

Abstract
Advancement in technology has improved recognition of genetic etiologies of disease, which has impacted diagnosis and management of rare disease patients in the pediatric pulmonary clinic. This review provides an overview of genetic conditions that are likely to present with pulmonary features and require extensive care by the pediatric pulmonologist. Increased familiarity with these conditions allows for improved care of these patients by reducing time to diagnosis, tailoring management, and prompting further investigation into these disorders.
Keywords: bronchiectasis and primary ciliary dyskinesia, cystic fibrosis (CF), genetics/Genome-Wide Association Studies (GWAS), immunology and immunodeficiency, surfactant biology and pathophysiology
1 |. INTRODUCTION
As primary care physicians are often comfortable handling more common diseases, referral to a pediatric pulmonologist is frequently based on the presence of unusual signs and symptoms or advanced progression of disease. In some cases, these uncommon presentations may be due to an underlying genetic etiology. Therefore, an understanding of genetic diseases that have a respiratory component is crucial for the pediatric pulmonologist.
Comfort with the principles of genetics and genetic counseling are important elements of caring for patients with genetic conditions; these key aspects are reviewed in the companion article by Hawley et al. The goal of this review is to provide a concise overview of monogenic diseases with pulmonary features that present in the pediatric setting, highlighting current understanding of these conditions. In this review, we focus on genetic diseases that cause acute respiratory distress of the newborn, bronchiectasis, pulmonary fibrosis, vascular abnormalities, pneumothorax, and central hypoventilation. Table 1 provides an overview of the genetic conditions discussed in this review. Many additional genetic conditions, such as neuromuscular disorders or chromosomal abnormalities, frequently require care by the pediatric pulmonologist but are outside of the scope of this review.
TABLE 1.
Overview of genetic pulmonary diseases presenting in childhood
| Condition | Associated genes | Inheritance pattern | Pulmonary features | Additional clinical features |
|---|---|---|---|---|
| Alveolar capillary dysplasia with misalignment of pulmonary veins | FOXF1 | AD | RDS, ACD, PH | Cardiovascular, gastrointestinal, and genitourinary malformations |
| Autosomal recessive pseudohypoaldosteronism-type 1 | SCNN1A, SCNN1B, SCNN1G | AR | BE, Rl | Salt wasting, failure to thrive, and muscle weakness |
| BHD syndrome | FLCN | AD | PX, C | Cutaneous hamartomas, renal cysts, and renal cancers |
| Brain-lung-thyroid syndrome | NKX2–1 | AD | RDS, ILD, PF, NEHI | Chorea, other neurological symptoms, hypothyroidism |
| Congenital central hypoventilation syndrome | PHOX2B | AD | CH | Hirschprung’s disease, neural crest-derived tumors, autonomic dysfunction |
| Cutis laxa | FBLN5, EFEMP2, LTBP4, ELN | AR, AD | PX, E, PH | Loose, saggy, inelastic skin, hernias, visceral diverticula, joint hypermobility |
| Cystic fibrosis | CFTR | AR | BE, Rl, PX | Pancreatic insufficiency and male infertility |
| Hereditary hemorrhagic telangiectasia | ACVRL1, ENG, GDF2, SMAD4 | AD | AVMs, PAH | Telangiectasias, hepatic and cerebral AVMs, juvenile polyposis syndrome (SMAD4) |
| FLNA-Associated periventricular heterotopia | FLNA | XL | E, ILD, PAH, PX | Intellectual disability, seizures, and cardiac valvular anomalies |
| Heritable pulmonary arterial hypertension | BMPR2, ACVRL1, BMPR1B, CAV1, CBLN2, EIF2AK4, ENG, KCNA5, KCNK3, SMAD9 | AD | PAH | |
| Hermansky-Pudlak syndrome | AP3B1, AP3D1, BLOC1S3, BLOC1S6, DTNBP1, HPS1, HPS3, HPS4, HPS5, HPS6 | AR | PF, RLD | Oculocutaneous albinism, bleeding diathesis, common in Puerto Rico |
| Immune deficiencies | Over 200 genes | AR, AD, XL | BE, ILD, PAP, Rl | Various additional features depending on underlying defect |
| Loeys-Dietz syndrome | TGFBR1, TGFBR2 | AD | PX, C, RLD | Aortic dilation, skeletal deformities, bifid uvula, velvety, translucent skin |
| Marfan syndrome | FBN1 | AD | PX, E, BE, PF | Tall stature, joint laxity, aortic dilation, arachnodactyly, and ocular findings |
| Primary ciliary dyskinesia | ARMC4, C21orf59, CCDC39, CCDC40, CCDC65, CCDC103, CCDC114, CCDC151, CCDC164, CCNO, CFAP298, DNAAF1, DNAAF2, DNAAF3, DNAAF4, DNAAF5, DNAH1, DNAH5, DNAH8, DNAH11, DNAI1, DNAI2, DNAJB13, DNAL1, DRC1, HEATR2, HYDIN, GAS8, LRRC6, MCIDAS, NME8, OFD1, PIH1D3, RPGR, RSPH1, RSPH3, RSPH4A, RSPH9, SPAG1, TTC25, TXNDC3, ZMYND10 | AR, XL | BE, RDS, Rl | Laterality defects, recurrent infections, and male infertility |
| Pulmonary alveolar proteinosis | CSF2RA, CSF2RB, MARS, SLC7A7, GATA2, 22qll.2 deletion | AR | PAP | MARS, SLC7A7, NPC2, GATA2, and 22qll.2 are syndromic conditions |
| Short telomere syndromes | TERC, TERT, DKC1, TINF2, RTEL1, ACD, CTC1, NOP10, NHP2, PARN, USB1, WRAP53 | AD, AR, XL | PF, RLD, AVMs | Nail and skin abnormalities, oral leukoplakia, short stature, premature graying, genitourinary abnormalities, brain abnormalities, bone marrow failure syndromes, and cirrhosis |
| Surfactant deficiencies | ABCA3, SFTPB, SFTPC | AD, AR | RDS, ILD, PAP, PF | Failure to thrive |
| Vascular Ehlers-Danlos syndrome | COL3A1 | AD | PX, C | Easy bruising, thin and translucent skin, and tissue fragility |
Abbreviations: ACD, alveolar capillary dysplasia; AVMs, arteriovenous malformations; BE, bronchiectasis; BHD, Birt-Hogg-Dubé; C, lung cysts; CCHS, congenital central hypoventilation syndrome; CH, central hypoventilation; E, emphysema; ILD, interstitial lung disease; LDS, Loeys-Dietz syndrome; NEHI, neuroendocrine cell hyperplasia of infancy; PAH, pulmonary arterial hypertension; PAP, pulmonary alveolar proteinosis; PF, pulmonary fibrosis; PH, pulmonary hypertension; PX, pneumothorax; RDS, respiratory distress syndrome; RI, recurrent infections; RLD, restrictive lung disease.
2 |. ACUTE RESPIRATORY DISTRESS OF THE NEWBORN/SURFACTANT DYSFUNCTION DISORDERS/OTHER INFANTILE FORMS OF DIFFUSE LUNG DISEASE
ABCA3, SFTPB, and SFTPC-related surfactant dysfunction
Brain-lung-thyroid syndrome
Pulmonary alveolar proteinosis
2.1 |. Clinical overview
Acute respiratory distress during the neonatal period, as characterized by hypoxemia and retractions, is expected in preterm infants due to immature lung development. However, if these signs and symptoms present in a full-term infant, or to a more severe degree than would be explained by prematurity alone, surfactant deficiency or dysfunction should be considered. Surfactant dysfunction disorders result from abnormal production or transport of the phospholipids and proteins that comprise the pulmonary surfactant complex. Surfactant is produced by alveolar type II epithelial cells before birth and is required to reduce surface tension in the alveolar space.1,2 Abnormal quantity or quality of surfactant results in alveolar collapse and insufficient gas exchange, leading to hypoxemia and hypercapnia. Infants with surfactant dysfunction display signs of respiratory distress often leading to respiratory failure in the newborn nursery.
In some cases, neonates may be well enough to be discharged home after delivery, but display chronic tachypnea, retractions, and failure to thrive. These infants are hypoxemic and often have crackles on exam. In milder disease, symptoms may present over time, occasionally even in adulthood. Symptoms include dyspnea and exercise intolerance with associated digital clubbing. These patients may have been diagnosed with interstitial lung disease or pulmonary fibrosis.2 Patients can present with recurrent respiratory infections (ie, infectious exacerbations of the underlying surfactant dysfunction), and crackles or wheezing can be heard on physical exam. Chest X-rays often demonstrate low lung volumes and diffuse homogenous granular opacities.3 Chest computed tomography (CT) findings include ground glass opacities, cysts, atelectasis, and fibrosis in older patients.4,5
Nongenetic causes of respiratory failure in a full-term newborn include infection, meconium aspiration, or aspiration due to an anatomic defect such as a tracheoesophageal fistula or laryngeal cleft must be considered before ordering genetic testing. The most common genetic causes, discussed in depth below, include surfactant deficiencies related to ABCA3, SFTPB, SFTPC, and NKX2-1 mutations. Pulmonary alveolar proteinosis (PAP), which also disrupts surfactant homeostasis and may have a genetic etiology, does not typically present in the newborn period, however, given its similarity in surfactant homeostasis dysfunction, it is also discussed below. Figure 1 shows cellular disease pathology of surfactant dysfunction, with corresponding genetic mutations.
FIGURE 1.
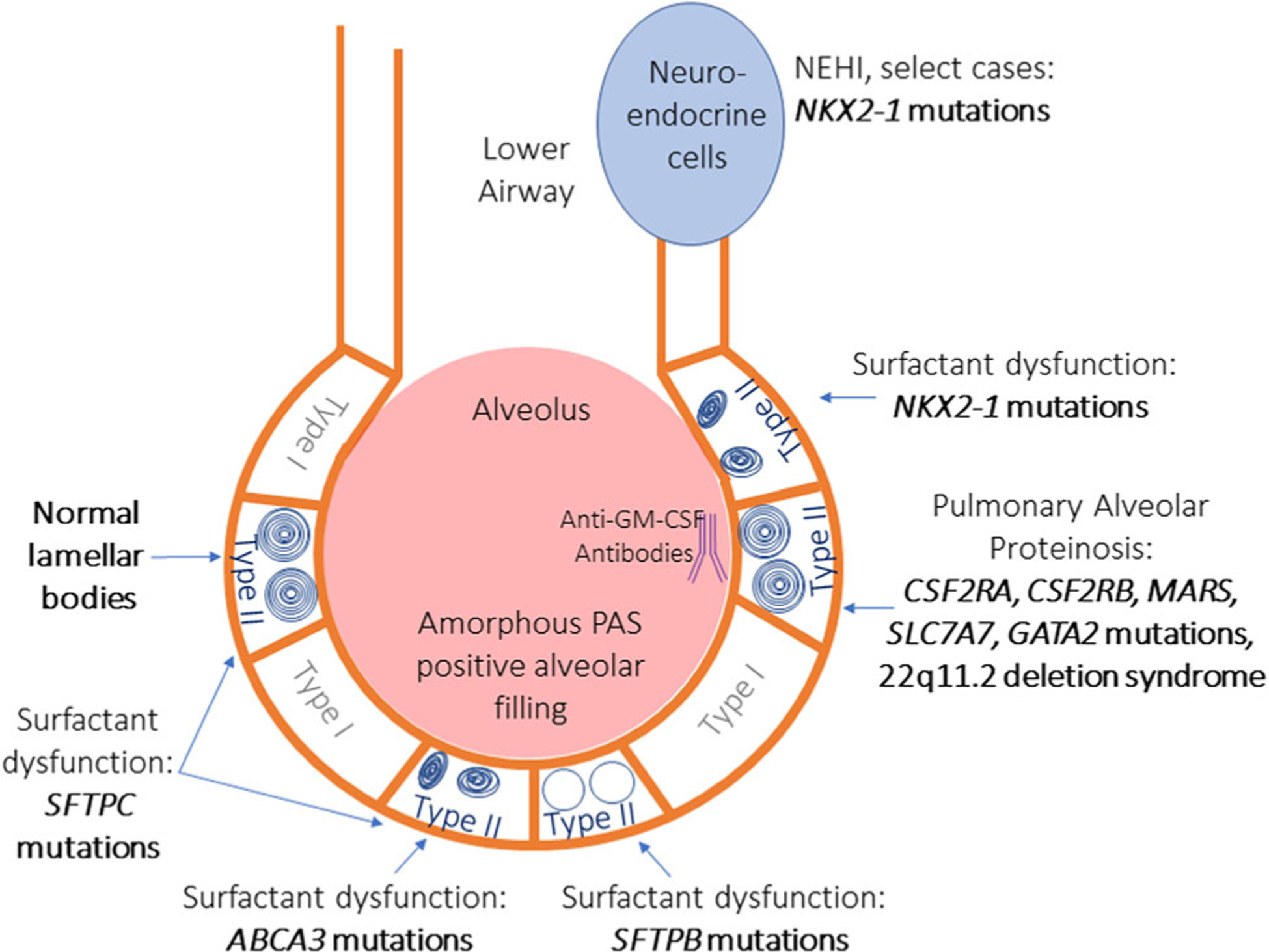
Cartoon overview of disorders of surfactant dysfunction. The lower airway and alveolus are represented. Type I and type II alveolar cells line the airway. Type II cells contain lamellar bodies that are responsible for generating surfactant. Normal type II alveolar cells have a circumferential organized appearance. SFTPC mutations cause milder surfactant dysfunction and may have normal or disorganized lamellar bodies. ABCA3 and NKX2–1 mutations can also have a range of severity of surfactant dysfunction and both have small disorganized lamellar bodies. Of note, neuroendocrine hyperplasia of infancy has also been reported in association with NKX2–1 mutations, which affects the lower airways rather than the alveolus. Mutations in SFTPB cause the most severe surfactant dysfunction, often lethal in the newborn period, and lamellar bodies are often decreased or absent. Mutations causing pulmonary alveolar proteinosis have normal lamellar bodies and normal surfactant secretions, rather, antibodies are generated to disrupt mechanisms of surfactant homeostasis. GM-CSF, granulocyte-macrophage colony-stimulating factor; PAS, periodic acid-Schiff
Other causes of neonatal interstitial lung disease, including neuroendocrine hyperplasia of infancy (NEHI) and pulmonary interstitial glycogenosis (PIG), should also be considered. NKX2-1 mutations have been described in NEHI, although this is a minority of cases, and no genetic mutations have been associated with PIG; further research into a likely genetic predisposition for these diseases is needed.6,7
Several additional genetic syndromes not included in this review that are associated with a disruption of surfactant homeostasis have been described, including interstitial lung and liver disease (associated with the MARS gene), lysinuric protein intolerance (associated with the SLC7A7 gene), and Niemann-Pick disease type C2 (associated with the NPC2 gene). Immunodeficiencies, specifically GATA2-deficiency and 22q11.2 microdeletion syndrome, also known as DiGeorge syndrome, can also exhibit surfactant abnormalities.8,9
Other genetic conditions can also cause respiratory distress in the full-term neonate, including primary ciliary dyskinesia (PCD) and alveolar capillary dysplasia with misalignment of pulmonary veins (ACD/MPV) which are discussed later in this review. Loss-of-function mutations in the FLNA gene which encodes filamin A, a key protein guiding cell shape and migration, can also result in lobar hyperinflation and respiratory failure in young infants.10 Loss-of- function mutations in FLNA have also been associated with pulmonary hypertension and pneumothorax in older age groups, which are discussed later in this review.10,11
2.2 |. Genetic conditions
2.2.1 |. Surfactant dysfunction disorders
Surfactant dysfunction most often results from mutations in the ABCA3, SFTPB, or SFTPC genes. Surfactant dysfunction is associated with various inheritance patterns and clinical presentations depending on the underlying genetic defect. ABCA3 and SFTPB-related surfactant deficiencies are inherited in an autosomal recessive pattern, while SFTPC-related surfactant dysfunction is inherited in an autosomal dominant pattern.
Mutations in the ABCA3 gene, which encodes a protein involved in surfactant production and transport, are the most common genetic cause of surfactant dysfunction. ABCA3-related surfactant dysfunction can present in infancy, childhood, or rarely in adulthood.2 Lung biopsy reveals PAP and desquamative interstitial pneumonitis in the neonatal period, and nonspecific interstitial pneumonitis in older children. On electron microscopy, dense abnormal lamellar bodies can be seen.12 Loss of function or null mutations in ABCA3 result in the complete absence of protein production and are associated with a more severe clinical presentation with lung transplantation or death within the first year of life being the usual outcome. Missense variants and other mutation types are associated with a more variable disease trajectory.13
Individuals with pathogenic changes in the SFTPB gene, which encodes surfactant protein B, can have a similar clinical course to those with ABCA3 mutations, typically experiencing acute respiratory failure and death in the newborn period. Lung biopsy also shows PAP desquamative interstitial pneumonitis, but absent lamellar bodies on electron microscopy.4
The SFTPC gene encodes surfactant protein C and is associated with autosomal dominant inheritance, variable severity, and reduced penetrance. Individuals with SFTPC mutations can exhibit neonatal respiratory distress, but more frequently present with interstitial lung disease at varying ages of onset.2,14,15 Lung biopsy shows differing degrees of PAP, fibrosis, and desquamative interstitial pneumonitis, and lamellar bodies may be normal or disorganized on electron microscopy.4 The majority of SFTPC mutations occur as de novo changes, while others are inherited, often from unaffected or mildly affected parents. Some mutations in SFTPC result in loss of function of the protein, while others, usually missense variants, resulting in the production of an abnormal protein product that disrupts function.2,16
2.2.2 |. Brain-lung-thyroid syndrome
Mutations in the NKX2-1 gene, previously known as the thyroid transcription factor-1 (TTF1) gene, are also associated with neonatal or childhood-onset interstitial lung disease. However, in contrast to ABCA3, SFTPB, and SFTPC-related surfactant deficiencies, individuals with NKX2-1 mutations often present with additional clinical features resulting from disruption of brain and thyroid development. About half of individuals exhibit involvement of all three organ systems, while the other half can manifest any combination of the associated features.17,18 Most individuals have some neurological involvement, with childhood onset of chorea being one of the hallmark features of the disease. Patients may also have intention tremor, dysarthria, facial apraxia, sensorineural hearing loss, hypotonia, motor delay, attention deficit hyperactivity disorder, learning disabilities, myoclonus, dystonia, ataxia, and structural brain abnormalities.19 Thyroid problems include congenital hypothyroidism, compensated hypothyroidism, and hypoplasia or aplasia.17,20
Lung disease is the leading cause of mortality in brain-lung-thyroid syndrome patients. Approximately, 50% of individuals with NKX2-1 mutations have some degree of pulmonary dysfunction, although clinical presentation and age of onset of lung disease are variable.5,17 Respiratory distress may occur in the neonatal period,18,21,22 while other patients can present with interstitial lung disease or pulmonary fibrosis at any age.5,17 CT findings in patients with lung disease can include mild to diffuse ground-glass opacities, cysts, atelectasis, and fibrosis. Data from lung biopsies in these patients are limited but can have an appearance typical of surfactant dysfunction disorders.5,22 NEHI, a form of interstitial lung disease characterized by tachypnea, crackles, retractions, and hypoxemia in infancy, has been reported in rare cases in association with NKX2-1 mutations.7,23 Most cases of NEHI do not have a clear genetic cause; diagnosis relies on typical CT patterns or lung biopsy.24 There have also been a few reports of individuals with NKX2-1 mutations developing pulmonary carcinoma, although the association of NKX2-1 related conditions with cancer remains unclear.5,25
Brain-lung-thyroid syndrome results from dysregulation of neuron development and migration in the brain, altered expression of surfactant genes in the lungs, and reduced hormone production in the thyroid gland. These functions all are attributable in part to the homeobox gene, NKX2-1, which encodes the Nkx-2.1 transcription factor.21,26 Mutations in the NKX2-1 gene resulting in reduced protein production or abnormal protein function are associated with brain-lung-thyroid disease and other NKX2-1 related conditions, such as benign hereditary chorea. Deletions of the entire NKX2-1 gene have also been reported. NKX2-1 related conditions are inherited in an autosomal dominant pattern with markedly variable expressivity among affected individuals.18
2.2.3 |. Pulmonary alveolar proteinosis
PAP also causes disruption of surfactant homeostasis. However, PAP usually presents in adolescents or adults, and rather than an absence of surfactant, there is an accumulation of surfactant in the alveolar space. Individuals with PAP may develop signs of interstitial lung disease, including dyspnea, exercise intolerance, and dry cough, with crackles on the exam and a “crazy paving” pattern on CT imaging of the chest. Bronchoalveolar lavage reveals copious frothy liquid, and lung biopsy is characterized by amorphous periodic acid-Schiff-positive fluid filling the alveolar space.27,28
Although PAP can occur following exposure to toxins or infection, autoimmune PAP is the most common cause of PAP, with autoantibodies targeting granulocyte-macrophage colony-stimulating factors (GM-CSF).27 Genetic causes of PAP are rare but should be considered, especially in children. Mutations in CSF2RA and CSF2RB genes, which encode the target receptors for GM-CSF, have been reported in autosomal recessive PAP with reduced penetrance.8
3 |. BRONCHIECTASIS
Cystic fibrosis
Primary ciliary dyskinesia
Immune deficiencies
Autosomal recessive pseudohypoaldosteronism-type 1
3.1 |. Clinical overview
Bronchiectasis is irreversible dilation of the bronchial lumen, with resulting reduced mucus clearance. Bronchiectasis can be focal and isolated, or it can be progressive and become diffuse. Nongenetic causes of bronchiectasis can include chronic aspiration, allergic hypersensitivity, severe lung infection, and inhalation injury.29 Areas of the lung affected by bronchiectasis are at increased risk for inflammation and infection. Clinical features include cough, sputum production, recurrent respiratory infections, and occasionally, hemoptysis. Genetic causes of bronchiectasis that should be considered in pediatric patients include cystic fibrosis (CF), PCD, immune defects, and pseudohypoaldosteronism (Figure 2). Bronchiectasis has also been reported as a rare complication of Marfan syndrome, which is discussed later in this review.30
FIGURE 2.
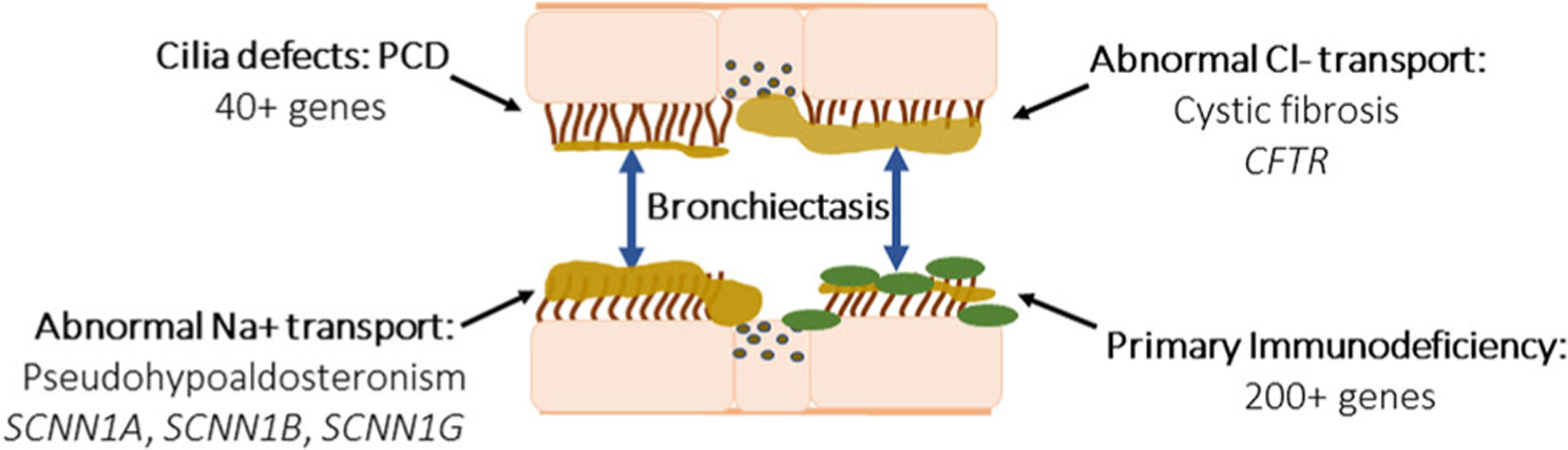
This cartoon represents the four main genetic categories of bronchiectasis, or dilation of the airway: (1) ciliary abnormalities, such as primary ciliary dyskinesia, which is caused by mutations in over 40 genes affecting cilia function or structure (2) abnormal epithelial Chloride (Cl−) transport, characteristic of cystic fibrosis, and mutations in CFTR, resulting build-up of thick tenacious airway secretions. (3) Abnormal sodium (Na+) transport as seen in pseudohypoaldosteronism due to mutations in SCNN1A, SCNN1B, and SCNN1G, which also cause thickened dehydrated airway secretions. (4) Primary immunodeficiencies, which comprise over 200 genetic conditions that cause impaired immune responses to infection, triggering increased mucus production and impaired airway clearance. CFTR, cystic fibrosis transmembrane conductance regulator; PCD, primary ciliary dyskinesia
3.2 |. Genetic conditions
3.2.1 |. Cystic fibrosis
CF, characterized by obstructive lung disease and pancreatic insufficiency, is one of the most common pediatric genetic conditions. It is an autosomal recessive condition associated with mutations in the CFTR gene, which encodes an epithelial chloride ion channel known as cystic fibrosis transmembrane conductance regulator (CFTR). Loss of CFTR expression leads to abnormal chloride transport which results in mucus obstruction in the pancreatic ducts and lungs.31,32
Recurrent infections in these patients can cause bronchiectasis, chronic cough, sputum production, dyspnea, airway obstruction, hemoptysis, and pneumothorax. Individuals with CF eventually progress to respiratory failure due to the accumulation of extensive lung damage, which is the principal cause of death in this patient population.31 CF-associated bronchiectasis generally presents within the first few years of life and is progressive. It is usually bilateral and can be saccular or cystic in appearance.33,34 Common infections in the CF population include various strains of Pseudomonas, Staphylococcus aureus, and Haemophilus influenzae. Additional manifestations include diabetes, sinusitis, gastrointestinal complications, and male infertility due to congenital absence of the vas deferens.32
CF is now universally screened for in the United States as a part of the newborn screening program; however, this screening does not identify all affected individuals, and clinical suspicion should guide additional diagnostic work-up. A positive newborn screen or high level of clinical suspicion at any age warrants quantification of sweat chloride through pilocarpine iontophoresis (“sweat test”), which should be completed at an accredited Cystic Fibrosis Center. Individuals found to have elevated sweat chloride levels (>60 mmol/L) should have full gene sequencing, with deletion and duplication analysis, at a clinical diagnostic laboratory.35
Pathogenic variants in CFTR are categorized into five classes based on the mechanism by which they cause disease. These mutation classes are associated with variable expressivity of symptoms. Mutations with a more severe impact result in the classical multisystemic presentation, while those at the mild end are associated with isolated congenital absence of the vas deferens or can be found in asymptomatic individuals.31,36 An online database known as CFTR2 (https://www.cftr2.org/) is maintained by CF experts and provides detailed information about specific variants, including whether a variant is disease-causing and the clinical features of variant carriers.
CF occurs in patients of all ethnicities but has the highest prevalence in the Caucasian population with carrier rates of approximately 1:28 in individuals of Northern European descent.37,38 The life expectancy of patients with CF has increased at a steady rate from 28 years in 1986 to 47.7 years in 2016 due to advances in treatment. The advent of CFTR modulators that can partially restore the function of the chloride channel in patients with specific mutations has been a major breakthrough for the treatment of patients with CF, particularly with the recent introduction of elexacaftor-tezacaftor-ivacaftor combination therapy that is indicated for nearly 90% of patients. These drugs, as well as other therapies under development, should have a substantial impact on overall life expectancy and quality of life for these patients in the coming years.32,39,40
CF-related metabolic syndrome, CF screen positive inconclusive diagnosis, and CF-related disorders are terms that are used to describe individuals who do not meet the full criteria for CF. These individuals may develop isolated features of CF, such as bronchiectasis, sinus disease, or absence of the vas deferens, or they may remain asymptomatic. They may have an intermediate sweat chloride (30–59 mmol/L) with one or no CF-causing mutation, or a normal sweat chloride (<30 mmol/L) despite two CFTR mutations.41,42 Although genotype-phenotype predictions are available for some combinations of mutations, additional factors, including environmental influences and genetic modifiers can impact disease expression. Therefore, this information is not predictive of clinical course on an individual level and patients with mild mutations should still receive periodic monitoring for clinical features of CF.43
3.2.2 |. Primary ciliary dyskinesia
PCD, previously known as immotile cilia syndrome or, if in conjunction with situs inversus, Kartagener’s syndrome, is one of a group of genetic conditions known as ciliopathies that are characterized by ciliary dysfunction.44 PCD results from abnormalities in structure or function of motile cilia, which line the respiratory tract, the ventricles of the central nervous system, the Fallopian tubes, and are required for sperm mobility. The most common ultrastructure defects in individuals with PCD result in absent or shortened dynein arms of the cilia, which are essential for a proper beating. The pulmonary symptoms associated with PCD, including recurrent respiratory tract infections and bronchiectasis, are caused by inadequate mucociliary clearance leading to mucus retention.45–47
Individuals with PCD often present during the newborn period with nasal congestion and as full-term neonates with respiratory distress requiring supplemental oxygen; however, the diagnosis of PCD is rarely made in infancy.48 PCD-associated respiratory distress is characterized by tachypnea, lobar collapse, hypoxemia, and atelectasis.49 Most individuals develop bronchiectasis involving the right middle lobe and lower lobes. Chronic cough, recurrent infections, digital clubbing, and obstructive lung disease are also observed in these patients.46,50
Extrapulmonary features include recurrent sinusitis and chronic otitis media that, if severe, can lead to conductive hearing loss. Approximately, 50% of individuals with PCD are reported to have laterality defects, including situs inversus totalis, heterotaxy with or without polysplenia, and congenital heart defects. However, there may be ascertainment bias in these reported rates due to the high clinical suspicion for PCD when these laterality defects are identified.50–52 Therefore, PCD should still be considered in the absence of laterality defects in individuals with consistent sino-pulmonary features. Most affected males are infertile due to abnormalities of sperm flagellar structure with the exception of those with CCDC114 mutations. Females are typically fertile but can experience issues due to the role of cilia in egg transport.47
The American Thoracic Society recently published guidelines regarding the diagnosis of PCD.53 Use of nasal nitric oxide (nNO) is suggested as the first line of testing, if possible, with reflex to genetic testing and/or ciliary biopsy to corroborate the diagnosis. When nNO is not possible, gene panel testing is recommended as the initial diagnostic study. Notably, negative gene panel does not definitively rule out PCD, as approximately 30% of individuals with high clinical suspicion have negative or inconclusive genetic testing results.52 Transmission electron microscopic analysis of ciliary ultrastructure, obtained by sampling the nasal or carinal epithelium, is not recommended as a first-line study as structural changes can be subtle and ciliary biopsy can even appear unremarkable in many individuals with PCD.54 However, ciliary ultrastructure analysis can be used to aid in clarifying a diagnosis, particularly in cases where genetic testing results are inconclusive.53
PCD is typically inherited in an autosomal recessive pattern, with biallelic pathogenic changes in the DNAI1, DNAH5, and DNAH11 genes as the most common causes of disease.55,56 Two genes, OFD1 and RPGR, are associated with syndromic X-linked recessive forms of PCD. To date, over 40 genes have been associated with PCD.49
3.2.3 |. Primary immunodeficiencies
Primary immunodeficiencies (PID) encompass over 180 monogenic disorders that cause an intrinsic defect in the immune system. PID can present with varying clinical severity or infection tendencies. The most severe types are lethal in infancy or early childhood, while milder types can cause minimal symptoms or be subclinical. Individuals with PID may develop recurrent and persistent infections caused by organisms that are rarely infectious in the general population, or they may develop inflammatory-mediated interstitial lung disease. The pulmonary phenotype of these conditions can be similar to what is observed in CF and PCD patients: bronchiectasis, asthma, chronic cough, and dyspnea.57–59
Severe combined immunodeficiency (SCID) is a rare severe condition affecting both cellular and humoral immunity, characterized by absent T lymphocytes with variable B and natural killer cell function that presents in infancy with life-threatening infections.60 The most common type of SCID is inherited in an X-linked recessive pattern and caused by mutations in the IL2RG gene. The most common autosomal recessive causes of SCID result from mutations in the ADA, CD3D, DCLRE1C, JAK3, RAG1, and RAG2 genes.61 SCID is lethal without effective treatment, usually a bone marrow transplant, and is, therefore, included as a part of the national newborn screening program in the United States and several other countries.62
Common-variable immune deficiency (CVID) is one of the most prevalent PIDs and is characterized by B cell dysfunction resulting in low immunoglobulin levels. CVID can often be mild and may not be recognized until adulthood.58,59 The majority of CVID cases occur in individuals with no apparent family history of the disorder and may have a multifactorial inheritance. When a genetic cause is identified, the most commonly mutated gene is TNFRSF13B (previously known as TACI). TNFRSF13B-associated CVID can be inherited in an autosomal dominant or recessive pattern.63,64
The majority of pediatric-onset PID is inherited in autosomal recessive or X-linked recessive patterns; however, in a large proportion of individuals with suspected immune deficiencies, underlying genetic causes are not identified. It is also important to note that several syndromic genetic conditions, including DiGeorge syndrome, trisomy 21, Noonan syndrome, ataxia-telangiectasia, and Wiskott-Aldrich syndrome, are also associated with immunodeficiency, and recurrent infections in these patient populations may warrant further immune work-up and treatment.62
Identification of unusual pathogens or recurrent/severe infections in a child should prompt an immune evaluation including measurement of immunoglobulin levels (including response to immunizations), complement function, neutrophil function, mitogen and antigen responses, and flow cytometry to measure immune cell populations, guided by the clinical suspicion of the underlying immune defect.
3.2.4 |. Autosomal recessive pseudohypoaldosteronism-type 1
Autosomal recessive pseudohypoaldosteronism-type 1 (PHA1) is characterized by salt wasting leading to hyponatremia, hyperkalemia, and severe dehydration. Systemic PHA1 often presents within the first two weeks of life, with varying severity. Symptoms include failure to thrive, fatigue, nausea, vomiting, and muscle weakness. Individuals with autosomal recessive PHA1 have high plasma levels of aldosterone and renin. Additional features include cardiac arrhythmias, skin eruptions, cholelithiasis, and short stature. Life-long salt supplementation is usually necessary; however, salt-wasting episodes tend to decrease in frequency and severity over time.65,66
Respiratory symptoms of autosomal recessive PHA1 are similar to CF: cough, chest congestion, excessive, thick airway mucus, and recurrent lower respiratory infections. In addition, patients with PHA1 can have positive sweat chloride testing, which can lead to misdiagnosis of CF.67,68 However, unlike the progressive clinical course of CF, respiratory symptoms in PHA1 tend to improve over time.65,66,69
Mutations in the SCNN1A, SCNN1B, and SCNN1G genes, encoding the alpha, beta, and gamma subunits of the epithelial sodium channel, respectively, result in autosomal recessive PHA1.70 Loss of function of these genes results in reduced or absent function of the epithelial sodium channel, which is found in the kidneys, lungs, and sweat glands, leading to a reduced stimulatory response to aldosterone.71 The autosomal dominant form of PHA1 is isolated to the renal manifestations.65
4 |. PULMONARY FIBROSIS
Short telomere syndromes
Hermansky-Pudlak syndrome
4.1 |. Clinical overview
Patients presenting with exercise intolerance, hypoxia, or desaturation with activity should prompt the consideration of pulmonary fibrosis. Crackles can be heard on auscultation of the lungs, and pulmonary function testing reflects restrictive patterns with a decreased diffusion capacity. Chest CT shows patchy peripheral reticular abnormalities with intralobular linear opacities, irregular septal thickening, subpleural honeycombing, and traction bronchiectasis. Although in the adult population pulmonary fibrosis may be multifactorial, resulting from a combination of genetic and environmental risk factors,72,73 it is exceedingly rare in the pediatric population and underlying causes are often identifiable, including genetic conditions, such as short telomere syndromes and Hermansky-Pudlak syndrome (HPS). Pulmonary fibrosis is also reported in several other conditions discussed in this review including, brain-lung-thyroid syndrome, Marfan syndrome, and surfactant deficiencies.
4.2 |. Genetic conditions
4.2.1 |. Short telomere syndromes
Eukaryotic chromosomes end in structures known as telomeres, which are repetitive sequences that protect the coding DNA and the integrity of the chromosome. With each mitotic cell division, telomere length normally shortens slightly. Once telomeres have shortened sufficiently, cell death occurs. While this is a natural part of aging, individuals with excessively short telomeres for age can have various clinical manifestations affecting multiple organ systems.74 Short telomere syndromes occur when there are defects in telomere elongation or maintenance, resulting in a significant reduction in telomere length. In general, shorter telomere length correlates with increased severity of the disease, disproportionately impacting tissue types with high rates of cell proliferation, such as bone marrow, skin, and immune cells.75–77
Short telomere syndromes exhibit variable expressivity, including a wide range of clinical features and ages of onset. Dyskeratosis congenita (DC), a childhood-onset syndromic condition, represents the more severe end of the disease spectrum. Symptoms include the diagnostic triad of oral leukoplakia, nail dystrophy, and reticulated pigmentation. Epiphora (excessive watering of the eyes due to lacrimal duct obstruction), lymphopenia, dental abnormalities, short stature, developmental delay, premature hair loss or graying, esophageal stricture or narrowing, hyperhidrosis, brain abnormalities, intrauterine growth restriction, and genitourinary abnormalities may also be seen.78,79 In rare cases, pulmonary arteriovenous malformations (AVMs) have been reported.80
Although pulmonary fibrosis can be seen in pediatric patients with DC, it is otherwise rare for children to develop pulmonary fibrosis as part of a short telomere syndrome. However, adult-onset pulmonary fibrosis may be the first life-threatening manifestation of short telomere syndromes, often presenting with comorbid aplastic anemia or liver cirrhosis.75,76,81–84 If a family member presents with symptoms of short telomere syndrome, genetic evaluation of family members could be considered.
Genetic testing can confirm a diagnosis of DC in children with clinical features. Measurement of leukocyte telomere length by automated multicolor flow cytometry fluorescence in situ hybridization can confirm the diagnosis of short telomere syndromes when an underlying genetic mutation cannot be identified.79,80 The most common genetic defects responsible for short telomere syndromes result in reduced activity of telomerase, the main enzyme involved in telomere elongation. TERC, TERT, and DKC1, which encode the proteins that make up the major subunits of the telomerase complex, are the most frequently reported mutations. Mutations in TNIF2, RTEL1, CTC1, ACD, NOP10, NHP2, PARN, USB1, and WRAP53 genes have also been identified as rare causes of short telomere syndromes.76,79,80
Families with TERC and TERT-related autosomal dominant forms of the disease can exhibit genetic anticipation, which means there may be increased severity of the clinical presentation with each subsequent generation. It is believed that the additive effect of inheriting a mutation that reduces telomere length in combination with inheriting cells with shorter baseline telomere length from an affected parent results in the increased severity of disease.20,76
4.2.2 |. Hermansky-Pudlak syndrome
HPS is an autosomal recessive disease characterized by oculocutaneous albinism combined with a deficiency of platelet delta granules, resulting in a bleeding diathesis.85 Individuals with HPS generally have lighter skin, hair, and eye color than other family members plus ocular abnormalities including nystagmus, photo-phobia, decreased visual acuity, and lack of visual attention. Individuals with HPS can also experience easy bruising, epistaxis, prolonged bleeding after minor procedures, postpartum hemorrhage, colonic bleeding, gingival bleeding, and prolonged menstruation.86,87
Pathogenic variants in AP3B1, AP3D1, BLOC1S3, BLOC1S6, DTNBP1, HPS1, HPS3, HPS4, HPS5, and HPS6 genes are associated with HPS. These genes encode proteins involved in the biogenesis of lysosome-related organelle complexes (BLOCs). BLOCs traffic lysosome-related organelles, including melanosomes and platelet dense granules. Defective BLOC function results in improper trafficking of these organelles, leading to albinism and bleeding diathesis.87,88
Some subtypes of HPS are associated with specific clinical features. The HPS1, HPS4, and AP3B1 genes are linked with the development of early-onset pulmonary fibrosis, typically occurring during the third decade of life, which can mimic idiopathic pulmonary fibrosis.85,89 HPS1 and HPS4 are also associated with granulomatous colitis, while the AP3B1 and AP3D1 genes are associated with immunodeficiencies and severe neutropenia as well as increased neurological involvement.85,90,91 Although HPS occurs across all ethnicities, there is an increased incidence of HPS1 and HPS3-related disease in individuals of northwest Puerto Rican descent due to founder mutations in that population.85
5 |. ABNORMALITIES OF THE PULMONARY VASCULATURE
Alveolar capillary dysplasia
Pulmonary arterial hypertension
Hereditary hemorrhagic telangiectasia
5.1 |. Clinical overview
Genetic conditions associated with abnormalities of the pulmonary vasculature should be considered when there is significant shortness of breath or hypoxemia without underlying pulmonary parenchymal or cardiac structural abnormalities. Genetic causes of pulmonary vascular disease may alter either the structure or function of pulmonary veins and arteries and are often associated with abnormalities of the vasculature in other parts of the body or additional syndromic features. ACD, pulmonary arterial hypertension (PAH), and hereditary hemorrhagic telangiectasia (HHT) are discussed in this review; however, many other genetic syndromes that are associated with vascular abnormalities can impact pediatric pulmonary patients.
5.2 |. Genetic conditions
5.2.1 |. Alveolar capillary dysplasia with misalignment of pulmonary veins
ACD/MVP is generally lethal within the neonatal period. Shortly after birth, infants develop severe hypoxemia and respiratory distress that is unresponsive to standard treatments, including mechanical ventilation and supplemental oxygen. An echocardiogram reveals persistent pulmonary hypertension that fails to improve with inhaled nitric oxide. Most individuals with ACD/MVP have extrapulmonary involvement.92,93 Cardiovascular, gastrointestinal, and genitourinary malformations, including atrial septal defects, intestinal malrotation, Hirschprung’s disease, imperforate anus, duodenal atresia, and omphalocele have all been identified. In rare cases with a milder phenotype, later onset of the disease has been reported.94–96
Respiratory issues result primarily from improperly positioned alveolar capillaries leading to the defective transfer of oxygen from the alveoli to the circulation. Histologically, lung tissue is characterized by reduced and malpositioned alveolar wall capillaries, abnormally located pulmonary veins, deficient pulmonary lobular development, and thickening of the muscle walls of small pulmonary arteries with possible pulmonary lymphangiectasis.92
Mutations in the FOXF1 gene cause autosomal dominant ACD/MVP. FOXF1 encodes a transcription factor that impacts the function of many genes, including those involved in the development of the pulmonary mesenchyme. Most pathogenic mutations result in loss of function of the FOXF1 gene. Almost all cases of FOXF1-related ACD/MPV occur in individuals with no family history of the disease as a result of de novo changes.97 In a few reported cases, variants in FOXF1 were found to be inherited from an unaffected mother, suggesting a possible imprinting effect.92 Some individuals have mutations within the FOXF1 gene, while others have a microdeletion at position 16q24.1 resulting in loss of the entire FOXF1 gene. Microdeletions in this region can encompass other nearby genes and be associated with additional features, such as heart defects.95
5.2.2 |. Heritable PAH
Patients who present with dyspnea, fatigue, syncope, chest pain, palpitations, and leg edema, and are found to have signs of pulmonary hypertension on echocardiogram or electrocardiogram, should be evaluated for hereditary PAH.98,99 PAH results from the narrowing and obstruction of the pulmonary arterioles, causing increased pulmonary arterial pressure and right ventricular overload.100 It can occur at any age and is often associated with a precipitous decline in health, with a mean survival of 2.8 years postdiagnosis.101
Most cases of hereditary PAH are caused by mutations in the BMPR2 gene and are inherited in an autosomal dominant pattern. Variants resulting in loss of function of the BMPR2 gene, leading to an absent or truncated protein product are the most common cause of hereditary PAH. BMPR2 encodes the bone morphogenetic protein receptor type 2, which is involved in the growth and differentiation of cartilage and bone; however, it remains unclear how pathogenic variants in this gene lead to the development of PAH.102
In rare cases, pathogenic variants in other genes have been identified in individuals with PAH. These genes include ACVRL1, BMPR1B, CAV1, CBLN2, EIF2AK4, ENG, KCNA5, KCNK3, and SMAD9. The ACVRL1 and ENG genes are also associated with HHT, which is described in more detail below.98,99 Additional genetic syndromes associated with PAH include FLNA-associated periventricular heterotopia, Gaucher disease, pulmonary veno-occlusive disease, and sickle cell anemia.10,103–106
Hereditary PAH is characterized by markedly reduced penetrance of approximately 20% and variable expressivity.107 Females are more likely to be affected than males; however, affected males generally have a more severe disease course.98,108 Pregnancy in individuals with hereditary PAH can be dangerous and is associated with high mortality.102
5.2.3 |. Hereditary hemorrhagic telangiectasia
Similar to PAH, HHT should be considered in patients with dyspnea or hypoxemia with exertion. Distinctive features of HHT include recurrent, potentially severe epistaxis, usually beginning in childhood, and the presence of telangiectasias.109,110 Telangiectasias occur on the mucocutaneous surfaces of the body, especially the lips, oral cavity, fingers, face, and chest. When present in the gastrointestinal mucosa, they can lead to gastrointestinal bleeding, a common complication in HHT.110,111
Larger AVMs occurring in the lungs, liver, and brain, can be life- threatening. Although pulmonary AVMs occur in only 30% to 50% of individuals with HHT, most individuals with pulmonary AVMs have HHT.112 Pulmonary AVMs generally increase in size over time and serious complications include stroke, brain abscess, and transient ischemic attacks from paradoxical emboli.113 They occasionally rupture causing hemoptysis and some individuals with pulmonary AVMs develop PAH.112,114 CT imaging of the chest, completed with IV contrast, reveals well-circumscribed areas of increased blood supply, occasionally with a feeding vessel visualized. Hepatic AVMs are also common in individuals with HHT, although they are frequently asymptomatic.115,116 Cerebral AVMs, which can lead to hemorrhage, occur in approximately 10% of individuals and are usually present from birth.110,117
HHT, inherited in an autosomal dominant pattern, is most often caused by pathogenic variants in the ACVRL1, ENG, and SMAD4 genes. Mutations in GDF2 have been identified as a rare cause of HHT. SMAD4-related HHT presents with the concurrence of HHT and juvenile polyposis syndrome, which is characterized by a predisposition to develop hamartomatous polyps in the gastrointestinal tract and an increased risk of gastrointestinal cancers.111 A significant portion of individuals meeting clinical criteria for a diagnosis of HHT do not have identifiable mutations in one of these genes, suggesting that additional genetic etiologies of HHT have yet to be discovered or that these individuals have another syndrome with overlapping features.111,118
The ACVRL1 and ENG genes encode proteins involved in the differentiation of blood vessels into veins and arteries and the coordination of smooth muscle cells in developing vessels. The SMAD4 gene encodes a protein involved in signaling in the transforming growth factor-β pathway. It functions as both a transcription factor and tumor suppressor, although the exact mechanism by which SMAD4 mutations cause HHT with juvenile polyposis is not well understood.110
6 |. PNEUMOTHORAX
Connective tissue diseases
Birt-Hogg-Dubé syndrome
6.1 |. Clinical overview
Although pneumothorax may develop spontaneously or following trauma, repeated occurrences of pneumothorax should prompt suspicion of an underlying genetic etiology, such as a connective tissue disorder or Birt-Hogg-Dubé (BHD), especially if there is a family history of pneumothorax. Additional clinical manifestations such as joint laxity, skin abnormalities, dysmorphic facial features, or the presence of lung blebs or cysts seen on chest imaging, should also prompt genetic evaluation.119
While recurrent pneumothorax may trigger genetic evaluation, it is not usually life-threatening. Associated extrapulmonary complications, namely the vascular issues common in connective tissue disorders and renal cancer associated with BHD are associated with high mortality if left undetected. Thus, efficient diagnosis is essential for proper screening and management of these patients. As the majority of the genetic conditions described below has an autosomal dominant inheritance and potentially life-threatening extrapulmonary features, it is important to consider the implications of these diagnoses for family members who may also need genetic testing and clinical screening.
Pneumothorax can also occur in conditions associated with chronic respiratory infections, such as CF and PCD,120,121 and in other genetic conditions not described in detail in this review, such as autosomal recessive polycystic kidney disease, FLNA-associated periventricular heterotopia, homocystinuria, and tuberous sclerosis.10,103,122–124
6.2 |. Genetic conditions
6.2.1 |. Connective tissue disorders
Connective tissue disorders are a large genetically heterogeneous group of conditions that are associated with cutaneous, skeletal, and vascular abnormalities.125 They typically result from mutations in genes involved in the structure or function of the primary proteins that comprise connective tissues including collagen, elastin, and glycosaminoglycans. A subset of connective tissue disorders is associated with the development of pneumothorax. Figure 3 highlights the overlap of clinical features and distinctions of the select connective tissue disorders described below.
FIGURE 3.
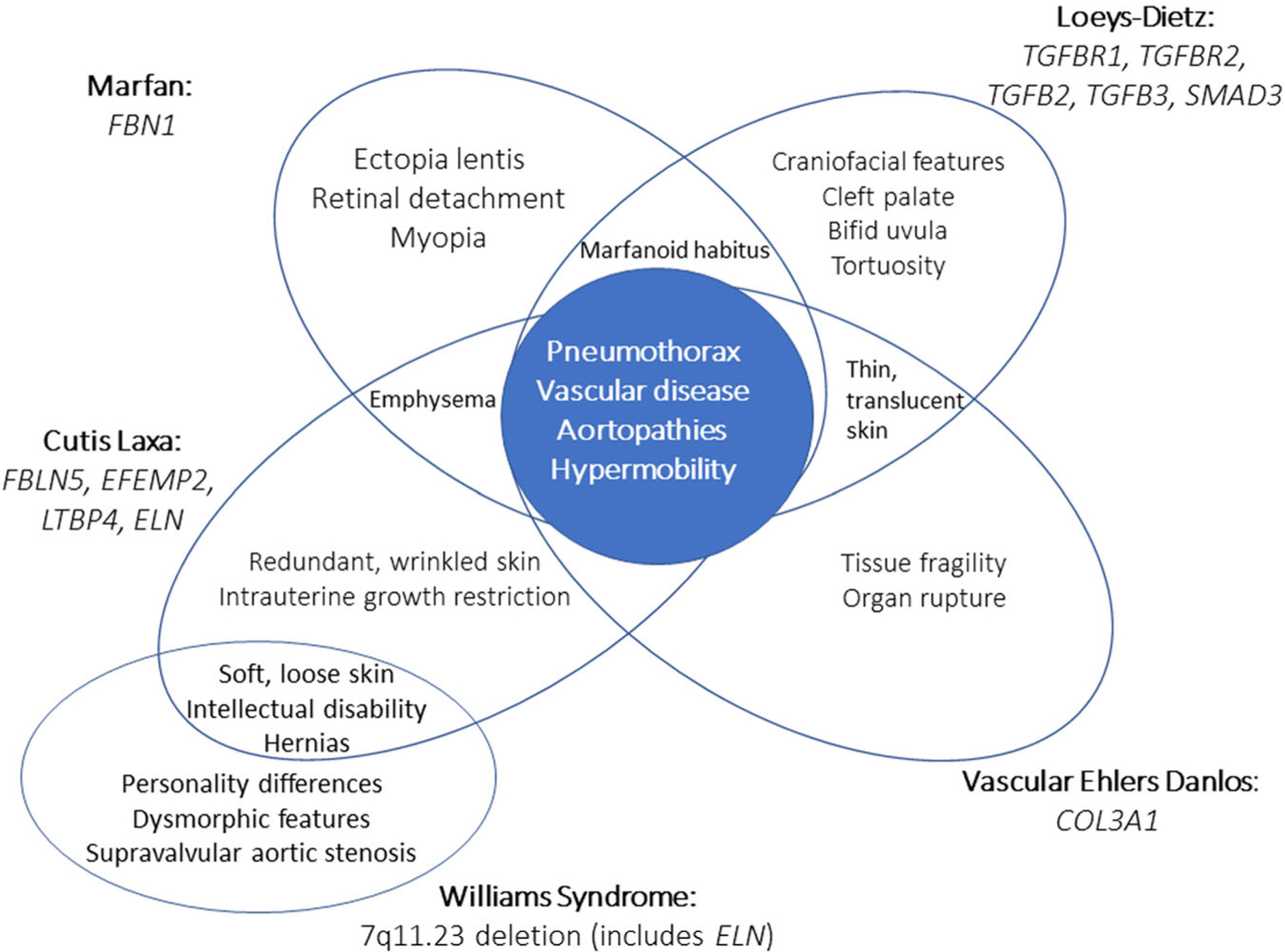
This Venn diagram illustrates the phenotypic overlap of connective tissue disorders associated with pneumothorax, Marfan syndrome (FBN1), Loeys-Dietz (TGFBR1 and TGFBR2), vascular Ehlers-Danlos syndrome (COL3A1), cutis laxa (FBLN5, EFEMP2, LTBP4, and ELN), and Williams syndrome (7q11.23 deletion, which includes ELN). Central features of these diseases, however, include pneumothorax, joint hypermobility, and vascular disease, including aortopathies
Marfan syndrome
Marfan syndrome is an autosomal dominant condition caused by mutations in the FBN1 gene, which encodes the fibrillin protein. Fibrillin is a structural protein that makes up part of the structure of microfibrils, which form elastic fibers found in skin ligaments and blood vessels and provide support in nonelastic tissues including bone.126
Marfan syndrome can have variable features, but classically involves characteristic phenotypic changes of the skeletal, ocular, and cardiovascular systems. Individuals with Marfan syndrome are often taller than unaffected family members with long extremities, arachnodactyly, pectus deformities, scoliosis, and joint laxity. Ocular manifestations include ectopia lentis, retinal detachment, and myopia. Cardiovascular complications include aortic dilation, mitral valve prolapse, tricuspid valve prolapse, and enlargement of the pulmonary artery root, which can be life-threatening.126,127
Pneumothorax occurs in approximately 5% to 10% of individuals with Marfan syndrome and has been attributed to various underlying causes in different individuals, including the presence of apical pulmonary blebs or bullae, connective tissue abnormalities in the lung parenchyma, and mechanical stress on the lung apices due to tall habitus.128 Individuals with Marfan syndrome can also have additional pulmonary manifestations including cystic changes, emphysema, bronchiectasis, congenital malformations, and fibrosis.30
Ehlers-Danlos syndromes
Ehlers-Danlos syndromes (EDS) are a group of 13 disorders characterized by joint hypermobility, skin hyperextensibility, and tissue fragility.129 Spontaneous pneumothorax has primarily been described in patients with the vascular subtype of EDS, which is an autosomal dominant condition caused by mutations in the COL3A1 gene. COL3A1 encodes type III collagen, which is found in the skin, lungs, intestinal walls, and vascular walls.130 Common features of the vascular form of Ehlers-Danlos syndrome (vEDS) include easy bruising, thin, translucent skin, characteristic facial features, and tissue fragility. Ruptures or dissection of arteries, gastrointestinal perforation, and organ rupture all lead to the high mortality associated with vEDS.130 Several other forms of EDS can have genetic causes and overlapping features of vEDS but have not yet been associated with pneumothorax.
Pneumothorax in vEDS can present in childhood or adulthood and can be spontaneous or recurrent. Hemothorax and hemopneumothorax have been reported in individuals with vEDS and are often associated with pulmonary blebs, cystic legions, or lung nodules.129 Individuals can also have hematomas and severe recurrent hemoptysis.131
Loeys-Dietz syndrome
Loeys-Dietz syndrome (LDS) is an autosomal dominant condition characterized by vascular, skeletal, craniofacial, and cutaneous features. Skeletal manifestations include pectus deformities, scoliosis, joint laxity, arachnodactyly, and talipes equinovarus. Vascular complications include aortic dilation, which is present in more than 95% of individuals with LDS and can lead to aortic dissection. Arterial aneurysms and tortuosity can also be present in other organs. Craniofacial features include hypertelorism, bifid uvula, cleft palate, and craniosynostosis, while cutaneous findings include velvety, translucent skin, easy bruising, and dystrophic scarring. The prevalence of pneumothorax and restrictive lung disease in LDS patients is unknown.132,133 Most cases of LDS are caused by mutations in the TGFBR1 and TGFBR2 genes with mutations in the SMAD2, SMAD3, TGFB2, and TGFB3 genes accounting for rare causes.132
Cutis laxa
Cutis laxa is characterized by loose, saggy, and inelastic skin, that is particularly noticeable around the neck, armpits, and groin. Individuals with cutis laxa may also have inguinal, umbilical, and diaphragmatic hernia, visceral diverticula, joint hypermobility, developmental issues, and cardiovascular involvement. Pulmonary manifestations include neonatal or childhood-onset emphysema, which can be severe, progressive, and lead to pneumothorax, laryngomalacia, tracheomalacia, bronchomalacia, bronchiolitis, and pulmonary hypertension.
The FBLN5, EFEMP2, LTBP4, and ELN genes are associated with cutis laxa and the development of emphysema. EFEMP2 and LTBP4 are associated with autosomal recessive cutis laxa; FBLN5 mutations can be inherited in an autosomal dominant or recessive pattern.134–136 Mutations in the ELN gene may cause autosomal dominant cutis laxa. Notably, Williams syndrome, which results from a microdeletion at 7q11.23 and is characterized by unique personality features, distinct facial features, and intellectual disability, may also result in deletion of the ELN gene, with ensuing connective tissue complications, including possible emphysema or pneumothorax.137
6.2.2 |. Birt-Hogg-Dubé syndrome
BHD syndrome is an autosomal dominant condition caused by mutations in the FLCN gene, which encodes the folliculin protein. Although the exact function of folliculin is unknown, it is hypothesized to function at least in part as a tumor suppressor and is expressed in the brain, heart, lungs, kidneys, skin, and testes.138 BHD primarily affects the skin, lungs, and kidneys. Although the penetrance of BHD is high, the clinical presentation and severity of the disease is variable, and BHD is thought in general to be underdiagnosed.138,139
Cutaneous hamartomas, including fibrofolliculomas, trichodiscomas, and acrochordons, are common and generally benign. Fibrofolliculomas, which are the most common skin finding, typically appear on the face, neck, and upper trunk and develop in adulthood.140
The majority of individuals with BHD has bilateral multifocal basal or medial lung cysts. Although these lung cysts are initially asymptomatic, these patients are at high risk to develop pneumothoraces that can be recurrent. Pneumothoraces primarily occur in adolescence or adulthood, and they can be the presenting feature of BHD.141,142
Importantly, individuals with BHD are at increased risk of developing renal tumors (an estimated 15%−30% lifetime risk), and thus the diagnosis of BHD should prompt kidney screening. Oncocytic hybrid tumors, chromophobe renal cell carcinoma, and renal oncocytomas are the most common tumors observed in patients with BHD. Clear cell renal cell carcinoma and papillary renal cell carcinoma have also been reported. While the renal tumors in BHD tend to be slow-growing, early detection in this high-risk population is essential to prevent malignancy. Many individuals with BHD are found to have renal cysts on imaging.138,143
7 |. CONGENITAL CENTRAL HYPOVENTILATION SYNDROME
7.1 |. Clinical overview
Congenital central hypoventilation syndrome (CCHS) is a disorder of the respiratory and autonomic nervous systems. CCHS classically presents in the newborn period with alveolar hypoventilation with increased episodes during non-rapid eye movement sleep. In severe cases, infants can have complete apnea during sleep as well as hypoventilation during wakefulness. Hypoventilation in individuals with CCHS often results in hypoxemia and hypercapnia and requires mechanical ventilation or placement of a diaphragmatic pacemaker. Some individuals require 24-hour mechanical ventilatory support. Although the respiratory issues associated with CCHS can be life- threatening, early diagnosis and aggressive management can significantly improve prognosis and quality of life.144–146
Individuals with CCHS have additional complications of autonomic nervous system dysfunction affecting multiple body systems. Symptoms include decreased heart rate variability, dysrhythmias, syncope, gastrointestinal issues, altered pain perception, ophthalmological abnormalities, temperature and sweating dysregulation, and urinary issues.146,147 In addition, some individuals have altered the development of neural crest-derived structures which can lead to Hirschprung’s disease or tumors of neural origin.148 While most individuals present in the newborn period, later onset has been reported. Patients with later onset disease usually have a milder phenotype.149
7.2 |. Genetic condition
CCHS is caused by mutations in the PHOX2B gene and is inherited in an autosomal dominant pattern. Most mutations in PHOX2B are expansions of the polyalanine repeat region in exon 3. Normally, individuals have 20 repeats of three nucleotides encoding the amino acid alanine. When this region expands to 25 or greater alanine repeats, individuals are affected by CCHS. Individuals with 24 repeats and some individuals with 25 repeats can have a mild clinical presentation. Due to the repetitive nature of the polyalanine region, these types of mutations can be difficult to detect by traditional sequencing methods and targeted fragment length analysis may be necessary to characterize these variations.144 Some individuals with CCHS have other types of mutations in the PHOX2B gene, primarily frameshift variants.149
The majority of repeat expansions occur as de novo mutations; however, a subset of asymptomatic parents have been found to have somatic mosaicism for the polyalanine repeat variations identified in their children.150,151 Somatic mosaicism can be difficult to detect, but this is an important finding because it suggests that the recurrence risk of CCHS in future children of these individuals may not be negligible as compared to most other conditions caused by primarily de novo mutations that are rarely known to recur in families.
PHOX2B encodes a homeobox domain transcription factor that is expressed during embryonic development and important for proper formation of the peripheral and central autonomic nervous systems. PHOX2B is essential for the development of hindbrain motor neurons and neural crest cells.149 Although mutations in PHOX2B are generally repeated expansion variations or frameshift variants, which usually have a loss-of-function impact on gene function, it is thought that pathogenic mutations in PHOX2B cause disease via a dominant-negative effect. This dominant-negative effect is hypothesized to occur when the abnormal protein product from the mutated allele dimerizes with the normal protein product of the wild type allele and disrupts its function.144,152
8 |. CONCLUSION
The genetic determinants for a growing number of pediatric pulmonary diseases have been identified. Recent advances in genetic testing technology combined with the reduced cost of genetic testing have made genetic evaluation a routine and integral component of the diagnostic work-up. It is important that pediatric pulmonologist be aware of the latest developments in pulmonary genetic disease and how to obtain genetic testing in the most effective manner.
Genetic evaluation and diagnosis is the first step in caring for a patient with a genetic condition. Individuals with a confirmed genetic condition should be connected with the appropriate registries and guideline-directed care should be followed. In the future, genetic therapies for an increasing number of these conditions will become available. In summary, genetics is a crucial component of pediatric pulmonary medicine and understanding and properly incorporating genetic evaluation in the pulmonary clinic is becoming a standard of care.
REFERENCES
- 1.Griese M Pulmonary surfactant in health and human lung diseases: state of the art. Eur Respir J. 1999;13(6):1455–1476. [DOI] [PubMed] [Google Scholar]
- 2.Whitsett JA, Wert SE, Weaver TE. Diseases of pulmonary surfactant homeostasis. Annu Rev Pathol. 2015;10:371–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liszewski MC, Stanescu AL, Phillips GS, Lee EY. Respiratory distress in neonates: underlying causes and current imaging assessment. Radiol Clin North Am. 2017;55(4):629–644. [DOI] [PubMed] [Google Scholar]
- 4.Hamvas A, Deterding RR, Wert SE, et al. Heterogeneous pulmonary phenotypes associated with mutations in the thyroid transcription factor gene NKX2–1. Chest. 2013;144(3): 794–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nattes E, Lejeune S, Carsin A, et al. Heterogeneity of lung disease associated with NK2 homeobox 1 mutations. Respir Med. 2017;129: 16–23. [DOI] [PubMed] [Google Scholar]
- 6.Cutz E, Chami R, Dell S, Langer J, Manson D. Pulmonary interstitial glycogenosis associated with a spectrum of neonatal pulmonary disorders. Hum Pathol. 2017;68:154–165. [DOI] [PubMed] [Google Scholar]
- 7.Nevel RJ, Garnett ET, Worrell JA, et al. Persistent lung disease in adults with NKX2.1 mutation and familial neuroendocrine cell hyperplasia of infancy. Ann Am Thorac Soc. 2016;13(8):1299–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Griese M Pulmonary alveolar proteinosis: a comprehensive clinical perspective. Pediatrics. 2017;140(2):e20170610. [DOI] [PubMed] [Google Scholar]
- 9.Nathan N, Borensztajn K, Clement A. Genetic causes and clinical management of pediatric interstitial lung diseases. Curr Opin Pulm Med. 2018;24(3):253–259. [DOI] [PubMed] [Google Scholar]
- 10.Pelizzo G, Collura M, Puglisi A, et al. Congenital emphysematous lung disease associated with a novel filamin A mutation. Case report and literature review. BMC Pediatr. 2019;19(1):86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kremer TM, Lindsay ME, Kinane TB, Hawley MH, Little BP, Mino-Kenudson M. Case 28–2019: a 22-year-old woman with dyspnea and chest pain. N Engl J Med. 2019;381(11):1059–1067. [DOI] [PubMed] [Google Scholar]
- 12.Doan ML, Guillerman RP, Dishop MK, et al. Clinical, radiological and pathological features of ABCA3 mutations in children. Thorax. 2008; 63(4):366–373. [DOI] [PubMed] [Google Scholar]
- 13.Kröner C, Wittmann T, Reu S, et al. Lung disease caused by ABCA3 mutations. Thorax. 2017;72(3):213–220. [DOI] [PubMed] [Google Scholar]
- 14.Turcu S, Ashton E, Jenkins L, Gupta A, Mok Q. Genetic testing in children with surfactant dysfunction. Arch Dis Child. 2013;98(7): 490–495. [DOI] [PubMed] [Google Scholar]
- 15.Kröner C, Reu S, Teusch V, et al. Genotype alone does not predict the clinical course of SFTPC deficiency in paediatric patients. Eur Respir J. 2015;46(1):197–206. [DOI] [PubMed] [Google Scholar]
- 16.Thurm T, Kaltenborn E, Kern S, Griese M, Zarbock R. SFTPC mutations cause sp-c degradation and aggregate formation without increasing er stress. Eur J Clin Invest. 2013;43(8):791–800. [DOI] [PubMed] [Google Scholar]
- 17.Gras D, Jonard L, Roze E, et al. Benign hereditary chorea: phenotype, prognosis, therapeutic outcome and long term follow-up in a large series with new mutations in the TTF1/NKX2–1 gene. J Neurol Neurosurg Psychiatry. 2012;83(10):956–962. [DOI] [PubMed] [Google Scholar]
- 18.Patel NJ, Jankovic J. In: Adam MP, Ardinger HH, Pagon RA et al. , eds. NKX2-1-Related Disorders. Seattle, WA: Genereviews; 1993. [Google Scholar]
- 19.de Gusmao CM, Kok F, Casella EB, Waugh JL. Benign hereditary chorea related to NKX2–1 with ataxia and dystonia. Neurol Genet. 2016;2(1):e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carre A, Szinnai G, Castanet M, et al. Five new TTF1/NKX2.1 mutations in brain-lung-thyroid syndrome: rescue by PAX8 synergism in one case. Hum Mol Genet. 2009;18(12):2266–2276. [DOI] [PubMed] [Google Scholar]
- 21.Guillot L, Carré A, Szinnai G, et al. NKX2–1 mutations leading to surfactant protein promoter dysregulation cause interstitial lung disease in “brain-lung-thyroid syndrome”. Hum Mutat. 2010;31(2): E1146–E1162. [DOI] [PubMed] [Google Scholar]
- 22.Galambos C, Levy H, Cannon CL, et al. Pulmonary pathology in thyroid transcription factor-1 deficiency syndrome. Am J Respir Crit Care Med. 2010;182(4):549–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Young LR, Deutsch GH, Bokulic RE, Brody AS, Nogee LM. A mutation in TTF1/NKX2.1 is associated with familial neuroendocrine cell hyperplasia of infancy. Chest. 2013;144(4):1199–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gomes VC, Silva MC, Maia Filho JH, et al. Diagnostic criteria and follow-up in neuroendocrine cell hyperplasia of infancy: a case series. J Bras Pneumol. 2013;39(5):569–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Willemsen MA, Breedveld GJ, Wouda S, et al. Brain-thyroid-lung syndrome: a patient with a severe multi-system disorder due to a de novo mutation in the thyroid transcription factor 1 gene. Eur J Pediatr. 2005;164(1):28–30. [DOI] [PubMed] [Google Scholar]
- 26.Nettore IC, Mirra P, Ferrara AM, et al. Identification and functional characterization of a novel mutation in the NKX2–1 gene: Comparison with the data in the literature. Thyroid. 2013;23(6): 675–682. [DOI] [PubMed] [Google Scholar]
- 27.Trapnell BC, Whitsett JA, Nakata K. Pulmonary alveolar proteinosis. N Engl J Med. 2003;349(26):2527–2539. [DOI] [PubMed] [Google Scholar]
- 28.Carrington JM, Hershberger DM. Pulmonary Alveolar Proteinosis. Treasure Island, FL: StatPearls; 2018. [PubMed] [Google Scholar]
- 29.Schafer J, Griese M, Chandrasekaran R, Chotirmall SH, Hartl D. Pathogenesis, imaging and clinical characteristics of CF and non-CF bronchiectasis. BMC Pulm Med. 2018;18(1):79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dyhdalo K, Farver C. Pulmonary histologic changes in Marfan syndrome: a case series and literature review. Am J Clin Pathol. 2011; 136(6):857–863. [DOI] [PubMed] [Google Scholar]
- 31.Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005; 352(19):1992–2001. [DOI] [PubMed] [Google Scholar]
- 32.Goetz D, Ren CL. Review of cystic fibrosis. Pediatr Ann. 2019;48(4): e154–e161. [DOI] [PubMed] [Google Scholar]
- 33.Sly PD, Gangell CL, Chen L, et al. Risk factors for bronchiectasis in children with cystic fibrosis. N Engl J Med. 2013;368(21): 1963–1970. [DOI] [PubMed] [Google Scholar]
- 34.Milliron B, Henry TS, Veeraraghavan S, Little BP. Bronchiectasis: mechanisms and imaging clues of associated common and uncommon diseases. Radiographics. 2015;35(4):1011–1030. [DOI] [PubMed] [Google Scholar]
- 35.Farrell PM, White TB, Ren CL, et al. Diagnosis of cystic fibrosis: consensus guidelines from the Cystic Fibrosis Foundation. J Pediatr. 2017;181S:S4.e1–S15.e1. [DOI] [PubMed] [Google Scholar]
- 36.Zeitlin PL. Pharmacologic restoration of delta F508 CFTR-mediated chloride current. Kidney Int. 2000;57(3):832–837. [DOI] [PubMed] [Google Scholar]
- 37.Hamosh A, FitzSimmons SC, Macek M, Knowles MR, Rosenstein BJ, Cutting GR. Comparison of the clinical manifestations of cystic fibrosis in black and white patients. J Pediatr. 1998;132(2):255–259. [DOI] [PubMed] [Google Scholar]
- 38.Zvereff VV, Faruki H, Edwards M, Friedman KJ. Cystic fibrosis carrier screening in a North American population. Genet Med. 2014; 16(7):539–546. [DOI] [PubMed] [Google Scholar]
- 39.Ren CL, Morgan RL, Oermann C, et al. Cystic fibrosis foundation pulmonary guidelines. Use of cystic fibrosis transmembrane conductance regulator modulator therapy in patients with cystic fibrosis. Ann Am Thorac Soc. 2018;15(3):271–280. [DOI] [PubMed] [Google Scholar]
- 40.Middleton PG, Mall MA, Dřevínek P, et al. Elexacaftor-tezacaftor-ivacaftor for cystic fibrosis with a single Phe508del allele. N Engl J Med. 2019;381(19):1809–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Levy H, Nugent M, Schneck K, et al. Refining the continuum of CFTR-associated disorders in the era of newborn screening. Clin Genet. 2016;89(5):539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ren CL, Borowitz DS, Gonska T, et al. Cystic fibrosis transmembrane conductance regulator-related metabolic syndrome and cystic fibrosis screen positive, inconclusive diagnosis. J Pediatr. 2017;181S: S45–S51.e41. [DOI] [PubMed] [Google Scholar]
- 43.Castellani C, Cuppens H, Macek M, et al. Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice. J Cyst Fibros. 2008;7(3):179–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reiter JF, Leroux MR. Genes and molecular pathways underpinning ciliopathies. Nat Rev Mol Cell Biol. 2017;18(9):533–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mitchison HM, Valente EM. Motile and non-motile cilia in human pathology: from function to phenotypes. J Pathol. 2017;241(2): 294–309. [DOI] [PubMed] [Google Scholar]
- 46.Zariwala MA, Knowles MR, Leigh MW. In: Adam MP, Ardinger HH, Pagon RA et al. , eds. Primary Ciliary Dyskinesia. Seattle, WA: Genereviews; 1993. [Google Scholar]
- 47.Damseh N, Quercia N, Rumman N, Dell SD, Kim RH. Primary ciliary dyskinesia: mechanisms and management. Appl Clin Genet. 2017;10: 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mullowney T, Manson D, Kim R, Stephens D, Shah V, Dell S. Primary ciliary dyskinesia and neonatal respiratory distress. Pediatrics. 2014; 134(6):1160–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Horani A, Ferkol TW. Advances in the genetics of primary ciliary dyskinesia: clinical implications. Chest. 2018;154:645–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Davis SD, Ferkol TW, Rosenfeld M, et al. Clinical features of childhood primary ciliary dyskinesia by genotype and ultrastructural phenotype. Am J Respir Crit Care Med. 2015;191(3):316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu L, Belmont JW, Ware SM. Genetics of human heterotaxias. Eur J Hum Genet. 2006;14(1):17–25. [DOI] [PubMed] [Google Scholar]
- 52.Knowles MR, Daniels LA, Davis SD, Zariwala MA, Leigh MW. Primary ciliary dyskinesia. Recent advances in diagnostics, genetics, and characterization of clinical disease. Am J Respir Crit Care Med. 2013; 188(8):913–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shapiro AJ, Davis SD, Polineni D, et al. Diagnosis of primary ciliary dyskinesia. An official american thoracic society clinical practice guideline. Am J Respir Crit Care Med. 2018;197(12):e24–e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kouis P, Yiallouros PK, Middleton N, Evans JS, Kyriacou K, Papatheodorou SI. Prevalence of primary ciliary dyskinesia in consecutive referrals of suspect cases and the transmission electron microscopy detection rate: a systematic review and meta-analysis. Pediatr Res. 2017;81(3):398–405. [DOI] [PubMed] [Google Scholar]
- 55.Zariwala MA, Omran H, Ferkol TW. The emerging genetics of primary ciliary dyskinesia. Proc Am Thorac Soc. 2011;8(5):430–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Horani A, Ferkol TW. Primary ciliary dyskinesia and associated sensory ciliopathies. Expert Rev Respir Med. 2016;10(5):569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boyle JM, Buckley RH. Population prevalence of diagnosed primary immunodeficiency diseases in the United States. J Clin Immunol. 2007;27(5):497–502. [DOI] [PubMed] [Google Scholar]
- 58.Nonas S Pulmonary manifestations of primary immunodeficiency disorders. Immunol Allergy Clin North Am. 2015;35(4):753–766. [DOI] [PubMed] [Google Scholar]
- 59.Picard C, Al-Herz W, Bousfiha A, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency 2015. J Clin Immunol. 2015;35(8):696–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chinn IK, Shearer WT. Severe combined immunodeficiency disorders. Immunol Allergy Clin North Am. 2015;35(4):671–694. [DOI] [PubMed] [Google Scholar]
- 61.Kwan A, Abraham RS, Currier R, et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA. 2014;312(7):729–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.King JR, Hammarstrom L. Newborn screening for primary immunodeficiency diseases: history, current and future practice. J Clin Immunol. 2018;38(1):56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Resnick ES, Cunningham-Rundles C. The many faces of the clinical picture of common variable immune deficiency. Curr Opin Allergy Clin Immunol. 2012;12(6):595–601. [DOI] [PubMed] [Google Scholar]
- 64.Sathkumara HD, De Silva NR, Handunnetti S, De Silva AD. Genetics of common variable immunodeficiency: role of transmembrane activator and calcium modulator and cyclophilin ligand interactor. Int J Immunogenet. 2015;42(4):239–253. [DOI] [PubMed] [Google Scholar]
- 65.Riepe FG. Clinical and molecular features of type 1 pseudohypoaldosteronism. Horm Res. 2009;72(1):1–9. [DOI] [PubMed] [Google Scholar]
- 66.Nur N, Lang C, Hodax JK, Quintos JB. Systemic pseudohypoaldosteronism type I: a case report and review of the literature. Case Rep Pediatr. 2017;2017:7939854–7939858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Amin N, Alvi NS, Barth JH, et al. Pseudohypoaldosteronism type 1: clinical features and management in infancy. Endocrinol Diabetes Metab Case Rep. 2013;2013:130010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mora-Lopez F, Bernal-Quiros M, Lechuga-Sancho AM, Lechuga-Campoy JL, Hernandez-Trujillo N, Nieto A. Novel mutation in the epithelial sodium channel causing type I pseudohypoaldosteronism in a patient misdiagnosed with cystic fibrosis. Eur J Pediatr. 2012; 171(6):997–1000. [DOI] [PubMed] [Google Scholar]
- 69.Tajima T, Morikawa S, Nakamura A. Clinical features and molecular basis of pseudohypoaldosteronism type 1. Clin Pediatr Endocrinol. 2017;26(3):109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hanukoglu I, Hanukoglu A. Epithelial sodium channel (ENAC) family: phylogeny, structure-function, tissue distribution, and associated inherited diseases. Gene. 2016;579(2):95–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chang SS, Grunder S, Hanukoglu A, et al. Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkalaemic acidosis, pseudohypoaldosteronism type 1. Nat Genet. 1996;12(3): 248–253. [DOI] [PubMed] [Google Scholar]
- 72.American thoracic society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the eUropean Respiratory Society (ERS). Am J Respir Crit Care Med. 2000;161(2 pt 1):646–664. [DOI] [PubMed] [Google Scholar]
- 73.Gross TJ, Hunninghake GW. Idiopathic pulmonary fibrosis. N Engl J Med. 2001;345(7):517–525. [DOI] [PubMed] [Google Scholar]
- 74.Herrmann M, Pusceddu I, Marz W, Herrmann W. Telomere biology and age-related diseases. Clin Chem Lab Med. 2018;56(8):1210–1222. [DOI] [PubMed] [Google Scholar]
- 75.Armanios M Syndromes of telomere shortening. Annu Rev Genomics Hum Genet. 2009;10:45–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Savage SA, Bertuch AA. The genetics and clinical manifestations of telomere biology disorders. Genet Med. 2010;12(12):753–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Alter BP, Rosenberg PS, Giri N, Baerlocher GM, Lansdorp PM, Savage SA. Telomere length is associated with disease severity and declines with age in dyskeratosis congenita. Haematologica. 2012; 97(3):353–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vulliamy TJ, Dokal I. Dyskeratosis congenita: the diverse clinical presentation of mutations in the telomerase complex. Biochimie. 2008;90(1):122–130. [DOI] [PubMed] [Google Scholar]
- 79.Barbaro PM, Ziegler DS, Reddel RR. The wide-ranging clinical implications of the short telomere syndromes. Intern Med J. 2016;46(4): 393–403. [DOI] [PubMed] [Google Scholar]
- 80.Savage SA. In: Adam MP, Ardinger HH, Pagon RA et al. , eds. Dyskeratosis Congenita. Seattle, WA: Genereviews; 1993. [Google Scholar]
- 81.Armanios MY, Chen JJL, Cogan JD, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007; 356(13):1317–1326. [DOI] [PubMed] [Google Scholar]
- 82.Patnaik MM, Kamath PS, Simonetto DA. Hepatic manifestations of telomere biology disorders. J Hepatol. 2018;69(3):736–743. [DOI] [PubMed] [Google Scholar]
- 83.Alter BP, Giri N, Savage SA, Rosenberg PS. Cancer in dyskeratosis congenita. Blood. 2009;113(26):6549–6557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Alder JK, Chen JJL, Lancaster L, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A. 2008;105(35):13051–13056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.El-Chemaly S, Young LR. Hermansky-Pudlak syndrome. Clin Chest Med. 2016;37(3):505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Huizing M, Helip-Wooley A, Westbroek W, Gunay-Aygun M, Gahl WA. Disorders of lysosome-related organelle biogenesis: clinical and molecular genetics. Annu Rev Genomics Hum Genet. 2008;9: 359–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Huizing M, Malicdan MCV, Gochuico BR, Gahl WA. In: Adam MP, Ardinger HH, Pagon RA et al. , eds. Hermansky-Pudlak Syndrome. Seattle, WA: Genereviews; 1993. [Google Scholar]
- 88.Di Pietro SM, Dell’Angelica EC. The cell biology of Hermansky- Pudlak syndrome: recent advances. Traffic. 2005;6(7):525–533. [DOI] [PubMed] [Google Scholar]
- 89.Vicary GW, Vergne Y, Santiago-Cornier A, Young LR, Roman J. Pulmonary fibrosis in hermansky-pudlak syndrome. Ann Am Thorac Soc. 2016;13(10):1839–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ammann S, Schulz A, Krägeloh-Mann I, et al. Mutations in ap3d1 associated with immunodeficiency and seizures define a new type of Hermansky-Pudlak syndrome. Blood. 2016;127(8):997–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.de Boer M, van Leeuwen K, Geissler J, et al. Hermansky-Pudlak syndrome type 2: aberrant pre-mRNA splicing and mislocalization of granule proteins in neutrophils. Hum Mutat. 2017;38(10):1402–1411. [DOI] [PubMed] [Google Scholar]
- 92.Bishop NB, Stankiewicz P, Steinhorn RH. Alveolar capillary dysplasia. Am J Respir Crit Care Med. 2011;184(2):172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Goel D, Oei JL, Shand AW, Mowat D, Loo C. Foxf1 gene mutation in alveolar capillary dysplasia associated with Hirschsprung’s disease and clinical review. J Paediatr Child Health. 2016;52(7):787–788. [DOI] [PubMed] [Google Scholar]
- 94.Antao B, Samuel M, Kiely E, Spitz L, Malone M. Congenital alveolar capillary dysplasia and associated gastrointestinal anomalies. Fetal Pediatr Pathol. 2006;25(3):137–145. [DOI] [PubMed] [Google Scholar]
- 95.Sen P, Yang Y, Navarro C, et al. Novel FOXF1 mutations in sporadic and familial cases of alveolar capillary dysplasia with misaligned pulmonary veins imply a role for its dna binding domain. Hum Mutat. 2013;34(6):801–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Goel D, Oei JL, Lui K, et al. Antenatal gastrointestinal anomalies in neonates subsequently found to have alveolar capillary dysplasia. Clin Case Rep. 2017;5(5):559–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nogee LM. Interstitial lung disease in newborns. Semin Fetal Neonatal Med. 2017;22(4):227–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Austin ED, Loyd JE, Phillips JA III. In: Adam MP, Ardinger HH, Pagon RA et al. , eds. Heritable Pulmonary Arterial Hypertension. Seattle, WA: Genereviews; 1993. [PubMed] [Google Scholar]
- 99.Dodson MW, Brown LM, Elliott CG. Pulmonary arterial hypertension. Heart Fail Clin. 2018;14(3):255–269. [DOI] [PubMed] [Google Scholar]
- 100.Hoeper MM, Bogaard HJ, Condliffe R, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 suppl):D42–D50. [DOI] [PubMed] [Google Scholar]
- 101.D’Alonzo GE. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991;115(5):343–349. [DOI] [PubMed] [Google Scholar]
- 102.Machado RD, Eickelberg O, Elliott CG, et al. Genetics and genomics of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(1 suppl):S32–S42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sasaki E, Byrne AT, Phelan E, Cox DW, Reardon W. A review of filamin A mutations and associated interstitial lung disease. Eur J Pediatr. 2019;178(2):121–129. [DOI] [PubMed] [Google Scholar]
- 104.Mistry PK, Sirrs S, Chan A, et al. Pulmonary hypertension in type 1 Gaucher’s disease: Genetic and epigenetic determinants of phenotype and response to therapy. Mol Genet Metab. 2002;77(1–2): 91–98. [DOI] [PubMed] [Google Scholar]
- 105.Montani D, Achouh L, Dorfmüller P, et al. Pulmonary veno-occlusive disease: clinical, functional, radiologic, and hemodynamic characteristics and outcome of 24 cases confirmed by histology. Medicine (Baltimore). 2008;87(4):220–233. [DOI] [PubMed] [Google Scholar]
- 106.Parent F, Bachir D, Inamo J, et al. A hemodynamic study of pulmonary hypertension in sickle cell disease. N Engl J Med. 2011; 365(1):44–53. [DOI] [PubMed] [Google Scholar]
- 107.Newman JH, Wheeler L, Lane KB, et al. Mutation in the gene for bone morphogenetic protein receptor ii as a cause of primary pulmonary hypertension in a large kindred. N Engl J Med. 2001;345(5): 319–324. [DOI] [PubMed] [Google Scholar]
- 108.Larkin EK, Newman JH, Austin ED, et al. Longitudinal analysis casts doubt on the presence of genetic anticipation in heritable pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186(9): 892–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Berg J, Porteous M, Reinhardt D, et al. Hereditary haemorrhagic telangiectasia: a questionnaire based study to delineate the different phenotypes caused by endoglin and ALK1 mutations. J Med Genet. 2003;40(8):585–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.McDonald J, Bayrak-Toydemir P, Pyeritz RE. Hereditary hemorrhagic telangiectasia: an overview of diagnosis, management, and pathogenesis. Genet Med. 2011;13(7):607–616. [DOI] [PubMed] [Google Scholar]
- 111.McDonald J, Pyeritz RE. In: Adam MP, Ardinger HH, Pagon RA et al. , eds. Hereditary Hemorrhagic Telangiectasia. Seattle, WA: Genereviews; 1993. [Google Scholar]
- 112.Ishiwata T, Terada J, Tanabe N, et al. Pulmonary arterial hypertension as the first manifestation in a patient with hereditary hemorrhagic telangiectasia. Intern Med. 2014;53(20):2359–2363. [DOI] [PubMed] [Google Scholar]
- 113.Kroon S, Snijder RJ, Faughnan ME, Mager HJ. Systematic screening in hereditary hemorrhagic telangiectasia: a review. Curr Opin Pulm Med. 2018;24(3):260–268. [DOI] [PubMed] [Google Scholar]
- 114.Dupuis-Girod S, Cottin V, Shovlin CL. The lung in hereditary hemorrhagic telangiectasia. Respiration. 2017;94(4):315–330. [DOI] [PubMed] [Google Scholar]
- 115.Ianora AA, Memeo M, Sabba C, Cirulli A, Rotondo A, Angelelli G. Hereditary hemorrhagic telangiectasia: multi-detector row helical ct assessment of hepatic involvement. Radiology. 2004;230(1):250–259. [DOI] [PubMed] [Google Scholar]
- 116.Buscarini E, Danesino C, Olivieri C, et al. Doppler ultrasonographic grading of hepatic vascular malformations in hereditary hemorrhagic telangiectasia—results of extensive screening. Ultraschall Med. 2004; 25(5):348–355. [DOI] [PubMed] [Google Scholar]
- 117.Kritharis A, Al-Samkari H, Kuter DJ. Hereditary hemorrhagic telangiectasia: diagnosis and management from the hematologist’s perspective. Haematologica. 2018;103(9):1433–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Richards-Yutz J, Grant K, Chao EC, Walther SE, Ganguly A. Update on molecular diagnosis of hereditary hemorrhagic telangiectasia. Hum Genet. 2010;128(1):61–77. [DOI] [PubMed] [Google Scholar]
- 119.Scott RM, Henske EP, Raby B, Boone PM, Rusk RA, Marciniak SJ. Familial pneumothorax: towards precision medicine. Thorax. 2018; 73(3):270–276. [DOI] [PubMed] [Google Scholar]
- 120.Hou J, Zhang Y, Gong R, Zheng X, Yang X. Primary ciliary dyskinesia presenting with spontaneous pneumothorax: case report and review of the literature. Respir Med Case Rep. 2017;21:167–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lord RW, Jones AM, Webb AK, Barry PJ. Pneumothorax in cystic fibrosis: beyond the guidelines. Paediatr Respir Rev. 2016;20(suppl): 30–33. [DOI] [PubMed] [Google Scholar]
- 122.Bergmann C, Senderek J, Windelen E, et al. Clinical consequences of PKHD1 mutations in 164 patients with autosomal-recessive polycystic kidney disease (ARPKD). Kidney Int. 2005;67(3):829–848. [DOI] [PubMed] [Google Scholar]
- 123.Boone PM, Scott RM, Marciniak SJ, Henske EP, Raby BA. The genetics of pneumothorax. Am J Respir Crit Care Med. 2019;199: 1344–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gupta N, Henske EP. Pulmonary manifestations in tuberous sclerosis complex. Am J Med Genet C Semin Med Genet. 2018;178(3):326–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Armon K, Bale P. Identifying heritable connective tissue disorders in childhood. Practitioner. 2012;256(1752):19–23. 12–13. [PubMed] [Google Scholar]
- 126.Dietz H In: Adam MP, Ardinger HH, Pagon RA, eds. Marfan Syndrome. Seattle, WA: Genereviews; 1993. [Google Scholar]
- 127.Judge DP, Dietz HC. Marfan’s syndrome. Lancet. 2005;366(9501): 1965–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hao W, Fang Y, Lai H, et al. Marfan syndrome with pneumothorax: case report and review of literatures. J Thorac Dis. 2017;9(12): E1100–E1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Malfait F, Francomano C, Byers P, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175(1):8–26. [DOI] [PubMed] [Google Scholar]
- 130.Pepin MG, Murray ML, Byers PH. In: Adam MP, Ardinger HH, Pagon RA, eds. Vascular Ehlers-Danlos Syndrome. Seattle, WA: Genereviews; 1993. [Google Scholar]
- 131.Hatake K, Morimura Y, Kudo R, Kawashima W, Kasuda S, Kuniyasu H. Respiratory complications of Ehlers-Danlos syndrome type IV. Leg Med. 2013;15(1):23–27. [DOI] [PubMed] [Google Scholar]
- 132.Loeys BL, Dietz HC. In: Adam MP, Ardinger HH, Pagon RA et al. , eds. Loeys-Dietz Syndrome. Seattle, WA: Genereviews; 1993. [Google Scholar]
- 133.MacCarrick G, Black JH 3rd, Bowdin S, et al. Loeys-Dietz syndrome: a primer for diagnosis and management. Genet Med. 2014;16(8): 576–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Berk DR, Bentley DD, Bayliss SJ, Lind A, Urban Z. Cutis laxa: a review. J Am Acad Dermatol. 2012;66(5):842.e1–842.e17. [DOI] [PubMed] [Google Scholar]
- 135.Urban Z, Davis EC. Cutis laxa: Intersection of elastic fiber biogenesis, TGFbeta signaling, the secretory pathway and metabolism. Matrix Biol. 2014;33:16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Vanakker O, Callewaert B, Malfait F, Coucke P. The genetics of soft connective tissue disorders. Annu Rev Genomics Hum Genet. 2015;16: 229–255. [DOI] [PubMed] [Google Scholar]
- 137.Duque Lasio ML, Kozel BA. Elastin-driven genetic diseases. Matrix Biol. 2018;71–72:144–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Toro JR. In: Adam MP, Ardinger HH, Pagon RA et al. , eds. Birt-Hogg-Dube Syndrome. Seattle, WA: Genereviews; 1993. [Google Scholar]
- 139.Gupta N, Sunwoo BY, Kotloff RM. Birt-Hogg-Dube syndrome. Clin Chest Med. 2016;37(3):475–486. [DOI] [PubMed] [Google Scholar]
- 140.Tong Y, Schneider JA, Coda AB, Hata TR, Cohen PR. Birt-Hogg-Dube syndrome: a review of dermatological manifestations and other symptoms. Am J Clin Dermatol. 2018;19(1):87–101. [DOI] [PubMed] [Google Scholar]
- 141.Toro JR, Wei MH, Glenn GM, et al. BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dube syndrome: a new series of 50 families and a review of published reports. J Med Genet. 2008;45(6):321–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Frohlich BA, Zeitz C, Matyas G, et al. Novel mutations in the folliculin gene associated with spontaneous pneumothorax. Eur Respir J. 2008;32(5):1316–1320. [DOI] [PubMed] [Google Scholar]
- 143.Gupta S, Kang HC, Ganeshan D, et al. The abcs of bhd: an in-depth review of Birt-Hogg-Dube syndrome. AJR Am J Roentgenol. 2017; 209(6):1291–1296. [DOI] [PubMed] [Google Scholar]
- 144.Weese-Mayer DE, Berry-Kravis EM, Ceccherini I, et al. An official ats clinical policy statement: congenital central hypoventilation syndrome: genetic basis, diagnosis, and management. Am J Respir Crit Care Med. 2010;181(6):626–644. [DOI] [PubMed] [Google Scholar]
- 145.Ramanantsoa N, Gallego J. Congenital central hypoventilation syndrome. Respir Physiol Neurobiol. 2013;189(2):272–279. [DOI] [PubMed] [Google Scholar]
- 146.Moreira TS, Takakura AC, Czeisler C, Otero JJ. Respiratory and autonomic dysfunction in congenital central hypoventilation syndrome. J Neurophysiol. 2016;116(2):742–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Weese-Mayer DE, Silvestri JM, Huffman AD, et al. Case/control family study of autonomic nervous system dysfunction in idiopathic congenital central hypoventilation syndrome. Am J Med Genet. 2001; 100(3):237–245. [DOI] [PubMed] [Google Scholar]
- 148.Berry-Kravis EM, Zhou L, Rand CM, Weese-Mayer DE. Congenital central hypoventilation syndrome: PHOX2B mutations and phenotype. Am J Respir Crit Care Med. 2006;174(10):1139–1144. [DOI] [PubMed] [Google Scholar]
- 149.Weese-Mayer DE, Marazita L, Rand CM, Berry-Kravis EM. In: Adam MP, Ardinger HH, Pagon RA et al. , eds. Congenital Central Hypoventilation Syndrome. Seattle, WA: Genereviews; 1993. [Google Scholar]
- 150.Radley JJ, Rocher AB, Rodriguez A, et al. Repeated stress alters dendritic spine morphology in the rat medial prefrontal cortex. J Comp Neurol. 2008;507(1):1141–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Rand CM, Yu M, Jennings LJ, et al. Germline mosaicism of PHOX2b mutation accounts for familial recurrence of congenital central hypoventilation syndrome. Am J Med Genet A. 2012;158A(9): 2297–2301. [DOI] [PubMed] [Google Scholar]
- 152.Bachetti T, Matera I, Borghini S, Di Duca M, Ravazzolo R, Ceccherini I. Distinct pathogenetic mechanisms for PHOX2B associated polyalanine expansions and frameshift mutations in congenital central hypoventilation syndrome. Hum Mol Genet. 2005;14(13):1815–1824. [DOI] [PubMed] [Google Scholar]