

Abstract
A maladaptive shift from fat to carbohydrate (CHO) oxidation during exercise is thought to underlie myopathy and exercise‐induced rhabdomyolysis in patients with fatty acid oxidation (FAO) disorders. We hypothesised that ingestion of a ketone ester (KE) drink prior to exercise could serve as an alternative oxidative substrate supply to boost muscular ATP homeostasis. To establish a rational basis for therapeutic use of KE supplementation in FAO, we tested this hypothesis in patients deficient in Very Long‐Chain acyl‐CoA Dehydrogenase (VLCAD). Five patients (range 17‐45 y; 4 M/1F) patients were included in an investigator‐initiated, randomised, blinded, placebo‐controlled, 2‐way cross‐over study. Patients drank either a KE + CHO mix or an isocaloric CHO equivalent and performed 35 minutes upright cycling followed by 10 minutes supine cycling inside a Magnetic Resonance scanner at individual maximal FAO work rate (fatmax; approximately 40% VO2max). The protocol was repeated after a 1‐week interval with the alternate drink. Primary outcome measures were quadriceps phosphocreatine (PCr), Pi and pH dynamics during exercise and recovery assayed by in vivo 31P‐MR spectroscopy. Secondary outcomes included plasma and muscle metabolites and respiratory gas exchange recordings. Ingestion of KE rapidly induced mild ketosis and increased muscle BHB content. During exercise at FATMAX, VLCADD‐specific plasma acylcarnitine levels, quadriceps glycolytic intermediate levels and in vivo Pi/PCr ratio were all lower in KE + CHO than CHO. These results provide a rational basis for future clinical trials of synthetic ketone ester supplementation therapy in patients with FAO disorders.
Trial registration: ClinicalTrials.gov. Protocol ID: NCT03531554; METC2014.492; ABR51222.042.14.
Keywords: fatty acid oxidation, in vivo 31P MRS, ketone ester, mitochondrial energy transduction, muscle, nutritional ketosis, very long‐chain acyl‐CoA dehydrogenase, VLCADD
1. INTRODUCTION
Very long‐chain acyl‐CoA dehydrogenase deficiency (VLCADD) is an inborn error of fatty acid oxidation (FAO) that is included in many newborn screening programs worldwide. Despite early diagnosis, the lack of therapeutic options may cause patients to present with a broad clinical spectrum, ranging from infant fatality to exercise intolerance and elevated risk of exertional rhabdomyolysis starting in childhood or adolescence.1, 2, 3, 4, 5, 6 The latter symptoms in patients with VLCADD have been attributed to an inability of carbohydrate oxidation (CHO) to fully compensate for the loss of mitochondrial FAO capacity without the risk of glycogen depletion and subsequent ATP depletion during prolonged exercise.7, 8, 9 We recently showed that this risk is aggravated by a higher ATP cost of muscle contraction10 likely resulting from the same adaptive phenotypic muscle remodelling observed in a mouse model for FAO deficiency.9, 11
The standard treatment for VLCADD patients has been dietary restriction of long‐chain triglycerides (LCT) and supplementation of medium‐chain triglycerides (MCT) to bypass defective long‐chain fatty acid oxidation as well as boost production of ketone bodies by the liver.5, 6, 12, 13, 14 However, this has proven to be unsuccessful to prevent rhabdomyolysis and muscle‐related complaints. The same problems apply for supplementation of triheptanoin, a 7‐carbon fatty acid.15, 16 Instead, supplementation of ketone bodies would be an attractive alternative dietary treatment strategy.17
The ketone bodies acetoacetate (AcAc) and beta‐hydroxybutyrate (BHB) are readily taken up by all tissues including cardiac and skeletal muscle, and, except for liver, exclusively used as oxidative substrate.17, 18, 19, 20 For example, studies in dogs have shown that the myocardium switches almost completely from fat oxidation to BHB oxidation upon infusion of the latter.20, 21 Moreover, in vivo 31P magnetic resonance spectroscopy recordings from the hearts of these dogs showed that this substrate switch was associated with an energetically favourable change in myocardial concentration ratio of inorganic phosphate (Pi) and phosphocreatine (PCr).21 This observation confirmed a previous proposition that the thermodynamic efficiency of intracellular energy transduction—that is, the transformation of chemical energy stored in oxidative substrates (CHO, FFA, BHB) to Gibbs free energy stored in a highly non‐equilibrium cytoplasmic concentration ratio of ATP and its hydrolysis products ADP and Pi22 — is higher for BHB oxidation than for fat or CHO oxidation.23 Skeletal muscle in these dogs was likewise found to switch completely to BHB oxidation, albeit that this observation was made in sedated animals and not in exercised state.20 Therapeutic use of BHB has been described in multiple acyl‐CoA dehydrogenase deficiency (MADD), a mitochondrial disorder that also involves impaired FAO, but with a more severe phenotype than fatty acid oxidation disorders (FAODs).24, 25, 26
The first described synthetic ketone ester (KE) was a glycerol mono‐ester of AcAc (“monoacetoacetin”) that allows circumvention of any problem of salt‐loading associated with parenteral or enteral administration of common commercially available sodium salt preparations of AcAc or BHB.27 The later synthesised (R,S)1,3‐butanediol AcAc esters18, 19 produced mild ketosis in pigs lasting several hours after enteral administration18, 19 demonstrating potential for clinical use in humans. Clarke and coworkers first tested and confirmed the potential of synthetic KE supplementation in healthy subjects using an alternative KE28 and later showed in elite athletes that mild ketosis resulted in leg muscle BHB uptake and glycogen sparing during exercise.29
On basis of these considerations, we hypothesised that single dose oral administration of KE prior to any exercise may protect VLCADD and other FAOD patients from glycogen depletion and rhabdomyolysis after exercise. However, the specific metabolic and physiological actions of nutritional ketosis (NK) during exercise in humans have only been tested in athletes,29 not in patients with a FAO disorder. Therefore, prior to embarking on any long‐term clinical trial, a number of questions need to be answered including: (a) is enteral delivery of the KE well tolerated by patients; (b) does a single dose of KE induce mild NK in blood with concomitant preservation of normoglycaemia in VLCADD patients; (c) do muscles of VLCADD patients take up and oxidise BHB during exercise rather than CHO in the presence of mild NK?
2. METHODS
2.1. Patients
A single‐centre, multi‐location, randomised, blinded, placebo‐controlled, 2‐way cross‐over trial with 5 VLCADD patients (median 22 years [range 17‐45]; 4 M/1F) were included. Diagnosis was confirmed by enzymatic analysis of lymphocytes and fibroblasts in combination with bi‐allelic mutations in the ACADVL gene (OMIM 609575). None of the patients had a history of abnormal cardiopulmonary function. Baseline characteristics and results of enzymatic and genetic studies in the five VLCADD patients are presented in Tables S1 and S2.
The study was approved by the medical ethics committee of the University Medical Centre Groningen (METC 14‐492). All patients provided written informed consent for participation in this study. All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000.
2.2. Study design
The study consisted of three exercise tests separated by at least 1 week (Figure 1). First, patients performed a graded cardio‐pulmonary exercise test (CPET)30 on an upright bicycle ergometer at location AMC. From the results of this test, the workload corresponding to maximal bodily fat oxidation (FATMAX) was determined for each subject. Details on materials and methods of this baseline visit are available as supplemental material. Exercise tests two and three including in‐magnet exercise were performed at location UMCG. Thirty minutes prior to exercise patients received either a drink containing 395 mg/kg of ketone ester (KE) + 54 g of dextrose or an isocaloric carbohydrate (CHO) drink containing only dextrose. Details on randomisation, blinding, drink preparation, and dietary standardisation are available as supplemental material.
Figure 1.
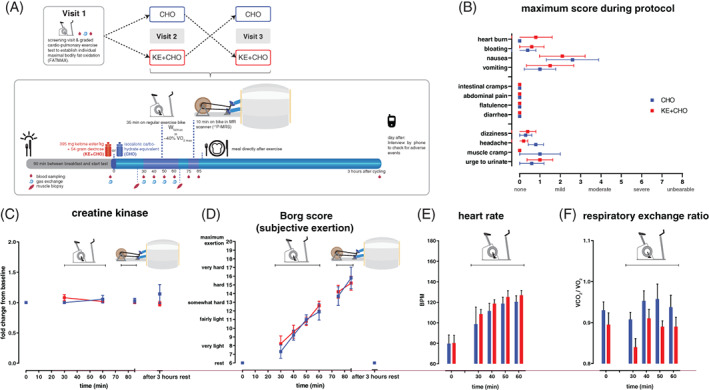
Study protocol and effects of dietary substrates during exercise on tolerability and cardiopulmonary exercise testing. A, Studyprotocol; B, Maximum scores of complaints during study protocol reported by patients; C, Concentration of creatine kinase in plasma after CHO (blue) or KE + CHO (red) ingestion; D, Subjective exertion score reported by patients after CHO (blue) or KE + CHO (red) ingestion; E and F, Heart rate (E) and respiratory exchange ratio measured during upright bicycling bout of protocol after CHO (blue) or KE + CHO (red) ingestion. N = 5, data are presented as mean ± SEM. Differences between groups were analysed with two‐way ANOVA for repeated measures with Bonferroni post‐hoc analysis
2.3. Primary and secondary outcome
Primary outcome measures were quadriceps phosphocreatine (PCr), Pi and pH dynamics during exercise and recovery assayed by in vivo 31P‐MR spectroscopy. Secondary outcomes were whole blood BHB and glucose levels, plasma lactate, creatine kinase, free fatty acids, insulin, and acylcarnitine species. Muscle metabolites (glycolysis and TCA intermediates and acylcarnitine species), respiratory gas exchange recordings and subjective exertion and physical complaints scores were also secondary outcomes.
2.4. Endurance exercise test ‐ second and third test day
The exercise protocol for the second and third test day was identical except for drink content that was allocated by randomisation. At t = 0 the patient consumed the test drink. At t = 30 minutes the patient initiated a 35‐minute exercise bout on an upright cycle ergometer (Lode Corival, Lode BV, Groningen, the Netherlands) at individual FATMAX. Pedalling frequency was maintained between 60 and 80 rpm. After completion of the upright exercise bout, the patient was fitted with bicycle race shoes equipped with binding cleats and transported to the MR scanner in a wheelchair for a final 10 minutes supine cycling exercise bout inside the MR‐scanner. Patients received the reverse drink during the third test day.
2.5. Blood sampling
At the start of days two and three of the protocol, a venous catheter was inserted percutaneously into an antecubital vein of the patient for blood sampling. Glucose and d‐bhb were directly measured on‐site in whole blood (Freestyle Precision, Abbot Diabetes Care, Alameda, California; Glucomen LX plus, Menarini Diagnostics, Florence, Italy). Blood samples were immediately stored on ice. At the end of the protocol, all samples were transferred to the biochemistry lab in another building, centrifuged (1200 rcf at 10 minutes) and stored at −80°C until further analysis. FFA were analysed by an enzymatic colorimetric method (NEFA C test kit [Wako Chemicals, Neuss, Germany]) as described previously.31
2.6. Pulmonary gas exchange recordings
Dynamic respiratory gas collection was performed on days two and three of the protocol. Patients were fitted with a mask and heart rate monitor (K4b2, Cosmed, Rome, Italy) and seated on the upright cycle ergometer. First, baseline recordings were collected for 10 minutes. During stationary upright bicycling at individual FATMAX respiratory gas samples were collected at t = 30, 40, 50, and 60 minutes. The duration of respiratory gas sampling was 2 minutes for each time point. Respiratory gas sampling was not possible during cycling inside the MR scanner.
2.7. Muscle biopsy
Muscle tissue was collected from patients 2‐5 using percutaneous needle biopsies from the lower third of the vastus lateralis muscle (Bard Monopty, Bard Biopsy, Tempe, Arizona) as described.29 No muscle biopsies were collected from patient 1. Biopsy samples were obtained from fresh incisions prior to and immediately following the 35 minutes exercise bout, respectively. Tissue was frozen immediately in liquid nitrogen and stored at −80°C until further analysis.
2.8. In vivo 31P MR spectroscopy
In vivo 31P MR spectra were acquired from the vastus lateralis muscle using a 3 Tesla Achieva MR scanner (Philips Healthcare, Best, the Netherlands). The exact positioning of the MR‐compatible bicycle ergometer inside the scanner tube and length of the hand straps were customised to the physical dimensions of the patient prior to the start of the study protocol on days two and three. Patients also performed a short bout of unloaded exercise to familiarise themselves with the MR study protocol. After 65 minutes of the study, a 6 cm surface coil (Philips Healthcare) was fastened over the vastus lateralis muscle of the leg that had not been used for muscle biopsy prior to and immediately after the preceding upright bicycle exercise bout. Scout MRI scans were acquired for image‐based shimming of the magnet. Dynamic 31P MR spectra during exercise and recovery were acquired with 8 seconds time resolution using an adiabatic half‐passage pulse (NSA 2, TR 4000) according to methods described elsewhere.10 The desired pedalling frequency (target setting: 80 rpm) was set by a metronome audible over the in‐magnet speaker system.32 Synchronisation of MRS data acquisition with pedalling during exercise was performed using custom‐built Labview software (National Instruments, Roscoe, Illinois) as described elsewhere.10 Details on processing of 31P MRS data are available as supplemental materials.
2.9. Acylcarnitine analysis
Plasma acylcarnitines were analysed using electrospray tandem mass spectrometry as described previously.33
2.10. Muscle metabolomics
Muscle samples were freeze dried, homogenised and approximately 1 mg of dry weight muscle tissue was prepared for mass spectrometry. After addition of internal standards (D3‐aspartic acid, D3‐serine, D5‐glutamine, D3‐glutamate, 13C3‐pyruvate, 13C6‐isoleucine, 13C6‐glucose, 13C6‐fructose‐1,6‐biphosphate, 13C6‐glucose‐6‐phosphate, adenosine‐15N5‐monophosphate, guanosine‐15N5‐monophosphate, adenosine‐15N5‐triphosphate, and guanosine‐15N5‐triphosphate (5 μM)), MilliQ was added to a total volume of 500 μL. Subsequently 500 μL Methanol and 1 mL chloroform were added. Samples were vortexed, sonicated, and centrifuged for 5 minutes at 14 000 rpm at 4°C. The “polar” top layer was dried in a vacuum concentrator in a new tube. Dried samples were dissolved in 100 μL methanol/water (6/4; v/v). A Thermo Scientific ultra‐high‐pressure liquid chromatography system (Waltman, Massachusetts) coupled to Thermo Q Exactive (Plus) Orbitrap mass spectrometer (Waltman, Massachusetts) was used for the analysis as described previously.34 Values were depicted as the ratio of peak area over internal standard (PA) and normalised for the sum of ATP + ADP + AMP (TAN) per sample.
2.11. Statistical analysis
Prism 6 software (GraphPad Software, Inc., La Jolla, California) was used for statistical analysis. Results are expressed as means ± SEM and significance were established a priori at P < .05. For plasma metabolites, heart rate, RER, Borg score a single paired t test was performed for every time point and a two‐way ANOVA for repeated measures with Bonferroni post‐hoc analysis for differences between test drinks throughout the protocol. For muscle metabolites, two‐way ANOVA with Bonferroni post‐hoc analysis for the effect of both treatment and exercise and paired t tests to compare the effect of exercise within treatment. For statistical differences in the 31P‐MRS data, a two‐tailed paired student's t test was used.
3. RESULTS
3.1. Ingestion of the KE drink was tolerated by all patients
On visits 2 and 3, patients ingested either the blinded isocaloric KE + CHO or CHO drink, (Figure 1A). Both drinks were taste‐matched (see Section 2) and were tolerated by all patients. Two patients complained of mild to moderate transient nausea following ingestion of the blinded KE drink (Figure 1B), with one patient regurgitating the KE drink after which the study was discontinued and repeated on a different day. These patients also reported a mild sensation of heart burn and bloating during upright bicycling. Lower intestinal complaints (eg, intestinal cramps, diarrhoea) were not reported. One patient reported moderate muscle cramps during upright bicycling, but was able to complete the entire protocol. The total of 45 minutes of submaximal exercise performed during these visits did not result in any muscle damage as evidenced by lack of any significant increase in individual basal plasma CK levels (Figure 1C). Subjective exertional (Borg) scores during bicycling at Wfatmax increased from 8 to 12 during upright bicycling (maximum: 20; Figure 1D). Heart rate increased moderately in both arms of the study (Figure 1F). The respiratory quotient (RQ) was typically higher in the CHO arm compared to KE + CHO (Figure 1G). In both arms, however, values remained close to 0.9.
3.2. Ingestion of the KE drink results in mild ketosis while blood glucose levels remain normal
Within 30 minutes of ingestion of the KE drink (KE + CHO) BHB concentrations in blood increased to 2.0 ± 0.2 mmol/L (Figure 2A). BHB concentrations remained significantly higher compared to values after isocaloric CHO ingestion for the entire protocol (P = .0002 for the effect of intervention, P < .0001 for time and P < .0001 for intervention × time). During the second bout of cycling exercise inside the scanner in the KE arm, BHB blood concentration significantly dropped 1.4‐fold (P < .05). Peak blood BHB concentrations in the patients were comparable to previous studies of NK after KE supplementation in healthy individuals.29, 35, 36 Four hours after ingestion of the KE drink, BHB blood concentrations had almost dropped to baseline (Figure 2A). Blood glucose in both arms of the study remained within normal values during the entire protocol (KE 5.7 ± 0.4 vs CHO 6.4 ± 0.7 mmol/L, respectively; mean ± SD) (Figure 2B). During the second bout of cycling exercise inside the scanner in the CHO arm, but not the KE + CHO arm, blood glucose dropped significantly from an average value of 7.0 to 5.0 mmol/L (P < .05) (Figure 2B). Plasma insulin levels increased transiently in response to each drink and returned to basal levels during the first bout of exercise in all patients (Figure 2C). Basal and peak insulin concentrations in patients 2‐5 were similar, but in patient 1 these concentrations were one order of magnitude higher. At the end of exercise as well as 3 hours after ingestion of the drink, plasma insulin in this patient were 2‐fold lower compared to baseline after ingestion of the KE + CHO drink, but remained similar after ingestion of the CHO drink (Figure 2C). Plasma lactate levels during the bout of upright cycling remained between 1 and 2 mmol/L in both arms. In the KE + CHO arm plasma lactate concentrations tended to be 1.3‐fold lower at the end of exercise than at onset (P = .07; Figure 2D).
Figure 2.
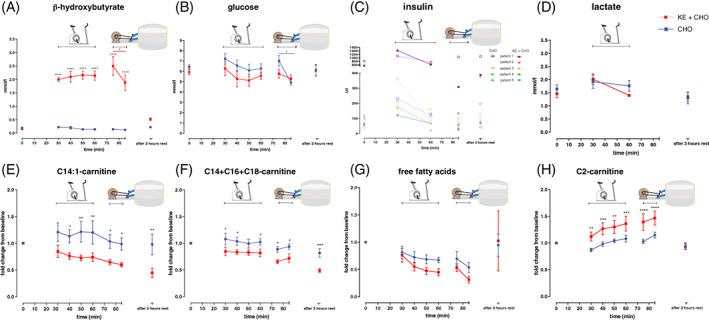
Effects of dietary substrates during exercise on plasma metabolites in VLCADD patients. A‐D, Concentration of beta‐hydroxybutyrate (A) glucose (B) in whole blood, insulin (C) and lactate (D) in plasma after CHO or KE + CHO ingestion. E‐H, Fold change from baseline concentration of C14:1‐carnitine (C), sum of C14 + C16 + C18‐carnitine (D), free fatty acids (E) and C2‐carnitine (F) in plasma. In A, C‐F n = 5 for CHO and KE + CHO. In B n = 5 for KE + CHO, n = 5 for CHO in t = 0 and 3 hours after exercise, n = 4 for CHO in t = 30‐85. Data are presented as mean ± SEM. Differences between groups were analysed with two‐way ANOVA for repeated measures with Bonferroni post‐hoc analysis. The red bar in graph A represents the differences between t = 75 and t = 85 for KE + CHO analysed with paired –t test. The blue bar in graph B represents the difference between t = 75 and t = 85 for CHO analysed with paired –t test. **** indicates P value <.001, *** indicates P value <.001, ** indicates P value <.01, and * indicates P value <.05
3.3. Ingestion of the KE drink reduces plasma long‐chain acylcarnitines
NK reduced the total concentration of long‐chain acylcarnitines in plasma at rest, including the VLCADD specific disease marker [35] C14:1‐carnitine (C14:1 carnitine P = .06 for the effect of intervention, P = .006 for time, and P = .09 for intervention × time; C14 + C16 + C18 carnitine P = .09 for the effect of intervention, P = .0001 for time, and P = .61 for intervention × time) (Figure 2E‐F) suggesting FAO in heart, liver, and kidney was reduced compared to CHO. Plasma long‐chain acylcarnitine concentrations during NK remained lower than with CHO for 4 hours including during exercise. Plasma concentrations of free fatty acids (FFA) fell after both KE + CHO and CHO ingestion (P = .3 for the effect of intervention, P = .001 for time, and P = .5 for intervention × time) (Figure 2G).The plasma concentration of acetylcarnitine during NK increased compared to CHO (P = .05 for the effect of intervention, P = .0001 for time, P = .0002 for intervention × time) (Figure 2H).
3.4. Ingestion of the KE drink results in muscular BHB uptake and appears to blunt glycolysis
Next, we analysed the impact of NK during exercise on intramuscular oxidative metabolism by performing semi‐targeted metabolomics in muscle biopsies. Specifically, we analysed muscle samples obtained from patients 2‐5. Because of highly unfavourable muscle‐to‐fat body composition ratio of patient #1 no muscle biopsy was taken from this patient. To control for variation in actual biopsy tissue composition between patients all results of muscle metabolomics analysis were normalised to total adenosine nucleotides (TAN): ATP + ADP + AMP. The muscle metabolomics results first showed that BHB was taken up by muscle during NK (Figure 3A). This confirmed that BHB was available as oxidative substrate to maintain intramuscular energy balance during exercise. Secondly, NK tended to blunt the increase of intramuscular glycolytic intermediates during exercise seen after CHO ingestion (Figure 3B). Specifically, the sum of intramuscular glycolytic intermediates normalised to TAN remained unchanged in NK but increased in three out of four patients after ingestion of the CHO drink (Figure 3B). This seems to be attributed to the change in fructose‐1,6‐bisphosphate (Figure 3C). Qualitatively different trends in the metabolic response to exercise for the CHO vs KE + CHO arms for other metabolites including the sum of tricyclic acid (TCA) cycle intermediates (Figure 3D) could not be statistically objectified. However, individual trends were in agreement with the notion that NK blunted intramuscular glycolysis during exercise. No significant differential effect of NK vs CHO ingestion on intramuscular fat oxidation was found on basis of metabolomics analysis of long‐, medium‐, and short‐chain acylcarnitines in biopsies (Figure 3E‐P).
Figure 3.
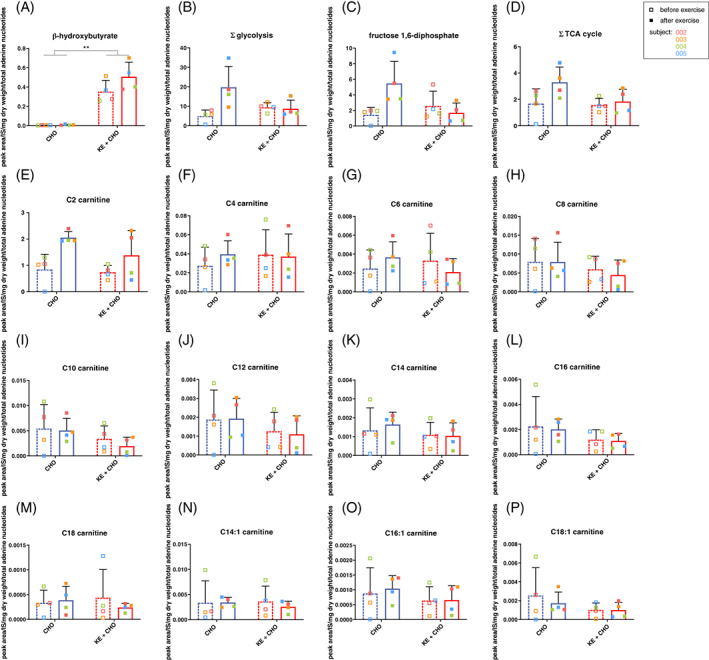
Effects of dietary substrates and exercise on muscle glucose and fat metabolism in VLCADD patients before and after exercise. A, Intramuscular concentrations of beta‐hydroxybutyrate before and after exercise after ingestion of CHO or KE + CHO. B, Sum of intramuscular concentrations of glycolytic intermediates (Hexose‐P, Fructose‐1,6‐diphosphate, Glyceraldehyde‐3P, 1,3‐Diphosphoglyceric acid, 2‐/3‐Phosphoglyceric acid and Phosphoenolpyruvate) before and after exercise after ingestion of CHO or KE + CHO. C, Intramuscular concentrations of fructose 1,6‐diphosphate before and after exercise after ingestion of CHO or KE + CHO. D, Sum of intramuscular concentrations of tricyclic acid cycle intermediates (citrate/isocitrate, α‐ketoglutarate, succinate, fumarate, malate) before and after exercise after ingestion of carbohydrates or ketone ester. E‐P, Intramuscular concentrations of acylcarnitine species before and after exercise after ingestion of CHO or KE + CHO. Values on the Y‐axis are the ratio of peak area over internal standard (PA), corrected for the total adenosine nucleotides (ATP + ADP + AMP) (TAN), per sample. Error bars are mean ± SD. N = 4 for all conditions. ** = P < .01 with 2 way ANOVA
3.5. Ingestion of the KE drink improves muscular energy balance during exercise
Lastly, we conducted in vivo MR image measurements on the leg muscles to investigate if NK during exercise caused a similar favourable change in the intramuscular concentration ratio of inorganic phosphate (Pi) and phosphocreatine (PCr) as previously reported in dogs infused with BHB.21 Scout T1‐weighted MR images of the legs confirmed that patients #2‐5 but not patient #1 had normal muscle‐to‐fat ratio (Figure 4A). In vivo 31P‐MR spectra obtained during stationary cycling exercise at a workload equivalent to FATMAX in each patient were of adequate quality to estimate the in vivo intramuscular Pi and PCr concentrations in working leg muscle (Figure 4B). Quantitative analysis of the spectra showed that in patients 3, 4, and 5 the Pi/PCr ratio in exercising leg muscle during NK was 40% % lower than in the CHO arm at one and the same normalised workload (Figure 4C). In patient 1 no change in Pi/PCr ratio was observed (Figure 4C). In patient 2, 31P‐MRS data recorded during the KE arm were lost due to technical problems. Post‐exercise, the rate of oxidative metabolic recovery indexed by the time constant of PCr recovery to basal level was identical for both arms (Figure 4D).
Figure 4.
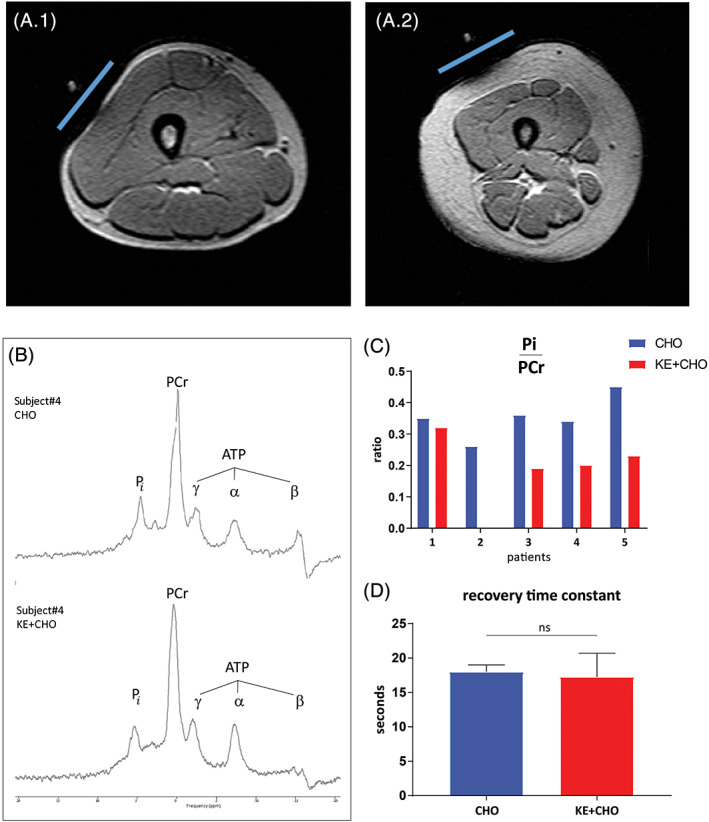
Effects of dietary substrates on in vivo muscle energetics during and after cycling in VLCADD patients. A, Transversal T1‐weighted MR images of the right upper leg of patients #4 (A.1) and #1 (A.2). Subjects were positioned feet‐first. The slightly flattened left side of the thigh image indicates the position of the 31P surface coil overlying the m. vastus lateralis. Note the large diameter of the subcutaneous fat layer surrounding the thigh muscles in female patient #1. B, in vivo 31P Magnetic Resonance spectra of the vastus lateralis muscle of patient ID4 recorded during stationary exercise at individual FATMAX workload after either CHO (top trace) or KE + CHO ingestion (bottom trace), respectively. ATP, adenosine triphosphate; Pi, inorganic phosphate; PCr, phosphocreatine. C and D, Mean in vivo concentration ratio of inorganic phosphate (Pi) and phosphocreatine (PCr) during stationary exercise (C) and mean recovery time constant (D) of the vastus lateralis muscle of 4 VLCADD patients after CHO vs KE + CHO ingestion, respectively. * indicates P value <.05; two tailed paired t test. Data are presented as mean ± SEM
4. DISCUSSION
Our study shows that clinical use of a BHB‐ (R) 1,3‐butanediol ketone ester in patients with VLCADD has significant potential to serve as a treatment. First, the tolerability of this particular synthetic ketone ester was acceptable. Secondly, ingestion of the ester resulted in all patients rapidly and reproducibly in mild ketosis in blood as well as significant BHB uptake by skeletal muscle, while blood glucose levels remained within normal range during the entire study protocol. The level of ketosis in blood was in close agreement with previous findings in healthy individuals.29, 35, 36 Thirdly, plasma concentrations of long‐chain acylcarnitines, disease‐specific biomarkers that are hallmarks of a FAO disorder, were significantly lower during exercise after use of the ketone ester. Finally, in vivo we observed that that the Pi/PCr ratio in leg muscle during exercise at FATMAX workload was lower in the majority of patients after use of the ketone ester. This indicates that intramuscular energy balance during exercise improved after use of the ketone ester.
The major complaint concerning the tolerability was nausea related to the large volume of the drink in combination with the acrid taste of the ester. This potential barrier to clinical use can, however, be overcome easily by adopting the shot‐like bolus ingestion strategy of the undiluted KE followed by a big volume of a tasty CHO drink.
Various findings in this study suggest that BHB in the bloodstream was indeed a major source of oxidative substrate in VLCADD muscle during exercise. As such, this study shows that nutritional ketosis by acute ingestion of KE is a viable new therapeutic option in clinical management of FAO disorders. Firstly, in the KE + CHO arm blood BHB significantly dropped during exercise. In contrast, in the CHO arm blood glucose dropped during exercise, suggesting that these substrates were oxidised during exercise. Secondly, in muscle, glycolytic flux was upregulated in the CHO arm, but not in the KE + CHO arm as evidenced by our metabolomics results. The tendency of plasma lactate to drop during exercise in the KE + CHO arm, but not the CHO arm, agrees with this scenario. Also, our finding of a 1.2‐fold higher rise of plasma acetylcarnitine (C2) during exercise in the KE + CHO arm compared to CHO arm fits with elevated oxidative substrate availability during NK.23, 37 In vivo, we observed that the intramuscular energy balance during exercise in the majority of patients improved in the KE + CHO arm compared to CHO analogous to previous findings on the impact of BHB vs CHO infusion on intracellular energy balance in working cardiac muscle.21
With respect to the magnitude of change in in vivo Pi/PCr ratio that we observed in leg muscle of VLCADD patients, it is unlikely that this was entirely due to the previously demonstrated favourable thermodynamic effect of BHB oxidation on mitochondrial energy transduction.21, 23 Specifically, Kim and coworkers found a 10% improvement in in vivo myocardial Pi/PCr ratio in dogs infused with BHB vs CHO21 whereas we observed a 40% improvement in in vivo Pi/PCr ratio in the working quadriceps muscle (Figure 4C). Less recruitment of fast‐twitch motor units of the quadriceps muscle to perform the voluntary exercise task in the NK arm vs the CHO arm would explain this finding. In this light, it is of interest to note that we previously found evidence that the quadriceps muscle of VLCADD patients exercising at individual FATMAX workload recruits more fast‐twitch motor units than healthy individuals to deliver the desired power‐output.10 As such, these findings would suggest that mild NK during exercise in VLCADD patients not only impacts metabolism but also mechanical performance of oxidative muscle fibres. This hypothesis remains to be tested. In the only available study of the effect of mild NK on physical performance, a minor but significant improvement was, however, found in elite athletes performing a maximal test (distance covered during a 30‐minute time trial).29
Long‐chain acylcarnitines are disease‐specific biomarkers for FAO disorders, for VLCADD in particular C14:1‐carnitine.38 Intramitochondrial accumulation of long‐chain acylcarnitines due to a defective FAO can have detrimental effects on cell function.39 Therefore, an additional beneficial effect of supplying patients with FAO disorders with an alternative oxidative substrate in the form of a KE drink, is that accumulation of these potentially toxic metabolites during exercise may be averted. Indeed, accumulation of plasma C14:1‐carnitine during exercise was almost halved in the KE + CHO arm compared to CHO.
The overall outcome of the present study in VLCADD patients suggests that a rational basis exists for the therapeutic use of synthetic KE supplementation prior to exercise in patients with a defective FAO. This is especially important considering the lack of satisfactory therapeutic options for these patients. Currently, MCT is widely used as an alternative oxidative substrate in patients, MCT ingestion prior to exercise induces mild ketosis, albeit at a significantly lower level (0.5‐1 mM)40 than found in the present study after a single‐dose KE ingestion. Moreover, the medium‐chain FA may serve as substrate for fatty acid synthesis by chain elongation, as was observed in FAO defective mice and patients' fibroblasts.41, 42, 43, 44 As such, the efficacy of MCT supplementation to provide alternative oxidative substrate for ATP synthesis in FAOD patients is questionable and is proven to be insufficient to prevent rhabdomyolysis completely in patients.15, 45, 46 The problem of rhabdomyolysis is also not resolved by triheptanoin treatment.15, 16 In the present study, one patient (subject 5) used MCT supplementation. The outcome in this particular patient did not differ significantly from the other patients. It could be interesting to test the potential complementary effect of MCT and KE or KE and triheptanoin supplementation prior to exercise in future studies.
The potential benefits of synthetic KE supplementation are probably not limited to FAO disorders such as VLCADD, but might very well be relevant for other inborn errors of metabolism, such as MADD patients that suffer from a defect in the reoxidation of all acyl‐CoA dehydrogenases and can therefore not oxidise MCT or triheptanoin. Ketone salt supplementation is the only treatment option in these patients, but long‐term use comes with adverse effects and alternative forms such as ketone acids do not seem to have a clinical benefit.47, 48 Inborn errors of metabolism that are treated with a ketogenic diet include glucose transporter type 1 (GLUT1) deficiency and pyruvate dehydrogenase complex (PDHc) deficiency49 which may also benefit from KE supplementation.
5. CONCLUSION
The results of the studies described in this paper provide strong evidence in favour of KE supplementation prior to exercise in patients with defective FAO at the level of VLCAD. Future studies should determine the clinical and therapeutic utility of NK in VLCADD and other metabolic myopathies.
CONFLICT OF INTEREST
The intellectual property and patents covering the uses of ketone bodies and esters are owned by BTG Ltd, The University of Oxford, the NIH, and TΔS Ltd. Should royalties ever accrue from these patents, K.C. and P.J.C. as named inventors may receive a share of royalties as determined by the terms of the respective institutions. K.C. is director of TΔS Ltd, a University of Oxford company with the aim of developing and commercialising products based on the ketone ester. P.J.C. is a former employee of TdeltaS. J.C.B., G.V., S.F., F.H.d.H., R.H.H., L.I., I.L.K., M.L., W.L.v.d.P., M.G.M.d.S.‐v.d.V., A.S.‐K., T.T., R.J.A.W., M.v.W., F.A.W., L.H.v.d.W., R.C.I.W., and J.A.L.J. declare that they have no conflict of interest.
AUTHOR CONTRIBUTIONS
J.C.B.: study design, clinical assessment of included patients, execution of protocol and data collection, metabolomics analysis and interpretation, preparation of manuscript
G.V.: study design, clinical assessment of included patients, execution of protocol and data collection, preparation of manuscript.
K.C.: providing of ketone ester and materials
S.F.: study design, interpretation of results
F.H.d.H.: facilitating CPET, analysis and interpretation of CPET results
R.H.H.: study design, metabolomics analysis and interpretation of results
L.I.: study design and analysis and interpretation of results
I.L.K.: execution protocol, collection of nutritional data
M.L.: study design and interpretation of results
W.L.v.d.P.: clinical assessment of included patients and interpretation of results
M.G.M.d.S.‐v.d.V.: metabolomics assays and interpretation of results
A.S.‐K.: execution of protocol, collection and analysis of 31P MRS data
T.T.: study design, facilitation and analysis of CPET results
R.J.A.W.: study design, interpretation of results
M.v.W.: analysis and interpretation of metabolomics assays
F.A.W.: Study design and interpretation of results
L.H.v.d.W.: facilitation of CPET, analysis and interpretation of CPET results
R.C.I.W.: metabolomics analysis and interpretation of results
P.J.C.: study design, providing ketone ester and materials, execution of protocol
J.A.L.J.: study design, execution of protocol, analysis and interpretation of 31P MRS data, preparation of manuscript, guarantor for the article who accepts full responsibility for the work and/or the conduct of the study, had access to the data, and controlled the decision to publish.
All authors critically reviewed the manuscript before submission.
Supporting information
Figure S1. CONSORT 2010 flow diagram.
Table S1. Baseline demographics, clinical history and results of maximal cardiopulmonary exercise test of VLCADD patients.
Table S2. Genetic and enzymatic studies in enrolled patients.
ACKNOWLEDGMENTS
We thank Brianna Stubbs for her assistance in setting up the protocol and Roos Oosterwijk, Sebastiaan van den Brink, and Rutger de Vries for their help with the indirect calorimetry. Harald Jorstad is thanked for assistance with CPET testing.
Funding: This study was supported by ESN, the Dutch Society for Inborn Errors of Metabolism (to J.C.B.), and donation by Stichting Spieren voor Spieren (to W.L.v.d.P.) and in part by a subcontract to NIH grant HL‐072011 (to J.A.L.J.).
Bleeker JC, Visser G, Clarke K, et al. Nutritional ketosis improves exercise metabolism in patients with very long‐chain acyl‐CoA dehydrogenase deficiency. J Inherit Metab Dis. 2020;43:787–799. 10.1002/jimd.12217
Pete J. Cox and Jeroen A. L. Jeneson are equal last authors.
Funding information ESN; National Institutes of Health, Grant/Award Number: 072011; Stichting Spieren voor Spieren
Communicating Editor: Manuel Schiff
Contributor Information
Gepke Visser, Email: gvisser4@umcutrecht.nl.
Jeroen A. L. Jeneson, Email: j.a.l.jeneson@umcg.nl, Email: j.a.jeneson@amc.nl.
REFERENCES
- 1. Baruteau J, Sachs P, Broue P, et al. Clinical and biological features at diagnosis in mitochondrial fatty acid beta‐oxidation defects: a French pediatric study of 187 patients. J Inherit Metab Dis. 2013;36:795‐803. [DOI] [PubMed] [Google Scholar]
- 2. Baruteau J, Sachs P, Broue P, et al. Clinical and biological features at diagnosis in mitochondrial fatty acid beta‐oxidation defects: a French pediatric study from 187 patients. Complementary data. J Inherit Metab Dis. 2014;37:137‐139. [DOI] [PubMed] [Google Scholar]
- 3. Bonnet D, Martin D, Pascale De L, et al. Arrhythmias and conduction defects as presenting symptoms of fatty acid oxidation disorders in children. Circulation. 1999;100:2248‐2253. [DOI] [PubMed] [Google Scholar]
- 4. Spiekerkoetter U. Mitochondrial fatty acid oxidation disorders: clinical presentation of long‐chain fatty acid oxidation defects before and after newborn screening. J Inherit Metab Dis. 2010;33:527‐532. [DOI] [PubMed] [Google Scholar]
- 5. Spiekerkoetter U, Lindner M, Santer R, et al. Management and outcome in 75 individuals with long‐chain fatty acid oxidation defects: results from a workshop. J Inherit Metab Dis. 2009a;32:488‐497. [DOI] [PubMed] [Google Scholar]
- 6. Spiekerkoetter U, Lindner M, Santer R, et al. Treatment recommendations in long‐chain fatty acid oxidation defects: consensus from a workshop. J Inherit Metab Dis. 2009b;32:498‐505. [DOI] [PubMed] [Google Scholar]
- 7. Hsu YD, Lee WH, Chang MK, Shieh SD, Tsao WL. Blood lactate threshold and type II fibre predominance in patients with exertional heatstroke. J Neurol Neurosurg Psychiatry. 1997;62:182‐187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tarnopolsky MA. Metabolic myopathies. Continuum (Minneap Minn). 2016;22:1829‐1851. [DOI] [PubMed] [Google Scholar]
- 9. Tucci S, Herebian D, Sturm M, Seibt A, Spiekerkoetter U. Tissue‐specific strategies of the very‐long chain acyl‐CoA dehydrogenase‐deficient (VLCAD−/−) mouse to compensate a defective fatty acid beta‐oxidation. PLoS One. 2012;7:e45429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Diekman EF, Visser G, Schmitz JP, et al. Altered energetics of exercise explain risk of rhabdomyolysis in very Long‐chain acyl‐CoA dehydrogenase deficiency. PLoS One. 2016;11:e0147818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tucci S, Mingirulli N, Wehbe Z, Dumit VI, Kirschner J, Spiekerkoetter U. Mitochondrial fatty acid biosynthesis and muscle fiber plasticity in very long‐chain acyl‐CoA dehydrogenase‐deficient mice. FEBS Lett. 2018;592:219‐232. [DOI] [PubMed] [Google Scholar]
- 12. Arnold GL, Van Hove J, Freedenberg D, et al. A Delphi clinical practice protocol for the management of very long chain acyl‐CoA dehydrogenase deficiency. Mol Genet Metab. 2009;96:85‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bach AC, Babayan VK. Medium‐chain triglycerides: an update. Am J Clin Nutr. 1982;36:950‐962. [DOI] [PubMed] [Google Scholar]
- 14. Bleeker JC, Kok IL, Ferdinandusse S, et al. Proposal for an individualized dietary strategy in patients with very long‐chain acyl‐CoA dehydrogenase deficiency. J Inherit Metab Dis. 2018;42:159‐168. [DOI] [PubMed] [Google Scholar]
- 15. Gillingham MB, Heitner SB, Martin J, et al. Triheptanoin versus trioctanoin for long‐chain fatty acid oxidation disorders: a double blinded, randomized controlled trial. J Inherit Metab Dis. 2017;40:831‐843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tucci S, Floegel U, Beermann F, Behringer S, Spiekerkoetter U. Triheptanoin: long‐term effects in the very long‐chain acyl‐CoA dehydrogenase‐deficient mouse. J Lipid Res. 2017;58:196‐207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Birkhahn RH, Long CL, Blakemore WS. New synthetic substrates for parenteral feeding. JPEN J Parenter Enteral Nutr. 1979;3:346‐349. [DOI] [PubMed] [Google Scholar]
- 18. Desrochers S, Dubreuil P, Brunet J, et al. Metabolism of (R,S)‐1,3‐butanediol acetoacetate esters, potential parenteral and enteral nutrients in conscious pigs. Am J Physiol. 1995a;268:E660‐E667. [DOI] [PubMed] [Google Scholar]
- 19. Desrochers S, Quinze K, Dugas H, et al. R, S‐1, 3‐butanediol acetoacetate esters, potential alternates to lipid emulsions for total parenteral nutrition. J Nutr Biochem. 1995b;6:111‐118. [Google Scholar]
- 20. Little JR, Goto M, Spitzer JJ. Effect of ketones on metabolism of FFA by dog myocardium and skeletal muscle in vivo. Am J Physiol. 1970;219:1458‐1463. [DOI] [PubMed] [Google Scholar]
- 21. Kim DK, Heineman FW, Balaban RS. Effects of beta‐hydroxybutyrate on oxidative metabolism and phosphorylation potential in canine heart in vivo. Am J Physiol. 1991;260:H1767‐H1773. [DOI] [PubMed] [Google Scholar]
- 22. Westerhoff H, Van Dam K. Thermodynamics and control of biological free energy transduction. 1987 Thermodynamics and Control of Biological Free Energy Transduction. 1987. Amsterdam, The Netherlands: Elsevier; 1987. [Google Scholar]
- 23. Sato K, Kashiwaya Y, Keon CA, et al. Insulin, ketone bodies, and mitochondrial energy transduction. FASEB J. 1995;9:651‐658. [DOI] [PubMed] [Google Scholar]
- 24. Gautschi M, Weisstanner C, Slotboom J, Nava E, Zurcher T, Nuoffer JM. Highly efficient ketone body treatment in multiple acyl‐CoA dehydrogenase deficiency‐related leukodystrophy. Pediatr Res. 2015;77:91‐98. [DOI] [PubMed] [Google Scholar]
- 25. Van Hove JL, Grunewald S, Jaeken J, et al. D,L‐3‐hydroxybutyrate treatment of multiple acyl‐CoA dehydrogenase deficiency (MADD). Lancet. 2003;361:1433‐1435. [DOI] [PubMed] [Google Scholar]
- 26. Van Rijt WJ, Heiner‐Fokkema MR, du Marchie Sarvaas GJ, et al. Favorable outcome after physiologic dose of sodium‐D,L‐3‐hydroxybutyrate in severe MADD. Pediatrics. 2014;134:e1224‐e1228. [DOI] [PubMed] [Google Scholar]
- 27. Birkhahn RH, Border JR. Intravenous feeding of the rat with short chain fatty acid esters. II. Monoacetoacetin. Am J Clin Nutr. 1978;31:436‐441. [DOI] [PubMed] [Google Scholar]
- 28. Clarke K, Tchabanenko K, Pawlosky R, et al. Kinetics, safety and tolerability of (R)‐3‐hydroxybutyl (R)‐3‐hydroxybutyrate in healthy adult subjects. Regul Toxicol Pharmacol. 2012;63:401‐408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cox PJ, Kirk T, Ashmore T, et al. Nutritional ketosis alters fuel preference and thereby endurance performance in athletes. Cell Metab. 2016;24:256‐268. [DOI] [PubMed] [Google Scholar]
- 30. Takken T, Mylius CF, Paap D, et al. Reference values for cardiopulmonary exercise testing in healthy subjects—an updated systematic review. Expert Rev Cardiovasc Ther. 2019;17:413‐426. [DOI] [PubMed] [Google Scholar]
- 31. Ter Horst KW, Gilijamse PW, Ackermans MT, et al. Impaired insulin action in the liver, but not in adipose tissue or muscle, is a distinct metabolic feature of impaired fasting glucose in obese humans. Metabolism. 2016;65:757‐763. [DOI] [PubMed] [Google Scholar]
- 32. van Brussel M, van Oorschot JW, Schmitz JP, et al. Muscle metabolic responses during dynamic in‐magnet exercise testing: a pilot study in children with an idiopathic inflammatory myopathy. Acad Radiol. 2015;22:1443‐1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ventura FV, Costa CG, Struys EA, et al. Quantitative acylcarnitine profiling in fibroblasts using [U‐13C] palmitic acid: an improved tool for the diagnosis of fatty acid oxidation defects. Clin Chim Acta. 1999;281:1‐17. [DOI] [PubMed] [Google Scholar]
- 34. Baardman J, Verberk SGS, Prange KHM, et al. A defective pentose phosphate pathway reduces inflammatory macrophage responses during hypercholesterolemia. Cell Rep. 2018;25(2044–2052):e2045. [DOI] [PubMed] [Google Scholar]
- 35. Stubbs BJ, Cox PJ, Evans RD, Cyranka M, Clarke K, de Wet H. A ketone ester drink lowers human ghrelin and appetite. Obesity. 2017;26:269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stubbs BJ, Cox PJ, Evans RD, et al. On the metabolism of exogenous ketones in humans. Front Physiol. 2017b;8:848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schroeder MA, Atherton HJ, Dodd MS, et al. The cycling of acetyl‐coenzyme A through acetylcarnitine buffers cardiac substrate supply: a hyperpolarized 13C magnetic resonance study. Circ Cardiovasc Imaging. 2012;5:201‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Knottnerus SJG, Bleeker JC, Wust RCI, et al. Disorders of mitochondrial long‐chain fatty acid oxidation and the carnitine shuttle. Rev Endocr Metab Disord. 2018;19:93‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McCoin CS, Knotts TA, Adams SH. Acylcarnitines—old actors auditioning for new roles in metabolic physiology. Nat Rev Endocrinol. 2015;11:617‐625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gillingham MB, Scott B, Elliott D, Harding CO. Metabolic control during exercise with and without medium‐chain triglycerides (MCT) in children with long‐chain 3‐hydroxy acyl‐CoA dehydrogenase (LCHAD) or trifunctional protein (TFP) deficiency. Mol Genet Metab. 2006;89:58‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jones PM, Butt Y, Messmer B, Boriak R, Bennett MJ. Medium‐chain fatty acids undergo elongation before beta‐oxidation in fibroblasts. Biochem Biophys Res Commun. 2006;346:193‐197. [DOI] [PubMed] [Google Scholar]
- 42. Tucci S, Behringer S, Spiekerkoetter U. De novo fatty acid biosynthesis and elongation in very long‐chain acyl‐CoA dehydrogenase‐deficient mice supplemented with odd or even medium‐chain fatty acids. FEBS J. 2015a;282:4242‐4253. [DOI] [PubMed] [Google Scholar]
- 43. Tucci S, Flogel U, Spiekerkoetter U. Sexual dimorphism of lipid metabolism in very long‐chain acyl‐CoA dehydrogenase deficient (VLCAD−/−) mice in response to medium‐chain triglycerides (MCT). Biochim Biophys Acta. 2015b;1852:1442‐1450. [DOI] [PubMed] [Google Scholar]
- 44. Tucci S, Primassin S, Ter Veld F, Spiekerkoetter U. Medium‐chain triglycerides impair lipid metabolism and induce hepatic steatosis in very long‐chain acyl‐CoA dehydrogenase (VLCAD)‐deficient mice. Mol Genet Metab. 2010;101:40‐47. [DOI] [PubMed] [Google Scholar]
- 45. Bleeker JC, Kok IL, Ferdinandusse S, et al. Impact of newborn screening for very‐long‐chain acyl‐CoA dehydrogenase deficiency on genetic, enzymatic, and clinical outcomes. J Inherit Metab Dis. 2019;42:414‐423. [DOI] [PubMed] [Google Scholar]
- 46. Pena LD, van Calcar SC, Hansen J, et al. Outcomes and genotype‐phenotype correlations in 52 individuals with VLCAD deficiency diagnosed by NBS and enrolled in the IBEM‐IS database. Mol Genet Metab. 2016;118:272‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fischer T, Elpers C, Och U, Fobker M, Marquardt T. Ketone body therapy with D/L‐beta‐hydroxybutyric acid solution in severe MADD. Mol Genet Metab Rep. 2019a;20:100491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fischer T, Och U, Marquardt T. Long‐term ketone body therapy of severe multiple acyl‐CoA dehydrogenase deficiency: a case report. Nutrition. 2019b;60:122‐128. [DOI] [PubMed] [Google Scholar]
- 49. Scholl‐Burgi S, Holler A, Pichler K, Michel M, Haberlandt E, Karall D. Ketogenic diets in patients with inherited metabolic disorders. J Inherit Metab Dis. 2015;38:765‐773. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. CONSORT 2010 flow diagram.
Table S1. Baseline demographics, clinical history and results of maximal cardiopulmonary exercise test of VLCADD patients.
Table S2. Genetic and enzymatic studies in enrolled patients.