

Abstract
Objectives
ABP 959 is a proposed biosimilar to eculizumab, a monoclonal antibody targeting the human C5 complement protein. The objective of this randomized, double‐blind, three‐arm, study was to demonstrate pharmacokinetic (PK) and pharmacodynamic (PD) similarity of ABP 959 relative to the eculizumab reference product (RP) in healthy adult male subjects.
Methods
Eligible subjects aged 18‐45 years were randomized to receive a 300‐mg IV infusion of ABP 959, or FDA‐licensed eculizumab (eculizumab US), or EU‐authorized eculizumab (eculizumab EU). Primary PK endpoint was area under the total serum concentration‐time curve from 0 to infinity (AUC0−∞); primary PD endpoint was area between the effect curve (ABEC) of CH50‐time data.
Results
The geometric mean of PK and PD parameters were similar between ABP 959 versus eculizumab US and eculizumab EU; PK and PD similarity was established based on 90% confidence intervals of the geometric mean ratio being within prespecified equivalence margin of 0.8 and 1.25. The incidence of treatment‐emergent adverse events was similar across groups. The incidence of binding anti‐drug antibodies was similar across treatments; no subjects developed neutralizing antibodies.
Conclusions
This study demonstrated PK and PD similarity of ABP 959 to eculizumab RP; safety and immunogenicity profiles were also similar.
Keywords: ABP 959, biosimilar, eculizumab, paroxysmal nocturnal hemoglobinuria
1. INTRODUCTION
ABP 959 is being developed as a biosimilar to eculizumab (Soliris®, Alexion), a recombinant humanized monoclonal immunoglobulin G2/4κ antibody that binds to the human C5 complement protein (C5). Eculizumab is approved for use in patients with paroxysmal nocturnal hemoglobinuria (PNH) to reduce hemolysis, in patients with atypical hemolytic uremic syndrome to inhibit complement‐mediated thrombotic microangiopathy, in adult patients with generalized myasthenia gravis who are anti‐acetylcholine receptor antibody positive, and in adult patients with neuromyelitis optica spectrum disorder who are anti‐aquaporin‐4 antibody positive. 1 , 2
Eculizumab is a terminal complement inhibitor. The primary mechanism of action of eculizumab is binding to C5 and preventing its cleavage into C5b, an essential component in the formation of the membrane attack complex that is the final effector pathway of complement activation. 3 By binding to C5, eculizumab inhibits the deployment of the terminal complement cascade including the formation of a membrane attack complex. In PNH, eculizumab blocks terminal complement‐mediated intravascular hemolysis. 1 , 2 , 3 Two multicenter phase 3 clinical studies in PNH, that is, the placebo‐controlled TRIUMPH study and the companion open‐label 52‐week SHEPHERD study, have demonstrated that terminal complement inhibition with eculizumab reduces intravascular reduction in hemolysis and leads to a reduction or elimination of the need for transfusion and clinical improvement of anemia and other PNH‐associated symptoms such as fatigue, pain, and difficulty in functioning. 1 , 2 , 4 , 5 In atypical hemolytic uremic syndrome, eculizumab treatment in the pivotal clinical trials resulted in a rapid and sustained reduction in complement‐mediated thrombotic microangiopathy. 2 , 6 , 7 , 8
Per regulatory definition, a biosimilar product is a highly similar entity to a licensed biologic that shows no clinically meaningful differences when compared to the originator reference product (RP) in terms of structure, purity, pharmacokinetics (PK), pharmacodynamics (PD), mechanism of action, potency, safety, and immunogenicity. 9 , 10 , 11 , 12 , 13 , 14 The regulatory pathway for biosimilar approval is rigorous and systematic, recommending a comparative stepwise totality of evidence approach to demonstrate similarity between the proposed biosimilar and the originator biologic. The foundation for the demonstration of biosimilarity is a comprehensive comparative analytical (structural and functional) characterization, followed by preclinical assessments, clinical PK and PD evaluations, and finally a confirmatory clinical trial to assess efficacy, safety, and immunogenicity in a representative indication using a sensitive population and sensitive endpoints. 9 , 10 , 11 , 12 , 13 , 14
The proposed biosimilar ABP 959 has the same pharmaceutical form and dosage strength as eculizumab, and evidence to date from an ongoing analytical program suggests that ABP 959 is analytically similar to eculizumab. The comparative analytical data indicate that ABP 959 is similar to eculizumab RP with respect to primary and higher‐order structure, product‐related substances and impurities, size variants, and biological activity (binding to C5 and FcRn). 15
The primary objectives of this study were to demonstrate PK equivalence and PD similarity of ABP 959 with eculizumab RP in healthy adult male subjects. The secondary objectives of the study were to determine the safety, tolerability, and immunogenicity of ABP 959 in healthy male subjects compared with eculizumab US and eculizumab EU. Here, we report the results of this study.
Eculizumab RP used in the present study was sourced from both the United States (US), the Food and Drug Administration (FDA)‐licensed eculizumab (eculizumab US) and the European Union (EU), the EU‐authorized eculizumab (eculizumab EU). This was done to comply with regulatory guidelines that define RP as that approved in the local jurisdiction (ie, the US and EU), thus mandating comparison of the proposed biosimilar to the locally sourced originator, that is, FDA‐licensed eculizumab and EU‐authorized eculizumab. Establishment of PK/PD equivalence between the two locally sourced RPs (ie, eculizumab US and eculizumab EU), along with previously established analytical similarity would complete the scientific bridge that would provide the rationale for using a single‐sourced eculizumab RP in future comparative clinical studies.
2. MATERIALS AND METHODS
2.1. Subjects
Eligible male subjects, ≥18 and ≤45 years of age at the time of screening, must have had a body weight of ≥50.0 and ≤90.0 kg, and negative urine drug and alcohol screens at screening and Day −1. All subjects were required to have a normal or clinically acceptable physical examination, clinical laboratory test values, urinalysis values, vital signs, ECGs (12‐lead ECG reporting heart rate and RR, PR, QRS, QT, and QTc intervals), and body weight, as determined by the investigator, at all predose assessments.
Subjects were excluded if they had known or suspected hereditary complement deficiency or had prior exposure to eculizumab or related compounds (ie, a monoclonal antibody that specifically binds to the complement protein C5), known or suspected sensitivity to products derived from mammalian cell lines or were receiving or had received any investigational drug within the 30 days or five half‐lives prior to receiving investigational product. Subjects with hypertension (defined as a systolic blood pressure >140 mm Hg and/or a diastolic blood pressure >90 mm Hg confirmed by a single repeat measurement that same day) or a history of hypertension requiring intervention, proteinuria (with a urine dipstick value of 2+ or above), or coagulation abnormalities at screening or check‐in were excluded. Also excluded were subjects with the presence or suspicion of active bacterial infection or a history of meningococcal infection or conditions known to interfere with the distribution, metabolism, or excretion of the test drugs. Patients with a positive test for human immunodeficiency virus (HIV) antibodies, hepatitis B surface antigen, or hepatitis C virus antibodies at screening were also excluded. Other exclusion criteria included men of reproductive potential who were unwilling to practice a highly effective method of birth control (such as sexual abstinence and vasectomy), had pregnant partners, or were unwilling to refrain from donating sperm during the study and for 6 months following treatment with study drug.
2.2. Study design
This was a randomized, double‐blind, single‐dose, three‐arm, parallel group study in healthy adult male subjects (Figure S1). The study was conducted at clinical pharmacology units (CPUs) in Australia.
Subjects were randomized in a ratio of 1:1:1 to receive a single intravenous (IV) dose (over a 35 ± 5‐min infusion) of 300 mg investigational product, that is, ABP 959, eculizumab US, or eculizumab EU, on Day 1.
Screening occurred within 28 days of dosing (−28 to −2 days). Eligible subjects were admitted to the CPU on Day −1, at which time the Day −1 assessments were performed. Results from both screening and Day −1 assessments were reviewed to confirm eligibility. Study drug administration occurred on Day 1. Subjects remained resident in the CPU for at least 24 hours after dosing for safety, PK, and PD assessments. They were discharged on Day 2 after study procedures were completed. Subjects returned to the CPU on Days 3, 5, 8, 11, 15, 22, 29, 36, 43, 50, and 57 (end of study [EOS] visit) for safety evaluations, PK, and PD assessments.
No concomitant medications (prescription, over‐the‐counter, or herbal) were allowed on the study, with the exception of acetaminophen (up to 2 g per day and not more than 4 g per week) unless these were prescribed by the investigator for treatment of specific clinical events. Subjects who were unable to show documentation of prior vaccination per local requirements were immunized with a Menactra® meningococcal vaccine ≥14 days prior to administering the study drug.
A Safety Data Review Team that included unblinded medical monitor(s) independently reviewed safety data through Day 2, at a minimum, from the initial 12 enrolled subjects. Blinded study data were monitored on an ongoing basis by the blinded clinical study team to ensure subjects’ safety.
2.3. Sample collection
Blood samples for PK analysis of serum eculizumab or ABP 959 concentrations were collected Day 1 (predose and postdose [35 minutes, 4, 8, 12 hours]), at each return visit (Days 2, 3, 5, 8, 11, 15, 22, 29, 36, 43, and 50), and at EOS (Day 57).
Blood samples for assessment of 50% total hemolytic complement activity (CH50), a PD marker, were collected predose (ie, Day −1 and Day 1), postdose (ie, 35 minutes, 4, 8, 12 hours), at each return visit to the CPU (Days 2, 3, 5, 8, 11, 15, 22, 29, 36, 43, and 50), and at EOS (Day 57).
Subjects were monitored throughout the study for adverse events (AEs); clinical laboratory tests, vital signs measurements, 12‐lead ECGs, and physical examinations were performed at specified time points. Blood samples for assessment of anti‐drug antibodies (ADA) were collected predose on Day 1 and on Day 11, Day 29, and at EOS (Day 57).
2.4. PK/PD methods
Serum concentrations of ABP 959 and eculizumab RP were determined using a single validated electrochemiluminescent assay on the same blood sample collected; the assay range was 0.5 to 160 µg/mL. Serum concentrations of CH50 total complement were determined by liposome immunoassay using the Wako Autokit CH50 on the Abbott ARCHITECT System; the analytical measuring range was 10 to 60 U/mL.
2.5. Immunogenicity methods
A single validated electrochemiluminescence bridging immunoassay using biotinylated and ruthenylated ABP 959 was used to detect and confirm binding antibodies in serum from patients administered ABP 959 or eculizumab RP. The assay sensitivity for binding ADAs was ~5 ng/mL with a drug tolerance of approximately 500 ng/mL of ADAs in the presence of 300 μg/mL drug. Samples positive for binding ADAs were tested in a single validated target binding assay using ABP 959 to detect neutralizing antibodies. The target binding assay sensitivity was 578 ng/mL with a drug tolerance of 5 μg/mL of ADA in the presence of 1 μg/mL drug. The magnitude of the positive binding and neutralizing antibody samples were reported as signal‐to‐noise (S/N) ratio and titer, respectively.
2.6. PK and PD endpoints
The primary PK endpoint was area under the serum concentration‐time curve from time 0 extrapolated to infinity (AUC0−∞). Secondary PK endpoints were AUC from time 0 to the time of the last observed quantifiable concentration (AUClast), the maximum observed serum concentration (C max), time of C max (t max), and terminal elimination half‐life (t 1/2).
The primary PD endpoint was area between the effect curve (ABEC) of CH50‐time data.
2.7. Safety endpoints
Safety endpoints included treatment‐emergent AEs (TEAEs), serious AEs (SAEs), findings of laboratory values, vital signs, physical examination, 12‐lead ECGs, and incidence of ADAs. Events of interest (EOIs; defined as noteworthy events for a particular product or class of products that the sponsor wished to monitor carefully) included infections, infusion reactions, meningococcal infection, sepsis, and hematologic abnormalities.
2.8. Statistical analysis
Approximately 210 healthy adult male subjects were to be enrolled in this study based on an assumed between‐subject coefficient of variation (CV) of 40% for AUC0−∞ and ABEC of CH50 with blinded assessments of the actual between‐subject CV to confirm the final sample size.
The PK parameters (AUC0−∞, AUClast, and C max) were loge‐transformed prior to modeling. For all comparisons, the point estimates and confidence interval (CI) for the mean difference in algorithmic PK parameters (AUC0−∞, AUClast, and C max) were estimated from an analysis of covariance model, using the PK parameter analysis set. The primary analysis population was the PK parameter analysis set, which consisted of all subjects with an evaluable eculizumab or ABP 959 serum concentration‐time profile. To establish PK similarity, the 90% CI for the geometric mean ratio (GMR) of test (ABP 959) and reference (eculizumab US or eculizumab EU) for AUC0−∞ was to fall within the (0.8, 1.25) equivalence margin. Pharmacokinetic variables were calculated from the serum concentration data using non‐compartmental methods (Phoenix WinNonlin®, Version 6.3, Pharsight Corp) and actual sampling times. Sensitivity analyses were performed for the PK parameters for the binding ADA‐negative subjects and neutralizing ADA‐negative subjects in the PK parameter population.
All CH50 analyses were performed using the PD population, which consisted of all subjects who received any amount of study drug and who had at least one reported CH50 value. If PK similarity between eculizumab EU and eculizumab US was established, eculizumab EU and eculizumab US arms were combined into a single eculizumab RP arm for the comparison of ABEC of CH50 between ABP 959 and RP. PD similarity was established if the 90% CI for GMR between ABP 959 and RP (eculizumab US and eculizumab EU, or the pooled eculizumab arm) fell within the bioequivalence criteria of 0.80 and 1.25. ABEC of CH50 were calculated from CH50 data using non‐compartmental methods (Phoenix WinNonlin®, Version 6.3, Pharsight Corp) and actual sampling times. In addition, percent reduction from baseline in CH50 was summarized descriptively by treatment and nominal time point. The mean percent reduction from individual baseline of CH50‐time data was also presented graphically by treatment.
Subject incidences of AEs, grade ≥ 3 AEs, fatal AEs, SAEs, AEs leading to discontinuation from investigational product or discontinuation from study, and incidence of ADAs were summarized using descriptive statistics. Findings of safety laboratory parameters and vital sign measurements were descriptively summarized.
3. RESULTS
3.1. Subject disposition and characterization
A total of 219 healthy adult male subjects were randomized (ABP 959, n = 71; eculizumab US, n = 74; eculizumab EU, n = 74). Of these, two subjects discontinued before investigational product dosing (Figure S2). Of the 217 who completed the infusion, 207 subjects completed the study (5 subjects withdrew consent and 5 subjects were lost to follow‐up [ABP 959, n = 3; eculizumab US, n = 5; eculizumab EU, n = 2]).
Demographics and baseline characteristics were well‐balanced between treatment groups (Table 1). In all randomized subjects, the majority of patients (88.5%) were not Hispanic or Latino; the mean age was 26.9 years (standard deviation: 6.10; range, 18‐45), and the mean body mass index was 23.33 kg/m2 (standard deviation: 2.38; range, 18.0‐29.1).
Table 1.
Demographics and baseline characterizations
|
ABP 959 (N = 71) |
Eculizumab US (N = 72) |
Eculizumab EU (N = 74) |
|
|---|---|---|---|
| Age, years | |||
| Mean (SD) | 27.0 (6.35) | 26.7 (5.52) | 26.9 (6.47) |
| Race, n (%) | |||
| Asian (first‐generation Japanese) | 4 (5.6) | 4 (5.6) | 4 (5.4) |
| Asian (other) | 11 (15.5) | 15 (20.8) | 19 (25.7) |
| Black or African American | 1 (1.4) | 3 (4.2) | 3 (4.1) |
| White | 51 (71.8) | 47 (65.3) | 45 (60.8) |
| Other | 4 (5.6) | 3 (4.2) | 3 (4.1) |
| Sex, n (%) | |||
| Male | 71 (100.0) | 72 (100.0) | 74 (100.0) |
| Weight (kg), Mean (SD) | 74.74 (8.93) | 71.67 (9.04) | 71.76 (7.98) |
| Height (cm), Mean (SD) | 177.3 (6.90) | 176.0 (5.99) | 176.1 (7.45) |
| Body mass index (kg/m2), Mean (SD) | 23.75 (2.33) | 23.12 (2.57) | 23.13 (2.23) |
Abbreviation: SD, standard deviation.
Overall, 49 (22.6%) subjects took a prestudy medication, and 78 (35.9%) subjects took a concomitant medication.
3.2. Pharmacokinetics
The mean concentration‐time profiles following a single IV infusion for all three treatment groups were similar and overlapped over the entire course of sampling in the overall patient population (Figure 1A).
Figure 1.
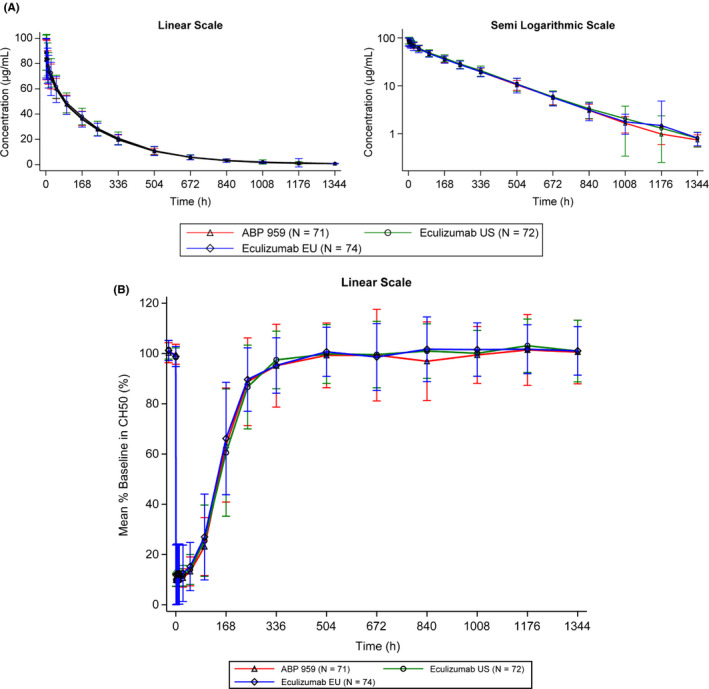
A, Mean (+/‐SD) Serum ABP 959 and Eculizumab RP Serum Total Concentration‐Time Profile. B, Mean (+/‐SD) Percent Baseline in CH50
Table 2 presents the results of the primary (AUC0−∞) and secondary (AUClast and C max) PK endpoints. The adjusted least squares GM of primary and secondary PK parameters (AUC0−∞, AUClast, and C max) following a single IV infusion of investigational product were similar across the three treatment groups. For comparisons between ABP 959 and eculizumab US and between ABP 959 and eculizumab EU, the 90% CIs of the ratios of the GMs were contained within the prespecified equivalence margin of 0.8 and 1.25 for all PK parameters (AUC0−∞, AUClast, and C max), thus establishing PK similarity. By the same criteria, PK similarity was also established between eculizumab US and eculizumab EU.
Table 2.
Ratio of least squares geometrical means of PK and PD parameters for ABP 959, eculizumab US, and eculizumab EU
|
Treatment and comparison |
AUC0→∞ (h•μg/mL) |
AUClast (h•μg/mL) |
C max (μg/mL) |
ABEC of CH50 (%*h) |
|---|---|---|---|---|
| Least squares GM [n] | ||||
| ABP 959 | 18 996.3 [70] | 18 760.1 [70] | 86.11 [71] | 17 724.5 [70] |
| Eculizumab US | 19 777.5 [68] | 19 522.9 [70] | 90.73 [72] | 16 549.4 [70] |
| Eculizumab EU | 18 921.1 [71] | 18 768.2 [72] | 85.85 [74] | 16 361.1 [73] |
| Ratio of least squares GM (90% CI) | ||||
|
ABP 959 vs Eculizumab US |
0.9588 (0.9129, 1.0070) | 0.9609 (0.9154, 1.0087) | 0.9489 (0.9096, 0.9899) | 1.0710 (0.9634, 1.1906) |
|
ABP 959 vs Eculizumab EU |
1.0022 (0.9547, 1.0520) | 0.9995 (0.9525, 1.0488) | 1.0025 (0.9613, 1.0455) | 1.0824 (0.9747, 1.2020) |
| Eculizumab US vs Eculizumab EU | 1.0453 (0.9954, 1.0976) | 1.0401 (0.9912, 1.0914) | 1.0564 (1.0131, 1.1016) | 1.0106 (0.9101, 1.1223)* |
Abbreviations: ABEC, area between the effect curve; AUC0→∞, area under the total serum concentration‐time curve (AUC) from time 0 extrapolated to infinity; AUClast, AUC from time 0 to the time of the last observed quantifiable concentration; CH50, 50% total hemolytic complement activity; CI, confidence interval; C max, maximum observed concentration; GM, geometric mean; n, number.
ABEC of CH50, ABP 959 vs. (Eculizumab US + Eculizumab EU) = 1.0768 [0.9830, 1.1796].
In addition, other PK parameters such as t max and t 1/2 were also similar across the three groups (Table 3). Across the three treatments, in the majority of the subjects, the time at which the peak total concentration was observed coincided with the end of infusion sample (35 minutes) or within 4 hours postdose. The terminal t 1/2 was estimated to be, on average, 8 days.
Table 3.
PK results: Other PK endpoints
| Treatment |
t max (h) Median [n] (Min‐Max) |
t 1/2 (h) Mean [n] (SD) |
|---|---|---|
| ABP 959 |
0.60 [71] (0.58‐8.00) |
189 [70] (18.8) |
| Eculizumab US |
0.62 [72] (0.58‐4.05) |
193 [68] (21.4) |
| Eculizumab EU |
0.58 [74] (0.58‐95.92) |
189 [71] (22.4) |
Abbreviations: Max, maximum; Min, minimum; n, number; SD, standard deviation; t 1/2, terminal elimination half‐life; t max, time of maximal concentration.
3.3. Pharmacodynamics
In the PD population, following a single IV infusion, the mean percent baseline in CH50 time profiles were similar and overlapped over the entire course of sampling for all three treatment groups (Figure 1B). Because the PK analysis demonstrated PK similarity between eculizumab US and eculizumab EU, eculizumab US and eculizumab EU were combined for the PD analysis per protocol specification.
For all comparisons (ABP 959 versus eculizumab US, ABP 959 versus eculizumab EU, and ABP 959 versus combined eculizumab RP), the 90% CI for the GMR were within the bioequivalence criteria of 0.80 to 1.25 (Table 2). These results confirmed PD similarity between ABP 959 and eculizumab RP.
3.4. Safety
The safety analysis was conducted on all 217 subjects that received investigational product (Table 4). The frequency of TEAEs was similar among groups; the most common TEAEs were headache, upper respiratory tract infection, back pain, rhinitis, abdominal pain, catheter site pain, oropharyngeal pain, and rhinorrhea. The majority of events were grade 1 or 2; there were no grade 4 or 5 events. A total of three subjects experienced six ≥grade 3 AEs; one subject each in the ABP 959 and eculizumab EU groups experienced one event, and one subject in the eculizumab US group experienced four events. The grade 3 events were viral infection, epistaxis, facial bones fractures, and headache; only headache was considered by investigators to be probably related to eculizumab EU. There were five subjects with eight SAEs, including viral infection, soft tissue injury, epistaxis, facial bone fracture, pericarditis, and headache; none except headache was considered probably related to study drug per investigators. The etiology of the grade 3 viral infection in the subject treated with ABP 959 could not be determined but was negative for Streptococcus pneumoniae. The subject was hospitalized with headache, tachycardia, elevated temperature, and decreased blood pressure; the symptoms resolved with treatment and subject was discharged in 1 day. In the subject with recurrent pericarditis (grade 2) treated with eculizumab EU, it is notable that the subject had a prior history of pericarditis, which was unknown at study enrollment and would have been exclusionary if known; the event was considered by investigators not to be related to study drug. No AEs led to investigational product or study discontinuation.
Table 4.
Overall safety results and TEAEs reported in ≥5% in any treatment group
| MedDRA preferred term | Number (%) of subjects † | ||
|---|---|---|---|
|
ABP 959 (N = 71) |
Eculizumab US (N = 72) |
Eculizumab EU (N = 74) |
|
| Any TEAE | 54 (76.1) | 46 (63.9) | 51 (68.9) |
| Any ≥ 3 Grade TEAE | 1 (1.4) | 1 (1.4) | 1 (1.4) ‡ |
| Any SAE | 1 (1.4) | 2 (2.8) | 2 (2.7) |
| Any EOI | 23 (32.4) | 25 (34.7) | 33 (44.6) |
| Infections | 20 (28.2) | 19 (26.4) | 29 (39.2) |
| Infusion reactions | 4 (5.6) | 8 (11.1) | 6 (8.1) |
| Infusion reactions with onset day coinciding on the day of study drug infusion or the day after infusion | 3 (4.2) | 0 (0.0) | 1 (1.4) |
| TEAEs Reported in ≥5% | |||
| Headache | 19 (26.8) | 18 (25.0) | 17 (23.0) |
| Upper respiratory tract infection | 14 (19.7) | 13 (18.1) | 22 (29.7) |
| Back pain | 4 (5.6) | 1 (1.4) | 6 (8.1) |
| Rhinitis | 5 (7.0) | 3 (4.2) | 3 (4.1) |
| Abdominal pain | 5 (7.0) | 1 (1.4) | 3 (4.1) |
| Catheter site pain | 4 (5.6) | 0 (0.0) | 4 (5.4) |
| Oropharyngeal pain | 2 (2.8) | 0 (0.0) | 4 (5.4) |
| Rhinorrhea | 1 (1.4) | 4 (5.6) | 1 (1.4) |
Abbreviations: EOI, event of interest; SAE, serious adverse event; TEAE, treatment‐emergent adverse event.
Subjects with multiple events in the same category were counted only once in that category. Subjects with events in more than 1 category were counted once in each of those categories.
1 subject experienced 4 events.
Events of interest including infections and infusion reactions were reported in 23 subjects (30 events) in the ABP 959 group, 25 subjects (30 events) in the eculizumab US group, and 33 subjects (43 events) in the eculizumab EU group (Table 4). Overall, infections occurred in 68 subjects (83 events) and infusion reactions in 18 subjects (20 events). Grade 1 and grade 2 infections (≥5 events) included upper respiratory tract infection, rhinitis, and pharyngitis; grade 3 event was viral infection. Infusion reactions coinciding with study drug infusion or the day after study drug infusion (n = 4) were grade 1 or 2 events of cough, pyrexia, skin reaction, and infusion‐related reaction. There were no reports of anaphylaxis or other hypersensitivity reactions.
3.5. Immunogenicity
Immunogenicity assessments were performed on all 217 subjects (Table 5). The incidence of binding ADA‐positive subjects was similar across the three treatments, and the frequencies for ABP 959 were comparable to those for eculizumab RP. At baseline (Day 1, predose), 7 (3.2%) subjects were positive for binding ADAs (ABP 959:4 [5.6%], eculizumab US: 2 [2.8%], eculizumab EU: 1 [1.4%]). Over the course of the study, 19 (8.8%) subjects developed binding ADAs (ABP 959:7 [9.9%], eculizumab US: 5 [6.9%], eculizumab EU: 7 [9.4%]). At EOS, only 4 (1.9%) subjects were positive for binding ADAs (ABP 959:1 [1.4%], eculizumab US: 2 [2.9%], eculizumab EU: 1 [1.4%]). All neutralizing ADA tests were negative.
Table 5.
Summary of binding ADA results
| Visit |
Subjects with binding ADA‐positive results n/N (%) |
||
|---|---|---|---|
|
ABP 959 (N = 71) |
Eculizumab US (N = 72) |
Eculizumab EU N = 74) |
|
| Day 1 (predose) | 4/71 (5.6) | 2/72 (2.8) | 1/74 (1.4) |
| Day 11 | 5/67 (7.5) | 4/64 (6.3) | 6/67 (9.0) |
| Day 29 | 1/67 (1.5) | 2/69 (2.9) | 0/70 (0.0) |
| Day 57 (EOS/ET) | 1/70 (1.4) | 2/68 (2.9) | 1/73 (1.4) |
| Positive at any time during the study | 7/71 (9.9) | 5/72 (6.9) | 7/74 (9.5) |
n = number of subjects with binding ADA‐positive results; N = number of subjects with binding ADA results.
Abbreviations: ADA, anti‐drug antibodies; EOS, end of study; ET, end of treatment.
3.6. PK‐ADA relationship
Sensitivity analyses performed in the subgroup of subjects with negative binding ADA status confirmed that the 90% CIs for all comparisons (ABP 959 versus eculizumab US, ABP 959 versus eculizumab EU, and eculizumab US versus eculizumab EU) were within the bioequivalence range of 0.80 to 1.25. Because no subjects were positive for neutralizing ADAs, results for the negative neutralizing ADA subjects were identical to those for the PK parameter population. Statistical comparisons of PK parameters were not performed in the binding ADA‐positive patient population due to small patient numbers.
4. DISCUSSION
Based on the results presented here, the current PK equivalence study in healthy adult male subjects demonstrated PK similarity, as assessed by AUC0−∞ for total concentration, of ABP 959 relative to that of eculizumab RP following a single infusion of study drugs. Additionally, PK similarity between ABP 959 and eculizumab RP was demonstrated for AUClast and C max. These conclusions of PK similarity for the PK parameters (AUC0−∞, AUClast, and C max) were based on 90% CI for the least squares GMRs meeting the prespecified equivalence margin of 0.8 and 1.25.
Pharmacodynamic similarity was also demonstrated for the PD endpoint, ABEC for CH50, for the comparison of ABP 959 to the combined eculizumab groups since the 90% CI for the GMRs fell within the bioequivalence margin of 0.80 to 1.25.
The present study also established PK and PD similarity between eculizumab US and eculizumab EU, thus, establishing a scientific bridge, which would allow using a single RP comparator in comparative clinical studies.
The safety analysis conducted on all subjects that received study drugs showed that single 300 mg IV infusions of ABP 959, eculizumab US, and eculizumab EU were safe and well tolerated. Overall, at the subtherapeutic dose used, the frequency, type, and severity of AEs were similar between treatment groups. In particular, the incidence of infections was similar across the groups; this was an EOI given the mechanism of action of eculizumab in blocking terminal complement activation and the potential for increased susceptibility to bacterial infections. Even though the dose in this study was a subtherapeutic single dose, the similarity of safety results between the treatment groups is important. At the therapeutic dose and schedule of eculizumab used in the TRIUMPH and SHEPHERD studies, the most common AEs were headache, back pain, nasopharyngitis, upper respiratory tract infection, and nausea. 4 , 5 In the TRIUMPH and SHEPHERD studies, most infection‐related AEs were considered unrelated to eculizumab; a few were considered to be possibly related, and none was definitely related. 4 , 5 In the present study, the grade 3 viral infection reported with the subtherapeutic dose was deemed by investigators not to be related to study drug.
The development of binding ADAs was similar across treatment arms; no neutralizing ADAs were reported. PK equivalence results using the binding ADA‐negative population were similar to those obtained in the primary PK analysis. PK equivalence analyses were not performed in the binding ADA‐positive population due to the small subject numbers. Overall, these results confirmed that there were no differences in immunogenicity between ABP 959 and eculizumab RP.
Based on the known PK of eculizumab RP, 16 the current study was conducted in healthy subjects to provide the most homogeneous population for a sensitive comparison of the PK of ABP 959 and eculizumab RP, which is consistent with the regulatory guidance for biosimilar development. Because the teratogenicity and effects of eculizumab RP and ABP 959 on reproductive capacity are unknown, only male subjects were enrolled in this study.
In the present study, a clinically subtherapeutic dose (300 mg) was used to reduce possible AE risks to subjects while providing sufficient PK and PD data for evaluation of similarity. Per label for patients with PNH, the recommended eculizumab dosage regimen is 600 mg weekly for the first 4 weeks, followed by 900 mg for the fifth dose 1 week later, followed by 900 mg every 2 weeks thereafter. 1 Based on the linear PK of eculizumab RP, 17 the 300 mg dose level is considered to be predictive of PK at the clinical dose level. At the subtherapeutic dose used in the present PK study, there were no safety signals of concern and the safety signals were consistent with those previously described in clinical studies of eculizumab RP. 4 , 5 , 6 , 7 , 8
5. CONCLUSIONS
This double‐blind, randomized PK/PD study established PK and PD similarity between ABP 959 and eculizumab RP in healthy adult male subjects. The overall safety and immunogenicity were similar between ABP 959 and eculizumab RP. Based on the results of this study, as well as the totality of evidence from analytical and functional similarity assessments, a comparative clinical study (NCT03818607) to assess the clinical (efficacy and safety) similarity of ABP 959 with eculizumab RP in patients with PNH is currently ongoing. The results of this clinical PK/PD study in healthy male subjects contribute toward the totality of evidence required for establishing similarity between ABP 959 and eculizumab RP.
MEDICAL WRITING ASSISTANCE
Medical writing assistance was provided by Sabby Muneer, PhD, Innovation Communications Group, New York, NY under the guidance of Monica Ramchandani, PhD, Amgen Inc.
CONFLICT OF INTEREST
Vincent Chow is an employee and stockholder of Amgen. Jean Pan is an employee and stockholder of Amgen. David Chien is an employee and stockholder of Amgen. Daniel T. Mytych is an employee and stockholder of Amgen. Vladimir Hanes is an employee and stockholder of Amgen.
AUTHOR CONTRIBUTIONS
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Supporting information
Fig S1
Fig S2
Chow V, Pan J, Chien D, Mytych DT, Hanes V. A randomized, double‐blind, single‐dose, three‐arm, parallel group study to determine pharmacokinetic similarity of ABP 959 and eculizumab (Soliris®) in healthy male subjects. Eur J Haematol. 2020;105:66–74. 10.1111/ejh.13411
Funding information
This study was funded by Amgen Inc., Thousand Oaks, CA, USA.
[Correction added on 8 May 2020, after first online publication: Table 2 has been corrected in this version.]
DATA AVAILABILITY STATEMENT
There is a plan to share data. This may include de‐identified individual patient data for variables necessary to address the specific research question in an approved data sharing request; also related data dictionaries, study protocol, statistical analysis plan, informed consent form, and/or clinical study report. Data sharing requests relating to data in this manuscript will be considered after the publication date and (a) this product and indication (or other new use) have been granted marketing authorization in both the United States and Europe, or (b) clinical development discontinues and the data will not be submitted to regulatory authorities. There is no end date for eligibility to submit a data sharing request for these data. Qualified researchers may submit a request containing the research objectives, the Amgen product(s) and Amgen study/studies in scope, endpoints/outcomes of interest, statistical analysis plan, data requirements, publication plan, and qualifications of the researcher(s). In general, Amgen does not grant external requests for individual patient data for the purpose of re‐evaluating safety and efficacy issues already addressed in the product labeling. A committee of internal advisors reviews requests. If not approved, a Data Sharing Independent Review Panel may arbitrate and make the final decision. Requests that pose a potential conflict of interest or an actual or potential competitive risk may be declined at Amgen's sole discretion and without further arbitration. Upon approval, information necessary to address the research question will be provided under the terms of a data sharing agreement. This may include anonymized individual patient data and/or available supporting documents, containing fragments of analysis code where provided in analysis specifications. Further details are available at the following: http://www.amgen.com/datasharing.
REFERENCES
- 1. Soliris® (eculizumab) prescribing information. Boston, MA: Alexion Pharmaceuticals, Inc.; 2019. [Google Scholar]
- 2. Soliris® (eculizumab) summary of product characteristics. Levallois‐Perret, France: Alexion Pharmaceuticals, Inc.; 2019. [Google Scholar]
- 3. Dubois EA, Cohen AF. Eculizumab. Br J Clin Pharmacol. 2009;68(3):318‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hillmen P, Young NS, Schubert J, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2006;355(12):1233‐1243. [DOI] [PubMed] [Google Scholar]
- 5. Brodsky RA, Young NS, Antonioli E, et al. Multicenter phase 3 study of the complement inhibitor eculizumab for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Blood. 2008;111(4):1840‐1847. [DOI] [PubMed] [Google Scholar]
- 6. Legendre CM, Licht C, Muus P. Terminal complement inhibitor eculizumab in atypical hemolytic‐uremic syndrome. N Engl J Med. 2013;368:2169‐2181. [DOI] [PubMed] [Google Scholar]
- 7. Licht C, Greenbaum LA, Muus P, et al. Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2‐year extensions of phase 2 studies. Kidney Int. 2015;87:1061‐1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fakhouri F, Hourmant M, Campistol JM, et al. Terminal complement inhibitor eculizumab in adult patients with atypical hemolytic uremic syndrome: a single‐arm, open‐label trial. Am J Kidney Dis. 2016;68(1):84‐93. [DOI] [PubMed] [Google Scholar]
- 9. Food and Drug Administration, Center for Drug Evaluation and Research . Guidance for industry: scientific considerations in demonstrating biosimilarity to a reference product. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf. Published April 2015. Accessed July 15, 2019.
- 10. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research . Quality considerations in demonstrating biosimilarity of a therapeutic protein product to a reference product. Guidance for industry. May 2019. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm291134.pdf. Accessed July 15, 2019.
- 11. Food and Drug Administration, Center for Drug Evaluation and Research . Guidance for industry: clinical pharmacology data to support a demonstration of biosimilarity to a reference product. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM397017.pdf. Published December 2016. Accessed July 15, 2019.
- 12. European Medicines Agency, Committee for Medicinal Products for Human Use . Guideline on similar biological medicinal products containing monoclonal antibodies – non‐clinical and clinical issues. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128686.pdf. Published 30 May 2012. Accessed July 15, 2019.
- 13. European Medicines Agency . Guideline on similar biological medicinal products containing biotechnology‐derived proteins as active substance: quality issues (revision 1). http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/06/WC500167838.pdf. Published 22 May 2014. Accessed July 15, 2019.
- 14. European Medicines Agency . Guideline on similar biological medicinal products. https://www.ema.europa.eu/en/documents/scientific‐guideline/guideline‐similar‐biological‐medicinal‐products‐rev1_en.pdf. Published 23 October 2014. Accessed July 15, 2019.
- 15. Hutterer K, Polozova A, Kuhns S, et al. Analytical and functional similarity of proposed Amgen biosimilar ABP 959 to eculizumab. Presented at 2019 PDA Biosimilars and Vaccines Conference: Lifecycle Similarities and Challenges, Long Beach, CA; May 9‐10, 2019.
- 16. Wijnsma KL, Ter Heine R, Moes DJAR, et al. Pharmacology, pharmacokinetics and pharmacodynamics of eculizumab, and possibilities for an individualized approach to eculizumab. Clin Pharmacokinet. 2019;58(7):859‐874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lathia C, Kassir N, Mouksassi S, et al. Modelling and simulations of eculizumab in paroxysmal nocturnal hemoglobinuria (PNH) and atypical hemolytic uremic syndrome (aHUS) patients: learning from one indication to the next. Presented at the American Society for Clinical Pharmacology and Therapeutics (ASCPT) annual meeting, Indianapolis, IN; March 5‐9, 2014.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Data Availability Statement
There is a plan to share data. This may include de‐identified individual patient data for variables necessary to address the specific research question in an approved data sharing request; also related data dictionaries, study protocol, statistical analysis plan, informed consent form, and/or clinical study report. Data sharing requests relating to data in this manuscript will be considered after the publication date and (a) this product and indication (or other new use) have been granted marketing authorization in both the United States and Europe, or (b) clinical development discontinues and the data will not be submitted to regulatory authorities. There is no end date for eligibility to submit a data sharing request for these data. Qualified researchers may submit a request containing the research objectives, the Amgen product(s) and Amgen study/studies in scope, endpoints/outcomes of interest, statistical analysis plan, data requirements, publication plan, and qualifications of the researcher(s). In general, Amgen does not grant external requests for individual patient data for the purpose of re‐evaluating safety and efficacy issues already addressed in the product labeling. A committee of internal advisors reviews requests. If not approved, a Data Sharing Independent Review Panel may arbitrate and make the final decision. Requests that pose a potential conflict of interest or an actual or potential competitive risk may be declined at Amgen's sole discretion and without further arbitration. Upon approval, information necessary to address the research question will be provided under the terms of a data sharing agreement. This may include anonymized individual patient data and/or available supporting documents, containing fragments of analysis code where provided in analysis specifications. Further details are available at the following: http://www.amgen.com/datasharing.