

Abstract
Background
EDP‐305 is a novel and potent farnesoid X receptor (FXR) agonist, with no/minimal cross‐reactivity to TGR5 or other nuclear receptors. Herein we report therapeutic efficacy of EDP‐305, in direct comparison with the first‐in‐class FXR agonist obeticholic acid (OCA), in mouse models of liver disease.
Methods
EDP‐305 (10 and 30 mg/kg/day) or OCA (30mg/kg/day) was tested in mouse models of pre‐established biliary fibrosis (BALBc.Mdr2‐/‐, n = 9‐12/group) and steatohepatitis induced by methionine/choline‐deficient diet (MCD, n = 7‐12/group). Effects on biliary epithelium were evaluated in vivo and in primary EpCAM + hepatic progenitor cell (HPC) cultures.
Results
In a BALBc.Mdr2‐/‐ model, EDP‐305 reduced serum transaminases by up to 53% and decreased portal pressure, compared to untreated controls. Periportal bridging fibrosis was suppressed by EDP‐305 at both doses, with up to a 39% decrease in collagen deposition in high‐dose EDP‐305. In MCD‐fed mice, EDP‐305 treatment reduced serum ALT by 62% compared to controls, and profoundly inhibited perisinusoidal ‘chicken wire’ fibrosis, with over 80% reduction in collagen deposition. In both models, treatment with 30mg/kg OCA reduced serum transaminases up to 30%, but did not improve fibrosis. The limited impact on fibrosis was mediated by cholestasis‐independent worsening of ductular reaction by OCA in both disease models; OCA but not EDP‐305 at therapeutic doses promoted ductular proliferation in healthy mice and favoured differentiation of primary HPC towards cholangiocyte lineage in vitro.
Conclusions
EDP‐305 potently improved pre‐established liver injury and hepatic fibrosis in murine biliary and metabolic models of liver disease, supporting the clinical evaluation of EDP‐305 in fibrotic liver diseases including cholangiopathies and non‐alcoholic steatohepatitis.
Keywords: cirrhosis, ductular reaction, EDP‐305, Farnesoid × receptor, hepatic progenitor cell
Abbreviations
- ALT
alanine aminotransferase
- ALKP
alkaline phosphatase
- AST
aspartate aminotransferase
- FGF
fibroblast growth factor
- FXR
farnesoid X receptor
- HPC
hepatic progenitor cell
- HSC
hepatic stellate cell
- MCD
methionine‐choline deficient diet
- Mdr2
multidrug resistance protein 2
- NAFLD
non‐alcoholic fatty liver disease
- NASH
non‐alcoholic steatohepatitis
- OCA
obeticholic acid
- PBC
primary biliary cholangitis
- PSC
primary sclerosing cholangitis
- TBIL
total bilirubin
- TGR5
Takeda G‐protein receptor 5
Key points.
We report that EDP‐305, novel FXR agonist, potently suppresses liver injury and fibrosis in mouse models of liver disease. Compared to first‐in‐class drug obeticholic acid, it demonstrates favourable therapeutic profile and does not negatively impact the biliary tree. These results support clinical trials with EDP‐305 in biliary diseases and non‐alcoholic steatohepatitis, and has important implications for novel drug design.
1. INTRODUCTION
Liver fibrosis, characterized by continuous and progressive deposition of extracellular matrix, leads to the formation of scar tissue in chronic liver diseases of many aetiologies, and is a major determinant of liver‐related morbidity and mortality. 1 , 2 Apart from liver transplantation, there is no effective treatment for the majority of non‐viral chronic liver diseases associated with progressive fibrosis, such as non‐alcoholic fatty liver disease (NAFLD) non‐alcoholic steatohepatitis (NASH) and cholangiopathies including primary biliary cholangitis (PBC), primary sclerosing cholangitis (PSC) and cystic fibrosis‐related liver disease (CFLD 3 ). Thus, the development of effective antifibrotic drugs that can halt or reverse progression of liver fibrosis is urgently needed. 4
The farnesoid X receptor (FXR, NR1H4) is nuclear receptor, which heterodimerizes with retinoid X receptor (NR2B1) upon binding to its endogenous ligand chenodeoxycholic acid to induce the expression of downstream genes, including FGF19 (fibroblast growth factor‐19), that regulate the biological processes of bile acid synthesis and transport, cholesterol metabolism and glucose homeostasis. 5 Upon activation by FXR in a disease state, the FXR/FGF19 system plays a crucial role in the pathogenesis of cholestatic disease, 6 NAFLD, 7 impaired liver regeneration, 8 liver fibrosis 9 and hepatocellular carcinoma. 10
Obeticholic acid (OCA; INT‐747), a potent first‐in‐class FXR agonist, is a 6α‐ethyl derivative of the natural FXR ligand chenodeoxycholic acid. In recent clinical phase 2 trials for NASH (FLINT 11 ) and PBC (POISE 12 ), OCA treatment resulted in rapid and robust improvement in relevant serum biochemistries (alanine aminotransferase (ALT) in NASH, alkaline phosphatase (ALKP) and bilirubin in PBC). The FLINT study successfully met its primary efficacy end point of histological improvement in NAFLD Activity Score on follow‐up biopsy without worsening of fibrosis. 11 These studies largely confirmed the therapeutic potential of OCA, and FXR agonism in general, as presumed from multiple preclinical studies. However, the optimism for the wide clinical use of OCA is hampered by several side effects reported in patients, such as drug‐induced pruritus in both NASH and PBC trials and increase in low density lipoprotein (LDL) cholesterol levels. A relatively narrow therapeutic window, with rare cases of severe hepatotoxity recorded in patients with advanced liver disease after accidental use of increased doses (https://www.fda.gov/Drugs/DrugSafety/ucm576656.htm), is another factor potentially limiting therapeutic use of OCA. At least some of these side effects can be theoretically ascribed to the bile acid‐like structure and behaviour of the OCA molecule, resulting in challenging pharmacokinetics due to enterohepatic circulation of OCA and its metabolites, and significant TGR5 agonistic properties 13 , 14 that are potentially responsible for drug‐related side effects such as pruritus. 15 , 16 Recent study also reports OCA may increase the risk of gallstone formation in susceptible patients. 17 Given these considerations, and the otherwise encouraging clinical results with OCA, targeting FXR in hepatobiliary disorders has generated high interest in the pharmaceutical industry, with multiple new non‐bile acid FXR agonists in various stages of preclinical and clinical development. 18 However, whether and to what extent these structurally new agents may offer significant advantages compared to OCA, either by improving therapeutic efficacy or avoiding the reported side effects, is currently unclear.
We hypothesized that the FXR agonists that are structurally different from bile acids may offer an improved efficacy and safety profile for chronic liver diseases. Here, we report detailed preclinical in vitro and in vivo efficacy data for EDP‐305, a novel non‐bile acid FXR agonist with single‐digit nanomolar FXR activity and minimal TGR5 reactivity, on liver injury and fibrosis in two mouse models representing biliary and metabolically induced liver disease, in direct comparison with the first‐in‐class FXR agonist OCA.
2. MATERIALS AND METHODS
2.1. Animal experiments
All animals were housed with a 12‐hour light‐dark cycle and permitted ad libitum consumption of water and a standard chow diet unless otherwise stated. All animal procedures were approved by the Institutional Animal Care and Use Committee at Beth Israel Deaconess Medical Center (protocol 010‐2015).
2.1.1. BALB/c.Mdr2‐/‐ mouse model of biliary liver fibrosis
Mdr2(abcb4)‐/‐ mice on the fibrosis‐susceptible BALB/c background, which spontaneously develop accelerated severe biliary fibrosis, early onset portal hypertension and liver cancer, were generated and characterized as reported previously 19 and bred in animal research facilities at BIDMC. Treatments started at 6 weeks of age, when advanced liver fibrosis was already established, and continued for the following 6 weeks.
2.1.2. Murine steatohepatitis model
Methionine and choline‐deficient diet (MCD) feeding of C57Bl/6 mice as a model of NASH was performed as reported previously. 20 Eight‐week‐old male C57Bl/6 mice (Jackson Labs, Bar Harbor, MA) were fed MCD (Research Diets, Inc) ad libitum for 8 weeks to induce progressive steatohepatitis. Treatments were started after 4 weeks of MCD feeding, when steatohepatitis and incipient fibrosis were already established, and continued for the following 4 weeks in parallel with ongoing MCD feeding.
2.2. FXR agonists
EDP‐305 is a novel and highly potent FXR agonist discovered by Enanta Pharmaceuticals, Inc which was characterized previously 21 with an EC50 value of 8 nM in a full‐length FXR reporter assay using Human Embionic Kidney 293 cells (compared to EC50 130 nM for OCA using the same assay). EDP‐305 has minimal activity against the G protein‐coupled bile acid receptor 1 (TGR5) with EC50 > 15 uM in a TGR5 activation assay in Chinese Hamster Ovarycells (compared to EC50 0.381 uM for OCA in the same assay). In the BALBc.Mdr2‐/‐ model, EDP‐305 and OCA were incorporated into D5001 rodent chow (Research Diets, Inc) at 71.4 mg/kg (10 mg/kg/day dose equivalent) and 214 mg/kg (30 mg/kg/day dose equivalent). The OCA dose was selected based on prior reports. 22 Medicated diets were fed ad libitum in parallel with a placebo control group receiving re‐pelleted D5001 base diet. In MCD model, EDP‐305 (Enanta Pharmaceuticals, Inc) and OCA (Enanta Pharmaceuticals, Inc) were administered via oral gavage at doses of 10 and/or 30 mg/kg in 0.5% methylcellulose once a day. Placebo controls received equivalent volumes of vehicle (0.5% methylcellulose).
2.2.1. Portal venous pressure measurement
At study end point, mice were anaesthetized with isoflurane (1.5% vol/vol) via high‐precision digital vaporizer (SomnoSuite from Kent Scientific, Braintree). After laparotomy, the portal vein was cannulated, and portal pressure was measured directly by inserting a 1.2‐Fr high‐fidelity pressure catheter (Scisense, London, ON, Canada). Pressure signals were recorded at 2 kHz for 5 minutes and analysed using PowerLab software (ADInstruments, Colorado Springs, CO), as described previously. 19
2.2.2. Immunohistochemistry and immunofluorescence
Connective tissue stain and immunohistochemistry were performed in formalin‐fixed paraffin‐embedded liver sections, and immunofluorescence were performed in acetone‐fixed EPCAM + hepatic progenitor cell (HPC) cultures, as described previously by us 23 and others. 24 For morphometric quantification of percent of collagen area (picrosirius red), positive area for immunohistochemistry staining in Mdr2‐/‐ and MCD‐fed mice was calculated using ImageJ software (NIH, Bethesda) in >10 random periportal high‐power fields (HPF). To quantify HPC activation in healthy wild‐type mice, CK19‐positive cells were counted in >10 randomly chosen portal tracts. 25 At least four randomly chosen individual mice/group were analysed at 200x magnification. Detailed information about primary antibodies is summarized in Table S1.
Additional methods, including isolation and culture of primary murine hepatic stellate cells (HSCs), hepatic progenitors, colony‐forming and differentiation assay and qRT‐PCR, can be found in the Supplementary Material.
2.3. Statistical analyses
Data are expressed as means ± SEM, and statistical analyses were performed using Microsoft EXCEL and GraphPad Prism version 5.00 (GraphPad Software, San Diego, CA). Multiple comparisons were performed by one‐way analysis of variance (ANOVA) with the Dunnett's post‐test. In vitro experiments were performed in triplicates and analyszed using ANOVA, or unpaired t‐test when appropriate. Two‐tailed p values lower than 0.05 were considered significant.
3. RESULTS
3.1. EDP‐305 inhibits primary murine HSC activation in vitro
To evaluate the direct effects of EDP‐305 on fibrogenic activation of HSCs, the major effector cells in liver fibrosis, we isolated primary murine HSC and incubated them with increasing concentrations of EDP‐305 (at 5‐500nM) for 24hours during their spontaneous activation in vitro. Stellate cell activation marker alpha‐smooth muscle actin (a‐SMA) expression was markedly reduced in the presence of EDP‐305 (50‐500nM) as assessed via immunofluorescence (Figure1A). Furthermore, EDP‐305 dose‐dependently inhibited serum‐induced stellate cell proliferation as assessed by MTT assay (Figure1B) and suppressed profibrogenic gene expression of procollagen α1(I), TGFβ1 and TIMP‐1 up to two‐ to three‐fold compared to controls at 500nM concentration of EDP‐305 (Figure1C). OCA, used as a comparator at highest equimolar concentration (500nM), had a less apparent effects on a‐SMA expression; HSC cell proliferation was not affected by OCA, and while average mRNA expression of procollagen α1(I), TGFβ1 and TIMP‐1 were moderately reduced by about 30%, none of these differences were statistically significant in multivariate analysis (Figure1). Similar results were observed in HSC‐X cell line (Figure1D,E). Both drugs at tested concentrations had no appreciable effect on cell viability, as assessed by trypan blue exclusion test (data not shown).
Figure 1.
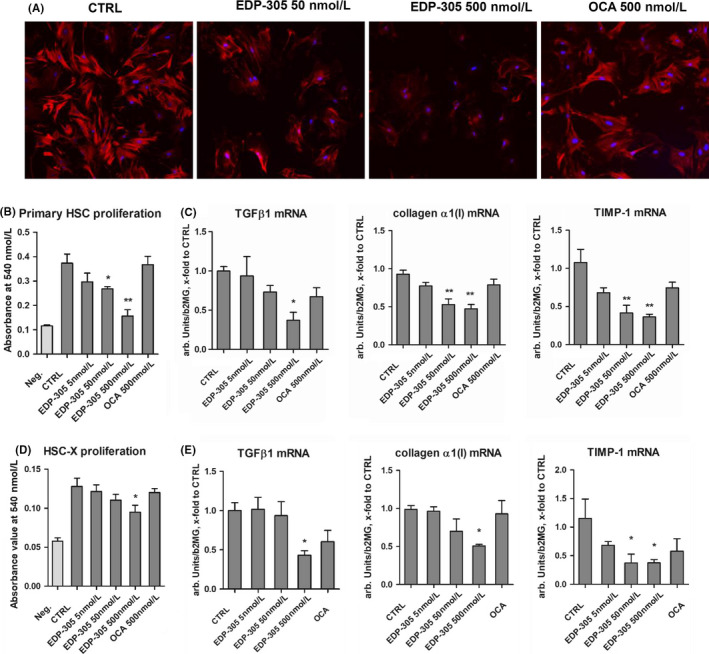
EDP‐305 potently and dose‐dependently inhibits hepatic stellate cells activation in vitro. (A) Representative immunofluorescence staining for αSMA and (B) cell proliferation in serum‐stimulated primary murine HSCs (n = 5, MTT assay). Neg. – negative control, untreated cells starved in 1% FBS; Ctrl – positive control, serum‐stimulated cells with 10% FBS for primary HSCs and vehicle (0.1%DMSO), all others were serum‐stimulated and supplemented with EDP‐305 or OCA at indicated concentrations. (C) Expression of profibrogenic transcript levels in primary murine HSC (procollagen α1(I), TGFβ1, TIMP‐1) as quantified by qRT‐PCR. (D). Serum‐induced cell proliferation of immortalized rat HSC‐X cells (MTT assay, n = 5, experimental design same as with primary HSC with the exception that reduced 2% FBS was used for stimulation) and (E) profibrogenic transcript levels in HSC‐X cells (procollagen α1(I), TGFβ1, TIMP‐1) as quantified by qRT‐PCR. Results are expressed as mean ± SEM, and in arbitrary units (fold to controls) relative to β2MG mRNA (n = 5). *P < .05 and **P < .01 compared to controls (ANOVA with Dunnett's post‐test)
3.2. EDP‐305 potently suppresses pre‐established biliary injury and fibrosis in BALBc.Mdr2‐/‐ mouse model
Next, we evaluated therapeutic efficacy of EDP‐305 in BALBc.Mdr2‐/‐ model of chronic progressive biliary liver disease. 19 EDP‐305 (10 mg/kg or 30 mg/kg) or OCA (30 mg/kg) was administered starting from 6 weeks of age (when advanced liver fibrosis was already pre‐established) for the following 6 weeks after which portal pressure, liver injury and fibrosis were assessed (Figure 2A). EDP‐305 was well tolerated at both doses, and no weight loss, deaths or other drug‐related toxicity events were observed (Table S3). In the OCA‐treated group, significant adverse effects were observed in BALBc.Mdr2‐/‐ mice in the first 2 weeks of treatment. Out of the 19 mice total, four died on day 10 of feeding, and three more lost 20% of their body weight, necessitating their early euthanasia at day 11 of feeding. Necropsy revealed white spotty macroscopic lesions throughout the liver parenchyma, which histologically presented as focal necrosis affecting up to 50% of hepatocytes (via H/E staining, not shown). The remaining 12 mice in the OCA‐treated group demonstrated transient weight loss between weeks 1 and 3 of treatment (Figure S2A), but survived to the end of the experiment and did not differ in their gross appearance, body weight or macroscopic appearance of the liver at the end of 6 weeks of treatment. Short‐term PK/PD study in Mdr2‐/‐ mice suggested that hepatotoxicity is likely attributable to the accumulation of taurine OCA conjugate in Mdr2‐/‐ mice (Figure S1 for details). Serum ALT and aspartate aminotransferase (AST) were significantly decreased by 30% and 34%, 53% and 66% in mice receiving 10 and 30 mg/kg of EDP‐305, respectively, and by 30% and 24% in OCA‐treated group, compared to placebo (Figure 2B , P < .05). Compared to placebo group, ALKP and total bile acid levels were significantly decreased by all treatments by 42% and 54.4% (10mg/kg EDP‐305), 39% and 45% (30mg/kg EDP‐305), and 43.6% and 54% (30 mg/kg OCA) respectively (P < .05). Serum levels of total bilirubin (TBIL) were significantly reduced by 41.8% and 33.2% lower at 10 and 30mg/kg dose of EDP‐305, respectively, but not impacted by 30 mg/kg OCA (Figure 2C). Histologically, bridging and perisinusoidal fibrosis were markedly suppressed in BALBc.Mdr2‐/‐ mice receiving EDP‐305, with 32% and 53% reduction in collagen area morphologically in low‐ and high‐dose groups respectively (Figure 2D). Similar results were observed with collagen type 1 immunohistochemistry (Figure S4). Biochemically, hepatic collagen levels (as assessed via hepatic hydroxyproline) were significantly elevated in the placebo group (12‐week‐old mice fed control diet for 6 weeks) compared to ‘start of treatment’ controls (6‐week‐old BALBc.Mdr2‐/‐, ‘6w start’ group) (Figure 2E). At the end of the experiment, treatment with EDP‐305 at both doses, but not OCA, reduced portal venous pressure compared to placebo control (by an average 1.26 mm Hg and 1.31 mm Hg by low‐ and high‐dose EDP‐305, respectively, P < .05, Figure 2F). EDP‐305 at 30 mg/kg dose inhibited collagen deposition by 52.8% compared to placebo controls (P > .05), while OCA at the same dose (30 mg/kg) did not. Furthermore, profibrogenic transcript levels of procollagen α1(I), TGFβ1 and TIMP‐1 were significantly reduced (up to two‐ to three‐fold) in the livers of mice treated with EDP‐305 as compared to placebo, while OCA did not have an effect (Figure 2G).
Figure 2.
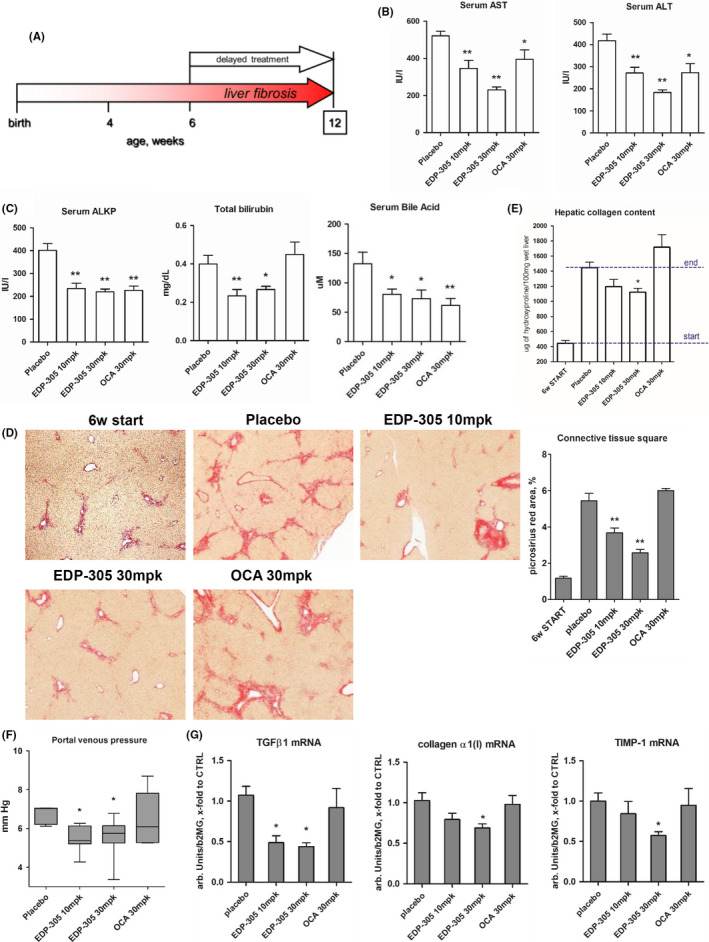
Delayed treatment with EDP‐305 potently suppresses liver injury and fibrosis in BALBc.Mdr2‐/‐ mouse model. (A) BALBc.Mdr2−/− mice with pre‐established advanced liver fibrosis were administered with EDP‐305 (10 mg/kg or 30 mg/kg), OCA (30 mg/kg) or repelleted base chow as placebo control, from 6 to 12 weeks of age (n = 9‐10). WT are healthy untreated wild‐type mice, 6w start group is a ‘start of treatment’ Mdr2‐/‐ controls. (B) Serum levels of transaminases (ALT and AST), and (C) Serum alkaline phosphatase test (ALKP), total bilirubin (TBIL) and bile acid. (D) Representative images of connective tissue (picrosirius red, 50x) staining and morphometric quantification of collagen. (E) Hepatic collagen content (determined biochemically via hydroxyproline content). (F) Portal venous pressure at study end‐point. (G). Profibrogenic transcript levels of procollagen α1(I), TGFβ1, TIMP‐1 as measured by TaqMan qRT‐PCR. Data are mean ± SEM, *P < .05; **P < .01 compared to placebo control group, #, P < .05 compared to start of treatment controls (ANOVA with Dunnett's post‐test)
3.3. HSC activation and ductular reaction in EDP‐305‐treated BALBc.Mdr2‐/‐ mice
Profibrogenic HSC activation and ductular reaction are key cellular processes driving liver fibrosis progression, and were analysed using immunohistochemistry for a‐SMA (HSC activation marker) and CK19 (ductal cell marker) followed by quantitative morphometry. EDP‐305 markedly and dose‐dependently suppressed HSC activation in Mdr2‐/‐ livers, with up to three‐fold reduction in the a‐SMA‐positive area at 30mg/kg dose (Figure 3A). Ductular reaction (CK19‐positive area morphometry) was similarly diminished by EDP‐305 treatment, with up to a three‐fold decrease at the 30mg/kg dose (Figure 3B). Surprisingly, and in stark contrast to EDP‐305, OCA treatment exerted dichotomous effects on stellate cell activation and ductular reaction in Mdr2‐/‐ livers. While OCA significantly reduced a‐SMA‐positive area, albeit to a lesser degree than EDP‐305 (by 21.5%, Figure 3A, P < .01), CK19‐positive area was increased by 26.4% in OCA‐treated livers compared to placebo (Figure 3B, P < .05). These results were corroborated by similar changes in SOX9 expression, a stem cell/progenitor marker which was reduced by 59.8% EDP‐305 at 30mg/kg, compared with placebo group while OCA had no effect. Dramatically expanded laminin expression, a critical extracellular hepatic progenitor niche component was decreased by 52.5% in Mdr2‐/‐ mice by EDP‐305 but not by OCA. (Figure 3C,D).
Figure 3.
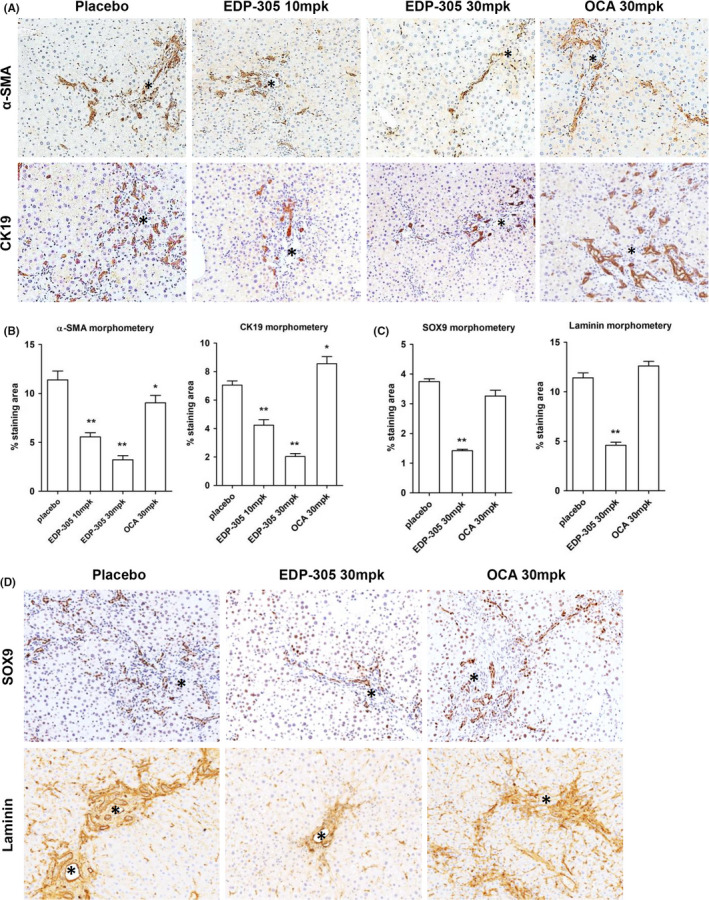
Histological assessment of hepatic stellate cell activation and ductular reaction in EDP‐305‐treated BALBc.Mdr2‐/‐ mice. Immunohistochemistry reveals that EDP‐305 markedly and dose‐dependently suppressed HSCs activation in vivo as assessed by α‐SMA (A) and ductular reaction (CK19, B) in BALB/c.Mdr2−/− livers. Representative images shown (original magnification, 200x, portal vein marked by asterisks). Morphometric area quantification of α‐SMA and CK19‐positive area (n = 5). 6w start group is a ‘start of treatment’ control. C‐D, Representative immunostaining for SOX9 and laminin (200×) with morphometric quantification. *P < .05; **P < .01 compared to placebo control group (ANOVA with Dunnett's post‐test)
3.4. EDP‐305 treatment suppresses liver injury and liver fibrosis in the pre‐established MCD‐induced steatohepatitis model
We next investigated the efficacy of EDP‐305 in a mechanistically different model of steatohepatitis due to MCD feeding, which is associated with a pericellular, ‘metabolic‐type’ liver fibrosis. Four‐week oral gavage treatment with vehicle only (‘placebo’), EDP‐305 (10 mg/kg or 30 mg/kg) or OCA (30 mg/kg) started after week 4 on MCD feeding, at the stage of incipient perisinusoidal fibrosis 20 (Figure 4A). All mice survived until the scheduled study termination with no apparent adverse effects or weight loss due to pharmacological treatments noted (Figure S2B). Serum transaminases were significantly decreased by all treatments, with the maximal response observed in the EDP‐305 30mg/kg group where transaminases were lowered by 62% and 37% for ALT and AST, respectively, in MCD‐fed mice compared to placebo controls (P < .05). Serum bilirubin showed a trend towards lower average values in 30mg/kg EDP‐305, but these changes were not statistically significant in multivariate analysis (Figure 4B). Connective tissue staining showed considerable progression of perisinusoidal fibrosis (‘chicken wire’) from minimal (week 4 on MCD, ‘MCD 4w start’ group) to significant (week 8 on MCD, ‘placebo group) in placebo‐treated, MCD‐fed mice (Figure 4C). Collagen area was reduced by 61% and 86% in low‐ and high‐dose EDP‐305‐treated groups respectively. Likewise, hepatic hydroxyproline levels were reduced in high and low doses of EDP‐305‐treated mice by 81.8% and 80.5% respectively (P < .05 vs placebo) (Figure 4D). OCA at 30mg/kg did not reduce connective tissue area or hepatic collagen content, which did not differ from the ‘placebo’ group (Figure 4C,D). EDP‐305, but not OCA, significantly and dose‐dependently reduced the immunopositivity for HSC activation marker, a‐SMA, and ductular reaction marker CK19 (by 51.3%, and 52.1% at 30mg/kg EDP‐305, respectively, P < .01, Figure 4E,F), SOX9 and laminin expression (Figure S3). Profibrogenic gene transcription levels of procollagen α1(I), TGFβ1, TIMP‐1 were decreased two‐ to three‐fold in all EDP‐305 treatment groups (Figure 4G). Although OCA did seem to reduce the average levels of profibrogenic mRNAs, these differences did not reach statistical significance.
Figure 4.
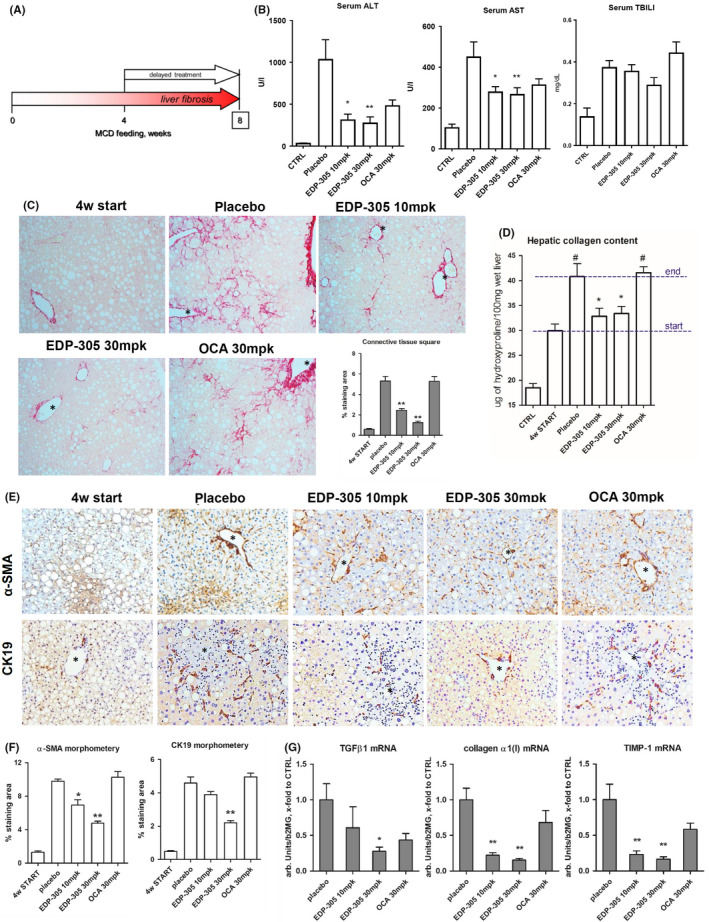
Delayed EDP‐305 treatment protects against MCD‐induced steatohepatitis and liver fibrosis. (A) Advanced steatohepatitis with liver fibrosis was induced in C57BL/6 mice by MCD feeding for 8 weeks. Delayed 4‐week treatment with EDP‐305 (10 mg/kg or 30 mg/kg), OCA (30 mg/kg) or vehicle was administered via daily oral gavage starting from week 4 on MCD. (n = 9‐10). CTRL are healthy untreated control mice (n = 6), 4w start group is a ‘start of treatment’ MCD‐fed controls (n = 10). (B) Serum levels of transaminases (ALT, AST), total bilirubin (TBIL) and (C) representative images of connective tissue staining (picrosirius red, 200×) and morphometric quantification of collagen area (n = 5). (D) Hepatic collagen content (via hydroxyproline levels). E. Immunohistochemistry for α‐SMA (upper row) and CK19 (lower row) with morphometric quantification (F, n = 5). Portal vein indicated by asterisk. (G) Profibrogenic transcript levels of procollagen α1(I), TGFβ1, TIMP‐1. Data are mean ± SEM, *P < .05; **P < .01 compared to placebo control group, #, P < .05 compared to start of treatment controls (ANOVA with Dunnett's post‐test)
3.5. Chronic administration of OCA but not EDP‐305 induces low‐grade ductular proliferation in the absence of liver injury
Based on the observation that OCA and EDP‐305 differentially impact ductular reaction in liver disease models, we hypothesized that OCA may have a direct effect on the biliary tree. In order to test that, we treated healthy wild‐type mice with OCA or EDP‐305 at 30 mg/kg (via diet) for up to 2 weeks. As expected, EDP‐305 or OCA had no influence on serum ALT, AST or bilirubin levels in the absence of liver disease (Figure 5A). Mild elevation of mean total bile acid levels was noted in the group treated with OCA for 2 weeks, but this was not statistically significant in the multivariate analysis when compared to placebo (P = .1441). However, immunochemistry staining for CK19 revealed that OCA (but not EDP‐305) induced a mild, but apparent increase in the numbers of duct‐like structures, with CK19‐positive cell clusters without a clearly defined lumen (pseudo‐ducts) and solitary progenitor‐like cells frequently extending into the liver lobule at week 2. Morphometrically, we observed a 48.6% increase in CK19‐positive cell numbers in OCA‐treated animals compared to controls (P = .001, Figure 5B,C). These ductular structures expressed SOX9 and αvβ6 integrin, a HPC surface marker that is functionally required for HPC expansion. 23 Enhanced expression of laminin around ductular proliferations was also noted in response to OCA but not to EDP‐305 in healthy wild‐type mice (Figure 5B). Furthermore, cholangiocyte/HPC markers CK19, EpCAM, Trop2 and Itgb6 23 were progressively elevated at mRNA level in OCA‐treated group up to two‐ to four‐fold compared to controls at 2 weeks, while EDP‐305 had no impact (Figure 5B,D). Finally, we analysed EDP‐305, OCA and its metabolites levels in the plasma, liver tissue and bile of these mice. Plasma EDP‐305 levels did not change between weeks 1 and 2 of dosing, and were about 10% of levels in liver or bile. In contrast, the taurine OCA conjugate was detected in plasma (683 ± 292 ng/mL) and liver (6337 ± 775 ng/g) after 1 week of dosing; OCA (160 ± 38 ng/mL) and glycine OCA conjugate (379 ± 57) were only found in bile, which also contained remarkably high levels of tauro‐OCA (2600 times and 300 times higher than plasma and liver levels respectively). OCA/metabolites showed a tendency to increase at week 2, except tauro‐OCA that remained at the same level (Table S6).
Figure 5.
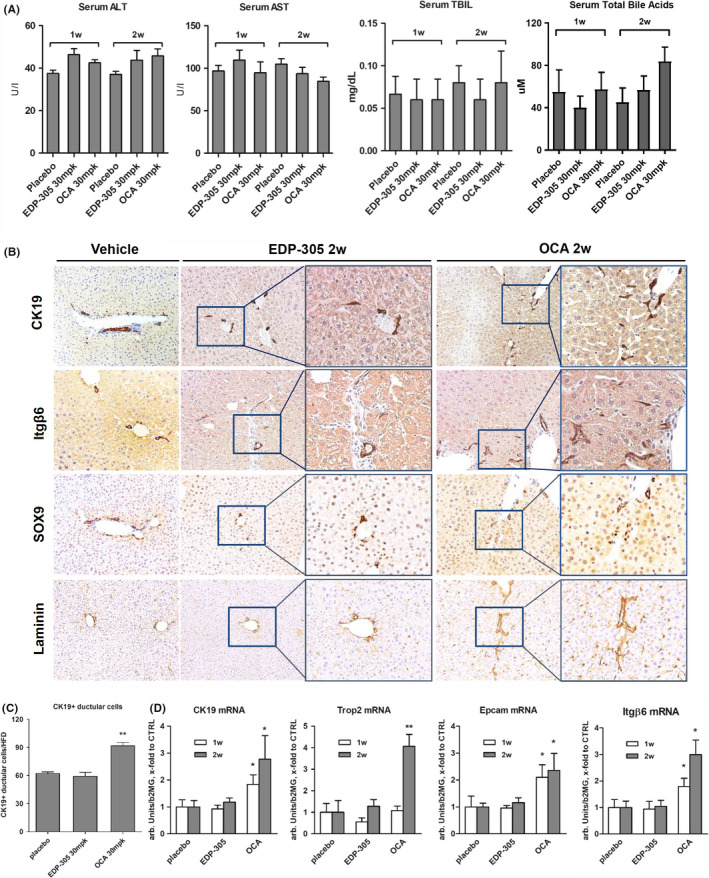
Chronic administration of OCA but not EDP‐305 induces mild ductular proliferation in livers of healthy wild‐type mice. Six‐week‐old wild‐type C57B16 mice received EDP‐305 or OCA‐supplemented diets (at 30 mg/kg/day dose equivalent) or control diet (Placebo) for 1 or 2 weeks. (A) Serum levels of transaminases (ALT, AST), TBIL and total bile acids. (B) Representative stainings for ductular markers CK19, integrin αvβ6, SOX9 and laminin (50x and x200 blow‐ups as indicated). (C) Quantification of CK19‐positive cells (n = 5). (D) Hepatic transcript levels of ductular/hepatic progenitor cell markers CK19, EpCAM, TROP2 and ITGB6, as measured by TaqMan qRT‐PCR. Data are mean ± SEM. *P < .05 compared to placebo control group (ANOVA with Dunnett's post‐test)
3.6. Differential effects of EDP‐305 and OCA on HPC lineage commitment in vitro
Activation of bipotent HPCs is driving ‘ductular reaction’ and is an almost universal finding in chronic liver disease. In order to investigate the direct effects of EDP‐305 and OCA on HPC biology, we freshly isolated EpCAM + cells from the chronically injured livers of Mdr2‐/‐ and cultured them in differentiation medium with equimolar concentrations (500 nM) of FXR agonists. The drug concentration of 500nM was chosen based on maximal biological activity in stellate cell cultures (Figure 1) and was well below the peak concentration of OCA/metabolites observed in the bile (Table S6). EpCAM + cells readily formed cell colonies demonstrating morphological features of ductular cells and hepatocytes. In the presence of OCA at 500nM, we noted that cells within colonies more frequently demonstrated spindle‐like or flattened morphology, and poorly formed monolayers, reminiscent of immature ductular cells (cholangiocyte lineage differentiation), compared to control or EDP‐305‐treated cells (Figure 6A). Quantification of the colony number and size revealed that neither EDP‐305 nor OCA affected colony‐forming ability of EpCAM + HPCs (Figure 6B). However, the expression of CK19, a cholangiocyte differentiation marker, was significantly upregulated in OCA‐treated HPC cultures (in situ immunofluorescence, Figure 6C). Transcript levels of CK19 and another HPC marker Trop2 was also significantly upregulated by OCA in total cell colony lysates, while EDP‐305 had no effect (Figure 6D). Conversely, hepatocyte lineage differentiation as assessed by a bona fide marker albumin secretion and albumin mRNA expression was diminished in OCA‐, but not EDP‐305‐treated HPC cultures (Figure 6E). HNF4α expression, a master hepatocyte differentiation factor, as well as LDL uptake (LDL, a functional hepatocyte marker) appeared to be modestly decreased only in OCA‐treated HPC cultures (Figure 6F,G).
Figure 6.
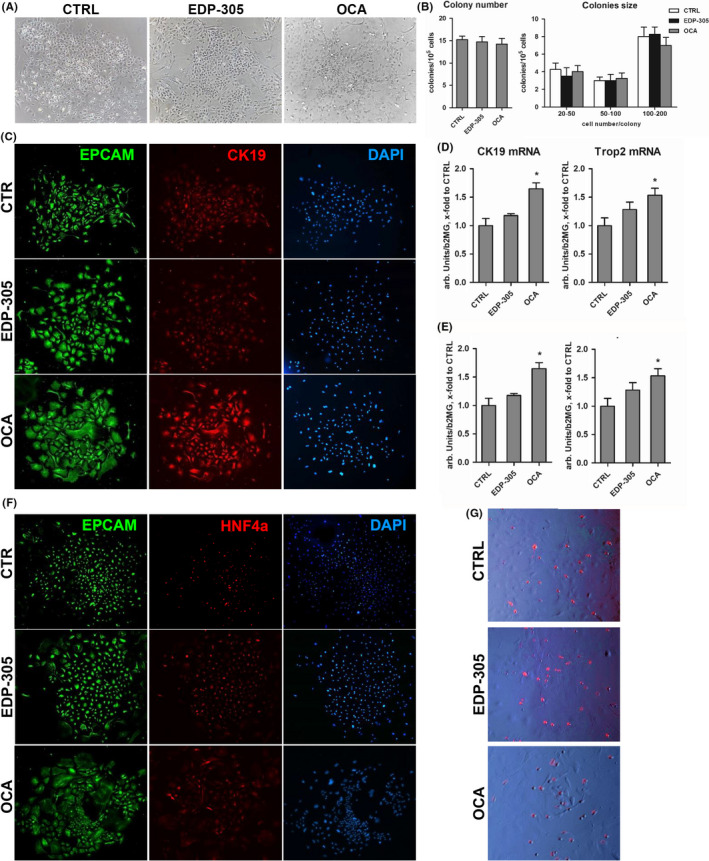
Differential effect of FXR agonists on cholangiocyte lineage commitment of EpCAM + hepatic progenitor cells in vitro. Primary EpCAM + progenitor cells were isolated from Mdr2‐/‐ mice and grown in differentiation medium for up to 14 days in the presence of EDP‐305 or OCA (both at 500nM, added 12h after cell plating). Representative morphology of HPC colonies (A) and their characteristics according to colony number and size (B). Cholangiocyte lineage differentiation (C) as assessed via immunofluorescence for EpCAM (green) and CK19 (red), DAPI (blue) as nuclear counterstain and mRNA levels of CK19 and Trop2 (D) in cell lysates. Hepatocyte differentiation as assessed via albumin at mRNA and secreted protein levels (E), HNF4α immunofluorescence (F, red) and LDL uptake using Dil‐AcLDL (G, red fluorescence overlayed on phase‐contrast image) in the EpCAM(+) cell cultures in the presence of FXR agonists. Representative images are shown (×200). Data are mean ± SEM (from three individual cell isolations). *P < .05 compared to untreated controls (t test)
4. DISCUSSION
In this study, we evaluated the therapeutic efficacy of EDP‐305, a potent and selective new generation FXR agonist, in direct comparison with the first‐in‐class FXR agonist OCA. Delayed (therapeutic) treatment with EDP‐305 and OCA significantly improved liver injury, as evidenced by robust serum transaminases responses, in biliary and metabolic models of liver disease in mice. However, hepatic fibrosis was suppressed in both models by only EDP‐305 but not by OCA. In‐depth investigation revealed that worsening of ductular reaction is linked to the limited effect of OCA on hepatic fibrosis. OCA, but not EDP‐305, directly promoted ductal proliferation in the absence of liver injury in vivo, and directed differentiation of primary HPCs towards cholangiocyte lineage in vitro.
Recent studies have shown that FXR is expressed in rodent and human HSCs, 9 the major fibrogenic effector cell in liver, and FXR agonism with OCA suppressed fibrogenic HSC activation in vitro and in vivo. 26 , 27 , 28 Our in vitro results with the structurally diverse FXR agonist EDP‐305 confirm previous reports and support the functional role of FXR in HSC activation. Compared to OCA, 26 EDP‐305 potently and dose‐dependently inhibited primary HSC activation, proliferation and fibrogenic factors expression at submicromolar concentration (Figure 1). Next, we performed formal, comprehensive efficacy testing of EDP‐305 in two preclinical murine models of chronic liver associated with progressive fibrosis, BALBc.Mdr2‐/‐ 19 (biliary‐type) and MCD‐induced steatohepatitis 20 (metabolic‐type). The effect on histology and all quantitative measures of liver fibrosis by EDP‐305 and OCA differed substantially (Figure 2). In BALBc.Mdr2‐/‐ model, EDP‐305 robustly and dose‐dependently reduced collagen deposition histologically (connective tissue square morphometry, up to 53% reduction) and biochemically (hepatic hydroxyproline content, up to 39% reduction), which are used as a ‘gold standard’ measure of the extent of fibrosis. 4 Hepatic mRNA expression of key profibrogenic factors, used to assess fibrogenesis activity, also decreased up to two‐fold. Importantly, EDP‐305 at both doses reduced portal venous pressure, a most reliable surrogate predictor of progression to cirrhosis in humans, by an average of approximately 2 mm Hg (Figure 2F). The observed antifibrotic efficacy was not model specific, since similar tests in the mechanistically different, MCD‐induced steatohepatitis model (that develops NASH‐like perisinusoidal, so called metabolic‐type fibrosis) produced even more pronounced EDP‐305 effect on fibrosis, exceeding 80% of collagen deposition reduction as measured via hydroxyproline, a ‘gold standard’ fibrosis measure (Figure 4). We observed no effect on fibrosis‐related parameters by OCA at a 30 mg/kg dose in parallel comparison groups, despite the obvious improvement in transaminases. Although the biochemical (serum ALT) response appeared to be more pronounced to EDP‐305 compared to OCA (both at 30mg/kg dose), small differences in liver injury improvement cannot fully explain the difference in fibrotic outcomes.
The profound differences in the effects of EDP‐305 and OCA on fibrosis were puzzling and this prompted an investigation into the potentially responsible mechanisms. In contrast to previous reports by others in liver disease models, we observed a considerable liver toxicity that resulted in morbidity and mortality in some OCA‐treated Mdr2‐/‐ mice at 30 mg/kg dose routinely used in previous preclinical studies. 22 This toxicity manifested itself during the second week of treatment only in the Mdr2‐/‐ model was characterized by weight loss, focal hepatocyte death and a spike in serum transaminases, ALKP and TBIL (Figure S1), and had a transient nature because surviving mice regained the weight by week 3 (Figure S2A), and did not show necrotic lesions and had instead reduced transaminases at the 6‐week study end point (Figure 2B).
Interestingly, Baghdasaryan et al previously reported worsening of liver disease and fibrosis in INT‐747/OCA‐fed Mdr2‐/‐, but observed no serious adverse effects for up to 4 weeks of treatment. 22 This can be explained by the markedly more severe disease phenotype in our Mdr2‐/‐ strain on fibrosis‐susceptible BALB/c genetic background 19 compared to relatively mild disease in FVB Mdr2‐/‐ strain used by Baghdasaryan et al It is tempting to speculate that such transient/low‐grade hepatotoxicity might help to explain the lack of OCA effect on fibrosis compared to EDP‐305 (Figures 2 and 4). One potential caveat of Mdr2‐/‐ experiment is incorporation of drugs into diet, the dosing regimen that is harder to precisely control. We did not test the efficacy of lower doses of OCA in our study which may have been more efficacious; in MCD model, where OCA was dosed precisely via daily oral gavage, no overt toxicity was observed. However, medicated diets were well tolerated in healthy mice, and in the MCD‐fed mice OCA still failed to significantly impact fibrosis (Figure 4C,D), which speaks against potential dosing issue in medicated diet experiment. Another limitation in our study is a potential confounding in Mdr2‐/‐ efficacy study due to exclusion of seven out of 19 mice that became moribund and were excluded from analysis. Thus, observed hepatotoxicity is most likely model specific due to previously reported remarkable susceptibility of Mdr2‐/‐ mice to bile acid‐induced liver injury, 29 , 30 and/or diminished liver function due to a very severe disease in our Mdr2‐/‐ strain 19 which has been shown to slow down OCA clearance. 31 Absence of any OCA‐induced liver injury in healthy wild‐type mice on two genetic backgrounds and dosed via same medicated diet supports this explanation (C57Bl6, Figure 5 and BALB/c, Figure S1). While this observation is unlikely to explain the differences in antifibrotic activity of OCA and EDP‐305, this may serve as a model to study the mechanism of FXR‐dependent liver toxicity, which is of practical importance given the recent reports of liver failure following the accidental OCA overdosing in human patients with end‐stage liver disease.
Another, and in our view more plausible, explanation for the remarkable differences we observed in FXR agonists’ effects on fibrosis in Mdr2‐/‐ and MCD models is linked to the different impact of these treatments on accompanying ductular reaction. Prominent paracrine profibrogenic role of reactive cholangiocytes/HPC, comprising ductular reaction, is well documented by us and others 23 , 32 and recently reviewed 33 , 34 ). Thus, activation of either progenitors or cholangiocyte proliferation (or both) may offset the direct therapeutic effect of FXR agonists on HSCs. In both our in vivo models, lack of improvement in ductular reaction by OCA was associated with rather modest decrease in HSC activation marker α‐SMA (20% decrease in Mdr2‐/‐ and no change in MCD model); this is in stark contrast with EDP‐305 which at the same dose of 30mg/kg decreased both ductular proliferation and HSC activation by 90% and 80% respectively (Figures 3 and 4). The expansion of SOX9 + undifferentiated progenitors is accompanied by laminin‐rich niche formation 35 ; in both Mdr2‐/‐ and MCD models, EDP‐305 robustly decreased laminin and SOX9 expression while OCA showed little effect (Figure 3C,D and Figure S3). These findings are generally consistent with our hypothesis that worsening of ductular reaction by OCA limits its antifibrotic activity. Since the effects of FXR agonism on ductular proliferation were not directly addressed in the existing literature, these intriguing observations prompted us to investigate it further.
Strikingly, even in the absence of underlying liver injury, 2 weeks of OCA treatment (but not EDP‐305) induced mild, but readily detectable expansion of CK19+ and SOX9 + ductular cells within laminin‐rich matrix in the livers of healthy mice, which exhibited morphological feature characteristic of HPC activation and progressive increase of cholangiocytes/HPC‐specific mRNA of CK19, EpCAM and Trop2 and ITGB6 23 in the livers of OCA‐treated mice, whereas EDP‐305 had no impact (Figure 5). These results together with either worsening or the lack of improvement in profibrogenic ductular reaction in OCA‐treated mice with two types of chronic liver injury (Figures 3B and 4E,F) strongly suggested that OCA exerts previously unrecognized effect on HPC activation, the major cell lineage giving rise to ‘ductular reaction’. Interestingly, differential effects on FXR agonists on ductular reaction were clearly independent of cholestasis since serum ALKP and total bile acid were similarly reduced by both FXR agonists (Figure 3C).
This prompted us to investigate the direct effects of FXR agonists on freshly isolated HPC in vitro. Colony formation assay revealed that neither EDP‐305 nor OCA had an obvious effect of ability of EpCAM + HPC cells to form colony or their size. However, HPC differentiation was impacted by OCA (but not EDP‐305), which promoted (profibrogenic) cholangiocyte lineage differentiation of HPC over beneficial (non‐fibrogenic) hepatocyte lineage commitment (Figure 6). Nonetheless, one must be aware of a caveat of direct in vitro comparison of OCA with other FXR agonists in our studies using primary stellate cells and HPC cultures, since in vivo two of its major metabolites (glyco‐ and tauro‐OCA) substantially contribute to the biological activity and therapeutic profile of OCA. 13 In contrast, all biological activity in vivo is largely attributed to the EDP‐305 molecule, and thus any in vitro differences in the activity of OCA and EDP‐305 should be interpreted with caution (Tables S5 and S6).
Taken together, these data suggest that OCA directly promotes cholangiocyte/HPC differentiation and ductular reaction. These in vitro results are generally consistent with substantial decrease in laminin‐rich HPC niche by EDP‐305 but not by OCA in vivo (Figure 3CD and Figure S3), a critical step precipitating hepatocyte differentiation of hepatic stem cells/progenitors. 24 , 35 Because structurally unrelated FXR agonist EDP‐305 at the same dose did not have any effect on ductular proliferation in vitro or in vivo, such effects appear to be OCA specific rather than general, FXR agonism‐related biological activity. An intriguing question that remains to be elucidated is whether such direct effects of OCA on biliary tree may explain enigmatic, paradoxical increase in ALKP reported in FLINT NASH trial 11 or exacerbation of gallstone disease in the subset of patients. 17
It is important to note that the biliary tree of healthy mice treated with OCA is predominantly exposed to high levels of bioactive OCA metabolite tauro‐OCA, which are about 2600 times greater than its plasma levels. These levels were two orders of magnitude higher than that of OCA itself, and tend to accumulate with time (Table S6), presumably due to enterohepatic circulation. Therefore, it is likely that tauro‐OCA primarily determines the efficacy and safety profile, as well as potential off‐target effects, in our model system. While OCA itself showed no toxicity in primary human hepatocyte cultures at concentration well above those observed in our in vivo experiments (100 µmol/L), both major bioactive metabolites (glyco‐ and tauro‐OCA) induced cell death at same concentration. 36 In the serum, both OCA and its major metabolite tauro‐OCA in our study were below reported toxic levels (Tables S5 and S6). However, in the bile, the tauro‐OCA levels reached 4.47 mM concentration (Table S6), at which direct toxic and/or off‐target effects can be expected. Based on all available preclinical and clinical pharmacokinetic data, 37 EDP‐305 does not generate any glycine‐ or taurine‐conjugated metabolites and thus, does not undergo intrahepatic circulation.
EDP‐305 molecule was specifically designed to avoid TGR5 engagement (EC50 > 15 µM). OCA itself is a weak TGR5 agonist (EC50 = 0.918 μM), whereas its glycine and especially taurine conjugates are considerably more potent TGR5 agonists, with reported EC50 values 0.315 and 0.205 μM respectively. 14 Because tauro‐OCA concentration reach low millimolar range in bile, there is little doubt that it will engage TGR5 receptors in biliary tree, and TGR5‐dependent mechanism may be responsible for differential effects on ductular reaction observed with FXR agonists (Figures 5 and 6). While the off‐target TGR5 reactivity may, in theory, increase the therapeutic efficacy of OCA, 22 it may as well be responsible for its common side effects such as pruritus 15 by mediating bile acid‐mediated activation of sensory nerves, 16 as well as for more subtle, subclinical effects on HPC/cholangiocyte liver compartment that we report here for the first time. It appears likely that recently reported OCA‐induced gallstone formation in subset of susceptible patients 17 is a TGR5‐mediated phenomenon and biliary obstruction may partially contribute to worsening of ductular reaction in our system. Although we did not directly interrogate this in our study, prior reports in knockout mice suggest that TGR5‐dependent mechanism is plausible, since TGR5 is required for bile acid‐induced cholangiocyte proliferation both in vivo and in vitro, 38 and for bile acid‐induced hepatic differentiation of mesenchymal stem cell. 39 A differential effects of FXR agonists on re‐expression of impaired tight junctions within biliary tree, as directly observed by Pradhan‐Sundd et al in different models of cholestatic and metabolic liver injury, 40 is an intriguing hypothesis that is worth exploring in the follow‐up studies.
A recent study by Schwabl et al 41 reported that another non‐bile acid FXR agonist PX20606 improved portal hypertension in both cirrhotic and non‐cirrhotic rats, substantially exceeding OCA efficacy. It should be noted that significant species variation in OCA metabolism should be carefully considered when studying OCA as comparator. For instance, most preclinical studies which demonstrated OCA efficacy on fibrosis and portal pressure and fibrosis were performed in rats 26 , 4241 whereas it was challenging to reproduce same results in a mouse system in earlier reports and in our present study. However, the OCA metabolism profile in mice is in fact closer to humans, with OCA conjugates representing the major fraction compared to OCA in both species; in contrast, unmetabolized OCA represents the major biologically active drug fraction in rats. 31
In conclusion, we report the comprehensive analysis of therapeutic efficacy of the FXR agonist EDP‐305 in mouse models of biliary and metabolic liver disease associated with fibrosis. In both models and by all studied parameters, EDP‐305 outperformed first‐in class FXR agonist OCA, especially in regard to the therapeutic effect on liver fibrosis. Our data further suggest that direct effects of OCA on the ductular reaction limits its antifibrotic activity, suggesting the biological activity towards biliary compartment should be taken into account when devising new FXR/FGF19 therapeutics. Our results are supporting the notion that structurally differentiated FXR agonists may offer a substantially improved therapeutic profile in liver disease. The encouraging efficacy and safety profile of EDP‐305 reported here warrants its further evaluation in clinical trials for biliary and metabolic liver diseases.
CONFLICT OF INTEREST
None to be declared. Shucha Zhang, Yang Li, Yat Sun Or, Li‐Juan Jiang are the employees of Enanta Pharmaceuticals who own Enanta stock options and/or stock.
Supporting information
Supplementary Material
An P, Wei G, Huang P, et al. A novel non‐bile acid FXR agonist EDP‐305 potently suppresses liver injury and fibrosis without worsening of ductular reaction. Liver Int. 2020;40:1655–1669. 10.1111/liv.14490
Handling Editor: Espen Melum
Funding information
This work was supported in part by research grants from Enanta Pharmaceuticals, PSC Partners for Cure Canada and an institutional grant from Department of Medicine, Beth Israel Deaconess Medical Center to YVP, and a fellowship by National Natural Science Foundation of China (81302131) to PA
REFERENCES
- 1. Dulai PS, Singh S, Patel J, et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta‐analysis. Hepatology. 2017;65:1557‐1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vesterhus M, Hov JR, Holm A, et al. Enhanced liver fibrosis score predicts transplant‐free survival in primary sclerosing cholangitis. Hepatology. 2015;62:188‐197. [DOI] [PubMed] [Google Scholar]
- 3. Lemoinne S, Friedman SL. New and emerging anti‐fibrotic therapeutics entering or already in clinical trials in chronic liver diseases. Curr Opin Pharmacol. 2019;49:60‐70. [DOI] [PubMed] [Google Scholar]
- 4. Popov Y, Schuppan D. Targeting liver fibrosis: strategies for development and validation of antifibrotic therapies. Hepatology. 2009;50:1294‐1306. [DOI] [PubMed] [Google Scholar]
- 5. Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev. 2009;89:147‐191. [DOI] [PubMed] [Google Scholar]
- 6. Liu Y, Binz J, Numerick MJ, et al. Hepatoprotection by the farnesoid X receptor agonist GW4064 in rat models of intra‐ and extrahepatic cholestasis. J Clin Invest. 2003;112:1678‐1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kong B, Luyendyk JP, Tawfik O, Guo GL. Farnesoid X receptor deficiency induces nonalcoholic steatohepatitis in low‐density lipoprotein receptor‐knockout mice fed a high‐fat diet. J Pharmacol Exp Ther. 2009;328:116‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huang W, Ma K, Zhang J, et al. Nuclear receptor‐dependent bile acid signaling is required for normal liver regeneration. Science. 2006;312:233‐236. [DOI] [PubMed] [Google Scholar]
- 9. Fickert P, Fuchsbichler A, Moustafa T, et al. Farnesoid X receptor critically determines the fibrotic response in mice but is expressed to a low extent in human hepatic stellate cells and periductal myofibroblasts. Am J Pathol. 2009;175:2392‐2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wolfe A, Thomas A, Edwards G, Jaseja R, Guo GL, Apte U. Increased activation of the Wnt/beta‐catenin pathway in spontaneous hepatocellular carcinoma observed in farnesoid X receptor knockout mice. J Pharmacol Exp Ther. 2011;338:12‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Neuschwander‐Tetri BA, Loomba R, Sanyal AJ, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non‐cirrhotic, non‐alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo‐controlled trial. Lancet. 2015;385:956‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nevens F, Andreone P, Mazzella G, et al. A placebo‐controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med. 2016;375:631‐643. [DOI] [PubMed] [Google Scholar]
- 13. Pellicciari R, Costantino G, Camaioni E, et al. Bile acid derivatives as ligands of the farnesoid X receptor. Synthesis, evaluation, and structure‐activity relationship of a series of body and side chain modified analogues of chenodeoxycholic acid. J Med Chem. 2004;47:4559‐4569. [DOI] [PubMed] [Google Scholar]
- 14. Tully DC, Rucker PV, Chianelli D, et al. Discovery of tropifexor (LJN452), a highly potent non‐bile acid FXR agonist for the treatment of cholestatic liver diseases and nonalcoholic steatohepatitis (NASH). J Med Chem. 2017;60:9960‐9973. [DOI] [PubMed] [Google Scholar]
- 15. Lieu TinaMarie, Jayaweera G, Zhao P, et al. The bile acid receptor TGR5 activates the TRPA1 channel to induce itch in mice. Gastroenterology. 2014;147:1417‐1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alemi F, Kwon E, Poole DP, et al. The TGR5 receptor mediates bile acid‐induced itch and analgesia. J Clin Invest. 2013;123:1513‐1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Al‐Dury S, Wahlström A, Panzitt K, et al. Obeticholic acid may increase the risk of gallstone formation in susceptible patients. J Hepatol. 2019;71(5):986‐991. [DOI] [PubMed] [Google Scholar]
- 18. Gege C, Hambruch E, Hambruch N, Kinzel O, Kremoser C. Nonsteroidal FXR ligands: current status and clinical applications. Handb Exp Pharmacol. 2019;256:167‐205. [DOI] [PubMed] [Google Scholar]
- 19. Ikenaga N, Liu SB, Sverdlov DY, et al. A new Mdr2(‐/‐) mouse model of sclerosing cholangitis with rapid fibrosis progression, early‐onset portal hypertension, and liver cancer. Am J Pathol. 2015;185:325‐334. [DOI] [PubMed] [Google Scholar]
- 20. Fisher FM, Chui PC, Nasser IA, et al. Fibroblast growth factor 21 limits lipotoxicity by promoting hepatic fatty acid activation in mice on methionine and choline‐deficient diets. Gastroenterology. 2014;147(1073–1083):e1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chau M, Li Y, Roqueta‐Rivera M, et al. Characterization of EDP‐305, a Highly Potent and Selective Farnesoid X Receptor Agonist, for the Treatment of Non‐alcoholic Steatohepatitis. Int J Gastroenterol. 2019;3:4‐16. [Google Scholar]
- 22. Baghdasaryan A, Claudel T, Gumhold J, et al. Dual farnesoid X receptor/TGR5 agonist INT‐767 reduces liver injury in the Mdr2‐/‐ (Abcb4‐/‐) mouse cholangiopathy model by promoting biliary HCO(‐)(3) output. Hepatology. 2011;54:1303‐1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Peng ZW, Ikenaga N, Liu SB, et al. Integrin alphavbeta6 critically regulates hepatic progenitor cell function and promotes ductular reaction, fibrosis, and tumorigenesis. Hepatology. 2016;63:217‐232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Carpino G, Cardinale V, Folseraas T, et al. Hepatic stem/progenitor cell activation differs between primary sclerosing and primary biliary cholangitis. Am J Pathol. 2018;188:627‐639. [DOI] [PubMed] [Google Scholar]
- 25. Ikenaga N, Peng Z‐W, Vaid KA, et al. Selective targeting of lysyl oxidase‐like 2 (LOXL2) suppresses hepatic fibrosis progression and accelerates its reversal. Gut. 2017;66:1697‐1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fiorucci S, Antonelli E, Rizzo G, et al. The nuclear receptor SHP mediates inhibition of hepatic stellate cells by FXR and protects against liver fibrosis. Gastroenterology. 2004;127:1497‐1512. [DOI] [PubMed] [Google Scholar]
- 27. Lee FY, Kast‐Woelbern HR, Chang J, et al. Alpha‐crystallin is a target gene of the farnesoid X‐activated receptor in human livers. J Biol Chem. 2005;280:31792‐31800. [DOI] [PubMed] [Google Scholar]
- 28. Fiorucci S, Rizzo G, Antonelli E, et al. A farnesoid x receptor‐small heterodimer partner regulatory cascade modulates tissue metalloproteinase inhibitor‐1 and matrix metalloprotease expression in hepatic stellate cells and promotes resolution of liver fibrosis. J Pharmacol Exp Ther. 2005;314:584‐595. [DOI] [PubMed] [Google Scholar]
- 29. Fickert P, Fuchsbichler A, Marschall H‐U, et al. Lithocholic acid feeding induces segmental bile duct obstruction and destructive cholangitis in mice. Am J Pathol. 2006;168:410‐422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Van Nieuwkerk CM, Elferink RP, Groen AK, et al. Effects of Ursodeoxycholate and cholate feeding on liver disease in FVB mice with a disrupted mdr2 P‐glycoprotein gene. Gastroenterology. 1996;111:165‐171. [DOI] [PubMed] [Google Scholar]
- 31. Roda A, Aldini R, Camborata C, et al. Metabolic profile of obeticholic acid and endogenous bile acids in rats with decompensated liver cirrhosis. Clin Transl Sci. 2017;10:292‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kuramitsu K, Sverdlov DY, Liu SB, et al. Failure of fibrotic liver regeneration in mice is linked to a severe fibrogenic response driven by hepatic progenitor cell activation. Am J Pathol. 2013;183:182‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fabris L, Spirli C, Cadamuro M, Fiorotto R, Strazzabosco M. Emerging concepts in biliary repair and fibrosis. Am J Physiol Gastrointest Liver Physiol. 2017;313:G102‐G116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sato K, Marzioni M, Meng F, Francis H, Glaser S, Alpini G. Ductular reaction in liver diseases: pathological mechanisms and translational significances. Hepatology. 2019;69(1):420‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lanzoni G, Cardinale V, Carpino G. The hepatic, biliary, and pancreatic network of stem/progenitor cell niches in humans: a new reference frame for disease and regeneration. Hepatology. 2016;64:277‐286. [DOI] [PubMed] [Google Scholar]
- 36. Zhang Y, Jackson JP, St. Claire RL, Freeman K, Brouwer KR, Edwards JE. Obeticholic acid, a selective farnesoid X receptor agonist, regulates bile acid homeostasis in sandwich‐cultured human hepatocytes. Pharmacol Res Perspect. 2017;5(4):e00329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ahmad A, Sanderson K, Dickerson D, Adda N. Pharmacokinetics, pharmacodynamics, and safety of EDP‐305, in healthy and presumptive NAFLD subjects. J Hepatol. 2018;68:S584. [Google Scholar]
- 38. Reich M, Deutschmann K, Sommerfeld A, et al. TGR5 is essential for bile acid‐dependent cholangiocyte proliferation in vivo and in vitro. Gut. 2016;65:487‐501. [DOI] [PubMed] [Google Scholar]
- 39. Sawitza I, Kordes C, Gotze S, Herebian D, Haussinger D. Bile acids induce hepatic differentiation of mesenchymal stem cells. Sci Rep. 2015;5:13320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pradhan‐Sundd T, Vats R, Russell JO, et al. Dysregulated bile transporters and impaired tight junctions during chronic liver injury in mice. Gastroenterology. 2018;155(1218–1232):e1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schwabl P, Hambruch E, Seeland BA, et al. The FXR agonist PX20606 ameliorates portal hypertension by targeting vascular remodelling and sinusoidal dysfunction. J Hepatol. 2017;66:724‐733. [DOI] [PubMed] [Google Scholar]
- 42. Verbeke L, Farre R, Trebicka J, et al. Obeticholic acid, a farnesoid X receptor agonist, improves portal hypertension by two distinct pathways in cirrhotic rats. Hepatology. 2014;59:2286‐2298. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material