

Abstract
Background
BT200, a pegylated form of the aptamer BT100, inhibits binding of von Willebrand factor (VWF) to platelet glycoprotein GPIb, preventing arterial thrombosis in cynomolgus monkeys. It is being developed for secondary prevention of arterial thrombosis such as stroke or myocardial infarction. Inhibition of thrombogenesis by BT200 is expected to provide a therapeutic benefit. However, there may be unexpected bleeding (eg, incidental trauma) in which a reversal agent is required. To address this need, BT101, a complementary aptamer, has been developed to specifically inhibit BT100 and BT200 function.
Objectives
To characterize the effects of BT101 both in vitro and in vivo.
Methods
The direct interaction between BT101 and the core aptamer BT100 was evaluated using polyacrylamide gel electrophoresis. The binding of BT200 to purified human VWF and inhibition of VWF activity was further characterized using enzyme‐linked immunosorbent assay. VWF‐dependent platelet function was measured by the platelet function analyzer and aggregometry in whole blood. In addition, both the in vivo pharmacokinetic profile of BT101 as well as its ability to reverse BT200 activity, were evaluated in cynomolgus monkeys.
Results
BT101 bound to the core aptamer BT100 at a 1:1 ratio, inhibited BT200 binding to purified human VWF, and reversed BT200‐induced inhibition of both VWF activity and VWF‐dependent platelet function in vitro. After intravenous injection to monkeys, BT101 reversed BT200‐induced effects on VWF activity and platelet function within minutes, without causing any adverse effects.
Conclusions
The results of this study demonstrate that BT101 is an effective reversal agent for BT200.
Keywords: glycoprotein Ib, platelet, reversal agent, thrombosis, von Willebrand factor
ESSENTIALS.
Platelet inhibitors increase the risk of bleeding; currently there are no antidotes available.
BT200 is an aptamer inhibitor of the von Willebrand factor A1 domain preventing arterial thrombosis.
A complementary aptamer, BT101, was developed as a rapid and effective reversal agent.
BT101 immediately antagonized BT200 in non‐human primates supporting its clinical development.
1. INTRODUCTION
Thrombus formation results from a confluence of two major physiologic pathways, one involving coagulation proteins and the other involving platelets. The latter, often referred to as platelet‐mediated thrombogenesis, is considered predominant in the arterial circulation. 1 , 2 The sequence of steps in platelet thrombogenesis is commonly described as adhesion followed by activation and then aggregation. 3 Adhesion begins when damaged or denuded vascular endothelium exposes subendothelial collagen to the bloodstream and shear force activates von Willebrand factor (VWF), enabling it to bridge between collagen and platelet glycoprotein GPIb.
BT100 and BT200 are synthetic RNA oligonucleotides belonging to the medicinal chemical family known as “aptamers.” 4 , 5 BT100 and BT200 (pegylated BT100) specifically block the A1 domain of human and non‐human primate VWF and thereby its binding to platelet GPIb. 6 This property of shear dependence confers a degree of anatomic specificity to the pharmacology of BT200, favoring the arterial over the venous circulatory system, and thromboembolism from arterial plaque rupture over cardioembolism from atrial fibrillation. Functional inhibition of platelet thrombogenesis by BT200 has been demonstrated via in vitro human pharmacology studies and in an arterial thrombosis model in nonhuman primates. 6 Targeted therapeutic indications for BT200 include secondary stroke prevention in high risk patients. This is supported by an abundance of literature highlighting the important role of VWF in stroke. 7 , 8 While these patient populations are expected to benefit from the inhibition of thrombogenesis by BT200, reversal strategies should be established for emergency situations such as life‐threatening major bleeding from trauma or non‐elective major surgery. 9
As aptamers are made of oligonucleotide sequences, they provide the code for their own complementary sequences that can be used to inhibit their function (ie, as specific reversal agents). 10 Consequently, a complementary control agent known as BT101 has been developed for BT200. In the present study, we evaluated the direct interaction between BT101 and the core aptamer of BT200 in vitro, the impact of that interaction on binding of BT200 to purified human VWF, and the effect of BT101 on BT200‐induced inhibition of platelet function in human whole blood samples. In addition, we hypothesized that BT101 would immediately reverse BT200‐induced inhibition of VWF activity and platelet function and studied the pharmacokinetic profile of BT101 in cynomolgus monkeys following intravenous infusion.
2. METHODS
2.1. Test substances
Oligonucleotides were synthesized using standard phosphoramidite solid‐phase chemistry. BT101 (mAmCmAmUmGmUmGmUmCmUmUmAmGmGmUmCmCmCmUmGmGmC‐idT) was synthesized by Boston Open Lab. Both BT100 (NH‐mGmCmCmAmGmGmGmAmCmCmUmAmAmGmAmCmAmCmAmUmGmUmCmCmCmUmGmGmC‐idT) and BT200, a 5′ 40K‐pegylated version of BT100 (PEG40K‐NH‐mGmCmCmAmGmGmGmAmCmCmUmAmAmGmAmCmAmCmAmUmGmUmCmCmCmUmGmGmC‐idT) were both manufactured by Suzhou Ribo Life Science Co. To prepare BT‐200, purified BT‐100 was mixed with N‐hydroxysuccinimide (NHS)‐activated branched 40K‐PEG (catalog number Y‐NHS‐40K, JenKem Technology USA Inc) at a molar ratio of BT‐100 to NHS‐activated branched 40K‐PEG of 1:2. The reaction proceeded overnight at room temperature. The resultant was purified through anion‐exchange and ultrafiltration to remove unconjugated 40K‐PEG and BT‐100, and BT‐200 was obtained.
2.2. In vitro binding of BT101 to BT200
The binding of BT101 to the core non‐PEGylated aptamer of BT100 was evaluated using polyacrylamide gel electrophoresis (PAGE). The core aptamer was used for this experiment as PEGylation would have prevented the ability to separate analytes using PAGE. BT100 and BT101 were each dissolved in 0.9% sodium chloride (physiological saline) at a range of concentrations to enable the mixing of the two compounds at different molar ratios. BT100:BT101 molar ratios tested were 0:1, 1:0, 1:2, 1:1, 2:1, and 5:1. Following mixing, the solutions were incubated at 37°C for 30 minutes prior to loading onto a 20 mL 16% PAGE‐urea gel composed of 8 mL 40% acrylamide, 4 mL deionized water, 8 mL 5xTBE (tris base, boric acid, ethylenediaminetetraacetic acid [EDTA]), 150 μL 10% ammonium persulfate, and 15 μL tetramethylethylenediamine. Following electrophoresis at 20 mA for 20 minutes followed by 40 mA for 60 minutes, the gel was immersed in a Gelred staining solution (GelredTM 10000X: Biotium; Cat:41003‐0.5 ml) for 10 minutes at room temperature. The gel was then visualized using Bio‐Rad Gel Doc XR.
2.3. Effect of BT101 on the binding of BT200 to purified human VWF
The affinity of BT200 for purified human VWF, with or without pre‐incubation with BT101, was evaluated using an enzyme‐linked immunosorbent assay (ELISA) method. In this study, purified human VWF protein (Sino Biological, Catalog No. 10973‐H08c) was dissolved in Dulbecco's phosphate buffered saline (dPBS) buffer at a concentration of 4 µg/mL and 100 µL of this solution was added to each well of a Nunc Maxisorp 96‐well plate. The plate was incubated overnight at 4°C. The following day, the plate was washed three times, followed by blocking with 5% bovine serum albumin (BSA) in dPBS at room temperature for 90 minutes. The blocked plate was then washed three times before adding either BT200 at concentrations of 0.3, 1, 3, 10, 30, 100, 300, 1000, 3000, and 10 000 ng/mL or a pre‐incubated 1:1 molar ratio of BT101:BT200 at the same concentrations. BT101 and BT200 were mixed and incubated at 37°C for 30 minutes prior to addition to the plate.
Plates were then incubated at 37°C for 2 hours. Each plate was then washed three times and 100 µL of anti‐PEG antibody at 1 µg/mL in 1% BSA/dPBS was added to each well and incubated for 1 hour at room temperature. Following an additional three washes, 100 µL of anti‐rabbit horseradish peroxidase (HRP) in dPBS with 1% BSA was added to each well for 60 minutes at room temperature. To detect HRP, 100 µL of Slow TMB solution was added to each well and incubated at room temperature for 30 minutes. To stop the reaction, 100 µL of 2 N H2SO4 was added to each well and the plate was then read at 450 nm for absorbance and the mean absorbance under each set of conditions was recorded.
2.4. Effect of BT101 on BT200‐induced inhibition of von Willebrand Factor activity
BT200 was incubated with citrated human plasma (obtained from Jiangsu Institute of Hematology) for 60 minutes at 37°C at a final concentration of 3 μg/mL. Subsequently, BT101 was added to the incubation at molar ratios of 0:1, 0.5:1, 1:1, and 2:1 and incubated with the plasma containing BT200 for an additional 30 minutes at 37°C. After completion of the incubation period, VWF activity was measured using the REAADS® VWF:Act assay (REAADS® Von Willebrand Factor Activity Test Kit, Corgenix Catalog No. 10826). 11
2.5. Effect of BT101 on BT100‐induced inhibition of VWF dependent platelet function
The effects of BT101 on BT100‐induced inhibition of platelet function were evaluated using a platelet function analyzer (PFA; Siemens) and multiple electrode aggregometry (MEA; Roche Diagnostics). The PFA assay measures the time required for occlusion of the aperture by platelet plugs, which is defined as closure time (CT). The instrument aspirates a blood sample under constant vacuum from the sample reservoir through a capillary and a microscopic aperture (147 μm) cut into the membrane, which leads to high shear induced platelet plug formation. The membrane coated with collagen/adenosine diphosphate (CADP) is sensitive to VWF levels: subnormal VWF levels (either congenitally or acquired 12 , 13 , 14 ) result in a prolonged CADP‐CT, whereas increased VWF activity shortens CADP‐CT. 15
For the PFA assay, whole blood samples were incubated with BT100 (25 or 100 nM) and either saline or 200 nM BT101 at 37°C for 15 minutes, after which platelet plug formation was measured by CADP‐CT and compared to an untreated control sample. Maximal CT measured by the PFA‐200 is 5 minutes and the instrument gives a result of > 300 seconds if this time is exceeded (for statistical purposes, a time of 301 seconds is used in these circumstances).
The MEA assay measures electrical impedance change due to adhesion and aggregation of platelets on two independent electrode‐set surfaces in a test cuvette containing whole blood.
For the MEA assay, whole blood was anticoagulated with hirudin (200 U/mL, Dynabyte) as recommended by the manufacturer and stored at room temperature (22°C) for 30 minutes. Afterward, 1:2 mixtures of 0.9% NaCl and hirudin‐anticoagulated blood were stirred at 37°C for 3 minutes in the test cuvettes. Whole blood was pre‐incubated with BT‐100 or mixtures of BT‐100 and BT‐101 at 37°C for 10 minutes prior to the addition of activating reagent, ristocetin. The increase in electrical impedance was recorded continuously for 6 minutes. The area under the curve (AUC) of the aggregation tracing was determined. The MEA instrument allows two ways to express the AUC: as arbitrary aggregation units (AUC*minutes) or as units (U). Ten AUC*minutes correspond to 1 U. AUC was measured in U, following the recommendation of the manufacturer. Low VWF activity levels reduce the AUC values. 16 , 17
2.6. Pharmacokinetics of BT101 following intravenous administration to cynomolgus monkeys
The pharmacokinetics of BT101 were evaluated following intravenous administration to cynomolgus monkeys. Three 3‐ to 5‐year‐old male cynomolgus monkeys (Guangzhou Aojun, Guangdong, China) with body weights ranging from 3.5 to 5 kg, were administered single intravenous bolus doses of BT101 in 0.9% saline at doses of 1 and 10 mg/kg and a dose volume of 1 mL/kg. There was a washout period of 14 days between the two dose levels.
Immediately prior to, and at 0.083, 0.25, 1, 2, 4, 8, and 24 hours after each administration, approximately 1 mL of whole blood was collected from each animal via an anterior cephalic vein into vacutainer tubes containing K2EDTA. Blood samples were mixed gently with the anticoagulant after collection and kept on wet ice until centrifugation at 2000 g for 10 minutes at 4°C within 1 hour after collection. Approximately 400 μL of plasma was harvested into microcentrifuge tubes which were frozen on dry ice temporarily until transferred to a freezer of approximately −65°C until analysis for BT101 concentrations. Plasma BT101 concentrations were measured with a high‐performance liquid chromatography‐ultraviolet (HPLC‐UV) method using an ion‐exchange HPLC DNAPac PA200 column (#063000, Thermo Fisher) with detection by UV260 nm and a lower limit of quantitation of 0.125 nmol/mL.
The concentration‐time curve was plotted for each animal and the following parameters were calculated using non‐compartmental models (WinNonlin 6.3) as data permitted: AUC from time zero to the last time point with measurable concentration (AUCt), the extrapolated plasma concentration at time 0 (C0), and the elimination half‐life (t½).
2.7. Effects of BT101 on BT200 pharmacokinetics and activity in cynomolgus monkeys
The effects of BT101 on BT200 pharmacokinetics and activity were evaluated following intravenous administration to cynomolgus monkeys. Twenty‐four hours prior to each BT101 dose, BT200 was administered at a dose level of 0.6 mg/kg by subcutaneous injection in 0.9% saline at a dose volume of 1 mL/kg. BT101 was administered intravenously in 0.9% saline at a dose volume of 1 mL/kg at escalating dose levels of 1, 3, and 10 mg/kg. There was a washout period of 21 days between the doses of BT101.
2.8. Pharmacokinetics
Immediately prior to, and at 0.083, 0.25, 1, 2, 4, 8, 24, 48, 168, and 336 hours after each BT101 administration, approximately 1 mL of whole blood was collected from each animal via an anterior cephalic vein into vacutainer tubes containing K2EDTA. Blood samples were mixed gently with the anticoagulant after collection and kept on wet ice until centrifugation at 2000g for 10 minutes at 4°C within 1 hour after collection. Approximately 400 μL of plasma was harvested into microcentrifuge tubes which were frozen on dry ice temporarily until transferred to a freezer of approximately −65ºC until analysis of BT101, BT200, and BT101/BT200 complex concentrations. Aptamers were measured with an HPLC‐UV method using an ion‐exchange HPLC DNAPac PA200 column (#063000, Thermo Fisher) with detection by UV260 nm and a lower limit of quantitation of 0.125 nmol/mL, 0.050 nmol/mL, and 0.125 nmol/mL for BT101, BT200, and BT101/BT200 duplex, respectively.
The concentration‐time curve was plotted for each animal for each analyte and the following parameters were calculated using non‐compartmental models (WinNonlin 6.3) as data permitted: AUC from time zero to the last time point with measurable concentration (AUCt), the extrapolated plasma concentration at time 0 (C0), and the elimination half‐life (t½).
2.9. Evaluation of von Willebrand factor activity
Immediately prior to, and at 0.083, 0.25, 1, 2, 4, 8, 24, 48, 168, and 336 hours after each BT101 administration, approximately 1 mL of whole blood was collected from each animal via an anterior cephalic vein into vacutainer tubes containing K2EDTA. Blood samples were mixed gently with the anticoagulant after collection and kept on wet ice until centrifugation at 2000 g for 10 minutes at 4°C within 1 hour after collection. Approximately 400 μL of plasma was harvested into microcentrifuge tubes which were frozen on dry ice temporarily until transferred to a freezer of approximately −65°C pending analysis for VWF activity (VWF:Act) using the REAADS® Von Willebrand Factor activity test kit (Corgenix, Inc).
2.10. Evaluation of platelet activity
The effects of BT101 on BT200‐induced inhibition of platelet function were evaluated using a PFA. Immediately prior to, and at 0.083, 0.25, 1, 2, 4, 8, 24, 48, 168, and 336 hours after each BT101 administration, approximately 1 mL of whole blood was collected from each animal via an anterior cephalic vein into vacutainer tubes containing sodium citrate. Blood samples were kept at room temperature and analyzed within 4 hours of sampling. Briefly, after incubation at 37°C for 15 minutes, platelet plug formation was measured by CADP‐CT with a PFA‐200 (Siemens, Marburg, Germany). Normal saline was used as a negative control. Maximal CT measured by the PFA‐200 is 5 minutes and the instrument gives a result of >300 seconds if this time is exceeded.
3. RESULTS
The nucleotide sequence and base pairing with BT200 are shown in Figure 1. Unlike BT200, BT101 does not contain PEG in order to limit its free concentrations in the systemic circulation and enable fine control of reversal activity.
Figure 1.

The sequence and depiction of the binding of BT200 to its reversal agent, BT101
3.1. BT101 binds to BT200 in vitro
BT101 bound to BT100, as evidenced by the formation of a duplex that was not seen under conditions in which BT100 or BT101 were loaded directly onto the gel without mixing them together (Figure 2). Under conditions in which BT100 and BT101 were incubated at a 1:1 ratio, only the duplex band was apparent, demonstrating nearly complete binding. No further formation of duplex was seen under conditions in which excess BT100 or excess BT101 was present, supporting the notion that the interaction between the two molecules is a 1:1 interaction.
Figure 2.
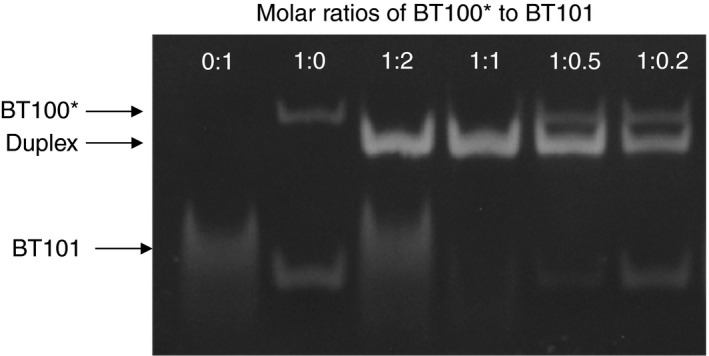
Interaction of BT101 with the core aptamer of BT200 (known as BT100) at molar ratios of 0:1, 1:0, 1:2, 1:1, 1:0.5, and 1:0.2 evaluated using polyacrylamide gel electrophoresis. The bands corresponding to BT101, BT100, and the duplex resulting from the binding of the two are indicated by the arrows.
3.2. BT101 inhibits the binding of BT200 to purified human VWF
BT200 bound to purified human VWF in a concentration‐dependent manner (Figure 3). The EC50 for the interaction between BT200 and human VWF was 33 nM. Pre‐incubation of BT200 with a 1:1 molar ratio of BT101 resulted in complete inhibition of the interaction between BT200 and VWF as no increase in optical density was noted at BT200 concentrations up to 10 μM under these conditions.
Figure 3.
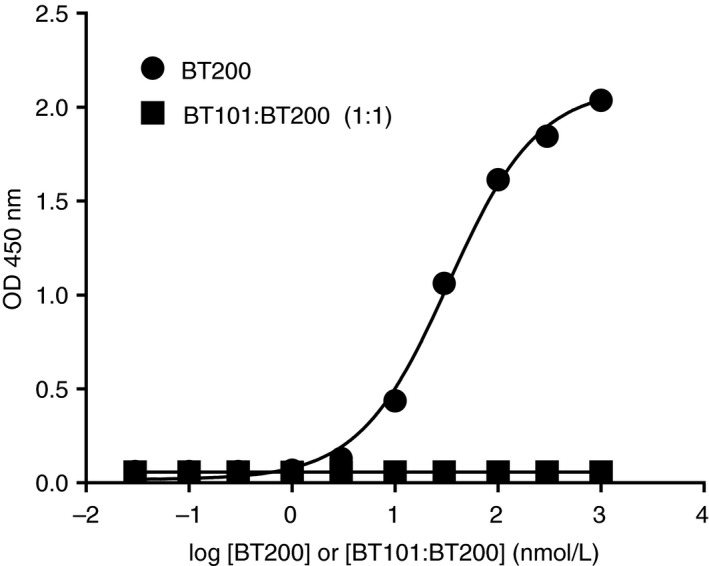
Binding of BT200 to purified human von Willebrand factor in the absence or presence of a 1:1 molar ratio of BT101 as measured by enzyme‐linked immunosorbent assay
3.3. BT101 reverses the inhibition of VWF activity by BT200 in human plasma
At a concentration of 3 μg/mL, BT200 reduced VWF activity in citrated human plasma to 28.5% of normal (Figure 4), consistent with our recently published results. 6 Incubation of the BT200‐treated plasma with increasing molar ratios of BT101 reversed the BT200‐induced reduction in VWF activity, with complete reversal at a 1:1 ratio and no further increase as the ratio of BT101 to BT200 was increased to 2:1.
Figure 4.
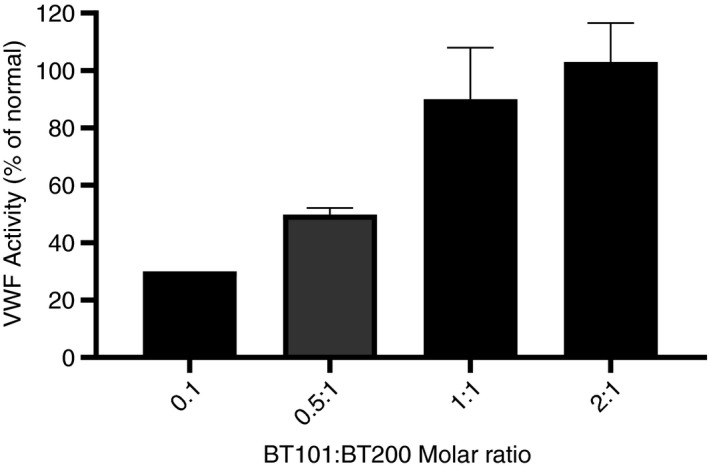
Effects of BT101 on BT200‐induced inhibition of von Willebrand factor activity measured using the REAADS® VWF:Act assay. BT200 was tested at a final concentration of 3 μg/mL with or without different molar ratios of BT101. VWF:Act is presented as relative percent concentration which was determined against a curve made from the reference plasma provided with the kit.
3.4. BT101 reverses the inhibition of VWF dependent platelet function by BT100
BT100 prolonged the time until platelet plug formation in a concentration‐dependent manner, reaching a maximum of 300 seconds at a concentration of 100 nM (Figure 5A), consistent with our recently published results. 6 Co‐incubation of the BT100‐treated whole blood with molar ratios of BT101 ≥ 1:1 completely antagonized the BT100‐induced prolongation of CADP‐CT.
Figure 5.
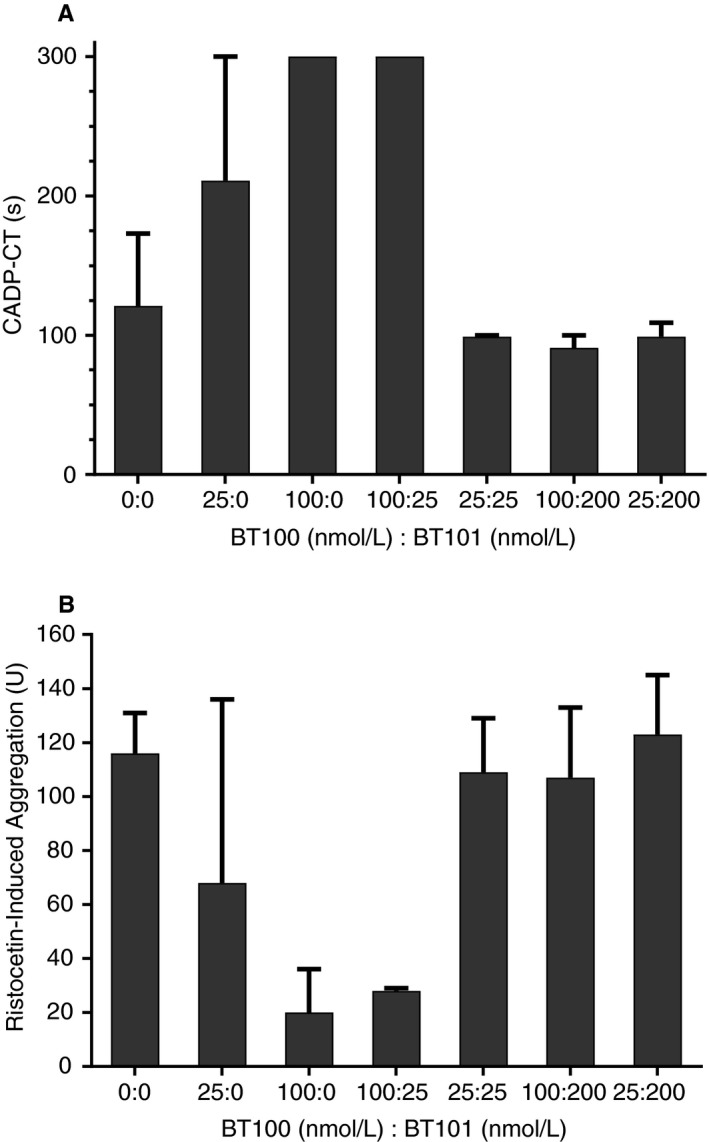
Effects of BT101 on BT100 (non‐PEGylated version of BT200)‐induced inhibition of platelet function. Platelet function was assessed as collagen/adenosine diphosphate induced closure time (CADP‐CT) in seconds (s) (A) and as ristocetin‐induced aggregation in U (B).
BT100 also concentration‐dependently decreased ristocetin‐induced platelet aggregation with a decrease of >80% at a 100 nM concentration (Figure 5B). As with the effects on CADP‐CT, co‐incubation of the BT100‐treated whole blood with molar ratios of BT101 ≥ 1:1 completely antagonized the BT100‐induced decrease in ristocetin‐induced aggregation.
3.5. BT101 has a short half‐life following intravenous infusion to monkeys
Plasma levels of BT101 following single intravenous infusions of 1 or 10 mg/kg BT101 are shown in Figure 6. Plasma concentrations fell below the limit of quantitation (<0.125 nmol/mL) by 1 and 4 hours post‐dose, respectively. Systemic exposure to BT101 as measured by C0 and AUCt increased with dose, and the increase was greater than dose proportional. The elimination half‐life (t½) could not be calculated following infusion of the lower dose due to an insufficient number of time points with measurable concentrations; mean t½, however, was estimated to be approximately 18 minutes at 10 mg/kg.
Figure 6.

Plasma concentrations of BT101 following intravenous administration to male cynomolgus monkey at 1 mg/kg (●) or 10 mg/kg (■). Only time points with concentrations exceeding the limit of quantitation (0.125 nmol/mL) are presented on the graph. Each concentration represents the mean of three animals ± standard error (SEM).
3.6. BT101 infusion affects BT200 pharmacokinetics and activity in monkeys
3.6.1. Pharmacokinetics
Plasma concentrations of BT200, BT101, and BT101/BT200 duplexes after subcutaneous injection of BT200 followed 24 hours later by intravenous injection of BT101 are shown in Figure 7. Mean BT200 concentrations ranged from 0.71 ± 0.17 nmol/mL to 0.89 ± 0.33 nmol/mL (Time = 0, Figure 7A) at 24 hours after subcutaneous injection of 0.6 mg/kg BT200 to cynomolgus monkeys (N = 3/group). Within 5 minutes (0.083 hours) following intravenous infusion of BT101 at dose levels of 1, 3, or 10 mg/kg, BT200 concentrations were below the limit of quantitation (BLQ, <0.05 nmol/mL). At 1 mg/kg BT101, BT200 concentrations remained BLQ for 0.25 hours post‐dose, after which they slowly increased through 48 hours post‐dose. At the higher dose levels of 3 and 10 mg/kg BT101, BT200 plasma concentrations remained BLQ for 2 and 4 hours, respectively. These decreases in BT200 plasma concentrations coincided with immediate increases in BT101/BT200 duplex concentrations that were noted by 5 minutes following BT101 infusion and peaked between 0.44 hours (at 1 mg/kg BT101) and 5.3 hours (at 10 mg/kg BT101) post‐dose (Figure 7B). After achieving peak concentrations, BT101/BT200 duplex concentrations slowly declined through 336 hours following BT101 infusion and were BLQ in the 1 and 3 mg/kg dose groups at that time. The mean elimination half‐life for BT101/BT200 duplexes ranged from 53.3 to 68.1 hours. BT101 plasma concentrations increased in a dose‐related manner from 1 to 10 mg/kg, peaking at 5 minutes following intravenous infusion and declining rapidly to BLQ by 1 hour (at 1 mg/kg) to 8 hours (at 10 mg/kg) post‐dose (Figure 7C). Overall, the pharmacokinetic profile of BT101 was similar to that seen following intravenous injection in the absence of BT200 pre‐treatment; however, slightly lower peak concentrations were noted, likely due to the interaction of BT101 with BT200 in the BT101/BT200 duplexes that were formed.
Figure 7.
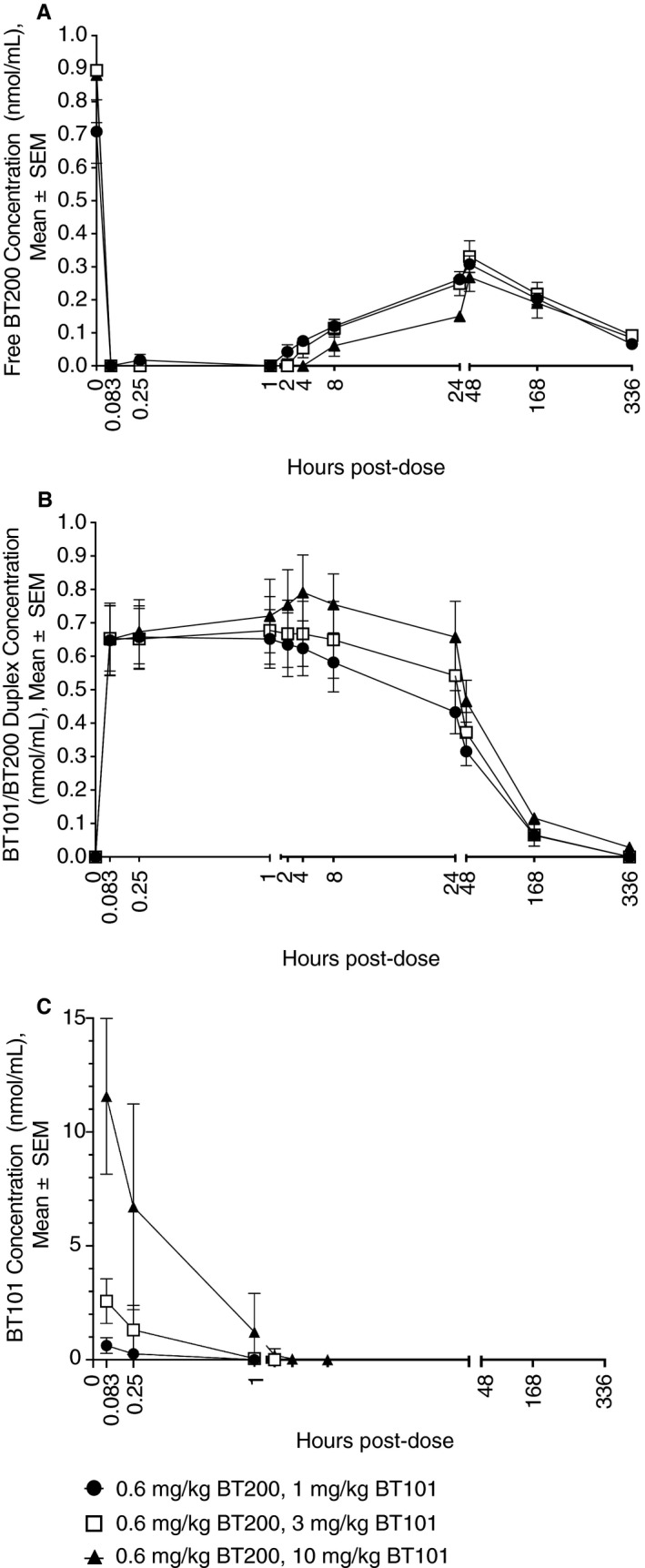
Plasma concentrations of BT200 (A), BT101/BT200 duplex (B), and BT101 (C) after subcutaneous administration of BT200 (0.6 mg/kg) followed 24 hours later by intravenous administration of BT101 at 1 mg/kg (●), 3 mg/kg (□), or 10 mg/kg (▲). For BT101, only time points with concentrations exceeding the limit of quantitation (0.125 nmol/mL) are presented on the graph. Each concentration represents the mean of three animals ± standard error (SEM).
3.7. Effect on BT200‐induced inhibition of VWF activity
VWF activity was evaluated using the REAADS® assay. Prior to subcutaneous injection of BT200 mean VWF activity values were 137 ± 12%. Injection of 0.6 mg/kg BT200 decreased mean VWF activity values to 5% after 24 hours (Time = 0, Figure 8A). Infusion of BT101 reversed the effects of BT200 within 5 minutes and VWF activity values returned to baseline. The onset of the effect on VWF activity coincided with the decline of BT200 plasma concentrations to BLQ values and the appearance of BT101/BT200 duplexes (Figure 7B). At all dose levels, VWF activities remained near baseline (pre‐BT200 administration) values for approximately 2 hours following BT101 infusion, after which they slowly decreased through 48 hours post‐dose with the rate of decrease being dose dependent. From 48 hours to 336 hours post‐dose VWF activity slowly increased, while BT200 concentrations slowly declined (Figure 7A) during this time period.
Figure 8.

(A) VWF activity as measured using the REAADS assay, or (B) platelet function as measured by collagen/adenosine diphosphate induced closure time (CADP‐CT) in seconds (s) after subcutaneous administration of BT200 (0.6 mg/kg) followed 24 hours later by intravenous administration of BT101 at 1 mg/kg (●), 3 mg/kg (□), or 10 mg/kg (▲). Each value represents the mean of three animals ± standard error (SEM).
3.8. Effect on BT200‐induced inhibition of VWF dependent platelet function
Platelet plug formation under high shear rates was evaluated by measuring closure times using a platelet function analyzer. Prior to subcutaneous injection of BT200, mean closure times were 78 ± 22 seconds. Injection of 0.6 mg/kg BT200 increased average closure times to a maximum value of 300 seconds (Figure 8B). Intravenous infusion of BT101 reversed the effects of BT200 within 5 minutes and closure times returned to baseline values. The onset of the effect coincided with the decline of BT200 plasma concentrations to BLQ values and the appearance of BT101/BT200 duplexes (Figure 7B). At all dose levels closure times remained near baseline (pre‐BT200 administration) values for approximately 8 hours following BT101 injection. When BT200 levels started to increase again, closure times also became prolonged and eventually normalized when BT200 was cleared from the circulation (Figure 7A).
3.9. Safety and tolerability of BT200 and BT101
There were no apparent safety or tolerability issues after injection of BT200 or bolus injection of BT100; in particular no bleeding or thrombotic events occurred.
4. DISCUSSION
The problem with the available therapeutic options for prevention of secondary stroke is that the risk/benefit profile of the potent platelet inhibitors, so well established in the domain of coronary disease, does not translate well into the domain of cerebrovascular disease. Aspirin, clopidogrel, or their combination are reasonably safe with respect to bleeding risk, but only modestly effective. 18 , 19 , 20 The more potent, next generation P2Y12 antagonist ticagrelor may moderately reduce the risk of thrombotic events by 11% as compared with aspirin. However, the pivotal trial failed to reach its primary endpoint. 21 Thus, it is only the results from the THALES trial, which investigated the combination of aspirin and ticagrelor versus aspirin alone, which are awaited. Dual antiplatelet therapy with clopidogrel and aspirin is increasingly used in management of stroke patients, and bleeding may be aggravated with any kind of dual anti‐platelet therapy. 22 This is especially problematic in the stroke population because of their inherent bleeding susceptibility. Thus, there is an unmet medical need for novel anti‐thrombotic pharmacotherapy with an improved risk/benefit profile for use in secondary stroke prevention.
BT200, an aptamer inhibitor of VWF A1 binding to platelet GPIb, was developed as an effective inhibitor of arterial thrombosis. 6 BT200 has entered clinical development with a primary indication of secondary stroke prevention. This is based on the role of VWF in thrombosis 23 , 24 , 25 and in particular the pathogenetic involvement of VWF in stroke. 7 , 8 , 26 Clinical experience with VWF inhibitors suggests that they, like other anti‐platelet agents, may also carry a bleeding risk. This is seen with the recently approved nanobody VWF inhibitor caplacizumab, which can cause mucocutaneous bleedings in patients with acquired thrombotic thrombocytopenic purpura. 27 Current prescribing information warns that there is a risk of severe bleeding, and interruption of treatment is recommended if acute bleeding does occur. A 7‐day interval prior to elective surgical procedures is recommended. Unfortunately, there is no direct reversal agent for caplacizumab. However, one of the patients treated in the phase III trial with caplacizumab received VWF concentrate as the only treatment for the resolution of a severe, serious adverse event of epistaxis. 28 Such VWF concentrates may not only be used to overcome the inhibitory action of caplacizumab but also that of BT200.
In order to maximize the potential therapeutic utility of BT200 and address the putative risk of bleeding, the direct reversal agent BT101 has been developed. The structure of aptamers made the generation of an effective antagonist relatively straightforward. Aptamer composition of oligonucleotide sequences provides the code for their own complementary sequences that can be used to develop inhibitory reversal agents. 10 Using this strategy BT101, a complementary aptamer, was developed to specifically inhibit BT200’s function. This study demonstrated the activity of the antidote in vitro, its pharmacokinetics alone, as well as the interactions with BT200, and rapid reversal of BT200‐induced inhibition of platelet function in cynomolgus monkeys. The results suggest that BT101 has potential as a direct reversal agent for BT200 in a clinical setting.
It is only recently that attempts have been made to develop reversal agents for various direct‐acting anticoagulants, many years after these agents have been marketed. These include idarucizumab against dabigatran, 29 ciraparantang or andexanet against oral FXa inhibitors, 30 and most recently an antibody against ticagrelor. 31 None of these antidotes were available during clinical development of the corresponding therapeutic agents. The REG1 system consisting of pegnivacogin, an aptameric factor IXa inhibitor, and its controlling agent anivamersen, had been in clinical development prior to discontinuation prior to market authorization. 10 Intravenous infusion of reversal agents is a common and necessary feature to allow for rapid onset of action, and this also holds true for BT101. In contrast to antibodies with an extended half‐life, free BT101 is rapidly cleared, a typical fate of unpeglated aptamers. 5 Due to its small size, BT101 can be cleared through renal filtration as evidenced by its short half‐life. BT200 is designed to have a long half‐life as the large size of 40K PEG protects it from renal filtration. In addition, BT200 is highly resistant to endonuclease and exonuclease activity with the combination of 5'‐end 40K PEG, 3'‐end inverted deoxythymidine and 2´‐O‐modified riboses. Potentially BT200, just like other pegylated aptamers and biologics, can be cleared through phagocytosis by macrophages, as is evidenced by the vacuolated macrophages in various tissues. 32 Details of the clearance mechanism of BT200 remain to be further studied. BT101, infused 24 hours after an injection of BT200, effectively lowered its free plasma concentrations for a minimum duration of 8 hours, restoring hemostatically effective VWF concentrations. Such a limited period appears ideally suited to the temporary restoration of VWF activity in case of mucocutaneous bleedings such as epistaxis, or peri‐interventionally/peri‐operatively. Continued resorption of BT200 from the subcutaneous depot still occurs 24 hours after its subcutaneous injection, because the average time to maximum plasma concentrations is 28 to 30 hours in non‐human primates. 6 This explains the observed increase in BT200 concentrations 8 to 48 hours after infusion of its reversal agent, because a single bolus infusion of BT101 is eliminated from the circulation within 2 hours. While we expect similar pharmacokinetics of BT101 in humans, different resorption kinetics of BT200 in humans may alter the duration of BT101 action, and this has to be investigated in properly designed human trials. If needed, longer lasting antagonism of BT200 can be achieved by repeated bolus or continuous infusion of BT101, and the applied platelet function tests can rapidly deliver information on existing reversal of BT200 action at bedside, if required.
In summary, the complementary aptamer BT101 rapidly reverses the VWF inhibitory actions of BT200 in vitro and in cynomolgus monkeys, which supports its further clinical development.
CONFLICTS OF INTEREST
Drs Zhu and Gilbert are employees of, and Drs Jilma and Tarantino are consultants to Guardian Therapeutics, Inc. Drs Zhu, Gilbert, and Jilma also have an equity position with Guardian. Drs Kang and Liang are employees of Suzhou Ribo Life Sciences, and Dr Liang also has an equity position.
AUTHOR CONTRIBUTIONS
All authors contributed to the preparation of the article and met the required conditions for authorship. All authors participated in the design, analysis, and interpretation of the data. All authors revised the article for critical content.
ACKNOWLEDGMENTS
The authors thank Dr R. Schaub for his editorial contributions and review of the manuscript.
Zhu S, Gilbert JC, Liang Z, et al. Potent and rapid reversal of the von Willebrand factor inhibitor aptamer BT200. J Thromb Haemost. 2020;18:1695–1704. 10.1111/jth.14822
Manuscript handled by: X. Long Zheng
Final decision: X. Long Zheng, 31 March 2020
Funding Information
These studies were funded by Guardian Therapeutics.
REFERENCES
- 1. Steinhubl SR, Moliterno DJ. The role of the platelet in the pathogenesis of atherothrombosis. Am J Cardiovasc Drugs. 2005;5:399‐408. [DOI] [PubMed] [Google Scholar]
- 2. Ruggeri ZM. Platelets in atherothrombosis. Nat Med. 2002;8:1227‐1234. [DOI] [PubMed] [Google Scholar]
- 3. Ruggeri ZM. Von Willebrand factor: looking back and looking forward. Thromb Haemost. 2007;98:55‐62. [PubMed] [Google Scholar]
- 4. Zhou J, Rossi J. Aptamers as targeted therapeutics: current potential and challenges. Nat Rev Drug Discov. 2017;16:181‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kovacevic KD, Gilbert JC, Jilma B. Pharmacokinetics, pharmacodynamics and safety of aptamers. Adv Drug Deliv Rev. 2018;134:36‐50. [DOI] [PubMed] [Google Scholar]
- 6. Zhu S, Gilbert JC, Hatala P, et al. The development and characterization of a long acting anti‐thrombotic von Willebrand factor (VWF) aptamer. J Thromb Haemost. 2020;10.1111/jth.14755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Buchtele N, Schwameis M, Gilbert JC, Schorgenhofer C, Jilma B. Targeting von Willebrand factor in ischaemic stroke: focus on clinical evidence. Thromb Haemost. 2018;118:959‐978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Denorme F, De Meyer SF. The VWF‐GPIb axis in ischaemic stroke: lessons from animal models. Thromb Haemost. 2016;116:597‐604. [DOI] [PubMed] [Google Scholar]
- 9. Christos S, Naples R. Anticoagulation reversal and treatment strategies in major bleeding: update 2016. West J Emerg Med. 2016;17:264‐270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Povsic TJ, Sullenger BA, Zelenkofske SL, Rusconi CP, Becker RC. Translating nucleic acid aptamers to antithrombotic drugs in cardiovascular medicine. J Cardiovasc Transl Res. 2010;3:704‐716. [DOI] [PubMed] [Google Scholar]
- 11. Diener JL, Daniel Lagasse HA, Duerschmied D, et al. Inhibition of von Willebrand factor‐mediated platelet activation and thrombosis by the anti‐von Willebrand factor A1‐domain aptamer ARC1779. J Thromb Haemost. 2009;7:1155‐1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jilma‐Stohlawetz P, Knobl P, Gilbert JC, Jilma B. The anti‐von Willebrand factor aptamer ARC1779 increases von Willebrand factor levels and platelet counts in patients with type 2B von Willebrand disease. Thromb Haemost. 2012;108:284‐290. [DOI] [PubMed] [Google Scholar]
- 13. Steinlechner B, Zeidler P, Base E, et al. Patients with severe aortic valve stenosis and impaired platelet function benefit from preoperative desmopressin infusion. Ann Thorac Surg. 2011;91:1420‐1426. [DOI] [PubMed] [Google Scholar]
- 14. Favaloro EJ. Utility of the platelet function analyser (PFA‐100/200) for exclusion or detection of von Willebrand disease: a study 22 years in the making. Thromb Res. 2020;188:17‐24. [DOI] [PubMed] [Google Scholar]
- 15. Fuchs I, Frossard M, Spiel A, Riedmuller E, Laggner AN, Jilma B. Platelet function in patients with acute coronary syndrome (ACS) predicts recurrent ACS. J Thromb Haemost. 2006;4:2547‐2552. [DOI] [PubMed] [Google Scholar]
- 16. Valarche V, Desconclois C, Boutekedjiret T, Dreyfus M, Proulle V. Multiplate whole blood impedance aggregometry: a new tool for von Willebrand disease. J Thromb Haemost. 2011;9:1645‐1647. [DOI] [PubMed] [Google Scholar]
- 17. Schmidt DE, Bruzelius M, Majeed A, Odeberg J, Holmstrom M, Agren A. Whole blood ristocetin‐activated platelet impedance aggregometry (Multiplate) for the rapid detection of Von Willebrand disease. Thromb Haemost. 2017;117:1528‐1533. [DOI] [PubMed] [Google Scholar]
- 18. Bath PM, Woodhouse LJ, Appleton JP, et al. Antiplatelet therapy with aspirin, clopidogrel, and dipyridamole versus clopidogrel alone or aspirin and dipyridamole in patients with acute cerebral ischaemia (TARDIS): a randomised, open‐label, phase 3 superiority trial. Lancet. 2018;391:850‐859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bhatt DL, Fox KA, Hacke W, et al. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med. 2006;354:1706‐1717. [DOI] [PubMed] [Google Scholar]
- 20. Diener HC, Bogousslavsky J, Brass LM, et al. Aspirin and clopidogrel compared with clopidogrel alone after recent ischaemic stroke or transient ischaemic attack in high‐risk patients (MATCH): randomised, double‐blind, placebo‐controlled trial. Lancet. 2004;364:331‐337. [DOI] [PubMed] [Google Scholar]
- 21. Johnston SC, Amarenco P, Albers GW, et al. Ticagrelor versus aspirin in acute stroke or transient ischemic attack. N Engl J Med. 2016;375:35‐43. [DOI] [PubMed] [Google Scholar]
- 22. Hackam DG, Spence JD. Antiplatelet therapy in ischemic stroke and transient ischemic attack. Stroke. 2019;50:773‐778. [DOI] [PubMed] [Google Scholar]
- 23. Swystun LL, Lillicrap D. Genetic regulation of plasma von Willebrand factor levels in health and disease. J Thromb Haemost. 2018;16:2375‐2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sonneveld MA, de Maat MP, Leebeek FW. Von Willebrand factor and ADAMTS13 in arterial thrombosis: a systematic review and meta‐analysis. Blood Rev. 2014;28:167‐178. [DOI] [PubMed] [Google Scholar]
- 25. Seaman CD, Yabes J, Comer DM, Ragni MV. Does deficiency of von Willebrand factor protect against cardiovascular disease? Analysis of a national discharge register. J Thromb Haemost. 2015;13:1999‐2003. [DOI] [PubMed] [Google Scholar]
- 26. Sanders YV, Eikenboom J, de Wee EM, et al. Reduced prevalence of arterial thrombosis in von Willebrand disease. J Thromb Haemost. 2013;11:845‐854. [DOI] [PubMed] [Google Scholar]
- 27. Knoebl P, Cataland S, Peyvandi F, et al. Efficacy and safety of open‐label caplacizumab in patients with exacerbations of acquired thrombotic thrombocytopenic purpura in the hercules study. J Thromb Haemost. 2020;18(2):479‐484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Scully M, Cataland SR, Peyvandi F, et al. Caplacizumab treatment for acquired thrombotic thrombocytopenic purpura. N Engl J Med. 2019;380:335‐346. [DOI] [PubMed] [Google Scholar]
- 29. Giustozzi M, Verso M, Agnelli G, Becattini C. Reversal of dabigatran‐associated bleeding using idarucizumab: review of the current evidence. J Thromb Thrombolysis. 2017;44:527‐535. [DOI] [PubMed] [Google Scholar]
- 30. Kaatz S, Bhansali H, Gibbs J, Lavender R, Mahan CE, Paje DG. Reversing factor Xa inhibitors ‐ clinical utility of andexanet alfa. J Blood Med. 2017;8:141‐149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bhatt DL, Pollack CV, Weitz JI, et al. Antibody‐based ticagrelor reversal agent in healthy volunteers. N Engl J Med. 2019;380:1825‐1833. [DOI] [PubMed] [Google Scholar]
- 32. Ivens IA, Achanzar W, Baumann A, et al. PEGylated biopharmaceuticals: current experience and considerations for nonclinical development. Toxicol Pathol. 2015;43:959‐983. [DOI] [PubMed] [Google Scholar]