

Abstract
Introduction
The efficacy and safety of recombinant factor VIII Fc fusion protein (rFVIIIFc) as an extended half‐life treatment for severe haemophilia A were demonstrated in the Phase 3 A‐LONG and Kids A‐LONG studies. Eligible subjects who completed A‐LONG and Kids A‐LONG could enrol in ASPIRE (NCT01454739), an open‐label extension study.
Aim
To report the long‐term safety and efficacy of rFVIIIFc in subjects with severe haemophilia A who enrolled in ASPIRE.
Methods
Previously treated subjects received one or more of the following regimens: individualized prophylaxis (IP), weekly prophylaxis, modified prophylaxis or episodic treatment. Subjects could switch treatment regimen at any time. The primary endpoint was inhibitor development.
Results
A total of 150 subjects from A‐LONG and 61 subjects from Kids A‐LONG enrolled in ASPIRE. Most subjects received the IP regimen (A‐LONG: n = 110; Kids A‐LONG: n = 59). Median (range) treatment duration in ASPIRE for subjects from A‐LONG and Kids A‐LONG was 3.9 (0.1‐5.3) years and 3.2 (0.3‐3.9) years, respectively. No inhibitors were observed (0 per 1000 subject‐years; 95% confidence interval, 0‐5.2) and the overall rFVIIIFc safety profile was consistent with prior studies. For subjects on the IP regimen, annualized bleed rates (ABR) remained low (median overall ABR for adults and adolescents was <1.0) and extended‐dosing intervals were maintained (median of 3.5 days) for the majority of subjects in ASPIRE.
Conclusion
ASPIRE results, which include up to 5 years of follow‐up data, confirm earlier reports on the consistent and well‐characterized safety and efficacy of rFVIIIFc treatment for severe haemophilia A.
Keywords: bleed rate, extended half‐life, individualized prophylaxis, perioperative haemostasis, rFVIIIFc
1. INTRODUCTION
Prophylactic replacement of clotting factor VIII (FVIII) is standard‐of‐care for people with severe haemophilia A. 1 Individualizing prophylactic regimens is essential to decrease the likelihood of spontaneous bleeds by adjusting plasma FVIII activity to meet individual and lifetime needs. 2 Long‐term bleed protection preserves joint health and maintains quality of life, which are increasingly important outcomes for people with haemophilia because life expectancies are now similar to those of the general population. 3 , 4 , 5
Prophylactic clotting factor regimens can be individualized by considering factors such as bleed phenotype, FVIII pharmacokinetics, physical activity and treatment preferences. 6 , 7 Even with individualized regimens, compliance with standard half‐life FVIII prophylaxis is challenging; the short half‐life necessitates frequent infusion, and limitations imposed by maximum trough levels do not assure complete bleed protection. 8 , 9 , 10 Consequently, extended half‐life (EHL) FVIII products have been designed with structural modifications to decrease FVIII clearance and reduce infusion frequency. 11
Recombinant FVIII Fc fusion protein (rFVIIIFc) is manufactured in a human cell line in an environment free of animal and human additives. It consists of a single monomeric molecule of recombinant FVIII fused to the Fc domain of immunoglobulin G1; the latter binds to the neonatal Fc receptor and extends FVIII half‐life via the natural Fc recycling pathway. 12 rFVIIIFc was the first EHL FVIII product approved by the United States Food and Drug Administration and European Medicines Agency (EMA) and is currently the only EMA‐approved EHL FVIII product for patients <12 years of age. rFVIIIFc is indicated for on‐demand treatment and control of bleed episodes, routine prophylaxis to reduce the frequency of bleeds and perioperative management in children and adults with haemophilia A. 13 , 14 rFVIIIFc has reduced treatment burden by extending the dosing interval and increasing treatment flexibility through individualized regimens, while maintaining or increasing bleed protection. 15 , 16 , 17 Two Phase 3, open‐label, global studies to assess rFVIIIFc treatment for severe haemophilia A in previously treated adults and adolescents (A‐LONG [NCT01181128]) 18 and children (Kids A‐LONG [NCT01458106]) 19 confirmed the safety, efficacy and prolonged activity of rFVIIIFc in all ages. As a single agent, rFVIIIFc enables comprehensive bleed protection across clinical scenarios (acute bleed treatment, prophylaxis and perioperative management). Benefits of rFVIIIFc include improvements in and protection of joint health, attributable to decreased swelling, increased strength and improved range of motion. 17
Eligible subjects completing A‐LONG or Kids A‐LONG could enrol in ASPIRE, the Phase 3 long‐term extension study. Interim analyses from ASPIRE provided initial evidence on the long‐term safety and efficacy of rFVIIIFc prophylaxis. 20 , 21 Here, we report final results from ASPIRE.
2. MATERIALS AND METHODS
2.1. Study design
ASPIRE (NCT01454739) was an open‐label, non‐randomized, global extension trial to assess long‐term safety and efficacy of rFVIIIFc for prevention and treatment of bleed episodes in previously treated adults (≥150 documented prior exposure days [ED]) and children (≥50 EDs) with severe haemophilia A (<1 IU/dL [<1%] endogenous FVIII activity). Eligible subjects who had completed a rFVIIIFc Phase 3 safety and efficacy trial (A‐LONG: subjects ≥12 years of age [NCT01181128]; Kids A‐LONG: subjects <12 years of age [NCT01458106]) 18 , 19 could enrol. Twenty‐nine subjects from two smaller safety and pharmacokinetic trials (NCT02083965; NCT02502149) were subsequently enrolled in ASPIRE but were excluded from this analysis due to their short duration of treatment in ASPIRE. Exclusion of these subjects did not affect the reported outcomes. Subjects with a history of anti‐FVIII‐neutralizing antibodies (inhibitors), hypersensitivity associated with any FVIII concentrate or intravenous immunoglobulin, or other coagulation disorders were excluded. ASPIRE was performed according to the Declaration of Helsinki, and all procedures were approved by local ethics committees. Written informed consent was obtained prior to enrolment from all subjects or subjects’ parent or legal guardian.
Details on study design and treatment have been published previously. 20 , 21 At enrolment, three prophylactic (individualized prophylaxis [IP], weekly prophylaxis [WP] and modified prophylaxis [MP]) regimens and one on‐demand (episodic treatment [ET]) regimen were available to adult and adolescent (≥12 years of age) subjects. Paediatric subjects (<12 years of age) were eligible for IP or MP but could switch to other regimens upon reaching 12 years of age. For all prophylactic regimens, the dose and interval were based on the subject's pharmacokinetic (if available) and clinical profile observed in the parent study and FVIII trough and peak (recovery) values during ASPIRE. The IP group received rFVIIIFc at a dose and interval to target a trough plasma FVIII activity ≤5%, with the lowest effective dose administered to target trough levels 1%‐3%. The IP group received rFVIIIFc at 25‐65 IU/kg every 3‐5 days, or twice weekly at 20‐65 IU/kg on Day 1 and 40‐65 IU/kg on Day 4; subjects <12 years of age received doses ≤80 IU/kg with dosing intervals ≥2 days. The WP group received 65 IU/kg rFVIIIFc weekly. If optimal prophylaxis could not be achieved with IP or WP treatment, subjects could switch to a MP regimen personalized by the investigator (Supporting Information). In the ET group, dose was based on individual bleed type and severity. During ASPIRE, subjects could switch between eligible regimens at any time.
Subjects were followed for ≥100 rFVIIIFc EDs across both parent and extension trials and could continue in the extension study for 4 years or until rFVIIIFc therapy became commercially available in their country. Study visits were scheduled at 6‐month (±2 weeks) intervals, with unscheduled visits occurring per the investigator.
2.2. Outcome measures
The primary endpoint was inhibitor development. Inhibitor testing occurred at each study visit or upon suspected inhibitor development. A positive inhibitor result was defined as a neutralizing antibody value ≥0.6 Bethesda units/mL confirmed by Nijmegen‐modified Bethesda assay within 2‐4 weeks of the initial occurrence. Secondary endpoints were annualized bleed rate (ABR; overall, spontaneous, traumatic, joint and spontaneous joint) per subject, total rFVIIIFc EDs, total weekly prophylactic dose and yearly consumption, physician's global assessment of response to a treatment regimen (excellent, effective, partially effective or ineffective [Supporting Information]) and the subject's self‐assessment of response to treatment of acute bleeds (4‐point scale: excellent, good, moderate or none). Additional endpoints included the incidence of adverse events (AE), investigator and surgeon assessment of haemostatic response to major surgery (defined previously 20 ), number of rFVIIIFc infusions and dose per infusion to maintain haemostasis during major surgery 22 and Hemophilia Joint Health Score (HJHS) or modified HJHS (mHJHS) for individuals <12 and ≥12 years of age, respectively. HJHS is a sensitive tool for detection of early signs of joint damage and is used to assess joint health in children. 17 , 23 Modifications in the mHJHS are minor and involve condensing response scales based on recommendations from the latest HJHS validation study (Supporting Information). 17 , 24 AEs were classified using the Medical Dictionary for Regulatory Activities system organ classes and preferred terms.
2.3. Statistical analyses
The safety analysis included data from all subjects who were exposed to ≥1 dose of rFVIIIFc during ASPIRE. The efficacy analysis included data from all subjects who received ≥1 dose of rFVIIIFc, but excluded data collected during surgical/rehabilitation periods and when >28 days elapsed between infusions for subjects receiving prophylaxis. Efficacy data were stratified by treatment group. Subjects were included in the summary efficacy analysis of each treatment group for the period they received that treatment during ASPIRE and, therefore, may be represented in ≥1 group in the summary analyses. Data were analysed separately for subjects from A‐LONG and Kids A‐LONG, and paediatric subjects were further stratified by age (<6 and 6 to <12 years) at the time of entry into Kids A‐LONG. All statistical analyses were descriptive in nature and no tests were performed on the efficacy endpoints.
3. RESULTS
3.1. Study population
A total of 211 previously treated male subjects from A‐LONG (n = 150) and Kids A‐LONG (<6 years of age: n = 30; 6 to <12 years of age: n = 31) enrolled in ASPIRE (Figure 1). Subject age ranged from 2 to 66 years (Table 1). All subjects received ≥1 dose of rFVIIIFc and 88% of subjects (186/211; 132 from A‐LONG and 54 from Kids A‐LONG) completed the study. During ASPIRE, 21 subjects from A‐LONG and 1 from Kids A‐LONG (IP to MP) switched to an on‐study regimen; most switches were to MP to allow a preventive dose before strenuous activity (Supporting Information). No subject switched regimens more than once.
FIGURE 1.
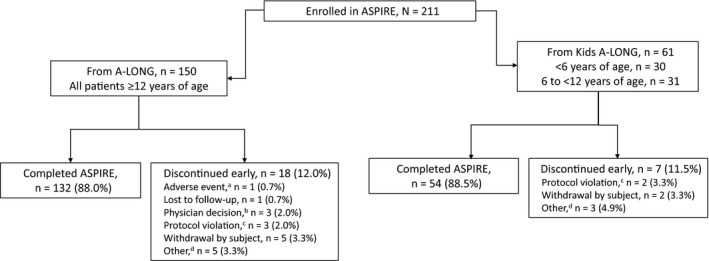
Subject disposition for ASPIRE extension study. aSubject was on an episodic treatment regimen and discontinued owing to a non‐serious adverse event of chronic renal failure that was considered unrelated to recombinant factor VIII Fc fusion protein. bSubjects were withdrawn because of the physician's decision for non‐compliance with the study (n = 3). cProtocol violations included non‐compliance with prophylactic dosing (n = 1), use of non‐study factor VIII under circumstances that were not an emergency or an accident (n = 2), non‐compliance with study procedures, including infusion timing and concomitant medications (n = 1), lost to follow‐up and incomplete end of study visit (n = 1). dIncludes product becoming commercially available in the subject's country (n = 3), commencing a different clinical trial (n = 2), inability to comply with the demands of the study (n = 1), early termination (n = 1) and incarceration (n = 1)
TABLE 1.
Subject demographics in ASPIRE by parent study and treatment regimen
| Parent study | A‐LONG a | Kids A‐LONG | ||||||
|---|---|---|---|---|---|---|---|---|
| Treatment regimen | IP (n = 110) | WP (n = 27) | MP (n = 21) | ET (n = 13) | Aged <6 y b | Aged 6 to <12 y | ||
| IP (n = 29) | MP (n = 2) | IP (n = 30) | MP (n = 1) | |||||
| Median (min‐max) age, y | 31 (13‒66) | 32 (19‒63) | 33 (14‒59) | 35 (14‒57) | 4 (2‒6) | 6 (6‒6) | 9 (6‒12) | 9 |
| Geographic location, n (%) | ||||||||
| Europe c | 29 (26.4) | 8 (29.6) | 2 (9.5) | 2 (15.4) | 16 (55.2) | 0 (0.0) | 15 (50.0) | 0 (0.0) |
| North America d | 40 (36.4) | 7 (25.9) | 4 (19.0) | 5 (38.5) | 3 (10.3) | 1 (50.0) | 6 (20.0) | 1 (100.0) |
| Australia e | 6 (5.5) | 0 (0.0) | 6 (28.6) | 1 (7.7) | 4 (13.8) | 1 (50.0) | 2 (6.7) | 0 (0.0) |
| Brazil | 3 (2.7) | 0 (0.0) | 0 (0.0) | 1 (7.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Asia f | 17 (15.5) | 10 (37.0) | 9 (42.9) | 4 (30.8) | 2 (6.9) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| South Africa | 15 (13.6) | 2 (7.4) | 0 (0.0) | 0 (0.0) | 4 (13.8) | 0 (0.0) | 7 (23.3) | 0 (0.0) |
| Race, n (%) | ||||||||
| White | 75 (68.2) | 18 (66.7) | 10 (47.6) | 10 (76.9) | 19 (65.5) | 2 (100.0) | 21 (70.0) | 1 (100.0) |
| Black | 7 (6.4) | 1 (3.7) | 0 (0.0) | 0 (0.0) | 4 (13.8) | 0 (0.0) | 4 (13.3) | 0 (0.0) |
| Asian | 23 (20.9) | 8 (29.6) | 10 (47.6) | 3 (23.1) | 3 (10.3) | 0 (0.0) | 1 (3.3) | 0 (0.0) |
| Other | 5 (4.5) | 0 (0.0) | 1 (4.8) | 0 (0.0) | 3 (10.3) | 0 (0.0) | 4 (13.3) | 0 (0.0) |
| Ethnicity, n (%) | ||||||||
| Hispanic or Latino | 6 (5.5) | 1 (3.7) | 0 (0.0) | 2 (15.4) | 1 (3.4) | 0 (0.0) | 1 (3.3) | 0 (0.0) |
| Not Hispanic or Latino | 104 (94.5) | 26 (96.3) | 21 (100.0) | 11 (84.6) | 28 (96.6) | 2 (100.0) | 29 (96.7) | 1 (100.0) |
Abbreviations: ET, episodic treatment; IP, individualized prophylaxis; max, maximum; min, minimum; MP, modified prophylaxis; WP, weekly prophylaxis.
Twenty‐one subjects switched treatment regimens once during the study.
One subject switched from IP to MP.
Includes Austria, Belgium, France, Germany, Ireland, Italy, Netherlands, Poland, Spain, Sweden, Switzerland and the United Kingdom.
Includes Canada and the United States.
Includes Australia and New Zealand.
Includes China (Hong Kong), India, Israel and Japan.
3.2. Duration and exposure
For subjects from A‐LONG, the median (range) cumulative treatment duration in A‐LONG and ASPIRE was 4.5 (0.7‐5.9) years, which includes 3.9 (0.1‐5.3) years in ASPIRE only. For paediatric subjects, corresponding values were 3.5 (0.4‐4.4) years and 3.2 (0.3‐3.9) years, respectively.
For subjects from A‐LONG, the median (range) cumulative number of EDs in A‐LONG and ASPIRE was 333 (36‐735) days, which includes 268 (8‐660) days in ASPIRE only. For paediatric subjects, corresponding values were 375 (42‐529) days and 332 (18‐467) days, respectively.
3.3. Safety
No subject developed an inhibitor during ASPIRE (0 per 1000 subject‐years; 95% confidence interval, 0‐5.2). rFVIIIFc was well tolerated with an AE pattern consistent with those expected for subjects with severe haemophilia A (Table 2). One subject from A‐LONG receiving ET discontinued ASPIRE owing to a non‐serious AE of chronic renal failure (elevated serum creatinine due to chronic kidney disease) that was considered unrelated to rFVIIIFc. No paediatric subjects discontinued treatment because of an AE. One subject from Kids A‐LONG developed urticaria, which was considered unrelated to study treatment, and discontinued ASPIRE owing to the use of non‐study FVIII. Three AEs in 2 A‐LONG subjects were considered related to rFVIIIFc treatment (headache and hot flush [n = 1]; chromaturia [n = 1]). These AEs were mild in severity and did not lead to study discontinuation. There were no treatment‐related serious AEs, deaths, anaphylaxis, serious hypersensitivity events or vascular thrombotic events.
TABLE 2.
AEs a during ASPIRE overall and by parent study
| Overall (N = 211) | A‐LONG (n = 150) | Kids A‐LONG (n = 61) | |
|---|---|---|---|
| ≥1 AE, n (%) | 184 (87.2) | 129 (86.0) | 55 (90.2) |
| ≥1 rFVIIIFc‐related AE, n (%) | 2 (0.9) | 2 (1.3) | 0 (0.0) |
| Most common AEs (≥10% per parent study population), n (%) | |||
| Nasopharyngitis | 43 (20.3) | 37 (24.7) | 6 (9.8) |
| Upper respiratory tract infection | 30 (14.2) | 17 (11.3) | 13 (21.3) |
| Fall | 30 (14.2) | 14 (9.3) | 16 (26.2) |
| Arthralgia | 26 (12.3) | 19 (12.7) | 7 (11.5) |
| Headache | 24 (11.4) | 13 (8.7) | 11 (18.0) |
| Diarrhoea | 20 (9.5) | 15 (10.0) | 5 (8.2) |
| Cough | 17 (8.1) | 9 (6.0) | 8 (13.1) |
| Haemophilic arthropathy | 15 (7.1) | 15 (10.0) | 0 (0.0) |
| Vomiting | 14 (6.6) | 6 (4.0) | 8 (13.1) |
| Seasonal allergy | 13 (6.2) | 5 (3.3) | 8 (13.1) |
| Tonsillitis | 13 (6.2) | 2 (1.3) | 11 (18.0) |
Abbreviations: AE, adverse event; rFVIIIFc, recombinant factor VIII Fc fusion protein.
Does not include AEs during major surgical or rehabilitation periods.
3.4. ABRs and joint health
ABRs remained low and stable throughout ASPIRE in the prophylaxis groups and were lowest in subjects from A‐LONG following the IP regimen (median for all ABR categories <1.0; Figure 2 and Supporting Information). Median ABR for spontaneous joint bleeds was 0.0 for subjects of all ages receiving IP. ABRs were also low for subjects with impaired joint health (≥1 target joint at entry into the parent trials; Supporting Information). In this group, median (interquartile range [IQR]) overall ABR during ASPIRE for subjects from A‐LONG was 0.7 (0.0‐2.8) for IP (n = 72), 2.2 (0.3‐5.2) for WP (n = 16), 5.0 (2.9‐11.0) for MP (n = 16) and 16.1 (0.0‐37.8) for ET (n = 11). Corresponding values for subjects with ≥1 target joint at entry into Kids A‐LONG were 1.1 (0.6‐2.2) for IP (n = 7) and 4.1 for MP (n = 1) during ASPIRE. From the beginning to the end of ASPIRE, the mean (standard deviation) changes in mHJHS for adult and adolescent subjects (n = 72) and HJHS for paediatric subjects (n = 35) were −2.5 (7.1) and −0.5 (1.7), respectively.
FIGURE 2.
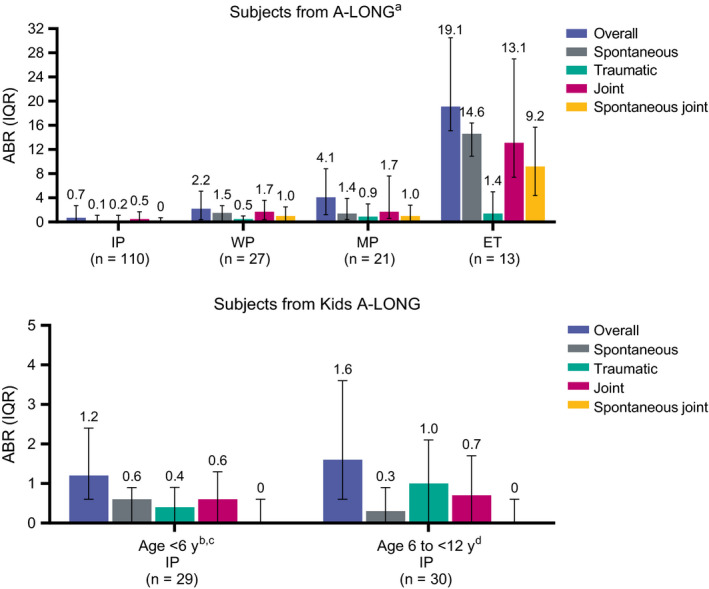
ABRs during ASPIRE by parent study and treatment regimen. ABR, annualized bleed rate; ET, episodic treatment; IP, individualized prophylaxis; IQR, interquartile range; MP, modified prophylaxis; WP, weekly prophylaxis. aTwenty‐one subjects switched treatment regimens once during the study. bOne subject switched from IP to MP. cThe two subjects <6 y of age receiving MP had overall, spontaneous, traumatic, joint and spontaneous joint ABR IQRs of 3.4‐4.1, 2.0‐3.1, 1.0‐1.3, 1.3‐4.1 and 1.3‐3.1, respectively. dThe single subject 6 to <12 y of age receiving MP had both an overall ABR and traumatic ABR of 1.0
3.5. Dosing interval, factor consumption and compliance
Median IP dosing interval was approximately 3.5 days for all age groups (Table 3). For subjects from A‐LONG, median WP and MP dosing intervals were 7.0 and 5.0 days, respectively (Table 3). Most subjects (A‐LONG: 71%; Kids A‐LONG: 89%) maintained dosing intervals achieved in the parent studies. For adults and adolescents, the dosing interval lengthened for 21% of subjects and shortened for 8% of subjects; 23% lengthened their dosing interval to >5 days. For paediatric subjects, corresponding values were 7% and 5%, respectively, and 3% (all <6 years of age) lengthened their dosing interval to >5 days.
TABLE 3.
Prophylactic dosing during ASPIRE by parent study and treatment regimen
| Parent study | A‐LONG a | Kids A‐LONG | |||
|---|---|---|---|---|---|
| Treatment regimen | IP | WP | MP b | Aged <6 y | Aged 6 to <12 y |
| IP | IP | ||||
| Number of subjects, n | 110 | 27 | 21 | 29 c , d | 30 e |
| Median (IQR) dosing interval, d | 3.5 (3.5‐5.0) | 7.0 (7.0‐7.1) | 5.0 (4.0‐6.9) | 3.5 (3.5‐3.5) | 3.5 (3.5‐3.5) |
| Median (IQR) weekly dose, IU/kg | 79.5 (73.7‐100.9) | 65.7 (61.9‐67.2) | 70.6 (62.3‐90.4) | 101.9 (88.7‐118.7) | 94.9 (81.7‐109.1) |
Abbreviations: IP, individualized prophylaxis; IQR, interquartile range; MP, modified prophylaxis; WP, weekly prophylaxis.
Twenty‐one subjects switched treatment regimens once during ASPIRE.
MP was not available in A‐LONG.
One subject switched from IP to MP.
The two subjects <6 y of age receiving MP had dosing intervals of 2.3‐5.5 d and weekly doses of 81.5‐118.7 IU/kg.
The single subject 6 to <12 y of age receiving MP had a dosing interval of 3.5 d and a weekly dose of 84.5 IU/kg.
There was no change in median (IQR) weekly factor consumption for adults and adolescents (n = 128) from the end of A‐LONG (75 [70‐91] IU/kg) to the end of ASPIRE (75 [70‐97] IU/kg) (Table 3). For paediatric subjects (n = 61), median (IQR) weekly factor consumption was higher in ASPIRE (95 [75‐116] IU/kg) than the end of Kids A‐LONG (75 [75‐105] IU/kg). Overall, 94% (190/202) and 95% (192/202) of subjects in a prophylactic regimen were dose compliant (within 80%‐125% of the prescribed dose) and interval compliant (within ±1 day of the prescribed interval), respectively.
3.6. Global assessment of response to prophylaxis
Over 99% of physicians’ assessments of responses at subject visits were excellent (87% [1464/1680]) or effective (13% [210/1680]). The remainder (0.4% [6/1680]) were graded as partially effective; no responses were graded as ineffective during ASPIRE.
3.7. Control of acute bleed episodes
Overall, >75% of acute bleed episodes were controlled by one rFVIIIFc infusion and >93% with ≤2 infusions (Table 4). The majority of first infusions (≥73%) were rated as having excellent or good responses by subjects.
TABLE 4.
Control of acute bleed episodes in ASPIRE by parent study and treatment regimen
| Parent study | A‐LONG | Kids A‐LONG | ||||
|---|---|---|---|---|---|---|
| Treatment regimen | IP (n = 110) | WP (n = 27) | MP (n = 21) | ET (n = 13) |
IP Aged < 6 y (n = 29) |
IP Aged 6 to <12 y (n = 30) |
| Episodes required ≤2 transfusions, % | 93.7 | 97.1 | 94.3 | 99.2 | 93.5 | 93.4 |
| Episodes required ≤1 transfusions, % | 82.5 | 91.5 | 85.7 | 97.9 | 79.9 | 75.8 |
| Median (IQR) total dose per bleed episode, IU/kg |
50.4 (30.4‐58.8) |
33.7 (26.7‐54.0) |
40.3 (31.5‐63.2) |
26.4 (20.4‐30.3) |
58.4 (33.8‐84.4) |
52.1 (42.4‐75.5) |
Abbreviations: ET, episodic treatment; IP, individualized prophylaxis; IQR, interquartile range; MP, modified prophylaxis; WP, weekly prophylaxis.
3.8. Perioperative management
During ASPIRE, 39 major and 69 minor surgeries were performed in 26 and 54 subjects, respectively. The most common major surgeries included unilateral knee, elbow and ankle arthroplasties; arthroscopy thoracotomy; spinal surgery; and ureteroscopy. Two of the 39 major surgeries were performed in A‐LONG, and the rehabilitative period extended into ASPIRE. Of the 37 major surgeries in ASPIRE, 33 were assessed for haemostatic response and all were rated as excellent (94% [31/33]) or good (6% [2/33]). For adults and adolescents, 74% required one rFVIIIFc infusion to maintain haemostasis during major surgery; 17% required two infusions, and specific infusion data were missing for the remaining subjects (9%) receiving surgery. For paediatric subjects, one rFVIIIFc infusion was sufficient to maintain haemostasis during both major surgeries. The median rFVIIIFc dose per infusion during surgery was 59.6 IU/kg for adults and adolescents and 51.8 IU/kg for paediatric subjects. Most major surgeries (92%) did not require red blood cell transfusions.
4. DISCUSSION
ASPIRE was a large international study enrolling subjects of all ages (range, 2‐66 years) with demographic diversity, long‐term (up to 5.3 years) follow‐up and flexibility in rFVIIIFc dosing with the option to switch treatment regimen at any time. With individualized dosing assuming an essential role in the management of severe haemophilia A, the unique design of ASPIRE approximated real‐world practice.
These final results of ASPIRE are consistent with those of the A‐LONG and Kids A‐LONG Phase 3 trials and an earlier interim analysis 17 , 19 , 20 and confirm the long‐term, well‐characterized safety and efficacy of rFVIIIFc in previously treated subjects with severe haemophilia A. rFVIIIFc was well tolerated across all age groups and did not lead to development of inhibitors or treatment‐related serious AEs. Most subjects received IP, which was associated with low ABRs in subjects of all ages. Median ABR (overall and all ABR subcategories) in adults and adolescents receiving IP was <1.0, with similar outcomes reported for children. ABR data for IP support rFVIIIFc dosing according to the pharmacokinetic profile of a subject. Perioperative rFVIIIFc infusion provided excellent haemostatic control during surgery for most subjects. Extended‐dose intervals achieved in the parent trials were lengthened or maintained for most subjects in ASPIRE.
Prophylaxis with FVIII should ideally be initiated before onset of repeated joint bleeds, to preserve long‐term joint function and prevent or diminish chronic pain and joint disability. 25 Median ABR for spontaneous joint bleeds was 0.0 for all subjects receiving IP. For subjects receiving IP that had ≥1 target joint at entry into A‐LONG, median overall ABR during ASPIRE was 0.7. Furthermore, mHJHS and HJHS decreased from the beginning to the end of ASPIRE. These improvements in joint health suggest that the clinical benefits of rFVIIIFc prophylaxis may go beyond ABR reduction.
The results in ASPIRE are complemented by reports describing real‐world use of prophylactic rFVIIIFc for people with severe haemophilia A. Wang and Young (2018) performed a retrospective review of medical records of 17 patients with severe haemophilia A receiving prophylaxis with recombinant FVIII who switched to rFVIIIFc. After switching, ABR and annualized joint bleed rate decreased from 2.3 and 1.8 to 1.3 and 0.7, respectively. rFVIIIFc dosing frequency ranged from twice weekly to once every 5 days, and weekly factor consumption decreased in 53% (9/17) of patients after starting rFVIIIFc prophylaxis. No patient developed inhibitors while on rFVIIIFc treatment (median [range] follow‐up of 230 [133‐329] days) and no treatment‐related AEs were reported. 26 Keepanasseril et al evaluated the real‐world experience with rFVIIIFc in Canada for the first 8 months after approval by Health Canada in 2014. There was a 19% decrease in factor consumption among 62 patients with severe haemophilia A who switched from prophylaxis with a standard half‐life product to rFVIIIFc. Reasons for switching included to improve quality of life, improve compliance and reduce bleed frequency. No patient receiving rFVIIIFc developed inhibitors during the follow‐up period. 27 Peyvandi et al conducted a real‐world survey to determine the efficacy of EHL products in Europe. After switching to an EHL product, 66% (15/23) of responding haemophilia treatment centres reported ≥30% reduction in the number of transfusions and 43% (9/21) reported a ≥20% reduction of bleeds. 9 As of 5 June 2019, the estimated patient exposure to rFVIIIFc is approximately 12 900 person‐years cumulatively since launch, based on commercial sales (excludes humanitarian sourced data). 28
In recent years, the haemophilia A treatment landscape has changed significantly. Prophylaxis and individualized care are increasingly becoming the global standard and non‐factor therapies are emerging. Still, factor‐based therapies remain fundamental and essential single‐agent treatments for the comprehensive management of adults and children with haemophilia across a wide range of clinical situations. Extensive clinical trial and real‐world evidence of safety and efficacy show that rFVIIIFc, as an EHL molecule, may provide bleed protection, resolve target joints and enhance joint protection without a burdensome dosing frequency. 17 rFVIIIFc provides effective treatment of acute bleeds, and prophylaxis with rFVIIIFc may intensify protection during high physical activity, provide effective perioperative management and improve quality of life. 17 , 18 , 19 , 20 , 21 Further, standard laboratory assays can be used to reliably monitor rFVIIIFc levels. 29 This study increases our understanding of the safety and efficacy profile of personalized prophylaxis with rFVIIIFc for adults and children with severe haemophilia A across clinical scenarios.
5. CONCLUSION
The results of the ASPIRE extension trial confirm findings from the Phase 3 A‐LONG and Kids A‐LONG studies that long‐term rFVIIIFc prophylaxis with an extended‐dose interval has a favourable safety profile, is well tolerated in previously treated subjects with severe haemophilia A and is not associated with inhibitor development. Prophylaxis was efficacious across all ages and was associated with low ABRs and improvements in joint health, an important goal for treatment of severe haemophilia A. The data from this long‐term follow‐up study demonstrate the value of rFVIIIFc for managing acute bleeds and perioperative haemostasis and for providing protection via personalized prophylaxis regimens.
DISCLOSURES
BN has been a study investigator for Sobi, Biogen/Bioverativ, a Sanofi company/Sanofi, CSL, Bayer and Sanofi; received honoraria from Sobi (honoraria donated to Irish Haemophilia Society); JM has received research grants from and has been on the advisory board for BioMarin, Baxalta, Catalyst Biosciences, CSL, Novartis, Novo Nordisk, Pfizer, Roche, Sanofi, Spark, Roche and Unique; and has been on the speaker's bureau for ISTH, Novo Nordisk, Pfizer, Roche, Sanofi, Takeda and the World Federation of Hemophilia; IP has received honoraria from and acted as a consultant for Sobi, CSL Behring, Bayer, Shire, Pfizer, Novo Nordisk and Biotest; GY has received honoraria from and/or has acted as a consultant for Bayer, Bioverativ, CSL Behring, Genentech/Roche, Grifols, Novo Nordisk, Spark, Takeda and Unique; BAK has acted as a paid consultant for BioMarin Pharmaceutical Inc, Genentech/Roche, Bioverativ/Sanofi and Spark Therapeutics; received research funding from Bioverativ/Sanofi, Shire/Takeda, Spark Therapeutics, Octapharma, Pfizer, Sangamo and uniQure; CB has acted as a paid consultant for Sanofi; KN, ES, LK, HY and JF have no competing interests; KJP has received honoraria from Sanofi, Sobi, Biotest, Octapharma, Novo Nordisk, Roche, Takeda, BioMarin and Catalyst Biosciences as a member of scientific advisory boards and symposia; BW is an employee of Sobi; DR is currently employed by Global Blood Therapeutics (GBT) with stock/options; is a past employee of Bioverativ and was provided with pay and stock at the time but no longer has any other financial interest; JO has received grants and personal fees from Bayer, Biotest, CSL Behring, Novo Nordisk, Octapharma and Shire, and personal fees from Chugai, Grifols, Pfizer, Roche and Sobi outside the submitted work. Personal fees were received for travel support, participation in advisory boards and participating in symposia as a chair or speaker.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
ASPIRE was sponsored by Sanofi (Cambridge, MA, USA) and Sobi (Stockholm, Sweden). Medical writing and editing support were provided by Rebecca Lawson, PhD, Francis Golder, BVSc, PhD, DACVAA and Jennifer Alexander, MSc, MBA, CMPP, of JK Associates Inc, a member of the Fishawack Health (Conshohocken, PA, USA) and was funded by Sanofi and Sobi.
Nolan B, Mahlangu J, Pabinger I, et al. Recombinant factor VIII Fc fusion protein for the treatment of severe haemophilia A: Final results from the ASPIRE extension study. Haemophilia. 2020;26:494–502. 10.1111/hae.13953
DATA AVAILABILITY STATEMENT
Qualified researchers may request access to patient level data and related study documents including the clinical study report, study protocol with any amendments, blank case report form, statistical analysis plan and dataset specifications. Patient level data will be anonymized and study documents will be redacted to protect the privacy of our trial participants. Further details on Sanofi's data sharing criteria, eligible studies and process for requesting access can be found at: https://www.clinicalstudydatarequest.com/.
REFERENCES
- 1. National Hemophilia Foundation . MASAC document 241: MASAC recommendations concerning Prophylaxis. https://www.hemophilia.org/Researchers‐Healthcare‐Providers/Medical‐and‐Scientific‐Advisory‐Council‐MASAC/MASAC‐Recommendations/MASAC‐Recommendation‐Concerning‐Prophylaxis. Accessed October 25, 2018.
- 2. Petrini P, Valentino LA, Gringeri A, Re WM, Ewenstein B. Individualizing prophylaxis in hemophilia: a review. Expert Rev Hematol. 2015;8:237‐246. [DOI] [PubMed] [Google Scholar]
- 3. Lambert T, Auerswald G, Benson G, et al. Joint disease, the hallmark of haemophilia: what issues and challenges remain despite the development of effective therapies? Thromb Res. 2014;133:967‐971. [DOI] [PubMed] [Google Scholar]
- 4. Oladapo AO, Epstein JD, Williams E, Ito D, Gringeri A, Valentino LA. Health‐related quality of life assessment in haemophilia patients on prophylaxis therapy: a systematic review of results from prospective clinical trials. Haemophilia. 2015;21:e344‐e358. [DOI] [PubMed] [Google Scholar]
- 5. Franchini M, Mannucci PM. Hemophilia A in the third millennium. Blood Rev. 2013;27:179‐184. [DOI] [PubMed] [Google Scholar]
- 6. Oldenburg J. Optimal treatment strategies for hemophilia: achievements and limitations of current prophylactic regimens. Blood. 2015;125:2038‐2044. [DOI] [PubMed] [Google Scholar]
- 7. Sørensen B, Auerswald G, Benson G, et al. Rationale for individualizing haemophilia care. Blood Coagul Fibronolysis. 2015;26:849‐857. [DOI] [PubMed] [Google Scholar]
- 8. Castaman G, Linari S. Prophylactic versus on‐demand treatments for hemophilia: advantages and drawbacks. Expert Rev Hematol. 2018;11:567‐576. [DOI] [PubMed] [Google Scholar]
- 9. Peyvandi F, Garagiola I, Boscarino M, Ryan A, Hermans C, Makris M. Real‐life experience in switching to new extended half‐life products at European haemophilia centres. Haemophilia. 2019;25(6):946‐952. [DOI] [PubMed] [Google Scholar]
- 10. Hacker MR, Geraghty S, Manco‐Johnson M. Barriers to compliance with prophylaxis therapy in haemophilia. Haemophilia. 2001;7:392‐396. [DOI] [PubMed] [Google Scholar]
- 11. Mahdi AJ, Obaji SG, Collins PW. Role of enhanced half‐life factor VIII and IX in the treatment of haemophilia. Br J Haematol. 2015;169:768‐776. [DOI] [PubMed] [Google Scholar]
- 12. Peters RT, Toby G, Lu Q, et al. Biochemical and functional characterization of a recombinant monomeric factor VIII‐Fc fusion protein. J Thromb Haemost. 2013;11:132‐141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bioverativ Therapeutics Inc . ELOCTATE® Prescribing Information. https://www.eloctate.com/_assets/pdf/eloctate_pi.pdf. Accessed February 04, 2019.
- 14. Swedish Orphan Biovitrum AB (publ) . Elocta: Summary of Product Characteristics. https://www.ema.europa.eu/en/documents/product‐information/elocta‐epar‐product‐information_en.pdf. Accessed September 06, 2019.
- 15. Berntorp E, Negrier C, Gozzi P, Blaas PM, Lethagen S. Dosing regimens, FVIII levels and estimated haemostatic protection with special focus on rFVIIIFc. Haemophilia. 2016;22:389‐396. [DOI] [PubMed] [Google Scholar]
- 16. Chowdary P, Fosbury E, Riddell A, Mathias M. Therapeutic and routine prophylactic properties of rFactor VIII Fc (efraloctocog alfa, Eloctate®) in hemophilia A. J Blood Med. 2016;7:187‐198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Oldenburg J, Kulkarni R, Srivastava A, et al. Improved joint health in subjects with severe haemophilia A treated prophylactically with recombinant factor VIII Fc fusion protein. Haemophilia. 2018;24:77‐84. [DOI] [PubMed] [Google Scholar]
- 18. Mahlangu J, Powell JS, Ragni MV, et al. Phase 3 study of recombinant factor VIII Fc fusion protein in severe hemophilia A. Blood. 2014;123(3):317‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Young G, Mahlangu J, Kulkarni R, et al. Recombinant factor VIII Fc fusion protein for the prevention and treatment of bleeding in children with severe hemophilia A. J Thromb Haemost. 2015;13:967‐977. [DOI] [PubMed] [Google Scholar]
- 20. Mahlangu J, Ragni M, Gupta N, et al. Long‐acting recombinant factor VIII Fc fusion protein (rFVIIIFc) for perioperative haemostatic management in severe haemophilia A. Thromb Haemost. 2016;116:1‐8. [DOI] [PubMed] [Google Scholar]
- 21. Nolan B, Mahlangu J, Perry D, et al. Long‐term safety and efficacy of recombinant factor VIII Fc fusion protein (rFVIIIFc) in subjects with haemophilia A. Haemophilia. 2016;22:72‐80. [DOI] [PubMed] [Google Scholar]
- 22. Srivastava A, Brewer AK, Mauser‐Bunschoten EP, et al. Guidelines for the management of hemophilia. Haemophilia. 2013;19:e1‐e47. [DOI] [PubMed] [Google Scholar]
- 23. Hilliard P, Funk S, Zourikian N, et al. Hemophilia joint health score reliability study. Haemophilia. 2006;12:518‐525. [DOI] [PubMed] [Google Scholar]
- 24. Feldman BM, Funk SM, Bergstrom B‐M, et al. Validation of a new pediatric joint scoring system from the International Hemophilia Prophylaxis Study Group: validity of the hemophilia joint health score. Arthritis Care Res (Hoboken). 2011;63:223‐230. [DOI] [PubMed] [Google Scholar]
- 25. Fischer K, Collins PW, Ozelo MC, Srivastava A, Young G, Blanchette VS. When and how to start prophylaxis in boys with severe hemophilia without inhibitors: communication from the SSC of the ISTH. J Thromb Haemost. 2016;14:1105‐1109. [DOI] [PubMed] [Google Scholar]
- 26. Wang C, Young G. Clinical use of recombinant factor VIII Fc and recombinant factor IX Fc in patients with haemophilia A and B. Haemophilia. 2018;24:414‐419. [DOI] [PubMed] [Google Scholar]
- 27. Keepanasseril A, Stoffman J, Bouskill V, et al. Switching to extended half‐life products in Canada ‐ preliminary data. Haemophilia. 2017;23:e365‐e367. [DOI] [PubMed] [Google Scholar]
- 28. Sanofi . Data on file.
- 29. Kitchen S, Jennings I, Makris M, Kitchen DP, Woods TAL, Walker ID. Clotting and chromogenic factor VIII assay variability in post‐infusion and spiked samples containing full‐length recombinant FVIII or recombinant factor VIII Fc fusion protein (rFVIIIFc). Int J Lab Hematol. 2019;41:176‐183. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
Qualified researchers may request access to patient level data and related study documents including the clinical study report, study protocol with any amendments, blank case report form, statistical analysis plan and dataset specifications. Patient level data will be anonymized and study documents will be redacted to protect the privacy of our trial participants. Further details on Sanofi's data sharing criteria, eligible studies and process for requesting access can be found at: https://www.clinicalstudydatarequest.com/.