

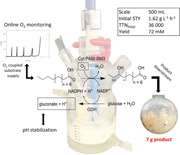
Abstract
Selective oxy‐functionalization of nonactivated C‐H bonds is a long‐standing “dream reaction” of organic synthesis for which chemical methodology is not well developed. Mono‐oxygenase enzymes are promising catalysts for such oxy‐functionalization to establish. Limitation on their applicability arises from low reaction output. Here, we showed an integrated approach of process engineering to the intensification of the cytochrome P450 BM3‐catalyzed hydroxylation of dodecanoic acid (C12:0). Using P450 BM3 together with glucose dehydrogenase for regeneration of nicotinamide adenine dinucleotide phosphate (NADPH), we compared soluble and co‐immobilized enzymes in O2‐gassed and pH‐controlled conversions at high final substrate concentrations (≥40mM). We identified the main engineering parameters of process output (i.e., O2 supply; mixing correlated with immobilized enzyme stability; foam control correlated with product isolation; substrate solubilization) and succeeded in disentangling their complex interrelationship for systematic process optimization. Running the reaction at O2‐limited conditions at up to 500‐ml scale (10% dimethyl sulfoxide; silicone antifoam), we developed a substrate feeding strategy based on O2 feedback control. Thus, we achieved high reaction rates of 1.86g·L−1·hr−1 and near complete conversion (≥90%) of 80mM (16g/L) C12:0 with good selectivity (≤5% overoxidation). We showed that “uncoupled reaction” of the P450 BM3 (~95% utilization of NADPH and O2 not leading to hydroxylation) with the C12:0 hydroxylated product limited the process efficiency at high product concentration. Hydroxylated product (~7g; ≥92% purity) was recovered from 500ml reaction in 82% yield using ethyl‐acetate extraction. Collectively, these results demonstrate key engineering parameters for the biocatalytic oxy‐functionalization and show their integration into a coherent strategy for process intensification.
Keywords: cytochrome P450 BM3, hydroxylation, mono‐oxygenase, process control, process engineering, process intensification
Cytochrome P450‐catalyzed oxy‐functionalization reactions of fatty acid substrates represent a class of synthetically important bio‐transformations. Here, the authors identified key engineering parameters for the hydroxylation of dodecanoic acid by soluble and immobilized forms of cytochrome P450 BM3 and demonstrated their integration into a coherent strategy for process intensification. Metrics of process performance (as shown in the figure) revealed a highly efficient enzymatic hydroxylation. Reaction scale up to 500 ml enabled production of gram amounts of purified product.
Abbreviations
- BM3
cytochrome P450 BM3
- C12:0
dodecanoic acid
- C12:0‐OH
monohydroxylated C12:0
- GDH
d‐glucose 1‐dehydrogenase
- STY
space‐time yield
- TTN
total turnover number
1. INTRODUCTION
Cytochrome P450 monooxygenases (P450s) are heme‐enzymes widely recognized in applied catalysis for the chemically challenging transformations they promote (Bernhardt & Urlacher, 2014; Girvan & Munro, 2016; Janocha, Schmitz, & Bernhardt, 2015; Julsing, Cornelissen, Bühler, & Schmid, 2008; Schmitz, Rosenthal, & Lütz, 2019; Urlacher & Girhard, 2012, 2019). Oxy‐functionalization, through selective insertion of oxygen from O2 into a nonactivated substrate C‐H bond, is an important type of P450 reaction of high synthetic interest (Hollmann, Arends, Bühler, Schallmey, & Bühler, 2011; Janocha et al., 2015; Urlacher & Girhard, 2012, 2019; Wei, Ang, & Zhao, 2018). The P450 BM3 from Bacillus megaterium represents a class of P450 mono‐oxygenases (family CYP102A) generally referred to as “self‐sufficient.” The enzyme is built from a single polypeptide chain that comprises the mono‐oxygenase and reductase domains required for the full mono‐oxygenase catalytic cycle (Scheme S1; Bernhardt & Urlacher, 2014; Munro et al., 2002). P450 BM3 catalyzes oxy‐functionalization of a broad variety of substrates with high specific activity, using nicotinamide adenine dinucleotide phosphate (NADPH) and O2 as the co‐substrates (Hammerer, Winkler, & Kroutil, 2018; Whitehouse, Bell, & Wong, 2012). Among the substrates utilized for hydroxylation, saturated long‐chain fatty acids (e.g., dodecanoic acid; C12:0) are of particular interest for large‐scale transformation (Biermann, Bornscheuer, Meier, Metzger, & Schäfer, 2011). The corresponding hydroxy fatty acids are broadly useful as commodities in chemical, food, and cosmetic industries (Cao & Zhang, 2013; Kim & Oh, 2013). There is further considerable interest in hydroxy fatty acids as central intermediates of synthetic pathways leading to valuable amino acids (Dennig, Blaschke, Gandomkar, Tassano, & Nidetzky, 2018) as well as amine or ester building blocks, for polymer synthesis, for example (Cha et al., 2020; Kadisch et al., 2016; Ladkau et al., 2016; Schrewe, Ladkau, Bühler, & Schmid, 2013).
Limitation on the applicability of the P450 oxy‐functionalization often arises due to low reaction output, especially at larger scale (Brummund et al., 2016; Bühler & Schmid, 2004; Kaluzna et al., 2016; Lundemo & Woodley, 2015; O'Reilly, Köhler, Flitsch, & Turner, 2011). Reaction intensification is a complex engineering task that involves analysis and optimization of a large parameter space defined by multiple interrelated process factors. Hydrophobic substrates such as C12:0 require solubilization in the aqueous phase to become accessible to the enzyme (Brummund et al., 2016; Kühnel et al., 2007; Maurer, Schulze, Schmid, & Urlacher, 2003). The O2 co‐substrate must likewise be supplied efficiently, typically from air or O2 gas (Brummund et al., 2016; Kaluzna et al., 2016). The NADPH used in the oxy‐functionalization needs to be regenerated (e.g. Valikhani, Bolivar, Dennig, & Nidetzky, 2018). The operational stability of the enzymes under multiphasic heterogeneous reaction conditions can be challenging (Brummund et al., 2016; Lundemo & Woodley, 2015). Reaction scale‐up based on engineering parameters critical for the process output (e.g., oxygen transfer rate; mixing) has only rarely been examined for P450 BM3‐catalyzed transformations (Brummund et al., 2016; Kaluzna et al., 2016; Solé, Caminal, Schürmann, Álvaro, & Guillén, 2018; for a general account on process engineering applied to oxy‐functionalization, see Bühler & Schmid, 2004).
Here, we demonstrate an integrated approach of reaction engineering to the intensification of the cytochrome P450 BM3‐catalyzed hydroxylation of dodecanoic acid (C12:0). Limitations encountered in previous studies (e.g., Kühnel et al., 2007) concerned the yield (≤68%; based on 50 mM substrate), the product concentration (≤6.8 g/L), and the space‐time yield (~0.1 g·L−1·hr−1). We use the P450 BM3 together with glucose dehydrogenase to regenerate NADPH from glucose. We compare soluble and co‐immobilized enzyme preparations in O2‐gassed and pH‐controlled conversions at final substrate concentrations of up to 80 mM. We identify O2 supply and substrate solubilization as the main process‐related bottlenecks on the biocatalytic transformation. An O2‐controlled procedure of feeding substrate to the reaction was developed to enable high product concentrations and avoid excessive foam formation from gassed substrate solution at the same time. Using optimized process operation, we obtain substantially intensified reaction rates of 1.86 g·L−1·hr−1 and near complete conversion (≥90%) of 80 mM (16 g/L) C12:0 with good selectivity (≤5% overoxidation). Importantly, we discovered that “uncoupled reaction” of the P450 BM3 (~95% utilization of NADPH and O2 not leading to hydroxylation) with the C12:0 hydroxylated product restricted the process efficiency at high product concentration. Overall, we demonstrate key engineering parameters for the biocatalytic oxy‐functionalization and show their integration into a coherent strategy for process intensification. Our findings have significance for oxygenase bio‐catalysis in general (e.g., Bühler & Schmid, 2004; Hammerer et al., 2018; Karande et al., 2015; Karande, Salamanca, Schmid, & Buehler, 2017; Liang, Wei, Qiu, & Jiao, 2018; Romero, Gómez Castellanos, Gadda, Fraaije, & Mattevi, 2018; Solé et al., 2019).
2. MATERIALS AND METHODS
2.1. Materials
Unless mentioned, the materials used were those from Valikhani et al. (2018).
2.2. Enzymes and their co‐immobilization
The wild‐type cytochrome P450 BM3 was used. It was obtained with His‐tag (his_BM3) or the cationic binding module Zbasic2 (Z_BM3) fused to its N‐terminus. Zbasic2 is a small (∼7 kDa) protein module previously reported for enzyme immobilization (Bolivar & Nidetzky, 2012; Valikhani et al., 2018). Zbasic2 folds into a three‐helical bundle. Due to multiple arginine residues exposed on one of its sides, Zbasic2 exhibits clustered positive charges on the surface (Graslund, Lundin, Uhlén, Nygren, & Hober, 2000). Thus, fused to the target protein, Zbasic2 can promote protein binding to anionic surfaces. The binding is noncovalent and usually involves reasonably defined orientation, with Zbasic2 interacting with the solid surface (Bolivar & Nidetzky, 2012). The Bacillus megaterium glucose dehydrogenase (isozyme IV) harboring N‐terminally placed Zbasic2 (Z_GDH) was used. Commercial preparation of B. megaterium GDH was kindly provided by DSM (Geleen, The Netherlands). Enzymes were expressed from a pET28(+) expression vector in E. coli BL21 (DE3). Production and purification of Z_BM3 and Z_GDH were done as described previously (Valikhani et al., 2018), with modifications noted in the Supporting Information Material. Expression and purification of his_BM3 were done as in Dennig, Lülsdorf, Liu, and Schwaneberg (2013), with modifications noted in the Supporting Information Material. The number of active sites in cytochrome P450 BM3 preparations was quantitated with CO titration (Supporting Information Material; Dennig et al., 2013). Enzymatic reactions were performed using lyophilized bacterial lysate or purified enzyme. E. coli expression culture (per liter) yielded ~27 mg Z_BM3 and ~76 mg his_BM3 in ~1.6 g lyophilized cell extract. Z_GDH was obtained in 2,200 U/g lyophilized cell extract.
Co‐immobilization of Z_BM3 and Z_GDH on ReliSorb SP400 beads at gram scale was done by loading each enzyme (solution prepared from lyophilized cell extract) in a single step, Z_BM3 (11 µM) before Z_GDH (66 U/ml). The immobilization yield was ~75% for both enzymes, giving a solid catalyst containing (per gram carrier) 81 nmol Z_BM3 and 480 U Z_BM3. Loading of Z_BM3 in three steps, similarly as in Valikhani et al. (2018) and offering 4 µM Z_BM3 each time, showed a similar binding (83 nmol/g) in 88% yield. However, enzyme loading in a single step is preferred for practical reasons.
2.3. Reactor set‐up, instrumentation, and characterization
A photograph of the instrumented reactor is shown in Figure S1. A Wheaton (Millville, NJ) glass reactor (100 ml) equipped with a heating jacket was used. The working volume was about 50 ml. For mixing, an IKA (Staufen, Germany) RCT basic hotplate stirrer was used with a magnetic stirrer bar or an IKA RW 20 digital overhead stirrer was used. A LAUDA‐Brinkmann (Königshofen, Germany) Ecoline RE104 Recirculating water bath was used for temperature control (25°C). An IsmaTec MCP‐CPF Process IP65 piston pump (Cole‐Parmer GmbH, Wertheim, Germany) was used for automated substrate feed. A TitroLine Alpha titration device (Mettler Toledo, Vienna, Austria) was used for automated pH control (pH 7.2) using KOH (5 M). Pure O2 gas was supplied under control of an EL‐FLOW select mass flow meter (Bronkhorst, AK Ruurlo, the Netherlands). A 200‐µl pipette tip was used as gas outlet. The O2 saturation in the liquid phase was measured with an OXROB10 (Pyroscience GmbH, Aachen, Germany) optical sensor coupled to a FireStingO2 (FSO2‐x) fiber‐optic oxygen meter. Upscaled reaction at 500 ml was done using the same instrumented set‐up, except that a glass beaker without external temperature control (room temperature; ~23°C) was used. For each reactor and reaction condition used, the O2 mass transfer coefficient (k L a) was determined using the gassing‐out method.
2.4. Biocatalytic conversions
Reactions were performed in 50mM potassium phosphate buffer (pH 7.5; automatically controlled to pH 7.2) at 25°C. Dodecanoic acid was added at once from stock solution in ethanol or dimethyl sulfoxide (DMSO) to a concentration of 40 mM. Alternatively, it was fed to the reaction in portions that were added manually or automatically to a concentration of up to 80 mM (see Section 3). Glucose was added at 200–300mM and catalase at 1mg/ml. Antifoam 204 (600µl; FLUKA, Munich, Germany) or silicone antifoam (2–20mg; Sigma Aldrich) was added. his_BM3 or Z_BM3 was present at 2µM active sites. The enzyme was used in soluble form from the cell extract or as immobilized preparation. Z_GDH or commercial GDH was added at an activity of 1.9–14U/ml. Pure O2 was supplied at a flow rate of 20–50ml/min. The O2 in solution was measured continuously. Stirring was at 250−500rpm. Samples were taken at suitable times and after extraction into ethyl‐acetate, fatty acid conversion was analyzed by gas chromatography with flame ionization detection (GC‐FID). Conversion of glucose into gluconic acid was analyzed in the aqueous phase using high‐performance liquid chromatography (HPLC).
2.5. Product isolation
Two‐phase extraction with ethyl‐acetate was used. If intended for reuse, solid particles with immobilized enzyme were centrifuged. Otherwise they were left in the suspension. The pH was lowered to ~1 (37% HCl), leading to enzyme precipitation. Substrate and hydroxylated products were extracted under vigorous mixing into the same volume of ethyl‐acetate. For analytics, 20 mM 1‐octanol was added as the internal standard. The organic phase was recovered by centrifugation (4°C, ≥3,000g; 2 min) and dried over anhydrous Na2SO4. For preparative isolation, the extraction was repeated three times. Substrate and products were analyzed as the respective methyl esters. To isolate the product at preparative scale, ethyl‐acetate was removed under vacuum. Water (two volumes) was added to the oily residue to form an emulsion which was then lyophilized.
2.6. Analytics
2.6.1. Gas chromatography
Sample preparation involved mixing of 120µl extract from the reaction with 60µl methanol into GC vials with 200‐µl inlets. Trimethylsilyl‐diazomethane (10–16µl) was added and the sample was used immediately for measurement of dodecanoic acid (C12:0) and ω‐hydroxy dodecanoic acids (C12:0‐OH). A Hewlett‐Packard Series II GC‐FID system was used. An Agilent HP‐5 column (30m×320µm; 0.25‐µm film) was used and H2 (GC‐FID) was the carrier gas. The Supporting Information Material summarizes the full protocol and shows the separation (Figure S2). Substrate and products were quantitated from peak areas relative to the peak area of the internal standard.
2.6.2. High‐performance liquid chromatography
Sample (50 µl) was mixed with 10% H2SO4 (50 µl), diluted 10‐fold with water and centrifuged (30 min; 16,000g). The supernatant (100 µl) was analyzed on a Merck Hitachi HPLC system equipped with a HPX‐87H column (22°C) and a UV detector (L‐7400). The flow rate was 0.6 ml/min and 5 mM H2SO4 was the eluent. Detection of gluconic acid was at 216 nm.
2.7. Enzyme kinetic study
2.7.1. Coupling efficiency
This is defined as mol hydroxylated product formed/ratio of mol NADPH (or mol O2) consumed. In reactions that used regeneration of NADPH, coupling efficiency is obtained as mol hydroxylated product/mol gluconic acid formed. Coupling efficiency is reported in percent.
2.7.2. Reaction with hydroxylated C12:0‐OH substrate
Enzymatic reaction rates were obtained in triplicates from measurements of O2 consumption at different substrate concentrations (0.1–10mM; 10% DMSO) using 500µM NADPH. NADPH consumption rates (absorbance change at 340nm) were additionally recorded at select substrate concentrations, using 250µM NADPH. Isolated C12:0‐OHs (≥95% purity) was used as substrate. C12:0 was used as reference. Potassium phosphate buffer (50mM; pH 7.5) was used at 22°C. The reaction volume was 1ml and stirring at 300rpm was used. Enzyme (his_BM3 and Z_BM3 from cell extract or in purified form; Z_BM3 co‐immobilized with Z_GDH) was added at 0.15–0.50µM of heme active sites. Kinetic parameters (k cat, K m) were determined from the data, using unweighted nonlinear least‐squares regression analysis with the program SigmaPlot13 (Systat Software, Erkrath, Germany). The k cat (=V max/E) is calculated from the maximum volumetric reaction rate (V max) and the molarity of heme active sites (E). Coupling efficiencies for reactions with C12:0‐OH substrate were determined from the remaining substrate and the NADPH consumed after overnight incubations.
3. RESULTS AND DISCUSSION
Soluble and co‐immobilized enzyme preparations were used in conversion studies. We noted that, upon storage in suspension (100 mg particles per ml; 50 mM potassium phosphate buffer, pH 7.5; 250 mM NaCl) at 4°C, the co‐immobilized enzyme preparation released all of the bound Z_BM3 within a week, resulting in activity for hydroxylation of C12:0 to be lost completely (Figure S3). Lyophilization of the carrier, and storage in the dried form, effectively preserved the activity.
3.1. Reaction set‐up and process control
To establish a suitable process set‐up that includes the required elements of process control, we performed the enzymatic hydroxylation of C12:0 (10 mM; 2% ethanol, by volume) according to the overall reaction shown in Scheme 1. We showed that pH control to avoid acidification from gluconic acid (Figure S4) and efficient supply of O2 from pressurized gas (bubble gassing; Figure S5) are important factors of reaction efficiency. Moreover, strategy to prevent foam formation is necessary when bubble gassing is used. Antifoam 204 (0.15%, by volume) was added to that end.
Scheme 1.
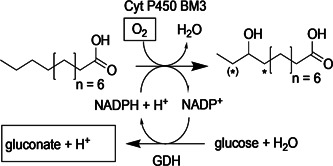
Biocatalytic reaction system for hydroxylation of C12:0 by cytochrome P450 BM3 using regeneration of nicotinamide adenine dinucleotide phosphate by glucose/glucose dehydrogenase (GDH). Under the conditions used, hydroxylation of C12:0 occurs predominantly at position ω‐2. Hydroxylation occurs additionally at position ω‐3 and to a small degree at position ω‐1, as indicated by asterisks (Valikhani et al., 2018). Substrate overoxidation can involve two hydroxylation events involving these positions. Parameters for process monitoring and control (pH, O2) are framed
To study the enzymatic conversion, we increased the C12:0 concentration to 40 mM and supplied O2 gas at 20 ml/min. Time courses of reactions catalyzed by free and co‐immobilized Z_BM3 (2 µM) and Z_GDH (6.8 Uml) are compared in Figure 1. It is immediately evident from the O2 concentration in liquid, that was effectively zero in the initial half of the conversion, that both reactions were largely limited by the supply of O2. The substrate was consumed completely in both reactions, about two‐fold faster when immobilized enzyme was used.
Figure 1.
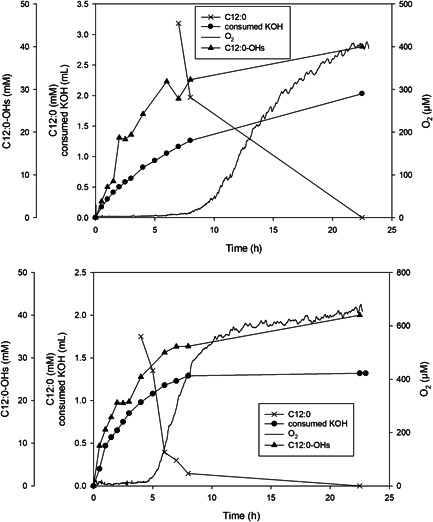
Hydroxylation of C12:0 by soluble (top panel) and co‐immobilized (bottom panel) Z_BM3 and Z_GDH. The reactions (45 ml) contained 2 µM Z_BM3 and 6.9 U/ml Z_GDH, 40 mM C12:0, 200 mM glucose, 200 µM NADP+, 1 mg/ml catalase, 2% EtOH (by volume), 50 mM potassium phosphate buffer (pH 7.5, 250 mM NaCl), and ~600 µl Antifoam 204. Reactions were operated at 20 ml/min O2 gassing and 250 rpm magnetic stirring. The temperature was 25°C. Note: the C12:0 substrate added to the reaction was partly insoluble in water and became only gradually dissolved as the conversion proceeded. Measurement of the change in the dissolved C12:0 concentration was relevant only in the later phase of the conversion, as shown in the figure
To explain the difference in the conversion rate for soluble and co‐immobilized enzymes, we determined the k L a in the absence (23.4 hr−1) and presence of solid particles (21.6 hr−1). Based on the equilibrium O2 concentration ([O2]* = 1.04 mM) under the conditions used, the O2 transfer rates (OTR = k L a [O2]*) are shown to have been similar in reactions with (22.5 mM/hr) and without (24.3 mM/hr) particles.
Further analysis revealed that reaction of the soluble enzymes consumed in total ~1.5‐fold more KOH for pH control than reaction of the co‐immobilized enzymes. This result indicated that relatively more gluconic acid was released for the same amount of C12:0 substrate converted when soluble compared to immobilized enzymes were used. The evidence thus suggested that the coupling efficiencies for reactions catalyzed by soluble and immobilized Z_BM3 are not the same. We therefore determined the coupling efficiency from the initial phase of the conversion when [O2] = 0 (Figure 1) and calculated it as the ratio of the product formation rate (soluble: 7.5 mM/hr; immobilized: 14.5 mM/hr) and the OTR. The coupling efficiency for the immobilized enzyme was higher (75%; consistent with Valikhani et al., 2018) than for the soluble enzyme (31%). Control experiments showed that lyophilized cell extract consumed both O2 and NADPH in the absence of the C12:0 substrate (data not shown). The co‐immobilized enzymes showed considerably lower activities (≤30%) toward utilization of O2 and NADPH under otherwise comparable conditions. Therefore, reaction rate and coupling efficiency of the soluble enzymes seem to be affected by the enzyme background of the cell extract, and the purification associated with immobilization results in improved performance. The result implies an important role of the enzyme expression level in E. coli when soluble enzymes are used as lyophilized cell extract. The his_BM3 is expressed to a ~three‐fold higher level than Z_BM3. When loaded in equal BM3 active‐site concentrations, as discussed later, the lyophilized cell extract showed largely decreased background activity for the his_BM3 preparation. The coupling efficiency (61%) of his_BM3 cell extract was comparable to that of the immobilized Z_BM3. Note: as shown later (Table 1), purified preparations of his_BM3 (92%) and Z_BM3 (79%) show high and comparable coupling efficiencies in the reaction with the C12:0 substrate. However, for conversion studies, the use of enzyme preparations from E. coli cell lysate was preferred for practical reason.
Table 1.
Kinetic parameters for reaction with C12:0 and C12:0‐OH, and coupling efficiencies and regioselectivities associated with the reactions
| Enzyme | ||||
|---|---|---|---|---|
| Substrate | Kinetic parameter a | his_BM3 | Z_BM3 | Immobilized Z_BM3 b |
| C12:0 | k cat (s−1) | |||
| Total O2 used | 2.9 | 4.6 | 3.9 | |
| NADPH used | 2.3 | n.d. | n.d. | |
| Substrate hydroxylated | 2.5 | 3.7 | 3.0 | |
| K m (µM) | 3.4 × 102 | 3.8 × 102 | 3.2 × 103 | |
| k cat/K m (mM−1·s−1) | ||||
| Total O2 used | 8.5 | 12 | 1.2 | |
| Substrate hydroxylated | 7.4 | 9.7 | 0.94 | |
| Coupling efficiency (%) | 92 | 79 | 77 | |
| Selectivity c % ω‐3/ω‐2/ω‐1 | 35/63/2 | 36/63/1 | 37/62/1 | |
| C12:0‐OH d | k cat (s−1) | |||
| Total O2 used | 1.0 | 2.1 | 0.7 | |
| NADPH used | 0.7 | n.d. | n.d. | |
| Substrate hydroxylated | 0.1 | 0.1 | 0.1 | |
| K m (µM) | 8.0 × 102 | 1.3 × 103 | 3.8 × 103 | |
| k cat/K m (µM−1·s−1) | ||||
| Total O2 used | 1.2 | 1.6 | 1.8 × 101 | |
| Substrate hydroxylated | 1.2 × 10−1 | 7.7 × 10−2 | 2.6 × 10−2 | |
| Coupling efficiency (%) | 6 | 5 | 12 | |
Abbreviations: NADPH, nicotinamide adenine dinucleotide phosphate; n.d., not determined.
Kinetic parameters are from nonlinear least‐squares regression analysis of the data and have relative SD of ≤15% (k cat), ≤25% (K m), and ≤15% (coupling efficiency).
The k cat of the immobilized Z_BM3 is determined based on the moles of enzyme active sitesbound from solution to the carrier.
From GC‐FID analysis, with peak assignment done according to Valikhani et al. (2018); data have relative SD of 10% and results are means from multiple samples (n ≥ 10). Results are from, and agree within, conversion experiments that used lyophilized cell extracts or purified enzymes.
Site selectivity was not determined with the C12:0‐OH substrate. However, from the product pattern in GC‐FID analysis, selectivity is the same for his_BM3, Z_BM3, and immobilized Z_BM3.
3.2. Immobilized enzyme stability
Considering recycling of the immobilized enzymes, we analyzed stability parameters of the solid catalyst. Unfortunately, activity for C12:0 conversion was lost completely after a single round of conversion (Figure 1, bottom). To identify reasons for the apparent inactivation, we showed that magnetic stirring caused grinding of the Relisorb particles. This could be avoided when an overhead Rushton turbine stirrer was used instead, but the enzyme inactivation occurred nonetheless. Using sodium dodecyl sulfate polyacrylamide gel electrophoresis, we analyzed the proteins in solution and on the solid carrier (Figure S6). We showed that Z_BM3 was released strongly from the carrier under all conditions used (Figure S6). Z_GDH was immobilized more stably by comparison. Partial absorption of C12:0 substrate by the Relisorb carrier (Figure S7), and surface/pore clogging effects associated with it, may have been an additional reason for the enzyme inactivation (Figure S8). In summary, therefore, despite the benefit of co‐immobilization of Z_BM3 and Z_GDH in terms of reaction efficiency (Figure 1), the relatively weak binding of Z_BM3 prevents the solid catalyst from being recycled. Release of immobilized Z_BM3 in a degree substantially larger than observed previously (Valikhani et al., 2018) is probably a combined effect of the enhanced agitation (≥250 rpm) and the O2 gassing used in the current study. Evidence that limitation on the space‐time yield of the reaction arises due to the OTR (Figure 1) emphasizes the need for a robust mono‐oxygenase catalyst that can tolerate the process conditions required for efficient OTR. Issues related to process robustness of cytochrome P450 BM3 have been noted in earlier studies (Brummund et al., 2016; Kaluzna et al., 2016; Valikhani et al., 2018). Different options are available for stabilizing the binding of Z_BM3 to the solid carrier (e.g. Valikhani, Bolivar, Pfeiffer, & Nidetzky, 2016). Here, since the focus was on intensification of the biocatalytic reaction, we left the aspect of immobilized enzyme recycling for consideration in the future. However, soluble and co‐immobilized enzymes were used in further experiments. Due to better expression in E. coli leading to higher coupling efficiency of enzyme preparations obtained as lyophilized cell lysate, the his_BM3 was used instead of the Z_BM3 when soluble enzymes were applied.
3.3. Fed‐batch reaction
During work‐up of reaction mixtures as from Figure 1, we noticed that Antifoam 204 (a polypropylene‐based polyether dispersion) was co‐extracted with the C12:0‐OH products into the ethyl‐acetate phase. The product thus obtained contained more than 50% (by mass) compounds from the Antifoam 204. In search of process conditions that enable high product concentrations and at the same time, prevent excessive foam formation without compromising the product isolation, we performed a series of experiments in which the co‐solvent (ethanol, DMSO), the stirring (stirrer speed, position of stirrer), and the liquid composition were varied. We found that foaming was correlated with dissolution of larger concentrations of C12:0. The C12:0‐OH product, interestingly, did not cause foaming. Under gassed conditions, stirring was mainly responsible for foam formation. In the absence of efficient mixing of the bulk liquid, however, the substrate precipitated (EtOH co‐solvent, no Antifoam 204) and due to its slow redissolution, the solid C12:0 was not accessible to the enzymatic transformation.
Since “co‐solvent optimization” was unlikely to resolve a fundamental engineering dilemma (C12:0 solubilization vs. foaming), we developed a fed‐batch strategy of supplying substrate in a controlled manner to the reaction. We took from Figure 1, that the O2 concentration in liquid started to rise when C12:0 had been depleted. This suggested that the substrate feed could be coupled to the on‐line monitored O2 concentration, as shown in Figure 2 (top). Thus, the C12:0 concentration was kept at a level (~8 mM) which, in combination with additional measures described below, enabled us to suppress the foam formation effectively.
Figure 2.
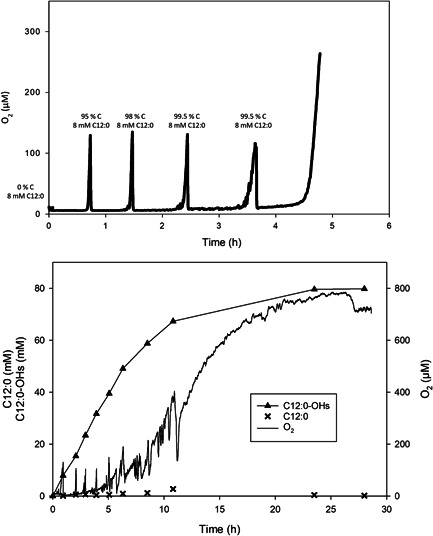
Development of a fed‐batch processing strategy for biocatalytic hydroxylation of C12:0. Top panel: Substrate feeding controlled by the steady‐state O2 concentration in bulk liquid. The reaction (50 ml) contained 2 µM his_BM3 and 1.9 U/ml commercial GDH, 8 mM C12:0, 200 mM glucose, 500 µM NADP+, 1 mg/ml catalase, 3.5% dimethyl sulfoxide (DMSO; v/v), 50 mM potassium phosphate buffer (pH 7.5), and silicone antifoam. The reaction was operated at 25 ml/min O2 gassing, 350 rpm magnetic stirring and automated pH control (pH 7.2). As indicated, new C12:0 (80 mg) dissolved in DMSO (1.25 ml) was added four times, resulting in a total concentration of 40 mM C12:0 and ~12% DMSO. Bottom panel: Fed‐batch reaction for hydroxylation of 80 mM C12:0. The reaction composition was identical to the reaction in the top panel, with the exception that 300 mM glucose and 2.8% DMSO (by volume) were used. Additionally, the feeding steps were adapted: five times 80 mg C12:0 in 0.92 ml DMSO and four times 100 mg C12:0 in 0.5 ml DMSO were added, resulting in a total concentration of 80 mM C12:0 and ~12% DMSO
DMSO was superior to ethanol in preventing precipitation of C12:0. It was applied at ~10% (concentration after final feed by volume) to ensure complete solubilization of the C12:0 used. Control of the maximum substrate concentration in the reaction notwithstanding, an antifoam strategy was necessary. The foam removes a substantial portion (up to 40%; data not shown) of C12:0 substrate from the reaction and so limits the maximum product output. The strategy was developed “backward,” considering first the compatibility with product isolation. We showed that silicone antifoam was not extracted into the ethyl‐acetate phase. It accumulated in a crud phase between the aqueous and organic phases and was conveniently removed from C12:0‐OH product. Being aware that silicone antifoam can affect the OTR (Brummund et al., 2016; Kawase & Moo‐Young, 1990), we showed that the k L a was decreased about three‐fold, from 30.6 to 10.4 hr−1 at a gas flow rate of 25 ml/min and stirrer speed of 350 rpm, when 0.3 mg/ml silicone were added. The [O2]* was also decreased by about 30% in the presence of silicone antifoam, leading to an overall ~four‐fold decrease in the maximum OTR and hence, the product formation rate (Figure S9). However, as stated above, use of antifoam was a practical necessity for high substrate conversion. Although not specifically examined here, negative effect of foam formation on enzyme stability can also be anticipated.
Figure 2 (bottom) shows the time course of a fed‐batch reaction with O2 controlled substrate supply, leading to the complete conversion of 80 mM C12:0 in 28 hr. The space‐time yield (STY) was 0.57 g·L−1·hr−1 overall and 1.86 g·L−1·hr−1 initially. The total turnover number (TTN) for the his_BM3 was ~40,000. The coupling efficiency was 61%. Accordingly, NADPH was recycled 343 times. Comparison with literature (Bernhardt & Urlacher, 2014; Kühnel et al., 2007; Maurer et al., 2003) shows that a highly efficient biocatalytic hydroxylation of C12:0 is here demonstrated. Soluble (his_BM3, Z_BM3) and immobilized (Z_BM3) enzyme preparations gave comparable reaction performances (Figure S10, Table S1).
3.4. Reaction scale‐up
To perform the hydroxylation of C12:0 at a ~10 g product scale, we increased the reaction volume 10‐fold to 500 ml. We applied constancy of OTR (k L a) as primary scale‐up criterion. To ensure gas supply throughout the liquid bulk, the gassing unit was changed to have three outlets (Figure S11). Thus, gas bubbles appeared to be homogeneously distributed in the bulk liquid (data not shown). The O2 gas flow rate and stirrer speed were increased to 50 ml/min and 500 rpm, respectively. Under these conditions, the k L a was 14.9 hr−1, which is comparable to the 10.4 hr−1 k L a of the 50 ml reactor. To minimize the enzyme amount initially loaded to the reactor, we determined the his_BM3 concentration required to decrease the steady‐state O2 concentration during reaction to effectively zero. In a titration experiment shown in Figure S12, we demonstrated that 1.1 µM his_BM3 are needed. Time course of the 500‐ml reaction using soluble enzymes is shown in Figure 3.
Figure 3.
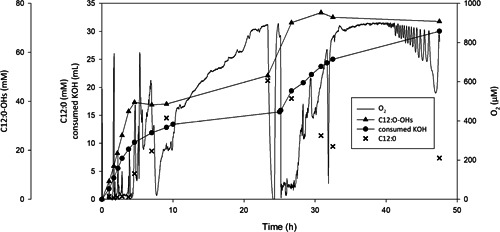
Hydroxylation of C12:0 in 500‐ml fed‐batch reaction. The reaction composition was identical to that in Figure 2b, except that initially 1 µM his_BM3 and 6U/ml Z_GDH were used. The amount of enzyme was increased to 2 µM his_BM3 (added in portions of 0.2 µM every 2 hr between 1 and 9 hr) and 14.8 U/ml Z_GDH (25.2 hr) and 0.6 U/ml commercial GDH (28.3 hr) in the course of the reaction. Note: the addition of fresh Z_GDH is the reason for the strong increase in conversion between 24 and 30 hr. Before the addition of the Z_GDH, the aeration was switched off for taking sample at 23.3 hr and switched on again at 24.2 hr, hence the sharp decrease and increase in the dissolved O2 concentration at these times. The reaction was operated at 50 ml/min O2 gassing up to 5 hr and at 5 ml/min later. Magnetic stirring (500 rpm) and automated pH control (pH 7.2) were used. The substrate was fed as five times 0.8 g C12:0 in 9.2 ml DMSO and four times 1 g C12:0 in 5 ml DMSO, resulting in a total concentration of 80 mM C12:0 and ~12% DMSO. DMSO, dimethyl sulfoxide
Overall, the 80 mM C12:0 substrate fed into the reaction was converted to 90%. Starting from a relatively low initial dosage of both his_BM3 and Z_GDH, fresh supplementation of each enzyme was necessary to promote the conversion. The his_BM3 was initially supplied at 1 µM and its concentration was increased in steps of 0.2 µM every 2 hours from 1 hr up until 9 hr of the reaction. After about 5 hr, the C12:0‐OH concentration (∼40 mM) hardly increased and despite decrease in the O2 gas flow rate from 50 to 5 ml/min, the dissolved O2 concentration increased to nearly 100% saturation at 23 hr (Figure 3). Sample from reaction showed that while the concentration of his_BM3 active sites was still sufficient (∼0.9 µM), the Z_GDH activity had been lost completely. Fresh Z_GDH (14.8 U/ml) was therefore added, leading the reaction to recommence. The C12:0‐OH concentration increased to ∼73 mM at 30 hr and the product formation leveled out at this point. Further addition of GDH (0.6 U/ml) did not promote the release of C12:0‐OH. The his_BM3 concentration measured at this point was still 0.6 µM. These results indicated that the GDH was less stable than the his_BM3 under the conditions used; and a process factor different from enzyme activity limited the product formation once high concentrations of the C12:0‐OH had been reached. We show later that uncoupled reaction of the P450 BM3 with C12:0‐OH is this relevant process factor.
Metrics of process efficiency from the 50‐ml reaction were largely retained at the larger scale. The STY was 0.30 g·L−1·hr−1 overall and 1.62 g·L−1·hr−1 initially. The TTN for the his_BM3 was ~36,000. The overall coupling efficiency was 47%, somewhat lower than in the 50‐ml reaction. The effect is explainable on account of different GDH preparations used in the 500‐ml (Z_GDH, lyophilized cell extract) and the 50‐ml reaction (commercial GDH) and the variable O2/NADPH consuming enzyme background that these preparations have. The NADPH was recycled 454 times in 500‐ml reaction. Scale up of the biocatalytic hydroxylation can thus be considered successful. Only few studies report on the scale‐up of cytochrome P450 BM3‐catalyzed reactions (Brummund et al., 2016; Kaluzna et al., 2016). A comparison of process performance data from a representative selection of studies is shown in Table S2.
Other P450 systems (e.g., enzymes of family CYP153, CYP52, and CYP94) have been applied in conversions of fatty acid substrates, including dodecanoic acid. As these P450 enzymes are typically not self‐sufficient and thus require redox partner proteins to perform the full mono‐oxygenase catalytic cycle, live whole cells (e.g., E. coli, Candida tropicalis, and Saccharomyces cerevisiae) expressing the complete enzymatic system have been used mostly. Recent review provides a comprehensive summary (Park et al., 2020). Compared to enzymes of family CYP102A that are typically not site‐selective in hydroxylating fatty acids (Dietrich, Do, Schmid, Pleiss, & Urlacher, 2009; Valikhani et al., 2018; Whitehouse et al., 2012), the alternative P450 systems (e.g., CYP153A enzymes) often provide selective oxidation at the terminal (ω) position (Jung et al., 2016; Scheps et al., 2013). However, limitations on the applicability of the whole cells can arise due to low uptake of the free fatty acids. Substrate is therefore often supplied as methyl ester. Using the outer membrane transporter AlkL (see Julsing et al., 2012) to facilitate methyl ester substrate uptake by engineered E. coli strain, production of 4g/L ω‐hydroxy dodecanoic acid in 28hr was demonstrated in the study of Scheps et al. (2013). A well‐known organism with which to carry out P450‐catalyzed transformations is Candida tropicalis (Park et al., 2020 and references therein). The organism contains CYP52A‐type P450 enzymes that are efficient in catalyzing ω‐hydroxylation of fatty acids. As shown in an important study of Gross and co‐workers (Lu et al., 2010), unless extensively engineered to eliminate enzyme background from additional P450s, fatty alcohol oxidases and alcohol dehydrogenases, C. tropicalis generally overoxidizes the fatty acid substrate to produce dicarboxylic acid product (e.g., Picataggio et al., 1992). The suitably engineered C. tropicalis strain showed an impressive production of 174g/L ω‐hydroxy tetradecanoic acid from 200g/L tetradecanoic acid methyl ester as the substrate (Lu et al., 2010). Substantial further intensification of the herein used enzymatic system would be required to boost the product concentration to a level comparable with Lu et al. (2010).
3.5. Product isolation
The downstream processing of the reaction mixture (500ml; see Figure 3) involved centrifugation of catalyst particles and other solid material, acidification and extraction into the same volume of ethyl‐acetate in three steps. Residual solid substrate was effectively removed in the centrifugation (Figure S13). The product was recovered in 82% yield (7.07 g) with a purity of 92%. About half of the impurity (3.5% of total) was due to overoxidized, doubly hydroxylated product (Figure S13). Product recoveries from 50‐ml reactions (n = 4) were similar (70–90% yield; 85–91% purity). Content of overoxidized species in total product varied between 2.5% and 5.0%, depending on C12:0 conversion and reaction time used. The total time for production of the ∼7 g product was 60 hr, which is distributed between 47hr for enzymatic production and ∼13 hr for the product isolation,(∼3 hr extraction and ∼10 hr lyophilization). Using conditions from Figure 3, production time could be shortened to about ∼30 hr.
3.6. Effect of substrate overoxidation on reaction performance
We examined the biochemical basis of substrate overoxidation to clarify a possible role of it in limiting the reaction output. Evidence supporting such role was that, upon fresh addition of C12:0 substrate (10–20 mM) and enzyme (his_BM3) to a reaction mixture containing ~40–60 mM C12:0‐OH product, the conversion proceeded only slowly and a pronounced decrease in the coupling efficiency was observed. We first performed a reaction that used the isolated C12:0‐OH product (20 mM; ~90% purity) as substrate for conversion by his_BM3. The overoxidation product identified from the C12:0 reaction was formed together with two additional products (Figure 4) at the expense of the C12:0‐OH substrate. Evidence from GC‐FID (elution pattern) was indicative of formation of doubly hydroxylated C12:0 species. The C12:0 overoxidation thus appears to be consisted in two consecutive steps of substrate hydroxylation. Reaction selectivity (single compared to double hydroxylation of C12:0) depends on the relative enzyme efficiencies for conversion of C12:0 and C12:0‐OH under the conditions emerging in the process. We therefore determined kinetic parameters of the purified enzymes (his_BM3, Z_BM3) for reaction with C12:0 and C12:0‐OH. The parameters are shown in Table 1 along with the corresponding coupling efficiencies of the reactions. The catalytic efficiency (k cat/K m) for reaction with C12:0‐OH was decreased 57‐fold (his_BM3) compared to reaction with C12:0, reflecting decrease in the catalytic constant (k cat) for hydroxylation by 25‐fold and increase in the Michaelis constant (K m) by 2.4‐fold. Reaction rates based on O2 consumption were largely consistent with reaction rates based on oxidation of NADPH (Table 1). The coupling efficiency was decreased dramatically to just ~5% when C12:0‐OH was compared to C12:0 as the substrate for hydroxylation (Table 1). The k cat for the total O2 consumption in hydroxylation and uncoupled reaction was only 2.9‐fold (his_BM3) decreased when C12:0‐OH instead of C12:0 was present. In terms of the apparent k cat/K m, enzymatic reaction with C12:0 is therefore preferred only about seven‐fold over the combined, coupled and uncoupled reaction with C12:0‐OH. These results support the idea that, as the conversion of C12:0 makes progress in the reaction, the C12:0‐OH product can compete with the original substrate to become involved in P450 BM3 catalysis. The results further suggest that a gradual shift in reaction toward utilization of the C12:0‐OH product will result in substantial uncoupling between O2 reduction and hydroxylation. Indeed, correlation between formation of the overoxidation product and decrease in coupling efficiency is suggested from detailed analysis of the reaction time course (Figure 4, bottom panel). The uncoupled reaction results in a futile cycle of NADPH usage and regeneration, leading to acid formation. Mechanistically, the uncoupled reaction can proceed along different paths, often referred to as autoxidation, peroxide, and oxidase shunt, that are indicated in Scheme S1. Reactive oxygen species are released in the autoxidation (O2 •−) and the peroxide (O2 2−) shunt (Hammerer et al., 2018; Munro et al., 2002). These oxygen species can destroy the enzyme activity and are potential factors of enzyme operational stability (e.g., Brummund et al., 2016). We thus show that substrate overoxidation can affect the process efficiency in important ways going beyond the effect on product purity.
Figure 4.
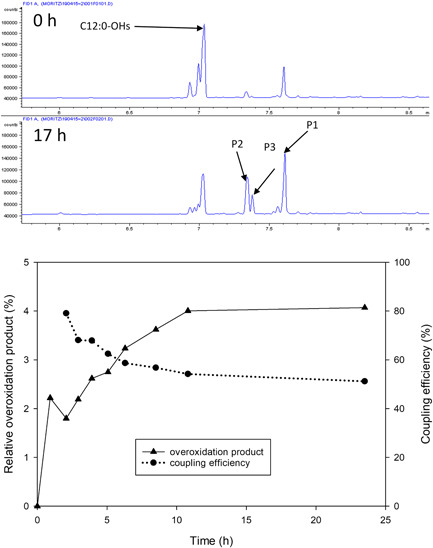
Hydroxylated C12:0 as substrate for P450 BM3‐catalyzed oxy‐functionalization. Top panel: gas chromatography traces from the reaction before and after incubation for 17 hr. The horizontal axis shows time (min) and the vertical axis flame ionization detection signal in arbitrary units. The reaction (10 ml) was composed of 10 µM his_BM3, 9.5 U/ml commercial GDH, 20 mM C12:0‐OHs (41.6 mg), 5% (by volume) DMSO, 200 mM glucose, 500 µM NADP+, 5 mg/ml catalase, and 50 potassium phosphate buffer (pH 7.5). The reaction was operated at 5 ml/min O2 gassing, 250 rpm magnetic stirring, and automated pH control (pH 7.2). Bottom panel: Correlation between overoxidized product formed and decrease in coupling efficiency. Data are from the reaction in Figure 2 (bottom panel). DMSO, dimethyl sulfoxide [Color figure can be viewed at wileyonlinelibrary.com]
4. CONCLUSIONS
Intensification (≥ three‐fold improvement compared to best benchmarks from literature; Bernhardt & Urlacher, 2014; Kühnel et al., 2007; Maurer et al., 2003) of cytochrome P450 BM3‐catalyzed hydroxylation of C12:0 was achieved to a product concentration of ~80 mM at complete substrate conversion, high initial reaction rate (1.86 g·L−1·hr−1) and useful overall STY (0.6 g·L−1·hr−1). The reaction was scaled up to 500 ml volume from which 7.1 g product was recovered directly via extraction, in 92% purity and 82% yield. The main engineering parameters of process output, namely O2 supply and substrate solubilization, were integrated into a coherent strategy of whole process operation, including the product isolation. An O2‐controlled procedure of feeding substrate to the reaction was developed to enable a high product concentration and avoid excessive foam formation from gassed substrate solution at the same time. With the main process‐related bottlenecks on the biocatalytic reaction identified and strategies for their removal developed, the hydroxylation of C12:0 reached a point where hitherto undiscovered features of P450 BM3 selectivity became limiting for process efficiency. The C12:0 hydroxylated product competed with the C12:0 substrate for the enzyme and triggered a largely uncoupled reaction (~95%). In the presence of glucose/GDH, this resulted in an overall futile cycle of O2 reduction by glucose via NADPH. Although efficient for one‐step purification and co‐immobilization of Z_BM3 and Z_GDH (this study; Valikhani et al., 2018), noncovalent binding of Z_BM3 to Relisorb SP400 carrier requires further stabilization to enable recycling of the solid mono‐oxygenase catalyst. Stabilized immobilization of the cytochrome P450 BM3 could be achieved by multivalent binding from two or more Zbasic2 modules present in the enzyme, as shown earlier for a different enzyme (sucrose phosphorylase; Valikhani et al., 2016), or by chemical crosslinking approaches (Hernandez & Fernandez‐Lafuente, 2011). In summary, our results make a strong case for the important role of biochemical engineering in achieving process intensification to biocatalytic oxy‐functionalization (Bernhardt & Urlacher, 2014; Bühler & Schmid, 2004; Hoschek, Bühler, & Schmid, 2017; Hoschek, Schmid, & Bühler, 2018).
Supporting information
Supporting information
ACKNOWLEDGMENTS
This study has been supported in part by the COMET Funding Program managed by the Austrian Research Promotion Agency FFG. Dr. Juan M. Bolivar (Institute of Biotechnology and Biochemical Engineering, Graz University of Technology) is thanked for discussion on enzyme immobilization. The thematic framework of the TU Graz Lead Project “PorousMaterials@Work” is gratefully acknowledged.
Buergler MB, Dennig A, Nidetzky B. Process intensification for cytochrome P450 BM3‐catalyzed oxy‐functionalization of dodecanoic acid. Biotechnology and Bioengineering. 2020;117:2377–2388. 10.1002/bit.27372
Moritz B. Buergler and Alexander Dennig contributed equally to this study.
REFERENCES
- Bernhardt, R. , & Urlacher, V. B. (2014). Cytochromes P450 as promising catalysts for biotechnological application: Chances and limitations. Applied Microbiology and Biotechnology, 98, 6185–6203. 10.1007/s00253-014-5767-7 [DOI] [PubMed] [Google Scholar]
- Biermann, U. , Bornscheuer, U. , Meier, M. A. R. , Metzger, J. O. , & Schäfer, H. J. (2011). Oils and fats as renewable raw materials in chemistry. Angewandte Chemie (International Edition in English), 50, 3854–3871. 10.1002/anie.201002767 [DOI] [PubMed] [Google Scholar]
- Bolivar, J. M. , & Nidetzky, B. (2012). Oriented and selective enzyme immobilization on functionalized silica carrier using the cationic binding module Zbasic2: Design of a heterogeneous D‐amino acid oxidase catalyst on porous glass. Biotechnology and Bioengineering, 109, 1490–1498. 10.1002/bit.24423 [DOI] [PubMed] [Google Scholar]
- Brummund, J. , Müller, M. , Schmitges, T. , Kaluzna, I. , Mink, D. , Hilterhaus, L. , & Liese, A. (2016). Process development for oxidations of hydrophobic compounds applying cytochrome P450 monooxygenases in‐vitro. Journal of Biotechnology, 233, 143–150. 10.1016/j.jbiotec.2016.07.002 [DOI] [PubMed] [Google Scholar]
- Bühler, B. , & Schmid, A. (2004). Process implementation aspects for biocatalytic hydrocarbon oxyfunctionalization. Journal of Biotechnology, 113, 183–210. 10.1016/j.jbiotec.2004.03.027 [DOI] [PubMed] [Google Scholar]
- Cao, Y. , & Zhang, X. (2013). Production of long‐chain hydroxy fatty acids by microbial conversion. Applied Microbiology and Biotechnology, 97, 3323–3331. 10.1007/s00253-013-4815-z [DOI] [PubMed] [Google Scholar]
- Cha, H. J. , Hwang, S. Y. , Lee, D. S. , Akula, R. K. , Kwon, Y. U. , Voß, M. , … Park, J. B. (2020). Whole‐cell photoenzymatic cascades to synthesize long‐chain aliphatic amines and esters from renewable fatty acids. Angewandte Chemie (International Edition in English), 59, 7024–7028. 10.1002/anie.201915108 [DOI] [PubMed] [Google Scholar]
- Dennig, A. , Blaschke, F. , Gandomkar, S. , Tassano, E. , & Nidetzky, B. (2018). Preparative asymmetric synthesis of canonical and non‐canonical α‐amino acids through formal enantioselective biocatalytic amination of carboxylic acids. Advanced Synthesis Catalysis, 361, 1348–1358. 10.1002/adsc.201801377 [DOI] [Google Scholar]
- Dennig, A. , Lülsdorf, N. , Liu, H. , & Schwaneberg, U. (2013). Regioselective o‐hydroxylation of monosubstituted benzenes by P450 BM3. Angewandte Chemie (International Edition in English), 52, 8459–8462. 10.1002/anie.201303986 [DOI] [PubMed] [Google Scholar]
- Dietrich, M. , Do, T. A. , Schmid, R. D. , Pleiss, J. , & Urlacher, V. B. (2009). Altering the regioselectivity of the subterminal fatty acid hydroxylase P450 BM‐3 towards gamma‐ and delta‐positions. Journal of Biotechnology, 139, 115–117. 10.1016/j.jbiotec.2008.10.002 [DOI] [PubMed] [Google Scholar]
- Girvan, H. M. , & Munro, A. W. (2016). Applications of microbial cyctochrome P450 enzymes in biotechnology and synthetic biology. Current Opinion in Chemical Biology, 31, 136–145. 10.1016/j.cbpa.2016.02.018 [DOI] [PubMed] [Google Scholar]
- Graslund, T. , Lundin, G. , Uhlén, M. , Nygren, P. Å. , & Hober, S. (2000). Charge engineering of a protein domain to allow efficient ion‐exchange recovery. Protein Engineering, Design and Selection, 13, 703–709. 10.1093/protein/13.10.703 [DOI] [PubMed] [Google Scholar]
- Hammerer, L. , Winkler, C. K. , & Kroutil, W. (2018). Regioselective biocatalytic hydroxylation of fatty acids by cytochrome P450s. Catalysis Letters, 148, 787–812. 10.1007/s10562-017-2273-4 [DOI] [Google Scholar]
- Hernandez, K. , & Fernandez‐Lafuente, R. (2011). Control of protein immobilization: Coupling immobilization and site‐directed mutagenesis to improve biocatalyst or biosensor performance. Enzyme and Microbial Technology, 48, 107–122. 10.1016/j.enzmictec.2010.10.003 [DOI] [PubMed] [Google Scholar]
- Hollmann, F. , Arends, I. W. C. E. , Bühler, K. , Schallmey, A. , & Bühler, B. (2011). Enzyme‐mediated oxidations for the chemist. Green Chemistry, 13, 226–265. 10.1039/C0GC00595A [DOI] [Google Scholar]
- Hoschek, A. , Bühler, B. , & Schmid, A. (2017). Overcoming the gas‐liquid mass transfer of oxygen by coupling photosynthetic water oxidation with biocatalytic oxyfunctionalization. Angewandte Chemie (International Edition in English), 56, 15146–15149. 10.1002/anie.201706886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoschek, A. , Schmid, A. , & Bühler, B. (2018). In situ O2 generation for biocatalytic oxyfunctionalization reactions. ChemCatChem, 10, 5366–5371. 10.1002/cctc.201801262 [DOI] [Google Scholar]
- Janocha, S. , Schmitz, D. , & Bernhardt, R. (2015). Terpene hydroxylation with microbial cytochrome P450 monooxgenases. Advances in Biochemical Engineering/Biotechnology, 148, 215–250. 10.1007/10_2014_296 [DOI] [PubMed] [Google Scholar]
- Julsing, M. K. , Cornelissen, S. , Bühler, B. , & Schmid, A. (2008). Heme‐iron oxygenases: Powerful industrial biocatalysts? Current Opinion in Chemical Biology, 12, 177–186. 10.1016/j.cbpa.2008.01.029 [DOI] [PubMed] [Google Scholar]
- Julsing, M. K. , Schrewe, M. , Cornelissen, S. , Hermann, I. , Schmid, A. , & Bühler, B. (2012). Outer membrane protein AlkL boosts biocatalytic oxyfunctionalization of hydrophobic substrates in Escherichia coli . Applied and Environmental Microbiology, 78, 5724–5733. 10.1128/AEM.00949-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, E. , Park, B. G. , Ahsan, Md. M. , Kim, J. , Yun, H. , Choi, K.‐Y. , & Kim, B.‐G. (2016). Production of ω‐hydroxy palmitic acid using CYP153A35 and comparison of cytochrome P450 electron transfer system in vivo. Applied Microbiology and Biotechnology, 100, 10375–10384. 10.1007/s00253-016-7675-5 [DOI] [PubMed] [Google Scholar]
- Kadisch, M. , Julsing, M. K. , Schrewe, M. , Jehmlich, N. , Scheer, B. , von Bergen, M. , … Bühler, B. (2016). Maximization of cell viability rather than biocatalyst activity improves whole‐cell ω‐oxyfunctionalization performance. Biotechnology and Bioengineering, 114, 874–884. 10.1002/bit.26213 [DOI] [PubMed] [Google Scholar]
- Kaluzna, I. , Schmitges, T. , Straatman, H. , van Tegelen, D. , Müller, M. , Schürmann, M. , & Mink, D. (2016). Enabling selective and sustainable P450 oxygenation technology. Production of 4‐hydroxy‐α‐isophorone on kilogram scale. Organic Process Research & Development, 20, 814–819. 10.1021/acs.oprd.5b00282 [DOI] [Google Scholar]
- Karande, R. , Debor, L. , Salamanca, D. , Bogdahn, F. , Engesser, K.‐H. , Buehler, K. , & Schmid, A. (2015). Continuous cyclohexane oxidation to cyclohexanol using a novel cytochrome P450 monooxygenase from Acidovorax sp. CHX100 in recombinant P. taiwanensis VLB120 biofilms. Biotechnology and Bioengineering, 113, 52–61. 10.1002/bit.25696 [DOI] [PubMed] [Google Scholar]
- Karande, R. , Salamanca, D. , Schmid, A. , & Buehler, K. (2017). Biocatalytic conversion of cycloalkanes to lactones using an in‐vivo cascade in Pseudomonas taiwanensis VLB120. Biotechnology and Bioengineering, 115, 312–320. 10.1002/bit.26469 [DOI] [PubMed] [Google Scholar]
- Kawase, Y. , & Moo‐Young, M. (1990). The effect of antifoam agents on mass transfer in bioreactors. Bioprocess engineering, 5, 169–173. 10.1007/BF00369581 [DOI] [Google Scholar]
- Kim, K.‐R. , & Oh, D.‐K. (2013). Production of hydroxy fatty acids by microbial fatty acid‐hydroxylation enzymes. Biotechnology Advances, 31, 1473–1485. 10.1016/j.biotechadv.2013.07.004 [DOI] [PubMed] [Google Scholar]
- Kühnel, K. , Maurer, S. C. , Galeyeva, Y. , Frey, W. , Laschat, S. , & Urlacher, V. B. (2007). Hydroxylation of dodecanoic acid and (2R,4R,6R,8R)‐tetramethyldecanol on a preparative scale using an NADH‐dependent CYP102A1 mutant. Advanced Synthesis & Catalysis, 349, 1451–1461. 10.1002/adsc.200700054 [DOI] [Google Scholar]
- Ladkau, N. , Assmann, M. , Schrewe, M. , Julsing, M. K. , Schmid, A. , & Bühler, B. (2016). Efficient production of the Nylon 12 monomer ω‐aminododecanoic acid methyl ester from renewable dodecanoic acid methyl ester with engineered Escherichia coli . Metabolic Engineering, 36, 1–9. 10.1016/j.ymben.2016.02.011 [DOI] [PubMed] [Google Scholar]
- Liang, Y. , Wei, J. , Qiu, X. , & Jiao, N. (2018). Homogeneous oxygenase catalysis. Chemical Reviews, 118, 4912–4945. 10.1021/acs.chemrev.7b00193 [DOI] [PubMed] [Google Scholar]
- Lu, W. , Ness, J. E. , Xie, W. , Zhang, X. , Minshull, J. , & Gross, R. A. (2010). Biosynthesis of monomers for plastics from renewable oils. Journal of the American Chemical Society, 132, 15451–15455. 10.1021/ja107707v [DOI] [PubMed] [Google Scholar]
- Lundemo, M. T. , & Woodley, J. M. (2015). Guidelines for development and implementation of biocatalytic P450 processes. Applied Microbiology and Biotechnology, 99, 2465–2483. 10.1007/s00253-015-6403-x [DOI] [PubMed] [Google Scholar]
- Maurer, S. C. , Schulze, H. , Schmid, R. D. , & Urlacher, V. B. (2003). Immobilisation of P450 BM‐3 and an NADP+ cofactor recycling system: Towards a technical application of heme‐containing monooxygenases in fine chemical synthesis. Advanced Synthesis & Catalysis, 345, 802–810. 10.1002/adsc.200303021 [DOI] [Google Scholar]
- Munro, A. W. , Leys, D. G. , McLean, K. J. , Marshall, K. R. , Ost, T. W. B. , Daff, S. , … Dutton, P. L. (2002). P450 BM3: The very model of a modern flavocytochrome. Trends in Biochemical Sciences, 27, 250–257. 10.1016/s0968-0004(02)02086-8 [DOI] [PubMed] [Google Scholar]
- O'Reilly, E. , Köhler, V. , Flitsch, S. L. , & Turner, N. J. (2011). Cytochromes P450 as useful biocatalysts: Addressing the limitations. Chemical Communications, 47, 2490–2501. 10.1039/c0cc03165h [DOI] [PubMed] [Google Scholar]
- Park, H. , Park, G. , Jeon, W. , Ahn, J. O. , Yang, Y. H. , & Choi, K. Y. (2020). Whole‐cell biocatalysis using cytochrome P450 monooxygenases for biotransformation of sustainable bioresources (fatty acids, fatty alkanes, and aromatic amino acids). Biotechnology Advances, 40, 107504 10.1016/j.biotechadv.2020.107504 [DOI] [PubMed] [Google Scholar]
- Picataggio, S. , Rohrer, T. , Deanda, K. , Lanning, D. , Reynolds, R. , Mielenz, J. , & Eirich, L. D. (1992). Metabolic engineering of Candida tropicalis for the production of long‐chain dicarboxylic acids. Bio/Technology, 10, 894–898. [DOI] [PubMed] [Google Scholar]
- Romero, E. , Gómez Castellanos, J. R. , Gadda, G. , Fraaije, M. W. , & Mattevi, A. (2018). Same substrate, many reactions: Oxygen activation in flavoenzymes. Chemical Reviews, 118, 1742–1769. 10.1021/acs.chemrev.7b00650 [DOI] [PubMed] [Google Scholar]
- Scheps, D. , Malca, S. H. , Richter, S. M. , Marisch, K. , Nestl, B. M. , & Hauer, B. (2013). Synthesis of ω‐hydroxy decanoic acid based on an engineered CYP153A fusion construct. Microbial Biotechnology, 6, 694–707. 10.1111/1751-7915.12073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz, L. M. , Rosenthal, K. , & Lütz, S. (2019). Recent advances in heme biocatalysis engineering. Biotechnology and Bioengineering, 116, 3469–3475. 10.1002/bit.27156 [DOI] [PubMed] [Google Scholar]
- Schrewe, M. , Ladkau, N. , Bühler, B. , & Schmid, A. (2013). Direct terminal alkylamino‐functionalization via multistep biocatalysis in one recombinant whole‐cell catalyst. Advanced Synthesis & Catalysis, 355, 1693–1697. 10.1002/adsc.201200958 [DOI] [Google Scholar]
- Solé, J. , Brummund, J. , Caminal, G. , Álvaro, G. , Schürmann, M. , & Guillén, M. (2019). Enzymatic synthesis of trimethyl‐ε‐caprolactone: Process intensification and demonstration on a 100 L Scale. Organic Process Resarch & Development, 23, 2336–2344. 10.1021/acs.oprd.9b00185 [DOI] [Google Scholar]
- Solé, J. , Caminal, G. , Schürmann, M. , Álvaro, G. , & Guillén, M. (2018). Co‐immobilization of P450 BM3 and glucose dehydrogenase on different supports for application as a self‐sufficient oxidative biocatalyst. Journal of Chemical Technology & Biotechnology, 94, 244–255. 10.1002/jctb.5770 [DOI] [Google Scholar]
- Urlacher, V. B. , & Girhard, M. (2012). Cytochrome P450 monooxygenases: An update on perspectives for synthetic application. Trends in Biotechnology, 30, 26–36. 10.1016/j.tibtech.2011.06.012 [DOI] [PubMed] [Google Scholar]
- Urlacher, V. B. , & Girhard, M. (2019). Cytochrome P450 monooxygenases in biotechnology and synthetic biology. Trends in Biotechnology, 37, 882–897. 10.1016/j.tibtech.2019.01.001 [DOI] [PubMed] [Google Scholar]
- Valikhani, D. , Bolivar, J. M. , Dennig, A. , & Nidetzky, B. (2018). A tailor‐made, self‐sufficient and recyclable monooxygenase catalyst based on co‐immobilized cytochrome P450 BM3 and glucose dehydrogenase. Biotechnology and Bioengineering, 115, 2416–2425. 10.1002/bit.26802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valikhani, D. , Bolivar, J. M. , Pfeiffer, M. , & Nidetzky, B. (2016). Multivalency effects on the immobilization of sucrose phosphorylase in flow microchannels and their use in the development of a high‐performance biocatalytic microreactor. ChemCatChem, 9, 161–166. 10.1002/cctc.201601019 [DOI] [Google Scholar]
- Wei, Y. , Ang, E. L. , & Zhao, H. (2018). Recent developments in the application of P450 based biocatalysts. Current Opinion in Chemical Biology, 43, 1–7. 10.1016/j.cbpa.2017.08.006 [DOI] [PubMed] [Google Scholar]
- Whitehouse, C. J. , Bell, S. G. , & Wong, L. L. (2012). P450BM3 (CYP102A1): Connecting the dots. Chemical Society Reviews, 41, 1218–1260. 10.1039/c1cs15192d [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information