

Abstract
Background and Aims
Biliary atresia (BA) is a devastating neonatal cholangiopathy that progresses to fibrosis and end‐stage liver disease by 2 years of age. Portoenterostomy may reestablish biliary drainage, but, despite drainage, virtually all afflicted patients develop fibrosis and progress to end‐stage liver disease requiring liver transplantation for survival.
Approach and Results
In the murine model of BA, rhesus rotavirus (RRV) infection of newborn pups results in a cholangiopathy paralleling human BA and has been used to study mechanistic aspects of the disease. Unfortunately, nearly all RRV‐infected pups succumb by day of life 14. Thus, in this study we generated an RRV‐TUCH rotavirus reassortant (designated as TR(VP2,VP4)) that when injected into newborn mice causes an obstructive jaundice phenotype with lower mortality rates. Of the mice that survived, 63% developed Ishak stage 3‐5 fibrosis with histopathological signs of inflammation/fibrosis and bile duct obstruction.
Conclusions
This model of rotavirus‐induced neonatal fibrosis will provide an opportunity to study disease pathogenesis and has potential to be used in preclinical studies with an objective to identify therapeutic targets that may alter the course of BA.
Abbreviations
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- AREG/AREGB
amphiregulin
- BA
biliary atresia
- CCL2
chemokine (C‐C motif) ligand 2
- CD
cluster of differentiation
- CK‐19
cytokeratin 19
- CXCL1
chemokine (C‐X‐C motif) ligand 1
- DOL
day of life
- GEO
Gene Expression Omnibus
- H&E
hematoxylin and eosin
- IL
interleukin
- ITGA2
integrin alpha 2
- LAMC2
laminin, gamma 2
- MMP‐7
matrix metalloproteinase 7
- NK
natural killer
- RRV
rhesus rotavirus
- SERPINE1
serpin peptidase inhibitor, plasminogen activator inhibitor type 1
- THBS1
thrombospondin 1
- TUCH
rotavirus named after the locations where the strain was isolated: Tulane National Primate Research Center and Cincinnati Children’s Hospital
- VCAN
versican
Biliary atresia (BA) is a devastating fibro‐obliterative cholangiopathy of the newborn that affects approximately 1:5,000‐18,000 infants. The inflammation results in end‐stage liver disease and, without surgical intervention, leads to death by 2 years of age.1, 2 Although Kasai portoenterostomy may restore biliary drainage in some infants, virtually all afflicted patients develop hepatic fibrosis associated with complications of portal hypertension and require liver transplantation for survival.3, 4 As a result, BA is the most common indication for pediatric liver transplantation, accounting for 50% of all pediatric transplants performed in the United States.5
The etiology of BA remains under investigation. In 1974, Landing6 suggested that a perinatal viral infection was the main cause of BA. The hypothesis has been supported by the detection of several viruses, including rotavirus group C,7 reovirus type 3,8 Epstein‐Barr virus,9 cytomegalovirus,10 and human papillomavirus11 in explanted livers of infants with BA. Additional support for a virus‐induced BA is the experimental perinatal infection of mice with rhesus rotavirus (RRV) that results in extrahepatic biliary obstruction paralleling human BA.12, 13 In this model 100% of newborn pups that are inoculated with RRV by day of life (DOL) 1 develop symptoms of biliary obstruction including growth retardation, jaundice, acholic stool, and hyperbilirubinemia, with a 90%‐100% mortality rate by DOL 14.13 Subsequent histopathological analysis of extrahepatic bile ducts in these mice has revealed obstruction of the bile duct lumen, similar to that seen in human BA.14
RRV is a double‐stranded RNA virus of the Reoviridae family composed of 11 gene segments encoding six structural (VP1‐VP4, VP6, and VP7) and six nonstructural (NSP1‐NSP6) proteins. It contains a triple‐layer protein capsid composed of VP6, VP4, and VP7.15 Reassortant viruses are created by coinfection of a cell with two parent strains that can produce progeny composed of various combinations of parental genes. Previously, using single‐gene reassortants derived from rotavirus strains RRV and TUCH (TUCH is another simian rotavirus strain that has tropism for murine cholangiocytes, but infection does not result in biliary obstruction; named after the locations where the strain was isolated: Tulane National Primate Research Center and Cincinnati Children’s Hospital), we established that the RRV’s VP4 is critical for the induction of the murine model of BA.16
Although the RRV‐induced model of murine BA has many parallels with human disease, it results in 80%‐90% mortality by DOL 14. As a result, studying late effects of biliary obstruction such as the mechanistic basis of fibrosis is not possible. While murine models of liver fibrosis using hepatotoxins17 and bile duct ligation18 have been described, a neonatal model of liver fibrosis as a sequela of perinatal biliary obstruction has not yet been established. In this study, we describe a model of neonatal hepatic fibrosis that resulted from experimental inoculation of a reassortant RRV strain inducing murine BA with lower mortality rates. In addition, we analyzed the similarities of this model to human BA and differences from CCl4 injection and duct ligation models.
Methods
Viruses and Cells
MA104 cells (Bio‐Whittaker, Walkersville, MD) were cultured as described.19 Two simian rotavirus strains were used: RRV (generously provided by H. Greenberg, Stanford University, Palo Alto, CA) and TUCH.20 Virus stocks were grown in MA104 cells as described and kept frozen at −80°C until thawed for use.14
Animal Breeding
Breeding pairs of BALB/c mice (Envigo, Indianapolis, IN) were kept in micro isolator cages in a virus‐free environment with free access to sterilized chow and water. Mice were bred and pups in litters of 6‐8 were used for experiments. All animal research was performed in accordance with protocols and regulations approved by the Institutional Animal Care and Use Committee at Cincinnati Children's Hospital Medical Center (protocol number IACUC2016‐0090), which adheres to the NIH OLAW regulation (Animal Assurance number A3108) and the Animal Welfare Act (certification number 31‐8‐001). All animals received humane care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals” prepared by the National Academy of Sciences and published by the NIH.
Generation of Reassortant Virus
Single‐gene and double‐gene reassortants were generated by coinfection of MA104 cells with parental RRV and TUCH rotavirus strains at varying multiplicities of infection. Progeny underwent plaque purification, followed by polyacrylamide gel electrophoresis and sequencing, as described,16 to determine genetic content. Reassortants were named for the parent (TUCH) strain first, followed by the RRV gene placed on TUCH. The TR(VP2) and TR(VP2,VP4) is based on the TUCH parent background strain with VP2 only and VP2 and VP4 genes originating from RRV, respectively.
Development of a Mouse Model of BA With Fibrosis
Newborn mice underwent intraperitoneal inoculation on DOL 4 with RRV, TR(VP2), or TR(VP2,VP4) reassortants at a dosage of 1.25 × 106 focus‐forming units/g body weight. Saline‐injected pups served as controls. Mice were monitored every other day for obstructive symptomatology including weight, jaundice, and acholic stool. The presence of bilirubin in urine was measured through commercially available urine dipsticks (Bayer Co., Elkhart, IN). Symptoms and survival rates were recorded. To perform histopathological analysis, mice were sacrificed at 28 days postinoculation, and livers and bile ducts were microdissected and harvested. A subset of mice (n = 6) from each group was sacrificed 7 days postinoculation for evaluation of infectious virus presence in bile ducts.
CCl4 Treatment of Mice
See Supporting Information.
Bile Duct Ligation of Mice
See Supporting Information.
Quantification of Virus Infectivity
See Supporting Information.
Histologic Analysis of Specimens
Livers were harvested from mice at 28 days postinoculation with viruses, saline, and CCl4 as well as 13 days post–bile duct ligation. The livers were then fixed in 10% formalin, embedded in paraffin, and sectioned at 5 μm along the length of the section as described.21 Bile ducts were harvested from identically treated animals at 14 days postinoculation. Slides were stained with hematoxylin and eosin (H&E) and sirius red using standard techniques. They were then blindly evaluated by a pathologist for bile duct morphology including the presence of ductular proliferation and severity of liver fibrosis consistent with Ishak scoring system (Supporting Table S1).
Handling of Human BA Samples
Deidentified, formalin‐fixed, paraffin‐embedded, exempt human tissue samples from patients with BA at the time of Kasai procedure were obtained from the Biobank pathology archives of Cincinnati Children’s Hospital Medical Center. Staining and histopathological analysis of samples were performed as described above.
Immunohistochemistry for the Detection of Cholangiocyte Proliferation
See Supporting Information.
Measurement of Serum Alanine Aminotransferase, Aspartate Aminotransferase, and Matrix Metalloproteinase 7
Enzymatic assays for the detection of alanine aminotransferase (ALT), aspartate aminotransferase (AST) (Bio Scientific, Austin, TX), and matrix metalloproteinase 7 (MMP‐7) (Aviva, San Diego, CA) were carried out in (1:100 phosphate‐buffered saline–diluted) serum samples collected at 2 weeks postinoculation. All assays were performed in duplicate in accordance with the manufacturer’s recommendations.
Flow‐Cytometric Analysis
See Supporting Information.
RNA Extraction and Real‐Time Quantitative PCR
BALB/c pups were inoculated with saline or reassortant virus as described above, and a subset of mice were sacrificed 2 weeks after, their livers were harvested, and total RNA was extracted using the Zymo Research Quick‐RNA MiniPrep RNA kit according to the manufacturer’s instructions (Irvine, CA).The mRNA expression of genes (chemokine [C‐C motif] ligand 2 [CCL2]; interleukin 6 [IL6]; chemokine [C‐X‐C motif] ligand 1 [CXCL1]; CXCL2; serpin peptidase inhibitor, plasminogen activator inhibitor type 1 [SERPINE1]; integrin alpha 2 [ITGA2]; versican [VCAN]; laminin, gamma 2 [LAMC2]; amphiregulin [AREG]; thrombospondin 1 [THBS1]; MMP‐7) was quantified by real‐time PCR. Total RNA (2 μg) was reverse‐transcribed using a high‐capacity RNA‐to‐complementary DNA (cDNA) kit (Fisher Scientific). cDNA pools were subjected to real‐time kinetic PCR on a CFX connect real‐time system (Bio‐Rad, Hercules, CA) using SYBR Green III (Agilent Technologies, Santa Clara, CA) to quantify mRNA expression of corresponding genes normalized to glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) using described techniques.22 Primers for the selected genes are detailed in Supporting Table S2.
Human liver mRNA expression levels were assessed from the publicly available microarray data sets GSE4696023 for patients with BA obtained from the National Center for Biotechnology Information’s (NCBI’s) Gene Expression Omnibus database (GEO). Patients described before23 were categorized into three groups: normal control group (n = 7), non‐BA group (n = 14), and BA group (n = 64). Data were analyzed using an online database, GEO2R.
Statistical Analysis
The graphs were created and statistical analyses were performed using GraphPad Prism, version 5.02 (GraphPad Prism Software, Inc., La Jolla, CA). Statistical comparisons between different groups were performed by one‐way analysis of variance (ANOVA) with Bonferroni’s post hoc test. Differences were considered significant at P < 0.05. Analysis of noncontinuous variables was performed by chi‐squared tests. Continuous variables were analyzed by ANOVA with post hoc t testing where appropriate.
Results
Generation of Reassortant Rotaviruses
Previously in our laboratory we generated a series of reassortant rotaviruses and demonstrated that the VP4 gene of RRV is required for BA pathogenesis.16 In generating the single‐gene viral reassortants, we generated a double‐gene reassortant TR(VP2,VP4) and a single‐gene reassortant TR(VP2) in which the VP2 and VP4 genes and the VP2 gene of TUCH were replaced with those of RRV. Reassortants were characterized and visualized by polyacrylamide gel electrophoresis (Supporting Fig. S1). When injected into newborn BALB/c mice, the TR(VP2,VP4) reassortant induced the BA phenotype with reduced mortality. We have reported16 that TR(VP2)‐injected mice do not develop symptoms of biliary obstruction and mortality, so we used these mice as disease/negative controls.
Reassortant Rotaviruses are Capable of Infecting the Mouse Extrahepatic Bile Duct
Extrahepatic bile ducts were microdissected 7 days postinoculation with wild‐type RRV and TR(VP2) and TR(VP2,VP4) reassortants. A focus‐forming assay performed on these bile ducts revealed that reassortants were capable of infecting and replicating within the bile ducts (Supporting Fig. S2); however, TR(VP2,VP4) replicated to a significantly lower titer than RRV.
The TR(VP2,VP4) Rotavirus Reassortant Produces Clinical Manifestations of BA With Reduced Mortality
We have reported that the murine model of BA is temporally dependent.22 In this model we tried using various doses of the virus (3.5 × 105 to 1.3 × 106/g body weight) at different days (DOL 0‐5) in multiple combinations and identified that a dose of 4.1 × 106 at DOL 4 resulted in the maximum number of mice exhibiting the symptoms of BA with reduced mortality. For this study 100 mice were inoculated with either TR(VP2,VP4) reassortant or RRV. Fifty mice were inoculated with the TR(VP2) reassortant and 20 with saline (control). RRV‐inoculated and TR(VP2,VP4)‐inoculated mice displayed symptoms of an obstructive cholangiopathy including jaundice, acholic stools, and bilirubinuria in 96% and 100% of mice, respectively, while only 40% of TR(VP2) reassortant–inoculated mice displayed such symptoms (Fig. 1A). Mice inoculated with TR(VP2,VP4) reassortant showed a 38% survival rate, while those inoculated with RRV only had a 20% survival. Conversely, both the TR(VP2) reassortant and saline controls had 100% survival (Fig. 1B). Likewise, mice inoculated with TR(VP2,VP4) and RRV experienced growth retardation (P < 0.05) compared to those inoculated with TR(VP2) and saline at 30 days (Fig. 2A,B). When dissected, mice inoculated with TR(VP2,VP4) and RRV appeared jaundiced with hepatic nodules (Fig. 2C).
Figure 1.
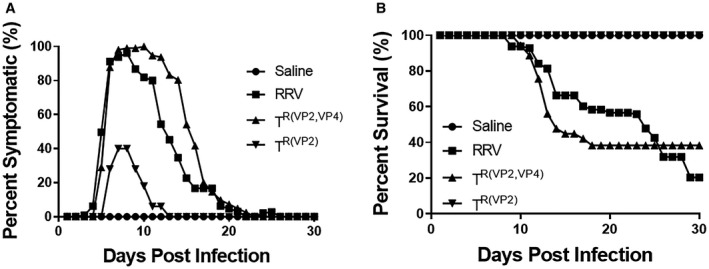
In vivo effect of reassortant virus in the murine model. Symptoms were recorded for different groups of mice for 30 days. One hundred percent of the mice injected with TR(VP2,VP4) developed symptoms compared to 96% in RRV and 40% in TR(VP2) (A). Survival of TR(VP2,VP4)‐injected mice was 38% compared to only 20% in RRV (B).
Figure 2.
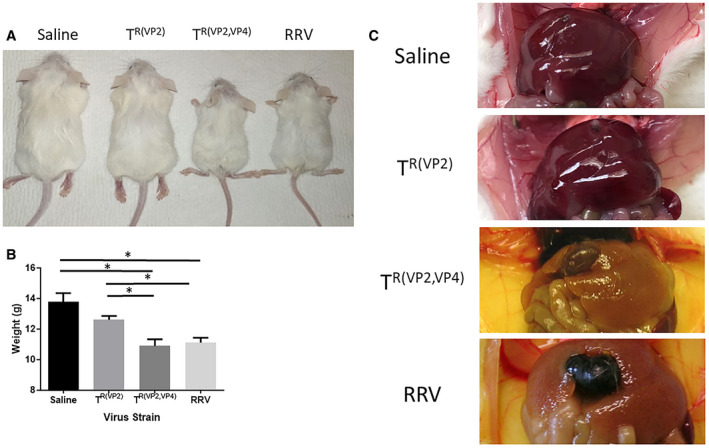
Body weight comparison of inoculated mice and gross morphology of the liver at 28 days. BALB/c pups inoculated with RRV (n = 47), TR(VP2,VP4) (n = 75), TR(VP2) (n = 50), or saline (n = 20) were quantitatively compared on DOL 30 (A, B). RRV‐injected and TR(VP2,VP4)‐injected pups were significantly smaller in size compared to both TR(VP2)‐injected and saline‐injected mice. Gross morphology of the livers in RRV‐injected and TR(VP2,VP4)‐injected mice were noticeably jaundiced with gross hepatic nodularity; by this time stools had already become pigmented (C). *P < 0.05.
TR(VP2,VP4) Induces Hepatic Fibrosis in Mice
Livers of mice harvested at 28 days postinjection with saline, TR(VP2), TR(VP2,VP4), and RRV were assessed for fibrosis. Compared to TR(VP2) (0%), TR(VP2,VP4)‐inoculated mice showed a higher frequency of Ishak 3‐5 stage fibrosis (63%) (P < 0.005). Interestingly TR(VP2,VP4)‐inoculated mice also had a significantly higher frequency of late‐stage fibrosis than RRV‐inoculated mice (40%) (P < 0.05) (Supporting Fig. S3). Histologically fibrosis was evenly distributed throughout the liver parenchyma and was composed of portal–portal as well as portal–central bridging. Multiple specimens also contained microscopic nodules that were not present in negative controls (Fig. 3). Because RRV‐inoculated mice had a 20% survival rate at 30 days postinoculation with a lower frequency of inducing stage 3‐5 fibrosis compared to those inoculated with TR(VP2,VP4) reassortant, we focused only on TR(VP2,VP4) for further analysis.
Figure 3.
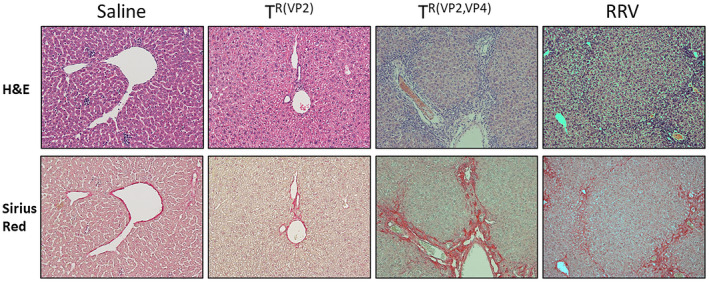
Histological assessment of hepatic fibrosis. Livers of mice inoculated with RRV, reassortants TR(VP2,VP4) and TR(VP2), and saline were harvested on DOL 28 and stained with H&E as well as sirius red to assess for fibrosis and inflammation (×10 magnification). All samples excluding saline displayed paucity of inflammatory cells. Saline and mice inoculated with TR(VP2) had no signs of collagen deposition on sirius red staining. However, mice inoculated with RRV and TR(VP2,VP4) had copious amounts of portal–portal and portal–central bridging fibrosis throughout the hepatic parenchyma.
TR(VP2,VP4)‐Inoculated Mice had Increased Mononuclear Cells in the Liver
In human BA, it has been shown that the predominant cellular immune response at diagnosis represents activated cluster of differentiation 4–positive (CD4+) and CD8+ T cells within portal tracts that produce T helper 1 cytokines (IL‐2, interferon‐gamma)24, 25, 26 and that in mice neonatal hepatic CD3+ cells that express interferon‐gamma induce the development of BA.27 We used flow cytometry to characterize the mononuclear cells harvested from the liver following inoculation with the different viruses. The mononuclear profile in livers harvested 12 days postinoculation with TR(VP2,VP4) mice exhibited a significantly higher number of CD4+ (24.3 ± 1.3) cells when compared to TR(VP2) and saline, respectively (9.7 ± 1.1, 13.1 ± 0.7) (Fig. 4A). In a similar fashion the percentage of CD8+ (19.7 ± 0.8) was increased over TR(VP2) (7.7 ± 0.5) and saline (4.1 ± 0.3) (Fig. 4B). Interestingly, TR(VP2) also had significantly increased CD8+ cells compared to saline but not to the same extent as TR(VP2,VP4). There was also a significant increase in natural killer (NK) cells in the TR(VP2,VP4)‐inoculated mice (15.9 ± 0.4) compared to both the TR(VP2) (9.9 ± 0.9) and saline (11.1 ± 0.3) groups (Fig. 4C).
Figure 4.
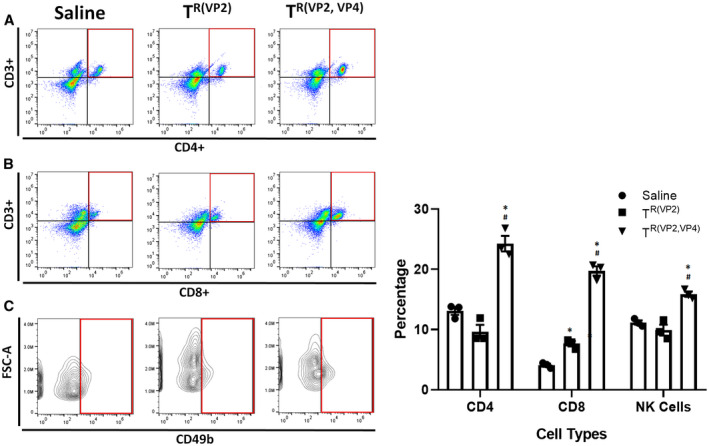
Liver immune profile of TR(VP2,VP4)‐inoculated mice. Flow cytometry of mononuclear cells harvested from 12‐day postinoculation pup livers illustrated a significant increase in the number of CD4+ (A), CD8+ (B), and NK cells (C) in TR(VP2,VP4)‐injected mice when compared to TR(VP2) and saline controls. *P < 0.05 versus saline, # P < 0.05 versus TR(VP2); n = 3.
Higher Serum ALT and AST Levels in TR(VP2,VP4)‐Inoculated Mice
The mean serum ALT levels at 28 days postinoculation were 60, 33, and 147 IU/L, while the mean serum AST levels were 322, 300, and 1,106 IU/L for saline‐inoculated, TR(VP2)‐inoculated, and TR(VP2,VP4)‐inoculated mice, respectively. The levels of ALT and AST in TR(VP2,VP4)‐inoculated mice were higher than those of other two groups, suggesting liver injury (P < 0.05 compared to TR(VP2) and P < 0.01 compared to saline) (Fig. 5A,B).
Figure 5.
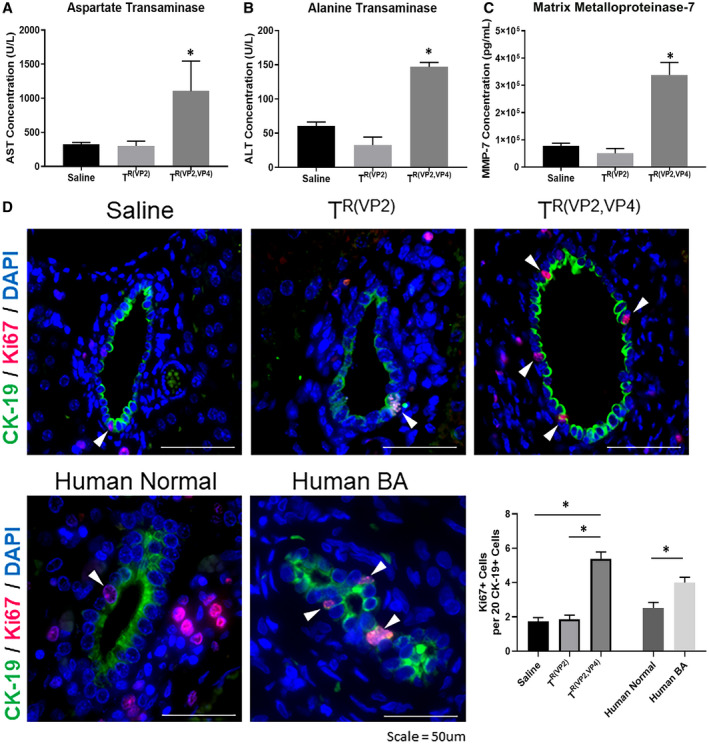
Quantification of ALT, AST, MMP‐7, and bile duct proliferation. Serum harvested from TR(VP2,VP4)‐injected mice at 2 weeks postinfection demonstrates significantly higher concentrations of AST (A), ALT (B), and MMP‐7 (C). *P < 0.05. Immunohistochemical staining on livers harvested 28 days postinoculation illustrated a significantly higher amount of CK‐19+ cells staining positive for Ki67 in TR(VP2,VP4)‐inoculated mice (D). Staining of human BA liver samples also demonstrated increased Ki67+ and CK‐19+ cells compared to healthy controls. The graph shows total number of Ki67+ cells per 20 CK‐19+ cells. *P < 0.05 versus saline and TR(VP2) and human BA versus healthy controls. Abbreviation: DAPI, 4′,6‐diamidino‐2‐phenylindole.
Increased MMP‐7 Serum Levels in TR(VP2,VP4)‐Inoculated Mice
The increased presence of MMP‐7 in human serum has been implicated in the pathogenesis of acute and chronic inflammatory conditions of the liver.28, 29 Thus, we measured the serum levels of MMP‐7 in three group of mice. At 2 weeks postinoculation higher levels of MMP‐7 (P < 0.0005) were detected in serum of TR(VP2,VP4)‐inoculated mice than saline‐inoculated controls (Fig. 5C).
TR(VP2,VP4)‐Inoculated Mice Display Cholangiocyte Proliferation in the Liver
Liver fibrosis and cholangiocyte proliferation are hallmarks of human BA.30, 31 To determine cholangiocyte proliferation, livers were harvested from mice 28 days postinoculation and stained for cytokeratin K19 (CK‐19), Ki67, and 4′,6‐diamidino‐2‐phenylindole. TR(VP2,VP4)‐injected mice exhibited a significantly higher number of cholangiocytes which were positive for both CK‐19 and Ki67 compared to TR(VP2) and saline (Fig. 5D), indicating that proliferation was occurring with obstruction. RRV‐injected mice also exhibited an increased number of proliferating cholangiocytes (Supporting Fig. S4). Similarly, liver harvested from human patients with BA at the time of Kasai showed higher cholangiocyte proliferation compared to healthy controls (Fig. 5D).
TR(VP2,VP4)‐Injected Mice Display Fibrosis Mimicking Human BA
Infants with BA exhibit portal tract expansion and bile duct proliferation. Ductular reaction was prominent in most liver tissues and consists of a proliferation of small ductules located at the periphery of the portal tracts (Fig. 6A). This finding reflects an obstructive process in the extrahepatic biliary tree that has been shown to be a key feature of BA. The dilated lumens are often accompanied by bile plugs. The ductular reaction is typically accompanied by an inflammatory cell infiltrate, especially neutrophils and macrophages. Similarly, BALB/c pups inoculated with TR(VP2,VP4) showed not only features of extrahepatic bile duct obstruction by DOL 28, including bile duct and ductular proliferation, but also a significant amount of portal fibrosis (Fig. 6B).
Figure 6.
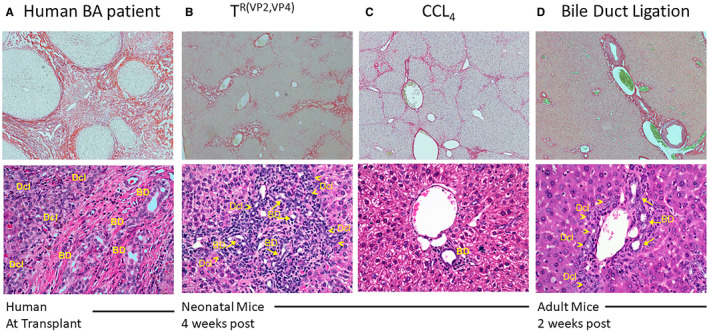
Liver histology in human and murine models of fibrosis. Sirius red staining (×4 magnification, upper panel) and H&E staining (×10 magnification, lower panel), was performed on liver samples of humans and mice. Sirius red section from human BA and mice inoculated with TR(VP2,VP4) and CCl4 showed extensive collagen deposition (A, B, and C, respectively) but less collagen deposition in bile duct–ligated mice (D). Bile duct proliferation was assessed by enumerating the number of bile ductules and ducts surrounding portal tracts. Typical histology of BA in infant liver at time of Kasai portoenterostomy, displaying marked increase in number of bile duct profiles and prominent ductular reaction at the portal tract periphery (A). Livers of TR(VP2,VP4) mice showed portal inflammation and a marked increase in the number of bile duct profiles (arrows) and bile ductular proliferation (arrowheads) at the portal tract edges (B). Mice treated with CCl4 showed normal portal tracts with one bile duct per portal tract and no signs of inflammation (C). Livers of 8‐week‐old mice that underwent bile duct ligation were harvested 15 days postoperatively and displayed a mildly increased number of bile duct profiles (arrows) and bile ductular proliferation at the edge of the portal tracts (arrowheads), consistent with obstruction of the extrahepatic biliary tract (D). Abbreviations: BD, bile duct; Dcl, ductular reaction.
TR(VP2,VP4)‐Induced Murine Fibrosis is Distinct From Existing Models of Murine Fibrosis
Although CCl4 and bile duct ligation–induced hepatic fibrosis are well‐established models using adult C57BL/6 mice,17, 18 these models do not reflect human BA. Through inoculation of BALB/c mice pups 3 times per week beginning at DOL 4 with CCl4, it was determined that 100% of mice developed Ishak stage 4‐5 fibrosis by DOL 32. This hepatotoxin‐induced fibrosis is histologically distinct from TR(VP2,VP4)‐induced fibrosis as it lacks bile duct proliferation (Fig. 6A‐C).
Bile duct ligation in adult C57BL/6 mice is another well‐characterized model used to study hepatic fibrogenesis through induction of cholestatic injury.18 In previous work,32 it has been shown that RRV inoculation of C57BL/6 newborn pups does not induce a BA phenotype. In order to compare the bile duct ligation–induced and rotavirus‐induced models of fibrosis, BALB/c mice were used. We found that in 8‐week‐old BALB/c mice in which bile duct ligation was performed, significant mortality was observed, with virtually all mice expiring by DOL 15 due to a large volume of ascites and hepatic necrosis. In the few mice (15%) that survived, fibrosis beyond Ishak stage 2 was not evident (Fig. 6D). The portal expansion and bile duct proliferation displayed in bile duct–ligated mice, however, was similar to that seen in the TR(VP2,VP4) model of fibrosis (Fig. 6B,D). The bile duct–ligated mice did not show bile duct infiltration by inflammatory cells, which is more prominent in TR(VP2,VP4)‐inoculated mice.
Extrahepatic Bile Ducts of Mice Inoculated With TR(VP2,VP4) Exhibit Partial Obstruction
Extrahepatic bile ducts obtained from TR(VP2,VP4)‐inoculated mice at 14 days postinoculation exhibited stenosis. In comparison with negative controls, histological analysis of these ducts from TR(VP2,VP4)‐inoculated mice revealed narrowing of the lumen (Fig. 7A) lined by reactive cholangiocytes with occasional foci of intraductal micropapillary projections, covered by hyperplastic reactive epithelium (Fig. 7B). The subepithelium exhibited a mild fibroblastic reaction with edematous stroma (Fig. 7A). A paucity of inflammatory cells consistent with human BA was also noted. Intraluminal epithelial cells were sloughed and replaced in areas with goblet cell–lined lumens. Strikingly, some of these findings, including the reactive fibroblastic proliferation and epithelial changes, were similar to observations in extrahepatic bile ducts of infants with BA at the time of Kasai procedure (Fig. 7C,D).
Figure 7.
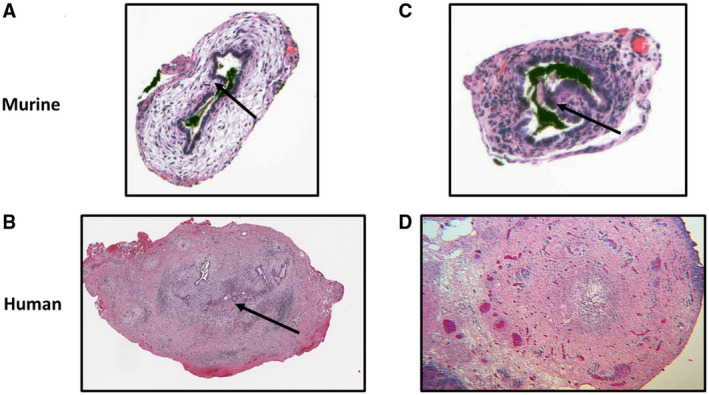
Bile duct histological features. Bile ducts of mice inoculated with TR(VP2,VP4) display multiple areas of partial obstruction, periductal edema, and hypertrophy (A). This is similar to human extrahepatic biliary remnants, which also display partial obstruction (B). Arrow points to area of obstruction. Mice inoculated with the TR(VP2,VP4) reassortant have papillary projections, contributing to partial obstruction (C) arrow exhibits papillary projection, similar to bile ducts of humans with BA (D).
Transcriptional Profiles of TR(VP2,VP4)‐Induced BA are Similar to Human BA
To identify the mRNA expression levels in human BA liver samples, we assessed the publicly available microarray data sets GSE46960 for patients with BA from the NCBI GEO database. Patients were categorized into three groups—normal control group, non‐BA group, and BA group—while data were reanalyzed using an online database, GEO2R. As reported before,23 the genes epithelial membrane protein 1; hyaluronan synthase 2; VCAN; IL6; IL8; CCL20; V‐set domain containing T cell activation inhibitor 1; ITGA2; SERPINE1 (serpin peptidase inhibitor, plasminogen activator inhibitor type 1); THBS1; CCL2; transmembrane 4 six family, member 1; LAMC2; AREG/AREGB; SLC2A3 solute carrier family 2, member 3; and MMP‐7 were found to be highly up‐regulated in human patients with BA compared to healthy controls and age‐matched disease controls (non‐BA, i.e., patients with intrahepatic cholestatic liver disease other than BA).23 RNA extraction and real‐time quantitative PCR were carried out in the liver samples from saline‐inoculated and reassortant virus–inoculated mice for comparison. Of the 15 genes up‐regulated in human BA, 11 genes (CCL2, IL6, IL8 [CXCL1, CXCL2, mouse homolog of IL8], SERPINE1, ITGA2, VCAN, LAMC2, AREG, THBS1, and MMP‐7) were significantly differentially expressed in the liver of TR(VP2,VP4)‐inoculated mice when compared to TR(VP2)‐inoculated and saline‐inoculated mice. A heat map shows the mean expression of all of the genes in humans and mice (Fig. 8A,B). The expression of individual genes is shown in Supporting Figs. S5A,B and S6.
Figure 8.
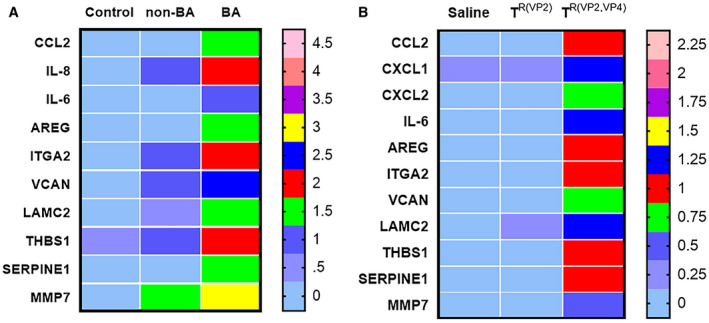
Heat map of functionally enriched genes in human BA liver compared to mouse. mRNA expression for CCL2, IL6, IL8, AREG, SERPINE1, ITGA2, VCAN, LAMC2, THBS1, and MMP‐7 in human livers (A) reanalyzed from GEO database GSE46960. mRNA expression of mouse livers (B) at 2 weeks after inoculation of saline, TR(VP2), and TR(VP2,VP4) normalized to internal Gapdh control. Heat map was generated by Prism. CXCL1 and CXCL2 are mouse homologs of IL8. For human samples, n = 7 (normal control), n = 17 (non‐BA, disease control), and n = 64 (BA); for mice, n = 3 samples per saline, n = 4 per TR(VP2), and n = 7 per TR(VP2,VP4).
Discussion
Through inoculation of newborn pups with RRV, the murine model of BA has helped us to elucidate some of the mechanistic aspects of the disease. Investigation of human BA progression to hepatic fibrosis, however, has not been feasible because nearly all mice expire by DOL 14 in the RRV‐induced model of BA. To our knowledge, there is only one study33 which used this model to demonstrate moderate fibrosis at 2 weeks as the mice did not survive beyond 21 days. Thus, it is of vital interest to develop a viral model of neonatal fibrosis which more closely mirrors the pathogenesis of human BA. Such a model would enable thorough examination of factors that play key roles in the development of the disease as well as those that may be targeted therapeutically to prevent or treat human fibrosis.
From a single‐gene reassortment study (between RRV and TUCH)16 and other studies,20 we have shown that the VP4 gene of RRV plays a critical role in BA pathogenesis.20 Besides the VP4 gene, VP2 also plays a minor role. Through substitution of the VP4 and VP2 genes from RRV onto a TUCH genome, the TR(VP2,VP4) reassortant was generated with the aim of reducing the mortality rate in mice compared to RRV. TR(VP2,VP4) reassortant when inoculated into mice at DOL 4 allowed survival beyond DOL 14 and disease progression to hepatic fibrosis. Using different doses of TR(VP2,VP4) reassortant and at a different day of inoculation after birth, we identified that when TR(VP2, VP4) was inoculated at DOL 4, it replicated to a lower titer within the bile ducts of pups compared to RRV, resulting in a higher survival rate. We initially started with the RRV, but due to low survival rate and lower frequency of inducing fibrosis, we continued with the TR(VP2,VP4) reassortant virus alone. Our TR(VP2,VP4) strain may be used to investigate hepatic fibrosis as a sequela of BA as inoculation of pups with this reassortant induces Ishak stage 3‐5 fibrosis in mice.
Liver cirrhosis has been described as the end phenotype in several diseases, including BA, nonalcoholic fatty liver disease (NAFLD), hepatitis C infection, hepatitis B infection, alcoholic liver disease, hemochromatosis, Wilson’s disease, autoimmune hepatitis and primary biliary cirrhosis.34 The etiology of liver cirrhosis in these diseases is multifactorial, while the pathophysiology of hepatocyte degeneration and necrosis followed by regeneration of tissue with fibrotic nodules is common to most.35 Myofibroblasts and hepatic stellate cells are believed to play an integral role in this fibrosis, through prolonged and exaggerated wound healing of liver injury, resulting in replacement of normal liver tissue with collagen scar.36 Cirrhosis arising from biliary tract disease, as in BA and primary biliary cirrhosis, is believed to differ from lesions associated with chronic hepatitis (such as hepatitis B or C). This is likely due to the initial portal tract damage developing into a jigsaw‐like pattern. These diseases have common histological findings, including early portal–portal bridging fibrosis, lack of isolated nodules, rosette formation of regeneration, and absence of central vein involvement until late in the disease course. In fibrosis associated with cholestatic disease, Mallory bodies may be seen around the portal tracts. Likewise, cholangiocyte proliferation around hepatic bile ducts is common to cirrhosis originating from bile duct disease.37 Interestingly, increased bile ductular proliferation has been implicated in worsened prognosis at the time of Kasai portoenterostomy.38 Histological similarities between both livers and extrahepatic bile ducts seen in TR(VP2,VP4)‐induced fibrosis and human BA, including key diagnostic features of bile duct obstruction, namely bile duct proliferation and ductular reaction, suggest that the murine model of fibrosis using the TR(VP2,VP4) reassortant is relevant in assessing human disease.
Several animal models of liver cirrhosis exist, including chemical‐induced models, dietary alterations, and mechanical obstruction of the bile duct. Chemically induced models use direct hepatotoxic agents such as CCl4,17 dimethyl nitrosamine,39 and thioacetamide,40 which initiate hepatic inflammation, activating hepatic stellate cells. NAFLD cirrhosis may arise in mice fed with methionine‐deficient or choline‐deficient41 or a high‐fat42 diet. Bile duct ligation creates a mechanical obstruction of the bile duct, inducing cholestasis, cholangiocyte and hepatocyte injury, and fibrosis.18 The hepatic injury resulting in fibrosis as a result of hepatotoxins, however, cannot be directly compared to cirrhosis resulting from BA as the cirrhosis of BA is believed to stem from cholestasis. Likewise, histological analysis of H&E‐stained tissues enumerating bile ducts and ductules around portal tracts has revealed that bile duct proliferation is not a key feature of this fibrosis, as seen in both human BA and murine TR(VP2,VP4) BA tissues. Bile duct ligation, on the other hand, produces an environment of cholestasis leading to fibrosis that has several limitations in the study of cirrhosis as a sequela of BA. The primary endpoint of bile duct ligation is bile duct proliferation.18 Although this may reflect the initial phenotype of liver fibrosis, portal–portal and portal‐central bridging as well as nodularity (seen in humans with BA) have not been described in mice with ligated bile ducts. This is likely due to the inability of mice to survive beyond Ishak stage 2 fibrosis. Likewise, another challenge in comparing bile duct ligation to cirrhosis of BA is that BA is a disease of infancy. Bile duct ligation of neonatal mice, however, is not technically feasible, thus demonstrating another limitation of that murine model. The TR(VP2,VP4) model of murine fibrosis as a sequela of BA is histologically similar to human BA, revealing bile duct proliferation, bridging, and nodularity, as a result of the cholestatic nature of the disease. Although the TR(VP2,VP4) virus does not induce complete extrahepatic bile duct obstruction, it does display skip lesions and partial obstruction, similar to that seen in remnant samples of patients with BA.43
In the liver, cholangiocytes are a mitotically dormant cell population, and an injury or insult might trigger ductular reaction. Cholangiocyte proliferation is seen in many different human cholangiopathies (i.e., primary biliary cirrhosis, primary sclerosing cholangitis, and biliary atresia).44 In the mouse model bile duct ligation also leads to cholangiocyte proliferation and liver fibrosis. Activation of biliary proliferation (ductular reaction) is thought to have a key role in the initiation and progression of liver fibrosis.45 These proliferating cholangiocytes can induce liver fibrosis either directly through epithelial–mesenchymal transition or indirectly through the activation of other liver cell types.45 At 4 weeks postinoculation, the TR(VP2,VP4)‐inoculated mice showed increased number of Ki67+ cholangiocytes in liver similar to human BA, which could indicate a role of the proliferating cholangiocytes in cholestatic fibrosis.
A recent study46 used proteomics to screen sera from infants with BA and showed a strong positive correlation to elevated levels of MMP‐7 and gamma‐glutamyltransferase. The MMP‐7 level is being used as a diagnostic biomarker for BA as initial findings were further confirmed by another study.47 Immunohistochemistry of liver samples with BA showed increased presence of MMP‐7 in extrahepatic bile duct cholangiocytes.46 MMP‐7 gene expression was also increased in the liver, and levels increased in intrahepatic cholangiocytes of patients with BA. Interestingly, in our mouse model of fibrosis, MMP‐7 levels were increased at 2 weeks postinoculation in the TR(VP2,VP4) group. Whether MMP‐7 contributes to the pathogenesis of BA remains to be determined and, if found to do so, might be considered as a therapeutic target.
Previous studies have shown that the mononuclear cell profile characterized in the livers of children with BA undergoes expansion of CD3+ cells (CD4+ and CD8+) and that this correlated with the RRV‐induced experimental model of BA.25, 27, 48 We found a similar result in the TR(VP2,VP4) model where an increased number of CD8+, CD4+, and NK cells occurred, paralleling the resemblance to human BA.
A detailed knowledge of human disease with appropriate animal models is required for preclinical studies. In this study, we used microarray data available from patients with BA and compared them to RNA expression data obtained from our mouse models of fibrosis to examine and compare the multiple pathophysiological processes involved in the different settings. Previously, it was shown that regulation of tissue development and morphology (organogenesis), extracellular matrix remodeling (wound healing), and inflammation processes are linked to pathogenesis of BA, which is evidenced from an increased gene expression pattern in humans and in our mouse model. The first set of up‐regulated genes, CCL2, IL6, and IL8 (CXCL1 and CXCL2 in mouse), are related to immunity; the second set of genes, SERPINE1, THBS1, VCAN, ITGA2, AREG, LAMC2, and MMP‐7, regulate tissue development and morphology and extracellular matrix remodeling.
In summary, we demonstrate that the TR(VP2,VP4) model of murine fibrosis can be used to study the mechanistic basis of and may give insight into human BA. This model is different from the CCl4 and bile duct ligation models and is relevant to human disease in that it is histologically and molecularly similar to human BA. Transcriptome analysis corroborated the pattern of gene expression in this mouse model and human BA. Currently, we are investigating the mechanism of fibrosis using defined gene knockout mouse models. Identifying the mechanistic basis of disease may facilitate the development of therapeutic targets in humans.
Supporting information
Supported by the National Institutes of Health (R01 DK‐091566, to G.T., S.K.M., and M.M.).
Potential conflict of interest: Nothing to report.
References
- 1. Balistreri WF, Grand R, Hoofnagle JH, Suchy FJ, Ryckman FC, Perlmutter DH, et al. Biliary atresia: current concepts and research directions. Summary of a symposium. Hepatology 1996;23:1682‐1692. [DOI] [PubMed] [Google Scholar]
- 2. Sokol RJ, Mack C. Etiopathogenesis of biliary atresia. Semin Liver Dis 2001;21:517‐524. [DOI] [PubMed] [Google Scholar]
- 3. Bezerra JA. Potential etiologies of biliary atresia. Pediatr Transplant 2005;9:646‐651. [DOI] [PubMed] [Google Scholar]
- 4. Bessho K, Bezerra JA. Biliary atresia: will blocking inflammation tame the disease? Annu Rev Med 2011;62:171‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Salzedas‐Netto AA, Chinen E, de Oliveira DF, Pasquetti AF, Azevedo RA, da Silva Patricio FF, et al. Grade IV fibrosis interferes in biliary drainage after Kasai procedure. Transplant Proc 2014;46:1781‐1783. [DOI] [PubMed] [Google Scholar]
- 6. Landing BH. Considerations of the pathogenesis of neonatal hepatitis, biliary atresia and choledochal cyst—the concept of infantile obstructive cholangiopathy. Prog Pediatr Surg 1974;6:113‐139. [PubMed] [Google Scholar]
- 7. Riepenhoff‐Talty M, Gouvea V, Evans MJ, Svensson L, Hoffenberg E, Sokol RJ, et al. Detection of group C rotavirus in infants with extrahepatic biliary atresia. J Infect Dis 1996;174:8‐15. [DOI] [PubMed] [Google Scholar]
- 8. Glaser JH, Balistreri WF, Morecki R. Role of reovirus type 3 in persistent infantile cholestasis. J Pediatr 1984;105:912‐915. [DOI] [PubMed] [Google Scholar]
- 9. Fjaer RB, Bruu AL, Nordbo SA. Extrahepatic bile duct atresia and viral involvement. Pediatr Transplant 2005;9:68‐73. [DOI] [PubMed] [Google Scholar]
- 10. Domiati‐Saad R, Dawson DB, Margraf LR, Finegold MJ, Weinberg AG, Rogers BB. Cytomegalovirus and human herpesvirus 6, but not human papillomavirus, are present in neonatal giant cell hepatitis and extrahepatic biliary atresia. Pediatr Dev Pathol 2000;3:367‐373. [DOI] [PubMed] [Google Scholar]
- 11. Drut R, Drut RM, Gomez MA, Cueto Rua E, Lojo MM. Presence of human papillomavirus in extrahepatic biliary atresia. J Pediatr Gastroenterol Nutr 1998;27:530‐535. [DOI] [PubMed] [Google Scholar]
- 12. Petersen C, Grasshoff S, Luciano L. Diverse morphology of biliary atresia in an animal model. J Hepatol 1998;28:603‐607. [DOI] [PubMed] [Google Scholar]
- 13. Riepenhoff‐Talty M, Schaekel K, Clark HF, Mueller W, Uhnoo I, Rossi T, et al. Group A rotaviruses produce extrahepatic biliary obstruction in orally inoculated newborn mice. Pediatr Res 1993;33:394‐399. [DOI] [PubMed] [Google Scholar]
- 14. Allen SR, Jafri M, Donnelly B, McNeal M, Witte D, Bezerra J, et al. Effect of rotavirus strain on the murine model of biliary atresia. J Virol 2007;81:1671‐1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li Z, Baker ML, Jiang W, Estes MK, Prasad BV. Rotavirus architecture at subnanometer resolution. J Virol 2009;83:1754‐1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang W, Donnelly B, Bondoc A, Mohanty SK, McNeal M, Ward R, et al. The rhesus rotavirus gene encoding VP4 is a major determinant in the pathogenesis of biliary atresia in newborn mice. J Virol 2011;85:9069‐9077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shi J, Aisaki K, Ikawa Y, Wake K. Evidence of hepatocyte apoptosis in rat liver after the administration of carbon tetrachloride. Am J Pathol 1998;153:515‐525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tag CG, Sauer‐Lehnen S, Weiskirchen S, Borkham‐Kamphorst E, Tolba RH, Tacke F, et al. Bile duct ligation in mice: induction of inflammatory liver injury and fibrosis by obstructive cholestasis. J Vis Exp 2015;96:e52438 10.3791/52438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Coulson BS, Londrigan SL, Lee DJ. Rotavirus contains integrin ligand sequences and a disintegrin‐like domain that are implicated in virus entry into cells. Proc Natl Acad Sci USA 1997;94:5389‐5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Walther A, Mohanty SK, Donnelly B, Coots A, Lages CS, Lobeck I, et al. Rhesus rotavirus VP4 sequence‐specific activation of mononuclear cells is associated with cholangiopathy in murine biliary atresia. Am J Physiol Gastrointest Liver Physiol 2015;309:G466‐G474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Walther AE, Mohanty SK, Donnelly B, Coots A, McNeal M, Tiao GM. Role of myeloid differentiation factor 88 in rhesus rotavirus–induced biliary atresia. J Surg Res 2013;184:322‐329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mohanty SK, Donnelly B, Bondoc A, Jafri M, Walther A, Coots A, et al. Rotavirus replication in the cholangiocyte mediates the temporal dependence of murine biliary atresia. PLoS ONE 2013;8:e69069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bessho K, Mourya R, Shivakumar P, Walters S, Magee JC, Rao M, et al. Gene expression signature for biliary atresia and a role for interleukin‐8 in pathogenesis of experimental disease. Hepatology 2014;60:211‐223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Davenport M, Gonde C, Redkar R, Koukoulis G, Tredger M, Mieli‐Vergani G, et al. Immunohistochemistry of the liver and biliary tree in extrahepatic biliary atresia. J Pediatr Surg 2001;36:1017‐1025. [DOI] [PubMed] [Google Scholar]
- 25. Mack CL, Tucker RM, Sokol RJ, Karrer FM, Kotzin BL, Whitington PF, et al. Biliary atresia is associated with CD4+ Th1 cell‐mediated portal tract inflammation. Pediatr Res 2004;56:79‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shinkai M, Shinkai T, Puri P, Stringer MD. Increased CXCR26 expression associated with CD3‐positive lymphocytes in the liver and biliary remnant in biliary atresia. J Pediatr Surg 2006;41:950‐954. [DOI] [PubMed] [Google Scholar]
- 27. Shivakumar P, Campbell KM, Sabla GE, Miethke A, Tiao G, McNeal MM, et al. Obstruction of extrahepatic bile ducts by lymphocytes is regulated by IFN‐gamma in experimental biliary atresia. J Clin Invest 2004;114:322‐329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Irvine KM, Wockner LF, Hoffmann I, Horsfall LU, Fagan KJ, Bijin V, et al. Multiplex serum protein analysis identifies novel biomarkers of advanced fibrosis in patients with chronic liver disease with the potential to improve diagnostic accuracy of established biomarkers. PLoS One 2016;11:e0167001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Prakobwong S, Charoensuk L, Hiraku Y, Pinlaor P, Pairojkul C, Mairiang E, et al. Plasma hydroxyproline, MMP‐7 and collagen I as novel predictive risk markers of hepatobiliary disease–associated cholangiocarcinoma. Int J Cancer 2012;131:E416‐E424. [DOI] [PubMed] [Google Scholar]
- 30. Bezerra JA, Wells RG, Mack CL, Karpen SJ, Hoofnagle JH, Doo E, et al. Biliary atresia: clinical and research challenges for the 21st century. Hepatology 2018;68:1163‐1173. 10.1002/hep.29905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cheung AC, Lorenzo Pisarello MJ, LaRusso NF. Pathobiology of biliary epithelia. Biochim Biophys Acta Mol Basis Dis 2018;1864:1220‐1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Leonhardt J, Kuebler JF, Turowski C, Tschernig T, Geffers R, Petersen C. Susceptibility to experimental biliary atresia linked to different hepatic gene expression profiles in two mouse strains. Hepatol Res 2010;40:196‐203. [DOI] [PubMed] [Google Scholar]
- 33. Keyzer‐Dekker CM, Lind RC, Kuebler JF, Offerhaus GJ, Ten Kate FJ, Morsink FH, et al. Liver fibrosis during the development of biliary atresia: proof of principle in the murine model. J Pediatr Surg 2015;50:1304‐1309. [DOI] [PubMed] [Google Scholar]
- 34. Zhou WC, Zhang QB, Qiao L. Pathogenesis of liver cirrhosis. World J Gastroenterol 2014;20:7312‐7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Anthony PP, Ishak KG, Nayak NC, Poulsen HE, Scheuer PJ, Sobin LH. The morphology of cirrhosis: definition, nomenclature, and classification. Bull World Health Organ 1977;55:521‐540. [PMC free article] [PubMed] [Google Scholar]
- 36. Elsharkawy AM, Oakley F, Mann DA. The role and regulation of hepatic stellate cell apoptosis in reversal of liver fibrosis. Apoptosis 2005;10:927‐939. [DOI] [PubMed] [Google Scholar]
- 37. Ferrell L. Liver pathology: cirrhosis, hepatitis, and primary liver tumors. Update and diagnostic problems. Mod Pathol 2000;13:679‐704. [DOI] [PubMed] [Google Scholar]
- 38. Kinugasa Y, Nakashima Y, Matsuo S, Shono K, Suita S, Sueishi K. Bile ductular proliferation as a prognostic factor in biliary atresia: an immunohistochemical assessment. J Pediatr Surg 1999;34:1715‐1720. [DOI] [PubMed] [Google Scholar]
- 39. Jezequel AM, Mancini R, Rinaldesi ML, Macarri G, Venturini C, Orlandi F. A morphological study of the early stages of hepatic fibrosis induced by low doses of dimethylnitrosamine in the rat. J Hepatol 1987;5:174‐181. [DOI] [PubMed] [Google Scholar]
- 40. Lukivskaya O, Patsenker E, Lis R, Buko VU. Inhibition of inducible nitric oxide synthase activity prevents liver recovery in rat thioacetamide‐induced fibrosis reversal. Eur J Clin Invest 2008;38:317‐325. [DOI] [PubMed] [Google Scholar]
- 41. Koca SS, Bahcecioglu IH, Poyrazoglu OK, Ozercan IH, Sahin K, Ustundag B. The treatment with antibody of TNF‐alpha reduces the inflammation, necrosis and fibrosis in the non‐alcoholic steatohepatitis induced by methionine‐ and choline‐deficient diet. Inflammation 2008;31:91‐98. [DOI] [PubMed] [Google Scholar]
- 42. Jia X, Naito H, Yetti H, Tamada H, Kitamori K, Hayashi Y, et al. Dysregulated bile acid synthesis, metabolism and excretion in a high fat‐cholesterol diet–induced fibrotic steatohepatitis in rats. Dig Dis Sci 2013;58:2212‐2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Moreira RK, Cabral R, Cowles RA, Lobritto SJ. Biliary atresia: a multidisciplinary approach to diagnosis and management. Arch Pathol Lab Med 2012;136:746‐760. [DOI] [PubMed] [Google Scholar]
- 44. Priester S, Wise C, Glaser SS. Involvement of cholangiocyte proliferation in biliary fibrosis. World J Gastrointest Pathophysiol 2010;1:30‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Glaser SS, Gaudio E, Miller T, Alvaro D, Alpini G. Cholangiocyte proliferation and liver fibrosis. Expert Rev Mol Med 2009;11:e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lertudomphonwanit C, Mourya R, Fei L, Zhang Y, Gutta S, Yang L, et al. Large‐scale proteomics identifies MMP‐7 as a sentinel of epithelial injury and of biliary atresia. Sci Transl Med 2017;9:eaan8462 10.1126/scitranslmed.aan8462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yang L, Zhou Y, Xu PP, Mourya R, Lei HY, Cao GQ, et al. Diagnostic accuracy of serum matrix metalloproteinase‐7 for biliary atresia. Hepatology 2018;68:2069‐2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mack CL, Tucker RM, Sokol RJ, Kotzin BL. Armed CD4+ Th1 effector cells and activated macrophages participate in bile duct injury in murine biliary atresia. Clin Immunol 2005;115:200‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials