

Abstract
Functionalization with C1‐building blocks are key synthetic methods in organic synthesis. The low reactivity of the most abundant C1‐molecule, carbon dioxide, makes alternative carboxylation reactions with CO2‐surrogates especially important. We report a photoredox‐catalyzed protocol for alkene carbamoylations. Readily accessible 4‐carboxamido‐Hantzsch esters serve as convenient starting materials that generate carbamoyl radicals upon visible light‐mediated single‐electron transfer. Addition to various alkenes proceeded with high levels of regio‐ and chemoselectivity.
Keywords: 1,4-dihydropyridines; carbamoyl radicals; Giese reaction; organic dyes; photoredox catalysis
Go photo! A photocatalytic carbamoylation of alkenes was developed under mild conditions with a metal‐free catalyst. The reaction utilizes easily available Hantzsch ester derivatives as precursors to carbamoyl radicals which readily add to electron‐deficient alkenes.

C1‐building blocks are among the most abundant and available chemicals (e.g. CO1‐2, COCl2, Me‐H/OH/Hal, HCN).1 However, the range of diverse C1‐building blocks is rather limited, hence access to a wide variety of methods is crucial to their implementation into various chemical syntheses. The use of nucleophilic carbamoyl radicals, as a formal amide synthon and one‐electron‐reduced CO2 surrogate, can provide one of the most valuable C1‐functional groups and carboxyl derivatives: carboxamides. Amide‐containing molecules are ubiquitous across every corner of chemical constituents, from proteins and pharmaceuticals, to polymers and agrochemicals.2 Classical condensation‐based protocols3 are readily contrasted with modern and non‐obvious retrosynthetic approaches, for example, by less‐explored radical strategies (Scheme 1, top). As such, the addition of organic carbon‐centered radicals to electron‐deficient olefins (Giese reaction) has become a versatile tool for the construction of C−C bonds in organic synthesis.4a, 4b, 4c Since its discovery, a variety of free radical species (alkyl,5a acyl,5b carbamoyl5c) have been employed as nucleophiles to furnish their corresponding homologues (alkanes, ketones, amides, respectively). While conventional Giese‐type protocols rely on harsh conditions for the initiation of the reaction (incl. toxic tin reagents, elevated temperatures, or high‐energy UV light), the advent of modern photoredox catalysis has provided milder routes of radical generation.6 Significant progress has been made towards photoredox‐catalyzed alkylations and acylations, whereas the convenient formation and wide utilization of free carbamoyl radicals has remained elusive and rather limited, in most of the cases, to the addition to (hetero)arenes in a Minisci‐type process.7 The first light‐mediated radical carbamoylation of olefins was reported by Elad and Rokach in 1964 via H‐atom abstraction of formamide to give low yields and low selectivity of the desired products.8 Photoredox‐catalyzed carbamoylations were recently reported by Donald9 via reductive decarboxylation of N‐oxyphthalimido oxamides and by Feng10 via a related oxidative decarboxylation of oxamic acids (Scheme 1 a). Both protocols involve noble‐metal containing photocatalysts and further reaction of the intermediate alkyl radicals onto arene substituents leading to dihydroquinolin‐2‐ones. Hantzsch ester derivatives have only very recently been discovered as convenient precursors to alkyl,11 acyl,12 and—during the course of this study—carbamoyl13 radicals via photoredox‐catalysis or direct photoexcitation. We surmised that the readily accessible 4‐carboxamido‐substituted Hantzsch esters (1) could be utilized for an unprecedented photocatalytic approach to mild carbamoyl‐radical additions to furnish functionalized carboxamides comprising 1,4‐dicarbonyl motifs (Scheme 1 b).
Scheme 1.
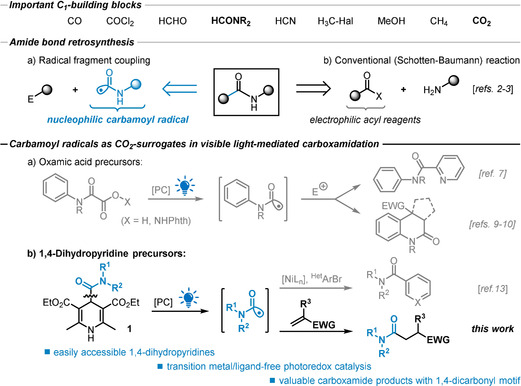
Carbamoyl radicals as CO2‐surrogates in the synthesis of carboxylate derivatives by a metal‐free, photocatalytic addition to alkenes.
We commenced our study with the reaction of the 4‐substituted Hantzsch ester derivative 1 a with benzylidene malonitrile 2 a under visible light irradiation (Table 1). We chose the organic dye 3DPAFIPN as a photocatalyst based on its oxidation capability (E 1/2(PC*/PC.−)=+1.09 V vs. SCE, CH3CN)14 which may enable single‐electron transfer (SET) from 1 a (E pa=+1.17 V vs. SCE, CH3CN) to deliver the carbamoyl radical upon aromatization‐driven fragmentation. Indeed, irradiation of a solution of 1 a and 2 a (1.3 equiv) with 2.5 mol % 3DPAFIPN in CH2Cl2 with blue light (λ max=455 nm) resulted in the formation of the desired dicyano‐propionamide 3 a in 80 % yield (Table 1, entry 1). Alternative organic photocatalysts such as the more reducing 3DPA2FBN (E 1/2(PC.+/PC*)=−1.60 V vs. SCE, CH3CN)14 and the more oxidizing Mes‐Acr+ (E red=+1.88 V vs. SCE, CH3CN)15 afforded lower yields (entries 2 and 3). The choice of solvent was crucial to the reaction (entry 4, see Supporting Information). Acetonitrile, N,N‐dimethyl‐formamide, and other chlorinated solvents gave lower yields. The influence of electron mediators (e.g. 1,4‐dicyanobenzene, [Ni(bipy)3](BF4)2, methyl viologen) was tested (entry 7, see Supporting Information). Slightly enhanced reactivity was observed in the presence of 1,4‐dicyanobenzene. Control experiments established the necessity of photocatalyst and light (entries 8 and 9). With the optimized conditions, we explored the scope of the photoredox‐catalyzed carbamoyl‐Giese reaction (Scheme 2). A diverse set of benzylidene malononitriles gave high yields of the adducts (3 ba–3 fa) whereas electron‐rich aryl substituents lowered the yield significantly. Heteroaryl motifs, such as pyridine (3 ga), thiophene (3 ia), and furan (3 ha), and alkylidene malononitriles (3 ja, 3 ka) were well‐tolerated. The reaction was also applied to alternative alkenes with good reactivity including Meldrum's acid, 1,3‐indandione, and 1,3‐dimethyl barbiturates (3 la–3 na). 1,1‐Diaryl‐ethylene derivatives were also competent radical acceptors (3 sa–3 wa). The scope of 4‐carboxamido substituents within the Hantzsch esters included acyclic and cyclic sec‐ and tert‐amides (3 ab–3 af).
Table 1.
Selected optimization and control experiments.[a]
|
| ||
|---|---|---|
|
Entry |
Deviations from standard conditions |
Yield of 3 aa [%][b] |
|
1 |
none |
80 |
|
2 |
3DPA2FBN instead of 3DPAFIPN |
78 |
|
3 |
Mes‐Acr+ instead of 3DPAFIPN |
65 |
|
4 |
CH3CN instead of CH2Cl2 |
65 |
|
5 |
405 nm, no 3DPAFIPN |
60 |
|
6 |
1 mol % 3DPAFIPN |
67 |
|
7 |
addition of 20 mol % 1,4‐dicyanobenzene |
89 |
|
8 |
in the dark |
0 |
|
9 |
no 3DPAFIPN |
8 |

[a] 0.1 mmol 1 a (0.1 m in DCM), 2 a (1.3 equiv), 16 h, at r.t. under irradiation of blue LED (455 nm). [b] 1H NMR yields vs. external 1,3,5‐trimethoxybenzene.
Scheme 2.

Scope of the photoredox‐catalyzed carbamoylation of alkenes. Isolated yields are given (0.3 mmol scales, 0.1 m in DCM).
We performed key experiments in an effort to probe the mechanism of this photoredox‐catalyzed carbamoyl addition reaction. The UV/Vis spectrum of the 4‐carboxamido‐Hantzsch ester 1 a (see Supporting Information) exhibited an absorption maximum at 350 nm. This excludes direct excitation and photolytic reactivity under irradiation with a blue LED (λ max=455 nm) where only the photocatalyst exhibits considerable absorption. Cyclic voltammetry (CV) studies supported the thermodynamic feasibility of the reaction as the oxidation potential of the Hantzsch ester 1 a (E pa=+1.17 V vs. SCE, CH3CN) matches that of the excited photocatalyst 3DPAFIPN* (E 1/2(PC*/PC.−)=+1.09 V vs. SCE, CH3CN).14 Fluorescence quenching studies and Stern–Volmer analysis documented that the rate of single‐electron transfer between 1 a and the photocatalyst operates at the diffusion limit (K q=9.22 1010 m −1 s−1), while the quenching with the benzylidene malononitrile 2 a is by more than a magnitude slower (K q=2.26 109 m −1 s−1). The standard reaction was completely inhibited in the presence of the radical scavenger (2,2,6,6‐tetra‐methylpiperidin‐1‐yl)oxyl (TEMPO, 1 equiv, see Supporting Information); the resultant carbamoyl‐TEMPO adduct 4 (Scheme 3) was detected by GC/MS. Based on our synthetic and spectroscopic observations and literature data, we propose the following reaction mechanism: Excitation of the organic dye 3DPAFIPN by blue light (λ max=455 nm) affords the strongly oxidizing species 3DPAFIPN* that undergoes single‐electron transfer (SET) from the electron‐rich Hantzsch ester derivative 1.
Scheme 3.

Top: Postulated reaction mechanism. Center: Key reaction intermediates. Bottom: Alternating light/dark experiments.
The resultant radical cation 1 .+ eliminates the carbamoyl radical I upon aromatization‐driven fragmentation. Addition of the nucleophilic radical I to the electron‐deficient alkene 2 furnishes an electrophilic radical II—via a formal Umpolung reaction—which can engage in a single‐electron reduction with the reduced photocatalyst PC.−(E 1/2(PC/PC.−)=−1.59 V vs. SCE, CH3CN).16 The stabilized anion III is protonated to give the formal hydro‐carboxamidation adduct 3. Deuteration experiments in the presence of D2O supported the formation of the anionic intermediate III. A hydrogen atom transfer was discarded based on the absence of deuterium incorporation in reactions performed in [D7]DMF and CD2Cl2, respectively (see Supporting Information). The operation of long radical chains was excluded by a light‐on/off experiment. Over a period of 7 h, steady conversion was recorded under irradiation while no turnover was detected in the dark (Scheme 3, bottom). Finally, we propose that the light‐driven reaction is very unlikely to proceed via energy transfer (EnT):17 The triplet energy (ET) of such dihydropyridine derivatives18 is ≈60 kcal mol−1 which is beyond the thermodynamic reach of the active photocatalysts (e.g. ET=44.7 kcal mol−1 for Mes‐Acr+).19
Hantzsch ester derivatives have been demonstrated to undergo direct photoexcitation in the absence of photocatalysts. The resultant highly reducing excited state species20 are capable of engaging in SET with a variety of π‐electrophiles to afford products of formal hydrogenation,21 alkylation or acylation.22 Such reactivity may be harnessed in catalyst‐free Giese reactions between suitable Hantzsch esters and alkenes, which was already observed in Table 1 (entries 5 and 10). We recorded the UV/Vis spectra of 1 a, 2 a, and of their equimolar combination (Figure 1 a). The formation of an electron donor‐acceptor (EDA) complex became obvious from the bathochromic shift observed when combining both reactants23 that exhibits considerable absorbance at 455 nm.
Figure 1.
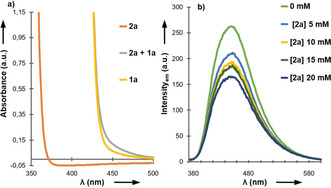
a) UV/Vis spectra of 2 a (orange line, 0.05 m in DCM), 1 a (yellow line, 0.05 m in DCM) and a 1:1 mixture of 1 a and 2 a (grey line, 0.05 m in DCM). b) Fluorescence quenching of 1 a in the prescence of increasing amounts of 2 a.
From the UV/Vis absorption and emission spectra and electrochemical data (see Supporting Information) we determined the redox potential Eox(1 a .+/1 a*) of the excited state 1 a* to be −1.9 V vs. SCE according to the Rehm–Weller approximation.24 This observation together with the fluorescence quenching experiments corroborated our hypothesis of effective SET between 1 a* and 2 a within the EDA complex (Figure 1 b).25a Consequently, reactions under irradiation at 405 nm (for better match with the absorption as the 455 nm irradiation led to only 8 % of yield) afforded good reactivity after short reaction times (Table 1, entry 5). A brief substrate scope evaluation showed that electron‐deficient alkenes fared better and afforded comparable yields to the photocatalytic conditions above (Scheme 4). However, the more electron‐rich 1,1‐diphenylethylene remained unreacted.25b
Scheme 4.

Catalyst‐free carbamoyl additions. 0.3 mmol 1 a; 1H NMR yields vs. external 1,3,5‐trimethoxybenzene. [a] Yield obtained under 455 nm irradiation (Table 1, entry 9).
In conclusion, we have reported a novel photoredox‐protocol that enables the facile generation of carbamoyl radicals from 4‐carboxamido‐1,4‐dihydropyridines and their rapid addition to various electron‐deficient and 1,1‐disubstituted alkenes. Notably, this synthetic strategy relies on the use of easily accessible and bench‐stable Hantzsch ester derivatives, the use of an inexpensive organic photocatalyst, and operation under mild conditions at room temperature with visible light. Mechanistic studies supported the notion of a rapid photoinduced SET between the excited photocatalyst and the Hantzsch ester. Finally, mechanistic studies were conducted to support the proposed reaction mechanism. Suitable combinations of Hantzsch esters and alkenes can be reacted through catalyst‐free direct excitation.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was generously supported by the University of Hamburg. M.O.K. thanks the Alexander von Humboldt Foundation for an individual research fellowship.
L. Cardinale, M. O. Konev, A. Jacobi von Wangelin, Chem. Eur. J. 2020, 26, 8239.
Contributor Information
Dr. Mikhail O. Konev, Email: mikhail.konev@uni-hamburg.de.
Prof. Dr. Axel Jacobi von Wangelin, Email: axel.jacobi@uni-hamburg.de.
References
- 1. van Leeuwen P. W. N. M., C1-building Blocks in Organic Synthesis Thieme, 2014. [Google Scholar]
- 2. Humphrey J. M., Chamberlin A. R., Chem. Rev. 1997, 97, 2243–2266. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Pattabiraman V. R., Bode J. W., Nature 2011, 480, 471–479; [DOI] [PubMed] [Google Scholar]
- 3b. Marcia de Figueiredo R., Suppo J.-S., Campagne J.-M., Chem. Rev. 2016, 116, 12029–12122. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Giese B., Angew. Chem. Int. Ed. Engl. 1983, 22, 753–764; [Google Scholar]; Angew. Chem. 1983, 95, 771–782; General reviews: [Google Scholar]
- 4b. Zhang W., Tetrahedron 2001, 57, 7237–7262; [Google Scholar]
- 4c. Srikanth G. S. C., Castle S. L., Tetrahedron 2005, 61, 10377–10441; [Google Scholar]
- 4d. Rowlands G. J., Tetrahedron 2009, 65, 8603–8655; [Google Scholar]
- 4e. Jasperse C. P. O., Curran D. P., Fevig T. L., Chem. Rev. 1991, 91, 1237–1286. [Google Scholar]
- 5.
- 5a. Giese B., Gonzalez-Gomez J. A., Witzel T., Angew. Chem. Int. Ed. Engl. 1984, 23, 69–70; [Google Scholar]; Angew. Chem. 1984, 96, 51–52; [Google Scholar]
- 5b. Chatgilialoglu C., Crich D., Komatsu M., Ryu I., Chem. Rev. 1999, 99, 1991–2069; [DOI] [PubMed] [Google Scholar]
- 5c. Bryon Gill G., Pattenden G., Reynolds S. J., J. Chem. Soc. Perkin Trans. 1 1994, 369–378. [Google Scholar]
- 6.
- 6a. Raviola C., Protti S., Ravelli D., Fagnoni M., Green Chem. 2019, 21, 748–764. For recent selected examples of photoredox-mediated Giese reactions, see: [Google Scholar]
- 6b. Millet A., Lefevre Q., Rueping M., Chem. Eur. J. 2016, 22, 13464–13468; [DOI] [PubMed] [Google Scholar]
- 6c. Ramirez N. P., Gonzalez-Gomez J. C., Eur. J. Org. Chem. 2017, 2154–2163; [Google Scholar]
- 6d. ElMarrouni A., Ritts C. B., Balsells J., Chem. Sci. 2018, 9, 6639–6646; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6e. Abadie B., Jardel D., Pozzi G., Toullec P., Vincent J.-M., Chem. Eur. J. 2019, 25, 16120–16127. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Coppa F., Fontana F., Lazzarini E., Minisci F., Heterocycles 1993, 36, 2687; [Google Scholar]
- 7b. Jouffroy M., Kong J., Chem. Eur. J. 2019, 25, 2217–2221; [DOI] [PubMed] [Google Scholar]
- 7c. Jatoi A. H., Pawar G. G., Robert F., Landais Y., Chem. Commun. 2019, 55, 466–469. [DOI] [PubMed] [Google Scholar]
- 8. Elad D., Rokach J., J. Org. Chem. 1964, 29, 1855–1859. [Google Scholar]
- 9. Petersen W. F., Taylor R. J. K., Donald J. R., Org. Biomol. Chem. 2017, 15, 5831–5845. [DOI] [PubMed] [Google Scholar]
- 10. Bai Q.-F., Jin C., He J.-Y., Feng G., Org. Lett. 2018, 20, 2172–2175. [DOI] [PubMed] [Google Scholar]
- 11. Wang P.-Z., Chen J.-R., Xiao W.-J., Org. Biomol. Chem. 2019, 17, 6936–6951. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Goti G., Bieszczad B., Vega-Peñaloza A., Melchiorre P., Angew. Chem. Int. Ed. 2019, 58, 1213–1217; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1226–1230; [Google Scholar]
- 12b. Bieszczad B., Perego L. A., Melchiorre P., Angew. Chem. Int. Ed. 2019, 58, 16878–16883; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 17034–17039. [Google Scholar]
- 13. Alandini N., Buzzetti L., Favi G., Schulte T., Candish L., Collins K. D., Melchiorre P., Angew. Chem. Int. Ed. 2020, 59, 5248–5253; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 5286–5291. [Google Scholar]
- 14. Speckmeier E., Fischer T. G., Zeitler K., J. Am. Chem. Soc. 2018, 140, 15353–15365. [DOI] [PubMed] [Google Scholar]
- 15. Fukuzumi S., Kotani H., Ohkubo K., Ogo S., Tkachenko N. V., Lemmetyinen H., J. Am. Chem. Soc. 2004, 126, 1600–1601. [DOI] [PubMed] [Google Scholar]
- 16. Capaldo L., Merli D., Fagnoni M., Ravelli D., ACS Catal. 2019, 9, 3054–3058. [Google Scholar]
- 17. Strieth-Kalthoff F., James M. J., Teders M., Pitzer L., Glorius F., Chem. Soc. Rev. 2018, 47, 7190–7202. [DOI] [PubMed] [Google Scholar]
- 18. Jimenez A. J., Fagnoni M., Mella M., Albini A., J. Org. Chem. 2009, 74, 6615–6622. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Fukuzumi S., Electron Transfer: Mechanisms and Applications, Wiley, 2020; [Google Scholar]
- 19b. Benniston A. C., Harriman A., Li P., Rostron J. P., van Ramesdonk H. J., Groeneveld M. M., Zhang H., Verhoeven J. W., J. Am. Chem. Soc. 2005, 127, 16054–16064. [DOI] [PubMed] [Google Scholar]
- 20. Jung J., Kim J., Park G., You Y., Cho E. J., Adv. Synth. Catal. 2016, 358, 74–80. [Google Scholar]
- 21.
- 21a. Hedstrand D. M., Kruizinga W. M., Kellogg R. M., Tetrahedron Lett. 1978, 19, 1255–1258; [Google Scholar]
- 21b. Zhang J., Jin M.-Z., Zhang W., Yang L., Liu Z.-L., Tetrahedron Lett. 2002, 43, 9687–9689; [Google Scholar]
- 21c. Konev M. O., Cardinale L., Jacobi von Wangelin A., Org. Lett. 2020, 22, 1316–1320. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Zhang J., Li Y., Xu R., Chen Y., Angew. Chem. Int. Ed. 2017, 56, 12619–12623; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12793–12797; [Google Scholar]
- 22b. Buzzetti L., Prieto A., Roy S. R., Melchiorre P., Angew. Chem. Int. Ed. 2017, 56, 15039–15043; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15235–15239; [Google Scholar]
- 22c. van Leeuwen T., Buzzetti L., Perego L. A., Melchiorre P., Angew. Chem. Int. Ed. 2019, 58, 4953–4957; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 5007–5011; [Google Scholar]
- 22d. Zheng C., Wang G.-Z., Shang R., Adv. Synth. Catal. 2019, 361, 4500–4505; [Google Scholar]
- 22e. Wu J., Grant P. S., Li X., Noble A., Aggarwal V. K., Angew. Chem. Int. Ed. 2019, 58, 5697–5701; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 5753–5757. [Google Scholar]
- 23.
- 23a. Mulliken R. S., J. Phys. Chem. 1952, 56, 801–822; [Google Scholar]
- 23b. Foster R., J. Phys. Chem. 1980, 84, 2135–2141. [Google Scholar]
- 24. Rehm D., Weller A., Isr. J. Chem. 1970, 8, 259–271. [Google Scholar]
- 25.
- 25a.Redox potentials of alkylidene malononitriles: L. Capaldo, D. Merli, M. Fagnoni, D. Ravelli, ACS Catal 2019, 9, 3054–3058. Redox potentials of styrenes:
- 25b. Corvaisier F., Schuurman Y., Fecant A., Thomazeau C., Raybaud P., Toulhoat H., Farrusseng D., J. Catal. 2013, 307, 352–361; [Google Scholar]
- 25c. Dahlén A., Nilsson Å., Hilmersson G., J. Org. Chem. 2006, 71, 1576–1580. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary