

Abstract
Fuel‐driven self‐assemblies are gaining ground for creating autonomous systems and materials, whose temporal behavior is preprogrammed by a reaction network. However, up to now there has been a lack of simple external control mechanisms of the transient behavior, at best using remote and benign light control. Even more challenging is to use different wavelengths to modulate the reactivity of different components of the system, for example, as fuel or building blocks. Success would enable such systems to navigate along different trajectories in a wavelength‐dependent fashion. Herein, we introduce the first examples of light control in ATP‐fueled, dynamic covalent DNA polymerization systems organized in an enzymatic reaction network of concurrent ATP‐powered ligation and restriction. We demonstrate concepts for light activation and modulation by introducing caged ATP derivatives and caged DNA building blocks, making it possible to realize light‐activated fueling, self‐sorting in structure and behavior, and transition across different wavelength‐dependent dynamic steady states.
Keywords: ATP, DNA, photocages, self-assembly, wavelength-orthogonal control
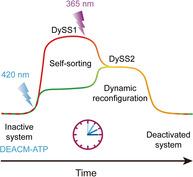
Tuning on multiple channels: Multiple components of the chemical reaction network in the ATP‐driven transient DNA polymerization systems can be caged by different photocleavable groups that respond to different colors of light. As a result, light can be used to modulate the non‐equilibrium behavior and multiple wavelength‐dependent dynamic steady states.
Introduction
Non‐equilibrium self‐assemblies that create transient structures with steady‐state dynamics are ubiquitous in nature to create unparalleled biological functions.1 The non‐equilibrium nature of these structures inspires scientists to strive for more life‐like dynamic synthetic self‐assemblies and functional materials that cannot be achieved from equilibrium‐type self‐assemblies, which after all dominate present soft materials research using stimuli‐responsive properties.2
In man‐made non‐equilibrium systems, van Esch and co‐workers reported the transient self‐assembly of carboxylate‐appended fiber‐forming molecules fueled by harsh methylating agents, which was recently extended to a milder activation by Boekhoven and co‐workers.3 Ulijn and co‐workers used α‐chymotrypsin to facilitate the formation of transient peptide hydrogels under biocatalytic control.4 Moreover, our group developed enzymatic and non‐enzymatic pH feedback systems that create programmable transient alkaline or acidic pH curves, allowing the precise temporal programming of a wide variety of pH‐responsive building blocks for different applications.5
In a biological context, ATP emerges as a very valuable fuel source. Prins and co‐workers demonstrated transient signal modulation in a self‐assembled nanosystem by time‐orchestrated degradation of environmental ATP.6 George's group reported a helical conformation transition of AxP coassemblies during the hydrolysis of ATP to ADP and AMP.7 Hermans’ group reported supramolecular polymers that change their structure by reversible phosphorylation and dephosphorylation.8 We recently reported an ATP‐driven dynamic covalent DNA assembly method that harnesses ATP's energy for transient structure formation.9 Subsequently, we demonstrated pathway complexity in this system by programming the stickiness and recognition of the sticky ends.10
In summary, although much progress has been made in fueled non‐equilibrium self‐assembly,1a, 1b simple external control mechanisms have not yet been reported, but will likely be of high importance for applications.11 In the present study, we develop strategies for remote light control in our ATP‐driven transient DNA polymerization systems by interfering with different subcomponents of the reaction networks. We demonstrate light activation, light modulation, and multiple wavelength‐dependent dynamic steady states (DySSs).
Results and Discussion
Design of the light‐controlled ATP‐driven DNA polymerization system crossing multiple wavelength‐dependent DySSs. The ATP‐dissipative dynamic covalent bond for our DNA polymerization systems is orchestrated by an enzymatic reaction network (ERN) of concurrent ATP‐fueled ligation using T4 DNA ligase and BamHI‐controlled restriction (Figure 1 a). In this ERN, ATP is used as chemical fuel to form a high‐energy phosphodiester bond in the relevant structural entity (DNA) of the system, which under the conditions designed by the ERN can be hydrolyzed with an energy release of −5.3 kcal mol−1.12 The concurrent ligation and restriction lead to the formation of a steady state with an average degree of ligation, in which constant bond shuffling occurs. The steady state structures are inherently dynamic as programmed through the enzyme concentrations and fuel level and this is hence termed a dynamic steady state (DySS).9
Figure 1.
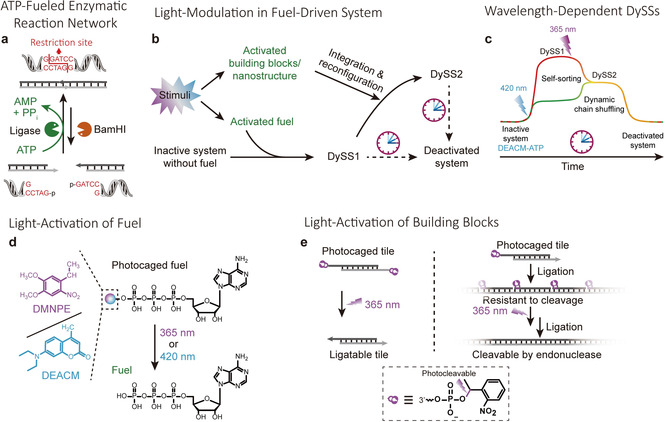
Light‐control mechanisms in ATP‐driven, dynamic covalent DNA assembly using multiple photocontrollers. a) General principle of an ATP‐fueled enzymatic reaction network, yielding a transient dynamic covalent DNA bond. The restriction path is given by the red line. b) Schematic illustration of light‐controlled non‐equilibrium systems with multiple DySSs. External stimuli are applied to activate the caged fuels or building blocks, rendering light modulation of the system. c) ATP‐driven DNA polymerization system with multiple wavelength‐dependent DySSs. Schematic illustration of the photocontrollers for the system including d) caged ATP derivatives, e) double‐side and single‐side caged DNA tiles.
To enable light modulation, we introduce photocaged ATP fuels and photocaged DNA building blocks (tiles) that carry different photocleavable caging groups (PCGs) susceptible to uncaging via remote light stimuli with specific wavelength. The stepwise activation of different PCGs allows for sequential light modulation of the system, which we will demonstrate to facilitate multiple wavelength‐dependent DySS structures and behavioral reconfiguration (Figure 1 b,c).
Figure 1 d,e gives an overview of all photocontrollers for the systems, including P3‐[7‐(diethylamino)coumarin‐4‐yl]methyl adenosine 5′‐triphosphate (DEACM‐ATP, Scheme S1, Figure S1, Supplementary Note 1), 3‐O‐(1‐(4,5‐dimethoxy‐2‐nitrophenyl)ethyl) adenosine‐5′‐triphosphate (DMNPE‐ATP), and double‐side and single‐side caged double‐stranded (ds) DNA tiles. Critically, the DEACM‐ATP and DMNPE‐ATP respond to 420 and 365 nm, respectively (UV/Vis in Figure S2a). The double‐side caged DNA tiles undergo polymerization only after photolysis, while the single‐side caged DNA tiles polymerize to DNA polymers that are, however, resistant to restriction by BamHI and only become cleavable upon removal of the photocages (discussed further below).
Light activation of photocaged ATPs for transient DNA polymerization. We started by investigating the kinetics for the photolysis of both caged ATP derivatives. The photolysis can be controlled by the illumination time, light power, and use of the proper wavelength (Figure S2b–e, Supplementary Note 2). Importantly, DMNPE‐ATP cannot be uncaged at 420 nm (Figure S3a). Thus, an orthogonal and stepwise irradiation can be realized to selectively photolyze DEACM‐ATP in the presence of DMNPE‐ATP. In preliminary experiments, we ensured successful polymerization of an α,ω‐telechelic bifunctional monomer (M1, Supplementary Table 1) by ATP released from both caged ATPs (Figure S3b,c, Supplementary Note 2).
Next, to realize the dissipative system, we implemented the caged ATPs with the full ERN (Figure 2 a). The systems were set to constant concentrations of M1 (0.05 mm), T4 DNA ligase (0.46 Weiss units (WU) μL−1), and BamH1 (10 units μL−1), and we first focus on the red‐shifted DEACM‐ATP (0.6 mm). We performed the photouncaging at 420 nm (27 mW) for different illumination times. Agarose gel electrophoresis (AGE) of time‐dependent aliquots of the transient systems visualizes the transient nature of the dynamically forming DNA polymers with regard to their length distributions (Figure 2 b; Figure S4, Supplementary Note 3). A quantification of the grayscale profiles extracted from AGE yields the average mass‐weighted chain length in base pairs from the mass‐weighted distributions.9 For instance, upon photolysis for 2 min, the chains rapidly grow and reach a DySS plateau of ca. 2000 bp after 1 h. After three days, the DySS DNA polymers are degraded and the system returns to its original state (Figure 2 b,c).
Figure 2.
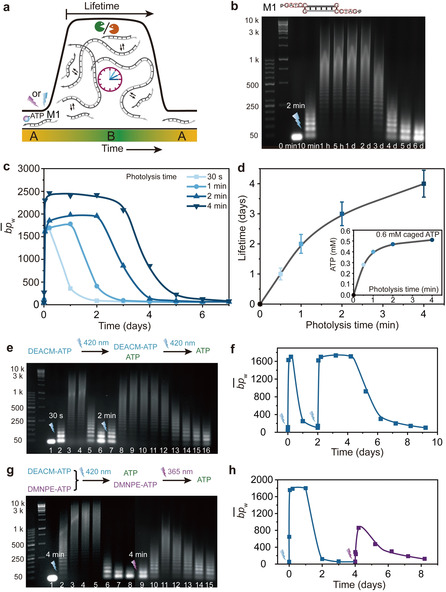
Light activation of the ATP‐driven DNA polymerization systems. a) Schematic illustration of transiently formed dynamic covalent DNA polymer state with lifetime control. b) AGE analysis of the DNA chain growth with time by 2 min uncaging of DEACM‐ATP (2 wt. % agarose gel, 105 V, 70 min). c) The development with time by varying uncaging time from 30 s to 4 min. d) Lifetime of the DNA assembly by different photolysis times; insert: ATP concentration as a function of illumination time by uncaging DEACM‐ATP at 420 nm at 27 mW. e) AGE analysis of the DNA polymerization with time by multiple uncagings of DEACM‐ATP. Lane assignment: 1: 0 min, 2: 10 min, 3: 1 h, 4: 5 h, 5: 1 d, 6: 2 d, 7: 10 min, 8: 1 h, 9: 5 h, 10: 1 d, 11: 2 d, 12: 3 d, 13: 4 d, 14: 5 d, 15: 6 d, 16: 7 d. f) Development of with time by repeatedly uncaging DEACM‐ATP. g) AGE analysis during the sequential activation of the assembling system using DEACM‐ATP and DMNPE‐ATP. Lane assignment: 1: 0 min, 2: 10 min, 3: 1 h, 4: 5 h, 5: 1 d, 6: 2 d, 7: 3 d, 8: 4 d, 9: 10 min, 10: 1 h, 11: 5 h, 12: 1 d, 13: 2 d, 14: 3 d, 15: 4 d. h) Development of with time for sequential activation using DEACM‐ATP and DMNPE‐ATP.
The development of the chain‐length distribution can be explained as follows. Initially, the ligation is favored by high concentrations of ATP and ligatable sticky ends, while the low quantity of restriction sites limits the speed for restriction. Thus, the chains rapidly grow, and the ligatable sticky ends are substantially consumed, and restriction sites are formed. As the reaction progresses, the mutual feedback of both processes in the ERN leads to a balance between ligation and cutting, which give rise to an average chain length in the DySS, where constant bond shuffling occurs. Once the system runs out of chemical fuel, the ligation ceases and the restriction dominates and leads to degradation.
Changing the uncaging time from 30 s to 4 min induces a shift of the in the DySS plateau from ca. 1600 bp to 2500 bp, and delayed recovery of the system to its monomer state (Figure 2 c). Higher is attributed to faster ligation by higher ATP concentration as ATP acts as enzyme cofactor. The corresponding lifetimes of the transient systems, as defined by the point where the decreases to half of the in the DySS plateau, are tunable from 1 to 4 days by changing the uncaging time from 30 s to 4 min corresponding to ATP concentrations from ca. 0.28 to 0.52 mm (Figure 2 d) as judged from photo‐uncaging studies (Figure S2d). When the light power is further decreased to 13.5 mW, a longer photolysis time is needed to achieve DNA polymers with comparable length as when using 27 mW (Figure S5, Supplementary Note 4).
In addition to DEACM‐ATP, we also investigated the blue‐shifted DMNPE‐ATP for a comprehensive understanding of this system, hoping to control the system in a wavelength‐orthogonal activation fashion. Overall, similar to DEACM‐ATP, the lifetimes of the transient DNA polymerization fueled by DMNPE‐ATP can be well regulated by the uncaging time (Figure S6). However, there are some further delays and longer lifetimes, which are due to enzyme inhibition by the byproduct (reactive nitroso compound) from photolysis (Supplementary Note 5). It is worth noting that the illumination does not show any significant effects on the enzymes (Figure S7, Supplementary Note 6).
Multiple and wavelength‐selective activation events by stored chemical energy in the system. One of the key advantages of caged fuels over classical chemical fuels is the possibility to store chemical energy inside the system and activate the system on demand through tailored light pulses.13 Herein, we repeatedly activated DEACM‐ATP to fuel the transient DNA assembly by multiple uncaging events (Figure 2 e,f). By storing 1 mm DEACM‐ATP, 30 s of uncaging (420 nm, 27 mW) for the first round rapidly promotes the polymerization of the monomers to DySS DNA polymers with a of ca. 1600 bp and a lifetime of ca. 1 day. This corresponds to ca. 0.3 mm ATP released, as judged by photo‐uncaging kinetics in Figure S2d, while the rest of the ATP remains stored in the system. After the DNA polymers are almost fully degraded by the ERN, a second round of uncaging at 420 nm at 27 mW for 2 min rapidly repopulates the DySS polymerization, leading to a second lifecycle with an assembly lifetime of ca. 4 days (ca. 0.47 mm ATP released). In comparison, the addition of fresh ATP to the second round can induce similar results as second round uncaging (Figure S8, Supplementary Note 7). However, DMNPE‐ATP failed to give repeated activations, which is attributed to the accumulated poisoning of the T4 DNA ligase by the byproduct from the photolysis (Figure S9, Supplementary Note 8).13
Critically, we integrated both caged ATPs that respond to different wavelengths for sequential and wavelength‐orthogonal activation. The dynamic transient DNA assembly can first be fueled by light‐induced uncaging of DEACM‐ATP at 420 nm, which leaves the DMNPE‐ATP intact and stored in the system. The DMNPE‐ATP can thereafter be activated by uncaging at 365 nm. Figure 2 g,h illustrates this behavior on a proof‐of‐principle level using 0.3 mm DEACM‐ATP and 0.3 mm DMNPE‐ATP. The first uncaging of DEACM‐ATP at 420 nm (4 min, 27 mW, 0.27 mm ATP released) induces a first lifecycle of the DNA polymers with ca. 1 day lifetime, which corresponds roughly to the solely DEACM‐ATP fueled system uncaged for 30 s with 0.28 mm ATP released for the fuel. After the system recovered to its original state, we illuminated the system at a lower wavelength (365 nm) for another 4 min to allow complete uncaging of the DMNPE‐ATP as judged by photo‐uncaging studies of DMNPE‐ATP (Figure S2b), leading to the second round of activation of the system with a lifetime of 1 day. The fueling of the system by 0.3 mm ATP released from DMNPE‐ATP leads to comparable lifetime with the other systems fueled by ca. 0.3 mm ATP (Figure 2 e, Figures S4a and S6b‐ii). The first round uncaging of DEACM‐ATP has very little effect on the DMNPE‐ATP. It is worth noting that more than 90 % DEACM‐ATP has been uncaged at the first round activation according to the photo‐uncaging studies and that the trace amount of the remained DEACM‐ATP cannot induce any obvious ligation of the DNA tiles in the second round activation at 365 nm. As a result, using sequential activation of two kinds of caged ATPs that respond to different wavelengths opens possibilities towards wavelength‐gated behavior and expands the possibilities beyond what is possible using a single photocaged ATP.
Light modulation of DySS trajectories via photocaged sticky ends on dsDNA tiles. To achieve light control over building blocks, we next turn to the use of photocaged DNA tiles in the form of nitrophenol (NP) caps, located at the phosphate groups at the 5′‐ends of the ssDNA overhangs. We will reveal that more complex DySS properties can be realized in multicomponent systems bearing selectively caged DNA tiles and containing tiles with sticky ends of different length. Such varied sticky end lengths (e.g. 3 or 5 nt vs. “standard” 4 nt) provide orthogonal molecular recognition in the initial ligation, but are reconfigured to “standard” 4 nt ssDNA overhangs during operation of the ERN since the restriction enzyme (BamHI) always cuts under the same principle. In general, PCGs have been previously applied to photocage oligonucleotides for light‐triggered hybridization,14 endonuclease restriction,15 DNA strand displacement,16 and DNA replication.17 In most of these cases, the PCGs are, however, either attached to the nucleobase or function as internal photocleavable linkers. PCGs have hardly been applied to cage sticky ends for light‐triggered DNA ligation.18
We first investigated the photo‐uncaging of NP‐caged dsDNA tiles. The photo‐uncaging of the double‐side caged dsDNA according to HPLC is displayed in Figure S10 and Supplementary Note 9, and we found 96 % and 98 % photo‐uncaging for the tiles illuminated for 2 min and 15 min at 365 nm and 135 mW, respectively. Next, we needed to understand whether only one side or both sides of a ligatable motif need to carry the PCG to deactivate the ligation. We studied this for dsDNA tiles bearing complementary 3 nt overhangs that were caged on either one or two sides. Indeed, the double‐side caged dsDNA tiles are completely deactivated for ligation/polymerization before photo‐uncaging, while the single‐side caged dsDNA tiles can still polymerize efficiently by formation of one phosphodiester bond connection at each ligation joint (Figure S11a, Supplementary Note 10).
Building on this, we designed the basic light‐activated transient system using a double‐side caged dsDNA tile (M2, 0.05 mm, Supplementary Table 1) and activated it using light irradiation at different ATP concentrations up to 0.9 mm (365 nm, 135 mW, 15 min, Figure 3 a). AGE of time‐dependent aliquots in Figure 3 b reveals the transient nature of the DySS DNA polymers fueled by 0.9 mm ATP regarding their length distribution. Clearly, the system polymerizes efficiently to long chains with 2600 bp for the (Figure 3 c), which confirms an efficient tile activation. The lifetimes of the autonomous systems are tunable from ca. 5 hours to 3 days by varying the ATP concentration from 0.1 to 0.9 mm (Figure S12). The for the DySS DNA polymers decreases by ca. 600 bp when the ATP concentration decreases from 0.9 to 0.3 mm (Figure 3 c). An even lower ATP concentration of 0.1 mm can no longer enable the build‐up of a DySS for the DNA polymers as the concentration of ATP is only double that of M2 (note that one full ligation requires two ATP molecules). Importantly, during operation of the ERN, the initial 3 nt sticky ends of M2 are reconfigured into the standard 4 nt sticky ends (M1); hence the start and end states are monomeric tiles with slightly different structure in the ssDNA overhang area (A, A*; Figure 3 a).
Figure 3.
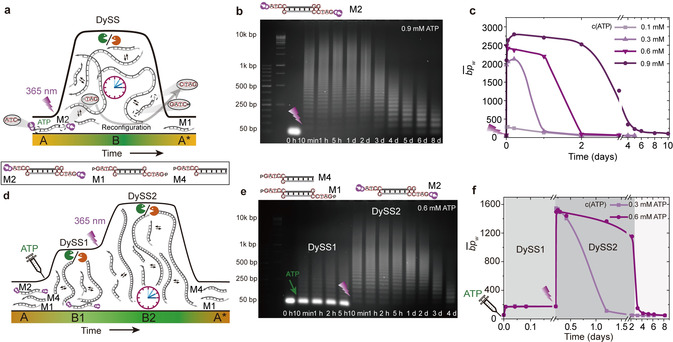
Caged DNA building blocks enable light modulations in ATP‐driven DNA polymerization system. a) Schematic illustration of light activation of caged building blocks for ATP‐driven transient polymerization. b) AGE analysis of the light‐activated DNA chain growth with time fueled by 0.9 mm ATP (2 wt. % agarose gel, 105 V, 70 min). c) The developments with time fueled by 0.1, 0.3, 0.6, and 0.9 mm ATP. d) Schematic illustration of light‐modulated multiple transient DySSs of the DNA assemblies. e) AGE analysis for light‐modulated two transient DySSs of DNA assemblies fueled with 0.6 mm ATP (2 wt.% agarose gel, 105 V, 70 min). f) The development over time fueled with 0.3 and 0.6 mm ATP.
A key advantage of using the caged tiles in the DySS DNA polymerization system is to enable on‐demand activation of the stored caged building blocks in a multicomponent system containing caged and uncaged species, leading to light modulation of the non‐equilibrium system in situ and thus switching of the DySS structures. To achieve this, it is important that there is no interference between these two parallel systems before photo‐uncaging. This requires distinct molecular recognition for each subsystem. Figure 3 d shows an overview of such a design. The system contains “standard” ligatable dsDNA M1 with two 4 nt ssDNA overhangs and “terminator” tiles bearing the 4 nt ssDNA overhang only on one side (M4, Supplementary Table 1). Additionally, we add double‐side photocaged dsDNA tiles with 3 nt ssDNA overhangs (M2). Critically, since the 3 nt ssDNA overhangs of M2 are not sufficiently complementary to the 4 nt ssDNA overhangs of M1 and M4, the M2 tiles are completely dormant and unreactive in the system. Yet, upon photo‐uncaging ligation and restriction set in, and a reconfiguration of M2 to M1 occurs. We use this system to showcase light modulation over multiple transient DySSs in ATP‐driven transient DNA polymerizations by in situ switching of the ratio of monofuntional terminator tile to active, difunctional polymerizing tile.
Experiments were performed using the standard ERN conditions and tile ratios of [M2]:[M4]:[M1]=0.03 mm:0.01 mm:0.01 mm, and various concentrations of ATP. By fueling with 0.6 mm ATP, before photo‐uncaging, only the non‐photocaged dsDNA tiles (M1, M4) undergo ATP‐powered DySS polymerization. This leads to short oligomers due to the presence of equimolar amounts of terminator ([M4]=[M1]). The strong band for the photocaged M2 at the lower end of the AGE clearly confirms its inert state before UV irradiation (Figure 3 e). We term this state DySS1. The oligomers therein have a of ca. 172 bp or 351 bp when considering or not considering the monomeric band. The latter reflects more accurately the distribution of the growing M1/M4 tile system (Figure 3 f).
Upon irradiation (365 nm, 135 mW, 15 min), the monomer band of the caged, inactive M2 immediately disappears, indicating fast integration of the released active M2 tiles into the DySS1 polymerization system (Figure 3 e). This proceeds, however, via an intermediate step. The photoactivated 3 nt sticky‐end M2 tiles first homopolymerize to long DNA polymers via distinct molecular recognition. Over time the 3 nt sticky ends are reconfigured to standard 4 nt sticky ends in the ERN due to BamHI cleavage. Consequently, the reconfigured M2 tiles become M1 tiles and integrate with the M1/M4 system, and the combined DySS DNA polymers in the system grow to a of ca. 1400 bp and enter DySS2 (Figure 3 f). Qualitatively, this clearly follows classical Carother's type step growth polymerization, in which the stoichiometry between dual functional monomers and monofunctional terminators controls the degree of polymerization.19 The effect is significant in the AGE profiles, because we intentionally provide a threefold excess of M2 compared to M1, so that the ratio of M1/M4 changes from 1:1 to 4:1. Once the ATP cofactor is increasingly consumed, the ligation process ceases and the kinetic balance shifts towards the degradation (ca. 3 days). By fueling with 0.3 mm ATP, the recovery of the system from DySS2 to its unassembled state is much faster and completed within less than 1.5 days (Figure 3 f; Figure S13). Thus, the lifetimes for the second state can be programmed by the ATP concentration.
Multicomponent self‐sorting and wavelength‐dependent multiple DySSs. Multicomponent self‐sorting in non‐equilibrium self‐assembly is unprecedented and light modulation of the behavior between the sorted structures offers an intriguing strategy to remotely tailor this property of multicomponent self‐assembling systems. Aside from the previously discussed light‐modulated multiple transient DySS structures, in this part we further discuss light modulation of ATP‐driven multicomponent self‐sorted structures in the ATP‐driven transient DNA polymerization system.
As a starting point, we already showed that single‐side photocaged tiles can still undergo ATP‐powered polymerization (Figure S11a). Interestingly, a previous study reported that one PCG in the restriction site can significantly inhibit BamHI restriction.15b Thus, we hypothesized that the DNA polymers formed from single‐side caged tiles are noncleavable by endonuclease restriction due to the presence of a PCG in the incompletely ligated restriction site. This would mean that such DNA polymers behave in a static fashion in the ERN. Moreover, it should be possible to activate the formed static structures via photo‐uncaging to enter into the DySS organized by the ERN.
Building on these considerations, we designed a system allowing for an in situ comparison. The system is composed of two “standard” tiles with 4 nt ssDNA overhangs—one dual functional (M6, Supplementary Table 1) and one monofunctional “terminator” (M5, Supplementary Table 1)—and one reconfigurable tile (M7, Supplementary Table 1) caged on one side and having an orthogonal 5 nt sticky‐end ssDNA overhang (Figure 4 a). The 5 nt overhang is intended for strong hybridization and ligation, and for distinct recognition without the chance of interfering with the M5 or M6 tiles bearing the 4 nt overhang. To clearly distinguish the different tiles, M6 and M7 are labeled with a green fluorophore (ATTO 488) and M7 is labeled with a red fluorophore (Cy5). Additionally, the “standard” 4 nt tile system is supplemented with a terminator tile (M5) to limit the molecular weight as discussed above (Figure 3 d). A T‐shaped tile structure was chosen for the ATTO 488‐labeled species, as ATTO 488 is only available as end‐group functionalization, and because we wanted to achieve a separation of the red and green oligomeric species in AGE.
Figure 4.
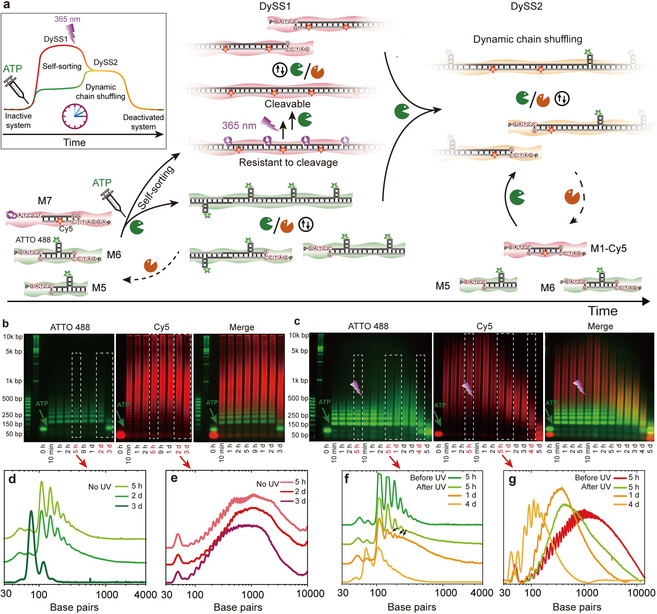
Light modulation of ATP‐driven multicomponent non‐equilibrium self‐sorting system. a) Schematic illustration of ATP‐powered multicomponent self‐sorting and light modulation of the non‐equilibrium behavior by programming molecular recognitions in the sticky ends and setting photocaging groups on restriction sites. b) Multicolor AGE analysis of self‐sorted structures in multicomponent systems (2 wt. % agarose gel, 90 V, 2.5 h). The green channel displays short oligomers from M5 and M6, while the red channel visualizes the long DNA polymers from M7. c) Multicolor AGE analysis of light modulation of multicomponent self‐sorting non‐equilibrium systems (2 wt. % agarose gel, 90 V, 2.5 h). d,e) and f,g) Grayscale plots showing oligomer distributions for the self‐sorted multicomponent systems without and with light modulation, respectively.
We first consider the ATP‐driven self‐sorting of the multicomponent system without UV illumination. Experiments were performed using tile ratios of [M5]:[M6]:[M7]=0.01 mm:0.005 mm:0.02 mm, in the presence of 0.46 WU μL−1 T4 DNA ligase, 15 units μL−1 BamHI, and 0.6 mm ATP. A higher BamHI concentration was used for more efficient cleavage and thus faster tile reconfiguration. AGEs of time‐dependent aliquots visualize the system behavior (Figure 4 b). Since the tiles bearing 4 nt and 5 nt sticky ends are labeled with ATTO 488 and Cy5, respectively, the narcissistically sorted structures arising from these species can be visualized by multicolor AGE (Figure 4 b). After the injection of ATP, T‐shaped tiles form short oligomers due to the presence of a twofold excess of terminator (M5, green channel), while the single‐side caged tiles, M7, directly polymerize to long DNA polymers (red channel) due to the absence of any matching terminator. The deliberately introduced separation in molecular length helps to clearly identify the self‐sorted nature of these structures (Figure 4 b, Merge).
It is important to note that these structures are not only self‐sorted in their chemical nature and structure (dye, tile shape), but moreover also self‐sorted in their behavior, that is the dynamics (dynamic vs. static). This can be seen by the fact that after three days incubation, there is only very limited change and degradation in the static red DNA polymers. This is due to the very slow cleavage of the caged DNA by BamHI that is severely inhibited by the presence of the PCG.15b Please note that the high‐molecular‐weight flank above 5 kbp in AGE is in fact not part of the distribution of the red species, but relates to some smearing artifacts (potentially related to Cy5 interaction with agarose) in AGE visible due to the high sensitivity of the instrument. In contrast, the green oligomers are nearly fully degraded to monomers after the ATP is consumed, indicating their highly dynamic nature in the ERN (Figure 4 b).
Grayscale plots in Figure 4 d,e further underscore the segregated nature of the sorted structures over time. The characteristic peaks for the gel bands of green short oligomers maintain their migration position in AGE during the active lifecycle in the ERN (Figure 4 d), which indicates that the green oligomers are isolated from the red tiles, because any dynamic chain shuffling between the green and red oligomers would lead to gel band shifts and thus new peaks in the grayscale plot. At the same time, the red DNA polymers also maintain the same migration distance in their oligomer distribution during the active lifetime of the green oligomers (Figure 4 e). Thus, the green and red species are well sorted from each other upon fueling with ATP, and there is no species interconversion between those two sorted structures.
Critically, the behavior of the sorted structures in the ERN can be drastically changed by photolyzing the PCGs on the red, static long polymer species formed by M7. The photolysis of the PCG promotes complete ligation at the restriction site and activates the restriction‐inert caged M7 polymers for BamHI restriction, whereupon the same 4 nt overhangs are created as those present in the M5 and M6 tiles. This switches M7 tiles from static to dynamic and also forces them to enter the DySS regime of M5/M6 with constant ligation and cutting (Figure 4 a).
To showcase the resulting behavior, we let a system evolve for 5 h in the ATP‐powered dynamic and static polymerization, and thereafter forced the reconfiguration and integration of the M7 tiles into the DySS by 15 min UV light exposure (135 mW, 365 nm). The successful dynamization of the PCG‐M7 system can be seen by the now occurring synchronous degradation of both polymers after ATP consumption (see red channel in Figure 4 c). More importantly, however, after the removal of PCGs, a randomization of the initially sorted structures appears. Thus, unlike the distinct green and red gel bands in the first sorted phase (before irradiation), color merging happens in the gel bands after UV illumination (Figure 4 c, Merge). Distinct red and green bands disappear and orange bands appear instead. This confirms that a mixed polymer structure has formed. Additionally, when one looks at the single channels, the green tiles shift to higher molecular weight range after UV illumination and the red tiles shift to lower molecular weight range (Figure 4 c,f). This is because the terminator M5 is now shared between all difunctional monomers (M6, M7), leading to an averaged degree of polymerization between the two originally sorted systems. Grayscale plots in Figure 4 f,g underscore this behavior. All of this confirms the light‐triggered dynamic exchange and randomization. It is worth noting that the light‐modulated system has a lifetime longer by 1 day compared to the system without light modulation (Figure 4 b,c). Similar to the influence of the DMNPE‐ATP, this can be attributed to the enzyme inhibitory effect of the nitroso compound from photolysis of the caged tiles. Nevertheless, the inhibition is very weak compared to the system fueled by DMNPE‐ATP, because the concentration of the nitroso compound from photolysis is very low (0.02 mm). Thus, the general DySS behavior is not obviously affected.
Combination of light‐activated fuel and light‐activated building blocks for wavelength‐orthogonal behavior. In this last part, we merge the understanding generated above and replace free ATP in the self‐sorting system with DEACM‐ATP to investigate wavelength‐orthogonal control of the system complexity (Figure 5 a).
Figure 5.
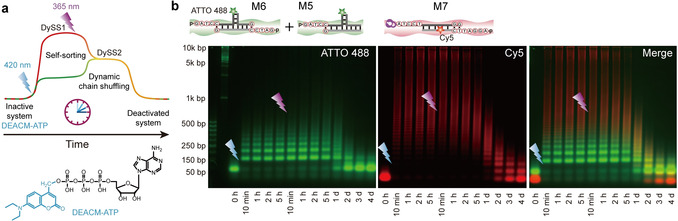
Integration of photocaged fuels and photocaged building blocks with wavelength‐selective activation. a) Schematic illustration of wavelength‐orthogonal control of system complexity. The first irradiation at 420 nm activates DEACM‐ATP to fuel self‐sorting of the multicomponent system into DySS short oligomers (from M5 and M6) and static long DNA polymers (from M7), and the following irradiation at 365 nm activates the static DNA polymers and thus promotes the dynamic chain shuffling between the sorted structures. b) Multicolor AGE analysis of orthogonal light‐controlled structure development of multicomponent self‐sorting non‐equilibrium systems (2 wt. % agarose gel, 90 V, 2.5 h).
The system has the same composition as in Figure 4, but now we use 0.6 mm DEACM‐ATP instead of ATP. For the first step, 420 nm LED light (27 mW, 4 min) was applied to uncage the DEACM‐ATP. This leads to self‐sorted DNA nanostructures (DySS1), where the green T‐shaped tiles form dynamic short oligomers and the red tiles homopolymerize to static long polymers (Figure 5 b). The species are clearly separated. After 5 h, 365 nm LED light (135 mW, 15 min) was further applied to uncage the NP‐caged static, red DNA polymers, enabling dynamic chain shuffling between the sorted DNA nanostructures (Figure 5 b). As a result, the green tiles shift to higher molecular weight range while the red tiles shift to lower molecular weight range. The distinct red and green bands disappear and orange bands gradually appear after 2 h reconfiguration of the tiles.
The results here are in accordance with the results of the experiment with 0.6 mm free ATP, while the shorter lifetime here is attributed to lower ATP concentration in the system (ca. 0.48 mm of 0.6 mm DEACM‐ATP released as judged from the photolysis study). Importantly, this last part shows a higher level of light control in this system. As a result, we achieve multiple wavelength‐dependent DySSs in the ATP‐fueled DNA polymerization system using the manipulation of different network components.
Conclusion
Taken together, we developed strategies to enable light activation and light modulation in ATP‐driven and dissipating dynamic covalent DNA polymerization system, and achieved light‐orthogonal control of system complexity by combining these two aspects. Incorporating caged ATPs into the system allows for programmable lifetimes of the system through variation of the uncaging time and light power. Our data shows that the selection of the correct caged ATP derivatives is in fact a crucial aspect. Compared to DMNPE‐ATP, DEACM‐ATP shows great advantages for multiple uncagings and activations. Furthermore, the incorporation of two kinds of caged ATPs that respond to different wavelengths makes it possible to sequentially activate the caged ATPs and repeatedly fuel the DNA assembly.
Light activation of photocaged DNA building blocks opens the door to remotely modulate the non‐equilibrium behavior of the system in a more complex fashion. Double‐side caged DNA tiles are fully dormant in a running non‐equilibrium system, while single‐side caged DNA tiles can polymerize, but are restriction inert. Through the use of light‐powered uncaging, such building blocks can be activated for integration into the full ERN, either to become DySS polymers, or to enter from a static polymer structure to a DySS polymer structure.
More importantly, the combination of such caged DNA species with uncaged DNA species, bearing orthogonal recognition length in the sticky‐end overhang area (3 nt, 4 nt, 5 nt) makes it possible to realize light‐modulated reconfiguration of self‐sorted structures, where we showed structural (oligomer/polymer length, T‐shaped vs. linear tile), chemical/functional (dye), and behavioral (static vs. dynamic) reconfiguration. Hence, the strategy of using photocaged building blocks opens fundamentally new avenues for the design of light‐modulated dynamic systems. Further combination of caged ATP and caged DNA, which respond to different colors of light, gives rise to multiple wavelength‐dependent DySSs in the non‐equilibrium DNA polymerization system. This serves as an inspiration for researchers to develop life‐like materials with wavelength‐dependent properties.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We acknowledge support by the European Research Council starting Grant (TimeProSAMat) Agreement 677960 and the MPG Minerva Foundation ARCHES Program. This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy—EXC‐2193/1‐390951807 via “Living, Adaptive and Energy‐Autonomous Materials Systems” (livMatS). We thank L. Heinen for introducing the ATP‐driven DNA polymerization system at the initial phase of this project and S. Löscher for help with HPLC measurements.
J. Deng, D. Bezold, H. J. Jessen, A. Walther, Angew. Chem. Int. Ed. 2020, 59, 12084.
References
- 1.
- 1a. Merindol R., Walther A., Chem. Soc. Rev. 2017, 46, 5588–5619; [DOI] [PubMed] [Google Scholar]
- 1b. van Rossum S. A., Tena-Solsona M., van Esch J. H., Eelkema R., Boekhoven J., Chem. Soc. Rev. 2017, 46, 5519–5535; [DOI] [PubMed] [Google Scholar]
- 1c. Henzler-Wildman K., Kern D., Nature 2007, 450, 964–972; [DOI] [PubMed] [Google Scholar]
- 1d. Needleman D., Dogic Z., Nat. Rev. Mater. 2017, 2, 17048. [Google Scholar]
- 2.
- 2a. Stuart M. A. C., Huck W. T., Genzer J., Müller M., Ober C., Stamm M., Sukhorukov G. B., Szleifer I., Tsukruk V. V., Urban M., Nat. Mater. 2010, 9, 101–113; [DOI] [PubMed] [Google Scholar]
- 2b. Kumar M., Ing N. L., Narang V., Wijerathne N. K., Hochbaum A. I., Ulijn R. V., Nat. Chem. 2018, 10, 696–703; [DOI] [PubMed] [Google Scholar]
- 2c. Mishra A., Korlepara D. B., Kumar M., Jain A., Jonnalagadda N., Bejagam K. K., Balasubramanian S., George S. J., Nat. Commun. 2018, 9, 1295; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2d. Green L. N., Subramanian H. K., Mardanlou V., Kim J., Hariadi R. F., Franco E., Nat. Chem. 2019, 11, 510–520; [DOI] [PubMed] [Google Scholar]
- 2e. Walther A., Adv. Mater. 2019, 10.1002/adma.201905111; [DOI] [Google Scholar]
- 2f. Agarwal S., Franco E., J. Am. Chem. Soc. 2019, 141, 7831–7841. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Boekhoven J., Brizard A. M., Kowlgi K. N., Koper G. J., Eelkema R., van Esch J. H., Angew. Chem. Int. Ed. 2010, 49, 4825–4828; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 4935–4938; [Google Scholar]
- 3b. Boekhoven J., Hendriksen W. E., Koper G. J., Eelkema R., van Esch J. H., Science 2015, 349, 1075–1079. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Toledano S., Williams R. J., Jayawarna V., Ulijn R. V., J. Am. Chem. Soc. 2006, 128, 1070–1071; [DOI] [PubMed] [Google Scholar]
- 4b. Debnath S., Roy S., Ulijn R. V., J. Am. Chem. Soc. 2013, 135, 16789–16792. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Heuser T., Weyandt E., Walther A., Angew. Chem. Int. Ed. 2015, 54, 13258–13262; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13456–13460; [Google Scholar]
- 5b. Heinen L., Walther A., Chem. Sci. 2017, 8, 4100–4107; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c. Heinen L., Heuser T., Steinschulte A., Walther A., Nano Lett. 2017, 17, 4989–4995; [DOI] [PubMed] [Google Scholar]
- 5d. Heuser T., Merindol R., Loescher S., Klaus A., Walther A., Adv. Mater. 2017, 29, 1606842; [DOI] [PubMed] [Google Scholar]
- 5e. Heuser T., Steppert A.-K., Molano Lopez C., Zhu B., Walther A., Nano Lett. 2015, 15, 2213–2219. [DOI] [PubMed] [Google Scholar]
- 6. Pezzato C., Prins L. J., Nat. Commun. 2015, 6, 7790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dhiman S., Jain A., Kumar M., George S. J., J. Am. Chem. Soc. 2017, 139, 16568–16575. [DOI] [PubMed] [Google Scholar]
- 8. Sorrenti A., Leira-Iglesias J., Sato A., Hermans T. M., Nat. Commun. 2017, 8, 15899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Heinen L., Walther A., Sci. Adv. 2019, 5, eaaw0590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Deng J., Walther A., J. Am. Chem. Soc. 2020, 142, 685–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Straight A. F., Cheung A., Limouze J., Chen I., Westwood N. J., Sellers J. R., Mitchison T. J., Science 2003, 299, 1743–1747; [DOI] [PubMed] [Google Scholar]
- 11b. Pellois J. P., Muir T. W., Angew. Chem. Int. Ed. 2005, 44, 5713–5717; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 5859–5863. [Google Scholar]
- 12. Dickson K. S., Burns C. M., Richardson J. P., J. Biol. Chem. 2000, 275, 15828–15831. [DOI] [PubMed] [Google Scholar]
- 13. Hess H., Clemmens J., Qin D., Howard J., Vogel V., Nano Lett. 2001, 1, 235–239. [Google Scholar]
- 14. Liu M., Jiang S., Loza O., Fahmi N. E., Šulc P., Stephanopoulos N., Angew. Chem. Int. Ed. 2018, 57, 9341–9345; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 9485–9489. [Google Scholar]
- 15.
- 15a. Vaníková Z., Hocek M., Angew. Chem. Int. Ed. 2014, 53, 6734–6737; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 6852–6855; [Google Scholar]
- 15b. Young D. D., Govan J. M., Lively M. O., Deiters A., ChemBioChem 2009, 10, 1612–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang F., You M., Han D., Xiong X., Liang H., Tan W., J. Am. Chem. Soc. 2013, 135, 7967–7973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tanaka K., Kuzuya A., Komiyama M., Chem. Lett. 2008, 37, 584–585. [Google Scholar]
- 18. Zhang K., Taylor J.-S., J. Am. Chem. Soc. 1999, 121, 11579–11580. [Google Scholar]
- 19. Zhao D., Moore J. S., J. Am. Chem. Soc. 2003, 125, 16294–16299. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary