

Abstract
Objective
Most of the patients with Down syndrome (DS) develop Alzheimer's disease (AD) neuropathology by age 40. Although this increased susceptibility to AD in DS is thought to be primarily due to triplication of the amyloid precursor protein located on chromosome 21, the precise molecular mechanisms are not well understood. Recent evidence has implicated defective protein sorting and trafficking secondary to deficiencies in retromer complex proteins in AD pathogenesis. Thus, the objective of the present study is to assess the retromer complex system in DS.
Methods
Human postmortem brain tissue and fibroblasts from subjects with DS and healthy controls were examined for the various retromer protein components using Western blot analysis and reverse transcription quantitative polymerase chain reaction (RT‐qPCR).
Results
Retromer recognition core proteins were significantly decreased in DS fibroblasts, and in both the hippocampi and cortices of young (age 15–40 years old) and aged (40–65 years old) subjects with DS compared with controls. Correlation analyses showed a significant inverse relationship between recognition core proteins and levels of soluble forms of Aβ 1–40 and 1–42 in both hippocampus (n = 33, Spearman = −0.59 to −0.38, p ≤ 0.03 for VPS35, VPS26, VPS29, and VPS26B) and cortex tissue (n = 57, Spearman = −0.46 to −0.27, p ≤ 0.04 for VPS35, VPS26, and VPS29) of the same patients.
Interpretation
We conclude that dysregulation of the retromer complex system is an early event in the development of the AD‐like pathology and cognitive decline in DS, and for this reason the system could represent a novel potential therapeutic target for DS. ANN NEUROL 2020 ANN NEUROL 2020;88:137–147
Down syndrome (DS) is a congenital condition resulting from a partial or complete triplication of chromosome 21 (HSA21) and is the leading cause of genetically defined intellectual disability. Individuals with DS have a significant increased risk of developing Alzheimer's disease (AD), with over 50% of all individuals with DS displaying a classical AD‐like neuropathology and cognitive decline. 1 This increased incidence of AD in DS cases, also known as AD‐DS, has primarily been attributed to gene overdosage of the amyloid precursor protein (APP), which is located on HSA21. However, the precise mechanisms that lead to the classical DS neuropathology remain unclear as the trisomy of HSA21 can induce plaque deposition independently of APP triplication. 2 Similar to AD, enlargement of APP‐positive endosomes occurs early in the pathogenesis of AD‐DS prior to amyloid beta (Aβ) deposition.3, 4 The endo‐lysosomal system is the first site of Aβ accumulation in individuals with DS, suggesting that the subcellular localization of APP may be critical during the early stages of AD‐DS pathogenesis.2, 5
In AD, endocytic pathway abnormalities have been linked to alterations in endosomal sorting and trafficking secondary to deficiencies in the vacuolar protein sorting system known as the retromer complex. VPS35 and VPS26, two components of the cargo recognition core of the retromer complex, are significantly decreased in the hippocampi of patients with AD, and variants of retromer components are associated with an increased risk of developing AD.6, 7, 8, 9 Loss of function of the retromer complex secondary to its deficiencies in its core components results in enhanced Aβ production, enlargement of early endosomes, and cognitive impairments,10, 11 whereas overexpression of VPS35 reduces AD pathology and cognitive impairments in a mouse model of AD. 12 Given that deficiencies in the retromer complex system are associated with AD pathogenesis, and early endosome enlargement is a key feature of both AD and DS, in the current study, we examined the retromer complex in DS using both postmortem brain tissues and patient‐derived fibroblasts from individuals with DS and unaffected controls (CTRs). For what we believe is the first time, we report that dysregulation of the retromer complex occurs in DS at an early stage in the pathogenesis and the onset of the AD‐like pathological phenotype.
Methods
Human Brain Samples
Postmortem cortex and hippocampus tissues from DS and unaffected matched CTR subjects were obtained from the University of Maryland Brain and Tissue Bank, the University of Miami Brain Endowment Bank, and the Brain Tissue Donation Program at the University of Pittsburg, all repositories of the National Institutes of Health (NIH) Neurobiobank. Both men and women were used in the study, and subjects ranged from 15 to 65 years of age. Patient information is listed in Table S1.
Cell Culture
Human fibroblasts from DS (AG04823) and CTR (AG08498) donors were obtained from Coriell Cell Repositories and cultured according to the providers protocols (https://catalog.coriell.org/). Cells were harvested at approximately 85 to 90% confluence and proteins were extracted using radioimmunoprecipitation assay (RIPA) buffer containing protease and phosphatase inhibitor cocktails (Santa Cruz Biotechnology, Dallas, TX, USA).
Immunoblot Analysis
Brain homogenates were extracted using RIPA buffer, as previously described.13, 14 Briefly, tissue samples were sonicated in RIPA buffer supplemented with protease and phosphatase inhibitor cocktails (Santa Cruz Biotechnology), ultracentrifuged at 90,000 RCF for 45 minutes, and supernatants used for Western blot analysis. Total protein concentration was determined by using a BCA Protein Assay Kit (Pierce, Rockford, IL, USA). Samples were separated by SDS‐PAGE using 10% Bis‐Tris gels, then transferred onto nitrocellulose membranes (Bio‐Rad). Membranes were incubated with primary antibodies overnight at 4°C, and subsequently with IRDye 800CW‐labeled or IRDye 680CW‐labeled secondary antibodies (LI‐COR Bioscience, Lincoln, NE, USA) at 22°C for 1 hour. Signals were developed with Odyssey Infrared Imaging Systems (LI‐COR Bioscience). Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) was used as an internal loading control. All proteins evaluated in this study are analyzed in the results. An internal control was always used in each immunoblot to allow for inter‐blot analysis. Additionally, samples from each age group were run on the same gel (with an internal control) to allow for comparison between groups. The following primary antibodies were used: APP (1:100; Abcam: ab32136), GAPDH (1:500, Cell Signaling Technology: 2118), CI‐MPR (1:1000, Abcam: ab124767), Sorla (1:100, Cell Signaling Technology: 79322), VPS26 (1:200; Abcam: ab23892), VPS26b (1:200, Proteintech Group, Chicago, IL, USA: 15915‐1‐AP), VPS29 (1:100, Santa Cruz: 398874), and VPS35 (1:200; Abcam: ab10099).
Real Time Quantitative Polymerase Chain Reaction
RNA was extracted and purified using the RNeasy mini kit (Qiagen, Valencia, CA, USA), as previously described. 15 Briefly, 1 μg of total RNA was used to synthesize cDNA in a 20 μl reaction using the RT2 First Strand Kit for real time‐quantitative polymerase chain reaction (RT‐qPCR) (Qiagen). Brain tissue and cells VPS35, VPS26, and VPS29 were amplified using the commercially available primers (Qiagen) and β‐actin was used as an internal control gene. One microliter of cDNA was added to 10 μl of SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) and RT‐qPCR) was performed using StepOnePlus Real‐Time PCR Systems (Applied Biosystems). Each sample was run in triplicate and analysis of relative gene expression was done by StepOne software version 2.1. (Applied Biosystems).
Enzyme‐Linked Immunosorbent Assay
Brain homogenates were extracted as previously described with RIPA buffer for the soluble Aβ 1–40 and 1–42 fractions, and formic acid was used for extraction of the insoluble fractions. Soluble and insoluble fractions were assayed using human Aβ 1–40 and 1–42 sandwich enzyme‐linked immunosorbent assay kits (Invitrogen, Carlsbad, CA, USA).
Data Analyses and Statistical Methods
Descriptive summary data were expressed as counts and percentages for categorical variables and mean ± SD/SEM and/or median (range) for continuous variables. Continuous variables that are skewed (eg, amyloid beta measurements) were transformed using the log function when the normality assumption is violated on the original scale. Spearman correlation coefficient was used for correlation analyses between the amyloid beta measurements and retromer protein levels. Pairwise group comparisons of retromer proteins and amyloid beta measurements between age (<40 vs. ≥40), brain region (cortex vs hippocampus), and type of subjects (DS vs controls) subgroups were performed under the framework of the multivariable mixed‐effects regression model approach for each variable of interest in order to take into account of the potential correlation between the observations of the two brain regions from the same subjects included in the study. In particular, all interaction terms among the age, brain region, and type of subjects were included in the model to account for possible heterogeneous effects across different subgroups. Multiple comparison adjusted p values and simultaneous 95% confidence intervals for the estimated group differences were derived via the Tukey–Kramer method from the multivariable mixed‐effects regression model. Note that cause of death differences between DS and CTRs, the former being largely related to the underlying disease (DS), whereas the latter not being DS‐related at all, could not be accounted for in the regression model due to the complete separation of this variable between the two groups. P values <0.05 were considered statistically significant. SAS version 9.4 (SAS Institute, Cary, NC, USA) and GraphPad Prism for Windows version 7.00 were used for all the data analyses.
Results
To assess the retromer complex system in DS, we evaluated retromer proteins in human postmortem cortex and hippocampus tissues from subjects with DS and unaffected matched CTRs (Table S1). To establish a temporal profile of retromer dysregulation within the context of AD‐DS, we examined the retromer core and associated proteins in young patients prior to significant plaque deposition (age 15–40 years), and older subjects (age 40–65 years) at a time when virtually all subjects with DS display neurofibrillary tangles and amyloid beta plaques. 16 Cortices from the younger subset of patients with DS already showed significant dysregulation of retromer recognition core proteins, with VPS26 and VPS29 significantly decreased compared with unaffected CTRs (Fig 1A, C). Because we observed changes in retromer core proteins, we next assessed additional proteins of the retromer complex system assembly implicated in neurodegenerative disease (see Fig 1A, C). We found that protein levels of Sorla, the endocytic receptor for APP, and cation‐independent mannose 6‐phosphate receptor (CI‐MPR), which transports the lysosomal protease Cathepsin D (CTSD), were reduced in cortices from young subjects with DS compared to CTRs, however, changes did not reach significance (see Fig 1A, C). In the older subset of patients VPS35 and VPS29 were also significantly decreased in the cortex, however, CI‐MPR and Sorla were unchanged (Fig 1B, D). Similar decreases in retromer complex proteins were observed in the hippocampi of DS subjects. In the young subset of patients, all retromer core proteins were reduced in the hippocampus of DS patients compared to controls, however, the differences failed to reach significance, possibly due to the smaller sample size of this group compared with cortex (Fig 2A, C). Significant reductions in all retromer recognition core proteins were also observed in the hippocampi of aged subjects with DS compared to matched CTRs (Fig 2B, D).
FIGURE 1.
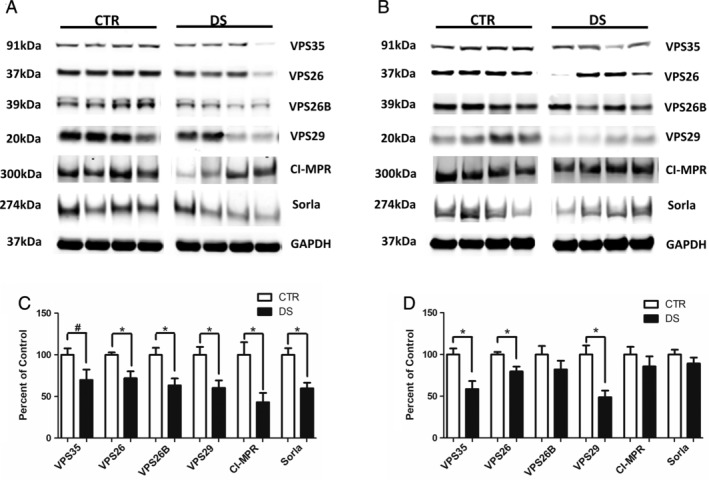
Retromer complex proteins are reduced in cortices of young and aged subjects with Down syndrome (DS) subjects. (A) Representative Western blot of retromer recognition core proteins (VPS35, VPS26, VPS26B, and VPS29) and cargo receptors (cation‐independent mannose 6‐phosphate receptors [CI‐MPR] and Sorla) in cortices from young subjects with DS and matched control (CTR) subjects (15–40 years old). (B) Representative Western blot of retromer recognition core proteins (VPS35, VPS26, VPS26B, and VPS29) and cargo receptors (CI‐MPR and Sorla) in cortices from aged subjects with DS and matched control subjects (40–65 years old). (C) Densitometry analysis of Western blots shown in panel A (CTR, n = 11; DS, n = 11). (D) Densitometry analysis of Western blots shown in panel B (CTR, n = 18; DS, n = 18). Values represent mean ± standard error of the mean (*p < 0.05, #p < 0.10). GAPDH, glyceraldehyde 3‐phosphate dehydrogenase.
FIGURE 2.
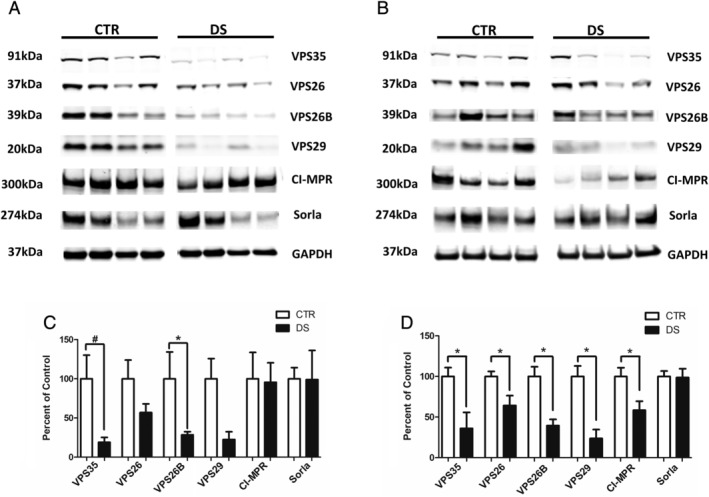
Retromer complex proteins are reduced in hippocampi of young and aged subjects with Down syndrome (DS). (A) Representative Western blot of retromer recognition core proteins (VPS35, VPS26, VPS26B, and VPS29) and cargo receptors (cation‐independent mannose 6‐phosphate receptors [CI‐MPRs] and Sorla) in hippocampus from young subjects with DS and matched control (CTR) subjects (15–40 years old). (B) Representative Western blot of retromer complex core proteins (VPS35, VPS26, VPS26B, and VPS29) and cargo receptors (CI‐MPR and Sorla) in hippocampus from old subjects with DS and CTR subjects (40–65 years old). (C) Densitometry analysis of Western blots shown in panel A (CTR, n = 5; DS, n = 4). (D) Densitometry analysis of Western blots shown in panel B (CTR, n = 14; DS, n = 10). Values represent mean ± standard error of the mean (*p < 0.05, #p < 0.10). GAPDH, glyceraldehyde 3‐phosphate dehydrogenase.
In order to examine differences in both brain regions and age between CTRs and subjects with DS, an additional analysis of retromer core proteins was performed between subject type (CTR vs DS), age group, and brain region (Fig 3, and Table S2). Interestingly, it was found that subject type was the largest determining factor for retromer dysregulation, with brain tissue from patients with DS having significantly less retromer core proteins than unaffected CTRs in both young and aged subjects. Brain region was also determined to be a significant factor for protein levels of all three retromer core components, whereas age was a more significant factor when assessing the cargo receptors CI‐MPR and Sorla. Although not all values reached significance, possibly due to the limitations of sample size in several groups, retromer deficiency was more pronounced in hippocampal tissue compared to cortical tissue for all three proteins examined.
FIGURE 3.
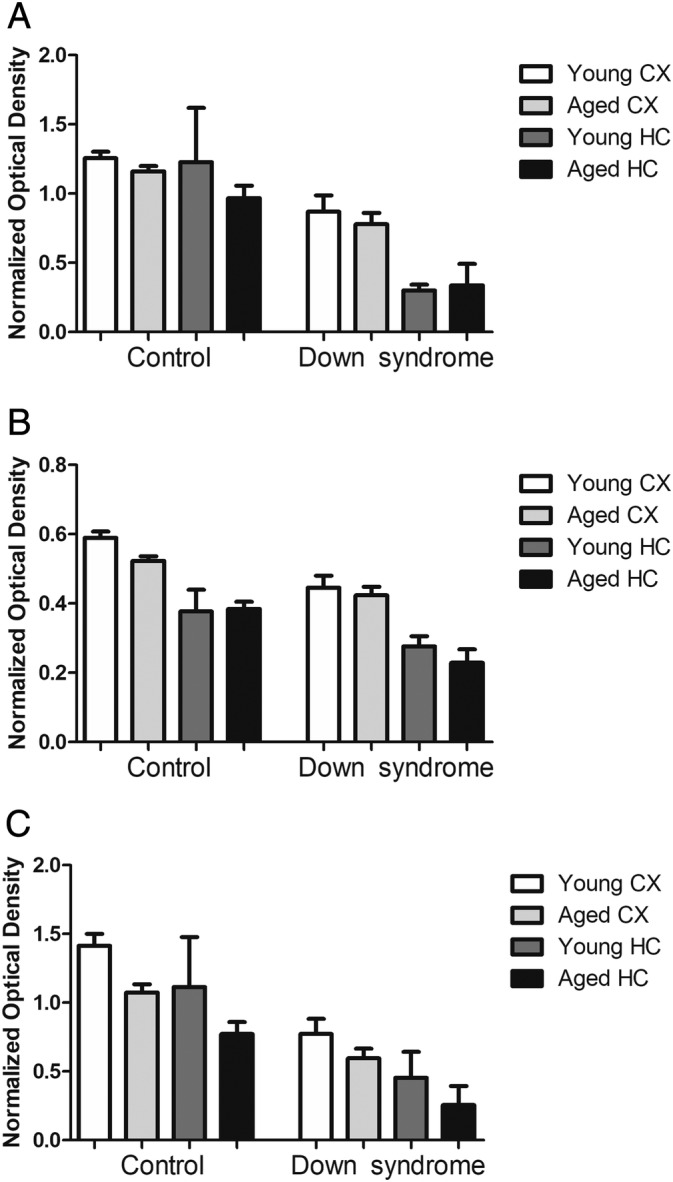
Retromer cargo recognition core; comparison among disorder, region, and age. (A) Densitometry analysis of VPS35 protein levels measured by Western blot analysis in brain tissue from Down syndrome (DS) and control donors. (B) Densitometry analysis of VPS26 protein levels measured by Western blot analysis in brain tissue from DS and control donors. (C) Densitometry analysis of VPS29 protein levels measured by Western blot analysis in brain tissue from DS and control donors (young CTR CX, n = 11; young DS CX, n = 11; aged CTR CX, n = 18; aged DS CX, n = 18; young CTR HC, n = 5; young DS HC n = 4; aged CTR HC, n = 14; and aged DS HC, n = 10). Values represent mean ± standard error of the mean. Multiple comparison adjusted p values displayed in Table S2. CX, cortex; HC, hippocampus.
Although virtually all patients with DS develop AD‐like Aβ neuropathology by age 40 years, not all subjects may conform to this paradigm. To address this, we examined both soluble and insoluble levels of Aβ peptides in cortex and hippocampus tissue from the subjects in the study. As expected, all forms of Aβ were elevated in cortices and hippocampi of aged subjects with DS compared to younger subjects with DS and all control subjects (Tables 1 and 2). Additionally, the relationship between retromer components and Aβ measurements was analyzed. Retromer core components showed significant inverse correlations with soluble forms of Aβ 1–40 and Aβ 1–42 in both hippocampus and cortex tissue (Table 3). On the other hand, several core components showed inverse correlations with the levels of insoluble forms of Aβ peptides, although not all correlations with insoluble forms reached significance (see Table 3). Representative plots showing Aβ levels as a function of VPS35 optical density in cortex tissue are shown in in Figure 4.
Table 1.
Levels of Abeta 1‐40 and Abeta 1‐42 peptides (on a Log Scale) in Cortices and Hippocampi of Down syndrome (DS) and Control Subjects (CTR).
| Soluble Aβ40 | Insoluble Aβ40 | Soluble Aβ42 | Insoluble Aβ42 | ||
|---|---|---|---|---|---|
| CTR CX < 40 | Mean | 2.106 | 1.359 | −0.227 | 1.093 |
| SD | 0.722 | 0.404 | 0.842 | 0.292 | |
| Median | 2.410 | 1.295 | −0.357 | 1.103 | |
| Range | 0.873, 2.933 | 0.755, 2.098 | −1.222, 1.612 | 0.623, 1.653 | |
| n | 11 | 11 | 10 | 11 | |
| CTR CX ≥ 40 | Mean | 2.104 | 1.231 | 0.156 | 0.834 |
| SD | 0.737 | 1.107 | 1.191 | 0.432 | |
| Median | 2.040 | 0.818 | −0.066 | 0.917 | |
| Range | 1.068, 4.492 | −0.099, 4.366 | −1.837, 2.745 | −0.018, 1.509 | |
| N | 18 | 18 | 17 | 18 | |
| CTR HC < 40 | Mean | 2.134 | 1.374 | −0.2612 | 0.8787 |
| SD | 0.416 | 0.680 | 0.712 | 1.274 | |
| Median | 2.112 | 1.206 | −0.393 | 1.197 | |
| Range | 1.655, 2.666 | 0.492, 2.241 | −1.057, 0.902 | −1.300, 2.069 | |
| N | 5 | 5 | 5 | 5 | |
| CTR HC ≥ 40 | Mean | 1.917 | 1.248 | −0.4851 | 0.7203 |
| SD | 0.285 | 1.017 | 0.676 | 0.643 | |
| Median | 1.897 | 0.861 | −0.596 | 0.956 | |
| Range | 1.440, 2.470 | 0.223, 3.441 | −1.882, 0.577 | −0.870, 1.254 | |
| N | 14 | 14 | 14 | 14 | |
| DS CX < 40 | Mean | 2.289 | 1.886 | 0.4354 | 1 |
| SD | 0.498 | 1.114 | 1.005 | 0.608 | |
| Median | 2.466 | 1.753 | 0.513 | 1.015 | |
| Range | 1.632, 3.114 | 0.602, 4.079 | −0.960, 1.885 | −0.193, 1.900 | |
| N | 10 | 10 | 9 | 10 | |
| DS CX ≥ 40 | Mean | 7.232 | 11.5 | 3.437 | 10.96 |
| SD | 1.755 | 2.432 | 1.612 | 1.929 | |
| Median | 6.804 | 11.479 | 3.512 | 11.516 | |
| Range | 4.586, 11.271 | 7.436, 15.908 | 0.035, 5.908 | 5.036, 12.901 | |
| n | 18 | 18 | 16 | 16 | |
| DS HC < 40 | Mean | 2.226 | 1.216 | 0.6308 | 1.374 |
| SD | 0.417 | 0.658 | 0.798 | 0.419 | |
| Median | 2.324 | 1.125 | 0.597 | 1.326 | |
| Range | 1.670, 2.587 | 0.539, 2.076 | −0.160, 1.490 | 0.945, 1.900 | |
| n | 4 | 4 | 4 | 4 | |
| DS HC ≥ 40 | Mean | 4.287 | 10.48 | 1.644 | 8.868 |
| SD | 1.068 | 2.685 | 1.136 | 3.425 | |
| Median | 4.309 | 10.270 | 1.778 | 10.056 | |
| Range | 2.091, 5.696 | 7.337, 14.983 | −0.313, 3.219 | 1.774, 12.028 | |
| n | 10 | 9 | 10 | 9 |
CTR = control; DS = Down syndrome; CX = cortex; HC = hippocampus. Age: <40 years; ≥40 years.
Table 2.
Group Comparisons between Down Syndrome (DS) and Controls (CTR) Subjects based on Mixed‐Effects Regression Model with Multiple Comparison Adjustments
| Group Comparisons | p‐value | |||
|---|---|---|---|---|
| CTR CX <40 vs. DS CX ≥40 | <.0001 | <.0001 | <.0001 | <.0001 |
| CTR CX <40 vs. DS HC ≥40 | 0.0003 | <.0001 | 0.0094 | <.0001 |
| CTR CX ≥40 vs. DS CX ≥40 | <.0001 | <.0001 | <.0001 | <.0001 |
| CTR CX ≥40 vs. DS HC ≥40 | 0.0002 | <.0001 | 0.0601 | <.0001 |
| CTR HC <40 vs. DS CX ≥40 | <.0001 | <.0001 | <.0001 | <.0001 |
| CTR HC <40 vs. DS HC ≥40 | <.0001 | <.0001 | 0.0169 | <.0001 |
| CTR HC ≥40 vs. DS CX ≥40 | <.0001 | <.0001 | <.0001 | <.0001 |
| CTR HC ≥40 vs. DS HC ≥40 | <.0001 | <.0001 | 0.0007 | <.0001 |
| DS CX <40 vs. DS CX ≥40 | <.0001 | <.0001 | 0.0002 | <.0001 |
| DS CX <40 vs. DS HC ≥40 | 0.0004 | <.0001 | 0.2261 | <.0001 |
| DS CX ≥40 vs. DS HC <40 | <.0001 | <.0001 | <.0001 | <.0001 |
| DS CX ≥40 vs. DS HC ≥40 | <.0001 | 0.0661 | 0.0219 | 0.0002 |
| DS HC <40 vs. DS HC ≥40 | 0.003 | <.0001 | 0.0543 | <.0001 |
CTR = control; DS = Down syndrome; CX = cortex; HC = hippocampus. Age = <40 years; ≥40 years.
Table 3.
Spearman correlations between amyloid beta measurements and retromer protein levelsa
| n | VPS35 | VPS26 | VPS29 | VPS26B | Sorla | CI‐MPR | |
|---|---|---|---|---|---|---|---|
| CX AB40 soluble | 57 | −0.38 | −0.27 | −0.43 | −0.11 | −0.16 | −0.35 |
| 0.003 | 0.041 | 0.001 | 0.40 | 0.25 | 0.008 | ||
| CX AB40 insoluble | 57 | −0.42 | −0.34 | −0.45 | −0.07 | −0.19 | −0.27 |
| 0.001 | 0.009 | 0.001 | 0.60 | 0.15 | 0.040 | ||
| CX AB42 soluble | 52 | −0.38 | −0.28 | −0.46 | −0.10 | −0.22 | −0.31 |
| 0.006 | 0.044 | 0.001 | 0.46 | 0.12 | 0.026 | ||
| CX AB42 insoluble | 55 | −0.33 | −0.20 | −0.35 | −0.12 | −0.15 | −0.20 |
| 0.013 | 0.15 | 0.008 | 0.37 | 0.29 | 0.14 | ||
| HC AB40 soluble | 33 | −0.42 | −0.39 | −0.47 | −0.38 | 0.01 | −0.35 |
| 0.016 | 0.026 | 0.005 | 0.030 | 0.98 | 0.043 | ||
| HC AB40 insoluble | 32 | −0.21 | −0.32 | −0.21 | −0.25 | 0.21 | −0.24 |
| 0.24 | 0.071 | 0.26 | 0.17 | 0.25 | 0.19 | ||
| HC AB42 soluble | 33 | −0.46 | −0.59 | −0.50 | −0.55 | 0.15 | −0.17 |
| 0.006 | 0.0003 | 0.003 | 0.001 | 0.39 | 0.33 | ||
| HC AB42 insoluble | 32 | −0.34 | −0.33 | −0.34 | −0.37 | 0.22 | −0.21 |
| 0.054 | 0.065 | 0.056 | 0.038 | 0.22 | 0.24 |
Table entry: First line = Spearman correlation coefficient, second line = p value.
CI‐MPR = cation‐independent mannose 6‐phosphate receptor; CX = cortex; HC = hippocampus.
FIGURE 4.
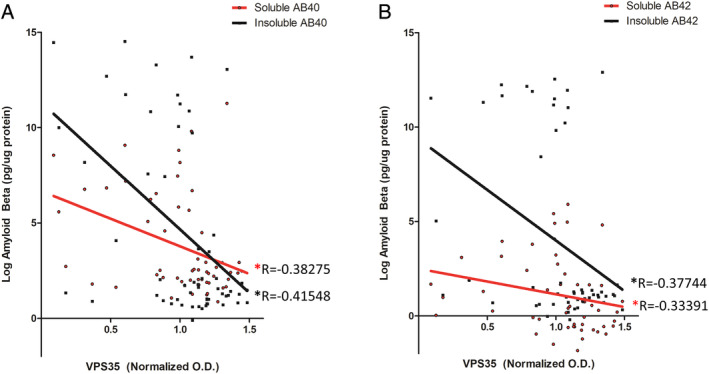
Relationship between Aβ measurements and VPS35 protein levels. (A) Soluble and insoluble Aβ 1–40 levels as a function of VPS35 optical density in cortices of Down syndrome (DS) and control subjects. (B) Soluble and insoluble Aβ 1–42 levels as a function of VPS35 optical density in cortices of subjects with DS and control subjects (insoluble and soluble Aβ 1–40 measurements, n = 57; soluble Aβ 1–42 measurements, n = 52; insoluble Aβ 1–42 measurements, n = 55). The R values represent Spearman correlation coefficients. Amyloid beta measurements are shown in Tables 1 and 2. Spearman correlation coefficients and p values are displayed in Table 3. [Color figure can be viewed at www.annalsofneurology.org]
Next, we examined protein levels of the retromer cargo recognition core in fibroblasts derived from human subjects with DS and unaffected controls. Compared to control 2N fibroblasts, DS fibroblasts had significant reductions in all three retromer recognition core proteins: VPS35, VPS26, and VPS29 (Fig 5A, B). Additionally, we found that protein levels of retromer cargo receptors Sorla and CI‐MPR were both significantly decreased in DS fibroblasts compared to 2N fibroblasts. As control, we confirmed that compared to the 2N fibroblast, the ones from patients with DS had a significant elevation of total APP (see Fig 5A, B).
FIGURE 5.
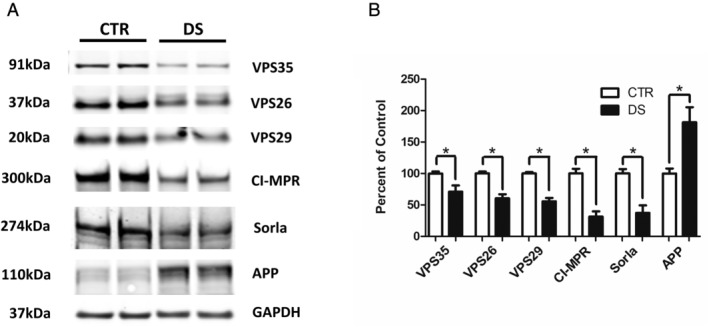
Retromer complex proteins are reduced in Down syndrome (DS) fibroblasts. (A) Representative Western blot of retromer cargo recognition core proteins, VPS35, VPS26, and VPS29, and cargo receptors cation‐independent mannose 6‐phosphate receptors (CI‐MPR) and Sorla in DS and control (CTR) 2N fibroblasts. (B) Densitometry analysis of Western blots shown in the previous panel (*p < 0.05). Values represent mean ± standard error of the mean (n = 3, in duplicate). APP, amyloid precursor protein; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase.
Because the retromer cargo recognition core proteins, VPS35, VPS29, and VPS26, were decreased in both fibroblasts and brain tissues from subjects with DS compared to controls, we next assessed mRNA levels of these proteins to determine whether the decrease was due to differences at the transcriptional regulation level. No differences for VPS35, VPS26, and VPS29 mRNA levels were observed when fibroblasts and cortices from aged subjects with DS were compared to controls (Fig 6).
FIGURE 6.
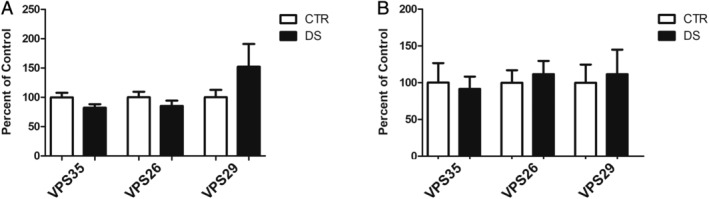
Retromer cargo recognition core mRNA levels do not differ between patients with Down syndrome (DS) and matched controls (CTRs). (A) VPS35, VPS26, and VPS29 mRNA levels measured by real time‐quantitative polymerase chain reaction (RT‐qPCR) in fibroblasts from DS and 2N CTR donors (n = 4, in duplicate). (B) VPS35, VPS26, and VPS29 mRNA levels measured by RT‐qPCR in cortex of DS and unaffected control subjects aged 40 to 65 years (CTR, n = 10; DS, n = 9). Values represent mean ± standard error of the mean.
Discussion
The retromer complex is a highly conserved multiprotein system involved in the sorting and trafficking of intracellular proteins from endosomes to the trans‐Golgi network or the cell membrane surface. The mammalian retromer is composed of several modules that define the different functions of the complex. However, the central cargo recognition core (CRC) composed of vacuolar sorting proteins VPS35, VPS26, and VPS29 remains conserved among modular assemblies and is considered the backbone of the complex as a whole. The CRC transiently associates with membrane targeting sorting nexin dimers, which bind and transport numerous intracellular cargoes, making the retromer CRC a key regulator of cellular proteostasis.
In recent years, studies have implicated dysregulation of the retromer complex in the pathogenesis of several neurodegenerative diseases.9, 17 In 2011, a VPS35 mutation causing altered trafficking and impaired alpha‐synuclein degradation was identified as the cause of dominant late‐onset Parkinson's disease. 18 The link to AD first materialized when a model‐guided microarray revealed decreased levels of VPS35 and VPS26 in the hippocampi of patients with AD. 6 Further support came from genetic studies associating VPS35 variants with increased risk of developing AD. 19 This loss of function of the retromer complex was proposed to directly influence AD pathogenesis, in part, by causing retention of the retromer cargo protein, APP, in the endosomes, which results in enhanced amyloidogenic cleavage of APP by β‐secretase (BACE‐1). 17 Several studies have confirmed this notion, showing that deficiency of VPS35 results in increased levels of Aβ and synaptic pathology, whereas VPS35 overexpression ameliorates the AD phenotype in mouse models of the disease.10, 12, 20
Despite evidence supporting the role of retromer dysregulation in AD pathogenesis, no data are available on retromer levels in DS. In the present study, we show, we believe for the first time, that the retromer complex is dysregulated in DS and may influence the development of the AD‐like amyloidotic phenotype, which is typically found in DS. Using both DS postmortem brain tissues and DS patient‐derived fibroblasts, we show that different retromer complex proteins are reduced in subjects with DS compared to unaffected matched controls. To start establishing a temporal profile of retromer dysregulation within the context of the evolution of the syndrome, we examined the retromer core and associated proteins in young patients prior to significant plaque deposition (age 15–30 years), and older subjects (age 40–65 years) at a time when virtually all subjects with DS display Aβ plaques. 16 This paradigm was confirmed in our patient cohort through measurements of both soluble and insoluble Aβ 1–40 and Aβ 1–42 levels. Retromer CRC proteins VPS35 and VPS29 were significantly decreased in the cortex, and all three CRC proteins were reduced in the hippocampus of the oldest subgroup of patients with DS compared to controls. Surprisingly, we found that retromer deficiency was not primarily aging related in DS. Dysregulation of the complex was also observed in younger patient subgroups, suggesting that retromer dysfunction may begin prior to full development of AD‐like pathology and cognitive symptoms in the DS population. Interestingly, in both young and aged subjects, retromer depletion was most evident in the hippocampus. This finding underlines the potential role of retromer reduction in AD‐like pathogenesis, as the hippocampus is one of the regions most severely affected by AD neuropathology in DS.21, 22 In contrast to retromer protein levels, retromer CRC mRNAs were unchanged, suggesting a post‐translational mechanism of retromer dysregulation in DS.
Additionally, we examined the relationship between retromer protein levels and both soluble and insoluble Aβ 1–40 and Aβ 1–42 levels for all subjects. Although reduction of retromer proteins seems to precede Aβ deposition, we found that the degree of retromer depletion for all retromer core proteins significantly and inversely correlates with the accumulation of both Aβ peptides in both the cortex and hippocampus. Whereas we recognize that this finding does not indicate causation, we believe that our observation further supports the evidence that retromer dysfunction and neurodegeneration are closely associated.
Although it is possible that APP elevations alone may contribute to retromer deficiency by overloading this trafficking system, it should also be considered that an independent mechanism may contribute to this dysfunction. Thus, HSA21 is home to several transcription factors and transcription factor binding sites that can lead to broad transcriptional changes.23, 24 Although transcription of retromer components seems to be unaffected in DS, proteins involved in retromer regulation or stability could be affected, ultimately affecting retromer protein levels. Additionally, at least 29 microRNAs reside on HSA21, many of which have uncharacterized targets. 25 It is possible that these or other regulatory components of this chromosome affect the retromer system leading to the observed decreases in protein.
Traditionally, it has been assumed that AD‐DS pathogenesis was principally a result of APP gene overdosage. However, recent studies showing that multiple HSA21 genes contribute to increased AD susceptibility independently of APP allow us to speculate that parallel mechanisms of AD pathogenesis may occur in DS.2, 26 The converging role of the retromer system in several neurodegenerative diseases suggests that dysfunction of this system may represent a vulnerability that allows for progression to one of several neurodegenerative pathways. In DS, it is conceivable that dysfunction of the retromer system is ultimately what allows for APP overdosage to translate into AD pathogenesis.
The overwhelming incidence of AD in the DS population provides a unique opportunity for the development of preventative treatments for AD. The complexity of AD‐DS pathogenesis necessitates whole cell approach, rather than current AD treatments, which have been found to be ineffective within the DS population. 1 In preclinical models of AD, enhancing retromer function has shown promise as an AD therapeutic. We recently demonstrated that restoration of VPS35 levels directly rescues the AD‐like phenotype in a mouse model of AD. 12 Similarly, stabilization of the retromer CRC via a small pharmacological chaperone reduces pathogenic tau phosphorylation independently of APP in a human stem cell model of AD, and a mouse model of AD.27, 28 Thus, it is possible that enhancing endocytic trafficking via retromer modulation may independently target both Aβ and tau pathology. Because we found that retromer dysregulation inversely correlates with overt AD‐like amyloid pathology in subjects with DS, thus, it is conceivable that targeting the retromer complex may have preventative implications in the treatment of AD‐DS.
In summary, our paper is the first, we believe, to report that compared with healthy controls retromer recognition core and other proteins of the complex system are significantly reduced in both cortices and hippocampi, and fibroblasts from subjects with DS. Our findings further support the importance of this sorting system in cellular proteostasis and provide new evidence that retromer dysfunction may play a functional role in the pathogenesis of AD‐DS. We conclude that targeting of the retromer by small pharmacological chaperones, which are known to stabilize and upregulate the complex, should be considered as a novel and viable therapeutic approach against the development of AD‐like amyloidotic neuropathology in subjects with DS.
Author Contributions
M.C. and D.P. contributed to study concept and design. M.C. and D.Y. contributed to data collection and analysis. M.C. and D.P. drafted the manuscript and figures. All the authors have seen and approved the final version of the manuscript.
Potential Conflicts of Interest
The authors have no conflicting financial interests to disclose.
Supporting information
Table S1 Patient information for donors of postmortem brain samples.
Table S2. Group comparisons based on mixed‐effects regression model for retromer proteins with multiple comparison adjustments.†
Table S3. Aβ 1 to 40 and Aβ 1 to 42 measurements (on log scale) and group comparisons based on mixed‐effects regression model with multiple comparison adjustments.
Acknowledgments
The authors would like to thank all the patients and families who by generously donating the brain tissues and fibroblasts made this research possible. We are also grateful to all the brain bank repositories that provided us the tissues. Domenico Praticò is the Scott Richards North Star Charitable Foundation Chair for Alzheimer's research. This study was supported in part by grants from the National Institute of Health (AG055707 and AG056689).
References
- 1. Alzheimer's Association . 2020 Alzheimer's disease facts and figures. Alzheimers Dement 2020;16:391–460. [Google Scholar]
- 2. Wiseman FK, Pulford LJ, Barkus C, et al. Trisomy of human chromosome 21 enhances amyloid‐beta deposition independently of an extra copy of APP. Brain 2018;141:2457–2474. 10.1093/brain/awy159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cataldo AM, Peterhoff CM, Troncoso JC, et al. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's disease and down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol 2000;157:277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cataldo AM, Mathews PM, Boiteau AB, et al. Down syndrome fibroblast model of Alzheimer‐related endosome pathology: accelerated endocytosis promotes late endocytic defects. Am J Pathol 2008;173:370–384. 10.2353/ajpath.2008.071053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cataldo AM, Petanceska S, Terio NB, et al. Abeta localization in abnormal endosomes: association with earliest Abeta elevations in AD and down syndrome. Neurobiol Aging 2004;25:1263–1272. 10.1016/j.neurobiolaging.2004.02.027. [DOI] [PubMed] [Google Scholar]
- 6. Small SA, Kent K, Pierce A, et al. Model‐guided microarray implicates the retromer complex in Alzheimer's disease. Ann Neurol 2005;58:909–919. 10.1002/ana.20667. [DOI] [PubMed] [Google Scholar]
- 7. Nicolas G, Charbonnier C, Wallon D, et al. SORL1 rare variants: a major risk factor for familial early‐onset Alzheimer's disease. Mol Psychiatry 2016;21:831–836. 10.1038/mp.2015.121. [DOI] [PubMed] [Google Scholar]
- 8. Vardarajan BN, Bruesegem SY, Harbour ME, et al. Identification of Alzheimer disease‐associated variants in genes that regulate retromer function. Neurobiol Aging 2012;33:2231 e2215–2231 e2230. 10.1016/j.neurobiolaging.2012.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vagnozzi AN, Pratico D. Endosomal sorting and trafficking, the retromer complex and neurodegeneration. Mol Psychiatry 2018;24:857–868. 10.1038/s41380-018-0221-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Muhammad A, Flores I, Zhang H, et al. Retromer deficiency observed in Alzheimer's disease causes hippocampal dysfunction, neurodegeneration, and Abeta accumulation. Proc Natl Acad Sci U S A 2008;105:7327–7332. 10.1073/pnas.0802545105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bhalla A, Vetanovetz CP, Morel E, et al. The location and trafficking routes of the neuronal retromer and its role in amyloid precursor protein transport. Neurobiol Dis 2012;47:126–134. 10.1016/j.nbd.2012.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li JG, Chiu J, Pratico D. Full recovery of the Alzheimer's disease phenotype by gain of function of vacuolar protein sorting 35. Mol Psychiatry 2019. 10.1038/s41380-019-0364-x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13. Di Meco A, Joshi YB, Lauretti E, Praticò D. Maternal dexamethasone exposure ameliorates cognition and tau pathology in the offspring of triple transgenic AD mice. Mol Psychiatry 2016;21:403–410. 10.1038/mp.2015.78. [DOI] [PubMed] [Google Scholar]
- 14. Li JG, Chu J, Barrero C, et al. Homocysteine exacerbates beta‐amyloid pathology, tau pathology, and cognitive deficit in a mouse model of Alzheimer disease with plaques and tangles. Ann Neurol 2014;75:851–863. 10.1002/ana.24145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li JG, Barrero C, Gupta S, et al. Homocysteine modulates 5‐lipoxygenase expression level via DNA methylation. Aging Cell 2017;16:273–280. 10.1111/acel.12550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Head E, Powell D, Gold BT, Schmitt FA. Alzheimer's disease in down syndrome. Eur J Neurodegener Dis 2012;1:353–364. [PMC free article] [PubMed] [Google Scholar]
- 17. Small SA, Petsko GA. Retromer in Alzheimer disease, Parkinson disease and other neurological disorders. Nat Rev Neurosci 2015;16:126–132. 10.1038/nrn3896. [DOI] [PubMed] [Google Scholar]
- 18. Vilarino‐Guell C, Wider C, Ross OA, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet 2011;89:162–167. 10.1016/j.ajhg.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lambert JC, Ibrahim‐Verbaas CA, Harold D, et al. Meta‐analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 2013;45:1452–1458. 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wen L, Tang FL, Hong Y, et al. VPS35 haploinsufficiency increases Alzheimer's disease neuropathology. J Cell Biol 2011;195:765–779. 10.1083/jcb.201105109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mullins D, Daly E, Simmons A, et al. Dementia in Down's syndrome: an MRI comparison with Alzheimer's disease in the general population. J Neurodev Disord 2013;5:19 10.1186/1866-1955-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hartley D, Blumenthal T, Carrillo M, et al. Down syndrome and Alzheimer's disease: common pathways, common goals. Alzheimers Dement 2015;11:700–709. 10.1016/j.jalz.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gardiner K, Costa AC. The proteins of human chromosome 21. Am J Med Genet C Semin Med Genet 2006;142C:196–205. 10.1002/ajmg.c.30098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cawley S, Bekiranov S, Ng HH, et al. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell 2004;116:499–509. 10.1016/s0092-8674(04)00127-8. [DOI] [PubMed] [Google Scholar]
- 25. Bras A, Rodrigues AS, Gomes B, Rueff J. Down syndrome and microRNAs. Biomed Rep 2018;8:11–16. 10.3892/br.2017.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Antonarakis SE, Lyle R, Dermitzakis ET, et al. Chromosome 21 and down syndrome: from genomics to pathophysiology. Nat Rev Genet 2004;5:725–738. 10.1038/nrg1448. [DOI] [PubMed] [Google Scholar]
- 27. Young JE, Fong LK, Frankowski H, et al. Stabilizing the retromer complex in a human stem cell model of Alzheimer's disease reduces TAU phosphorylation independently of amyloid precursor protein. Stem Cell Reports 2018;10:1046–1058. 10.1016/j.stemcr.2018.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li J, Chiu J, Ramanjulu M, et al. A pharmacological chaperone improves memory by reducing Aβ and tau neuropathology in a mouse model with plaques and tangles. Mol Neurodegener 2020;15:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Patient information for donors of postmortem brain samples.
Table S2. Group comparisons based on mixed‐effects regression model for retromer proteins with multiple comparison adjustments.†
Table S3. Aβ 1 to 40 and Aβ 1 to 42 measurements (on log scale) and group comparisons based on mixed‐effects regression model with multiple comparison adjustments.