

Abstract
The strong boron Lewis acid tris(pentafluorophenyl)borane B(C6F5)3 is known to catalyze the dehydrogenative coupling of certain amines and hydrosilanes at elevated temperatures. At higher temperature, the dehydrogenation pathway competes with cleavage of the C−N bond and defunctionalization is obtained. This can be turned into a useful methodology for the transition‐metal‐free reductive deamination of a broad range of amines as well as heterocumulenes such as an isocyanate and an isothiocyanate.
Keywords: amines, boron, defunctionalization, reduction, silanes
I mean defunct. Combinations of the boron Lewis acid B(C6F5)3 and hydrosilanes enable the reductive cleavage ofC−N bonds in various, mainly benzylic amines and heterocumulenes. By this, these functionalized substrates can be converted into the corresponding hydrocarbons in moderate to good yields.

Selective defunctionalization of synthetic intermediates is a key strategy in organic synthesis.1 Reductive alcohol deoxygenation is an important example of it2 and can be achieved by the reaction of alcohols or derivatives thereof and hydrosilanes in the presence of catalytic amounts of B(C6F5)3 (Scheme 1, top left).3 The ionic mechanism involves the thermodynamically favorable formation of Si−O bonds and [HB(C6F5)3]−,4 and these reactions are ultimately borohydride reductions. Employing various boron Lewis acid/hydrosilane combinations, Gagné showcased the impressive chemo‐ and regioselectivity of this methodology when applied to complex molecules.5 Morandi6 and our laboratory7 also contributed to this field. We asked ourselves whether reductive deamination of 1°, 2°, and 3° amines would be possible in the same manner (Scheme 1, top right). Based on a 45‐year‐old report on the reductive cleavage of disulfonimides with NaBH4,8 we anticipated that in situ generation of disilazanes by dehydrogenative Si‐N coupling9 followed by another reaction with a B(C6F5)3/hydrosilane pair would also result in the displacement of the amino group (Scheme 1, bottom). Existing protocols for reductive deamination either rely on preceding conversion of the amino group into a better leaving group followed by hydride substitution8, 10 or on transition‐metal‐catalyzed hydrogenolysis.11 We disclose here an alternative to these methods, thereby expanding the toolbox of B(C6F5)3 chemistry.12
Scheme 1.

B(C6F5)3‐catalyzed reductive defunctionalization with hydrosilanes. R1, R2, and R=alkyl or aryl; n=1–3. HMPA=hexamethylphosphoric triamide.
We began our investigation with optimizing the reductive deamination of 1‐(naphthalen‐2‐yl)ethan‐1‐amine (1 aa→2 a; Table 1). The reaction of 1 aa with 4.0 equiv of PhSiH3 in the presence of 20 mol % of B(C6F5)3 in benzene at 120 °C furnished β‐ethylnaphthalene (2 a) in 48 % yield after 24 h (Entry 1). Among several solvents screened, 1,2‐C6H4F2 was the best (Entries 2–8). The high reaction temperature of 120 °C was essential, and there was hardly any conversion at 80 °C (Entry 9). Lower catalyst loadings were not detrimental, and 5.0 mol % of B(C6F5)3 was sufficient to reach quantitative yield (Entries 10–12). For this, the reaction time of 24 h was needed (see the Supporting Information for a yield/time analysis). Less PhSiH3 resulted in a lower yield (Entry 13). Using MePhSiH2 or Me2PhSiH instead of PhSiH3 as reductants also gave excellent yields (Entries 14 and 15). Other tertiary hydrosilanes were however too unreactive under these reaction conditions (see the Supporting Information for details). In the case of Me2PhSiH the byproduct bis(dimethyl(phenyl)silyl)amine was detected in 65 % yield, suggesting the intermediacy of the aforementioned disilazane.
Table 1.
Selected examples of the optimization of the B(C6F5)3‐catalyzed reductive deamination.[a]

|
Entry |
B(C6F5)3 [mol %] |
Hydrosilane [equiv] |
Solvent |
T [°C] |
Yield [%][b] |
|---|---|---|---|---|---|
|
1 |
20 |
PhSiH3 (4.0) |
benzene |
120 |
48 |
|
2 |
20 |
PhSiH3 (4.0) |
toluene |
120 |
47 |
|
3 |
20 |
PhSiH3 (4.0) |
1,4‐xylene |
120 |
52 |
|
4 |
20 |
PhSiH3 (4.0) |
C6H5CF3 |
120 |
57 |
|
5 |
20 |
PhSiH3 (4.0) |
C6H5F |
120 |
58 |
|
6 |
20 |
PhSiH3 (4.0) |
C6H5Cl |
120 |
59 |
|
7 |
20 |
PhSiH3 (4.0) |
1,2‐C6H4Cl2 |
120 |
91 |
|
8 |
20 |
PhSiH3 (4.0) |
1,2‐C6H4F2 |
120 |
98 |
|
9 |
20 |
PhSiH3 (4.0) |
1,2‐C6H4F2 |
80 |
1 |
|
10 |
10 |
PhSiH3 (4.0) |
1,2‐C6H4F2 |
120 |
98 |
|
11 |
5.0 |
PhSiH3 (4.0) |
1,2‐C6H4F2 |
120 |
99 (92) |
|
12 |
2.5 |
PhSiH3 (4.0) |
1,2‐C6H4F2 |
120 |
89 |
|
13 |
5.0 |
PhSiH3 (2.0) |
1,2‐C6H4F2 |
120 |
58 |
|
14 |
20 |
MePhSiH2 (4.0) |
1,2‐C6H4F2 |
120 |
99 |
|
15 |
20 |
Me2PhSiH (4.0) |
1,2‐C6H4F2 |
120 |
96 |
[a] All reactions were performed on a 0.10–0.20 mmol scale in a sealed tube. [b] Yields determined by NMR spectroscopy using mesitylene as an internal standard; isolated yield in parenthesis.
The optimized setup was then applied to mainly benzylic amine derivatives (Scheme 2). Aside from model compound 1 aa with a β‐naphthyl group, an α‐naphthyl and a benzofuran‐5‐yl substituent as in 1 ba and 1 ca at the reactive site also worked. The functional‐group tolerance was assessed with 1‐phenylethan‐1‐amine derivatives 1 da–oa decorated with various electron‐donating or ‐withdrawing substituents on the aryl group (gray box). Yields were generally moderate to good, and all halo groups were compatible (1 ia–la). Substrate 1 ma bearing a methyl ester underwent exhaustive defunctionalization to yield 2 h rather than 2 m with the carboxy group fully reduced to a methyl substituent. Similarly, the deamination of 1 na containing a methyl ether resulted in demethylation3b but when employing Me2PhSiH instead of PhSiH3 the methoxy group remained intact, affording 2 n in 94 % yield along with 4‐methoxystyrene in 6 % yield (β‐elimination). In turn, trifluoromethyl‐substituted substrate 1 oa was too unreactive, and 2 o and 2 h were obtained in 3 % and 5 % yields, respectively. The opposed reactivities of 1 na (X=4‐OMe) and 1 oa (X=4‐CF3) already indicate that this reductive deamination may involve the intermediacy of a benzylic carbocation. This was further substantiated by another observation. Benzylic amine 1 ta with an α‐tert‐butyl group gave the expected hydrocarbon 2 t in 48 % yield; however, a rearranged product generated by a [1,2]‐migration of one of the methyl groups of the tert‐butyl residue to the assumed benzylic carbocation did form in 19 % yield. Other α‐alkyl‐ and α‐aryl‐substituted benzylic amines 1 ra, 1 sa, and 1 ua reacted in good and high yields, respectively. Primary amines 1 va‐ya with a fully substituted α‐carbon atom (benzylic or aliphatic) participated equally well; these transformations are likely to pass through tertiary carbenium ions as intermediates. Conversely, a simple primary amine such as 1 za furnished α‐methylnaphthalene (2 z) in mediocre yield.
Scheme 2.

Scope I: B(C6F5)3‐catalyzed reductive deamination of 1°, 2°, and 3° amines. [a] 5.0 mmol scale. [b] B(C6F5)3 (20 mol %) was used. [c] B(C6F5)3 (10 mol %) was used. [d] B(C6F5)3 (30 mol %) was used. [e] Me2PhSiH (4.0 equiv) was used. [f] 19 % of (3‐methylbutan‐2‐yl)benzene was formed. [g] PhSiH3 (3.0 equiv) was used. [h] PhSiH3 (2.0 equiv) was used. [i] 22 % of 1‐methyl‐3‐ethylindoline was formed.
The reductive deamination was successfully extended to secondary and tertiary benzylic amines (Scheme 2, lower). The yields were generally in range of those obtained for the corresponding primary amines. The deamination of 1 a′d afforded the desired indole derivative 2 a′ in 56 % yield but the corresponding indoline was also formed in 22 % yield. Two drug molecules were subjected to the reductive deamination (Scheme 3): The antidepressant Sertraline (1 b′b) was degraded to tetralin 2 b′ in 87 % yield and the antihistamine Meclizine (1 c′f) furnished 1‐benzyl‐4‐chlorobenzene (2 c′) in 99 % yield.
Scheme 3.

Scope II: B(C6F5)3‐catalyzed reductive deamination for the degradation of drug molecules.
To assess the relative reactivity of the different types of amines, we conducted several control experiments in the presence of 30 mol % of B(C6F5)3 (Scheme 4). We reacted 1.0 equiv of an equimolar mixture of benzylamines with different degrees of substitution at the α‐carbon atom with 3.0 and 6.0 equiv of PhSiH3 (top). With 3.0 equiv, cumene (2 w) formed exclusively in 42 % yield; increasing the amount of the trihydrosilane to 6.0 equiv, 2 w was formed next to ethylbenzene (2 d) in yields of 68 % and 90 %, respectively. The formation of toluene (2 d′) was not detected. These results show that a primary amine with a tertiary alkyl group is more reactive than those with secondary and primary alkyl groups. This trend is in accordance with the stability of their corresponding benzylic carbocations.13 Reactions were routinely run at 120 °C (cf. Table 1) where there was little dependence of conversion on the number of substituents at the nitrogen atoms in benzylic amine derivatives 1 d (bottom). However, when performing the same set of reactions at 80 °C, the reductive deamination was reasonably chemoselective in favor of the secondary amine 1 db. Hence, 1 db is more reactive than the tertiary amine 1 dd and the parent compound 1 da in the B(C6F5)3‐catalyzed reductive deamination.
Scheme 4.

Reactivity study.
To gain further insight into the reaction mechanism, several stoichiometric experiments were performed (Scheme 5).14 As part of the reaction optimization we had already found that 24 h is necessary to achieve full conversion of the primary amine 1 aa. Mixing this amine and B(C6F5)3 in equimolar ratio led to quantitative formation of the Lewis adduct 3 aa within 10 min at room temperature (top left). The subsequent reaction of 3 aa with PhSiH3 (4.0 equiv) at 80 °C generated a new Lewis pair 4 aa in 90 % yield after 8 h; no further reaction of 3 aa and PhSiH3 occurred at room temperature (top right). 4 aa is formally composed of amine 1 aa and Piers’ borane HB(C6F5)2 and can also be synthesized in quantitative yield within a few minutes at room temperature by the reaction of 1 aa and 1.0 equiv of HB(C6F5)2 (not shown). Apparently, B(C6F5)3 and PhSiH3 underwent the known exchange of substituents,15 and the formation of C6F5(Ph)SiH2 (5) was confirmed spectroscopically; the 86 % yield of 5 was estimated by 19F NMR spectroscopy (see the Supporting Information for details). The deaminated product 2 a did only form in 2 % yield, and we showed that 20 mol % of Piers’ borane cannot promote the deamination of 1 aa at 120 °C with 4.0 equiv PhSiH3 as the reductant (not shown). The degradation of the prevalent Lewis pair 1 aa⋅B(C6F5)3 by excess trihydrosilane to catalytically inactive 1 aa⋅HB(C6F5)2 therefore causes catalyst deactivation and in part explains the long reaction times at relatively high catalyst loadings of 5.0 mol % or more. Premixing 1 aa and 4.0 equiv of PhSiH3 followed by the addition of 1.0 equiv of B(C6F5)3 again generated Lewis pair 3 aa in 86 % yield along with its FLP‐type adduct with PhSiH3 in approximately 10 % yield (bottom). Silylammonium borohydrides such as 6 aa are kinetically stable and do not rapidly release dihydrogen9a, 16 which is in agreement with the high reaction temperature. Heating this reaction mixture at 80 °C for 8 h brings about the same outcome as the reverse order of addition. We actually believe that the dehydrogenative Si‐N coupling of the bissilylammonium borohydride intermediate is in competition with its dissociation into the corresponding benzylic carbocation and disilazane (Scheme 6).17 The carbocation is then captured by the borohydride to afford the defunctionalized product.18 However, the hydride source cannot be designated with certainty because trihydrosilane can also deliver one of its hydridic hydrogen atoms as verified in a control experiment. The reductive deamination can be initiated with the trityl cation; for example, 20 mol % of Ph3C+[B(C6F5)4]− converted amine 1 aa into hydrocarbon 2 a in 42 % yield under otherwise identical reaction conditions (4.0 equiv of PhSiH3 in 1,2‐C6H4F2 at 120 °C for 24 h; not shown).
Scheme 5.

Stoichiometric experiments with stepwise addition of the reactants (Ar=β‐naphthyl).
Scheme 6.
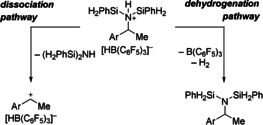
Mechanistic proposal for disilazane as leaving group (monosilazane omitted for clarity and trisilazane not considered for steric reasons): competing dehydrogenation (right) and dissociation pathways (left).
In summary, we have developed a B(C6F5)3‐catalyzed, efficient reductive deamination of 1°, 2°, and 3° mainly benzylic amines with hydrosilanes as the stoichiometric reducing agent. The method extends to other C−N bonds, and an isocyanate, isothiocyanate, and thionyl imide such as 7–9 were shown to undergo the defunctionalization using the same standard protocol (Scheme 7, left). An imine such as 10 does also react (Scheme 7, right). Application of this methodology in organic synthesis is currently ongoing in our laboratory.
Scheme 7.

Scope III: B(C6F5)3‐catalyzed reductive C−N bond cleavage in heterocumulenes and in an imine. [a] B(C6F5)3 (20 mol %) was used.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
H.F. gratefully acknowledges the Alexander von Humboldt Foundation for a postdoctoral fellowship (2018–2020). M.O. is indebted to the Einstein Foundation (Berlin) for an endowed professorship.
H. Fang, M. Oestreich, Angew. Chem. Int. Ed. 2020, 59, 11394.
Contributor Information
Dr. Huaquan Fang, http://www.organometallics.tu‐berlin.de.
Prof. Dr. Martin Oestreich, Email: martin.oestreich@tu-berlin.de.
References
- 1.
- 1a. McCombie S. W. in Comprehensive Organic Synthesis, Vol. 8 (Eds.: B. M. Trost, I. Fleming), Pergamon, Oxford, 1991, pp. 811–833; [Google Scholar]
- 1b. Modak A., Maiti D., Org. Biomol. Chem. 2016, 14, 21–35. [DOI] [PubMed] [Google Scholar]
- 2.For reviews, see:
- 2a. Deuss P. J., Barta K., Coord. Chem. Rev. 2016, 306, 510–532; [Google Scholar]
- 2b. Herrmann J. M., König B., Eur. J. Org. Chem. 2013, 7017–7027; [Google Scholar]
- 2c. Hartwig W., Tetrahedron 1983, 39, 2609–2645. [Google Scholar]
- 3.
- 3a. Gevorgyan V., Liu J.-X., Rubin M., Benson S., Yamamoto Y., Tetrahedron Lett. 1999, 40, 8919–8922; [Google Scholar]
- 3b. Gevorgyan V., Rubin M., Benson S., Liu J.-X., Yamamoto Y., J. Org. Chem. 2000, 65, 6179–6186; [DOI] [PubMed] [Google Scholar]
- 3c. Nimmagadda R. D., McRae C., Tetrahedron Lett. 2006, 47, 5755–5758; [Google Scholar]
- 3d. Yang W., Gao L., Lu J., Song Z., Chem. Commun. 2018, 54, 4834–4837. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Parks D. J., Blackwell J. M., Piers W. E., J. Org. Chem. 2000, 65, 3090–3098; [DOI] [PubMed] [Google Scholar]
- 4b. Rendler S., Oestreich M., Angew. Chem. Int. Ed. 2008, 47, 5997–6000; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 6086–6089; [Google Scholar]
- 4c. Sakata K., Fujimoto H., J. Org. Chem. 2013, 78, 12505–12512; [DOI] [PubMed] [Google Scholar]
- 4d. Houghton A. Y., Hurmalainen J., Mansikkamäki A., Piers W. E., Tuononen H. M., Nat. Chem. 2014, 6, 983–988; [DOI] [PubMed] [Google Scholar]
- 4e. Fallon T., Oestreich M., Angew. Chem. Int. Ed. 2015, 54, 12488–12491; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 12666–12670. [Google Scholar]
- 5.
- 5a. Adduci L. L., Bender T. A., Dabrowski J. A., Gagné M. R., Nat. Chem. 2015, 7, 576–581; [DOI] [PubMed] [Google Scholar]
- 5b. Bender T. A., Dabrowski J. A., Gagné M. R., ACS Catal. 2016, 6, 8399–8403; [Google Scholar]
- 5c. Bender T. A., Payne P. R., Gagné M. R., Nat. Chem. 2018, 10, 85–90; [DOI] [PubMed] [Google Scholar]
- 5d. Seo Y., Gagné M. R., ACS Catal. 2018, 8, 81–85. [Google Scholar]
- 6.
- 6a. Drosos N., Morandi B., Angew. Chem. Int. Ed. 2015, 54, 8814–8818; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 8938–8942; [Google Scholar]
- 6b. Drosos N., Cheng G.-J., Ozkal E., Cacherat B., Thiel W., Morandi B., Angew. Chem. Int. Ed. 2017, 56, 13377–13381; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 13562–13566; [Google Scholar]
- 6c. Cheng G.-J., Drosos N., Morandi B., Thiel W., ACS Catal. 2018, 8, 1697–1702. [Google Scholar]
- 7.
- 7a. Chatterjee I., Porwal D., Oestreich M., Angew. Chem. Int. Ed. 2017, 56, 3389–3391; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 3438–3441; see also: [Google Scholar]
- 7b. Richter S. C., Oestreich M., Chem. Eur. J. 2019, 25, 8508–8512. [DOI] [PubMed] [Google Scholar]
- 8. Hutchins R. O., Cistone F., Goldsmith B., Heuman P., J. Org. Chem. 1975, 40, 2018–2020. [Google Scholar]
- 9.
- 9a. Hermeke J., Mewald M., Oestreich M., J. Am. Chem. Soc. 2013, 135, 17537–17546; see also: [DOI] [PubMed] [Google Scholar]
- 9b. Greb L., Tamke S., Paradies J., Chem. Commun. 2014, 50, 2318–2320 (aniline derivatives and nitrogen heterocycles). [DOI] [PubMed] [Google Scholar]
- 10.Selected examples of transformations of the amino group into a better leaving group followed by hydride substitution, see:
- 10a. Nickon A., Sinz A., J. Am. Chem. Soc. 1960, 82, 753–754; [Google Scholar]
- 10b. Bumgardner C. L., Martin K. J., Freeman J. P., J. Am. Chem. Soc. 1963, 85, 97–99; [Google Scholar]
- 10c. Nickon A., Hill A. S., J. Am. Chem. Soc. 1964, 86, 1152–1158; [Google Scholar]
- 10d. Doldouras G., Kollonitsch J., J. Am. Chem. Soc. 1978, 100, 341–342; [Google Scholar]
- 10e. Katritzky A. R., Horvath K., Plau B., J. Chem. Soc. Perkin Trans. 1 1980, 2554–2560; [Google Scholar]
- 10f. Bumgardner C. L., Desai V. R., J. Fluorine Chem. 1987, 36, 307–312; for a short summary, see: [Google Scholar]
- 10g. Wirth T., Rüchardt C., Chimia 1988, 42, 230–231. [Google Scholar]
- 11.Selected examples of transition-metal-catalyzed hydrogenolysis, see:
- 11a. Maier W. F., Grubmüller P., Thies I., Stein P. M., McKervey M. A., von Ragué Schleyer P., Angew. Chem. Int. Ed. Engl. 1979, 18, 939–940; [Google Scholar]; Angew. Chem. 1979, 91, 1004–1005; [Google Scholar]
- 11b. Guttieri M. J., Maier W. F., J. Org. Chem. 1984, 49, 2875–2880; [Google Scholar]
- 11c. Lemaire-Audoire S., Savignac M., Genêt J. P., Bernard J.-M., Tetrahedron Lett. 1995, 36, 1267–1270; [Google Scholar]
- 11d. Kanai M., Yasumoto M., Kuriyama Y., Inomiya K., Katsuhara Y., Higashiyama K., Ishii A., Org. Lett. 2003, 5, 1007–1010; [DOI] [PubMed] [Google Scholar]
- 11e. Nugent T. C., Negru D. E., El-Shazly M., Hu D., Sadiq A., Bibi A., Umar M. N., Adv. Synth. Catal. 2011, 353, 2085–2092; [Google Scholar]
- 11f. Yap A. J., Chan B., Yuen A. K. L., Ward A. J., Masters A. F., Maschmeyer T., ChemCatChem 2011, 3, 1496–1502. [Google Scholar]
- 12.
- 12a. Oestreich M., Hermeke J., Mohr J., Chem. Soc. Rev. 2015, 44, 2202–2220; see also: [DOI] [PubMed] [Google Scholar]
- 12b. Weber D., Gagné M. R. in Organosilicon Chemistry: Novel Approaches and Reactions (Eds.: T. Hiyama, M. Oestreich), Wiley-VCH, Weinheim, 2019, pp. 33–85. [Google Scholar]
- 13.This order of reactivity is generally opposite to that of the deoxygenation where tertiary alcohols do not react[3b] or do not fully convert into the hydrocarbon.[3c,d] However, trityl alcohol underwent smooth deoxygenation,[3b] likely following an SN1 mechanism. The formation of stabilized carbenium ions as intermediates is crucial in the deamination as disilazanes are poorer leaving groups than disiloxanes. Hence, less substituted oxonium ions can also participate in SN2 displacements with the borohydride while related ammonium ions do not, even at 120 °C.
- 14.1,2-C6D4Cl2 was used instead of 1,2-C6D4F2 as we did not have access to this deuterated solvent.
- 15. Parks D. J., Piers W. E., Yap G. P. A., Organometallics 1998, 17, 5492–5503. [Google Scholar]
- 16. Sumerin V., Schulz F., Nieger M., Leskelä M., Repo T., Rieger B., Angew. Chem. Int. Ed. 2008, 47, 6001–6003; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 6090–6092. [Google Scholar]
- 17.Si-N coupling is observed with substrates less likely to form carbenium ions.[13] For example, the corresponding bissilylamine was found as the major product in the reaction of 1-cyclohexylethan-1-amine and PhSiH3 (cf. Ref. [9a]).
- 18. Chatterjee I., Qu Z.-W., Grimme S., Oestreich M., Angew. Chem. Int. Ed. 2015, 54, 12158–12162; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 12326–12330. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary