

Abstract
The ability to precisely measure and monitor temperature at high resolution at the nanoscale is an important task for better understanding the thermodynamic properties of functional entities at the nanoscale in complex systems, or at the level of a single cell. However, the development of high‐resolution and robust thermal nanosensors is challenging. The design, assembly, and characterization of a group of thermal‐responsive deoxyribonucleic acid (DNA) joints, consisting of two interlocked double‐stranded DNA (dsDNA) rings, is described. The DNA nanojoints reversibly switch between the static and mobile state at different temperatures without a special annealing process. The temperature response range of the DNA nanojoint can be easily tuned by changing the length or the sequence of the hybridized region in its structure, and because of its interlocked structure the temperature response range of the DNA nanojoint is largely unaffected by its own concentration; this contrasts with systems that consist of separated components.
Keywords: DNA catenanes, DNA nanomachines, DNA nanotechnology, Interlocked DNA nanostructures, Temperature sensors
A precise thermal sensing DNA nanojoint comprised of two interlocked DNA rings switches reversibly between a static and mobile state at different temperatures. The temperature response range is tuned by changing the length or sequence of the hybridizing region between two rings, which, unlike non‐interlocked systems, is independent from its concentration.
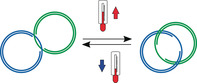
Deoxyribonucleic acid (DNA) has been applied as a versatile building material for constructing various nanoarchitectures in a bottom‐up approach because of its unique self‐recognition and its suitability to be chemically modified. The flexibility and programmability provided by DNA allows dynamic functional architectures to be established that can respond to different stimuli, such as pH,1 oligo(deoxy)nucleotides (ODNs),2 light,3 and temperature.4 Among them, thermoresponsive systems that fully depend on the thermodynamic stability of DNA have been reported in living organisms,5 illustrating their high potential for biomedical applications. In recent years, thermoresponsive DNA architectures have been widely used as sensors,2, 3, 4 DNA nanothermometers,4c for cargo‐delivery,4b, 4d, 4f and as temperature‐sensitive hydrogel.4e However, the thermoresponsive mechanism of DNA architectures relies on the breakage of hydrogen bonds between nucleobases and the unwinding of double‐stranded DNA (dsDNA) at specific temperatures, which often leads to their partial or complete disassembly4a, 4c, 4e, 4f and requires special annealing methods to reverse the process and yield the original nanosystems.4c, 4e
DNA catenanes are stable mechanically interlocked structures that provide an ideal platform for overcoming these limitations. In recent years, DNA catenanes have been widely used as switchable joints,6 reversible logic circuits,1c and molecular motors7 because of their mobility. Unlike static DNA architectures, DNA catenanes consist of two or more individual DNA rings that are connected through mechanical bonds that permit high rotational degrees of freedom and allow them to move independently relative to each other.7a, 8 Previous studies showed that DNA catenanes can be reversibly switched between a static hybridized (Cathyb) state and a mobile non‐hybridized mechanically interlocked (Catmec) state through different stimuli, such as ions6a and pH changes,6a or by means of strand displacement.1b, 8 However, thermal stimuli have not yet been used for controlling the conversion from a Cathyb to a Catmec by selectively melting only the hybridizing portion between the rings constituting the hybridized and mechanically interlocked nanostructure, instead of dehybridizing the complete catenane structure. Herein, we present the design, synthesis, and characterization of a group of DNA nanostructures with thermal‐responsive junctions. The described thermal‐responsive DNA [2]catenanes consist of two interlocked dsDNA rings that are hybridized with each other through a pair of single‐stranded regions (ss‐regions) on each DNA ring and can reversibly interchange between the static and the mobile state at different temperatures.
The assembly of the catenanes is illustrated in Figure 1 a. In the first step, a dsDNA ring (ring A) with two ss‐regions (a1 and a2) is assembled from ODNs by thermal annealing. To achieve the mechanically interlocked structure, an ODN from the second dsDNA ring (ring B) is threaded through ring A and hybridizes to it through the complementary ss‐region a1 and b1. The remaining ODNs of ring B are added to form the second dsDNA ring (ring B) also containing two ss‐regions (b1 and b2), resulting in a Cathyb. To enhance the stability of the whole structure, all ODNs are enzymatically ligated using T4 DNA ligase. The rings are designed9 to increase the threading of the ODN through ring A; nevertheless, some will hybridize without threading through the ring and will result in hybridized but not mechanically interlocked [2]catenanes. To separate these from the properly assembled catenanes, a so‐called “release ODN” (R‐ODN) is added, which has a complementary sequence to the ss‐region a1 of ring A and can separate ring B from ring A through toehold‐mediated strand displacement. Consequently, the Cathyb is converted into a Catmec. Only in mechanically interlocked catenanes, the two rings will stay interlocked while all the non‐interlocked two‐ring hybrids will disassemble into the two rings. The Catmec can be separated from dsDNA rings by weak anion‐exchange high‐performance liquid chromatography (HPLC) purification (Supporting Information, Table S1). After purification, a so‐called “reverse ODN” (RR‐ODN), which is complementary to R‐ODN, is added to remove R‐ODN from the ss‐region a1, allowing ring B to hybridize again to ring A and restore the Cathyb (for sequences see the Supporting Information, Figure S1, Tables S2 and S3).
Figure 1.
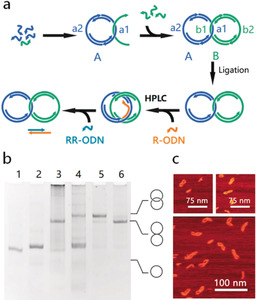
Assembly and characterization of the Cathyb. a) Schematic for a Cathyb assembly. b) Analysis of Cathyb assembly with 6 % native PAGE. Lane 1, ring A; lane 2, ring B; lane 3, catenane after ligation; lane 4, catenane after addition of R‐ODN; lane 5, catenane purified by weak anion exchange HPLC; lane 6, catenane after adding RR‐ODN. c) AFM images of the hybridized [2]catenane.
The assembly process and switching between the Cathyb and Catmec was characterized by 6 % native polyacrylamide gel electrophoresis (PAGE; Figure 1 b). After the R‐ODN was added (Figure 1 b, lane 4), the migration of the [2]catenane became slower, indicating the Cathyb to Catmec conversion.8a, 10 Furthermore, the band of DNA rings became more intense, suggesting that the hybridized but non‐interlocked DNA rings have been separated into the two individual DNA rings by the addition of the R‐ODN. After RR‐ODN was added to the HPLC‐purified product (Figure 1 b, lane 6), the electrophoretic mobility of the [2]catenane was restored to that of the [2]catenane prior to the addition of R‐ODN (Figure 1 b, lane 3) indicating that the Catmec had been converted into the Cathyb. Tapping mode atomic force microscopy (AFM) analysis in liquid also confirmed the formation of the hybridized catenane (Figure 1 c; Figure S2).
The thermal switching behavior of the Cathyb is controlled by the melting temperature (T m) of the hybridized region (HR) between ss‐regions a1 and b1. T m can be defined as the temperature at which half of the HR is dissociated to single‐stranded DNA (ssDNA), and in this case generates the Catmec. To examine the switching process of the Cathyb, we labeled two ODNs from the DNA architecture with the fluorophore (6‐FAM) on one ring and the quencher (BHQ®‐1) on the other ring (Figure 2 a). Below the T m, the HR largely remains in the hybridized state with the fluorophore and quencher in close proximity, leading to a low fluorescence intensity. As temperature increases HR begins to melt and the Cathyb is converted into the Catmec. In that state, fluorophore and quencher separate, an increase of fluorescence intensity results. As a proof of concept, we first designed a Cathyb with a nine base pairs hybridization sequence in its HR (named G9, the sequence is shown in Table 1).
Figure 2.
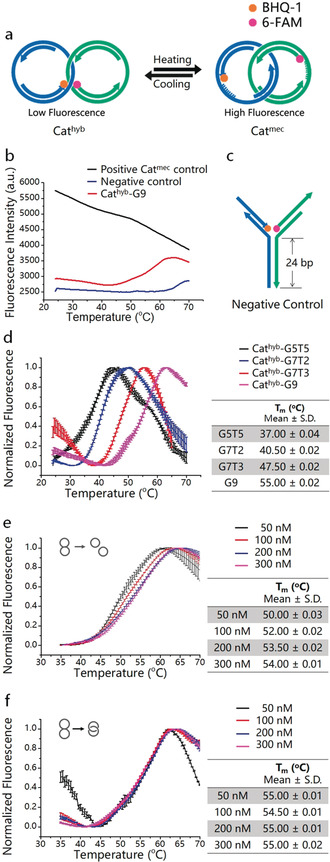
Thermal switching between Cathyb and Catmec. a) Schematic depiction of the mechanism of thermal switching of the Cathyb to Catmec. b) Temperature‐dependent fluorescence intensity of 20 μL of 100 nm Cathyb‐G9 with an initially quenched FAM (red), 20 μL of 100 nm Catmec positive control that has a non‐quenched FAM (black), and 20 μL of 100 nm of the Y‐shaped negative control (blue). c) Structure of the Y‐shaped negative control construct (for structures and sequences see the Supporting Information, Figure S3 and Table S2). d) Temperature‐dependent fluorescence intensity of Cathyb‐G5T5 (black), Cathyb‐G7T2 (blue), Cathyb‐G7T3 (red), and Cathyb‐G9 (pink; S.D., n=3). e) Temperature‐dependent fluorescence intensity of hybridized but non‐interlocked ring A and ring B with HR of G9 at 50 nm (black), 100 nm (red), 200 nm (blue), and 300 nm (pink; S.D., n=3). f) Temperature‐dependent fluorescence intensity of the Cathyb‐G9 at 50 nm (black), 100 nm (red), 200 nm (blue), and 300 nm (pink; S.D., n=3).
Table 1.
Sequences of different HRs in the Cathyb.
|
HR domains |
Sequence |
Length [nt] |
GC content |
T m [°C] |
|---|---|---|---|---|
|
G5T5 |
a1: 5′‐AAAAAGCGCC‐3′ b1: 3′‐TTTTTCGCGG‐5′ |
10 |
50.0 % |
37.00±0.04 |
|
G7T2 |
a1: 5′‐CGCAGAGCC‐3′ b1: 3′‐GCGTCTCGG‐5′ |
9 |
77.8 % |
40.50±0.02 |
|
G7T3 |
a1: 5′‐CCTGCTGCTC‐3′ b1: 3′‐GGACGACGAG‐5′ |
10 |
70.0 % |
47.50±0.02 |
|
G9 |
a1: 5′‐CGCCGGGCC‐3′ b1: 3′‐GCGGCCCGG‐3′ |
9 |
100.0 % |
55.00±0.02 |
As shown in Figure 2 b (red curve), in the DNA catenane buffer (DC buffer: 10 mm Tris‐HCl, 1 mm ethylenediaminetetraacetic acid (EDTA), 25 mm MgCl2, pH 8.0) the fluorescence intensity of the Cathyb‐G9 increased as the temperature rose from 42 °C to 62 °C, indicating the melting of the HR and conversion of the static Cathyb‐G9 to the mobile Catmec. The red curve indicates that the Cathyb‐G9 is fully converted into Catmec at 62 °C. However, above 62 °C the fluorescence intensity of the Cathyb‐G9 began to decrease with further rising temperature, which is consistent with a phenomenon that was previously observed to be a temperature‐dependent characteristic of 6‐FAM.11 Similarly, we observed an influence of temperature on the emitted non‐quenched fluorescence signal also with a different fluorophore (Figure S4) or in a different buffer system (Figure S5). As expected, the fluorescence intensity of a Catmec positive control that has a non‐quenched 6‐FAM, also decreased with increasing temperature (Figure 2 b, black curve). This curve allows distinguishing the temperature‐dependent fluorescence of FAM alone from the overall fluorescence changes that are caused by the conversion from Cathyb into the mechanically interlocked DNA nanojoint. The fluorescence intensity of the “Y”‐shaped negative control (Figure 2 b, blue curve) containing a 24 bp HR (Figure 2 c; Figure S3), remained unchanged when the temperature rose from 42 °C to 62 °C, indicating constant FAM quenching because of the stable fluorophore and quencher arrangement in this structure.
The temperature‐response range of the Cathyb is tunable by changing the length and sequence of HR without having to modify the whole nanostructure. As a proof of concept, we synthesized three other Cathyb ODNs—G5T5, G7T2, and G7T3—with different HR sequences that differ either in length or in their guanine–cytosine (GC) content (Table 1) and measured their melting curves. As shown in Figure 2 d, in the DC buffer the melting region of the Cathyb at 100 nm can change from low temperature (black curve, Cathyb‐G5T5, 24.50±0.01 °C–45.00±0.02 °C, S.D., n=3) to high temperature (pink curve, Cathyb‐G9, 42.00±0.01 °C–62.00±0.01 °C, S.D., n=3) by increasing the GC content or by increasing the number of base pairs in HR. Additionally, the melting regions of the Cathyb ODNs are comparable to the T m of DNA systems that consist of non‐interlocked dsDNA strands with the same sequences and the same concentration (Figure S6).
In general, the T m and melting curves are strongly influenced by strand concentration when two ssDNA strands interact.12 However, in an interlocked structure, the dsDNA rings will remain interlocked during the conversion of Cathyb into the Catmec, which means that the ss‐gap on the DNA rings could only hybridize with their counterparts in the same interlocked structure because of the catenation of the rings. This characteristic of DNA catenanes greatly reduces the influence of the strand concentration in the melting process. The T m and melting region of hybridized but non‐interlocked ring A and ring B (Figures 2 e,f; sequences are presented in Table S4) with HR of G9 shifted when the concentration of the dsDNA rings increased from 50 nm to 300 nm (Figure 2 d); however. the T m and melting range of the Cathyb‐G9 remained largely unchanged when the concentration of the Cathyb‐G9 increased from 50 nm to 300 nm (Figure 2 e). This result indicates that the catenanes are well suited as robust and sensitive nanothermometers since their response to temperature can be finely tuned by changing only the HR and is independent from the concentration of the structure itself.
Subsequently, we studied the thermal reversibility of the conversion of Cathyb to Catmec. Since the HR consists of only two short ss‐regions, the Cathyb can be easily restored to its original form at temperatures below T m. We measured the time‐dependent change of fluorescence intensity when the Catmec‐G9 was cooled from 65 °C down to 24 °C, and found that the fluorescence intensity decreased rapidly in the first 20–25 minutes (Figure 3 a, red), which was similar to the mixture of ring A‐G9 and ring B‐G9 (Figure 3 a, black). This finding indicates that Catmec can be converted into Cathyb without requiring special annealing processes. The thermal reversibility test was performed by a temperature cycling between 42 °C and 60 °C with a fast rate of 3 °C s−1, and the sample was incubated at 42 °C for 20 minutes and at 60 °C for 5 minutes in each cycle (Figure 3 b). The fluorescence change of the Cathyb‐G9 showed no indication of fatigue after 10 temperature cycles (Figure 3 c; Figure S7). The negative control construct with its longer HR that did not melt at 60 °C remains unchanged during the temperature cycles (Figure 3 c, blue). Although each dsDNA ring in the Cathyb‐G9 contains two different ss‐regions (Figure 1 a), and hybridization to the wrong ss‐region would not result in signal quenching, the fluorescence intensity of the Cathyb‐G9 at 42 °C in each temperature cycle remained the same, which indicates that the two dsDNA rings hybridize correctly when the Catmec is cooled and converts into Cathyb‐G9 because of the specificity of Watson–Crick base pairing.
Figure 3.
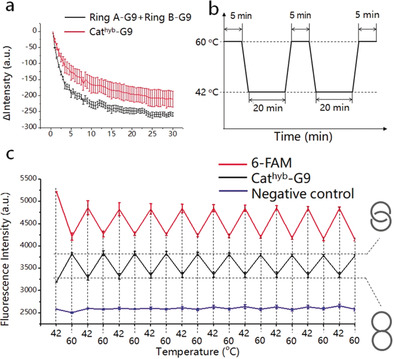
Thermal reversibility of the Cathyb to Catmec interconversion. a) Time‐dependent change of fluorescence intensity of the mixture of ring A‐G9 and ring B‐G9 (black, 100 nm) and the Catmec‐G9 (red, 100 nm) in DC buffer (S.D., n=3). b) Diagram of the temperature cycling between 42 °C and 60 °C. c) Fluorescence intensity of 6‐FAM (red), Cathyb‐G9 (black), and the quenched negative control (blue) at 42 °C and 60 °C (S.D, n=3).
In conclusion, we have demonstrated design, assembly, characterization by gel electrophoresis, AFM analysis, and fluorescence detection of a new class of thermal responsive nanojoints. The described DNA nanostructures consist of two interlocked dsDNA rings that are connected through a HR and can reversibly switch between the static and mobile state at different temperatures through the melting and rehybridization of its HR. The temperature response range of the nanojoint can be easily fine‐tuned by changing the length and/or the GC content of the HR sequence. In comparison with previously reported DNA thermoresponsive systems, these nanojoints provide serval unique features suitable for creating functional temperature‐sensitive DNA nanostructures. First, the nanojoint switches from the static state to the mobile state instead of disassembling at high temperature, which provides controlled and confined mobility between different components while keeping the whole nanostructure intact. Second, the nanojoint can be easily reversed without a special annealing process. Finally, unlike systems that consist of separated components, the temperature response range of the interlocked nanojoint remains essentially unaffected by its concentration because of its interlocked structure.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research was supported by the Max‐Planck‐Society and University of Bonn through a Max‐Planck‐Fellowship and the Alexander von Humboldt Foundation. Open access funding enabled and organized by Projekt DEAL.
Y. Ma, M. Centola, D. Keppner, M. Famulok, Angew. Chem. Int. Ed. 2020, 59, 12455.
In memory of Professor Rolf Huisgen
References
- 1.
- 1a. Hu Y., Ren J., Lu C.-H., Willner I., Nano Lett. 2016, 16, 4590–4594; [DOI] [PubMed] [Google Scholar]
- 1b. Li F., Gao Q., Yang M., Guo W., Langmuir 2018, 34, 14932–14939; [DOI] [PubMed] [Google Scholar]
- 1c. Li T., Lohmann F., Famulok M., Nat. Commun. 2014, 5, 4940; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1d. Patino T., Porchetta A., Jannasch A., Lladó A., Stumpp T., Schäffer E., Ricci F., Sánchez S., Nano Lett. 2019, 19, 3440–3447; [DOI] [PubMed] [Google Scholar]
- 1e. Wang C., Huang Z., Lin Y., Ren J., Qu X., Adv. Mater. 2010, 22, 2792–2798; [DOI] [PubMed] [Google Scholar]
- 1f. Modi S., Nizak C., Surana S., Halder S., Krishnan Y., Nat. Nanotechnol. 2013, 8, 459; [DOI] [PubMed] [Google Scholar]
- 1g. Modi S., Swetha M., Goswami D., Gupta G. D., Mayor S., Krishnan Y., Nat. Nanotechnol. 2009, 4, 325–330; [DOI] [PubMed] [Google Scholar]
- 1h. Wang W., Yang Y., Cheng E., Zhao M., Meng H., Liu D., Zhou D., Chem. Commun. 2009, 45, 824–826; [DOI] [PubMed] [Google Scholar]
- 1i. Kuzuya A., Watanabe R., Yamanaka Y., Tamaki T., Kaino M., Ohya Y., Sensors 2014, 14, 19329–19335; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1j. Green L. N., Amodino A., Subramanian H. K. K., Ricci F., Franco E., Nano Lett. 2017, 17, 7283–7288; [DOI] [PubMed] [Google Scholar]
- 1k. Fong F. Y., Oh S. S., Hawker C. J., Soh H. T., Angew. Chem. Int. Ed. 2016, 55, 15258–15262; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 15484–15488. [Google Scholar]
- 2.
- 2a. Andersen E. S., Dong M., Nielsen M. M., Jahn K., Subramani R., Mamdouh W., Golas M. M., Sander B., Stark H., Oliveira C. L., Nature 2009, 459, 73; [DOI] [PubMed] [Google Scholar]
- 2b. Centola M., Valero J., Famulok M., J. Am. Chem. Soc. 2017, 139, 16044–16047; [DOI] [PubMed] [Google Scholar]
- 2c. Goodman R. P., Heilemann M., Doose S., Erben C. M., Kapanidis A. N., Turberfield A. J., Nat. Nanotechnol. 2008, 3, 93; [DOI] [PubMed] [Google Scholar]
- 2d. Rahbani J. F., Hariri A. A., Cosa G., Sleiman H. F., ACS Nano 2015, 9, 11898–11908; [DOI] [PubMed] [Google Scholar]
- 2e. Zhou C., Yang Z., Liu D., J. Am. Chem. Soc. 2012, 134, 1416–1418; [DOI] [PubMed] [Google Scholar]
- 2f. Amodio A., Zhao B., Porchetta A., Idili A., Castronovo M., Fan C., Ricci F., J. Am. Chem. Soc. 2014, 136, 16469–16472; [DOI] [PubMed] [Google Scholar]
- 2g. Zhang D. Y., Winfree E., J. Am. Chem. Soc. 2009, 131, 17303–17314; [DOI] [PubMed] [Google Scholar]
- 2h. Teichmann M., Kopperger E., Simmel F. C., ACS Nano 2014, 8, 8487–8496. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Haydell M. W., Centola M., Adam V., Valero J. N., Famulok M., J. Am. Chem. Soc. 2018, 140, 16868–16872; [DOI] [PubMed] [Google Scholar]
- 3b. Liang X., Nishioka H., Takenaka N., Asanuma H., ChemBioChem 2008, 9, 702–705; [DOI] [PubMed] [Google Scholar]
- 3c. Lohmann F., Ackermann D., Famulok M., J. Am. Chem. Soc. 2012, 134, 11884–11887; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3d. Škugor M., Valero J., Murayama K., Centola M., Asanuma H., Famulok M., Angew. Chem. Int. Ed. 2019, 58, 6948–6951; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 7022–7025; [Google Scholar]
- 3e. Tam D. Y., Dai Z., Chan M. S., Liu L. S., Cheung M. C., Bolze F., Tin C., Lo P. K., Angew. Chem. Int. Ed. 2016, 55, 164–168; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 172–176; [Google Scholar]
- 3f. Yang Y., Endo M., Hidaka K., Sugiyama H., J. Am. Chem. Soc. 2012, 134, 20645–20653; [DOI] [PubMed] [Google Scholar]
- 3g. Veetil A. T., Chakraborty K., Xiao K., Minter M. R., Sisodia S. S., Krishnan Y., Nat. Nanotechnol. 2017, 12, 1183–1189; [DOI] [PubMed] [Google Scholar]
- 3h. Dutta P. K., Varghese R., Nangreave J., Lin S., Yan H., Liu Y., J. Am. Chem. Soc. 2011, 133, 11985–11993; [DOI] [PubMed] [Google Scholar]
- 3i. You M., Chen Y., Zhang X., Liu H., Wang R., Wang K., Williams K. R., Tan W., Angew. Chem. Int. Ed. 2012, 51, 2457–2460; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 2507–2510; [Google Scholar]
- 3j. Liu M., Jiang S., Loza O., Fahmi N. E., Šulc P., Stephanopoulos N., Angew. Chem. Int. Ed. 2018, 57, 9341–9345; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 9485–9489. [Google Scholar]
- 4.
- 4a. Dias J. T., Moros M., del Pino P., Rivera S., Grazú V., de la Fuente J. M., Angew. Chem. Int. Ed. 2013, 52, 11526–11529; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 11740–11743; [Google Scholar]
- 4b. Franch O., Iacovelli F., Falconi M., Juul S., Ottaviani A., Benvenuti C., Biocca S., Ho Y.-P., Knudsen B. R., Desideri A., Nanoscale 2016, 8, 13333–13341; [DOI] [PubMed] [Google Scholar]
- 4c. Gareau D., Desrosiers A., Vallée-Bélisle A., Nano Lett. 2016, 16, 3976–3981; [DOI] [PubMed] [Google Scholar]
- 4d. Juul S., Iacovelli F., Falconi M., Kragh S. L., Christensen B., Frøhlich R., Franch O., Kristoffersen E. L., Stougaard M., Leong K. W., ACS Nano 2013, 7, 9724–9734; [DOI] [PubMed] [Google Scholar]
- 4e. Xing Y., Cheng E., Yang Y., Chen P., Zhang T., Sun Y., Yang Z., Liu D., Adv. Mater. 2011, 23, 1117–1121; [DOI] [PubMed] [Google Scholar]
- 4f. Yu Z., Li N., Zheng P., Pan W., Tang B., Chem. Commun. 2014, 50, 3494–3497; [DOI] [PubMed] [Google Scholar]
- 4g. Zuidema J. M., Bertucci A., Kang J., Sailor M. J., Ricci F., Nanoscale 2020, 12, 2333–2339; [DOI] [PubMed] [Google Scholar]
- 4h. Ke G., Wang C., Ge Y., Zheng N., Zhu Z., Yang C. J., J. Am. Chem. Soc. 2012, 134, 18908–18911; [DOI] [PubMed] [Google Scholar]
- 4i. Zhang K., Zhu X., Jia F., Auyeung E., Mirkin C. A., J. Am. Chem. Soc. 2013, 135, 14102–14105; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4j. Jeon S., Turner J., Granick S., J. Am. Chem. Soc. 2003, 125, 9908–9909; [DOI] [PubMed] [Google Scholar]
- 4k. Ebrahimi S., Akhlaghi Y., Kompany-Zareh M., Rinnan Å., ACS Nano 2014, 8, 10372–10382. [DOI] [PubMed] [Google Scholar]
- 5. Klinkert B., Narberhaus F., Cell. Mol. Life Sci. 2009, 66, 2661–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Lu C.-H., Cecconello A., Elbaz J., Credi A., Willner I., Nano Lett. 2013, 13, 2303–2308; [DOI] [PubMed] [Google Scholar]
- 6b. Lu C.-H., Cecconello A., Qi X.-J., Wu N., Jester S.-S., Famulok M., Matthies M., Schmidt T.-L., Willner I., Nano Lett. 2015, 15, 7133–7137. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Schmidt T. L., Heckel A., Nano Lett. 2011, 11, 1739–1742; [DOI] [PubMed] [Google Scholar]
- 7b. Valero J., Pal N., Dhakal S., Walter N. G., Famulok M., Nat. Nanotechnol. 2018, 13, 496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Lohmann F., Valero J., Famulok M., Chem. Commun. 2014, 50, 6091–6093; [DOI] [PubMed] [Google Scholar]
- 8b. Lu C. H., Qi X. J., Cecconello A., Jester S. S., Famulok M., Willner I., Angew. Chem. Int. Ed. 2014, 53, 7499–7503; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 7629–7633. [Google Scholar]
- 9. Valero J., Centola M., Ma Y., Škugor M., Yu Z., Haydell M. W., Keppner D., Famulok M., Nat. Protoc. 2019, 14, 2818–2855. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Ackermann D., Schmidt T. L., Hannam J. S., Purohit C. S., Heckel A., Famulok M., Nat. Nanotechnol. 2010, 5, 436–442; [DOI] [PubMed] [Google Scholar]
- 10b. Weigandt J., Chung C. L., Jester S. S., Famulok M., Angew. Chem. Int. Ed. 2016, 55, 5512–5516; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 5602–5606; [Google Scholar]
- 10c. Lohmann F., Weigandt J., Valero J., Famulok M., Angew. Chem. Int. Ed. 2014, 53, 10372–10376; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10540–10544. [Google Scholar]
- 11. Liu W.-T., Wu J.-H., Li E. S.-Y., Selamat E. S., Appl. Environ. Microbiol. 2005, 71, 6453–6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Giesen U., Kleider W., Berding C., Geiger A., Ørum H., Nielsen P. E., Nucleic Acids Res. 1998, 26, 5004–5006; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Owczarzy R., Vallone P. M., Gallo F. J., Paner T. M., Lane M. J., Benight A. S., Biopolymers Orig. Res. Biomol. 1997, 44, 217–239. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary