

Abstract
Objective
Muscle inflammation is a feature in myositis and Duchenne muscular dystrophy (DMD). Autoimmune mechanisms are thought to contribute to muscle weakness in patients with myositis. However, a lack of correlation between the extent of inflammatory cell infiltration and muscle weakness indicates that nonimmune pathologic mechanisms may play a role. The present study focused on 2 microRNA (miRNA) sets previously identified as being elevated in the muscle of patients with DMD—an “inflammatory” miRNA set that is dampened with glucocorticoids, and a “dystrophin‐targeting” miRNA set that inhibits dystrophin translation—to test the hypothesis that these miRNAs are similarly dysregulated in the muscle of patients with myositis, and could contribute to muscle weakness and disease severity.
Methods
A major histocompatibility complex class I–transgenic mouse model of myositis was utilized to study gene and miRNA expression and histologic features in the muscle tissue, with the findings validated in human muscle biopsy tissue from 6 patients with myositis. Mice were classified as having mild or severe myositis based on transgene expression, body weight, histologic disease severity, and muscle strength/weakness.
Results
In mice with severe myositis, muscle tissue showed mononuclear cell infiltration along with elevated expression of type I interferon and NF‐κB–regulated genes, including Tlr7 (3.8‐fold increase, P < 0.05). Furthermore, mice with severe myositis showed elevated expression of inflammatory miRNAs (miR‐146a, miR‐142‐3p, miR‐142‐5p, miR‐455‐3p, and miR‐455‐5p; ~3–40‐fold increase, P < 0.05) and dystrophin‐targeting miRNAs (miR‐146a, miR‐146b, miR‐31, and miR‐223; ~3–38‐fold increase, P < 0.05). Bioinformatics analyses of chromatin immunoprecipitation sequencing (ChIP‐seq) data identified at least one NF‐κB consensus element within the promoter/enhancer regions of these miRNAs. Western blotting and immunofluorescence analyses of the muscle tissue from mice with severe myositis demonstrated reduced levels of dystrophin. In addition, elevated levels of NF‐κB–regulated genes, TLR7, and miRNAs along with reduced dystrophin levels were observed in muscle biopsy tissue from patients with histologically severe myositis.
Conclusion
These data demonstrate that an acquired dystrophin deficiency may occur through NF‐κB–regulated miRNAs in myositis, thereby suggesting a unifying theme in which muscle injury, inflammation, and weakness are perpetuated both in myositis and in DMD.
INTRODUCTION
The idiopathic inflammatory myopathies are a heterogeneous group of systemic connective tissue diseases characterized by chronic muscle inflammation and weakness. This group of diseases, which includes dermatomyositis (DM), polymyositis, and inclusion body myositis (IBM) (collectively referred to as myositis), affects both adults and children. Although rare, there is evidence that disease incidence is increasing 1. Histologic features of myositis include the following: 1) endomysial, perimysial, or perivascular infiltration by inflammatory cells, consisting predominantly of macrophages and T cells 2, 3; 2) myofiber‐specific expression of major histocompatibility complex (MHC) class I molecules; and 3) bouts of muscle degeneration/regeneration, fibrosis, and fat deposition 4. Of these, the most striking feature is the overexpression of MHC class I in the muscle of patients with myositis 5, 6.
The role of the innate and adaptive immune systems in myositis is well known, but results recently reported suggest that nonimmune mechanisms also contribute to the disease (for review, see refs 7 and 8), as there is not a strong correlation between the extent of inflammatory cell infiltration and muscle weakness in patients with myositis 9. Interestingly, myositis muscle seems primed for inflammatory signaling events, since it inappropriately expresses Toll‐like receptors (TLRs) 10, 11 and MHC class I 5, 6, which act as receptors that are normally confined to immune cells. Stimulation of TLR‐7 and MHC class I activates the proinflammatory transcription factor NF‐κB. In addition, TLR‐7 is a potent inducer of type I interferon (IFN) genes 12; this gene expression signature correlates with disease severity both in patients with DM 13, 14, 15, 16 and in those with juvenile DM 15, 17.
NF‐κB is a key regulator of inflammation in immune cells; however, it is also active in skeletal muscle under inflammatory conditions. Not surprisingly, in the muscle of patients with myositis, NF‐κB is highly activated and its activation corresponds to disease severity 18. Classic NF‐κB activation is triggered via inflammatory cytokines such as tumor necrosis factor (TNF), which is found at high levels in the muscle of patients with myositis 19. Nonclassic NF‐κB activation occurs through endoplasmic reticulum stress and autophagy; both of these processes/pathways are increased in myositis 20, 21. It has been previously shown that muscle‐specific MHC class I expression activates the NF‐κB pathway and turns on classic NF‐κB target genes such as TNF and TLR7, exacerbating inflammatory signaling 22. As further confirmation of its importance in disease pathogenesis, downstream gene targets of NF‐κB are highly up‐regulated in myositis muscle 19, 23.
At the molecular and histologic levels, there is significant overlap between inflammatory myopathies and other muscle disorders, in particular, Duchenne muscular dystrophy (DMD), Becker muscular dystrophy (BMD), and manifesting DMD female carriers 24, 25. Similar to myositis, muscles of patients with DMD show increased TLR‐7 expression 26, elevated NF‐κB activation 18, 27, and overexpression of muscle‐specific MHC class I 28. Previously, we described 2 distinct sets of microRNAs (miRNAs) that are elevated in DMD muscle. The first is an “inflammatory set” of miRNAs whose expression is increased in mdx mice and is reduced by administration of 2 different antiinflammatory drugs 29. The second set of miRNAs is a “dystrophin‐targeting” set (termed dystrophin‐targeting miRNAs [DTMs]). DTMs are elevated both in DMD and in BMD, and are prohibitory to dystrophin protein production in BMD and to exon‐skipping–mediated dystrophin rescue in a mouse model of DMD 30. Some of these miRNAs show increased expression in inflammatory connective tissue disorders that overlap with myositis, including scleroderma, rheumatoid arthritis, and Sjögren's syndrome, suggesting that inflammatory miRNAs and DTMs play a broader role in regulating chronic inflammation.
In mice, skeletal muscle–specific conditional overexpression of MHC class I induces myositis 3. Following MHC class I up‐regulation, female mice develop early histologic and functional changes mirroring human myositis, while male mice develop a more mild disease 3. In the present study, we tested the hypothesis that inflammatory miRNAs and DTMs contribute to the pathologic processes of myositis by reducing dystrophin in muscle fibers.
MATERIALS AND METHODS
Mice
All mouse studies were performed with adherence to the NIH Guide for the Care and Use of Laboratory Animals. All experiments were conducted according to protocols within the guidelines of and with the approval of the Institutional Animal Care and Use Committee of Children's National Medical Center. Animals were housed at room temperature in a 12‐hour/12‐hour light/dark cycle. Genotyping was carried out in all mice at age 3–4 weeks, as previously described 21. The mouse model of myositis used in this study is known as the HT mouse model of myositis, using (TRE)‐H‐2Kb (H) and mCK‐tTA (T) mice, as has been described previously 3. For this study, transgene expression was induced by withdrawal of doxycycline from the drinking water of the mice at age 5 weeks, followed by analyses at ages 9–13 weeks. Female mice were used for all studies, as the disease manifestations are more severe in female mice.
Human muscle biopsy tissue
Muscle biopsy tissue samples from human subjects were obtained from a member of the Telethon Network of Genetic Biobanks (project no. GTB12001). The samples obtained included skeletal muscle, peripheral nerve samples, DNA samples, and cell lines. Further details can be found in the Supplementary Materials and Methods (available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.41215/abstract).
Quantitative reverse transcription–polymerase chain reaction (qRT‐PCR) analyses of miRNAs and messenger RNAs (mRNAs)
Analyses of miRNA and mRNA expression by qRT‐PCR were performed in muscle tissue samples in a manner as previously reported 29, 30. Details can be found in the Supplementary Materials and Methods (http://onlinelibrary.wiley.com/doi/10.1002/art.41215/abstract).
Western blot analysis
Proteins from muscle were collected using a lysis buffer containing 75 mM Tris HCl (pH 6.8), 10% sodium dodecyl sulfate, 10 mM EDTA, and 5% 2‐mercaptoethanol. Protein samples (25 μg) were separated using a 3–8% Tris–acetate gel (Bio‐Rad) and then transferred onto a nitrocellulose membrane (20V at 4°C) overnight, followed by a 1‐hour pulse (80V at 4°C). Antibodies used included the DYS‐1 mouse monoclonal antibody against dystrophin (1:100; Novacastra), and a rabbit polyclonal antibody against vinculin (1:1,000, clone N3C1; GenTex). Horseradish peroxidase–conjugated anti‐mouse immunogloblulin or anti‐rabbit goat immunoglobulin (1:5,000; Millipore) were used as secondary antibodies. Enzyme chemiluminescence (Pierce) was used for detection of the proteins. The blots were scanned digitally with a ChemiDoc Touch Imager.
Hematoxylin and eosin (H&E) staining and histologic grading of the muscle tissue sections
Sections of muscle tissue (8 μM) were cut from frozen mouse quadriceps muscles or from frozen human muscle biopsy tissue. Sections were air‐dried for 1 hour on SuperFrost Plus slides and then stained with H&E. Stained slides were imaged on a VS120 Virtual Slide microscope (Olympus) at 20× magnification. Details on the grading of histopathologic features of the muscle can be found in the Supplementary Materials and Methods (http://onlinelibrary.wiley.com/doi/10.1002/art.41215/abstract).
Immunofluorescence
Frozen muscle sections (8 μM) were dried for 1 hour on SuperFrost Plus slides and then stored at −80°C. For staining, slides were thawed for 1 hour at room temperature and then fixed in either ice‐cold acetone (4°C) for 10 minutes or 4% paraformaldehyde for 10 minutes at room temperature. Further details on the primary and secondary antibody dilutions and incubation times can be found in the Supplementary Materials and Methods (http://onlinelibrary.wiley.com/doi/10.1002/art.41215/abstract).
Evaluation of force contractions on isolated skeletal muscle
Extensor digitorum longus (EDL) muscle was isolated from live anesthetized mice prior to their euthanization, and contractile properties of the skeletal muscle were then measured ex vivo. Details can be found in the Supplementary Materials and Methods (http://onlinelibrary.wiley.com/doi/10.1002/art.41215/abstract).
Bioinformatics analysis
We examined each miRNA gene promoter by bioinformatics analysis, to gain insight into the mechanisms of response to treatment, as has been previously reported 29. Details on the identification of NF‐κB/RelA binding sites can be found in the Supplementary Materials and Methods (http://onlinelibrary.wiley.com/doi/10.1002/art.41215/abstract).
RESULTS
Variable disease severity in mice with myositis
In the present study, we utilized female mice that overexpress MHC class I, and we performed our studies after withdrawal of doxycycline from the drinking water of the mice at age 5 weeks. At 4–8 weeks after doxycycline withdrawal, we observed variable disease severity among the mice, allowing us to investigate the molecular factors driving disease progression.
We initially observed that mice with a lower body weight at the time of euthanization appeared to have a more aggressive disease. To determine whether the body weight of mice is related to transgene expression, we plotted the weight of the mice at the time of euthanization against the extent of transgenic MHC class I expression in the most severely affected muscle (quadriceps). As shown in Figure 1A, body weight and transgene expression showed a strong inverse correlation (Spearman's rho = −0.9146), thus corroborating our observations that mice with a lower body weight develop a more severe disease, which can likely be attributed to increased MHC class I expression.
Figure 1.
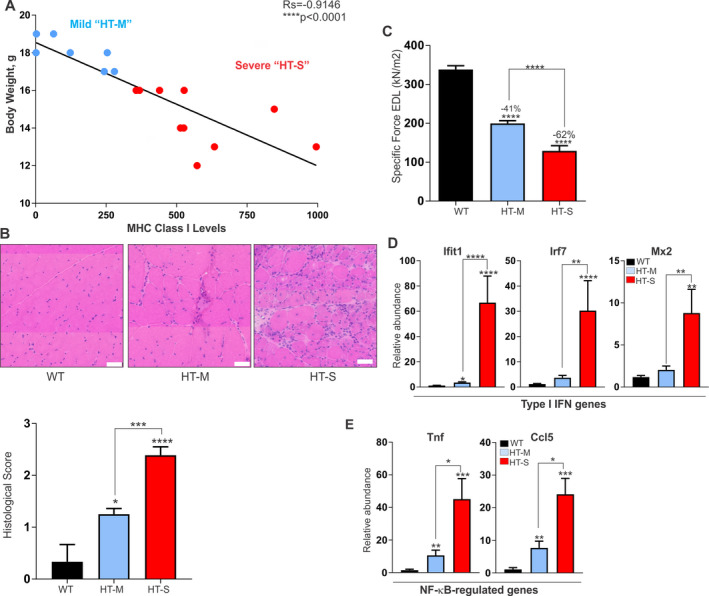
Variable disease severity in the HT mouse model of myositis. A, Classification system used for grouping HT mice according to the severity of myositis, based on body weight plotted against the extent of major histocompatibility complex (MHC) class I transgene expression. HT‐M = mice with mild myositis, body weight of >16 grams, and MHC class I expression of ≤300‐fold relative to wild‐type (WT) mice. HT‐S = mice with severe myositis, body weight of <16 grams, and MHC class I expression of >300‐fold relative to WT mice. Correlations were determined using Spearman's correlation coefficients. B, Representative images of histologic staining with hematoxylin and eosin (top) and histologic scores of staining (bottom) of the quadriceps muscle from WT mice and mice in the mild or severe myositis groups (n = 5 WT, n = 7 HT‐M, and n = 10 HT‐S). Bars = 50 μM. C, Isolated measurements of specific force contractions of the extensor digitorum longus (EDL) muscle in each group of mice (n ≥ 10 per group). D and E, Type I interferon (IFN) gene expression levels (genes for IFN‐induced protein with tetratricopeptide repeats 1 [Ifit], IFN regulatory factor 7 [Irf7], and MX dynamin like GTPase 2 [Mx2]) (D) and NF‐κB–induced gene expression (genes for tumor necrosis factor [Tnf] and C–C motif chemokine ligand 5 gene [Ccl5]) (E) in the quadriceps muscle of WT, HT‐M, and HT‐S mice (n = 5 WT, n = 7 HT‐M, and n = 10 HT‐S). Data were normalized to the values for 18S ribosomal RNA. Results in B–E are the mean ± SEM. * = P < 0.05; ** = P < 0.01; *** = P < 0.001; **** = P < 0.0001 for the indicated comparisons or versus WT mice, by analysis of variance.
The scatter plot of the correlation between body weight and transgene expression revealed 2 distinct mouse phenotypes: HT mice with mild myositis (HT‐M) and HT mice with severe myositis (HT‐S). Mice classified in the HT‐S myositis group weighed 12–16 grams at the time of euthanization, and their expression of muscle‐specific MHC class I was at a level of >300‐fold relative to wild‐type (WT) mice. Mice classified in the HT‐M myositis group weighed 17–20 grams and showed MHC class I transgene expression at a level of ≤300‐fold relative to WT mice.
To determine whether differences in transgene expression were reflected in the muscle pathology, we used a grading system on H&E‐stained muscle tissue from the HT‐M and HT‐S groups of mice. The muscle of mice with severe myositis showed 1) higher endomysial inflammation with invasion into the myofibers, 2) more highly variable muscle fiber size, and 3) increased necrosis and degeneration, with a mean histologic grade of 2.3 (on a scale of 0–3) (P < 0.0001 versus WT). In contrast, the muscle of mice with mild myositis showed 1) less endomysial inflammation with no invasion into fibers, 2) less fiber size variation, and 3) little‐to‐no necrosis and degeneration, with a mean histologic grade of 1.3 (P < 0.001 versus HT‐S) (Figure 1B).
To determine whether the differences in molecular and histologic features between the HT‐S and HT‐M groups of mice resulted in functional differences, we assessed the muscle strength of isolated EDL muscles ex vivo. Specific force in the EDL muscle was reduced by 62% in mice with severe myositis and was reduced by 41% in mice with mild myositis (each P < 0.0001 versus WT mice) (Figure 1C). Collectively, these data show that mice classified as HT‐S indeed display a more severe disease as compared to mice classified as classified as HT‐M.
Type I IFN signature and NF‐κB activation in mice with severe myositis
Results of recent studies have suggested that molecules induced by type I IFNs play a mechanistic role in the pathogenesis of myositis 13, 14, 15, 16; this is most commonly observed in adult and juvenile patients with DM 15, 17. To determine whether type I IFN gene expression was related to disease severity in our study, we performed qRT‐PCR on 3 type I IFN signature genes, the genes for IFN‐induced protein with tetratricopeptide repeats 1 (Ifit), IFN regulatory factor 7 (Irf7), and MX dynamin like GTPase 2 (Mx2). Muscle tissue from mice with severe myositis showed significant increases (~10–80‐fold) in the relative expression of all 3 genes as compared to WT mice (P < 0.0001 for Ifit and Irf7, and P < 0.01 for Mx2) (Figure 1D). In comparison, muscle tissue from mice with mild myositis showed modest increases in Ifit1, Irf7, and Mx2, with only the levels of Ifit1 reaching a statistically significant difference compared to WT mice (P < 0.05) (Figure 1D).
We have previously shown that NF‐κB is highly activated in human inflammatory myopathies and mouse models of myositis 22. To determine whether NF‐κB activation is associated with disease severity, we assessed the NF‐κB–activated genes Tnf and C–C motif chemokine ligand 5 gene (Ccl5). Both Tnf and Ccl5 followed a similar pattern as that of type I IFN signature genes. In the muscle of mice in the HT‐S group, the levels of both Tnf and Ccl5 were highly increased over the levels in WT mice (P < 0.001) (Figure 1E). In the HT‐M group of mice, NF‐κB–induced gene expression in the muscle also showed significant elevations compared to WT mice (P < 0.01), but to a lesser extent.
In response to their cognate ligands, TLRs induce both type I IFN and inflammatory (NF‐κB) gene expression 31. Investigators have recently observed myofiber‐specific expression of TLR‐7 in several muscle disorders 10, 26, 32. Because we observed elevated expression of both types of gene expression signatures in mice with severe myositis, we next assessed TLR‐7 expression in both the HT‐M and HT‐S myositis groups of mice. Using immunofluorescence, we observed an almost complete absence of TLR‐7 from WT mouse muscle (Figure 2A). In contrast, the muscle of mice with mild myositis showed modest TLR‐7 staining, while the muscle of mice with severe myositis showed strong TLR‐7 staining (Figure 2A).
Figure 2.
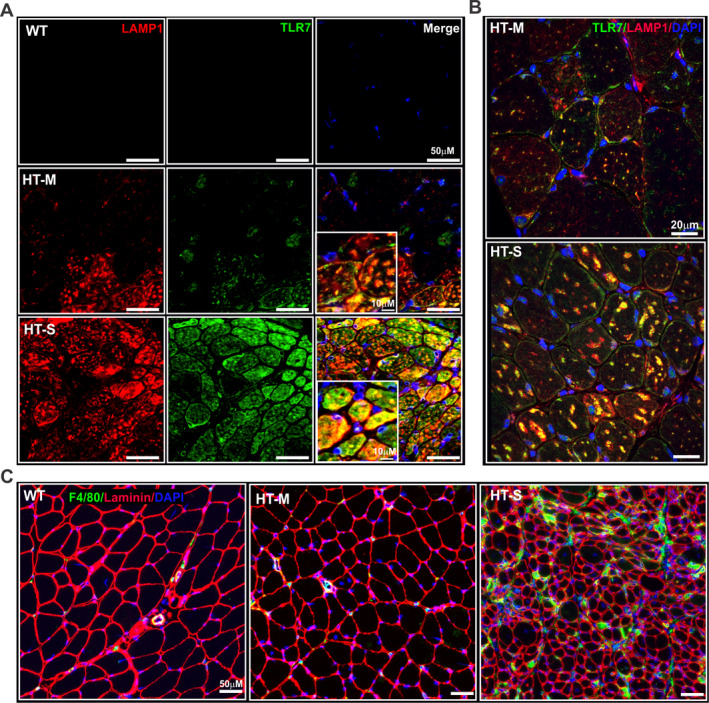
Increased Toll‐like receptor 7 (TLR‐7) staining and macrophage infiltration in the muscle of mice with severe myositis. A and B, Representative images of the quadriceps muscle of a WT mouse, mouse with mild myositits, and mouse with severe myositis, immunolabeled with an antibody against TLR‐7 (green) and lysosome‐associated membrane protein 1 (LAMP‐1) (red). DAPI counterstaining was used to visualize nuclei (blue). Images in A were obtained with a VS‐120 scanning microscope at 20× magnification, and images in B are from a second set of immunostained muscle tissue sections visualized using confocal microscopy. In both A and B, colocalization of TLR‐7 and LAMP‐1, a marker of late endosomes/early lysosomes, is an indication that TLR‐7 is localized to the endosomes and is in an activated state. C, Muscle tissue from HT‐M and HT‐S mouse quadriceps stained with an antibody against F4/80, which recognizes a glycoprotein expressed by murine macrophages (green), and with an antibody against laminin (red), which confirms the integrity of the muscle tissue, with DAPI counterstaining of the nuclei (blue). See Figure 1 for other definitions.
Using confocal microscopy, we observed colocalization of TLR‐7 and lysosome‐associated membrane protein 1 (LAMP‐1), a marker of late endosomes/early lysosomes, in the mouse muscle (Figure 2B; positive controls are shown in Supplementary Figure 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.41215/abstract), indicating that TLR‐7 was in an activated state 33. Analyses by qRT‐PCR corroborated the immunofluorescence data, showing elevated levels of Tlr7 in the muscle of mice with severe myositis (see Supplementary Figure 2A, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.41215/abstract). In addition, qRT‐PCR analyses indicated that Lamp1 gene expression was increased in the muscle of mice with severe myositis; however, the difference compared to WT mice did not reach significance (see Supplementary Figure 2B [http://onlinelibrary.wiley.com/doi/10.1002/art.41215/abstract]).
Signaling through TLR‐7 promotes macrophage recruitment and invasion into muscle fibers. Immunofluorescence analyses of mouse macrophage marker F4/80 showed increased macrophage infiltration in the muscle of mice with severe myositis as compared to WT mice or mice with mild myositis (Figure 2C). This observation was confirmed by qRT‐PCR analyses of a macrophage‐specific marker, endothelial growth factor–like module‐containing mucin‐like hormone receptor–like 1 (Emr1). Analyses by qRT‐PCR showed that the muscle of mice with severe myositis expressed elevated levels of Emr1 (P < 0.05) (see Supplementary Figure 2C [http://onlinelibrary.wiley.com/doi/10.1002/art.41215/abstract]). Taken together, these data suggest that progression of myositis involves distinct changes in the molecular signature of myofibers, which begin to take on an innate immune–like state via expression of type I IFN signature genes, NF‐κB–regulated genes, and myofiber‐specific expression of TLR‐7, all of which may result in increased macrophage recruitment.
Association between muscle levels of inflammatory and dystrophin‐targeting miRNAs with the severity of myositis
To further characterize the molecular changes that drive myositis disease progression, we analyzed 2 defined sets of miRNAs in the mouse quadriceps muscle. The first set encompassed 9 miRNAs (termed inflammatory miRNAs) that we previously found to be elevated in dystrophic mice. This set of miRNAs are regulated by NF‐κB and their expression decreases in response to antiinflammatory treatment 29. Of these, the levels of 5 miRNAs (miR‐146a, miR‐142‐3p, miR‐142‐5p, miR‐455‐3p, and miR‐455‐5p) were significantly elevated in the muscle of mice with severe myositis compared to WT mice (~4–6‐fold increase, P < 0.05) (Figure 3A). In the muscle of mice with mild myositis, the levels of miR‐142‐3p and miR‐146a were also significantly elevated compared to WT mice, but to a lesser extent.
Figure 3.
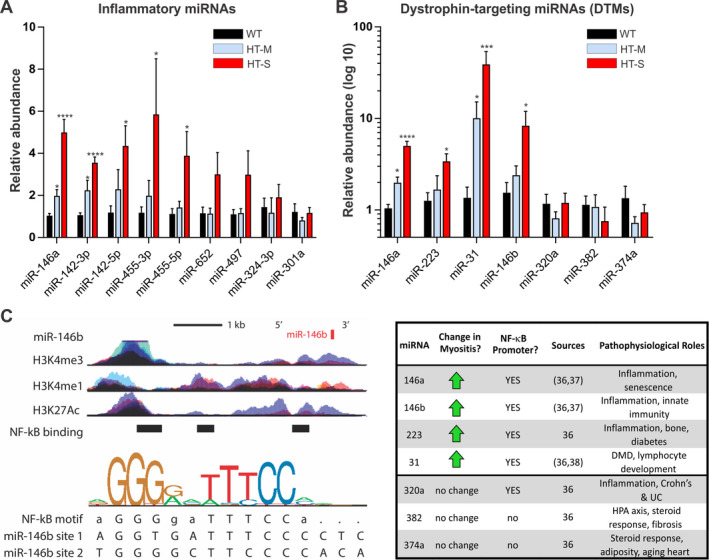
Expression of inflammatory microRNAs (miRNAs) and dystrophin‐targeting miRNAs (DTMs) in the quadriceps muscle of mice classified as having severe or mild myositis disease. A and B, The relative abundance of inflammatory miRNAs (A) and DTMs (B) was analyzed by quantitative reverse transcription–polymerase chain reaction in the quadriceps muscle from each group of mice. Data were normalized to the values for sno202. Results are the mean ± SEM (n = 5 WT, n = 6 HT‐M, and n = 8 HT‐S). * = P < 0.05; *** = P < 0.001; **** = P < 0.0001 versus WT mice, by analysis of variance. C, Transcription factor (NF‐κB) binding sites and histone (H3) modifications that mark regulatory regions were examined using chromatin immunoprecipitation sequencing data from ENCODE. Binding motifs for each transcription factor were identified through the Factorbook repository. Left Top, Schematic diagram of the gene locus for miR‐146b, illustrating the binding sites of 3 neighboring DNA loci bound directly by NF‐κB. Epigenetic modification maps show the location of histone modifications associated with active promoters (H3K4me3) and poised/active enhancers (H3K4me1/H3K27Ac). Left Bottom, Sequence logo pictogram of the base frequency at NF‐κB binding sites with the consensus NF‐κB motif. Two representative NF‐κB binding site sequences near miR‐146b are also shown. Right, Summary of promoter analysis and literature data for all DTMs, indicating each miRNA and known factors associated with its transcriptional regulation. DMD = Duchenne muscular dystrophy; UC = ulcerative colitis; HPA = hypothalamic–pituitary–adrenal axis (see Figure 1 for other definitions).
We next assayed a second defined set of miRNAs (termed DTMs), which we previously reported to be up‐regulated both in patients with DMD and in those with BMD. These miRNAs bind to the dystrophin 3′‐untranslated region (3′‐UTR) to inhibit its translation. Analyses by qRT‐PCR showed that the expression levels of 4 of 7 DTMs (miR‐146a, miR‐223, miR‐31, and miR‐146b) were highly elevated (~3–40‐fold increase, P < 0.05) in the quadriceps muscle of mice with severe myositis compared to WT mice (Figure 3B). We also observed significantly elevated levels of miR‐146a and miR‐31 in the muscle of mice with mild myositis compared to WT mice, but to a lesser extent.
Because we observed elevated TLR‐7 expression in myositis muscles, we also assessed expression of 3 miRNAs known to activate TLR‐7: miR‐21 34, miR‐29a 34, and let‐7 35. In our analyses, miR‐21 was undetectable in all samples assessed, while the expression of miR‐29a and let‐7 showed no significant differences between WT, HT‐M, and HT‐S mouse muscles (see Supplementary Figure 3, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.41215/abstract).
DTMs (miR‐146a and miR‐223) are induced by TNF in dystrophic myotubes, suggesting that they are regulated by NF‐κB 30. To gain insight into DTM regulation in myositis, we performed bioinformatics analyses on chromatin immunoprecipitation sequencing (ChIP‐seq) data at the sites of each miRNA promoter/enhancer region. Bioinformatics analyses showed that all 4 elevated DTMs possessed sites that are directly bound by NF‐κB at consensus elements within their promoter/enhancer regions (Figure 3C) 36. These elements overlapped with corresponding ChIP‐seq data indicating the presence of histone modifications, all of which corresponded to active regions of transcriptional regulation (H3K4me3, H3K27Ac, and H3K4me1). These findings are supported by previous studies in which miR‐146a 37, miR‐146b 37, miR‐223 38, and miR‐31 39 were found to be regulated by NF‐κB. Taken together, these data suggest that both defined sets of miRNAs play a key role in the progression of myositis, and that NF‐κB regulates their expression.
Reduction in dystrophin expression linked to the severity of myositis
Our laboratory has demonstrated that DTMs inhibit the translation of dystrophin, and that DTM levels inversely correlate with the levels of dystrophin in the muscle of patients with BMD and in the exon‐skipping–treated muscle of mdx mice in the model of DMD 30. To determine whether elevated levels of DTMs reduce dystrophin in myositis muscle, we performed dystrophin immunostaining in the quadriceps muscle of WT mice and mice in the HT‐M and HT‐S groups. Dystrophin expression was markedly reduced in the muscle of mice with severe myositis as compared to mice with mild myositis, most notably in areas of inflammation as shown by macrophage (F4/80) staining (Figure 4A; arrowheads indicating macrophage staining alongside myofibers showing reduced dystrophin).
Figure 4.
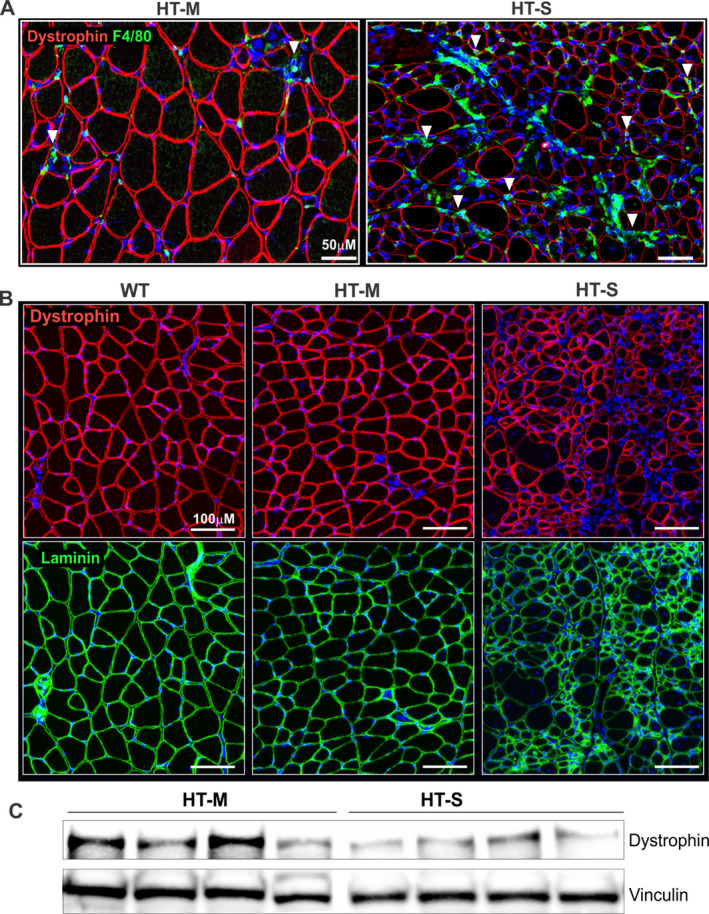
Reduced dystrophin levels in mice with severe myositis. A, Representative images of the quadriceps muscle of mice in the mild or severe myositis groups, immunolabeled with an antibody against dystrophin (red) and macrophage staining (F4/80; green). Arrowheads indicate regions with high macrophage infiltration (F4/80; green) where neighboring myofibers are observed to show diminished dystrophin levels (red). B, Representative images of the quadriceps muscle of mice in the mild or severe myositis groups compared to WT mice, using immunolabeling with an antibody against dystrophin (red). An anti‐laminin antibody (green) was used as a control to show sarcolemnal integrity of the muscle tissue. DAPI counterstaining was used to visualize nuclei (blue). C, Western blotting for dystrophin, using protein extracts from the tibialis anterior muscle of mice in the mild or severe myositis groups (n = 4 per group). Vinculin was used as a loading control and was run on the same Western blot. See Figure 1 for definitions.
We next performed immunostaining for both dystrophin and laminin to better visualize the muscle fibers lacking dystrophin. In the HT‐S group of mice, muscle fibers that lacked dystrophin stained positively for laminin, indicating the presence of intact muscle fibers (Figure 4B). To corroborate the immunofluorescence findings, we also performed Western blotting on muscle tissue extracts from the HT‐M and HT‐S groups of mice. The results similarly showed that dystrophin levels were reduced in muscle from mice with severe myositis (Figure 4C).
To determine the relevance of these findings to human disease, we obtained human muscle biopsy tissue from patients with DM and those with IBM as well as muscle biopsy tissue from healthy control subjects. Analysis by H&E staining revealed different levels of histologic severity in the muscle tissue based on the extent of inflammation and muscle fiber size variability. According to the histologic findings, we partitioned the patients into “histologically mild” or “histologically severe” cohorts. Two biopsy samples from patients with DM and 1 from a patient with IBM were grouped as “histologically mild,” and the remaining 3 biopsy samples (from patients with IBM) were grouped as “histologically severe.” Representative images are shown in Figure 5A.
Figure 5.
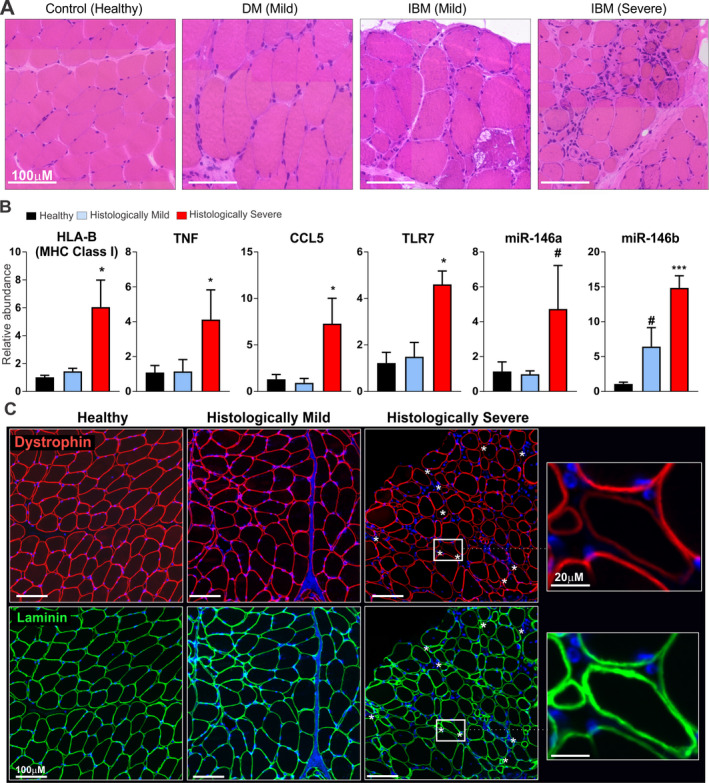
Elevations in major histocompatibility complex (MHC) class I expression, NF‐κB–driven gene expression, and dystrophin‐targeting microRNAs (miRNAs) and reduction in dystrophin levels in histologically severe human muscle biopsy tissue. A, Representative images of hematoxylin and eosin–stained human muscle biopsy tissue obtained from a healthy control, a patient with dermatomyositis (DM) classified as having histologically mild muscle disease, and 2 patients with inclusion body myositis (IBM), of whom 1 was classified as having histologically mild muscle disease and 1 as having histologically severe muscle disease. B, Gene and miRNA expression levels in human muscle biopsy tissue from each group (n = 3 per group). Data for mRNAs were normalized to the values for Hprt, and data for miRNAs were normalized to the values for RNU48. # = P < 0.10; * = P < 0.05; *** = P < 0.001 versus healthy controls, by Student's t‐test. C, Representative images of human muscle biopsy tissue from a healthy control, a patient with histologically mild muscle disease, and a patient with histologically severe muscle disease, using immunolabeling with an antibody against dystrophin (red). An anti‐laminin antibody (green) was used as a control to show sarcolemnal integrity of the muscle tissue. Asterisks in the histologically severe muscle highlight muscle fibers in which laminin staining is uniform, but dystrophin staining is either reduced, discontinuous, or absent. DAPI counterstaining was used to visualize nuclei (blue). TNF = human gene for tumor necrosis factor; TLR7 = human gene for Toll‐like receptor 7.
We next assessed MHC class I (HLA–B) gene expression in human muscle biopsy tissue. Those in the histologically severe group showed a ~6‐fold elevation in MHC class I expression (P < 0.05), whereas muscle tissue from the histologically mild group showed no significant elevation in MHC class I expression above the levels in healthy controls. Muscle with histologically severe disease showed elevated levels of NF‐κB–driven gene expression, with evidence of increased expression of TNF and CCL5 (each P < 0.05 versus healthy controls), whereas muscle with histologically mild disease showed no appreciable up‐regulation in gene expression (Figure 5B). We also observed elevated expression of TLR7 in histologically severe muscle (P < 0.05 versus healthy controls) (Figure 5B), consistent with our observations in the muscle of mice with severe myositis.
We assessed type I IFN gene expression and found no significant differences between the healthy control and histologically mild or histologically severe groups of human muscle biopsy tissue. We did, however, observe elevated expression of type I IFN genes in 1 of 2 muscle biopsy samples from patients with DM (data not shown), consistent with previous studies in which it was shown that this gene expression signature is a hallmark of DM, particularly in muscles that have perifascicular atrophy 15, 17.
We next assessed levels of NF‐κB–regulated DTMs, which were found to be elevated in the human muscle tissue with histologically severe disease. We observed that the expression of both miR‐146a (P < 0.10) and miR‐146b (P < 0.001) was increased in histologically severe muscle biopsy tissue compared to healthy controls, while that from the histologically mild group showed no differences in miR‐146a expression, but did show slightly higher miR‐146b levels in comparison to healthy muscles (P < 0.10) (Figure 5B).
We then performed immunofluorescence analysis of dystrophin expression in the human muscle biopsy samples, using sarcolemmal‐localized laminin as a control for the integrity of the muscle fibers. Whereas staining for dystrophin in the healthy control and histologically mild muscle tissue was uniform, we found areas of reduced and discontinuous dystrophin staining in histologically severe muscle tissue (Figure 5C).
Collectively, our data suggest a mechanism whereby increased TLR‐7 signaling activates inflammation and NF‐κB–driven miRNAs to promote muscle weakness through reduced expression of dystrophin (refer to model in Figure 6). Furthermore, elevated levels of DTMs may be responsible for the reduced and discontinuous dystrophin expression observed in myositis muscle, both in the HT mouse model and in patients with idiopathic inflammatory myopathies.
Figure 6.
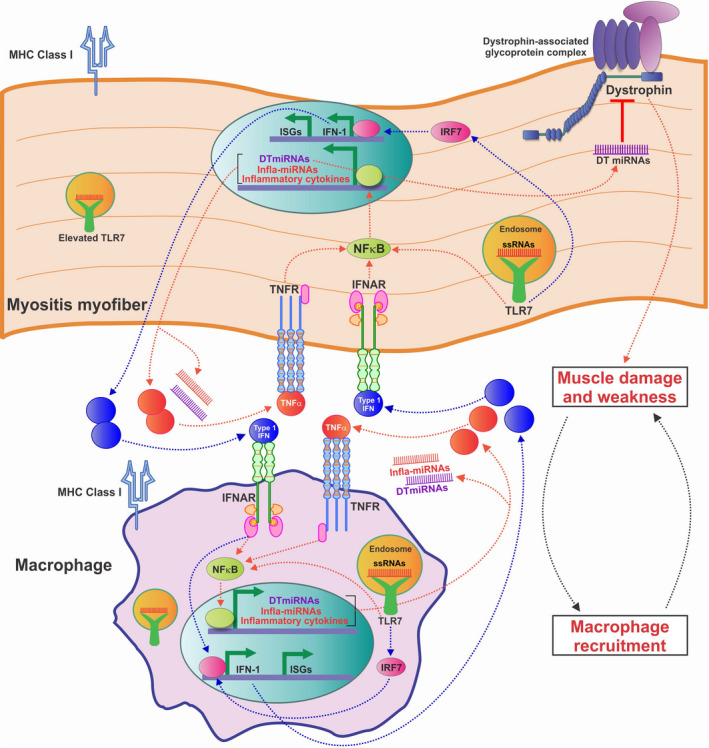
Model of the hypothesized self‐amplifying feedback loop between macrophages and myofibers in myositis. Progression of myositis is driven by myofibers that adopt a more immune‐like state, as evidenced by inappropriate expression of Toll‐like receptor 7 (TLR‐7) and major histocompatibility complex (MHC) class I. TLR‐7 activates NF‐κB–driven gene expression, eliciting the release of inflammatory cytokines and promoting macrophage recruitment. Activation of NF‐κB–driven genes such as tumor necrosis factor (TNF) and interferon‐α/β (IFN) genes leads to a feed–forward loop in which these cytokines activate their receptors (TNFR and IFNAR, respectively), thereby enhancing NF‐κB activation. TLR‐7 additionally activates the IFN regulatory factor 7 gene (IRF7), driving production of IFN‐1, which in turn activates IFNAR and expression of type I IFN–stimulated genes (ISGs), which includes IRF7. NF‐κB also triggers expression of inflammatory and dystrophin‐targeting microRNAs (miRNAs) (infla‐miRNA and DT‐miRNA, respectively), leading to reduction in the levels of dystrophin and muscle weakness, which in turn causes increased release of proinflammatory signals. All of these components contribute to a positive, self‐amplifying inflammatory feedback loop. ssRNAs = single‐stranded RNAs.
DISCUSSION
Herein, we utilized a mouse model of myositis using HT mice with variable disease severity to better understand the molecular and cellular pathways that contribute to myositis. Previous reports have described the common occurrence of a chronic disease progression in these mice, in which reduced locomotor activity is seen as early as 1 month after doxycycline withdrawal, and clinical and histopathologic signs are observable at ~2 months, which thereafter become profound and severe by 4–6 months 3. Due to the sensitivity of our assay, the analyses performed herein in mice at 1–2 months after being taken off doxycycline enabled us to identify early molecular aberrations and additionally to measure weakness in isolated EDL mouse muscles ex vivo. The cohort of HT mice in our study possessed variable rates of disease progression. Thus, because of the progressive nature of the model, we predict that the disease in HT mice with mild myositis will become more severe through time.
We classified mice into 2 distinct cohorts: HT‐M mice with low transgenic expression of muscle‐specific MHC class I, and HT‐S mice with high MHC class I transgene expression. Comparing these 2 groups and age‐matched WT littermates, we found that mice in the HT‐S group developed a more severe disease, as shown by a lower body weight, histologic severity of the disease in the muscle tissue, and reduced muscle strength/weakness, as well as elevated levels of type I IFN and NF‐κB gene signatures. Furthermore, mice in the HT‐S group showed inappropriate expression of endolysosomal‐localized TLR‐7 within myofibers and increased macrophage infiltration as visualized by F4/80 staining. Correspondingly, the muscle of mice in the HT‐S group displayed markedly increased levels of miRNAs from 2 defined sets: an inflammatory set previously described in a DMD mouse model 29, and a dystrophin‐targeting set previously described both in patients with BMD and in exon‐skipping–treated muscle of mdx mice that model DMD 30. All of the miRNAs that showed elevated expression in severe myositis muscle from these sets possessed at least one NF‐κB regulatory element within their promoter or enhancer regions.
Using both immunofluorescence and Western blot analyses, we demonstrated that dystrophin was reduced in the muscle of mice with severe myositis. To corroborate our data in the HT mouse model, we additionally analyzed human muscle biopsy samples from patients with myositis, and found that histologically severe muscles had elevated levels of MHC class I, elevated NF‐κB–driven gene expression, and high levels of miR‐146a and miR‐146b. Human muscle with histologically severe myositis also showed patchy/reduced levels of dystrophin immunostaining. Importantly, the muscle biopsy samples classified as severe were from patients with IBM, whereas the muscle biopsy samples from patients with DM were classified as mild. This is consistent with previous studies in which MHC expression was shown to be more prominent in the muscle of patients with IBM 40. Collectively, our data suggest a model whereby progression of myositis is driven by myofibers adopting a more immune‐like state, as evidenced by the inappropriate expression of TLR‐7 and MHC class I. TLR‐7 activates NF‐κB–driven gene expression, eliciting release of inflammatory cytokines and leading to macrophage recruitment. NF‐κB–driven gene expression also triggers an NF‐κB feed‐forward loop, in which production of inflammatory cytokines, including TNF, further turns on NF‐κB. TLR‐7 activation also leads to production of type I IFN, which, in turn, triggers expression of NF‐κB and type I IFN–stimulated genes. NF‐κB additionally activates dystrophin‐targeting miRNAs, which down‐regulate dystrophin protein levels. Taken together, these findings indicate that this combination of events appears to contribute to a self‐amplifying inflammatory feedback loop of muscle damage and inflammation. As dystrophin is required for maintenance of mechanical stability in the muscle, our observations suggest that reduced dystrophin levels in myositis muscle may contribute to muscle weakness (Figure 6).
Many signaling pathways described herein are similarly overexpressed or overactive in DMD. For instance, in muscles from patients with DMD there is strong activation of multiple components of the innate immune system before the onset of clinical symptoms, including elevated signaling from TLRs (TLR‐4 and TLR‐7), NF‐κB activation, and expression of MHC class I molecules. It is recognized that there is substantial overlap between the histopathologic presentation of myositis and muscular dystrophies, specifically DMD, BMD, and manifesting DMD female carriers. Transcriptional profiling also shows shared pathways between these diseases 41. In a few case studies, DMD female carriers were found to be initially misdiagnosed as having myositis, further illustrating the multitude of confounding factors that blur the boundaries between the 2 diseases 25, 42. In the present study, we describe miRNAs whose levels were elevated in both diseases. These similarities give rationale for the development of therapeutic strategies that could target shared pathways in DMD and myositis.
Recent studies have demonstrated inappropriate expression of TLR‐7 in myofibers from myositis patients 10, 11, and have shown that this increase in expression exacerbates disease 43. Similarly, TLR‐7 levels are elevated in the myofibers of patients with DMD at an early age (<1 year of age) 27. Because of the high TLR‐7 levels observed in DMD muscle, it is postulated that myofiber‐derived RNA molecules may be the most potent of all of the contributing damage‐associated molecular patterns (DAMPs), as they serve as the natural ligand for TLR‐7 44. In support of this, we have previously found that a more common feature of DMD and BMD muscle is miRNA up‐regulation as opposed to miRNA down‐regulation 30. Further, recent studies have suggested that the overabundance of specific miRNAs may trigger activation of TLR‐7 34, 35. Collectively, high levels of miRNAs in muscle disease and the misexpression of TLR‐7 in myofibers seem to be alluding to a mechanism whereby diseased muscle itself is primed to activate a strong inflammatory response.
TLR‐7 is trafficked to the endolysosomal compartment when activated 45. In the present study we observed elevated levels of endosomally localized TLR‐7 in muscle affected with severe myositis. We also observed elevated LAMP‐1 expression, which may be indicative of impaired autophagy 21, 46. It has been reported that autophagy is required for the activation of NF‐κB 47, and that TLR‐7 and its ligands regulate autophagy 48, 49. Thus, these seemingly intertwined pathways make attractive targets for myositis and perhaps for other inflammation‐associated muscle disorders.
MicroRNAs are responsible for fine‐tuning gene expression in all cell and tissue types, including muscle and immune cells. When the balance of miRNAs is shifted in a specific tissue type, gene expression programs are altered, driving tissue from a healthy state to a diseased state. In HT mice with severe myositis, we found significant up‐regulation of 5 of the 9 miRNAs from a predefined “inflammatory” set (miR‐146a, miR‐142‐3p, miR‐142‐5p, miR‐455‐3p, and miR‐455‐5p) and 4 of the 7 miRNAs from a predefined “dystrophin‐targeting” set (miR‐146a, miR‐223, miR‐31, and miR‐146b). We additionally found elevated levels of miR‐146a and miR‐146b in human muscle biopsy tissue with a classification of histologically severe disease. All of these miRNAs are regulated by the inflammatory transcription factor NF‐κB 36, and some, in turn, regulate key components of the NF‐κB signaling pathway. This highlights the importance of NF‐κB signaling in the pathogenesis of myositis. We will discuss the significance of these miRNAs below.
Levels of miR‐146a are highly up‐regulated in mice with severe myositis. Moreover, miR‐146a is induced by NF‐κB in immune cells 37 and is expressed directly in muscle 30. In diseases in which chronic inflammation is present, miR‐146a levels are highly elevated both in the serum and in the tissue affected by disease 50, 51, 52, 53, 54, 55, 56, 57. Similarly, miR‐146b is overexpressed in severe myositis, is induced by TNF/NF‐κB 37, 58, and is elevated in the serum of patients with inflammatory disorders such as inflammatory bowel disease 57. Both miR‐146a and miR‐146b are defined as DTMs, and we have shown that they down‐regulate dystrophin by binding to its 3′‐UTR 30. The miR‐142 family is highly expressed in monocytes 59 and lymphocytes 60. We and others have reported increased miR‐142 levels in DMD as well as in other genetic muscular dystrophies 29, 61. The miR‐142 locus possesses 13 NF‐κB binding sites within its promoter/enhancer region, illustrating the interdependence of its expression and inflammatory signaling. Increased expression of miR‐31 has been reported in DMD patients, BMD patients, and mdx mice 30, 62, 63, 64. In addition, miR‐31 regulates dystrophin expression by targeting its 3′‐UTR 30, 65, and its expression is induced by NF‐κB 39. Collectively, the miRNAs described herein seem to be uniquely primed to shift the balance toward a proinflammatory phenotype as they become increasingly expressed in diseased muscle.
A few early studies noted reduced and discontinuous dystrophin staining in human muscle biopsy tissue from patients with myositis 66, 67. Herein, we investigated dystrophin levels in a mouse model of myositis and in human muscle biopsy tissue after we observed elevated levels of DTMs in HT mice with severe myositis. Immunofluorescence analysis revealed that the quadriceps muscle of mice affected with severe myositis possessed a reduced and mosaic pattern of dystrophin staining that partly resembled the pattern that has been previously observed in the muscles of patients with BMD or the muscles from DMD female carriers. Reduced dystrophin expression was associated, in part, with the extent of inflammation, as shown by macrophage (F4/80) staining. Given that DTMs are regulated by inflammation, it is plausible that DTMs within the muscle microenvironment could dictate the local expression of dystrophin. As disease progresses, both inflammation and dystrophin reduction could “spread” through the muscle, as patches of damaged muscle communicate through DAMPs to healthy areas, in turn activating inflammatory signaling, further reducing dystrophin levels, and causing muscle weakness. Interestingly, reduced levels of dystrophin have been reported in other muscle disorders in which dysregulated NF‐κB signaling and chronic inflammation are present 67, 68, 69. Loss of dystrophin could provide a possible mechanism for muscle weakness in myositis which, to date, is not fully understood. It will be particularly important to study dystrophin deficiency in other muscle disorders, especially those characterized by high inflammation, to determine whether a secondary dystrophin deficiency could contribute to disease.
In this study, we have provided novel insights into the mechanisms driving muscle weakness and disease progression in myositis, using a murine model and human muscle biopsy tissue. Future work will be aimed at determining 1) whether the microRNAs described herein also play a role in other muscle/inflammatory disorders, and 2) whether these miRNAs could serve as therapeutic targets in muscle disease.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Drs. Nagaraju and Fiorillo had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Kinder, Heier, Tully, Fiorillo.
Acquisition of data
Kinder, Heier, Tully, Van der Muelen, Fiorillo.
Analysis and interpretation of data
Kinder, Heier, Tully, Hoffman, Nagaraju, Fiorillo.
ADDITIONAL DISCLOSURE
Author Hoffman is the Treasurer, Secretary, and Vice President of Research of ReveraGen BioPharma as well as the President and CEO of AGADA Biosciences. Author Nagaraju is the President and CEO of ReveraGen BioPharma as well as the Founder and Vice President of AGADA Biosciences.
Supporting information
Supplementary
Supported by the Foundation to Eradicate Duchenne, the Clark Charitable Foundation, the Department of Defense, and the NIH. Dr. Kinder's work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases, NIH (grant F31‐AR‐065362). Dr. Heier's work was supported by the NIH (grants K99‐HL‐130035, R00‐HL‐130035, L40‐AR‐068727, and T32‐AR‐056993). Dr. Nagaraju’s work is supported by grants from the National Institute of Allergy and Infectious Diseases, NIH (1R21AI128248‐01), the National Institute of Neurological Disorders and Stroke, NIH (1R56NS097229‐01), and the Myositis Foundation. Dr. Fiorillo’s work was supported by the Department of Defense (grant W81XWH‐17‐1‐047), the NIH (grants L40‐AR‐070539 and T32‐AR‐056993), the Foundation to Eradicate Duchenne, and the Clark Charitable Foundation. The microscopic analysis for this study, conducted at the Children's Research Institute Light Microscopy and Image Analysis Core, is supported by the Children's Research Institute and Eunice Kennedy Shriver National Institute of Child Health and Human Development, NIH (Intellectual and Developmental Disabilities Research Center award U54‐HD‐090257).
1Travis B. Kinder, PhD (current address: National Center for Advancing Translation Sciences, NIH, Bethesda, Maryland), Christopher R. Heier, PhD, Alyson A. Fiorillo, PhD: George Washington University and Children's National Hospital, Washington, DC; 2Christopher B. Tully, BS, Jack H. Van der Muelen, PhD: Children's National Hospital, Washington, DC; 3Eric P. Hoffman, PhD, Kanneboyina Nagaraju, PhD, DVM: Binghamton University, Binghamton, New York, and ReveraGen BioPharma, Rockville, Maryland.
Dr. Hoffman owns stock or stock options in ReveraGen BioPharma and Agada Biosciences. Dr. Nagaraju owns stock or stock options in ReveraGen BioPharma and Agada Biosciences. Dr. Fiorillo holds a patent for methods and agents to increase therapeutic dystrophin expression in muscle. No other disclosures relevant to this article were reported.
Contributor Information
Kanneboyina Nagaraju, Email: nagaraju@binghamton.edu.
Alyson A. Fiorillo, Email: afiorillo@childrensnational.org.
References
- 1. Meyer A, Meyer N, Schaeffer M, Gottenberg JE, Geny B, Sibilia J. Incidence and prevalence of inflammatory myopathies: a systematic review. Rheumatology (Oxford) 2015;54:50–63. [DOI] [PubMed] [Google Scholar]
- 2. Li CK, Knopp P, Moncrieffe H, Singh B, Shah S, Nagaraju K, et al. Overexpression of MHC class I heavy chain protein in young skeletal muscle leads to severe myositis: implications for juvenile myositis. Am J Pathol 2009;175:1030–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nagaraju K, Casciola‐Rosen L, Rosen A, Thompson C, Loeffler L, Parker T, et al. The inhibition of apoptosis in myositis and in normal muscle cells. J Immunol 2000;164:5459–65. [DOI] [PubMed] [Google Scholar]
- 4. Dalakas MC. Inflammatory muscle diseases. N Engl J Med 2015;373:393–4. [DOI] [PubMed] [Google Scholar]
- 5. Karpati G, Pouliot Y, Carpenter S. Expression of immunoreactive major histocompatibility complex products in human skeletal muscles. Ann Neurol 1988;23:64–72. [DOI] [PubMed] [Google Scholar]
- 6. Li CK, Varsani H, Holton JL, Gao B, Woo P, Wedderburn LR. MHC class I overexpression on muscles in early juvenile dermatomyositis. J Rheumatol 2004;31:605–9. [PubMed] [Google Scholar]
- 7. Coley W, Rayavarapu S, Nagaraju K. Role of non‐immune mechanisms of muscle damage in idiopathic inflammatory myopathies [review]. Arthritis Res Ther 2012;14:209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rayavarapu S, Coley W, Kinder TB, Nagaraju K. Idiopathic inflammatory myopathies: pathogenic mechanisms of muscle weakness [review]. Skelet Muscle 2013;3:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nyberg P, Wikman AL, Nennesmo I, Lundberg I. Increased expression of interleukin 1α and MHC class I in muscle tissue of patients with chronic, inactive polymyositis and dermatomyositis. J Rheumatol 2000;27:940–8. [PubMed] [Google Scholar]
- 10. Tournadre A, Lenief V, Miossec P. Expression of Toll‐like receptor 3 and Toll‐like receptor 7 in muscle is characteristic of inflammatory myopathy and is differentially regulated by Th1 and Th17 cytokines. Arthritis Rheum 2010;62:2144–51. [DOI] [PubMed] [Google Scholar]
- 11. Cappelletti C, Baggi F, Zolezzi F, Biancolini D, Beretta O, Severa M, et al. Type I interferon and Toll‐like receptor expression characterizes inflammatory myopathies. Neurology 2011;76:2079–88. [DOI] [PubMed] [Google Scholar]
- 12. Kawai T, Akira S. The role of pattern‐recognition receptors in innate immunity: update on Toll‐like receptors. Nat Immunol 2010;11:373–84. [DOI] [PubMed] [Google Scholar]
- 13. Greenberg SA. Type 1 interferons and myositis [review]. Arthritis Res Ther 2010;12 Suppl 1:S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wenzel J, Scheler M, Bieber T, Tüting T. Evidence for a role of type I interferons in the pathogenesis of dermatomyositis. Br J Dermatol 2005;153:462–3. [DOI] [PubMed] [Google Scholar]
- 15. Greenberg SA, Pinkus JL, Pinkus GS, Burleson T, Sanoudou D, Tawil R, et al. Interferon‐α/β‐mediated innate immune mechanisms in dermatomyositis. Ann Neurol 2005;57:664–78. [DOI] [PubMed] [Google Scholar]
- 16. De Paepe B. Interferons as components of the complex web of reactions sustaining inflammation in idiopathic inflammatory myopathies. Cytokine 2015;74:81–7. [DOI] [PubMed] [Google Scholar]
- 17. Salajegheh M, Kong SW, Pinkus JL, Walsh RJ, Liao A, Nazareno R, et al. Interferon‐stimulated gene 15 (ISG15) conjugates proteins in dermatomyositis muscle with perifascicular atrophy. Ann Neurol 2010;67:53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Monici MC, Aguennouz M, Mazzeo A, Messina C, Vita G. Activation of nuclear factor‐κB in inflammatory myopathies and Duchenne muscular dystrophy. Neurology 2003;60:993–7. [DOI] [PubMed] [Google Scholar]
- 19. Lundberg I, Ulfgren AK, Nyberg P, Andersson U, Klareskog L. Cytokine production in muscle tissue of patients with idiopathic inflammatory myopathies. Arthritis Rheum 1997;40:865–74. [DOI] [PubMed] [Google Scholar]
- 20. Vattemi G, Engel WK, McFerrin J, Askanas V. Endoplasmic reticulum stress and unfolded protein response in inclusion body myositis muscle. Am J Pathol 2004;164:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alger HM, Raben N, Pistilli E, Francia DL, Rawat R, Getnet D, et al. The role of TRAIL in mediating autophagy in myositis skeletal muscle: a potential nonimmune mechanism of muscle damage. Arthritis Rheum 2011;63:3448–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nagaraju K, Casciola‐Rosen L, Lundberg I, Rawat R, Cutting S, Thapliyal R, et al. Activation of the endoplasmic reticulum stress response in autoimmune myositis: potential role in muscle fiber damage and dysfunction. Arthritis Rheum 2005;52:1824–35. [DOI] [PubMed] [Google Scholar]
- 23. Lundberg I, Brengman JM, Engel AG. Analysis of cytokine expression in muscle in inflammatory myopathies, Duchenne dystrophy, and non‐weak controls. J Neuroimmunol 1995;63:9–16. [DOI] [PubMed] [Google Scholar]
- 24. Nirmalananthan N, Holton JL, Hanna MG. Is it really myositis? A consideration of the differential diagnosis. Curr Opin Rheumatol 2004;16:684–91. [DOI] [PubMed] [Google Scholar]
- 25. Ng KP, Smith CR, Isenberg DA. Muscular dystrophy mimicking refractory idiopathic inflammatory myositis: a trio of cases [letter]. Rheumatology (Oxford) 2007;46:1618–9. [DOI] [PubMed] [Google Scholar]
- 26. Henriques‐Pons A, Yu Q, Rayavarapu S, Cohen TV, Ampong B, Cha HJ, et al. Role of Toll‐like receptors in the pathogenesis of dystrophin‐deficient skeletal and heart muscle. Hum Mol Genet 2014;23:2604–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen YW, Nagaraju K, Bakay M, McIntyre O, Rawat R, Shi R, et al. Early onset of inflammation and later involvement of TGFβ in Duchenne muscular dystrophy. Neurology 2005;65:826–34. [DOI] [PubMed] [Google Scholar]
- 28. Appleyard ST, Dunn MJ, Dubowitz V, Rose ML. Increased expression of HLA ABC class I antigens by muscle fibres in Duchenne muscular dystrophy, inflammatory myopathy, and other neuromuscular disorders. Lancet 1985;1:361–3. [DOI] [PubMed] [Google Scholar]
- 29. Fiorillo AA, Tully CB, Damsker JM, Nagaraju K, Hoffman EP, Heier CR. Muscle miRNAome shows suppression of chronic inflammatory miRNAs with both prednisone and vamorolone. Physiol Genomics 2018;50:735–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fiorillo AA, Heier CR, Novak JS, Tully CB, Brown KJ, Uaesoontrachoon K, et al. TNF‐α‐induced microRNAs control dystrophin expression in Becker muscular dystrophy. Cell Rep 2015;12:1678–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kawai T, Akira S. Signaling to NF‐κB by Toll‐like receptors. Trends Mol Med 2007;13:460–9. [DOI] [PubMed] [Google Scholar]
- 32. Cappelletti C, Salerno F, Canioni E, Mora M, Mantegazza R, Bernasconi P, et al. Up‐regulation of Toll‐like receptors 7 and 9 and its potential implications in the pathogenic mechanisms of LMNA‐related myopathies. Nucleus 2018;9:398–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Petes C, Odoardi N, Gee K. The toll for trafficking: Toll‐like receptor 7 delivery to the endosome [review]. Front Immunol 2017;8:1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fabbri M, Paone A, Calore F, Galli R, Gaudio E, Santhanam R, et al. MicroRNAs bind to Toll‐like receptors to induce prometastatic inflammatory response. Proc Natl Acad Sci U S A 2012;109:E2110–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lehmann SM, Krüger C, Park B, Derkow K, Rosenberger K, Baumgart J, et al. An unconventional role for miRNA: let‐7 activates Toll‐like receptor 7 and causes neurodegeneration. Nat Neurosci 2012;15:827–35. [DOI] [PubMed] [Google Scholar]
- 36. Wang J, Zhuang J, Iyer S, Lin X, Whitfield TW, Greven MC, et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res 2012;22:1798–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF‐κB‐dependent induction of microRNA miR‐146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A 2006;103:12481–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kumar V, Palermo R, Talora C, Campese AF, Checquolo S, Bellavia D, et al. Notch and NF‐kB signaling pathways regulate miR‐223/FBXW7 axis in T‐cell acute lymphoblastic leukemia. Leukemia 2014;28:2324–35. [DOI] [PubMed] [Google Scholar]
- 39. Yan S, Xu Z, Lou F, Zhang L, Ke F, Bai J, et al. NF‐κB‐induced microRNA‐31 promotes epidermal hyperplasia by repressing protein phosphatase 6 in psoriasis. Nat Commun 2015;6:7652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Emslie‐Smith AM, Arahata K, Engel AG. Major histocompatibility complex class I antigen expression, immunolocalization of interferon subtypes, and T cell‐mediated cytotoxicity in myopathies. Hum Pathol 1989;20:224–31. [DOI] [PubMed] [Google Scholar]
- 41. Hoffman EP, Rao D, Pachman LM. Clarifying the boundaries between the inflammatory and dystrophic myopathies: insights from molecular diagnostics and microarrays. Rheum Dis Clin North Am 2002;28:743–57. [DOI] [PubMed] [Google Scholar]
- 42. Yoon J, Kim SH, Ki CS, Kwon MJ, Lim MJ, Kwon SR, et al. Carrier woman of Duchenne muscular dystrophy mimicking inflammatory myositis. J Korean Med Sci 2011;26:587–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sciorati C, Monno A, Doglio MG, Rigamonti E, Ascherman DP, Manfredi AA, et al. Exacerbation of murine experimental autoimmune myositis by Toll‐like receptor 7/8. Arthritis Rheumatol 2018;70:1276–87. [DOI] [PubMed] [Google Scholar]
- 44. Rosenberg AS, Puig M, Nagaraju K, Hoffman EP, Villalta SA, Rao VA, et al. Immune‐mediated pathology in Duchenne muscular dystrophy [review]. Sci Transl Med 2015;7:299rv4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sasai M, Linehan MM, Iwasaki A. Bifurcation of Toll‐like receptor 9 signaling by adaptor protein 3. Science 2010;329:1530–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nogalska A, D'Agostino C, Terracciano C, Engel WK, Askanas V. Impaired autophagy in sporadic inclusion‐body myositis and in endoplasmic reticulum stress‐provoked cultured human muscle fibers. Am J Pathol 2010;177:1377–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Criollo A, Chereau F, Malik SA, Niso‐Santano M, Mariño G, Galluzzi L, et al. Autophagy is required for the activation of NFκB. Cell Cycle 2012;11:194–9. [DOI] [PubMed] [Google Scholar]
- 48. Delgado MA, Deretic V. Toll‐like receptors in control of immunological autophagy. Cell Death Differ 2009;16:976–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll‐like receptors control autophagy. EMBO J 2008;27:1110–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Eisenberg I, Eran A, Nishino I, Moggio M, Lamperti C, Amato AA, et al. Distinctive patterns of microRNA expression in primary muscular disorders. Proc Natl Acad Sci U S A 2007;104:17016–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Panguluri SK, Bhatnagar S, Kumar A, McCarthy JJ, Srivastava AK, Cooper NG, et al. Genomic profiling of messenger RNAs and microRNAs reveals potential mechanisms of TWEAK‐induced skeletal muscle wasting in mice. PloS One 2010;5:e8760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Olivieri F, Lazzarini R, Recchioni R, Marcheselli F, Rippo MR, Di Nuzzo S, et al. MiR‐146a as marker of senescence‐associated pro‐inflammatory status in cells involved in vascular remodelling. Age (Dordr) 2013;35:1157–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lu J, Yan M, Wang Y, Zhang J, Yang H, Tian FF, et al. Altered expression of miR‐146a in myasthenia gravis. Neurosci Lett 2013;555:85–90. [DOI] [PubMed] [Google Scholar]
- 54. Vasa‐Nicotera M, Chen H, Tucci P, Yang AL, Saintigny G, Menghini R, et al. MiR‐146a is modulated in human endothelial cell with aging. Atherosclerosis 2011;217:326–30. [DOI] [PubMed] [Google Scholar]
- 55. Lukiw WJ. Micro‐RNA speciation in fetal, adult and Alzheimer's disease hippocampus. Neuroreport 2007;18:297–300. [DOI] [PubMed] [Google Scholar]
- 56. Ma X, Zhou J, Zhong Y, Jiang L, Mu P, Li Y, et al. Expression, regulation and function of microRNAs in multiple sclerosis. Int J Med Sci 2014;11:810–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Heier CR, Fiorillo AA, Chaisson E, Gordish‐Dressman H, Hathout Y, Damsker JM, et al. Identification of pathway‐specific serum biomarkers of response to glucocorticoid and infliximab treatment in children with inflammatory bowel disease. Clin Transl Gastroenterol 2016;7:e192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shi C, Zhu L, Chen X, Gu N, Chen L, Zhu L, et al. IL‐6 and TNF‐α induced obesity‐related inflammatory response through transcriptional regulation of miR‐146b. J Interferon Cytokine Res 2014;34:342–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fordham JB, Naqvi AR, Nares S. Regulation of miR‐24, miR‐30b, and miR‐142‐3p during macrophage and dendritic cell differentiation potentiates innate immunity. J Leukoc Biol 2015;98:195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kramer NJ, Wang WL, Reyes EY, Kumar B, Chen CC, Ramakrishna C, et al. Altered lymphopoiesis and immunodeficiency in miR‐142 null mice. Blood 2015;125:3720–30. [DOI] [PubMed] [Google Scholar]
- 61. Israeli D, Poupiot J, Amor F, Charton K, Lostal W, Jeanson‐Leh L, et al. Circulating miRNAs are generic and versatile therapeutic monitoring biomarkers in muscular dystrophies. Sci Rep 2016;6:28097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Greco S, De Simone M, Colussi C, Zaccagnini G, Fasanaro P, Pescatori M, et al. Common micro‐RNA signature in skeletal muscle damage and regeneration induced by Duchenne muscular dystrophy and acute ischemia. FASEB J 2009;23:3335–46. [DOI] [PubMed] [Google Scholar]
- 63. Roberts TC, Blomberg KE, McClorey G, El Andaloussi S, Godfrey C, Betts C, et al. Expression analysis in multiple muscle groups and serum reveals complexity in the microRNA transcriptome of the mdx mouse with implications for therapy. Mol Ther Nucleic Acids 2012;1:e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Roberts TC, Coenen‐Stass AM, Wood MJ. Assessment of RT‐qPCR normalization strategies for accurate quantification of extracellular microRNAs in murine serum. PloS One 2014;9:e89237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cacchiarelli D, Incitti T, Martone J, Cesana M, Cazzella V, Santini T, et al. MiR‐31 modulates dystrophin expression: new implications for Duchenne muscular dystrophy therapy. EMBO Rep 2011;12:136–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sewry CA, Clerk A, Heckmatt JZ, Vyse T, Dubowitz V, Strong PN. Dystrophin abnormalities in polymyositis and dermatomyositis. Neuromuscul Disord 1991;1:333–9. [DOI] [PubMed] [Google Scholar]
- 67. Voit T, Stuettgen P, Cremer M, Goebel HH. Dystrophin as a diagnostic marker in Duchenne and Becker muscular dystrophy: correlation of immunofluorescence and western blot. Neuropediatrics 1991;22:152–62. [DOI] [PubMed] [Google Scholar]
- 68. Acharyya S, Butchbach ME, Sahenk Z, Wang H, Saji M, Carathers M, et al. Dystrophin glycoprotein complex dysfunction: a regulatory link between muscular dystrophy and cancer cachexia. Cancer Cell 2005;8:421–32. [DOI] [PubMed] [Google Scholar]
- 69. Vainzof M, Passos‐Bueno MR, Canovas M, Moreira ES, Pavanello RC, Marie SK, et al. The sarcoglycan complex in the six autosomal recessive limb‐girdle muscular dystrophies. Hum Mol Genet 1996;5:1963–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary