

Abstract
Residual dipolar couplings (RDCs) offer additional information for structure elucidation by NMR spectroscopy. They are measured in anisotropic media, such as lyotropic liquid crystalline phases of polypeptides. Today, some suitable polypeptides are known. Nevertheless, structural influences of these polypeptides on the alignment properties are not really understood. Thus, which influence a chiral side chain has on enantiodiscrimination and whether we can improve the enantiodifferentiation significantly by adding an additional chiral center in the side chain are questions of interest. Therefore, new diastereomeric polypeptide‐based alignment media with an additional chiral center in the side chain derived from perillyl alcohol were synthesized and their properties were investigated (secondary structure, liquid crystallinity, etc.). The enantiomers of isopinocampheol and β‐pinene were used as model analytes for the study of enantiodiscrimination. Additionally, the usage of 1H–1H‐RDCs to improve the alignment tensor quality is demonstrated.
Keywords: alignment medium, chirality, NMR spectroscopy, peptides, polymers
Influences on enantiodifferentiation: The influence of an additional chiral center in the side chain of a polypeptide‐based alignment medium was investigated. Therefore, new diastereomeric alignment media were synthesized and the enantiomers of IPC and β‐pinene were used as analytes. The results are unexpected. Furthermore, 1H–1H‐RDCs (residual dipolar couplings) are used to improve the quality of alignment tensors.

Introduction
NMR spectroscopy is one of the most popular methods in structure elucidation of organic molecules. Apart from various routinely used methods, residual dipolar couplings (RDCs) as anisotropic NMR parameters provide helpful and complementary structural information. Over the last decades they have been receiving a lot of attention.1, 2 This is due to their utility for the determination of the spatial structure as they yield complementary information to those of established approaches, like NOE3, 4, 5 or J‐couplings.6, 7 RDCs are anisotropic NMR parameters. To get access to anisotropic NMR parameters, an anisotropic environment which induces weak alignment is necessary.8 Two major concepts for measurements of non‐water soluble analytes found their way into application: There are alignment media based on either anisotropically swollen gels or lyotropic liquid crystals (LLCs).9, 10, 11 Apart from a few exceptions12, 13, 14 most of the gels are not chiral. In contrast to that, the LLCs are mainly based on helical mesogens15, 16, 17 or other helical constructs18, 19 with a defined screw sense. In the case of polypeptides,20 which include the most prominent lyotropic liquid crystalline alignment media (poly‐γ‐benzyl‐l‐glutamate (PBLG) and poly‐γ‐ethyl‐l‐glutamate (PELG)),15, 21, 22, 23, 24, 25 the screw sense is determined by the configuration of the amino acid building up the backbone. For other examples, like polyacetlylenes,25, 26 polyguanidines27 and polyisocyanides17, 28 the chiral information for the helical sense is derived from the side chain. But no matter what determines the helical sense, if the helices are homochiral or rather if they have a preferred handedness they allow for enantiodifferentiation.22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33 This is one of the most interesting characteristics of the investigation of alignment properties.
Which properties of alignment media are mainly responsible for the extent of the enantiodifferentiation between enantiomers is so far not really understood. Nevertheless, this question is of interest as some predictions about the alignment process are needed. This is especially important concerning the theoretical aspect of the determination of the absolute configuration in the future.34 Some alignment media based on polypeptides are known and have been investigated with respect to enantiodiscrimination and solute–polymer interaction.15, 20, 29, 30, 31, 35, 36, 37, 38, 39, 40 In these investigations, it became clear that the enantiodifferentiation induced by these polypeptides depends, among other things, on their side chain. Hence, the question came up, whether the chirality of the side chain in polypeptide‐based media would have an influence on the alignment process.
In the case of a polypeptide with an achiral side chain the enantiodifferentiation is caused by diastereomorphous interactions between the analyte and the homochiral helix of the polypeptide backbone. Hence, different orientations are obtained for two enantiomers measured in a homochiral polypeptide. In contrast to that, the same orientation results if one enantiomer is measured in the polypeptide with the l‐amino acid and the other enantiomer is measured in the polypeptide with the d‐amino acid. This is due to the enantiomorphous relation between these systems. As a result, only two different orientations are obtained by comparing the four possible combinations [analyte (+ or −) and polypeptide (l or d)]. The situation becomes more complex if an additional stereogenic element is added in the polypeptide side chain, as the polymers then become diastereomeric. If one could treat the helix and the chiral side chain as separate entities an additional site for diastereomorphous interactions (DI) is provided (Figure 1). It is assumed that this additional interaction has a significant influence on the induced analyte orientation. The resulting enantiodifferentiation should thus depend on the helical sense and on the chiral information from the side chain. If the polypeptide is available with the amino acid in the l and d configuration (as glutamic acid is available both in l‐ and d‐configuration), four systems [analyte (+ or −) and polypeptide (S l or S d)] can be prepared (as the side chain is available only in S‐configuration, see Figure 1). Systematic considerations of the orientations induced by different combinations of analyte configuration, side chain configuration and backbone sense should thus allow for the determination of the influence of the side chain's chirality by permuting the stereogenic elements.
Figure 1.
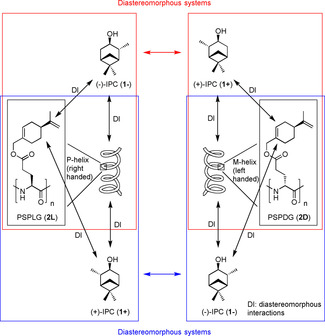
Possible diastereomorphous interactions (DI) between analytes (1−, 1+) and polypeptide based alignment media (2L, 2D) with chiral side chains. The relation of the combinations in the two red boxes is diastereomorphous with respect to each other (the same is true for the blue combinations). This is in contrast to polypeptides with achiral side chains in which these combinations would be enantiomorphous.
Poly‐γ‐S‐2‐methylbutyl‐l‐glutamate and poly‐γ‐S‐2‐methylbutyl‐d‐glutamate have previously been used to determine the enantiodifferentiation between (+)‐isopinocampheol [(+)‐IPC] and (−)‐isopinocampheol [(−)‐IPC], respectively.36 The results indicated, that there is a “matched” and a “mismatched” situation. This means that the chiral information from the side chain works in one case hand in hand with the chiral backbone (matched), resulting in a higher enantiodifferentiation relative to the other case, in which the enantiodifferentiation is lower (mismatched). Nevertheless, the difference between the enantiodifferentiation of the two situations is rather small. The influence of the chiral backbone seemed to be higher than the influence of the chiral side chain. Thus, the idea to introduce a bulkier chiral side chain, which should have a bigger influence, came up. This should not only contribute to increase the amount of available highly enantiodifferentiating alignment media but also allow for further investigations concerning influences on enantiodifferentiation and the alignment process.
In the present study, we synthesized the novel polymers poly‐γ‐S‐perillyl‐l‐glutamate 2L and poly‐γ‐S‐perillyl‐d‐glutamate 2D which are diastereomers to each other. After the synthesis, we were able to prepare liquid crystalline phases of these polymers and we investigated their alignment properties concerning enantiodifferentiation and side chain influence. Furthermore, the usage of 1H–1H‐RDCs to improve the quality of alignment tensors is demonstrated.
Results and Discussion
Synthesis of poly‐γ‐S‐perillyl‐l‐glutamate 2L and poly‐γ‐S‐perillyl‐d‐glutamate 2D
In order to provide excellent spectral quality polypeptides of high molecular weight are necessary. It has already been shown for PBLG that the resulting analyte orientation does not depend on the molecular weight of the polypeptide.41 The same is assumed to be valid here. Nevertheless, the macromolecular properties of the two diastereomeric polymers should be comparable to avoid any disturbing influences thus asking for a controlled (ring‐opening) polymerization of N‐carboxyanhydrides (NCAs), which are directly obtained from the corresponding amino acid esters.
Our first step in that direction consists of synthesizing the amino acid ester 3L (Scheme 1). In the case of glutamic acid (4L), there are two carboxyl groups. The regioselectivity of the esterification must thus be controlled to take place exclusively at the γ‐carboxyl group. Different methods for regioselective γ‐esterification of the carboxyl group are known.42, 43, 44, 45, 46, 47 We tried different procedures (using N‐phthalyl protection,42 using tetrafluoroboric acid45 and using alkylboranes46, 47) to get access to 3L. None of them were successful, thus we ended up using the improved copper(II) strategy developed by Van Heeswijk.44 Glutamic acid 4L is converted into complex 5L by reaction with copper(II) acetate in excellent yields. The isolated complex 5L is then used in the esterification step, which starts with a change of the counterion by N,N,N′,N′‐tetramethylguanidine (TMG) to improve the solubility of the reactants. The following esterification proceeds regioselectively due to the complexation of the α‐carboxyl group by copper(II). The alkenyl bromide 6 used is derived from the corresponding alcohol 7 by an Appel reaction. The copper is successfully removed from the complex with a freshly prepared ethylenediaminetetraacetic acid solution (EDTA) in a following step.
Scheme 1.
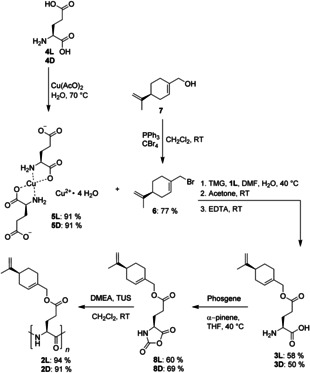
Synthesis of PSPLG 2L via regioselective γ‐esterification of 4L to 3L, following conversion to 8L using phosgene and polymerisation to yield 2L. Yields for the synthesis of PSPDG 2D, which follows the same route, are given underneath.
The glutamic acid ester 3L is then converted to the corresponding NCA 8L by reacting the ester 3L under an argon atmosphere with a solution of phosgene in toluene in the presence of α‐pinene. α‐pinene is added to avoid side‐reactions due to HCl, which stems from the conversion of phosgene.48 This is not only important to avoid side reactions that might lead to ring opening of the NCA48 but also to avoid hydrohalogenation of the double bonds in product 8L. After the reaction the solution becomes clear (indicating completed reaction), the product 8L is isolated by transferring the solution into n‐hexane. The white crystals obtained are recrystallized twice from tetrahydrofuran/n‐hexane by adding a n‐hexane layer slowly on top of a tetrahydrofuran layer containing the NCA by the use of a syringe pump. This strategy, which allows very slow crystallization, was used by our group for the purification of other NCAs before and is highly recommendable to obtain pure NCAs.41, 49 Last but not least we use the combination of N,N‐dimethyl ethanolamine (DMEA) and N,N′‐bis[3,5‐bis(trifluoromethyl)phenyl]thiourea50 (TUS) as initiation system to prepare the polypeptides.51 A monomer to initiator ratio of 500:1 is used to ensure that the desired polypeptides 2L and 2D are long enough for their use as alignment media. The synthesized polypeptides 2L and 2D have narrow molecular weight distributions (Table 1) and decent polydispersity indices (PDI). Furthermore, the two batches synthesized are comparable in terms of MW and PDI demonstrating the reproducibility of the polymerization.
Table 1.
GPC results for the synthesized polymer batches.
|
Polymer[a] |
M/I |
Mn [b] |
Mw [b] |
PDI |
|---|---|---|---|---|
|
S l 1 |
500 |
2.97×105 |
3.76×105 |
1.27 |
|
S d 1 |
500 |
2.87×105 |
3.69×105 |
1.29 |
|
S l 2 |
500 |
2.35×105 |
2.87×105 |
1.22 |
|
S d 2 |
500 |
2.06×105 |
2.81×105 |
1.36 |
[a] Configuration of side chain and amino acid followed by batch number. [b] Mn is the number average molar mass and Mw is the mass average molar mass. Expressed in g mol−1. Determined relative to polystyrene standards.
An important fact, which should be mentioned, is that the polymers were found not to be bench stable for prolonged periods of time. They become insoluble if they are stored at room temperature. We believe that this is due to the reactivity of the double bonds. This problem can be easily solved by storage in a freezer (−30 °C) in the dark. This is thus highly recommended. The polymers stored under these conditions are stable over months.
Secondary structures, liquid crystalline behavior and alignment properties
To determine the secondary structure of the polypeptides 2L and 2D synthesized, their chiroptical properties were investigated by the measurement of CD‐spectra (Figure 2) in 1,1,2,2‐tetrachloroethane (TCE) and THF (see Supporting Information). THF does not show a solvent cut off in the region of interest for α‐helical structures in polypeptides (about 220 nm). Thus, CD measurements in THF are straightforward. TCE is chosen as solvent as it is halogenated like chloroform (used for NMR). The issue with the solvent cutoff is circumvented by using a 0.01 mm cuvette. As expected, a negative Cotton effect was observed for the l‐polymer 2L and a positive Cotton effect was observed for the d‐polymer 2D. Additionally, the typical two (negative and positive) maxima for α‐helices in the region of 220 nm are observed. This indicates α‐helical secondary structures, being right and left handed, respectively, for the two polypeptides (2L and 2D). All these findings are in agreement with literature data for other polypeptides based on glutamic acid.36, 37 Despite the fact that the polymers are diastereoisomers, the curves are approximately mirror images with respect to each other. This behavior indicates the enantiomeric character of the backbone of the polypeptides. The chiral side chain has obviously no significant influence on the CD‐spectra. This finding is also in agreement with the literature.36 Additionally CD‐spectra of a 1:1 mixture of both polypeptides were measured. The resulting curve shows only a very minor amplitude confirming the “racemic” mixture of right‐ and left‐handed backbones. Additionally, the “racemic” mixtures are prepared by weighing, which is never perfectly exact.
Figure 2.
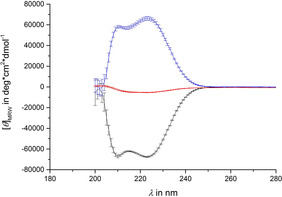
CD‐spectra of poly‐γ‐S‐perillyl‐l‐glutamate 2L (black), poly‐γ‐S‐perillyl‐d‐glutamate 2D (blue) and a 1:1 mixture (red) of them in TCE. [θ]MRW is the mean residue molar ellipticity.
In addition to the question of helicity, the desired ability to form lyotropic liquid crystalline phases was of interest and indicates whether the polypeptides are useful as alignment media. Therefore, samples of the polypeptides were prepared and viewed under crossed polarization filters (Figure 3). The birefringent behavior of these samples provide evidence for their liquid crystalline behavior.
Figure 3.

Samples of the polypeptides 2L (left) and 2D (right) in CDCl3 between crossed polarization filters. Birefringence is observed for the polypeptide samples in contrast to the isotropic water reference (middle).
Whether these phases are suitable as alignment media becomes clear only if quadrupolar splittings of the solvent signal are observed in the 2H‐spectra of the polypeptides in bulk samples of CDCl3 (see Figure 4). It is highly remarkable that different sizes of quadrupolar splitting are observed for the l‐polymer 2L and d‐polymer 2D with respect to each other despite identical concentrations and similar molecular weights. An analogous behavior is observed for all samples independent of analyte (see Supporting Information). It would thus be interesting to investigate whether the signs of the quadrupolar splittings of the solvent are different or identical in these diastereomeric media. The signs of the quadrupolar splittings of the solvent have been evaluated based on the known fact that positive one‐bond dipolar couplings (1 D CD) are related to negative quadrupolar splittings for the same bond (see corresponding equation in literature52). 1 T CD (was measured to be ca. 37 Hz in all samples), as determined from the 13C‐satellites in the 2H‐spectra and 1 J CD (32 Hz) of CDCl3 were used to calculate 1 D CD (1 T CD=1 J CD+21 D CD) which is thus positive in all samples. Hence, a negative sign results for the quadrupolar splitting of CDCl3 for all samples. Nevertheless, the different sizes of quadrupolar splitting suggest that this is caused by the diastereomeric relationship between the two polymers and was considered very promising in terms of enantiodiscrimination and the investigation of the influence of the side chain on the orientational process. The investigation of the enantiodifferentiation caused by each of the polypeptides, which might potentially be influenced as well, was therefore of interest as a next step.
Figure 4.
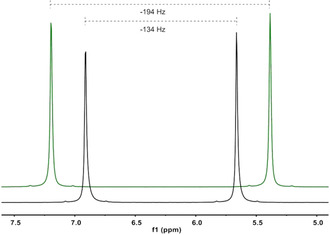
2H‐spectra (107 MHz, 300 K) of CDCl3 within LLC phases of 2L (black, 10.5 % w/w) and 2D (green, 10.6 % w/w) with (+)‐IPC, respectively. The 2H signal of [D6]acetone in the added capillary was used for chemical shift referencing. Note, that the shift of CDCl3 is affected by the presence of the polypeptides.
Enantiodifferentiation and influence of the side chain
For the investigation of the influence of the side chain′s chirality the enantiodifferentiation of a pair of enantiomers in both of the diastereomeric polypeptides has to be considered. It would be expected that a different extent of enantiodifferentiation, with respect to the two polymers, is observed. We decided to use the enantiomers of IPC as well as the enantiomers of β‐pinene as analytes due to the fact that they are rigid and both enantiomers of each of them are commercially available. Additionally their structure is well known and they were already used within RDC analyses.23, 37, 41 According to that, samples for all combinations ((+)‐ and (−)‐IPC in l‐polymer 2L and d‐polymer 2D and (+)‐ and (−)‐β‐pinene in l‐polymer 2L and d‐polymer 2D) in CDCl3 were prepared. The resulting CLean In Phase (CLIP)‐HSQC53 spectrum of one anisotropic sample is shown and compared to the isotropic one in Figure 5.
Figure 5.
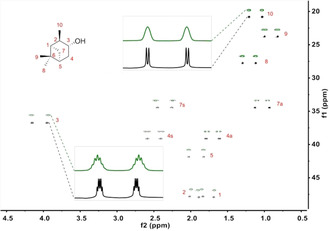
CLIP‐HSQC53 spectrum (700 MHz, 300 K) of (−)‐IPC in an anisotropic phase of PSPLG/CDCl3 (green, 10.6 % w/w) and (−)‐IPC in CDCl3 (black). The descriptors a (antiperiplanar) and s (synperiplanar) describe the orientation of the diastereotopic protons relative to the dimethyl bridge. Chemical shift referencing was done with respect to the isotropic measurement. The spectrum from the anisotropic measurement is shifted in order to obtain the stacked plot.
Narrow line shapes demonstrate the high quality of the spectra. Total and scalar coupling constants are extracted by reading out specific traces, following phase correction and fitting them to a copy of themselves using an established approach.54 The errors are determined using the same fitting approach, also reported in the literature.54 Residual dipolar couplings are calculated using the expression 1 T CH=1 J CH+21 D CH. Order tensors are determined for all data sets using the software RDC@hotFCHT.55, 56, 57 The eigenvectors of the Saupe tensors are shown in Figure 6.
Figure 6.
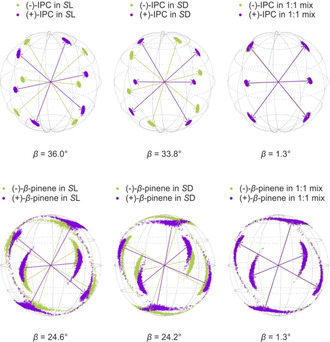
Eigenvectors of IPC (upper row) and β‐pinene (lower row) in both of the polymers (S l 2L and S d 2D) and in their 1:1 mixture. The corresponding β angles are given below. The arrows represent the eigenvectors of the best‐fitting SVD‐solution, while the scattered points show the distribution of the eigenvectors determined by Monte Carlo‐boot‐strapping within the range of experimental RDC uncertainties.
For IPC and β‐pinene, respectively, enantiodifferentiation is observed in both polypeptides. Enantiodifferentiation, in the case of RDC analysis, is usually quantified and discussed in terms of the generalized angle β 58, 59 which represents the “5D”‐angle between two alignment tensors. The cosine of this angle yields the normalized scalar product between them. Thus, a β‐angle of 90° means that the alignment tensors are perpendicular to each other, whereas a β‐angle of 0° means that they are collinear. Using the angle β to quantify enantiodifferentiation is straightforward as it does not only include considerations of the orientations but also considerations of the shapes of the compared tensors.16, 58, 59 The β‐angles determined for IPC in the l‐polymer 2L and in the d‐polymer 2D, respectively, are both above 30°, indicating comparatively high enantiodiscrimination. One would have expected different β‐angles for the two diastereomeric polymers, though. Surprisingly, these are very similar. This observation indicates that no significant influence of the side chain′s chirality on the orientation of IPC can be observed. This is the result expected for enantiomorphous systems, but not for diastereomeric polymers. For the β‐angles obtained from the measurements of β‐pinene analogous findings are made although tensor orientations are much less well defined. This is indicated by the rather large spread of points in Figure 6.
The vanishing difference in β between diastereomeric polymers was not expected and is not in accordance with the literature.36 Therefore further samples, containing both polymers in a 1:1 mixture, were prepared. These samples can be considered as containing a “racemic” backbone and shall thus demonstrate the influence of the side chain′s chirality. In the case of β‐pinene as well as for IPC, β‐angles of about 1.3° were obtained. These results are thus in accordance with the findings discussed above and seem to indicate that no influence of the bulky chiral side chain on enantiodifferentiation can be detected. In order to consolidate our findings, we tried to improve the quality of the β‐pinene tensors as a next step.
The use of 1H–1H‐RDCs to improve tensor quality
The alignment tensors obtained for the β‐pinene samples are not well defined, as shown by the point distribution obtained from the Monte Carlo output of our calculations. In order to improve the mathematical treatment of data and thus get more reliable tensor orientations we used 1H–1H‐RDCs. Their absolute values (in contrast to 13C–1H RDCs, the signs are not known a priori) are extracted from TSE‐PSYCHEDELIC60, 61 measurements. In order to use the 1H–1H‐RDCs additionally in combination with the 13C–1H RDCs and even on their own to calculate tensors, their signs have to be determined. For this purpose, P.E.HSQMBC62 experiments and the results from COSY based experiments, a method, which was described separately,61 are used in combination. The absolute signs obtained from the P.E.HSQMBC experiments are used as starting point to handle the relative information obtained by the COSY type experiments. All of the signs needed can be determined successfully using this approach. The resulting alignment tensors using the 1H–1H‐RDCs alone or in combination with the 13C–1H RDCs are much better defined than before. Their quality is improved enormously, as indicated by the much narrower point distribution in the Monte Carlo boot‐strapping (see Figure 7). The β‐angles from the l‐polymer 2L and from the d‐polymer 2D, respectively, are more or less the same, as compared to the 13C–1H‐only‐tensors. This is true for both calculations (1H–1H‐RDCs only and 1H–1H+13C–1H RDCs). Furthermore, the data obtained with the 1H–1H RDC measurements of the 1:1 polymer mixture show the same results as before. The β‐angles obtained for these are even closer to zero than before. Thereby, it is not only shown that the alignment tensor quality of β‐pinene can be improved by using 1H–1H RDC, but also that we have not drawn any wrong conclusions because of the lower quality of the 13C–1H‐only‐alignment tensors determined for β‐pinene.
Figure 7.
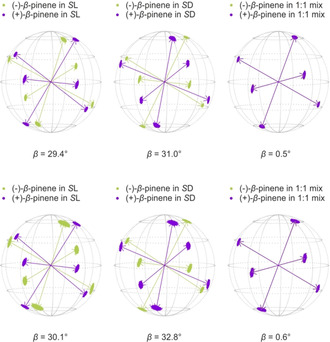
Eigenvectors of β‐pinene in both of the polymers (S l 2L and S d 2D) and in their 1:1 mixture. Calculated with 1H–1H RDCs only (upper row) and with 13C–1H+1H–1H RDCs (lower row). The corresponding β angles are given below. The arrows represent the eigenvectors of the best‐fitting SVD‐solution, while the scattered points show the distribution of the eigenvectors determined by Monte Carlo‐boot‐strapping within the range of experimental RDC uncertainties.
Discussion of the influence of the side chain chirality
All of our observations indicate that the influence of the side chain's chirality is rather negligible for the samples investigated here. Nevertheless, the extent of the enantiodifferentiation obtained is high. In comparison to other alignment media based on glutamic acid it is the highest enantiodifferentiation, obtained under comparable experimental conditions, for IPC up to now.23, 36, 37 Over the years of research on the field of alignment media based on polypeptides, a few speculations have been made. It seemed as if side chains like the benzyl group in PBLG lower the enantiodifferentiation relative to smaller ones like the ethyl group in PELG. These findings were explained by more or less steric demand of the side chain, influencing the accessibility to the chiral backbone.23, 36 The results from this work clearly show that high enantiodifferentiation is possible even if there are bulky side chains and this contradicts previous findings. These results are in accordance with the data of another new alignment medium with biphenyl side chains.37 We were able to synthesize a novel alignment medium with a chiral bulky side chain which shows high enantiodifferentiation. This seems, however, not to be due to the chirality of the side chain alone but rather by the performance of the whole polymer. This result is very interesting and it shows that the factors controlling or rather guiding the interactions during the alignment process are very complex and far from being understood. The speculation that the chirality of the side chain always induces significant enantiodifferentiation and must lead to a matched/mismatched situation is disproven. The question which factors contribute to the enantiodifferentiation remains open. The data available up to now do not show a clear trend between induced enantiodifferentiation and chemical or steric properties of the side chain. To get some more hints and to be able to predict some of the interactions, which are highly important for the resulting orientation, further investigations have to be carried out.
Conclusions
Two polypeptides based on glutamic acid with chiral side chains, which are diastereomeric with respect to each other were successfully synthesized. They have α‐helical secondary structures, as indicated by CD spectra. Furthermore they are able to form lyotropic liquid crystalline phases which can be used as alignment media within the RDC approach. The enantiomers of IPC and β‐pinene were used for investigations concerning the alignment properties. The quality of the alignment tensors determined for β‐pinene was significantly improved by the use of 1H–1H‐RDCs. For both analytes (IPC and β‐pinene) high enantiodifferentiation was observed. Furthermore, the enantiodifferentiation for each analyte pair was compared between the diastereomeric LLC phases. Additionally, 1:1‐mixtures of the diastereomeric polypeptides were used to determine the resulting enantiodifferentiation in order to get information about the influence of the side chain′s chirality. This influence was determined to be negligible. Nevertheless, the extent of enantiodifferentiation is really high in comparison with other media. These results are interesting and demonstrate that the factors leading to high enantiodifferentiation are still not understood. The purposive design and the synthesis of highly promising alignment media is thus still very challenging.
Experimental Section
Synthesis of S‐perillyl‐bromide
The synthesis of S‐perillyl‐bromide was carried out following a published procedure.63 S‐perillyl alcohol (7) (50.057 g, 328.8 mmol, 1.0 equiv) and tetrabromomethane (121.138 g, 365.3 mmol 1.1 equiv) were dissolved in dichloromethane (150 mL). Triphenylphosphine (94.924 g, 361.9 mmol 1.1 equiv) was added under cooling, using an ice bath. The reaction solution was stirred for 18 h at room temperature. The solvent was removed under reduced pressure. The obtained residue was poured into 300 mL of n‐hexane. The resulting suspension was filtered using silica gel. Additional n‐hexane was used to wash. The solvent was removed under reduced pressure. The remaining oil was distilled under vacuum. Finally, the product 6 (54.601 g, 77.3 %) was obtained as a colorless to light yellow oil.
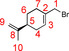
=−63.30° (c=1, CHCl3); 1H NMR (700 MHz, 300 K, CDCl3): δ=1.48–1.55 (m, 1 H, 6‐Ha), 1.74 (s, 3 H, 10‐H), 1.85–1.90 (m, 1 H, 6‐Hb), 1.93–2.00 (m, 1 H, 4‐Ha), 2.12–2.21 (m, 2 H, 4‐Hb, 5‐H), 2.21–2.25 (m, 2 H, 7‐H), 3.93–3.98 (m, 2 H, 1‐H), 4.70–4.72 (m, 1 H, 9‐Ha) 4.73–4.75 (m, 1 H, 9‐Hb), 5.88–5.91 ppm (m, 1 H, 3‐H); 13C NMR (175 MHz, 300 K, CDCl3): δ=20.9 (10‐C), 27.0 (7‐C), 27.4 (6‐C), 31.0 (4‐C), 39.3 (1‐C), 40.7 (5‐C), 109.1 (9‐C), 127.8 (3‐C), 134.5 (2‐C), 149.5 ppm (8‐C). The NMR data are in accordance with the literature.64, 65
Synthesis of γ‐S‐perillyl‐l‐glutamate
The synthesis of γ‐S‐perillyl‐l‐glutamate was performed following a literature procedure.44 Glutamic acid (4L) (29.366 g, 166.6 mmol, 1.0 equiv) was suspended in 750 mL of water. A solution of copper(II) acetate monohydrate (41.286 g, 206.8 mmol, 1.2 equiv) in water (750 mL) was added dropwise to this suspension at 70 °C. After addition, the solution was allowed to cool down. To ensure complete crystallization, the solution was kept at room temperature for 2 d. Filtration, washing steps with water, ethanol and diethyl ether and drying in high vacuum yielded the complex 5L (44.556 g, 91.2 %) as a blue fine powder. This l‐glutamic acid copper(II) complex (5L) (29.486 g, 60.3 mmol, 1.0 equiv) and glutamic acid (4L) (17.759 g, 120.7 mmol, 2.0 equiv) were suspended in a mixture of N,N‐dimethylformamide (DMF, 107 mL) and water (17.3 mL). N,N,N′,N′‐tetramethylguanidine (30 mL, 283.7 mmol, 4.7 equiv) was added subsequently. The mixture was stirred for 2 h, so that a homogenous solution was obtained. Additional DMF (86 mL) and the alkenyl bromide 6 (54.602 g, 253.8 mmol, 4.2 equiv) were added. The resulting mixture was stirred at 40 °C for 56 h. After this time, 1.45 L of acetone were added. A fine precipitate resulted after 3 h of stirring and was isolated by filtration. The resulting powder was suspended in a freshly prepared solution of ethylenediaminetetraacetic acid (34.455 g, 117.9 mmol 2.0 equiv.) and sodium hydrogen carbonate (19.430 g, 231.3 mmol, 3.8 equiv.) in water (280 mL). The suspension was stirred for 16 h. A following filtration and several wash cycles using water, yielded a white to light blue solid. The product 3L (39.883 g, 58.8 %) was obtained as a white solid by recrystallization, using a water/ethanol (2:1) mixture.
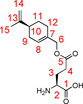
=−38.40° (c=0.25, MeOH); 1H NMR (700 MHz, 300 K, D2O+DCl): δ=1.32–1.40 (m, 1 H, 11‐Ha), 1.63 (s, 3 H, 15‐H), 1.70–1.75 (m, 1 H, 11‐Hb), 1.81–1.88 (m, 1 H, 9‐Ha), 1.95–2.00 (m, 2 H, 12‐H), 2.01–2.08 (m, 2 H, 9‐Hb, 10‐H), 2.13–2.23 (m, 2 H, 3‐H), 2.53–2.63 (m, 2 H, 4‐H), 4.06 (t, 1 H, 2‐H, 3J2,3=6.7 Hz), 4.42 (s, 2 H, 6‐H), 4.63 (s, 2 H, 14‐H), 5.69 ppm (s, 1 H, 8‐H); 13C NMR (100 MHz, 300 K, D2O+DCl): δ=20.3 (15‐C), 25.0 (3‐C), 25.9 (12‐C), 27.0 (11‐C), 29.7 (4‐C), 30.2 (9‐C), 40.5 (10‐C), 52.0 (2‐C), 69.0 (6‐C), 108.4 (14‐C), 125.9 (8‐C), 132.4 (7‐C), 150.2 (13‐C), 171.3 (1‐C), 173.8 ppm (5‐C); MS (ESI): m/z=calcd: 282.16998 [C15H24NO4]+ found: 282.17012 [M+H]+.
Synthesis of γ‐S‐perillyl‐d‐glutamate
The d‐ester 3D and the corresponding complex 5D (91.7 %) were synthesized according to the procedure described for the l‐ester 3L. The product 3D was obtained as a white solid (50.3 %).
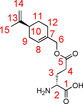
=−49.20° (c=0.25, MeOH); 1H NMR (600 MHz, 300 K, D2O+DCl): δ=1.31–1.40 (m, 1 H, 11‐Ha), 1.62 (s, 3 H, 15‐H), 1.70–1.75 (m, 1 H, 11‐Ha), 1.80–1.88 (m, 1 H, 9‐Ha), 1.94–2.00 (m, 2 H, 12‐H), 2.00–2.08 (m, 2 H, 9‐Hb, 10‐H), 2.13–2.24 (m, 2 H, 3‐H), 2.53–2.64 (m, 2 H, 4‐H), 4.06 (t, 1 H, 2‐H, 3J2,3=6.7 Hz), 4.38–4.45 (m, 2 H, 6‐H), 4.62 (s, 2 H, 14‐H), 5.69 ppm (s, 1 H, 8‐H); 13C NMR (100 MHz, 300 K, D2O+DCl): δ=20.3 (15‐C), 24.9 (3‐C), 25.9 (12‐C), 27.0 (11‐C), 29.7 (4‐C), 30.2 (9‐C), 40.4 (10‐C), 51.9 (2‐C), 69.2 (6‐C), 108.4 (14‐C), 126.0 (8‐C), 132.4 (7‐C), 150.4 (13‐C), 171.1 (1‐C), 173.8 ppm (5‐C); MS (ESI): m/z=calcd: 282.16998 [C15H24NO4]+ found: 282.16993 [M+H]+.
Synthesis of γ‐S‐perillyl‐l‐glutamic acid N‐carboxyanhydride
The synthesis of the NCA 8L was done following literature procedures.48, 66 The amino acid ester 3L (10.004 g, 35.6 mmol, 1.0 equiv.) and α‐pinene (12.071 g, 88.6 mmol, 2.5 equiv.) were added, under argon, to absolute tetrahydrofuran (THF, 90 mL). A solution of 20 % phosgene in toluene (22.4 mL, 48.4 mmol, 1.4 equiv.) was added to this suspension. The resulting mixture was stirred at 40 °C for 2 h. The clear reaction solution was then added to absolute n‐hexane (360 mL) using a syringe filter (0.45 μm, PTFE). The precipitate obtained was isolated by filtration under inert conditions, washed with 50 mL absolute n‐hexane and dried under high vacuum. The crystals were redissolved in absolute THF (3.5 mL g−1) and recrystallized, by layering with n‐hexane (10.5 mL g−1) using a syringe pump. Filtration, washing and drying were repeated and followed by a second recrystallization using the diffusion controlled crystallisation again. The product 8L (6.640 g, 60.8 %) was finally obtained as white crystals by filtration, washing and drying.
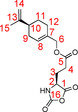
1H NMR (600 MHz, 300 K, [D8]THF): δ=1.44–1.52 (m, 1 H, 11‐Ha), 1.73 (s, 3 H, 15‐H), 1.81–1.86 (m, 1 H, 11‐Hb), 1.92–2.00 (m, 1 H, 9‐Ha), 1.97–2.03 (m, 1 H, 3‐H), 2.06–2.11 (m, 2 H, 12‐H), 2.11–2.17 (m, 3 H, 3‐H, 9‐Hb, 10‐H), 2.46–2.50 (m, 2 H, 4‐H), 4.37–4.40 (m, 1 H, 2‐H), 4.42–4.49 (m, 2 H, 6‐H), 4.69–4.71 (m, 2 H, 14‐H), 5.72–5.75 (m, 1 H, 8‐H) 7.91 ppm (s, 1 H, N‐H); 13C NMR (150 MHz, 300 K, [D8]THF): δ=20.7 (15‐C), 26.9 (12‐C), 27.8 (3‐C), 28.1 (11‐C), 29.7 (4‐C), 31.1 (9‐C), 41.7 (10‐C), 57.1 (2‐C), 68.7 (6‐C), 109.0 (14‐C), 125.9 (8‐C), 133.7 (7‐C), 150.1 (13‐C), 152.4 (16‐C), 171.2 (1‐C), 172.0 ppm (5‐C).
Synthesis of γ‐S‐perillyl‐d‐glutamic acid N‐carboxyanhydride
The synthesis of the d‐derivative 8D was achieved using the same procedure as described for the l‐NCA 8L. The product 8D (69.1 %) was obtained as white crystals.
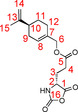
1H NMR (700 MHz, 300 K, [D8]THF): δ=1.44–1.51 (m, 1 H, 11‐Ha), 1.73 (s, 3 H, 15‐H), 1.81–1.86 (m, 1 H, 11‐Hb), 1.93–1.99 (m, 1 H, 9‐Ha), 1.97–2.03 (m, 1 H, 3‐H), 2.06–2.11 (m, 2 H, 12‐H), 2.11–2.17 (m, 3 H, 3‐H, 9‐Hb, 10‐H), 2.48 (t, 2 H, 4‐H, 3J3,4=7.5 Hz), 4.37–4.40 (m, 1 H, 2‐H), 4.42–4.49 (m, 2 H, 6‐H), 4.69–4.71 (m, 2 H, 14‐H), 5.73–5.75 (m, 1 H, 8‐H) 7.92 ppm (s, 1 H, N‐H); 13C NMR (175 MHz, 300 K, [D8]THF): δ=20.7 (15‐C), 26.9 (12‐C), 27.8 (3‐C), 28.1 (11‐C), 29.7 (4‐C), 31.1 (9‐C), 41.7 (10‐C), 57.1 (2‐C), 68.7 (6‐C), 109.0 (14‐C), 125.9 (8‐C), 133.7 (7‐C), 150.1 (13‐C), 152.4 (16‐C), 171.2 (1‐C), 172.0 ppm (5‐C).
Synthesis of poly‐γ‐S‐perillyl‐l‐glutamate
The synthesis of the polypeptide 2L was done following a literature procedure for PBLG.51 The corresponding NCA 8L (2.000 g, 6.5 mmol) was dissolved in degassed absolute dichloromethane (40 mL) inside of a glovebox. 670 μL of a stock solution, containing 50.3 mg TUS per 5.2 mL (9.67 mg mL−1) degassed absolute dichloromethane, were added. Subsequently, 160 μL of a stock solution, containing 40 μL DMEA per 5ml (7.12 mg mL−1) degassed absolute dichloromethane, were added. The resulting reaction mixture was stirred for 116 h. The reaction progress was controlled using ATR‐IR spectroscopy. Isolation of the polypeptide was achieved by precipitation in n‐hexane. Drying in high vacuum was followed by a second precipitation from dichloromethane/n‐hexane. The finally high vacuum dried product 2L (1.627 g, 94.9 %) was obtained as a white rubberlike solid. The NMR spectra of the polypeptide 2L showed broad signals as common for polymers. Multiplicity was beyond recognition and signals of the backbone or near to it could not be observed in 13C spectra.
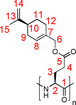
=−47.50° (c=0.1, THF); 1H NMR (700 MHz, 300 K, CDCl3): δ=1.46 (br, 1 H, 11‐Ha), 1.71 (br, 3 H, 15‐H), 1.82 (br, 1 H, 11‐Hb), 1.93 (br, 1 H, 9‐Ha), 2.05 (br, 2 H, 12‐H), 2.12 (br, 2 H, 9‐Hb, 10‐H), 2.37 (br, 2 H, 3‐H), 2.72 (br, 2 H, 4‐H), 3.99 (br, 1 H, 2‐H), 4.37 (br, 1 H, 6‐Ha), 4.54 (br, 1 H, 6‐Hb), 4.69 (br, 1 H, 14‐Ha), 4.70 (br, 1 H, 14‐Hb), 5.71 ppm (br, 1 H, 8‐H); 13C NMR (175 MHz, 300 K, CDCl3): δ=20.9 (15‐C), 26.5 (12‐C), 27.5 (11‐C), 30.6 (9‐C), 41.0 (10‐C), 68.3 (6‐C), 108.9 (14‐C), 125.3 (8‐C), 132.8 (7‐C), 149.7 (13‐C), 172.3 ppm (5‐C).
Synthesis of poly‐γ‐S‐perillyl‐d‐glutamate
The synthesis of the d‐polypeptide 2D was done according to the procedure used for the l‐polypeptide 2L. The resulting product 2D (91.4 %) was obtained as a white rubberlike solid. The NMR spectra of the polypeptide 2D showed broad signals as common for polymers. Multiplicity was beyond recognition and signals of the backbone or near to it could not be observed in 13C spectra.
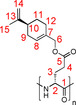
=−60.50° (c=0.1, THF); 1H NMR (700 MHz, 300 K, CDCl3): δ=1.46 (br, 1 H, 11‐Ha), 1.71 (br, 3 H, 15‐H), 1.82 (br, 1 H, 11‐Hb), 1.93 (br, 1 H, 9‐Ha), 2.05 (br, 2 H, 12‐H), 2.13 (br, 2 H, 9‐Hb, 10‐H), 2.37 (br, 2 H, 3‐H), 2.73 (br, 2 H, 4‐H), 3.98 (br, 1 H, 2‐H), 4.38 (br, 1 H, 6‐Ha), 4.53 (br, 1 H, 6‐Hb), 4.69 (br, 1 H, 14‐Ha), 4.70 (br, 1 H, 14‐Hb) 5.71 ppm (br, 1 H, 8‐H); 13C NMR (175 MHz, 300 K, CDCl3): δ=20.9 (15‐C), 26.5 (12‐C), 27.5 (11‐C), 30.6 (9‐C), 41.0 (10‐C), 68.3 (6‐C), 108.9 (14‐C), 125.3 (8‐C), 132.8 (7‐C), 149.7 (13‐C), 172.3 ppm (5‐C).
NMR samples and data
Detailed information on sample preparation as well as the collection of NMR data are given in the supporting information.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors thank the Fonds der chemischen Industrie (FCI) for a Ph.D. scholarship and financial support. We would like to thank Davy Sinnaeve for being able to use the pulse sequences for 1H–1H‐RDC measurements prior to publication and Julian Ilgen for the help with setup and evaluation of the resulting data. Furthermore, we acknowledge Volker Schmidts for the support concerning the software RDC@hotfcht and Max Hirschmann for advice concerning the 0.01 mm cuvettes for CD measurements.
M. Alcaraz Janßen, C. M. Thiele, Chem. Eur. J. 2020, 26, 7831.
References
- 1. Schmidts V., Magn. Reson. Chem. 2017, 55, 54–60. [DOI] [PubMed] [Google Scholar]
- 2. Kummerlöwe G., Luy B., TrAC Trends Anal. Chem. 2009, 28, 483–493. [Google Scholar]
- 3. Overhauser A. W., Phys. Rev. 1953, 92, 411–415. [Google Scholar]
- 4. Anet F. A. L., Bourn A. J. R., J. Am. Chem. Soc. 1965, 87, 5250–5251. [Google Scholar]
- 5. Kaiser R., J. Chem. Phys. 1965, 42, 1838–1839. [Google Scholar]
- 6. Karplus M., J. Chem. Phys. 1959, 30, 11–15. [Google Scholar]
- 7. Haasnoot C. A. G., de Leeuw F. A. A. M., Altona C., Tetrahedron 1980, 36, 2783–2792. [Google Scholar]
- 8. Tjandra N., Bax A., Science 1997, 278, 1111. [DOI] [PubMed] [Google Scholar]
- 9. Thiele C. M., Eur. J. Org. Chem. 2008, 5673–5685. [Google Scholar]
- 10. Luy B., J. Indian Inst. Sci. 2010, 90, 119–132. [Google Scholar]
- 11. Böttcher B., Thiele C. M. in EMagRes, Wiley, 2012. [Google Scholar]
- 12. Kummerlöwe G., Kiran M. U., Luy B., Chem. Eur. J. 2009, 15, 12192–12195. [DOI] [PubMed] [Google Scholar]
- 13. Schmidt M., Sun H., Leonov A., Griesinger C., Reinscheid U. M., Magn. Reson. Chem. 2012, 50, S38–S44. [DOI] [PubMed] [Google Scholar]
- 14. Montag T., Thiele C. M., Chem. Eur. J. 2013, 19, 2271–2274. [DOI] [PubMed] [Google Scholar]
- 15. Aroulanda C., Sarfati M., Courtieu J., Lesot P., Enantiomer 2001, 6, 281–287. [PubMed] [Google Scholar]
- 16. Schwab M., Schmidts V., Thiele C. M., Chem. Eur. J. 2018, 24, 14373–14377. [DOI] [PubMed] [Google Scholar]
- 17. Li G.-W., Cao J.-M., Zong W., Hu L., Hu M.-L., Lei X., Sun H., Tan R. X., Chem. Eur. J. 2017, 23, 7653–7656. [DOI] [PubMed] [Google Scholar]
- 18. Leyendecker M., Meyer N.-C., Thiele C. M., Angew. Chem. Int. Ed. 2017, 56, 11471–11474; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 11629–11632. [Google Scholar]
- 19. Knoll K., Leyendecker M., Thiele C. M., Eur. J. Org. Chem. 2019, 720–727. [Google Scholar]
- 20. Lesot P., Aroulanda C., Berdagué P., Meddour A., Merlet D., Farjon J., Giraud N., Lafon O., Prog. Nucl. Magn. Reson. Spectrosc. 2020, 116, 85–154. [DOI] [PubMed] [Google Scholar]
- 21. Meddour A., Canet I., Loewenstein A., Pechine J. M., Courtieu J., J. Am. Chem. Soc. 1994, 116, 9652–9656. [Google Scholar]
- 22. Marx A., Schmidts V., Thiele C. M., Magn. Reson. Chem. 2009, 47, 734–740. [DOI] [PubMed] [Google Scholar]
- 23. Hansmann S., Larem (née Montag) T., Thiele C. M., Eur. J. Org. Chem. 2016, 1324–1329. [Google Scholar]
- 24. Thiele C. M., J. Org. Chem. 2004, 69, 7403–7413. [DOI] [PubMed] [Google Scholar]
- 25. Meyer N.-C., Krupp A., Schmidts V., Thiele C. M., Reggelin M., Angew. Chem. Int. Ed. 2012, 51, 8334–8338; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 8459–8463. [Google Scholar]
- 26. Krupp A., Reggelin M., Magn. Reson. Chem. 2012, 50, S45–S52. [DOI] [PubMed] [Google Scholar]
- 27. Arnold L., Marx A., Thiele C. M., Reggelin M., Chem. Eur. J. 2010, 16, 10342–10346. [DOI] [PubMed] [Google Scholar]
- 28. Reller M., Wesp S., Koos M. R. M., Reggelin M., Luy B., Chem. Eur. J. 2017, 23, 13351–13359. [DOI] [PubMed] [Google Scholar]
- 29. Sarfati M., Courtieu J., Lesot P., Chem. Commun. 2000, 1113–1114. [Google Scholar]
- 30. Lesot P., Sarfati M., Courtieu J., Chem. Eur. J. 2003, 9, 1724–1745. [DOI] [PubMed] [Google Scholar]
- 31. Lafon O., Lesot P., Rivard M., Chavarot M., Rose-Munch F., Rose E., Organometallics 2005, 24, 4021–4028. [Google Scholar]
- 32. Sarfati M., Lesot P., Merlet D., Courtieu J., Chem. Commun. 2000, 2069–2081. [Google Scholar]
- 33. Lesot P., Berdagué P., Meddour A., Kreiter A., Noll M., Reggelin M., ChemPlusChem 2019, 84, 144–153. [DOI] [PubMed] [Google Scholar]
- 34. Berger R., Courtieu J., Gil R. R., Griesinger C., Köck M., Lesot P., Luy B., Merlet D., Navarro-Vázquez A., Reggelin M., Reinscheid U. M., Thiele C. M., Zweckstetter M., Angew. Chem. Int. Ed. 2012, 51, 8388–8391; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 8512–8515. [Google Scholar]
- 35. Schwab M., Herold D., Thiele C. M., Chem. Eur. J. 2017, 23, 14576–14584. [DOI] [PubMed] [Google Scholar]
- 36. Hansmann S., Schmidts V., Thiele C. M., Chem. Eur. J. 2017, 23, 9114–9121. [DOI] [PubMed] [Google Scholar]
- 37. Jeziorowski S., Thiele C. M., Chem. Eur. J. 2018, 24, 15631–15637. [DOI] [PubMed] [Google Scholar]
- 38. Hirschmann M., Schwab M., Thiele C. M., Macromolecules 2019, 52, 6025–6034. [Google Scholar]
- 39. Serhan Z., Aroulanda C., Lesot P., J. Phys. Chem. A 2016, 120, 6076–6088. [DOI] [PubMed] [Google Scholar]
- 40. Lesot P., Lafon O., Aroulanda C., Dong R. Y., Chem. Eur. J. 2008, 14, 4082–4092. [DOI] [PubMed] [Google Scholar]
- 41. Marx A., Thiele C., Chem. Eur. J. 2009, 15, 254–260. [DOI] [PubMed] [Google Scholar]
- 42. Yamamoto H., Kondo Y., Hayakawa T., Biopolymers 1970, 9, 41–52. [Google Scholar]
- 43. Schumann I., Boissonnas R. A., Nature 1952, 169, 154–155. [Google Scholar]
- 44. Van Heeswijk W. A. R., Eenink M. J. D., Feijen J., Synthesis 1982, 744–747. [Google Scholar]
- 45. Albert R., Danklmaier J., Hönig H., Kandolf H., Synthesis 1987, 635–637. [Google Scholar]
- 46. Nefkens G. H. L., Zwanenburg B., Tetrahedron 1983, 39, 2995–2998. [Google Scholar]
- 47. Albericio F., Nicolas E., Rizo J., Ruiz-Gayo M., Pedroso E., Giralt E., Synthesis 1990, 119–122. [Google Scholar]
- 48. Smeets N. M. B., van der Weide P. L. J., Meuldijk J., Vekemans J. A. J. M., Hulshof L. A., Org. Process Res. Dev. 2005, 9, 757–763. [Google Scholar]
- 49. Kramer J. R., Deming T. J., Biomacromolecules 2010, 11, 3668–3672. [DOI] [PubMed] [Google Scholar]
- 50. Lippert K. M., Hof K., Gerbig D., Ley D., Hausmann H., Guenther S., Schreiner P. R., Eur. J. Org. Chem. 2012, 5919–5927. [Google Scholar]
- 51. Zhao W., Gnanou Y., Hadjichristidis N., Polym. Chem. 2015, 6, 6193–6201. [Google Scholar]
- 52. Emsley J. W., Lesot P., Merlet D., Phys. Chem. Chem. Phys. 2004, 6, 522–530. [Google Scholar]
- 53. Enthart A., Freudenberger J. C., Furrer J., Kessler H., Luy B., J. Magn. Reson. 2008, 192, 314–322. [DOI] [PubMed] [Google Scholar]
- 54. Kummerlöwe G., Schmitt S., Luy B., Open Spectrosc. J. 2010, 4, 16–27. [Google Scholar]
- 55. Berger R., Fischer C., Klessinger M., J. Phys. Chem. A 1998, 102, 7157–7167. [Google Scholar]
- 56. Thiele C. M., Schmidts V., Böttcher B., Louzao I., Berger R., Maliniak A., Stevensson B., Angew. Chem. Int. Ed. 2009, 48, 6708–6712; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 6836–6840. [Google Scholar]
- 57.V. Schmidts, Ph.D. Thesis, TU Darmstadt, 2013.
- 58. Sass J., Cordier F., Hoffmann A., Rogowski M., Cousin A., Omichinski J. G., Löwen H., Grzesiek S., J. Am. Chem. Soc. 1999, 121, 2047–2055. [Google Scholar]
- 59. Kramer F., Deshmukh M. V., Kessler H., Glaser S. J., Concepts Magn. Reson. Part A 2004, 21, 10–21. [Google Scholar]
- 60. Sinnaeve D., Foroozandeh M., Nilsson M., Morris G. A., Angew. Chem. Int. Ed. 2016, 55, 1090–1093; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 1102–1105. [Google Scholar]
- 61. Sinnaeve D., Ilgen J., Di Pietro M. E., Primozic J. J., Schmidts V., Thiele C. M., Luy B., Angew. Chem. Int. Ed. 2020, 59, 5316–5320; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 5354–5358. [Google Scholar]
- 62. Saurí J., Nolis P., Castañar L., Virgili A., Parella T., J. Magn. Reson. 2012, 224, 101–106. [DOI] [PubMed] [Google Scholar]
- 63. Baughman T. W., Sworen J. C., Wagener K. B., Tetrahedron 2004, 60, 10943–10948. [Google Scholar]
- 64. Barrero A. F., Herrador M. M., Quílez del Moral J. F., Arteaga P., Arteaga J. F., Diéguez H. R., Sánchez E. M., J. Org. Chem. 2007, 72, 2988–2995. [DOI] [PubMed] [Google Scholar]
- 65. Wu X., Wang H.-J., Huang Y.-S., Li W.-D. Z., Org. Lett. 2018, 20, 1871–1874. [DOI] [PubMed] [Google Scholar]
- 66.T. Montag, Ph.D. Thesis, TU Darmstadt, 2013.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary