− and Ir.+‐POMMn .− according to the spectroelectrochemical results (Figures S2 and S3). These spectra were arbitrarily scaled to integrate into the Figure. In (i) an enlarged amplitude (by a factor of 2.8) of the infinite component is shown for comparison.
Ir‐POM Co and Ir‐POM Mn initially (i.e. at 0.4 ps after photoexcitation) display similar transient absorption features as Ir, that is, a rather broad but red‐shifted ESA band in the visible region (Figure 2 d, g). However, between 0.4 and 10 ps the dyads reveal spectral changes different to Ir: For one, the evolution of the ESA band in the visible region is faster in the dyads than in Ir. At 1.0 ps the band sharpens compared to Ir, that is, the FWHM (full width at half maximum) of the spectrum is reduced by 5, 16 and 11 % compared to the data at 0.4 ps for Ir, Ir‐POM Co and Ir‐POM Mn, respectively. Meanwhile the spectral intensity already reaches a maximum (Figure 2 d, g). Additionally, the ESA band in the UV region decreases instead of increasing as observed for Ir (Figure 2 a, d and g). This is seen directly from the kinetic trace at 375 nm (blue lines in Figures 2 b, e, h), which reflects the formation of a charge‐separated state, that is, Ir.+‐POMM .−. The one‐electron‐reduced POMM (i.e. POMM .−) has a negative absorption below 400 nm (Figure S3 a, c, e), which apparently compensates the positive absorption signal of bpy.− (Figure 2 c, orange dashed line). At delay times longer than 100 ps, a pronounced decay of the overall TA signal is observed (Figure 2 e, h) for both Ir‐POM Co and Ir‐POM Mn. However, within the experimentally accessible delay time range of 1.8 ns the decay is not complete, that is, a long‐lived component is apparent in the fs data.
The quantitative interpretation of the fs TA data is based on a global fit.9 For all compounds, two decay components and an offset are sufficient to describe the experimental data. The decay‐associated spectra (DAS, see ESI for a description on the DAS) and the corresponding characteristic time constants are given in Figure 2 c, f, i. The kinetic components obtained for Ir are in agreement with previous work by Lochbrunner on the same complex:7b, 7c Following Lochbrunner's work, we assign the component associated with τ 1=1.1 ps (Figure 2 c) to vibrational relaxation within the 3MLCT manifolds involving both ppy and bpy ligands. This is followed by ILET from a 3MLCTppy to a 3MLCTbpy state. This process is characterized by τ 2=11 ps. The long‐lived species represents the long‐lived thermalized 3MLCTbpy state, the decay of which to the electronic ground state is beyond the experimentally accessible delay‐time range.
Considering the DAS‐analysis of the fs TA data of the dyads, the fastest kinetic component is associated with a characteristic decay time τ 1=0.5 ps. The corresponding spectral changes resemble the spectral shape of τ 1 in Ir, nonetheless comprising a slight red‐shift compared to the spectra of the photosensitizer only (Figure 2 f, i and Figure S1 c; Figure S5 a, c, e). Furthermore, DAS(τ 1) reveals a broader negative feature compared to the DAS(τ 1) in Ir. Specifically, in the dyads an increase of the transient absorption at 550 nm is associated with the τ 1‐component (Figure 2 f, i), which is not present in Ir. This ESA increase is due to the absorption of the reduced form of POMM (i.e. POMM .−, Figure S4) combined with possible contributions from the oxidized IrIII center (i.e. Ir.+, Figure S2 a. Note that the Ir photosensitizer in the dyads has additional substituents on the bpy ligand compared to reference Ir which might cause slight spectral difference to the MLCT and charge‐separated states). This indicates that in Ir‐POMM electron transfer from the photo‐excited IrIII complex to the POMM occurs rapid and in concert with vibrational relaxation of 3MLCT states. The fast formation of Ir.+‐POMM .−, which indicates effective electronic coupling between the initially excited MLCT state of the photosensitizer and the POMM, is corroborated by the difference spectrum of the DAS(τ 1) in Ir and Ir‐POMM (see Figure S5). Notably, the intensities of the TA signal at 550 nm in DAS (τ1), which accounts for the charge‐separation process, vary within the set of dyads (see Figure 3). Namely, ΔAbs at 550 nm in Ir‐POM Mn is roughly 2 times as high as that in Ir‐POM Co (Figure 3). This indicates different yields of Ir.+‐POMM .− being generated in the dyads, that is, the yield of Ir.+‐POMMn .− is (two times) higher than that of Ir.+‐POMCo .−.
Figure 3.
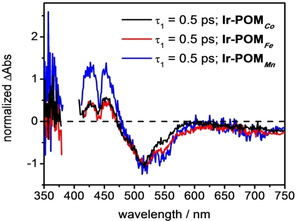
Comparison of the normalized (at 517 nm) decay‐associated spectrum (DAS, associated with τ1) in Ir‐POMM.
The 11‐ps‐component related to ILET in Ir is absent in the dyads. Instead, a hundreds of ps component is apparent, that is, τ 2=540 and 290 ps for Ir‐POM Co and Ir‐POM Mn, respectively. This component reflects the decay of the charge‐separated state Ir.+‐POMM .−, whose absorption has been simulated based on UV/Vis spectroelectrochemical data (Figure 2 f, i, S2 and S3). In addition to τ 1 and τ 2, all dyads reveal a long‐lived state, whose decay extends beyond the experimentally accessible delay‐time window (∼1.8 ns). The corresponding infinite component observed for Ir‐POM Co shows nearly identical features to that in Ir (Figure 2 f), that is, a strong band below 400 nm and a moderate band (asymmetric and flat) in the visible region. This may indicate a decay of the 3MLCTbpy state in Ir‐POM Co. In contrast, the infinite component in Ir‐POM Mn (Figure 2 i) and Ir‐POM Fe (Figure S1 d) displays a rather distinct absorption at 550 nm. This points to a different long‐lived state in Ir‐POM Mn and Ir‐POM Fe compared to Ir‐POM Co.
The nature of the long‐lived state was assessed by ns TA spectroscopy (Figure 4). The ns TA spectra match the features of the infinite component in fs TA data. For Ir‐POM Co, similar to Ir (Figure S6), the ns TA signal decays mono‐exponentially with a characteristic time constant of 120 ns (Figure 4 c, d). For Ir‐POM Mn and Ir‐POM Fe, the shoulder at 550 nm disappears after 30 ns as revealed by the ns data in Figures 4 b and S10 a, while the overall decay of the differential absorption is slower. A global fit of the ns TA data yields a comparably short‐lived component, τ ns1=30 ns, and a relatively long‐lived one, τ ns2=120 ns (Figure S10 b and S12 b). The 120 ns‐component in Ir‐POMM resembles the key spectral features of the 100 ns‐component in Ir (Figure 4 c and S14). Furthermore, the changes of its spectra and lifetime upon changing the solvent polarity (Figures S16–S20) correlate with the corresponding changes observed for Ir. Thus, we assign the 120 ns‐component to the decay of the 3MLCTbpy state. The significant increase of τ ns2 upon exclusion of oxygen from the solution (120 vs. 640 ns in aerated and deaerated DMF, see Figure 4 d, Figures S9, S11, S13 and S15) corroborate this assignment.10
Figure 4.
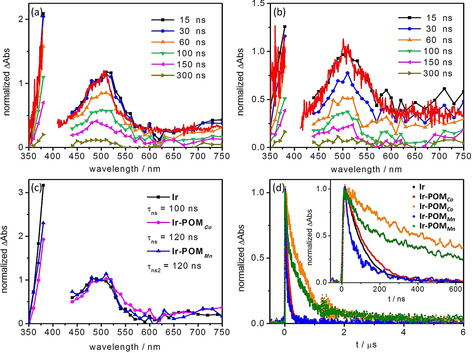
ns transient absorption spectra of (a) Ir‐POM Co and (b) Ir‐POM Mn collected upon excitation at 410 nm in aerated DMF. The spectrum of the infinite component in fs TA data (red line) is added for comparison. The fs TA spectrum and ns TA spectrum (at 15 ns) were normalized at 510 nm. (c) Normalized (at 500 nm) global fit results of the ns TA data of Ir, Ir‐POM Co and Ir‐POM Mn. Only the long‐lived species in Ir‐POM Mn is incorporated. (d) Normalized integrated kinetic traces (between 450 and 750 nm of the ns TA spectra) in aerated (black, red and blue for Ir, Ir‐POM Co and Ir‐POM Mn, respectively) and deaerated DMF (yellow and green for Ir‐POM Co and Ir‐POM Mn, respectively). Inset: The enlargement of the time region up to 640 ns.
Considering the 30 ns component, the ns TA results in DMF and DMSO indicate that it is a bright state whose lifetime is independent of solvent polarity (τ ns1=30 ns in DMF and τ ns1=40 ns in DMSO, Figures S10 d, S12 d, S18 d and S19 d). These properties exclude the possibility of a CSS being formed. On the contrary, τ ns1 doubles upon exclusion of oxygen from DMF (inset in Figure 4 d; Figure S11 d and S13 d). Furthermore, its energetic position appears to be drastically shifted when changing the solvent from DMF (emission at 550 nm) to DMSO (emission at 680 nm). Nevertheless, the nature of the 30 ns component is beyond the scope of this study because the different HER activity of Ir‐POMM is not impacted by the presence of this state: Both dyads, which reveal this component, Ir‐POM Mn and Ir‐POM Fe display rather different HER activity (TON=80 vs. 20, respectively).6b Ir‐POM Co, whose ns‐decay does not reveal the 30‐ns component, displays an intermediate TON of 34, which is 70 % higher than that of Ir‐POM Fe.6b
Relaxation model
Scheme 1 summarizes the photophysical picture of Ir‐POMM as it emerges from the time‐resolved spectroscopy. According to the fs TA results, photoinduced electron transfer in the dyads occurs concertedly with vibrational energy dissipation, indicating that electron transfer is most efficient from vibrationally hot 3MLCT states (Scheme 1). Considering electron transfer from the individual 3MLCT states intrinsic to the system, the driving force for electron transfer can be estimated by the Rehm–Weller equation ΔG CS = e(E D+/d − E A/A−) − E 00 − (e2 / 4πϵ0ϵRDA).11 As the 3MLCTppy state is energetically higher than the 3MLCTbpy state (i.e. a larger E 00),7 a more negative driving force will be induced. Hence, we cannot exclude the direct through‐space electron transfer from the hot 3MLCTppy to the POM (Scheme 1). The presence of this direct electron‐transfer pathway explains the absence of ILET in the dyads Ir‐POMM as compared to Ir, where ILET dominates the 11‐ps process. Besides, ultrafast electron transfer from the initially excited hot 1MLCT states (see resonance Raman data in Figure S21), which would have more favorable driving forces, might be operative as well. However, due to the temporal resolution (∼110 fs) of the fs TA setup such ultrafast process cannot be explored.
Scheme 1.
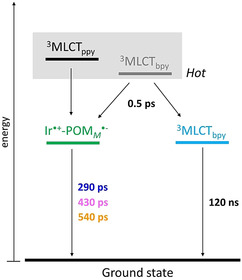
Simplified energy‐level diagrams of Ir‐POM Co (orange), Ir‐POM Fe (pink) and Ir‐POM Mn (blue). The color code indicates the corresponding time constants for each compound. The same time constants for dyads are shown in black. According to the electrochemical results, the energetic level of Ir.+‐POMM .− is estimated to be 2.25∼2.35 eV in DMF.6 The energetic level of the thermalized 3MLCTbpy state can be estimated from the steady‐state emission spectrum of [Ir(ppy)2(bpy)]+. At 77 K, the emission maximum is at 532 nm (2.33 eV) for [Ir(ppy)2(bpy)]+ (3MLCTbpy is the lowest state) in frozen 2‐MeTHF.12 Note: due to the temporal resolution (∼110 fs) of the fs TA setup and our data‐processing (a temporal window of 200 fs around time‐zero was excluded to avoid contributions of coherent artifacts13) any ultrafast processes taking place within 200 fs, for example, intersystem crossing or possible 1MLCT→POM electron transfer, cannot be probed. Hence, the photophysical model derived from the data presented in this manuscript focuses on the decay of the 3MLCT states.
τ 2 in the dyads is attributed to charge recombination yielding the decay of Ir.+‐POMM .−. The lifetime of Ir.+‐POMM .− depends on the central metal ion M, that is, Ir.+‐POMMn .− (290 ps)<Ir.+‐POMFe .− (430 ps)<Ir.+‐POMCo .− (540 ps). In hydrogen evolution experiment investigating the Ir‐POMM dyads studied here, the concentration of the sacrificial donor (1 m) was 10 000 higher than that of the dyads (0.1 mm).6b Under these conditions, the diffusion‐limited rate constant of the sacrificial donor for quenching is about 7.6×109 s−1 (calculated by multiplying the diffusion‐limited rate constant in DMF, 7.6×109 m −1 s−1, with the concentration of sacrificial donor, 1 m). This rate corresponds to a characteristic time constant of 126 ps.14, 15 This indicates that the characteristic lifetimes of the Ir.+‐POMM .− (τ 2=290−540 ps) provides a sufficiently large temporal window for the interaction with the sacrificial donor, yielding Ir‐POMM .− as an essential intermediate in the photocatalytic cycle. Based on the time constants for diffusion and charge recombination (τ 2), the quenching efficiency by the sacrificial donor is estimated to be 70 and 80 % in Ir‐POM Mn and Ir‐POM Co, respectively. However, the HER activity of Ir‐POM Mn (TON=80) was reported to be 2.4 times as high as that of Ir‐POM Co (TON=34).6b Therefore, we conclude that the HER activity of the dyads as observed under conventionally employed experimental conditions6b seems not governed by the lifetimes of the charge‐separated state Ir.+‐POMM .−. Instead, this study suggests that the yield of Ir.+‐POMM .− plays an important role in the catalytic capacity of Ir‐POMM. As discussed above, the yield of Ir.+‐POMMn .− is roughly twice the yield of Ir.+‐POMCo .−. This finding is consistent with the ratio of the HER activity of the respective dyads. Furthermore, from the perspective of photostability, more efficient hot 3MLCT→POM electron transfer results in a reduced population of the long‐lived thermalized 3MLCT state. Thereby, 3MLCT→POM electron transfer reduces the potential for destructive side‐reactions taking place from the high‐energy 3MLCT state, which might lead to decomposition of the Ir‐POMM dyad under catalytic conditions. Hence, an increased yield of the CSS might benefit the catalytic HER activity twofold by increasing the yield of a critical charge‐transfer intermediate of the catalytic cycle and by diminishing the potential for detrimental side reactions. Our work points out that for a good performance of the catalysts the photoinduced CSS should be sufficiently long‐lived for the subsequent reactions (i.e. to react with external reactants according to the specific reaction schemes). When the decay dynamics of the CSS itself are comparable to the rates of subsequent reactions, extending the lifetimes of the CSS would not improve the function of the catalysts. At this point, the yields of CSS have to be considered. In this respect, proper molecular design for fast charge separation (e.g. by inducing a larger driving force or a stronger electronic coupling), which can compete with the decay of the excited state itself (e.g. the cooling process in Scheme 1), could promote the yields of CSS.
Conclusions
The photoinduced electron transfer dynamics underlying the distinct catalytic capacity (Ir‐POM Mn>Ir‐POM Co>Ir‐POM Fe) of three covalently linked Ir‐POMM dyads were investigated spectroscopically. The central metal ion M of POMM, that is, [MMo6O24]n−, was changed from Mn3+ to Co3+ to Fe3+ to modify the redox properties of the POMM acceptors.6b As a consequence of this modification, different photophysical properties of the Ir‐POMM dyads can be expected. Irrespective of the central metal ion M, however, ultrafast charge separation was observed, which takes place concertedly with vibrational energy dissipation. The yields and the lifetimes of the charge‐separated state, that is, Ir.+‐POMM .−, vary with the nature of the POMM. The yields of Ir.+‐POMM .− decrease in the order Ir‐POM Mn>Ir‐POM Fe>Ir‐POM Co. The lifetimes of the CSS show the inverse, that is, Ir.+‐POMMn .− (290 ps)<Ir.+‐POMFe .− (430 ps)<Ir.+‐POMCo .− (540 ps). Considering the diffusion‐limited rate constant for intermolecular quenching process with the lifetimes of Ir.+‐POMM .−, the sub‐ns lifetime is sufficient to allow for efficient capture of the Ir.+‐POMM .− by collision with a sacrificial donor under catalytic conditions (70–80 %). We thus conclude that the yield of Ir.+‐POMM .− plays an important role in affecting the catalytic capacity of Ir‐POMM under the given kinetic conditions. Thus, considering the yield of the formation of the charge‐separated states presents and additional photophysical metric—next to the conventionally considered lifetime of charge separation—to target in order to device catalytically efficient PS‐POM dyads. To improve the yields of the photoinduced charge‐separated states in the Ir‐POMM system, it might be useful to increase either the driving force or the electronic coupling between the IrIII complex and the POMM to accelerate the charge‐separation process. For this purpose, decreasing the distance between PS and POMM, or using a different POM which is easier to be reduced would be promising choices.
Experimental Section
General
Steady‐state UV/Vis absorption spectra were recorded in a quartz cell with 1 mm path length (for fs transient absorption experiment, JASCO V‐670 spectrophotometer) and with 1 cm path length (for ns transient absorption experiment, Cary 5000 UV/Vis spectrometer, Varian, USA). For all time‐resolved experiments, the stability of samples was ensured by recording the steady‐state UV/Vis absorption spectra before and after every measurement. The steady‐state emission spectra were recorded in a quartz cell with 1 cm path length on a FLS980 spectrofluorimeter (Edinburgh).
Electrochemistry and spectroelectrochemistry
Cyclic voltammetry (CV) and spectroelectrochemistry (SEC) measurements were performed in a home‐built three‐electrode thin‐layer cell with a path length of 1 mm. The three‐electrode system consists of a glassy carbon working electrode, a platinum wire counter electrode and an Ag/AgCl reference electrode. CV and potential‐controlled monitoring were performed using a computer‐controlled potentiostat (VersaSTAT 3, Princeton Applied Research). All potentials given in the manuscript refer to the ferrocene/ferrocenium couple as internal standard. The corresponding UV/Vis spectra were recorded on a single‐beam spectrometer (Avantes, Avalight‐DH‐S‐BAL) at room temperature.
Time‐resolved spectroscopy
Femtosecond (fs) transient absorption spectra were collected by using a previously reported home‐built pump‐probe laser system which is based on an amplified Ti: Sapphire oscillator (Libra, Coherent Inc.).16 All compounds were excited by pump pulse centered at 400 nm (TOPASwhite, Lightconversion Ltd.) with a duration of 110 fs. The power of the pump beam was kept at 0.4 mW and the beam diameter of the pump was 145 μm at the sample position. This corresponds to 0.95×1020 photons m−2 per pulse. A white light supercontinuum generated by focusing a fraction of the fundamental in a rotating CaF2 plate is used to probe the samples in a wide spectral range (340 to 750 nm). The probe beam is delayed in time with respect to the pump beam by means of an optical delay line and the polarization between probe and pump is set at the magic angle (54.7°). Each solution (optical density ca. 0.2 at the excitation wavelength) was kept in a 1 mm quartz cuvette. Transient absorption data were displayed after chirp correction. The transient absorption data was analyzed by a global multi‐exponential fit after exclusion of a temporal window of 200 fs around time‐zero in order to avoid contributions of the coherent‐artifact region to the data analysis.13 Furthermore, a spectral band of 20 nm around the pump‐wavelength is omitted from the data analysis due to pump‐scatter in this spectral range.
Nanosecond (ns) transient absorption spectra16c were collected to study the long‐lived species in the fs transient absorption data. The pump pulses centered at 410 nm were produced by a Continuum OPO Plus which is pumped by an continuum surelite Nd:YAG laser system (pulse duration 5 ns, repetition rate 10 Hz). The probe light is provided by a 75 W xenon arc lamp. Spherical concave mirrors are used to focus the probe beam into the samples and then send the beam to the monochromator (Acton, Princeton Instruments) and detected by a photomultiplier tube (Hamamatsu R928). The signal is amplified and processed by a commercially available detection system (Pascher Instruments AB). For all measurements, the power of the pump beam was kept at 0.35 mJ. ns TA spectra were recorded by using a bandpass (325–385 nm) and a long pass filter (435 nm) to eliminate the pump scattering. Time‐resolved emission spectra were collected with the use of a long pass filter (435 nm) as well. Each sample was freshly prepared and the optical density (ca. 0.3) at the excitation wavelengths 410 nm was kept the same. All measurements were performed in 1 cm path length fluorescence cuvettes. Oxygen‐free solutions were obtained by at least five freeze‐pump‐thaw cycles.
Resonance Raman spectroscopy
Resonance Raman (RR) spectra were recorded through excitation by a 405 nm diode laser (TopMode‐405‐HP, Toptica, Germany) and detected by an IsoPlane 160 spectrometer (Princeton Instruments, USA) with an entrance slit width of 0.05 mm, a focal length of 750 mm, and grating 2400 grooves mm−1. The excitation energy was attenuated to around 8 mW. The Raman signals were recorded by a thermoelectrically cooled PIXIS eXcelon camera (Princeton Instruments, USA). The Raman spectra were initially baseline corrected and normalized with respect to a solvent band, that is, to the signal at 1404 cm−1 for DMF.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research is supported by the CataLight CRC/TRR 234 (project number 364549901, projects A1 and A4 and Z2) funded by the Deutsche Forschungsgemeinschaft (DFG). S.M. thanks the Deutscher Akademischer Austauschdienst DAAD for a Ph.D. fellowship.
Y. Luo, S. Maloul, S. Schönweiz, M. Wächtler, C. Streb, B. Dietzek, Chem. Eur. J. 2020, 26, 8045.
References
- 1.
- 1a. Nocera D. G., Acc. Chem. Res. 2017, 50, 616–619; [DOI] [PubMed] [Google Scholar]
- 1b. Dalle K. E., Warnan J., Leung J. J., Reuillard B., Karmel I. S., Reisner E., Chem. Rev. 2019, 119, 2752–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Fukuzumi S., Ohkubo K., Suenobu T., Acc. Chem. Res. 2014, 47, 1455–1464; [DOI] [PubMed] [Google Scholar]
- 2b. Gilbert M., Albinsson B., Chem. Soc. Rev. 2015, 44, 845–862; [DOI] [PubMed] [Google Scholar]
- 2c. Rudolf M., Kirner S. V., Guldi D. M., Chem. Soc. Rev. 2016, 45, 612–630; [DOI] [PubMed] [Google Scholar]
- 2d. La Porte N. T., Martinez J. F., Chaudhuri S., Hedström S., Batista V. S., Wasielewski M. R., Coord. Chem. Rev. 2018, 361, 98–119; [Google Scholar]
- 2e. Zhang B., Sun L., Chem. Soc. Rev. 2019, 48, 2216–2264; [DOI] [PubMed] [Google Scholar]
- 2f. Neumann S., Kerzig C., Wenger O. S., Chem. Sci. 2019, 10, 5624–5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Flamigni L., Collin J.-P., Sauvage J.-P., Acc. Chem. Res. 2008, 41, 857–871; [DOI] [PubMed] [Google Scholar]
- 3b. Wenger O. S., Coord. Chem. Rev. 2009, 253, 1439–1457; [Google Scholar]
- 3c. Wenger O. S., Coord. Chem. Rev. 2015, 282–283, 150–158; [Google Scholar]
- 3d. Barthelmes K., Winter A., Schubert U. S., Dalton Trans. 2016, 45, 14855–14882; [DOI] [PubMed] [Google Scholar]
- 3e. Luo Y., Barthelmes K., Wächtler M., Winter A., Schubert U. S., Dietzek B., Chem. Eur. J. 2017, 23, 4917–4922; [DOI] [PubMed] [Google Scholar]
- 3f. Luo Y., Barthelmes K., Wächtler M., Winter A., Schubert U. S., Dietzek B., J. Phys. Chem. C 2017, 121, 9220–9229. [Google Scholar]
- 4.
- 4a. Walsh J. J., Bond A. M., Forster R. J., Keyes T. E., Coord. Chem. Rev. 2016, 306, 217–234; [Google Scholar]
- 4b. Izzet G., Volatron F., Proust A., Chem. Rec. 2017, 17, 250–266; [DOI] [PubMed] [Google Scholar]
- 4c. Cameron J. M., Wales D. J., Newton G. N., Dalton Trans. 2018, 47, 5120–5136; [DOI] [PubMed] [Google Scholar]
- 4d. Chen L., Chen W.-L., Wang X.-L., Li Y.-G., Su Z.-M., Wang E.-B., Chem. Soc. Rev. 2019, 48, 260–284; [DOI] [PubMed] [Google Scholar]
- 4e. Kibler A. J., Newton G. N., Polyhedron 2018, 154, 1–20. [Google Scholar]
- 5.
- 5a. Elliott K. J., Harriman A., Le Pleux L., Pellegrin Y., Blart E., Mayer C. R., Odobel F., Phys. Chem. Chem. Phys. 2009, 11, 8767–8773; [DOI] [PubMed] [Google Scholar]
- 5b. Matt B., Coudret C., Viala C., Jouvenot D., Loiseau F., Izzet G., Proust A., Inorg. Chem. 2011, 50, 7761–7768; [DOI] [PubMed] [Google Scholar]
- 5c. Matt B., Xiang X., Kaledin A. L., Han N., Moussa J., Amouri H., Alves S., Hill C. L., Lian T., Musaev D. G., Izzeta G., Proust A., Chem. Sci. 2013, 4, 1737–1745; [Google Scholar]
- 5d. Black F. A., Jacquart A., Toupalas G., Alves S., Proust A., Clark I. P., Gibson E. A., Izzet G., Chem. Sci. 2018, 9, 5578–5584; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5e. Yokoyama A., Kojima T., Ohkubo K., Shiro M., Fukuzumi S., J. Phys. Chem. A 2011, 115, 986–997; [DOI] [PubMed] [Google Scholar]
- 5f. Zhao C., Huang Z., Rodríguez-Córdoba W., Kambara C. S., O'Halloran K. P., Hardcastle K. I., Musaev D. G., Lian T., Hill C. L., J. Am. Chem. Soc. 2011, 133, 20134–20137; [DOI] [PubMed] [Google Scholar]
- 5g. Luo Y., Wächtler M., Barthelmes K., Winter A., Schubert U. S., Dietzek B., Chem. Commun. 2018, 54, 2970–2973; [DOI] [PubMed] [Google Scholar]
- 5h. Luo Y., Wächtler M., Barthelmes K., Winter A., Schubert U. S., Dietzek B., Phys. Chem. Chem. Phys. 2018, 20, 11740–11748; [DOI] [PubMed] [Google Scholar]
- 5i. Allain C., Schaming D., Karakostas N., Erard M., Gisselbrecht J.-P., Sorgues S., Lampre I., Ruhlmann L., Hasenknopf B., Dalton Trans. 2013, 42, 2745–2754; [DOI] [PubMed] [Google Scholar]
- 5j. Proust A., Matt B., Villanneau R., Guillemot G., Gouzerh P., Izzet G., Chem. Soc. Rev. 2012, 41, 7605–7622; [DOI] [PubMed] [Google Scholar]
- 5k. Hampson E., Cameron J. M., Amin S., Kyo J., Watts J. A., Oshio H., Newton G. N., Angew. Chem. Int. Ed. 2019, 58, 18281–18285; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 18449–18453. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Schönweiz S., Rommel S. A., Kübel J., Micheel M., Dietzek B., Rau S., Streb C., Chem. Eur. J. 2016, 22, 12002–12005; [DOI] [PubMed] [Google Scholar]
- 6b. Schönweiz S., Heiland M., Anjass M., Jacob T., Rau S., Streb C., Chem. Eur. J. 2017, 23, 15370–15376. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Pomarico E., Silatani M., Messina F., Braem O., Cannizzo A., Barranoff E., Klein J. H., Lambert C., Chergui M., J. Phys. Chem. C 2016, 120, 16459–16469; [Google Scholar]
- 7b. Tschierlei S., Neubauer A., Rockstroh N., Karnahl M., Schwarzbach P., Junge H., Bellerd M., Lochbrunner S., Phys. Chem. Chem. Phys. 2016, 18, 10682–10687; [DOI] [PubMed] [Google Scholar]
- 7c. Bevernaegie R., Marcélis L., Moreno-Betancourt A., Laramée-Milette B., Hanan G. S., Loiseau F., Sliwa M., Elias B., Phys. Chem. Chem. Phys. 2018, 20, 27256–27260. [DOI] [PubMed] [Google Scholar]
- 8. Bokarev S. I., Hollmann D., Pazidis A., Neubauer A., Radnik J., Kühn O., Lochbrunner S., Junge H., Beller M., Brückner A., Phys. Chem. Chem. Phys. 2014, 16, 4789–4796. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Siebert R., Winter A., Schubert U. S., Dietzek B., Popp J., Phys. Chem. Chem. Phys. 2011, 13, 1606–1617; [DOI] [PubMed] [Google Scholar]
- 9b. Reichardt C., Sainuddin T., Wächtler M., Monro S., Kupfer S., Guthmuller J., Gräfe S., McFarland S., Dietzek B., J. Phys. Chem. A 2016, 120, 6379–6388. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Kawaoka K., Khan A. U., Kearns D. R., J. Chem. Phys. 1967, 46, 1842–1853; [Google Scholar]
- 10b. Abdel-Shafi A. A., Worrall D. R., Ershov A. Y., Dalton Trans. 2004, 30–36. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Rehm D., Weller A., Isr. J. Chem. 1970, 8, 259–271; [Google Scholar]
- 11b. Göransson E., Boixel J., Fortage J., Jacquemin D., Becker H.-C., Blart E., Hammarström L., Odobel F., Inorg. Chem. 2012, 51, 11500–11512. [DOI] [PubMed] [Google Scholar]
- 12. Ladouceur S., Fortin D., Zysman-Colman E., Inorg. Chem. 2010, 49, 5625–5641. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Kovalenko S. A., Dobryakov A. L., Ruthmann J., Ernsting N. P., Phys. Rev. A 1999, 59, 2369–2384; [Google Scholar]
- 13b. Dietzek B., Pascher T., Sundström V., Yartsev A., Laser Phys. Lett. 2007, 4, 38–43. [Google Scholar]
- 14.Note: the calculated time constant for intermolecular quenching only considered the rate of encounter between the photo-excited dyad and the sacrificial donor. In reality this process would kinetically compete with backward electron transfer within the solvent cage to reform the original ground-state species. These processes together with the escape of the redox species out of the cage into bulk solution determine the yield of the Ir-POMM .− intermediate. But we do not expect that the intermolecular kinetics would depend drastically on the nature of the reduced state of Ir-POMM.
- 15.
- 15a. Georgopoulos M., Hoffman M. Z., J. Phys. Chem. 1991, 95, 7717–7721; [Google Scholar]
- 15b. Hötzer K. A., Klingert A., Klumpp T., Krissinel E., Bürssner D., Steiner U. E., J. Phys. Chem. A 2002, 106, 2207–2217. [Google Scholar]
- 16.
- 16a. Karnahl M., Kuhnt C., Ma F., Yartsev A., Schmitt M., Dietzek B., Rau S., Popp J., ChemPhysChem 2011, 12, 2101–2109; [DOI] [PubMed] [Google Scholar]
- 16b. Kübel J., Schroot R., Wächtler M., Schubert U. S., Dietzek B., Jäger M., J. Phys. Chem. C 2015, 119, 4742–4751; [Google Scholar]
- 16c. Barthelmes K., Kübel J., Winter A., Wächtler M., Friebe C., Dietzek B., Schubert U. S., Inorg. Chem. 2015, 54, 3159–3171. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary