

Abstract
Over the last two decades it has become clear that well-defined structure is not a requisite for proteins to properly function. Rather, spectra of functionally competent, structurally disordered states have been uncovered requiring canonical paradigms in molecular biology to be revisited or reimagined. It is enticing and oftentimes practical to divide the proteome into structured and unstructured, or disordered, proteins. While function, composition, and structural properties largely differ, these two classes of protein are built upon the same scaffold, namely, the protein backbone. The versatile physicochemical properties of the protein backbone must accommodate structural disorder, order, and transitions between these states. In this review, we survey these properties through the conceptual lenses of solubility and conformational populations and in the context of protein-disorder mediated phenomena (e.g., phase separation, order–disorder transitions, allostery). Particular attention is paid to the results of computational studies, which, through thermodynamic decomposition and dissection of molecular interactions, can provide valuable mechanistic insight and testable hypotheses to guide further solution experiments. Lastly, we discuss changes in the dynamics of side chains and order–disorder transitions of the protein backbone as two modes or realizations of “entropic reservoirs” capable of tuning coupled thermodynamic processes.
Graphical Abstract
INTRODUCTION
The sequence–structure–function paradigm provided what was believed to be a universal framework capable of describing, predicting, and mechanistically rationalizing protein function and dysfunction or disease.1 The notion that a well-defined three-dimensional structure of a protein was explicitly determined by its primary sequence was first proposed over 120 years ago2,3 and pioneering experiments and discoveries over the century to follow further cemented this paradigm in molecular biophysics and chemistry. Sparked by seminal papers at the turn of the 21st century,1,4 overwhelming evidence has amassed in the last two decades indicating that significant numbers of eukaryotic proteins rely on a lack or absence of structure to carry out critical cellular functions.5–8 Such intrinsically disordered proteins (IDPs) or regions (IDRs) within proteins populate a dynamic ensemble of structures, necessitating a statistical description of morphology (e.g., collapsed, extended, coil), one which is dictated by amino acid sequence and environment, among other factors.9–12
The biological functions of IDR-containing proteins are numerous and beyond the scope of this paper. We refer to van der Lee10 for a very thorough discussion and classification of IDR-mediated functions. Generally, though, proteins enriched with disorder facilitate and regulate cellular signaling networks most notably associated with transcription, translation, and the cell cycle.13,14 Underpinning these functions are the ability of IDRs to bind targets with high specificity but low affinity,1,4,14 or with a high dissociation constant15 due to the energetic and/or entropic cost of folding upon binding (necessary for rapid response to changing cellular signals); the fact that multiple, short, disordered recognition motifs for various targets can be embedded within the same protein (i.e., multivalency);1,14,16,17 the ability of the same recognition motif to bind different targets (i.e., one-to-many);18 regulation of function through disorder-mediated allostery/cooperativity19–23 and facilitating or contributing to the supramolecular assemblies of nonenveloped organelles (i.e., liquid–liquid phase separation);24–26 among others. Key to these processes is the ability of IDRs to undergo structural transitions (order → disorder, disorder → order, disorder remodeling) to different functional states in response to a number of factors including protein/ligand binding, post-translational modifications, and environmental changes.10,12,17,21,22,27–29 Additionally, it is important to note that eukaryotic proteins, particularly nuclear receptor transcription factors, have evolved to use a combination of functionally (i.e., thermodynamically)-coupled structured and disordered domains to enable fine spatial and temporal regulation of (transcription) activity.19,30–32
Given that proteins enriched in or functionally relying on disorder are involved in the regulation of critical cellular functions, it is not surprising that IDPs/IDRs have been associated with a range of diseases,33,34 most notably Alzheimer’s and Huntington’s. Such diseases are, at the most fundamental level, manifestations of an altered or aberrant protein conformational landscape. While pathological effects of mutations in well-structured proteins often lead to unfolding or misfolding and loss-of-function is readily justified, such a clear distinction is lacking with regard to the effects of mutations on the distribution of functionally competent, conformational states of IDRs. Physical mechanisms of intrinsic protein disorder and how it couples sequence to function remain to be discovered. This fundamental knowledge, analogous to the sequence–structure–function paradigm, will be critical to successfully targeting drugs to IDRs/IDPs or genetically engineering IDRs/IDPs with tunable, therapeutic properties.
Below we briefly describe the thermodynamic framework within which we consider the biophysical mechanisms of protein disorder and how it can be used to shed light on experimentally observable phenomena like IDR-mediated collapse, aggregation, specific and nonspecific interface formation, and allosteric regulation. Following, we pay particular attention to the important, active role the protein backbone plays in determining the physicochemical properties of IDRs. In this way, side chains, post-translational modifications, and environmental changes, among others, can be viewed as perturbations to the innate, structural propensities of the protein backbone, which must accommodate both order and disorder in proteins as well as transitions between the two states.
THERMODYNAMIC VIEW OF PROTEIN DISORDER
Conformational Landscape.
IDRs are highly dynamic and flexible and lack stable, persistent secondary or tertiary structures. Conceptually, the ensemble of disordered conformations, or structural heterogeneity, is best characterized by a relatively flat, rugged free energy landscape wherein the probability of a particular conformation or conformational state (i) is proportional to the free energy:11,17,19,23
| (1) |
where β is proportional to the inverse temperature and Ai is the free energy of state i. More amenable to a physical chemistry understanding of the mechanisms that give rise to the free energy surface, Ai can be further decomposed into energetic (U) and entropic (S) components as
| (2) |
where the subscripts “u” and “v” denote the solute and solvent, respectively, and T is the absolute temperature. For simplicity, we drop the subscript i that references a conformational state in the second line of eq 2 but note the implicit dependence of U and S on the conformational state. Solute and solvent entropy include momentum, translational, rotational, and conformational contributions (see 35 for a rigorous discussion on this decomposition of entropy). Computational approaches grounded in molecular mechanics are reasonably well-suited to study or probe the individual energetic and entropic terms in eq 2 associated with certain processes or thermodynamic cycles and also permit further energetic decomposition into electrostatic and van der Waals components.
A simplified illustration of the free energy surface is shown in Figure 1. Characteristically different than well-structured proteins, for which the free energy landscape is depicted as a deep funnel with a global energetic minimum,36,37 IDRs rapdily transition between conformations, suggesting a lack of substantial energetic barriers.11,12,17,38 In other words, IDRs are able to populate a number of energetically similar but conformationally distinct states, indicative of high conformational entropy (i.e., “widths” of the states). Highlighted are three hypothetical conformational states (Figure 1B) of interest that may differ, for example, by their extent of compactness or collapse (e.g., the abscissa represents the continuum of collapsed to expanded in the positive direction) and/or function. The three states are in thermodynamic equilibrium. Equation 2 indicates that their free energies, and thus populations, depend on the balance or competition between solute–solute, solute–solvent, and solvent–solvent interactions in addition to solute and solvent entropies. While the individual terms in eq 2 have been separated, it is important to note the nontrivial coupling among terms. For example, an IDR that collapses due to strong, intrachain interactions (Uuu) may minimize its exposed surface area, limiting its potential interactions with solvent (Uuv), which in turn limits the disruption of the solvent network, allowing the solvent to potentially occupy more thermodynamic states (Sv). With respect to conformational transitions, binding, post-translational modifications, or changes in the environment alter the IDR free energy landscape (Figure 1C) with the change of populations indicative of the free energy required to elicit such a remodeling of the ensemble. High conformational entropy of IDRs manifests as the width or breadth over the iso-energetic states,17,39 the extent of which represents the energetic penalty that must be paid, for example, when an IDR folds upon binding.
Figure 1.

Free energy landscape of a well-structured protein (left) and an IDR (right). The ordinate represents the conformational free energy and the abscissa as some hypothetical, structural reaction coordinate. The free energy landscape for a well-structured protein (A) is often depicted as a rugged funnel (one reaction coordinate) with folding driven down the funnel to a stable conformation with a global, energetic minima. The free energy landscape of an IDR (B) is comparatively flat and rugged with smaller energetic barriers between conformational populations, permitting IDRs to interconvert between them. We have highlighted three hypothetical conformational states, numbered i = 1–3, and their associated probabilities as Pi. The fraction of conformers populating each state is given by eqs 1 and 2. Ligand binding or changes in the environment, for example, can remodel the free energy landscape of an IDR (C) and reapportion the conformational populations enabling the propagation of an allosteric signal through the disordered ensemble.
Solubility.
As briefly mentioned previously and discussed in further detail below, IDRs are enriched in proteins contained within membrane-less organelles and have been found to facilitate multivalent intramolecular interactions leading to such supramolecular assemblies or liquid–liquid phase separations (LLPS).24–26,40 The concept of solubility lends a useful framework to consider the physicochemical properties of protein mixtures and to rationalize the biophysical mechanisms driving IDR-mediated LLPS. A well-defined and measurable chemical property of a multi-component system,41 the equilibrium solubility or solubility limit, s, is the concentration of the saturated solution where excess solute has phase separated (e.g., molecular condensates). As seen in the thermodynamic cycle depicted in Figure 2, it is related both to the free energy of solution and the free energy required to remove a molecule from the concentrated phase (i.e., vaporization free energy). In a saturated solution we have
| (3) |
where γi is the activity coefficient, Vm is the molar volume of the molecule in the aggregated state, ΔGsol is the difference in free energy between the saturated solution and the aggregated form (liquid or solid), ΔGhyd is the free energy of hydration to transfer an isolated molecule in the gas phase to a concentrated solution, and ΔGvap is the free energy of transfer from the aggregated state to the gas/vapor state. Often times the transfer to solution is approximated as an ideal system with a unit activity coefficient, standard state, and set of concentration units, such that ΔGsol = −RT ln s0Vm where s0 is considered the intrinsic solubility. However, it has been shown that polypeptide systems42,43 and even smaller molecules44 are most often nonideal in solution. Considering solvation free energies at infinite dilution, while informative with respect to probing a molecule’s interactions with solvent and effects on solvent structure, does not provide sufficient information to make any inferences or generalizations with respect to phase behavior because, by design, intersolute interactions are absent. Note, the left-hand terms in eq 3 may be decomposed in a manner similar to that in eq 2 wherein the different states, i, can represent one of the phases in the thermodynamic cycle in Figure 2. This explict representation highlights the importance of intra- and intermolecular interactions as well as solute and solvent entropy in contributing to or determining phase separation.
Figure 2.

Thermodynamic cycle often used in solubility calculations using a saturated solution reference.
Approaches To Study Disorder.
Numerous experimental methods or approaches can be used to detect disorder and characterize properties of the conformational ensemble of IDRs.1,4,9,45–50 NMR is well suited for this purpose as it reports on interatomic distances, motions and structural heterogeneity of individual residues and provides constraints on the available structural states of IDRs.29,46,51 The structural signatures measured by NMR, as well as other classical techniques like CD and SAXS, represent population averages over an ensemble of protein structures in solution, which may mask dynamic structural features of functional importance.49,52 Single-molecule techniques (e.g., smFRET, smFCS) circumvent such problems associated with ensemble averaging and provide highly resolved, spatial and temporal information on individual proteins, thus permitting, for example, the discrimination of conformational subpopulations of IDRs or the observation of rare structural transitions, among others.49 However, these methods often yield structural metrics (e.g., hydrodynamic radius and distance distributions) that are one-dimensional projections of the complex conformational space. Furthermore, solution methods in general can be hampered by the tendency of IDRs and IDPs to aggregate or undergo LLPS at the necessary concentrations.1,53
Computational modeling and molecular simulations directly yield a model of the IDR structural ensemble at atomic resolution. Not limited in the same way by experimental constraints like solubility, molecular simulations can provide insight into the thermodynamic and structural mechanisms underlying IDR function otherwise inaccessible to traditional methodologies. However, results from molecular simulations, especially for IDRs/IDPs, are dependent on the underlying potential energy models (i.e., force fields).54–57 Force field development for IDR/IDP simulations is an active field.55,58,59 Nonetheless, simulations are useful in the design of targeted experiments, testable hypotheses and interpretations of biophysical phenomena. Much of the discussions to follow will highlight experimentally observable IDR-mediated phenomena along with a survey of results from computational studies that may shed light on biophysical mechanisms underlying such phenomena.
MECHANISMS OF COLLAPSE AND AGGREGATION
Collapse of IDRs.
An understanding of how primary sequence specifies the disordered ensemble (i.e., structural properties) is key to understanding not only how disorder mediates function but also how alterations in the primary sequence aberrantly propagate through the ensemble to disrupt function. Generally, IDRs are characterized by low-complexity amino acids sequences devoid of “order-promoting”, nonpolar residues (e.g., Cys, Trp, Tyr, Ile, Leu, Phe) and enriched with “disorder-promoting”, polar, and charged amino acids (e.g Pro, Glu, Asp, Gln, Ser, Lys, …).1,5,51,60 Disorder (i.e., conformational (in)stability) is achieved through a complex balance of interactions both within an IDR (Uuu) and between an IDR and solvent (Uuv), and the spatial organization of side chains along the protein backbone dictates gross (statistical) morphology (e.g., collapsed, extended, coiled).61–70 On the basis of classical hydrophobicity scales71 and the observed burial of nonpolar residues that are expected to stabilize well-structured proteins,72 one would expect IDRs composed of tracts of polar residues (e.g., glutamine, asparagine, serine, etc.) to favor expanded coil conformations. However, molecular simulation and solution biophysics (e.g., smFRET and smFCS) studies find that polyglutamine,66,68,73 glutamine/asparagine-rich,74 repetitive glycine-serine blocks,75–77 and oligoglycine polypeptides57,64,67,76,78 all collapse in solution (relative to a random coil) and maintain a heterogeneous, disordered ensemble, despite the predicted large, favorable solvation free energies of their constituent residues.65,79
Systems of oligoglycine (Gly) peptides serve as a model IDR and a means to investigate the contributions of the protein backbone to IDR collpase. We have found the solvation free energy of oligoglycine to be favorable and to decrease with increasing chain length from simulations with multiple protein force fields.57,78 Results from a rigorous decomposition of solvation free energies (ΔG) of Gly2–5 into enthalpic and entropic components as well as their van der Waals (vdw) and electrostatic (elec) contributions finds that the entropic penalty (Sv) of solvating successively longer oligoglycines is more than compensated by a favorable electrostatic solvation enthalpy that decreases with chain length.65 ΔGelec and ΔGvdw, to a much lesser extent, decrease with increasing solvent exposed surface area, indicating that solvation favors extended, exposed conformers.63,64 What, then, drives the chain length-dependent collapse of oligoglycine? Simulation suggests that collapse is largely be due to the formation of favorable intrapeptide interactions (Uuu) between CO dipoles along the backbone chain rather than well-formed hydrogen bonds;64,80 however, this is an active area of debate as others have suggested intrapeptide hydrogen bonds to be the primary driver of collapse of oligoglycine and IDRs in general.67,81 In either case, favorable intrapeptide interactions alter the balance of Uuu and Uuv to favor collapse, but not to an extent that preferentially stabilizes a single conformation.
To further examine when intramolecular interactions overcome hydration to drive collapse transition in Gly15, we considered the change in free energy as a function of the radius of gyration (Rg) conditioned on end-to-end distance (r), −kBT ln P(Rg|r).70 The free energy change along these coordinates was found to vary more gently compared to the corresponding variation in the excess hydration free energy. Using this observation within a multistate generalization of the potential distribution theorem, we calculated a tight upper bound on the hydration free energy of Gly15 for a given r. On this basis, hydration greatly favored the expanded state of the chain while the net free energy of collapse was found, as anticipated, to be a delicate balance between opposing intrapeptide and hydration effects, with intrapeptide contributions increasingly favoring collapse with increasing chain length.
The preference of polar polypeptide tracts and IDRs in general to form collapsed globules may be explained, in part, by the innate ability of the polypeptide backbone (i.e., oligoglycine) to collapse.64,65,67,82 As we have seen for oligoglycines, Uversky et al. found that the extent of compaction of IDRs increases with increasing chain length.83 Rather than viewing this phenomena through the classical lens of hydrophobicity, which at times is inconsistent with experimental observations of IDRs,62 it is perhaps better explained by solubility. The effective, local concentration of residues in the vicinity of an IDR is orders of magnitude beyond their solubility limits due to the fact that they are covalently bonded together (Figure 3). As chain length increases, so does the number of potential favorable intrapeptide (backbone and side chain) interaction sites, and at some point these interactions saturate within a specified volume to drive collapse or compaction.
Figure 3.
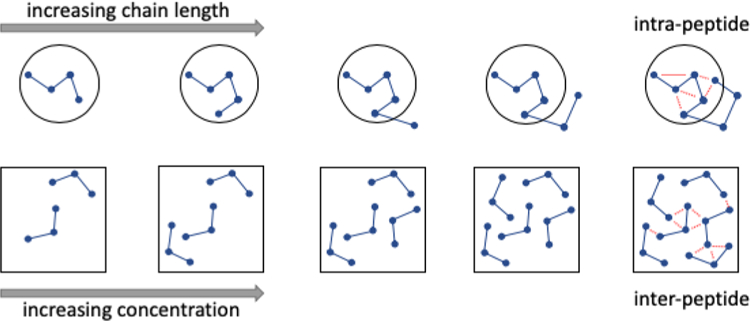
Collapse and aggregation of polypeptides and IDRs as analogous, solubility driven events. (Top) The effective, local concentration of residues increases with chain length in a manner analogous to the increasing total number of molecules (global concentration) in solution (bottom). Despite the fact that solvation free energies of IDRs may be favorable and decrease with chain length, at some local or global concentration intrapeptide interactions (dashed, red lines) saturate to drive collapse or aggregation.
Dimensions of IDRs with More Sequence Complexity.
The influence of charged amino acids on the structural propensities of IDRs is complex and nontrivial. From a combined molecular simulation and smFCS study of various protamines, Mao et al.84 found that the net charge per residue (NCPR) is positively correlated with radius of gyration. They observed a globule-to-coil transition at a critical NCPR. Other studies have also shown that a higher net charge leads to the expansion of polyelectrolyte IDRs.62,69,85 In this situation, the repulsion of like charges (i.e., unfavorable Uuu) in concert with favorable chain–solvent interactions (Uuv) opposes the collapse of the protein backbone.69,82 A majority of IDR sequences are polyampholytes86 with a distribution of positively and negatively charged residues. The charge distribution can result in a zero net charge. Whether an IDR forms a collapsed globule or an expanded coil depends on the spatial patterning of charged residues across the IDR.86 For example, blocks of residues with the same charges separated in the primary sequence would repel one another while a disperse arrangement of opposite charges could have a compensatory effect. This spatial distribution of charges results in a complicated balance between Uuu and Uuv, making a prediction of IDR structural properties challenging. Despite our nascent understanding of the mechanisms that encode disordered ensembles within primary sequences, we are beginning to see early evidence of the potential “tunability” of the structural propensities of IDRs through point mutations. For example, Munshi et al.87 modulated the dimensions of CytR DNA-binding domain, a high sequence-complexity IDP, through the introduction of rationally positioned point mutations. These mutations resulted in a maximal reduction in the Stokes radius of ∼3.5 Å of the disordered CytR ensemble, corresponding to 40% reduction in occupied volume compared to the mutant with the most extended set of conformers. They note that certain combinations of mutations (e.g., A29V & R28Q) exhibit cooperative or emergent behaviors in that their apparent effects on the structural properties requires the presence of both mutations simultaneously.
Aggregation and Disorder.
A variety of nonenveloped (i.e., membrane-less) organelles exist in cells as aggregates, which can best be characterized as dense liquid droplets that have phase-separated from the cytosol,24,25,88–90 with P granules being perhaps one of the most notable.91 These dynamic macromolecular assemblies have evolved to provide a mode of regulation through higher-order functional organization of DNA, RNA, and protein components.25,90 Such regulatory modes are achieved, for example, by the establishment of concentration gradients in the cell,92 by sequestration of proteins that carry out their functions in cytosol,93 and oppositely, by increasing chemical activity through the rise in effective concentration of proteins within the condensed phase.94 Evidence continues to mount that it is the misregulation of these biomolecular condensates that underlies the pathogenesis of diseases like Alzheimer’s, amyotrophic lateral sclerosis, and Huntington’s, among others.34,90,95,96
Membraneless organelles have an apparent propensity to incorporate proteins with intrinsic disorder,24,40 and experimental observations suggest that these disordered protein regions facilitate, at least in part, phase separation.95,96 As a prime example, the RNA-binding protein FUS has been found to fuse and shuttle between liquid compartments in the nucleus and isolated FUS phase separates in vitro.95 A single point mutation, G156E, observed in patients with amyotrophic lateral sclerosis (ALS) in the disordered, prion-like domain of FUS was shown to markedly accelerate the transition of FUS liquid droplets to less dynamic, fibrous formations.95 Although we lack a detailed mechanistic understanding, which may be needed to design targeted interventions, it is widely accepted that conformational flexibility/heterogeneity and multivalency are key features of IDRs that drive phase separation.90 Flexibility can allow for a more rich set of interactions (Uuu) leading to enthalpic stabilization (i.e., nonspecific protein interface).64,80 Note, though, that this stabilization is likely the result of rapidly reorganizing, numerous nonspecific interactions (e.g., dipole–dipole, electrostatic, van der Waals, hydrogen bonding) within and between the side chains and backbones of proteins in the condensates and not the selection of a stable conformational complex.90 Such properties of IDRs may have evolved to achieve the thermodynamic control necessary for liquid–liquid phase transitions97,98 and may be the same mechanisms underlying the collapse of IDRs.
Phase separation and interface formation are free energy and solubility driven events where local concentration and effective interactions play a central role (Figure 3). Molecular simulations of supersaturated systems afford the precise characterization of model chemical principles leading to phase separation through, for example, the thermodynamic decomposition of the process (eq 2) and teasing out the contributions of the protein backbone and side chains. The choice of the underlying atomic model (i.e., force field)99–102 is a persistent concern as intramolecular distributions and solubility have been seen to depend on the choice of force field.61,80
The free energy of solvation at infinite dilution alone does not predict the liquid–liquid phase transition (Figure 2). Previously we computed the first solubility limit for a Gly5 peptide by all-atom computer simulations.61 More recently, we set out to consider peptide solubility limits for systems with a variety of side chains and to take into account the role of backbone and side chain interactions. The solubility limit, or concentration of free solute at saturation (S, eq 3), was calculated for a variety of GGXGG pentapeptides via molecular dynamics simulations in water.80 The order of pentapeptides in terms of solubility limits followed that reported for amino acid monomers from experiment (R > D > G > V > Q > N > F) but was different from most hydrophobicity scales.103 The order does, however, correlate with the fluctuations of waters in the first solvation shell, which is in line with the predictors of hydrophobicity from the theory of Lum, Chandler, and Weeks.104 Investigation of dynamical properties of the peptides showed that the time spent by the peptide in aggregated clusters was inversely related to the solubility limit (i.e., a higher solubility limit led to a decrease in cluster residence time). We demonstrated that fluctuations in conformation and hydration number of a monomeric peptide are correlated with its solubility limit. Furthermore, our results confirmed that CO–CO dipole interactions more than interbackbone hydrogen bonds are important for the nonspecific interactions in phase separation for these systems, just as we had observed for the collapse of long oligoglycine at infinite dilution.64 The physicochemical properties of the protein backbone are well suited to mediate phase separation and the addition of certain side chains perturbs these innate properties, shifting its chemical potential and solubility limits resulting in systems with different phase behaviors.
In addition to the balance of or competition between intra- and intermolecular interactions, changes in the protein conformational landscape (Figure 1) and solvent structure can play important roles in the formation of membraneless organelles. The complex interplay among these aforementioned thermodynamic components in phase separation is apparent in a recent study by Majumdar and co-workers105 in which they investigated the phase separation behavior of tau K18, an amyloidogenic 129-residue fragment of human tau.106,107 After covalently labeling two cysteine residues with pyrene, they monitored the fluorescence of the probes during phase separation and found that tau K18 formed compact, disordered globules in the monomeric state yet adopted more extended, coil-like conformations in the phase-separated state. This remodeling of the conformational landscape, perhaps attributed to inter-Uuu outcompeting intra-Uuu as more interaction surface is exposed, was also accompanied by an increase in backbone torsional mobility as measured by time-resolved fluorescence anisotropy. The authors hypothesize that this may indicate a favorable increase in the conformational entropy of the protein backbone (Su), which helps promote the phase separation. Calculation of the changes in conformational entropy associated with an order-to-disorder transition of the protein backbone suggests this to be significant source of free energy capable of playing important roles in the thermodynamics of IDR-mediated phenomena.108
Interestingly, from an additional set of fluorescence experiments, the accessibility of water to the protein chain was found to be higher in the condensed phase compared to the monomeric state wherein tau K18 forms collapsed conformations. That is, it appears that extended tau K18 conformers recruit water into the condensed phase and that any unfavorable change in solvent entropy (Sv) may be more than compensated by peptide–solvent (Uuv) and/or solvent–solvent (Uvv) enthalpic interactions. Lastly, the authors note the abundance of glycines found in PGGG motifs along the primary sequence, which they hypothesize imparts flexibility of the protein backbone such that its increased exposure in the condensed phase enables backbone–π interactions, subsequently promoting phase separation. This idea is consistent with our results from simulations of supersaturated solutions of oligoglycine and glycine-rich pentapeptides as well simulations of long oligoglycines in infinite dilution, albeit with differences in the types of interactions driving these phenomena.
Structural Transitions: Remodeling the Disordered Ensemble.
The pliability of the disordered conformational ensemble or free energy landscape is a chemical feature of IDRs that enables their functions, particularly as it pertains to mechanisms of molecular recognition and allosteric regulation. IDRs and IDPs are capable of undergoing structural transitions, which may proceed to more ordered or disordered states or result in a population redistribution (Figure 1), in response to protein/ligand binding, environmental changes, and post-translational modifications, among others.10,12,17,21,22,27–29 The contributions of such structural transitions to the thermodynamics of the associated mechanisms remains to be understood, but it is widely accepted that conformational entropy (Su) of IDRs plays a critical role.17,21,109,110 As demonstrated in the simple relationships in eqs 1 and 2, conformational entropy can either stabilize or destabilize populations of conformers and conformational entropy changes, ΔSu, brought on by structural transitions can either promote or oppose thermodynamic processes. It is also possible for an isentropic redistribution of populations that is accompanied by a change in functional state.110 We refer to refs 22 and 27 for detailed reviews of experimentally observed structural transitions of IDRs but highlight some representative examples below.
Keul and co-workers111 provide rigorous evidence for the ability of an IDR to (de)stabilize populations in the conformational free energy landscape as a means to regulate protein activity. The protein hUGDH, which assembles into a hexamer (three dimers), catalyzes the oxidation of substrate UDP-α-d-glucose and, through binding at the same active site, is allosterically inhibited by UDP-α-D-Xylose (UDP-Xyl). hUGDH contains a 30-residue, intrinsically disordered C-terminus that becomes structurally constrained at the three interfaces formed when the hexamer assembles. Through kinetic experiments, the authors show that the disordered tail increases affinity of UDP-Xyl (ΔΔG = −1.39 kcal mol−1) relative to a hUGDH mutant with the tail removed (hUGDHΔIDR) and that this mechanism depends on the length of the IDR but is independent of primary structure. A series of thermal denaturation and hydrogen/deuterium exchange studies found the high affinity hUGDH dimer to be less stable than low affinity (hUGDHΔIDR) dimer. That is, the IDR effectively destabilizes the low affinity state/complex, the effect of which is a shift in the conformational ensemble to high affinity, structurally distinct states. The authors propose that the energetic source of this destabilization is due to the disordered tail being confined to a much smaller volume, constraining the number of available conformations (i.e., entropy), by formation of the various interfaces in the hUGDH complex. They estimate the entropically driven free energy cost of confining the disordered tail using a state counting approach to be 2.4 kcal mol−1, which is sufficiently strong enough to explain the increased binding affinity of hUGDH relative to hUGDHΔIDR.
The above example demonstrates how structural transitions of IDRs may be thermodynamically coupled to other processes, like binding, to realize disorder-mediated modes of regulation through remodeling of the conformational landscape. This disorder → less-disorder transition results in a decrease in conformational entropy. However, by maintaining some level of disorder, the entropic cost may be tuned or mitigated to maintain pliability of the conformational free energy landscape (i.e., preventing the creation of large energetic barriers or stable minima). This is analogous to the formation of “fuzzy complexes” between IDRs and target proteins wherein IDRs maintain a level of disorder in the bound state, thereby limiting unfavorable decreases in conformational entropy.112 At one extreme, IDRs may undergo disorder → order transitions to well-structured states, which is often seen in IDRs responsible for directly interfacing target proteins.10,113–115 This represents a significant decrease in conformational entropy and redistribution of the free energy landscape. Heller et al. refer to this as “entropic collapse”.110 Oppositely, small molecule or ligand binding and changes in environment can prompt order → disorder transitions in residues near and far to the binding site bringing with it an increase in conformational entropy.12,27,28,116 When coupled, this “entropic expansion”110 may help drive binding or, in allosteric regulatory mechanisms, the increased disorder may alter stability and poise the protein to respond to subsequent signals. The population-based framework presented by Heller et al.110 and depicted in Figure 4 nicely illustrates the effects of structural transitions (i.e., ensemble modulation) and its associate entropic coupling with thermodynamic processes.
Figure 4.
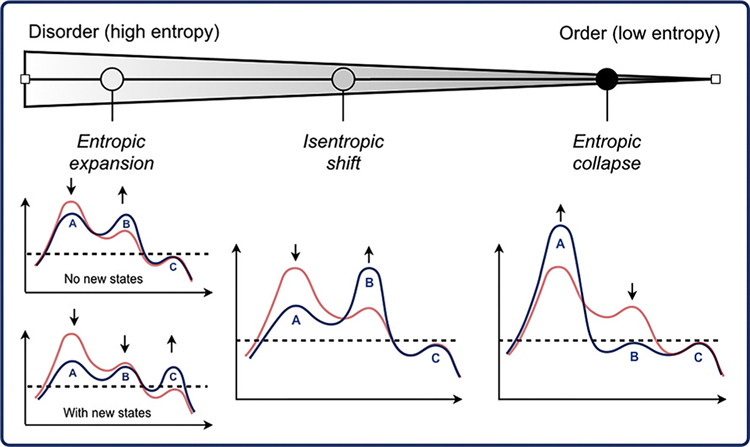
Population-based view of the relationship between modulation of the structural ensemble of disordered proteins and conformational entropy. Figure originally appeared in Heller et al.110 and reproduced here under the terms of Creative Commons CC-BY license and with permission from the author. In that article, the red line represented an apo-ensemble or population while blue represented a structural ensemble modulated or redistributed as a result of small-molecule binding. Here, we conceptually generalize the notions of entropic expansion, shift, and collapse to be the result of any process as means to couple the thermodynamics (i.e., entropy) associated with IDR structural transitions. Reprinted with permission from ref 110. Copyright 2018 Elsevier.
Quantification or calculation of conformational entropy is notoriously challenging and attempting to catalog it across the sequence space of IDRs is prohibitive. In an attempt to bound or provide an upper limit on conformational entropy changes of disordered polypeptides undergoing structural transitions, we have calculated the absolute backbone conformational entropy of oligoglycines (Gly3–15) from trajectories of backbone ϕ and ψ dihedral angles sampled using molecular dynamics simulations with all-atom protein force fields.108 calculated with a mutual information expansion approach117 (assuming different levels of approximation for short/long-range correlations of motion) was found to scale linearly with chain length with slopes of 3.86–4.75 cal mol−1 K−1 residue−1, or ∼1.2–1.4 kcal mol−1 residue−1 at a temperature of 300 K. These estimates are consistent with the loss of entropy upon folding of well-structured proteins reported from a number of different experimental and computational studies.118–120 From a large-scale analysis of 807 structured proteins in the Dynameomics MD database, Towse et al.120 similarly approximated a linear scaling of backbone conformational entropy with respect to chain length, possibly suggesting that this linear relationship may be robust in terms of chain length and sequence space.
Assuming an isothermal process initiates an order → disorder or disorder → order transition, the change in conformational entropy is likely dominated by (i.e., “soft” degrees of freedom) as, in many cases, it may be reasonable to assume equilibrium bond and angle vibrations are not significantly perturbed. So we view, then, absolute estimates of of the protein backbone models considered here as an upper bound on the amount of conformational entropy gained or lost, which even for the relatively short Gly10 is significant . This suggests that the protein backbone is capable of releasing or storing a significant amount of free energy as conformational entropy but it also indicates the presence of compensating sources of energy, for example the loss of considerable, favorable enthalpic interactions. In its native state, though, the backbone largely resists folding or ordering. Patterning of side chains along the backbone can remodel its conformational free energy landscape to achieve different properties of the structural ensemble or promote folding.
In an approach similar to but distinct from Keul,111 we calculated the ensemble average change in free energy to confine oligoglycines of various lengths to substates defined in terms of backbone dihedral angles that were visited during MD simulations. These confinement free energies, which implicitly account for intrachain and chain–solvent interactions, also scaled linearly with chain length with slopes of 0.94–0.99 kcal mol−1 residue−1. While similar to those estimated from absolute conformational entropies, these slopes are slightly less because, by definition, each conformational substate maintains some internal entropy or disorder.108 London and co-workers121 found that polypeptide–protein interfaces utilize significantly more main-chain/main-chain hydrogen bonds than larger protein–protein complexes, suggesting that the protein backbone is equipped to provide compensating enthalpic interactions, at least in part.
Last, we highlight another apparent property of the protein backbone we hypothesize might play important roles in IDR stability/solubility and disorder-mediated allostery. From structurally constrained simulations with multiple force fields,108 of Gly15 was found to be largely independent of end-to-end distance and radius of gyration (Rg), two global parameters often used to describe the structural ensemble of IDRs. This intrinsic property of the protein backbone may help ensure the “flatness” of the IDR conformational landscape and enable (i.e., not inhibit) IDRs to rapidly sample conformations capable of forming transient but stabilizing interactions (e.g., the example of tau K18 discussed in the previous section). Greater entropic responses are likely elicited by structural constraints that alter an individual residue’s dihedral angle populations brought on, for example, by volume exclusion like that seen for the IDR in hUGDH.111
Protein Backbone and Side Chains: Entropy Reservoirs.
Wand and others proposed that residual conformational entropy may serve as an entropic (free energy) reservoir which can be thermodynamically coupled with binding events and utilized through concomitant changes in the structural fluctuations of side chains.122–126 The development and calibration of the so-called “entropy meter”127,128 (an NMR-based technique in which methyl side chain dynamics serve as a proxy for conformational entropy) has provided valuable experimental data in support of their hypothesis. Through a combination of this entropy meter and isothermal titration calorimetry, they performed a detailed decomposition of the binding free energies for 28 protein–ligand complexes125 and found, among other things, that changes in the fast side chain motions elicited conformational entropic responses that favored, disfavored, or had no impact on binding free energy (Figure 5). In some situations, conformational entropy largely drove the binding thermodynamics.
Figure 5.
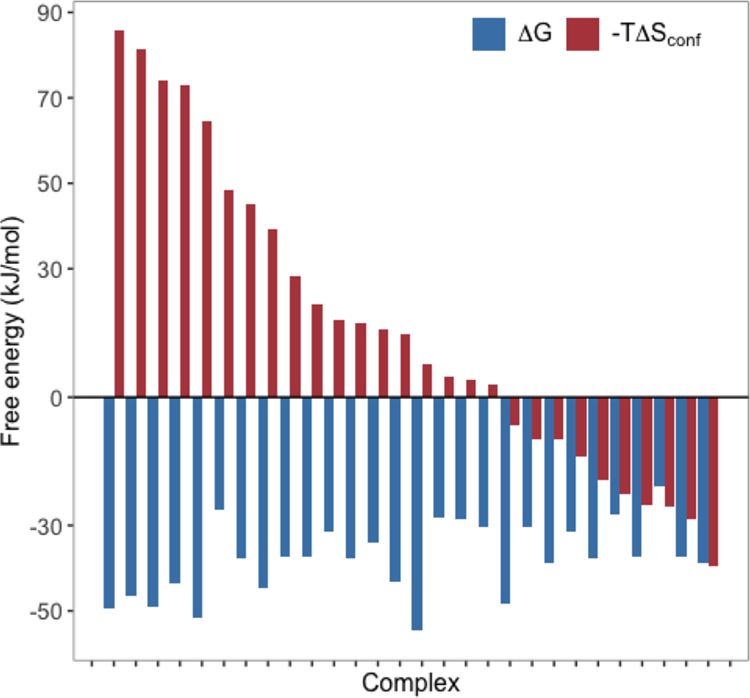
Binding free energy (blue) and diverse conformational entropy signatures (red) for 28 protein–ligand complexes.125 Side chain conformational entropies were estimated using the NMR-based entropy meter approach.127,128 The figure was recreated from supplemental data tables in Caro et al.125 and corresponds to Figure 2 in that article.
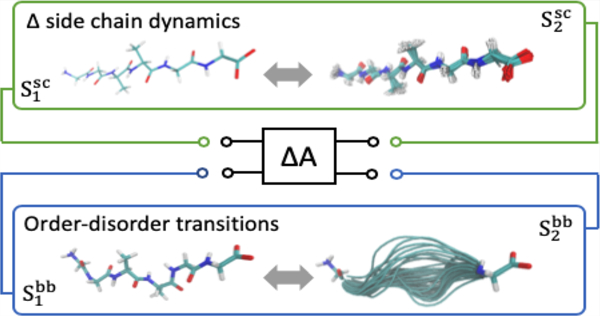
Mechanisms appear to have evolved to modulate the extent with which the dynamics of side chains may respond to binding events, thereby enabling a mode of entropically driven allosteric regulation for structured proteins.129 For example, homodimeric CzrA (chromosomal zinc-regualted repressor) binds target DNA and an observed increase in side chain motions is estimated to significantly contribute to the total, favorable change in entropy.126 When bound, zinc prevents this favorable change in entropy by preventing the increased side chain dynamics when binding to DNA. This ultimately destabilizes the CzrA:DNA complex and decreases binding affinity. The structural integrity of the protein backbone is required to maintain the overall fold, and subsequently function, for well-structured proteins. It necessarily lacks the capacity to alter its dynamics to the extent observed for side chains in mechanisms of binding/recognition and allosteric regulation. Side chains represent an entropic (free energetic) reservoir that can be effectively utilized (i.e., thermodynamically coupled) through changes in side chain dynamics (i.e., structural population shifts). Analogously, structural transitions in IDRs provide a mechanism to thermodynamically couple the conformational entropy of the protein backbone to events such as binding or altering the allosteric poise of a protein to respond to downstream signals. Here the protein backbone can be considered an entropic reservoir from which free energy may be extracted or deposited.17,108,109 These different modes of allosteric regulation are graphically depicted in Figure 6.
Figure 6.

Illustration of the entropic reservoir concept as mediated by changes in the structural fluctuations or dynamics of protein side chains (sc) (top) and backbone (bb) (bottom). These regulatory modes are expected to couple significantly to thermodynamic processes through conformational entropy changes of the solute. However, it is important to note that the effects of conformational fluctuations will also propagate to solute–solvent, solvent–solvent, and solvent–solvent interaction energies and structural network of the solvent.
CONCLUSION
Experiments and simulations demonstrate the important active role the protein backbone plays in protein folding/unfolding, IDR collapse, and phase separation. Concepts of solubility/stability, rather than classical arguments of hydrophobicity, and population-based descriptions of the disordered conformational ensemble provide a consistent framework to study the physicochemical properties of the protein backbone and to consider how such properties may contribute to those of more sequence-complex IDRs. The free energy landscape of IDRs (the result of which is a delicate, complex balance between intrapeptide, peptide–solvent, and solvent–solvent interactions as well as solute and solvent entropies) and its capacity to be remodeled underlies their versatility in terms of mediating a variety of biophysical phenomena. A quantitative understanding of these thermodynamic properties that enable disorder and their dependence on amino acid composition is critical to establishing the first leg of a sequence-disorder-function paradigm19 or, perhaps better yet, a unified sequence-ensemble-function paradigm incorporating that for structured proteins. Such a robust picture will reveal not only the biophysical mechanisms that enable disorder-mediated functions but also strategies to exploit those mechanisms for engineering purposes.
ACKNOWLEDGMENTS
We gratefully acknowledge the Robert A. Welch Foundation (H-0013), the National Science Foundation (CHE-1152876), the National Institutes of Health (GM-037657), and the National Center for Supercomputing Applications Blue Waters Graduate Research Fellowship program for partial support of this work. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1548562 and the Blue Waters sustained petascale computing project, which was supported by the National Science Foundation (award numbers OCI-0725070 and ACI-1238993). The authors also acknowledge the Texas Advanced Computing Center (TACC) at The University of Texas at Austin for providing HPC resources that have contributed to some of the research results reported within this paper. URL: http://www.tacc.utexas.edu
Biographies
Justin A. Drake is currently a Research Associate in the Health Analytics Group at the Texas Advanced Computing Center and Assistant Professor at the Dell Medical School, University of Texas at Austin. He received his Ph.D. in Computational Biophysics from the University of Texas Medical Branch where his research focused on the mechanisms that drive the collapse and aggregation of model disordered polypeptides. He was particularly interested in the calculation of protein backbone conformational entropy and its changes or contributions to order–disorder transitions of disordered polypeptides. Dr. Drake’s current research lies at the interface of ubiquitous computing and medicine.
B. Montgomery Pettitt is the Robert A. Welch Distinguished University Professor of Chemistry at the University of Texas Medical Branch. He obtained bachelors degrees in chemistry and mathematics, and a Ph.D. in chemistry from the University of Houston. His recent work concerns the solution physical chemistry of proteins and nucleic acids. The laboratory’s work uses liquid state theory and computation to consider the mechanisms of molecular recognition.
Footnotes
The authors declare no competing financial interest.
Contributor Information
Justin A. Drake, Sealy Center for Structural Biology and Molecular Biophysics, University of Texas Medical Branch, Galveston 77555, Texas, United States; Texas Advanced Computing Center, University of Texas at Austin, Austin 78712, Texas, United States
B. Montgomery Pettitt, Sealy Center for Structural Biology and Molecular Biophysics, University of Texas Medical Branch, Galveston 77555, Texas, United States.
REFERENCES
- (1).Dunker A; et al. Intrinsically disordered protein. J. Mol. Graphics Modell 2001, 19, 26–59. [DOI] [PubMed] [Google Scholar]
- (2).Fischer E Einfluss der Configuration auf die Wirkung der Enzyme. Ber. Dtsch. Chem. Ges 1894, 27, 2985–2993. [Google Scholar]
- (3).Lemieux RU; Spohr U How Emil Fischer was led to the lock and key concept for enzyme specificity. Adv. Carbohydr. Chem. Biochem 1994, 50, 1–20. [PubMed] [Google Scholar]
- (4).Wright PE; Dyson H Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J. Mol. Biol 1999, 293, 321–331. [DOI] [PubMed] [Google Scholar]
- (5).Vucetic S; Brown CJ; Dunker AK; Obradovic Z Flavors of protein disorder. Proteins: Struct., Funct., Genet 2003, 52, 573–584. [DOI] [PubMed] [Google Scholar]
- (6).Ward J; Sodhi J; McGuffin L; Buxton B; Jones D Prediction and Functional Analysis of Native Disorder in Proteins from the Three Kingdoms of Life. J. Mol. Biol 2004, 337, 635–645. [DOI] [PubMed] [Google Scholar]
- (7).Peng Z; Yan J; Fan X; Mizianty MJ; Xue B; Wang K; Hu G; Uversky VN; Kurgan L Exceptionally abundant exceptions: comprehensive characterization of intrinsic disorder in all domains of life. Cell. Mol. Life Sci 2015, 72, 137–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Piovesan D; et al. DisProt 7.0: a major update of the database of disordered proteins. Nucleic Acids Res 2017, 45, D219–D227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Uversky VN Natively unfolded proteins: A point where biology waits for physics. Protein Sci 2002, 11, 739–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).van der Lee R; et al. Classification of Intrinsically Disordered Regions and Proteins. Chem. Rev 2014, 114, 6589–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Forman-Kay JD; Mittag T From Sequence and Forces to Structure, Function, and Evolution of Intrinsically Disordered Proteins. Structure 2013, 21, 1492–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Uversky VN Unusual biophysics of intrinsically disordered proteins. Biochim. Biophys. Acta, Proteins Proteomics 2013, 1834, 932–951. [DOI] [PubMed] [Google Scholar]
- (13).Dyson HJ; Wright PE Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol 2005, 6, 197–208. [DOI] [PubMed] [Google Scholar]
- (14).Wright PE; Dyson HJ Intrinsically Disordered Proteins in Cellular Signaling and Regulation. Nat. Rev. Mol. Cell Biol 2015, 16, 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zhou H-X Intrinsic disorder: signaling via highly specific but short-lived association. Trends Biochem. Sci 2012, 37, 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Dunker AK; Cortese MS; Romero P; Iakoucheva LM; Uversky VN Flexible nets. FEBS J 2005, 272, 5129–5148. [DOI] [PubMed] [Google Scholar]
- (17).Flock T; Weatheritt RJ; Latysheva NS; Babu MM Controlling entropy to tune the functions of intrinsically disordered regions. Curr. Opin. Struct. Biol 2014, 26, 62–72. [DOI] [PubMed] [Google Scholar]
- (18).Hsu W-L; Oldfield CJ; Xue B; Meng J; Huang F; Romero P; Uversky VN; Dunker AK Exploring the binding diversity of intrinsically disordered proteins involved in one-to-many binding. Protein Science: A Publication of the Protein Society 2013, 22, 258–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hilser VJ; Thompson EB Intrinsic disorder as a mechanism to optimize allosteric coupling in proteins. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 8311–8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Liu Y; Matthews KS; Bondos SE Multiple Intrinsically Disordered Sequences Alter DNA Binding by the Homeodomain of the Drosophila Hox Protein Ultrabithorax. J. Biol. Chem 2008, 283, 20874–20887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Motlagh HN; Wrabl JO; Li J; Hilser VJ The ensemble nature of allostery. Nature 2014, 508, 331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Tompa P Multisteric Regulation by Structural Disorder in Modular Signaling Proteins: An Extension of the Concept of Allostery. Chem. Rev 2014, 114, 6715–6732. [DOI] [PubMed] [Google Scholar]
- (23).Nussinov R Introduction to Protein Ensembles and Allostery. Chem. Rev 2016, 116, 6263–6266. [DOI] [PubMed] [Google Scholar]
- (24).Kato M; et al. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 2012, 149, 753–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Mitrea DM; Kriwacki RW Phase separation in biology; functional organization of a higher order. Cell Commun. Signaling 2016, DOI: 10.1186/s12964-015-0125-7. [DOI] [PMC free article] [PubMed]
- (26).Seydoux G; Smith J; Calidas D; Schmidt H; Rasoloson D Organelles without membranes: intrinsically-disordered proteins bring order to the cytoplasm. FASEB J 2017, DOI: 10.1096/fasebj.31.1_-supplement.525.2. [DOI]
- (27).Jakob U; Kriwacki R; Uversky VN Conditionally and Transiently Disordered Proteins: Awakening Cryptic Disorder To Regulate Protein Function. Chem. Rev 2014, 114, 6779–6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Zea DJ; Monzon AM; Gonzalez C; Fornasari MS; Tosatto SCE; Parisi G Disorder transitions and conformational diversity cooperatively modulate biological function in proteins. Protein Sci 2016, 25, 1138–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).DeForte S; Uversky VN Order, Disorder, and Everything in Between. Molecules 2016, 21, E1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Minezaki Y; Homma K; Kinjo AR; Nishikawa K Human Transcription Factors Contain a High Fraction of Intrinsically Disordered Regions Essential for Transcriptional Regulation. J. Mol. Biol 2006, 359, 1137–1149. [DOI] [PubMed] [Google Scholar]
- (31).Liu J; Perumal NB; Oldfield CJ; Su EW; Uversky VN; Dunker AK Intrinsic disorder in transcription factors. Biochemistry 2006, 45, 6873–6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Bondos SE; Tan X-X; Matthews KS Physical and Genetic Interactions Link Hox Function with Diverse Transcription Factors and Cell Signaling Proteins. Mol. Cell. Proteomics 2006, 5, 824–834. [DOI] [PubMed] [Google Scholar]
- (33).Uversky VN; Oldfield CJ; Dunker AK Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu. Rev. Biophys 2008, 37, 215–246. [DOI] [PubMed] [Google Scholar]
- (34).Uversky VN Wrecked regulation of intrinsically disordered proteins in diseases: pathogenicity of deregulated regulators. Frontiers in Molecular Biosciences 2014, DOI: 10.3389/fmolb.2014.00006. [DOI] [PMC free article] [PubMed]
- (35).Hnizdo V; Gilson MK Thermodynamic and Differential Entropy under a Change of Variables. Entropy 2010, 12, 578–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Onuchic JN; Luthey-Schulten Z; Wolynes a. P. G. THEORY OF PROTEIN FOLDING: The Energy Landscape Perspective. Annu. Rev. Phys. Chem 1997, 48, 545–600. [DOI] [PubMed] [Google Scholar]
- (37).Fisher CK; Stultz CM Constructing ensembles for intrinsically disordered proteins. Curr. Opin. Struct. Biol 2011, 21, 426–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Papoian GA Proteins with weakly funneled energy landscapes challenge the classical structure-function paradigm. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 14237–14238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Onuchic JN; Wolynes PG Theory of protein folding. Curr. Opin. Struct. Biol 2004, 14, 70–75. [DOI] [PubMed] [Google Scholar]
- (40).Meng F; Na I; Kurgan L; Uversky VN Compartmentalization and Functionality of Nuclear Disorder: Intrinsic Disorder and Protein-Protein Interactions in Intra-Nuclear Compartments. Int. J. Mol. Sci 2016, 17, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).IUPAC. Compendium of Chemical Terminology; Blackwell Scientific Publications: Oxford, 1997. [Google Scholar]
- (42).Auton M; Bolen DW Additive transfer free energies of the peptide backbone unit that are independent of the model compound and the choice of concentration scale. Biochemistry 2004, 43, 1329–1342. [DOI] [PubMed] [Google Scholar]
- (43).Auton M; Holthauzen LMF; Bolen DW Anatomy of energetic changes accompanying urea-induced protein denaturation. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 15317–15322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Thompson JD; Cramer CJ; Truhlar DG Predicting aqueous solubilities from aqueous free energies of solvation and experimental or calculated vapor pressures of pure substances. J. Chem. Phys 2003, 119, 1661–1670. [Google Scholar]
- (45).Uversky VN; Oldfield CJ; Dunker AK Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J. Mol. Recognit 2005, 18, 343–384. [DOI] [PubMed] [Google Scholar]
- (46).Dyson HJ; Wright PE Unfolded Proteins and Protein Folding Studied by NMR. Chem. Rev 2004, 104, 3607–3622. [DOI] [PubMed] [Google Scholar]
- (47).Miyagi A; Tsunaka Y; Uchihashi T; Mayanagi K; Hirose S; Morikawa K; Ando T Visualization of Intrinsically Disordered Regions of Proteins by High-Speed Atomic Force Microscopy. ChemPhysChem 2008, 9, 1859–1866. [DOI] [PubMed] [Google Scholar]
- (48).Bernadó P; Svergun DI Analysis of intrinsically disordered proteins by small-angle X-ray scattering. Methods in Molecular Biology (Clifton, N.J.) 2012, 896, 107–122. [DOI] [PubMed] [Google Scholar]
- (49).Brucale M; Schuler B; Samorì B Single-Molecule Studies of Intrinsically Disordered Proteins. Chem. Rev 2014, 114, 3281–3317. [DOI] [PubMed] [Google Scholar]
- (50).Chong S-H; Chatterjee P; Ham S Computer Simulations of Intrinsically Disordered Proteins. Annu. Rev. Phys. Chem 2017, 68, 117. [DOI] [PubMed] [Google Scholar]
- (51).Oldfield CJ; Dunker AK Intrinsically Disordered Proteins and Intrinsically Disordered Protein Regions. Annu. Rev. Biochem 2014, 83, 553–584. [DOI] [PubMed] [Google Scholar]
- (52).Bustamante C singulo Biochemistry: When Less Is More. Annu. Rev. Biochem 2008, 77, 45–50. [DOI] [PubMed] [Google Scholar]
- (53).Crescenzi O; Tomaselli S; Guerrini R; Salvadori S; D’Ursi AM; Temussi PA; Picone D Solution structure of the Alzheimer amyloid β-peptide (1–42) in an apolar microenvironment. Eur. J. Biochem 2002, 269, 5642–5648. [DOI] [PubMed] [Google Scholar]
- (54).Karplus M; McCammon JA Molecular dynamics simulations of biomolecules. Nat. Struct. Biol 2002, 9, 646–652. [DOI] [PubMed] [Google Scholar]
- (55).Palazzesi F; Prakash MK; Bonomi M; Barducci A Accuracy of Current All-Atom Force-Fields in Modeling Protein Disordered States. J. Chem. Theory Comput 2015, 11, 2–7. [DOI] [PubMed] [Google Scholar]
- (56).Fluitt AM; de Pablo JJ An Analysis of Biomolecular Force Fields for Simulations of Polyglutamine in Solution. Biophys. J 2015, 109, 1009–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Drake JA; Pettitt BM Force field-dependent solution properties of glycine oligomers. J. Comput. Chem 2015, 36, 1275–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Huang J; Rauscher S; Nawrocki G; Ran T; Feig M; de Groot BL; Grubmü H; MacKerell AD Jr CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Robustelli P; Piana S; Shaw DE Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. U. S. A 2018, 115, E4758–E4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Romero P; Obradovic Z; Li X; Garner EC; Brown CJ; Dunker AK Sequence complexity of disordered protein. Proteins: Struct., Funct., Genet 2001, 42, 38–48. [DOI] [PubMed] [Google Scholar]
- (61).Karandur D; Wong K-Y; Pettitt BM Solubility and aggregation of Gly(5) in water. J. Phys. Chem. B 2014, 118, 9565–9572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Marsh JA; Forman-Kay JD Sequence Determinants of Compaction in Intrinsically Disordered Proteins. Biophys. J 2010, 98, 2383–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Harris RC; Drake JA; Pettitt BM Multibody correlations in the hydrophobic solvation of glycine peptides. J. Chem. Phys 2014, 141, 22D525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Karandur D; Harris RC; Pettitt BM Protein collapse driven against solvation free energy without H-bonds. Protein Sci 2016, 25, 103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Drake JA; Harris RC; Pettitt BM Solvation Thermodynamics of Oligoglycine with Respect to Chain Length and Flexibility. Biophys. J 2016, 111, 756–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Walters RH; Murphy RM Examining Polyglutamine Peptide Length: A Connection between Collapsed Conformations and Increased Aggregation. J. Mol. Biol 2009, 393, 978–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Teufel DP; Johnson CM; Lum JK; Neuweiler H Backbone-Driven Collapse in Unfolded Protein Chains. J. Mol. Biol 2011, 409, 250–262. [DOI] [PubMed] [Google Scholar]
- (68).Crick SL; Jayaraman M; Frieden C; Wetzel R; Pappu RV Fluorescence correlation spectroscopy shows that monomeric polyglutamine molecules form collapsed structures in aqueous solutions. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 16764–16769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Müller-Späth S; Soranno A; Hirschfeld V; Hofmann H; Rüegger S; Reymond L; Nettels D; Schuler B Charge interactions can dominate the dimensions of intrinsically disordered proteins. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 14609–14614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Asthagiri D; Karandur D; Tomar DS; Pettitt BM Intramolecular Interactions Overcome Hydration to Drive the Collapse Transition of Gly15. J. Phys. Chem. B 2017, 121, 8078–8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Uversky VN; Gillespie JR; Fink AL Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins: Struct., Funct., Genet 2000, 41, 415–427. [DOI] [PubMed] [Google Scholar]
- (72).Nick Pace C; Scholtz JM; Grimsley GR Forces stabilizing proteins. FEBS Lett 2014, 588, 2177–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Wang X; Vitalis A; Wyczalkowski MA; Pappu RV Characterizing the conformational ensemble of monomeric polyglutamine. Proteins: Struct., Funct., Genet 2006, 63, 297–311. [DOI] [PubMed] [Google Scholar]
- (74).Mukhopadhyay S; Krishnan R; Lemke EA; Lindquist S; Deniz AA A natively unfolded yeast prion monomer adopts an ensemble of collapsed and rapidly fluctuating structures. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 2649–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Finnegan ML; Bowler BE Scaling Properties of Glycine-Rich Sequences in Guanidine Hydrochloride Solutions. Biophys. J 2012, 102, 1969–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Tran HT; Mao A; Pappu RV Role of Backbone–Solvent Interactions in Determining Conformational Equilibria of Intrinsically Disordered Proteins. J. Am. Chem. Soc 2008, 130, 7380–7392. [DOI] [PubMed] [Google Scholar]
- (77).Daidone I; Neuweiler H; Doose S; Sauer M; Smith JC Hydrogen-Bond Driven Loop-Closure Kinetics in Unfolded Polypeptide Chains. PLoS Comput. Biol 2010, 6, No. e1000645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Hu CY; Lynch GC; Kokubo H; Pettitt BM Trimethylamine N-oxide influence on the backbone of proteins: An oligoglycine model. Proteins: Struct., Funct., Genet 2009, 78, 695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).König G; Bruckner S; Boresch S Absolute Hydration Free Energies of Blocked Amino Acids: Implications for Protein Solvation and Stability. Biophys. J 2013, 104, 453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Sarma R; Wong K-Y; Lynch GC; Pettitt BM Peptide Solubility Limits: Backbone and Side-Chain Interactions. J. Phys. Chem. B 2018, 122, 3528–3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Bolen DW; Rose GD Structure and Energetics of the Hydrogen-Bonded Backbone in Protein Folding. Annu. Rev. Biochem 2008, 77, 339–362. [DOI] [PubMed] [Google Scholar]
- (82).Mao AH; Lyle N; Pappu RV Describing sequence-ensemble relationships for intrinsically disordered proteins. Biochem. J 2013, 449, 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Uversky VN; Santambrogio C; Brocca S; Grandori R Length-dependent compaction of intrinsically disordered proteins. FEBS Lett 2012, 586, 70–73. [DOI] [PubMed] [Google Scholar]
- (84).Mao AH; Crick SL; Vitalis A; Chicoine CL; Pappu RV Net charge per residue modulates conformational ensembles of intrinsically disordered proteins. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 8183–8188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Johansen D; Trewhella J; Goldenberg DP Fractal dimension of an intrinsically disordered protein: small-angle X-ray scattering and computational study of the bacteriophage λ N protein. Protein Sci 2011, 20, 1955–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Das RK; Pappu RV Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 13392–13397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Munshi S; Rajendran D; Ramesh S; Subramanian S; Bhattacharjee K; Kumar MR; Naganathan AN Controlling Structure and Dimensions of a Disordered Protein via Mutations. Biochemistry 2020, 59, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Han TW; Kato M; Xie S; Wu LC; Mirzaei H; Pei J; Chen M; Xie Y; Allen J; Xiao G; McKnight SL Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell 2012, 149, 768–779. [DOI] [PubMed] [Google Scholar]
- (89).Martin EW; Mittag T The relationship of sequence and phase separation in protein low-complexity regions. Biochemistry 2018, 57, 2478–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Boeynaems S; Alberti S; Fawzi NL; Mittag T; Polymenidou M; Rousseau F; Schymkowitz J; Shorter J; Wolozin B; Van Den Bosch L; Tompa P; Fuxreiter M Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Biol 2018, 28, 420–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Brangwynne CP; Eckmann CR; Courson DS; Rybarska A; Hoege C; Gharakhani J; Jülicher F; Hyman AA Germline P Granules Are Liquid Droplets That Localize by Controlled Dissolution/Condensation. Science 2009, 324, 1729–1732. [DOI] [PubMed] [Google Scholar]
- (92).Li P; Banjade S; Cheng H-C; Kim S; Chen B; Guo L; Llaguno M; Hollingsworth JV; King DS; Banani SF; Russo PS; Jiang Q-X; Nixon BT; Rosen MK Phase transitions in the assembly of multivalent signalling proteins. Nature 2012, 483, 336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Matera AG; Izaguire-Sierra M; Praveen K; Rajendra T Nuclear Bodies: Random Aggregates of Sticky Proteins or Crucibles of Macromolecular Assembly? Dev. Cell 2009, 17, 639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Case LB; Zhang X; Ditlev JA; Rosen MK Stoichiometry controls activity of phase-separated clusters of actin signaling proteins. Science 2019, 363, 1093–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Patel A; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [DOI] [PubMed] [Google Scholar]
- (96).Aguzzi A; Altmeyer M Phase Separation: Linking Cellular Compartmentalization to Disease. Trends Cell Biol 2016, 26, 547–558. [DOI] [PubMed] [Google Scholar]
- (97).Tompa P; Schad E; Tantos A; Kalmar L Intrinsically disordered proteins: emerging interaction specialists. Curr. Opin. Struct. Biol 2015, 35, 49–59. [DOI] [PubMed] [Google Scholar]
- (98).Uversky VN Intrinsically disordered proteins in overcrowded milieu: Membrane-less organelles, phase separation, and intrinsic disorder. Curr. Opin. Struct. Biol 2017, 44, 18–30. [DOI] [PubMed] [Google Scholar]
- (99).Brooks CL III; Karplus M; Pettitt BM In Proteins: A Theoretical Perspective of Dynamics, Structure, and Thermodynamics; Prigogine I, Rice S, Eds.; John Wiley & Sons, Inc.: New York, NY, 1988. [Google Scholar]
- (100).Best RB; Zheng W; Mittal J Balanced Protein-Water Interactions Improve Properties of Disordered Proteins and Non-Specific Protein Association. J. Chem. Theory Comput 2014, 10, 5113–5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Lindorff-Larsen K; Piana S; Palmo K; Maragakis P; Klepeis JL; Dror RO; Shaw DE Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins: Struct., Funct., Genet 2010, 78, 1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Best RB Computational and theoretical advances in studies of intrinsically disordered proteins. Curr. Opin. Struct. Biol 2017, 42, 147–154. [DOI] [PubMed] [Google Scholar]
- (103).Wimley WC; White SH Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Mol. Biol 1996, 3, 842–848. [DOI] [PubMed] [Google Scholar]
- (104).Lum K; Chandler D; Weeks JD Hydrophobicity at Small and Large Length Scales. J. Phys. Chem. B 1999, 103, 4570–4577. [Google Scholar]
- (105).Majumdar A; Dogra P; Maity S; Mukhopadhyay S Liquid–Liquid Phase Separation Is Driven by Large-Scale Conformational Unwinding and Fluctuations of Intrinsically Disordered Protein Molecules. J. Phys. Chem. Lett 2019, 10, 3929–3936. [DOI] [PubMed] [Google Scholar]
- (106).Elbaum-Garfinkle S; Rhoades E Identification of an Aggregation-Prone Structure of Tau. J. Am. Chem. Soc 2012, 134, 16607–16613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Eschmann NA; Georgieva ER; Ganguly P; Borbat PP; Rappaport MD; Akdogan Y; Freed JH; Shea J-E; Han S Signature of an aggregation-prone conformation of tau. Sci. Rep 2017, 7, 44739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Drake JA; Pettitt BM Thermodynamics of Conformational Transitions in a Disordered Protein Backbone Model. Biophys. J 2018, 114, 2799–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Pritisǎnac I; Vernon RM; Moses AM; Forman Kay JD Entropy and Information within Intrinsically Disordered Protein Regions. Entropy 2019, 21, 662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Heller GT; Bonomi M; Vendruscolo M Structural Ensemble Modulation upon Small-Molecule Binding to Disordered Proteins. J. Mol. Biol 2018, 430, 2288–2292. [DOI] [PubMed] [Google Scholar]
- (111).Keul ND; Oruganty K; Bergman ETS; Beattie NR; McDonald WE; Kadirvelraj R; Gross ML; Phillips RS; Harvey SC; Wood ZA The entropic force generated by intrinsically disordered segments tunes protein function. Nature 2018, 563, 584–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Tompa P; Fuxreiter M Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci 2008, 33, 2–8. [DOI] [PubMed] [Google Scholar]
- (113).Van Roey K; Uyar B; Weatheritt RJ; Dinkel H; Seiler M; Budd A; Gibson TJ; Davey NE Short Linear Motifs: Ubiquitous and Functionally Diverse Protein Interaction Modules Directing Cell Regulation. Chem. Rev 2014, 114, 6733–6778. [DOI] [PubMed] [Google Scholar]
- (114).Oldfield CJ; Cheng Y; Cortese MS; Romero P; Uversky VN; Dunker AK Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry 2005, 44, 12454–12470. [DOI] [PubMed] [Google Scholar]
- (115).Mohan A; Oldfield CJ; Radivojac P; Vacic V; Cortese MS; Dunker AK; Uversky VN Analysis of Molecular Recognition Features (MoRFs). J. Mol. Biol 2006, 362, 1043–1059. [DOI] [PubMed] [Google Scholar]
- (116).Amemiya T; Koike R; Fuchigami S; Ikeguchi M; Kidera A Classification and annotation of the relationship between protein structural change and ligand binding. J. Mol. Biol 2011, 408, 568–584. [DOI] [PubMed] [Google Scholar]
- (117).Killian BJ; Kravitz JY; Gilson MK Extraction of Configurational Entropy from Molecular Simulations via an Expansion Approximation. J. Chem. Phys 2007, 127, 024107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Thompson JB; Hansma HG; Hansma PK; Plaxco KW The Backbone Conformational Entropy of Protein Folding: Experimental Measures from Atomic Force Microscopy. J. Mol. Biol 2002, 322, 645–652. [DOI] [PubMed] [Google Scholar]
- (119).Baxa MC; Haddadian EJ; Jumper JM; Freed KF; Sosnick TR Loss of conformational entropy in protein folding calculated using realistic ensembles and its implications for NMR-based calculations. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 15396–15401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Towse C-L; Akke M; Daggett V The Dynameomics Entropy Dictionary: A Large-Scale Assessment of Conformational Entropy across Protein Fold Space. J. Phys. Chem. B 2017, 121, 3933–3945. [DOI] [PubMed] [Google Scholar]
- (121).London N; Movshovitz-Attias D; Schueler-Furman O The Structural Basis of Peptide-Protein Binding Strategies. Structure 2010, 18, 188–199. [DOI] [PubMed] [Google Scholar]
- (122).Tzeng S-R; Kalodimos CG Protein activity regulation by conformational entropy. Nature 2012, 488, 236–240. [DOI] [PubMed] [Google Scholar]
- (123).Wand AJ The dark energy of proteins comes to light: conformational entropy and its role in protein function revealed by NMR relaxation. Curr. Opin. Struct. Biol 2013, 23, 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Sharp KA; O’Brien E; Kasinath V; Wand AJ On the relationship between NMR-derived amide order parameters and protein backbone entropy changes. Proteins: Struct., Funct., Genet 2015, 83, 922–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Caro JA; Harpole KW; Kasinath V; Lim J; Granja J; Valentine KG; Sharp KA; Wand AJ Entropy in molecular recognition by proteins. Proc. Natl. Acad. Sci. U. S. A 2017, 114, 6563–6568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Capdevila DA; Braymer JJ; Edmonds KA; Wu H; Giedroc DP Entropy redistribution controls allostery in a metalloregulatory protein. Proc. Natl. Acad. Sci. U. S. A 2017, 114, 4424–4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Marlow MS; Dogan J; Frederick KK; Valentine KG; Wand AJ The role of conformational entropy in molecular recognition by calmodulin. Nat. Chem. Biol 2010, 6, 352–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Wand AJ; Moorman VR; Harpole KW A surprising role for conformational entropy in protein function. Top. Curr. Chem 2013, 337, 69–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Capdevila DA; Huerta F; Edmonds KA; Le MT; Wu H; Giedroc DP Tuning site-specific dynamics to drive allosteric activation in a pneumococcal zinc uptake regulator. eLife 2018, 7, No. e37268. [DOI] [PMC free article] [PubMed] [Google Scholar]