

Abstract
The present review aims to summarize the pharmacological profile of 1,2,4-triazole, one of the emerging privileged scaffold, as antifungal, antibacterial, anticancer, anticonvulsant, antituberculosis, antiviral, antiparasitic, analgesic and anti-inflammatory agents, etc. along with structure-activity relationship. The comprehensive compilation of work carried out in the last decade on 1,2,4-triazole nucleus will provide inevitable scope for researchers for the advancement of novel potential drug candidates having better efficacy and selectivity.
Keywords: 1,2,4-Triazole; Molecular interactions; Enzyme inhibitors; Hybrid compounds; Pharmacological activities; Structure-activity relationship
Graphical abstract
1. Introduction
Among the various azaheterocyclic systems, azoles, in general, and 1,2,4-triazoles in particular are the focus of renewed interest among organic and medicinal chemists since several novel hybrids with broader spectrum have been synthesized based on molecular hybridization approach [1]. Among the azoles, triazoles are the most stable compounds and are difficult to cleave. 1,2,4-Triazole having molecular formula C2H3N3 acts as isosteres of amide, ester and carboxylic acid. It may be formally derived from pyrazole by substitution of a carbon at position-4 by nitrogen atom. 1,2,4-Triazole exists in two tautomeric forms A and B in which 1H-1,2,4-triazole (A) is more stable than 4H-1,2,4-triazole (B) as depicted in Fig. 1 [2].
Fig. 1.
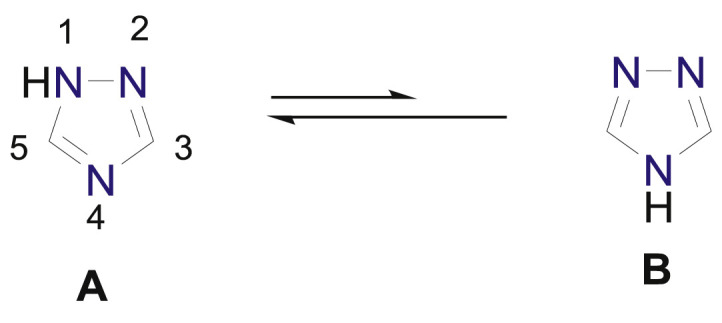
Tautomeric forms of 1,2,4-triazole.
1,2,4-Triazoles act as important pharmacophores by interacting with the biological receptors with high affinity owing to their dipole character, hydrogen bonding capacity, rigidity and solubility. This motif is an integral part of a variety of drugs available in clinical therapy including antifungal (fluconazole, itraconazole, posaconazole, voriconazole, ravuconazole) anxiolytic, anticonvulsant and hypnotic (estazolam, alprazolam), anxiolytic and skeletal muscle relaxant (etizolam), antimigraine (rizatriptan), antiplatelet (trapidil), antidepressant (trazodone), anticancer (anastrozole), aromatase inhibitor (letrozole), antiviral (ribavirin) and anticonvulsant (loreclezole) (Fig. 2 ) [3,4]. Some commercial plant protection fungicides contain the triazole moiety, such as prothioconazole, triadimefon, metconazole, propiconazole, tebuconazole, epoxyconazole, triadimenol and cyproconazole [5].
Fig. 2.

Clinically used drugs having 1,2,4-triazole scaffold.
1,2,4-Triazoles and their fused heterocyclic derivatives have been reported to possess a wide range of bioactivities such as neuroprotectant [6], antioxidant [7], antimalarial [8], antileishmanial [9], anti-urease [10], antiviral [11,12], anticonvulsant [13], cannabinoid CB1 receptor antagonists [14], PDE4A inhibitors [15] and γ-aminobutyric acid-A (GABA-A) α-2, α-3 and α-5 containing receptor antagonists [16]. Moreover, they have applications in ionic liquids, corrosion inhibitors, agrochemicals, polymers, supramolecular and material science [[17], [18], [19], [20], [21]].
Owing to the mounting importance of 1,2,4-triazoles in emerging domains, it is envisaged to bring out a comprehensive review of this privileged framework from 2010 onwards on the medicinal profile.
2. Biological activities of 1,2,4-triazole derivatives
2.1. Antifungal agents
The emergence of multidrug-resistant pathogens impelled the researchers to develop novel broad spectrum triazoles having high impact, ease of administration and low toxicity to conquer the resistance. The triazole antifungal drugs potently act by inhibiting the activity of cytochrome P450-dependent enzyme, the lanosterol 14α-demethylase (CYP51), which is the key enzyme in ergosterol biosynthesis of fungi [22].
Design, synthesis and antifungal activities of large number of 1-(2-(2,4-difluorophenyl)-2-hydroxy-3-(1H-1,2,4-triazol-1-yl)propyl)-4-substituted derivatives 1–15 as fluconazole or voriconazole or ravuconazole analogues have been carried out by Chinese group(s) (Fig. 3 ) [[23], [24], [25], [26], [27], [28], [29], [30], [31], [32], [33], [34], [35]]. Compound 1n (MIC80: 0.0156 μg/mL) exhibited 16 fold more antifungal activity than fluconazole against Candida albicans [23]. Docking study of compound 2 revealed the significance of 1,2,3-triazole group and the substituted benzyl as side chains for antifungal activity [24,25]. Compound 3 having R1 = CF3 group displayed broad antifungal spectrum with MIC80 values in the range of 0.00097–0.0156 μg/mL against human pathogenic fungi (C. albicans, Candida parapsilosis, Candida tropicalis, Cryptococcus neoformans, Trichophyton rubrum, Fonsecaea compacta and Microsporum gypseum) [26]. It exhibited 64 fold more potency than reference drugs fluconazole and voriconazole against Aspergillus fumigatus (MIC80: 1 μg/mL). Molecular docking studies of 4 (R = 3-Cl) in active site of CACYP51 showed multiple molecular interactions of difluorophenyl group and terminal triazolone side chain with hydrophobic region as well as coordinate bond formation of triazole ring with iron of heme group [27]. Lengthening of the side chain by a double bond influences the spatial orientations of compounds 5 in target enzyme leading to low antifungal activities.
Fig. 3.


SAR and antifungal activity profiles of 1-(2-(2,4-difluorophenyl)-2-hydroxy-3-(1H-1,2,4-triazol-1-yl)propyl)-4-substituted derivatives.
Compounds 6 demonstrated good antifungal activity (MIC: 0.0625–1 μg/mL) for C. albicans [28]. Among compounds 7 and 8, analogue 7a (R1 = Br and R2 = H) displayed excellent potency (MIC: 0.0313–1 μg/mL) against all tested fungal strains [29]. Triazole derivatives 9 and 10 having heterocycle-benzene bioisosteric replacement showed excellent antifungal activity with improved oral absorption. SAR study revealed that substituted piperazine derivatives 10 were comparable or superior to the corresponding N-methyl derivatives 9 and heterocyclic substitutions influenced the activity differently in compounds 9 and 10 [30]. The MIC80 values of compounds 11a-m against C. albicans were ranged in nanomole levels (0.009–0.480 nmol/mL) [31].
Dithiocarbamate derivatives of fluconazole 12 exhibited high activity (MIC80: <0.125–2 μg/mL) against C. albicans, C. neoformans, C. parapsilosis and Candida glabrata [32]. SAR indicated that among compounds 13, two compounds 13e and 13f having R = 2-Cl and R = 3-Cl, respectively displayed the highest activity against C. albicans with MIC80 of 0.0039 μg/mL and were 16-, 64-, 128-, and 2051-fold more potent than voriconazole, itraconazole, fluconazole, and ketoconazole, respectively [33]. Isoxazole containing triazole analogues of ravuconazole 14a-c displayed superior activity than ravuconazole against 8 fungal isolates [34]. Wu et al. synthesized and evaluated voriconazole analogues 15 having substituted amines or heterocycles as side chain for their in vitro and in vivo antifungal activity against several human pathogenic fungi [35]. From screening results and docking experiment, it was observed that compound having morpholine moiety exhibited the strongest activity to inhibit the growth of ten fungal pathogens (MIC80: 0.0156–0.5 μg/mL).
In another concomitant study, a series of triazole alcohols having 4-(substituted-1H-indol-3-ylmethyl)-piperazinyl side chain 16 were synthesized and evaluated for antifungal activity against C. albican, C. neoformans, C. krusei, and A. fumigatus by Young Min Na [36]. SAR study revealed that multihalogenated indole derivatives of triazole were 4-fold more active against C. Albicans, A. fumigatus and C. krusei (Fig. 4 ). Several triazoles with fused-heterocycle nuclei were designed and synthesized by Cao et al. [37], among which the most potent compound 17 (Fig. 4) displayed excellent activity against Candida, Cryptococcus, Aspergillus species and selected fluconazole-resistant strains. Shrestha et al. [38] synthesized a series of alkylated-fluconazole derivatives 18 which exhibited low hemolytic activity, low cytotoxicity and good activity against C. albicans, non-albicans Candida and Aspergillus strains (Fig. 4).
Fig. 4.

SAR and antifungal activity profiles of 1-(2-(2,4-difluorophenyl)-2-hydroxy-3-(1H-1,2,4-triazol-1-yl)propyl)-4-substituted derivatives 16–20.
Several carbazole-triazole conjugates 19 (Fig. 4) were synthesized and screened for their antifungal activities against C. albicans, C. tropicalis, C. parapsilosis and A. fumigatus by Zhang et al. [39]. Preliminary mechanistic study revealed that the most active compound 19 having 3,6-dibromocarbazole could depolarize fungal membrane potential and intercalate into DNA to exhibit antifungal action. Coumarin-substituted triazole antifungals 20 were screened against a panel of Candida pathogens by Elias et al. [40] and live-cell imaging revealed that fluorescent 7-diethylaminocoumarin-based triazoles localized to the fungal cell endoplasmic reticulum (Fig. 4).
Luo et al. synthesized a series of 1,3,4-thiadiazole derivatives bearing 1,2,4-triazolo[1,5-a]pyrimidine moiety 21 (Fig. 5 ) and evaluated their antifungal activities against Fusarium oxysporum f.sp. vasinfectum, Gibberella sanbinetti, Cercospora beticola Sacc, Physaclospora piricola and Rhizoctonia solani [41]. SAR studies showed that compounds (21d, 21f, 21h, 21i, 21k, 21o, 21t and 21u) having electron-withdrawing groups (Cl, Br, F, NO2) at position 2 and 4 of the benzene ring exhibited better activity than others against P. piricola. Among them, compound 21t bearing two electron-withdrawing F atoms at position 2 and 4 displayed best activity with 86% inhibition against P. piricola which was found to be more than carbendazim (74%).
Fig. 5.

1,2,4-Triazole derivatives with antifungal activity.
A series of triazole-oxadiazole derivatives 22 (Fig. 5) was synthesized and evaluated for antifungal and apoptotic activities against C. albicans, C. parapsilosis, C. krusei and C. glabrata by Çavusoglu et al. [42]. The study unveiled that compound 22i was equipotent to ketoconazole against C. albicans and C. glabrata and exhibited antifungal effect via apoptotic pathway. Among the synthesized quinoline based benzothiazolyl-1,2,4-triazoles 23 (Fig. 5), compounds 23f and 23j (MIC: 6.25 μg/mL) were 2-fold more potent than standard fluconazole (MIC: 12.5 μg/mL) against C. albicans while compounds 23g and 23i (MIC: 6.25 μg/mL) exerted high activities against Aspergillus niger and were equipotent to fluconazole (MIC: 6.25 μg/mL) [43].
Lin et al. have reported the synthesis and in vitro antifungal activity of a series of myrtenal derivatives bearing 1,2,4-triazole moiety 24 at 50 μg/mL [44]. The study revealed that most of the compounds showed enhanced activities than that of myrtenal, indicating that the incorporation of 1,2,4-triazole-thioether moiety into the myrtenal molecule was beneficial to the increase of antifungal activity (Fig. 5). Some of the compounds exhibited excellent activity against P. piricola with an inhibitory rate 90–98% comparable to commercial fungicide azoxystrobin 96%.
1,2,4-Triazole Schiff base 25 (EC50: 0.0087–0.0309 g/L) exhibited higher antifungal activity than triadimefon (EC50: 0.0195–0.0620 g/L) against Gibberlla nicotiancola and Gibberlla saubinetii (Fig. 5) [45]. Zoumpoulakis et al. have reported the synthesis and antifungal activity of sulfonamide-1,2,4-triazole derivatives 26 (MIC: 0.01–0.27 μmol/mL) against several fungal strains (Fig. 5) [46]. With certain fungi (e.g. A. niger, Trichoderma viride, and Aspergillus flavus) this activity was 10–70 times higher than the commercial antifungal agents bifonazole and ketoconazole. A series of amide derivatives of 1,2,4-triazole 27 (Fig. 5) was reported to exhibit moderate to high antifungal activity against Gibberella azeae, Fusarium oxysporum, Cytospora mandshurica, Pellicularia sasakii, and Phytophthora infestans at 50 mg/L by Tang et al. [47]. SAR study revealed the significance of R group as shown in Fig. 5.
2.2. Antibacterial agents
Most of the synthesized clinafloxacin-triazole hybrids 28 (MIC: 0.25–2 μg/mL) endowed with good antibacterial and antifungal activities were comparable or more potent than the reference drugs chloramphenicol, clinafloxacin and fluconazole [48]. SAR studies revealed that compound 28g with a 2,4-difluoro at phenyl ring exhibited most potent antimicrobial efficacy (MIC: 0.25–1 μg/mL) particularly against methicillin-resistant Staphylococcus aureus (MRSA) among the tested compounds as displayed in Fig. 6 .
Fig. 6.

1,2,4-Triazole-quinolone hybrids with antibacterial activity.
Most of the ciprofloxacin-1,2,4-triazole-5(4H)-thione hybrids 29 (MIC: 0.046–3.11 μM) were tested against a panel of pathogens and were found to have higher potency against MRSA than the references vancomycin (MIC: 0.68 μM) and ciprofloxacin (MIC: 2.96 μM) [49,50]. SAR analysis of hybrids 29 (Fig. 6) divulged that phenyl groups at C-3 position played crucial role in exerting high activity and electron-donating groups, particularly –OH on the phenyl ring favored the activity; while substituted phenyl group on N-4 position of the 1,2,4-triazole-5(4H)-thione moiety was not essential for activity; the length of the alkyl chain on position N-4 had influence on the activity and the longer alkyl chain decreased the activity significantly.
Mermer et al. synthesized quinolone-triazole hybrids 30 (Fig. 6) and evaluated for their antibacterial, DNA gyrase and topoisomerase IV inhibitory activities [51]. Among them, compounds 30a and 30b displayed the highest antibacterial activity (MIC: 0.125–8 μg/mL) against S. aureus, Enterococcus faecalis, Escherichia coli, Pseudomonas aeruginosa, Klebsiella pneumoniae and Acinetobacter haemolyticus.
Two set of quinolone triazoles 31 and 32 (Fig. 6) were screened for their antimicrobial activities against a panel of bacterial and fungal strains in which 31d having trifluoromethyl group at phenyl ring (MIC: 1–8 μg/mL) exhibited broader bioactive spectrum against all bacterial strains (Micrococcus luteus, MRSA, S. aureus, P. aeruginosa, E. coli, Shigella dysenteriae and Eberthella typhosa) than norfloxacin and chloromycin. Compound 31b exhibited excellent antifungal activities against A. flavus, C. albicans and B. yeast (MIC: 0.5, 2 and 4 μg/mL, respectively) in comparison with fluconazole (MIC: 256, 1 and 16 μg/mL, respectively) [52].
Triazole-fused fluoroquinolones 33 with a functional Mannich-base moiety at the C-8 position (Fig. 6) exhibited considerable antibacterial activities [53]. Nalidixic acid based 1,2,4-triazolo[3,4-b] [1,3,4]thiadiazole derivatives 34 were evaluated for their antimicrobial activity against two Gram-positive bacteria (S. aureus and Bacillus subtilis), three Gram-negative bacteria (P. aeruginosa, E. coli and K. pneumoniae) and two fungi (A. niger and F. oxysporum) by Aggarwal et al. [54]. SAR study revealed that compound 34b with MIC of 16 μg/mL was found to possess comparable antibacterial properties to streptomycin (MIC: 2–15 μg/mL) against all tested microorganisms, while 34e with nitro on phenyl was detrimental to the activity (Fig. 6).
Antimicrobial activity of 1,2,4-triazole-naphthyridinone hybrids 35 and 36 as structural surrogates of nalidixic acid (Fig. 6) against resistant strains of Gram-positive, Gram-negative and Mycobacterium phlei indicated that hybrids 35a, 35f, 35g, 36a and 36d (MIC: 3.68–5.30 μM/mL) showed remarkable selectivity against B. subtilis, which was resistant to nalidixic acid [55]. Further study revealed that the compounds 35c and 36d (IC50: 3.67 and 3.21 μg/mL, respectively) elicited more potent inhibitory activity against E. coli DNA gyrase.
Prakash et al. synthesized dihydroindeno and indeno[1,2-e] [1,2,4]triazolo[3,4-b] [1,3,4]thiadizines (37 and 38) (Fig. 7 ) and profiled them for their antibacterial activity against S. aureus, B. subtilis, E. coli and P. aeruginosa and antifungal activity against two fungal strains namely, A. niger and A. flavus [56]. Compounds 37g, 37i and 37k showed most potent inhibitory effect (MIC: 2–32 μg/mL) on tested bacteria. Moreover, compounds 37a-l possessed more potent antibacterial activity than compounds 38a-l.
Fig. 7.

SAR and antibacterial activity studies of fused 1,2,4-triazole derivatives.
1,2,4-Triazolo[3,4-b] [1,3,4]thiadiazines 39 (Fig. 7) were screened for their antibacterial activity against S. aureus, E. coli, P. aeruginosa and Bacillus cereus bacterial strains by Sumangala et al. [57]. Among the tested compounds, 39c and 39h (MIC: 3.125 μg/mL) showed excellent antibacterial activity against E. coli and P. aeruginosa, respectively. 1,2,4-Triazolo[3,4-b] [1,3,4]thiadiazine derivatives 40 (Fig. 7) at concentration 100 μg/mL exhibited moderate to good antibacterial activity against four human pathogenic bacteria (E. coli, K. pneumonia, S. dysenteriae and Shigella flexnei) [58]. Among them, compound 40d with zone of inhibition more than standard neomycin and equal to streptomycin demonstrated potential inhibitory activities against all the bacteria.
Thiourea derivatives 41 having triazolopyrimidines core (Fig. 7) demonstrated moderate to high antimicrobial activities against various bacteria such as S. aureus, B. subtilis, P. aeruginosa and E. coli and fungi such as A. fumigatus, Geotrichum candidum, C. albicans and Syncephalastrum racemosum [59]. 1,2,4-Triazolo[1,5-a]pyrimidines containing quinazoline thioether moiety 42 (Fig. 7) possessed significant activities against the tested phytopathogenic bacteria, among which compound 42a was found to be most active and it was 12-fold more potent against Xanthomonas oryzae pv. oryzae with EC50 value of 7.2 μg/mL than bismerthiazol (EC50: 89.8 μg/mL) [60].
In vitro antibacterial activity of 1,2,4-triazolo[3,4-b] [1,3,4]thiadiazoles 43a-h (Fig. 7) indicated high activity towards both drug-sensitive and drug-resistant Gram-positive bacteria, which was up to 16 times more than ampicillin [61]. Thiouracil derivatives containing a triazolo-thiadiazole moiety 44a-l (Fig. 7) displayed good to potent activity against Bacillus amyloliquefaciens, S. aureus and B. subtilis [62]. Interestingly, compound 44d exhibited inhibitory activity against SecA ATPase.
Barbuceanu et al. reported the synthesis and antibacterial activity of mercapto-1,2,4-triazoles bearing diphenylsulfone 45 against S. aureus, B. cereus, E. coli, Enterobacter cloacae, Acinetobacter baumannii and P. aeruginosa [63]. Among them, one of the compounds having bromo diphenylsulfone moiety at position-3 and 3,4,5-trimethoxyphenyl fragment at the nitrogen atom N-4 of triazole ring, exhibited the strongest action against B. cereus (MIC: 8 μg/mL) (Fig. 8 ).
Fig. 8.

Mercapto/thione/thio-substituted 1,2,4-triazole derivatives with antibacterial activity.
A series of Schiff bases of 1,2,4-triazole 46–47 (Fig. 8) were synthesized and evaluated for in vitro antimicrobial potential against bacteria (S. aureus, B. subtilis, E. coli and P. aeruginosa) and fungus (C. albicans) by Mange et al. [64]. All synthesized compounds 46–47 (MIC: 3.125 μg/mL) were equipotent with standard drug ceftriaxone against S. aureus whereas compounds 46a and 47d were more potent (MIC: 3.125 μg/mL) than ceftriaxone against C. albicans.
A series of 5-(2-aminothiazol-4-yl)-4-substituted phenyl-4H-1,2,4-triazole-3-thiols 48 and 49 (Fig. 8) were synthesized and assessed for their antibacterial activity against S. aureus, B. subtilis, E. coli and P. aeuroginosa by Hassan et al. [65]. SAR indicated that the compound 48g having phenoxy moiety at para-postion of the phenyl ring exhibited broad spectrum antibacterial activity (MIC: 0.5–1 μM) which was comparable to gentamicin and ciprofloxacin.
Yang and Bao synthesized 1,2,4-triazole derivatives 50 bearing quinazolinylpiperidinyl moiety and N-(substituted phenyl)acetamide unit (Fig. 8) and evaluated them for their antimicrobial activities [66]. Compounds 50e, 50g, 50i, 50l and 50n (EC50: 34.5–47.5 μg/mL) had better bactericidal activity than control bismerthiazol (85.6 μg/mL) against phytopathogenic bacterium X. oryzae pv. oryzae. SAR study presented the significance of strongly electron-withdrawing substituents (such as 2,4-di-F, 3-F, 3-NO2, 3-COCH3, 2-NO2, 2-CF3 and 4-COCH3) and their positions on the benzene ring for enhancing antibacterial activity. 1,2,4-Triazole-pyrimidine hybrids 51a and 51b (MIC: 1.8–4.7 μM, Fig. 8) displayed excellent activity against S. aureus and E. coli [67]. Moreover, compounds 51a (MIC: 0.75 μg/mL) and 51b (MIC: 0.43 μg/mL) were found to be 10->1600 fold more effective than most clinically used antibiotics against MRSA strain.
Coumarin-based 1,2,4-triazoles 52 and 53 (Fig. 9 ) were tested for their in vitro antibacterial activity against four Gram-positive (S. aureus, MRSA, B. subtilis and M. luteus) and four Gram-negative bacteria (E. coli, Proteus vulgaris, Salmonella typhi and S. dysenteriae) and antifungal activity against C. albicans, Saccharomyces cerevisiae and A. fumigatus by Shi and Zhau [68]. It was proposed that incorporation of triazole to coumarin enhanced the activity. The SAR of coumarin triazoles 52a-c and 53a-c (MIC: 1–32 μg/mL) indicated that the compounds with alkyl-substituent as spacer were more active than the analogues with aralkyl spacer 52d and 53d (MIC: 32–64 μg/mL). Further, bis-triazoles 53 displayed better antimicrobial activities than mono-triazoles 52.
Fig. 9.

1,2,4-Triazoles derivatives 52–56 with antibacterial activity.
Antimicrobial evaluation of bis-1,2,4-triazole derivatives 54 revealed that triazole derivative with 3,4-dichlorobenzyl group showed more potent antibacterial activity against B. proteus (MIC: 0.5 μg/mL) than standard drugs norfloxacin and chloramphenicol [69]. SAR study showed that dihalobenzyl groups are more helpful for increasing antibacterial and antifungal efficacy in comparison with the monohalobenzyl ones (Fig. 9).
A series of isopropanol-bridged carbazole triazoles 55 (Fig. 9) were evaluated for their antibacterial activity against E. faecalis, S. aureus and E. coli by Zhang et al. [70]. Among them, compound 55a (Fig. 9) exhibited highest potency against E. faecalis (MIC: 2 μg/mL) which might be due to intercalation into DNA. Among the α-triazolyl chalcones 56, compound 56a emerged as a promising candidate which exhibited excellent activity (MIC: 4 μg/mL) against MRSA and M. luteus than chloromycin (Fig. 9) [71].
2.3. Anticancer agents
Anticancer chemotherapeutic agents can exert diverse action mechanisms such as cell cycle arrest, enzyme inhibitors, tubulin modulators, angiogenesis inhibitors, DNA intercalators and groove binders, transcription regulators and gene regulators etc. [72]. A large number of chemical entities having 1,2,4-triazole motifs have emerged as promising anticancer agents such as vorozole, letrozole, and anastrozole.
2.3.1. Enzyme inhibitors
2.3.1.1. Kinase inhibitors
Kinases are a class of enzymes that catalyze activation of many proteins by phosphorylation of mostly serine, threonine, or tyrosine amino acids. Deregulation of kinases may lead to growth of cancer. Kinase inhibitors are being explored as antitumor agents due to their target specific action. PIM kinase family (PIM-1, PIM-2 and PIM-3), a class of serine/threonine kinase, are key molecular targets for the development of selective inhibitors having therapeutics potential in cancer treatment.
Martínez-González et al. reported synthesis of a series of novel triazolo[4,3-b]pyridazin-3-yl-quinoline derivatives 57 (Fig. 10 ) as PIM inhibitors [73]. Lead optimization techniques identified compound 57q as a selective PIM-1/3 inhibitor (IC50: 7 nM/70 nM) and antiproliferative agent against several tumor cells lines with GI50 values of 1.48–25.4 μM.
Fig. 10.

1,2,4-Triazole derivatives as PIM inhibitors.
Han et al. reported synthesis and antiproliferative evaluation of a series of 1,2,4-triazole containing hydrazide-hydrazones 58 (Fig. 11 ) derived from (S)-naproxen [74]. Compound 58a showed best activity with IC50 values of 26.0, 34.5, and 48.8 μM against the prostate cancer cell lines PC-3, DU-145 and LNCaP, respectively. Molecular docking studies of 58a on human methionine aminopeptidase-2 presented H-bonds and halogen interactions (Fig. 11). Molecular mechanism of anticancer potential of 58a in PC-3 cells is revealed by reduction of EGFR, Akt phosphorylation and PI3K phosphorylation.
Fig. 11.

(a) 1,2,4-Triazole derivatives as EGFR inhibitors. (b) 3D interactions of compound 58a with amino acid residues in the catalytic channel of MetAP2 enzyme.
Batran et al. reported VEGFR-2 and p38α MAPK inhibitory activity of pentacyclic coumarinyltriazolopyrimidine derivatives 59a-c (Fig. 12 ) along with antiproliferative activity [75]. Among these, compound 59a was documented to exhibit most potent inhibitory activity against VEGFR-2 (94% inhibition at 117 ng/mL) and anticancer activity against MCF-7 cancer cells with IC50 value of 7.9 μg/mL than tamoxifen (IC50: 8.38 μg/mL). Docking studies showed that compound 59a binds to the active site of VEGFR-2 through H-bonds, arene-cation and hydrophobic interactions.
Fig. 12.

(A) 1,2,4-Triazole derivatives as VEGFR inhibitors. (B) Predicted binding mode of compound 59a in the active site of VEGFR-2 which shows H-bond between C O group and Lys868.
Qin et al. reported synthesis of 2-(4-(2-(dimethylamino)ethyl)-4H-1,2,4-triazol-3-yl)pyridine derivatives 60 and 61 (Fig. 13 ) along with antitumor activity [76]. Compound 60g, displayed higher cytotoxicity against MKN-45, H460 and HT-29 cells with IC50 values of 51, 72 and 130 nM, respectively, which were 45.5, 30.4 and 27.8 folds more potent than sorafenib against these cell lines. SAR study revealed that the dimethylaminoethyl group was essential for high activity.
Fig. 13.

1,2,4-Triazole derivatives as tyrosine kinases inhibitors.
In another study, a series of diarylurea derivatives bearing a triazole moiety 62 (Fig. 13) were evaluated for antitumor activity [77]. The most potent compound 62i exhibited significant inhibition (>80%) of tyrosine kinases including c-Kit, RET and FLT3 and antiproliferative activity against HT-29, H460 and MDA-MB-231 cancer cells, with IC50 values of 0.90, 0.85 and 1.54 μM, respectively. It was more potent than the reference sorafenib (IC50: 2.25–3.37 μM) and also significantly induced apoptosis of HT-29 cells.
Liu et al. synthesized 41 compounds containing 1,2,4-triazolone moiety 63 (Fig. 14 ) and studied their cytotoxic activity [78]. Selected compounds 63a-k exhibited excellent inhibitory activity against c-Met kinase (IC50: 1.57–31.52 nM). Compound 63g showed moderate selectivity (306.03 fold) to VEGFR-2 kinase and significant cytotoxicity against HT-29, H460, A549 and MKN-45 cell lines with IC50 values of 0.08 μM, 0.14 μM, 0.11 μM and 0.031 μM, respectively. Antitumor activity of 63g was 1.1–2.3 folds higher than foretinib. SAR studies showed that the introduction of electron-withdrawing groups on the terminal phenyl rings enhanced the antitumor activity.
Fig. 14.

1,2,4-Triazole derivatives as c-MET kinase inhibitors.
Xu et al. synthesized a novel [1,2,4]triazolo[4,3-b][1,2,4,5]tetrazine derivative 64 (Fig. 14) which was found to be potent antiproliferative agent (IC50: 1.30 μM for Bewo, 1.45 μM for HL-60 and 2.24 μM for MCF-7) and inhibited c-Met kinase (IC50: 11.77 μM) [79]. The docking analysis rationalized the binding of compound 64 to c-Met kinase through three hydrogen bonding interactions.
Egile et al. reported the triazolopyridazine derivative 65 SAR125844 (Fig. 14) which strongly inhibited the kinase activity of wild-type MET enzyme with IC50 value of 4.2 nmol/L, as well the H1094Y, Y1235D, M1250T, L1195V, and D1228H kinase domain mutants with IC50 values of 0.22, 1.7, 6.5, 65, and 81 nmol/L, respectively [80]. It also inhibited the growth of tumor in MET-amplified xenograft model, autophosphorylation of AXL and cell proliferation of TPM-NTRK1-overexpressing KM12 cell line with IC50 values of 110 and 1400 nmol/L, respectively.
Zhan et al. synthesized a series of CH2-/CF2-linked triazolotriazine derivatives among which compound 66 (Fig. 14) displayed the most potent inhibition with IC50 value of 0.24 nM against c-Met kinase and with IC50 value of 0.85 nM against EBC-1 cancer cell line [81]. Further, compound 66 exhibited excellent in vivo efficacy with 97.1% of tumor growth inhibition in EBC-1 xenograft mice model at dose of 25 mg/kg. X-ray crystallography revealed that compound 66 binds at the ATP-binding site of c-Met with a U shape.
Compound 67 (AMG 337) (Fig. 14) is identified as selective inhibitor of c-Met kinase (IC50: 1 nM) which displayed exquisite selectivity profile over 402 kinases and sustained inhibition of MET phosphorylation in a mouse liver pharmacodynamic model [82]. Moreover, AMG 337 at dose of 3 and 10 mg/kg exhibited >90% tumor growth inhibition in the NIH-3T3/TPR-Met xenograft model.
Gu et al. synthesized a series of 2-substituted-4-(2-fluorophenoxy)pyridine derivatives 68 (Fig. 14) bearing pyrazolone and triazole moieties as dual c-Met/VEGFR-2 inhibitors [83]. Compound 68d showed the most potent inhibition with IC50 values of 0.11 μM and 0.19 μM for c-Met and VEGFR-2, respectively. Various 8-fluorotriazolopyridines/triazolo[4,3-b]pyridazine derivatives were synthesized as inhibitors of c-Met activity [84,85].
2.3.1.2. Thymidine phosphorylase inhibitors
Shahzad et al. synthesized a series of 3-mercapto-1,2,4-triazole analogues 69 and 3-mercapto-1,2,4-triazole carboxylic acids 70 (Fig. 15 ) as thymidine phosphorylase (TP) inhibitors [86]. Compounds 70b-g revealed a good inhibitory potential with IC50 in the range of 43.86–163.43 μM and angiogenic potential of compound 70c was elicited using the chick chorionic allantoic membrane (CAM) assay.
Fig. 15.

1,2,4-Triazole derivatives as thymidine phosphorylase inhibitors.
Various synthesized 1,2,4-triazolo[1,5-a] [1,3,5]triazine derivatives were evaluated for their inhibitory effects on TP by Bera et al. [87]. Compounds 71 (IC50: 10.84 μM) and 72 (IC50: 2.95 μM) (Fig. 15) displayed the most promising activity as mixed-type inhibitors of TP.
2.3.1.3. Topoisomerase inhibitors
Eissa et al. synthesized two set of triazoloquinoxalines 73 and 74 (Fig. 16 ) and studied their cytotoxic activity against HepG2, Hep-2, and Caco-2 cancer cell lines [88]. Most promising compound 73d significantly induced apoptosis in HepG2 cells via downregulating the Bcl-2 levels and arrested G2/M cell cycle. Results also indicated that compounds 73d and 73e exhibited potent topoisomerase II inhibitory activity (IC50: 0.97 and 1.10 μM, respectively).
Fig. 16.

1,2,4-Triazole derivatives as topoisomerase inhibitors.
Ibrahim et al. synthesized new series of 1,2,4-triazolo[4,3-a]quinoxaline 75 and bis 1,2,4-triazolo[4,3-a:3′,4′-c]quinoxaline derivatives 76 (Fig. 16) and evaluated their inhibitory effects on topoisomerase II and cytotoxic effects against HepG2, Hep-2 and Caco-2 [89]. SAR indicated that bis 1,2,4-triazolo[4,3-a:3′,4′-c]quinoxaline derivatives 76a, 76g, and 76h improved the activity than 1,2,4-triazolo[4,3-a]quinoxaline derivatives 75. Compounds 75f-h, 76a, 76g, and 76h displayed good topoisomerase-II inhibitory activity (IC50: 0.68–1.22 μM) and induced DNA intercalation significantly. Treatment of Caco-2 cells with 76g induced apoptosis and resulted in G2/M cell cycle arrest.
2.3.1.4. Methionine aminopeptidase type II inhibitors
Hou et al. synthesized 1,2,4-triazole derivatives containing 1,4-benzodioxane fragment 77 (Fig. 17 ) and evaluated their methionine aminopeptidase type II (MetAP2) inhibitory activity in an enzyme assay [90]. From biological study of tested compounds it was observed that most of the compounds exhibited potent MetAP2 inhibitory effect and 77k most effectively inhibited the growth of HepG2 cells and MetAP2.
Fig. 17.

1,2,4-Triazole derivatives as methionine aminopeptidase type II inhibitors.
2.3.1.5. COX inhibitors
Cui et al. explored diaryl-1,2,4-triazole-caffeic acid hybrids as COX-2/5-LOX dual inhibitors for cancer therapy [91]. The anticancer SAR of hybrids 78 (Fig. 18 ) indicated that amide derivatives 78e-g with superior COX-2 inhibition activities were less potent (IC50: 16.37–26.14 μM) than ester derivatives (78d, IC50: 9.52–11.16 μM) against A549, Caco-2, PC-3 and B16–F10 cancer cell lines. Introduction of electron-withdrawing groups at para-position of N-1 phenyl ring (R1) improved the antiproliferative activity. Most potent compound 78j (IC50: 6.78–9.05 μM) also demonstrated significant inhibition on tumor growth in vivo. The preliminary mechanism studies revealed that hybrid 78d arrested the cell cycle in G2 phase and induced apoptosis in A549 cells in a dose-dependent manner.
Fig. 18.

1,2,4-Triazole derivatives as COX-2 inhibitors.
A series of non-carboxylic naproxen analogues, bearing triazole ring 79–81 (Fig. 18) was synthesized by El-Husseiny et al., among which arylidene derivatives 81b-c exhibited potent antitumor activities against cell lines MCF-7, MDA-231, HeLa, and HCT-116, with IC50 in the range of 4.83–12.07 μM [92]. Compound 81c also exhibited the most potent COX-2 inhibitory activity with IC50 value of 0.40 μM and selectivity index (SI) value of >62.50 and showed strong interactions at the COX-2 binding site.
Sever et al. studied cytotoxic effects of 1,2,4-triazolo[3,4-b] [1,3,4]thiadiazine derivatives 82 (Fig. 18) against T98 human glioma cell line [93]. Study revealed that the most potent compound 82h exhibited dose-dependent anticancer effect via inhibition of COX-2 mRNA levels and similar binding pattern as indomethacin in active site of COX-2 enzyme.
2.3.1.6. Carbonic anhydrase inhibitors
SitaRam et al. synthesized a series of novel benzenesulfonamide bearing 1,2,4-triazole scaffolds 83–85 (Fig. 19 ) and studied their inhibitory activity against four isomers of the α-class of carbonic anhydrases (CAs, EC 4.2.1.1), comprising hCAs I and II (cytosolic, ubiquitous isozymes) and hCAs IX and XII (transmembrane, tumor associated isozymes) [94]. Compounds 83d, 83f and 84f displayed excellent inhibitory potential against all of the four isozymes hCA I, II, IX and XII with Ki values in the range of 2.8–170 nM, 1.3–132 nM and 3–89 nM, respectively even better than the standard drug acetazolamide (Ki: 5.7–250 nM).
Fig. 19.

1,2,4-Triazole derivatives as carbonic anhydrase inhibitors.
2.3.1.7. Aromatase inhibitors
Song et al. synthesized 4-N-nitrophenyl substituted amino-4H-1,2,4-triazole derivatives 86 (Fig. 20 ) as aromatase inhibitors [95]. SAR study revealed that the compounds containing substituted benzyl group on amine have improved aromatase inhibitory activities. Compound 86g was the most active one with an IC50 of 9.02 nM.
Fig. 20.

1,2,4-Triazole derivatives as aromatase inhibitors.
2.3.1.8. Lysine-specific histone demethylase 1 (LSD1/KDM1A) inhibitors
Wang et al. designed and synthesized pyrazolo[1,5-a]pyrimidine derivatives as potent LSD1/KDM1A inhibitors [96]. Compounds 87a-c and 88a-b (Fig. 21 ) selectively inhibited growth of A549 cells with IC50 in the range of 3.23–10.58 μM. Compounds 87d and 87e were highly potent inhibitor of LSD1 (IC50: 0.154 and 1.19 μM, respectively). Further, compound 87d significantly inhibited migration of A549 and PC-9 cells in a concentration-dependent manner. Following this work, Wang et al. designed new LSD1 inhibitors 89 (Fig. 21). From series, compound 89a having selectivity over MAO-A/B, reversibly inhibited LSD1 (IC50: 1.72 μM) and significantly inhibited migration of A549 cells. Docking studies presented that 89a displayed FAD-competitive binding toward LSD1 (Fig. 21) [97].
Fig. 21.

1,2,4-Triazole derivatives as LSD1/KDM1A inhibitors and binding of compound 89a and FAD (colored in green and yellow, respectively) in the active site of LSD1. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
2.3.1.9. Tankyrases (TNKSs) inhibitors
Liscio et al. designed and synthesized a series of 6,8-disubstituted triazolo[4,3-b]pyridazines 90 (Fig. 22 ) as tankyrases (TNKSs) inhibitors [98]. SAR study revealed that one of the compounds (R1 = CH3 and R2= (CH2)2-Ph-4-OH) showed full inhibition of TNKS-1 and 82% of TNKS-2 at 1 μM. Replacement of hydroxyl group by benzoyl or amine resulted in the loss of activity. All the derivatives bearing 4-hydoxyphenyl in the side chain, were found to be the most potent TNKS inhibitor and assessed further at a concentration of 10 μM on several members of the PARP superfamily (PARP 1–3, 6–8, 10–12), exhibiting clean selectivity toward PARP-1 and 2 compared with AZD22816 (Olaparib).
Fig. 22.

1,2,4-Triazole derivatives as TNKS inhibitors.
2.3.2. Transcription regulators and gene regulators
Bromodomain-containing protein 4 (BRD4), a transcriptional and epigenetic regulator, recognises acetylated lysine residues in histones and has been emerged as key target for cancer therapy. A series of 4,5-dihydro-[1,2,4]triazolo[4,3-f]pteridine derivatives 91 (Fig. 23 ) were designed and synthesized as BRD4 inhibitors by Bi et al. [99], among which the most potent compound 91r exhibited antiproliferative activity against MV4; 11 (biphenotypic B myelomonocytic leukemia) with an IC50 of 1.53 μM through inducing apoptosis by downregulating c-Myc.
Fig. 23.

1,2,4-Triazole derivatives as BRD4 inhibitors.
2.3.3. Tubulin modulators
Tubulin and microtubules are prime molecular targets for cancer chemotherapy which play fundamental role in mitosis and cell division. Saez-Calvo et al. reported anti-mitotic effect of 1,2,4-triazolo[1,5-a]pyrimidines 92 against A549 lung carcinoma cells [100]. It was unveiled that compounds act as vinca-site microtubule-stabilizing agents that mediate longitudinal tubulin contacts and are not affected by p-glycoprotein overexpression. Binding of compound 92a to the vinblastine site is close to the bound GDP nucleotide of the β1-tubulin subunit as shown in Fig. 24 .
Fig. 24.

1,2,4-Triazole derivatives 92–99 as tubulin polymerization inhibitors.
Alswah et al. designed and synthesized novel chalcone derivatives bearing triazolo[4,3-a]quinoxaline moiety 93 (Fig. 24) as antiproliferative agents with dual inhibitory activity on EGFR kinase and tubulin polymerization effects [101]. Compound 93g was the most active against MCF-7, HCT-116 and HepG2 cell lines with IC50 value of 1.65, 3.61 and 8.58 μM, respectively. Molecular docking analysis of 93g demonstrated diverse interactions in the colchicine binding pocket of tubulin. Triazoloquinazolinone 94a (Fig. 24) showed potential tubulin polymerization inhibitory activity (IC50: 0.15 μM) and exhibited cytotoxic activity against human cancer cell lines panel including on HL-60(TB), NCI–H522, MDA-MD-435 and OVCAR-3 with GI50 values in the nanomolar range [102]. Molecular docking studies indicated that N-methylated amide group in compound 94a could form hydrophobic contact with Leu248, which was responsible for its potent antitubulin activity.
El-Sherief et al. synthesized new 1,2,4-triazole scaffolds 95–99 (Fig. 24) and most of the tested compounds exhibited noteworthy antiproliferative effects against a panel of cancer cell lines with IC50 values < 2.0 μM [103]. SAR studies revealed that compounds 95–97 bearing free NH2 group at triazole ring were more effective than compounds 98 and 99 in which 5-amino group was substituted with N-acyl and isothiocyanate, respectively. Mechanistic study against Tubulin, EGFR and BRAFV600E kinase enzymes showed that two compounds 95c and 95d have a capability to strongly inhibit tublin (957 and 872, respectively), EGFR (IC50: 3.6 and 4.6 μM, respectively), and BRAFV600E (IC50: 1.9 and 1.8 μM, respectively).
Yang et al. synthesized triazolylthioacetamides containing 3,4,5-trimethoxyphenyl moiety 100 (Fig. 25 ) and ten selected compounds were evaluated as tubulin polymerization inhibitors [104]. Compounds 100c and 100f displayed most promising anticancer activity against MCF-7, HeLa and HT-29 cell lines with IC50 in the range 0.05–26.83 μM. SAR studies indicated that the substitution of N-4 and the N-substituted acetamide moiety at 3-position on the 1,2,4-triazole ring have considerable role in potency. Compound 100f could induce significant cell cycle arrest at the G2/M phase in HeLa cell lines and have antitubulin activity with an IC50 value of 5.9 μM.
Fig. 25.

1,2,4-Triazole derivatives 100–104 as tubulin polymerization inhibitors.
Mustafa et al. synthesized new combretastatin A4 analogues containing 1,2,4-triazole 101–102 (Fig. 25) and evaluated for their anticancer activity against different cancer lines including leukemia, lung, colon, CNS, melanoma, ovarian, renal, prostate and breast cancers [105]. Compounds 101a, 102a, and 102c showed the highest promising anticancer activities and compound 102c arrested cell cycle at G2/M phase in HepG2 cells. Selected compounds 101a, 101b, 101e, 102a and 102c also displayed in vitro tubulin polymerization inhibitory activity displaying almost similar binding feature towards tubulin as CA-4.
Romagnoli et al. synthesized a series of regioisomeric 1,5-diaryl-1,2,4-triazole derivatives 103 (Fig. 25) [106]. Among them, compounds 103e (IC50: 5–100 nM) and 103h (IC50: 3–20 nM) were found to have highest antiproliferative activity against six tumor cell lines namely HeLa, A549, HL-60, Jurkat, K562 and MCF-7. SAR study revealed the significance of the substituent pattern on the phenyl ring at the 5-position of the 1,2,4-triazole ring on inhibition of tubulin polymerization and antiproliferative activities. Compounds 103e and 103h induced arrest in G2/M phase in Jurkat cells and induced apoptosis by activating caspase-3 and downregulating Bcl-2.
1-(3′,4′,5′-Trimethoxybenzoyl)-5-amino-1,2,4-triazoles 104 were evaluated for their anticancer effect against five human cancer cell lines, Jurkat, RS4; 11, HeLa, HT29 and MCF-7 [107]. SAR study revealed the effects of different substituents and their position on the phenyl ring on antiproliferative activity. Only four compounds 104a-d (Fig. 25) exhibited potent antiproliferative activity (IC50 < 1 μM) against selected cancer cells. Compounds 104b and 104c act as more potent inhibitors of tubulin polymerization with IC50 value of 0.66 μM and 0.97 μM, respectively than CA-4 (IC50: 1.2 μM).
2.3.4. Antiproliferatives
Wang et al. synthesized a series of [1,2,4]triazolo[1,5-a]pyridinylpyridines 105–106 (Fig. 26 ) and studied their anticancer activities against three human cancer cell lines- HCT-116, U-87 MG and MCF-7 [108]. Among the tested series, compound 105d (IC50: 0.84–1.82 μM) and 106d (IC50: 0.82–1.77 μM) exhibited potential activity and could inhibit the PI3K/AKT/mTOR pathway. Compound 105d also exhibited in vivo inhibitory effect on tumor growth in mice bearing sarcoma S-180 model.
Fig. 26.

Fused 1,2,4-triazole derivatives 105–113 as anticancer agents.
Xu et al. carried out three dimensional quantitative structure-activity relationship (3D-QSAR) on [1,2,4]triazolo[4,3-b][1,2,4,5]tetrazine derivatives with antitumor activities against MCF-7 cell [109]. The results of CoMFA (q2: 0.716, r2: 0.985) and CoMSIA (q2: 0.723, r2: 0.976) generated models with good predictive abilities. Compounds 107 and 108 (Fig. 26) showed significant potency against MCF-7, Bewo and HL-60 cells with IC50 values in 0.63–13.12 μM.
Fares et al. synthesized a series of pyrido[2,3-d] [1,2,4]triazolo[4,3-a]pyrimidines 109–111 (Fig. 26) and studied their in-vitro antiproliferative activities against PC-3 and A549 cell lines using the Sulfo-rhodamine B (SRB) colorimetric assay [110]. SAR studies of 3-un/substituted derivatives 109–110 revealed that lipophilic group (thio and methyl) at C-3 position on the 1,2,4-triazole ring significantly diminished antitumor activity. Among 1,3-disubstituted triazolo derivatives 111, compounds 111a, 111c, 111d and 111f with acetyl moiety at 3-position of 1,2,4-triazole ring were more potent than corresponding analogues 111g-j having 3-ethyl carboxylate moiety and introduction of sulphonamide group on N-1 phenyl ring increased the activity. Mechanistic study revealed that compound 111f exhibited good profile as apoptosis inducer via caspase-3 dependent pathway and arrested cell cycle at G1 phase in PC-3 cells line.
A series of novel 7-amino- [1,2,4]triazolo[4,3-f]pteridinone derivatives 112–113 (Fig. 26) was designed, synthesized and evaluated for their antitumor activity by Hou et al. [111]. SAR revealed that the presence of different hydrophilic amino groups on phenyl ring at C-7 position had a significant influence on potency. Of these 28 compounds, compound 113g with 2,6-dimethylpiperazine displayed the most potent antiproliferative activity against A549, PC-3, HCT116, MCF-7 and MDA-MB-231 cell lines with IC50 values of 0.16 μM, 0.30 μM, 0.51 μM, 0.30 μM, and 0.70 μM, respectively. Molecular docking and enzymatic studies demonstrated that compound 113g inhibited PLK1 (86.4%) and cancer cell growth by inducing a great decrease in mitochondrial membrane potential leading to apoptosis and arresting G1 phase of A549 cells.
Kandeel et al. synthesized compounds 114 containing both chromenes and triazolopyrimidine moieties (Fig. 27 ) and evaluated their cytotoxic activity (IC50: 0.007–0.039 μM) against MCF-7 cell line [112]. Most active compounds 114c (IC50: 0.007 μM), 114g and 114h (each having IC50: 0.008 μM) displayed 1.5–2 folds superior activity than that of colchicine (IC50: 0.013 μM). Further, anticancer activity of thieno[3,2-e]triazolo[4,3-c]pyrimidine derivatives 115 (Fig. 27) was evaluated against a panel of 59 human tumor cell lines, representing leukemia, melanoma and cancers of lung, colon, central nervous system (CNS), ovary, kidney, prostate as well as breast [113]. Among them, compound 115c endowed with broad spectrum anticancer activity (GI50: 0.495–5.57 μM) against 56 human cancer cell lines was highly selective against T-47D and MDA-MB-468 cell lines with GI50 0.495 and 0.568 μM, respectively. Molecular mechanisms illustrated that compound 115c could induce cell cycle arrest at G2/M phase and show accumulation of cells in pre-G1 phase in MDA-MB-468 cell line.
Fig. 27.

Fused 1,2,4-triazole derivatives 114–122 as anticancer agents.
Botros et al. synthesized of a series of substituted benzothieno[3,2-e][1,2,4]triazolo[4,3-a]pyrimidines 116–119 (Fig. 27) and some selected compounds were screened for their in vitro cytotoxic activity against two human cancer cell lines, PC-3 and HCT-116 [114]. Two compounds 116l and 119c were found to be the most active against HCT-116 cell line with IC50 values of 6.56 and 6.12 μM as compared to doxorubicin (IC50: 15.82 μM) and one of the compound 117c (IC50: 5.48 μM) showed highest activity against PC-3 cell line. SAR study illustrated the significance of phenylpiperazine moiety (R) and extending side chain (X) on bioactivity.
Recently our group synthesized a series of 6-chloro-3-substituted-[1,2,4]triazolo[4,3-b]pyridazines 120 (Fig. 27) and evaluated them for their antitumor activities [115]. Among the tested series, three compounds 120a-c exhibited potential activity and 2–9 folds selectivity against SB-ALL and NALM-6 cell lines compared to MCF-7 cells. Further, these compounds efficiently induced apoptosis of NALM-6 cells via caspase 3/7 activation. SAR study revealed the significance of para-substituted phenyl group over ortho- and meta-derivative.
Issa et al. reported 1,2,4-triazolo[4,3-a]quinoxalines derivatives 121 (Fig. 27) as antiproliferative agents [116]. Among these, the most active compound 121b was further evaluated against 60 human cell lines and exhibited significant antitumor activity against leukemia SR, non-small cell lung cancer HOP-92, NCI–H460, colon cancer HCT-116, HCT-15, CNS cancer U251, melanoma LOX IMVI, renal cancer A498, prostate cancer PC-3, and breast cancer MDA-MB-468 cell lines (GI50: 3.91, 3.45, 3.49, 3.21, 1.96, 5.18, 3.69, 1.80, 5.19, and 5.55 μM, respectively). Several 6-aryl-benzo[H] [1,2,4]-triazolo[5,1-b]quinazoline-7,8-diones 122 (Fig. 27) were screened for their cytotoxicity against human gastric carcinoma cell line SCG7901 and hepatoma cell line HepG2 by Wu et al. [117]. SAR study revealed that ortho-quinone moiety and presence of electron-rich aromatic ring at the C-6 position improved the cytotoxicity.
Xue et al. synthesized a series of 1,2,4-triazolo[3,4-a]phthalazine derivatives 123 (Fig. 28 ) and studied their cytotoxic activities (IC50: 1.7–124.5 μM) against four human cancer cell lines (MGC-803, EC9706, HeLa and MCF-7) [118]. Among the series, the most active compound 123h was more potent than 5-fluorouracil and exhibited cytotoxicity via induction of apoptosis in EC-9706 cells.
Fig. 28.

Fused 1,2,4-triazole derivatives 123–126 as anticancer agents.
Husain et al. have screened a series of triazolothiadiazoles 124 and triazolothiadiazines 125 (Fig. 28) for their in vitro anticancer activity for 60 cell line panel representing full nine human systems as leukemia, melanoma and cancers of lung, colon, brain, breast, ovary, kidney and prostate [119]. One compound, 124i exhibited remarkable anticancer activity against all tested cell lines (IC50: 0.20–2.58 μM) and emerged as promising lead with broad spectrum of anticancer activities against tumor cell lines.
Hu et al. synthesized novel 2,4-diaminopyrimidine derivatives possessing triazolopiperazine scaffolds 126 (Fig. 28) and screened them against a panel of kinases (CDK4, JAK2, VEGFR2, PI3Kα and FLT3) and four tumor cell lines [120]. Among them, compound 126k exhibited most potent antitumor activities against A549, HCT-116, PC-3 and MCF-7 cell lines with IC50 values of 2.14 μM, 3.59 μM, 5.52 μM and 3.69 μM, respectively. Significance of R group on phenyl ring and amino groups (R1) of terminal aniline on the pyrimidine core is shown in Fig. 28. Furthermore, mechanistic studies revealed that compound 126k could suppress the migration of tumor cells, induce apoptosis, and prolonged the A549 cell cycle distribution, representing blockage at the G2-M phase and accumulation at the S phase.
Zhang et al. designed a series of 1,2,4-triazolo[3,4-b] [1,3,4]thiadiazines 127 containing furan and thiophene nuclei (Fig. 29 ) and evaluated them for antitumor activity [121]. Among the series, compound 127a presented eleven-, three-, and two-fold improvement compared to positive control fluorouracil in inhibiting HepG2, PC-3, and A549 cell proliferation with IC50 values of 5.09, 3.70 and 12.74 μM, respectively and arrested G2/M cell cycle in PC-3 cells.
Fig. 29.

Fused 1,2,4-triazole derivatives 127–132 as anticancer agents.
Zhao et al. have reported the synthesis and anticancer activity of 3,4-disubstituted-5-(3,4,5-trimethoxyphenyl)-4H-1,2,4-triazoles 128 and some novel 5,6-dihydro-[1,2,4]triazolo[3,4-b] [1,3,4]thiadiazoles 129 (Fig. 29) [122]. Among them, compound 129b showed promising activity against HepG2 cell line with an IC50 value of 0.58 μM, whereas compound 129c (IC50: 3.17–13.79 μM) exhibited broad spectrum of antitumor activity against HepG2, MCF-7 and MKN45. SAR studies revealed that 1,2,4-triazoles fused with thiadiazole ring 129 were highly potent as compared to 1,2,4-triazole derivatives 128 containing iminoacyl moiety on the 4-position.
Kamel and Abdo have reported the synthesis and anticancer activity of a series of N-substituted-3-mercapto-1,2,4-triazoles 130, triazolo[3,4-b][1,3,4]thiadiazoles 131, and triazolo[3,4-b][1,3,4]thiadiazines 132 (Fig. 29) against six human cancer cell lines gastric cancer (NUGC), human colon cancer (DLD1), human liver cancer (HA22T and HepG2), nasopharyngeal carcinoma (HONE1), human breast cancer (MCF) and normal fibroblast cells (WI38) [123]. Seven of the tested compounds (130a, 130b, 131d, 131g-i and 132a) showed remarkable activity with IC50 values < 800 nM. Compound 131d displayed equivalent cytotoxic effect to the standard CHS 828 against gastric cancer cell line.
Pharmacological evaluation of synthesized 2-(4H-1,2,4-triazole-3-ylthio)acetamide derivatives 133 (Fig. 30 ) was carried out against the full panel of 60 human cancer cell lines. Results demonstrated that compounds 133a, 133b and 133c exhibited antiproliferative activity against PC-3 cells (IC50: 5.96 μM), A549/ATCC cells (IC50: 7.90 μM) and K-562 cells (IC50: 7.71 μM), respectively [124]. Compounds 133a-c revealed significant increase in caspase-3 activity in a dose-dependent manner and decreased the mitochondrial membrane potential and the expression of Bcl-2.
Fig. 30.

Thio-substituted 1,2,4-triazole derivatives as anticancer agents.
Zhao et al. synthesized a series of isoindoline-1,3-diones containing 1,2,4-triazole moiety and three representative compounds 134a-c (Fig. 30) exhibited more potent antitumor activities against four human cancer cell lines (HepG2, A549, PC-3M and MKN45) than the reference 5-fluorouracil [125]. Notably, the flow-activated cell sorting analysis revealed that compound 134b dose-dependently inhibit the proliferation of HepG2 cells via inducing apoptosis. Further, a series of novel 3-alkylsulfanyl-4-amino-1,2,4-triazoles 135 (Fig. 30) was designed and evaluated for antitumor activity [126]. Compound 135d was found to have the highest potency with IC50 values of 0.37, 2.94 and 31.31 μM against HCT116, HeLa and PC-3, respectively. Mechanistic studies demonstrated that it not only induces cell cycle arrest in a dose-dependent manner in HeLa cells at G2/M phase but also induced apoptosis.
Wang et al. synthesized nonsymmetrical disulfides bearing 1,2,4-triazole moiety 136–137 (Fig. 30) and evaluated their antiproliferative activity against human cancer cell lines SMMC-7721, HeLa, A549, and normal cell lines L929 by CCK-8 assay [127]. Most of the tested compounds exhibited better activity than positive control 5-fluorouracil. Compound 136d exhibited the best inhibition against A549 cells (IC50: 2.79 μM) and 137c was found to be the most potent against SMMC-7721 cells (IC50: 2.97 μM).
Tokala et al. reported that out of twenty five 1,2,4-triazole-linked urea and thiourea conjugates screened for anticancer activity against five cancer cell lines, compounds 138a-h (Fig. 30) displayed good cytotoxicity (IC50: <50 μM) against breast (MCF-7, MDA-MB-231), lung (A549) prostate (DU-145) and one mouse melanoma (B16–F10) cell line [128]. SAR revealed that the thiourea congeners 138d-h were comparatively more potent than the urea derivatives 138a-c. Compound 138g (IC50: 4.51–11.75 μM) was found to have significant activity against all cell lines and was more potent than 5-fluorouracil. Moreover, compound 138g induced apoptosis of MCF-7 cells, inhibited colony formation in MCF-7 cells and arrested tumor cell cycle at the G0/G1 phase.
Mavrova et al. synthesized thieno[2,3-d]pyrimidin-4(3H)-ones containing 1,2,4-triazoles 139 (Fig. 30) and evaluated for their cytotoxicity against four human cancer cell lines (HT-29, MDA-MB-231, HeLa, HepG2) and normal diploid cell (Lep3) [129]. Amongst them, compound 139c with IC50 9.5 X 10−4 μM was found to be most toxic to HeLa cells.
Boraei et al. designed, synthesized and evaluated indolyl-triazole hybrids 140–142 (Fig. 31 ) for antitumor activity [130]. N-unsubstituted triazole 140 showed higher activity with IC50 of 3.58 μg/mL and 4.53 μg/mL against HepG2 and MCF-7, respectively than corresponding N-2 substituted analogues 141 (10.8->100 μg/mL) and N-1 substituted analogues 142 (11.5–16.6 μg/mL). Hybrid 141b (IC50: >100 μg/mL) demonstrated lower activity than its analogues and positional isomer 142c, implying sugar fragment at N-1 position was deterimental for the activity. Molecular docking analysis rationalized that hybrid 140 inhibit EGFR through the hydrogen bonding with Asp 855 and several interactions with the key amino acids in the EGFR active site (Fig. 31).
Fig. 31.

Thio-substituted 1,2,4-triazole derivatives as anticancer agents. Compound 140 interaction with key residues in the active site of EGFR enzyme.
A series of triazole-pyrazolylcoumarin derivatives 143 (IC50: 0.42–4.54 μm) (Fig. 32 ) displayed growth inhibitory effect against human prostate cancer cell lines LNCaP and PC-3 [131]. Compound 143g exhibited more potent activity as inhibitor of 5α-reductase with ED50 of 0.15 μm than anastrozole (ED50 of 1.09 μm). Antitumor activity of coumarin-triazole hybrids 144 (IC50: 3.1–37.9 μg/mL) was evaluated against four cancer cell lines (BT-20, SK-Mel-128, DU-145 and A549, MTT assay) by Kahveci et al. [132]. Hybrids 144d and f (Fig. 32) showed better selectivity index value (SI: 5.2 and 2.7) against BT-20 cell line than cisplatin (SI: 2).
Fig. 32.

(a) Coumarin-1,2,4-triazole derivatives as anticancer agents. (b) Stereoview of compound 145i docked into the active site of EGFR-TKD which shows two hydrogen binding interaction of oxygen atom of carbonyl group at coumarin ring with hydrogen atom of the amino acid residue Lys721.
Coumarin-3-yl-thiazol-3-yl-1,2,4-triazolin-3-ones 145 (IC50: 0.16–1.12 μM) (Fig. 32) showed promising activity against MDA-MBA-231, A549, K562 and HeLa cancer cell lines [133]. SAR studies reveal that electron-donating group at R1 position and electron-withdrawing group at R2 position highly enhanced the potency as evident in case of compound 145f (IC50: 0.16–0.31 μg/mL). Docking studies of compounds 145i into the active site of EGFR-TKD revealed polar and hydrophobic interactions (Fig. 32).
Among the synthesized 4-(1H-1,2,4-triazol-1-yl)benzoic acid hybrids 146–148 (Fig. 33 ), compounds 148b (IC50: 15.6 μM) and 148c (IC50: 23.9 μM) displayed potent activity against MCF-7 and HCT-116 cancer cell lines, respectively when compared with doxorubicin (IC50: 19.7 and 22.6 μM, respectively) [134]. A mechanistic study illustrated that compounds 146b and 148b induced apoptosis in MCF-7 cells.
Fig. 33.

4-(1H-1,2,4-Triazol-1-yl)benzoic acid hybrids as anticancer agents.
2.4. Anticonvulsant agents
Deng et al. have repoted synthesis of several triazolo[1,5-a]pyrimidin-5(4H)-ones 149 (Fig. 34 ) as anticonvulsant agents [135]. SAR study indicated the significance of position of halogen on phenyl ring on the anticonvulsant activity. Compound 149i displayed most promising activity in maximal electroshock test (MES) with ED50 value of 19.7 mg/kg and protective index (PI) value of 34.8 via inhibiting voltage-gated ion channels and modulating GABAergic activity against several chemically induced seizures.
Fig. 34.

Fused 1,2,4-triazole derivatives as anticonvulsant agents.
Biological assessment of 1,2,4-triazolo[1,5-a]pyrimidinones 150 as new agonists of benzodiazepine receptors indicated that most of the compounds have higher affinity for benzodiazepine binding site in radioligand receptor binding assay than diazepam [136]. Particularly, compound 150c (Fig. 34) with highest binding affinity (Ki: 0.42 nM and IC50: 0.68 nM) exhibited substantial hypnotic and weak anticonvulsant activities with no impairment on learning and memory in vivo.
Anticonvulsant activity of 2,5-disubstituted [1,2,4]-triazolo[1,5-a]pyrimidine-7(4H)-one derivatives 151 (Fig. 34) as positive modulators of GABAA1 was evaluated by MES and pentylenetetrazole (PTZ) and rotarod neurotoxicity test by Huang et al. [137]. Results revealed that compounds 151a and 151b showed significant anticonvulsant activities in PTZ-induced epilepsy model with ED50 values at 31.81 mg/kg and 40.95 mg/kg, respectively. Both compounds displayed higher PI value of 17.22 and 9.09 than four standard drugs.
Several 10-alkoxy-5,6-dihydro-triazolo[4,3-d]benzo[f][1,4]oxazepines 152 and 8-alkoxy-4,5-dihydrobenzo[b][1,2,4]triazolo[4,3-d][1,4]thiazepine derivatives (153 and 154) (Fig. 34) were synthesized and their in vivo anticonvulsant activity were evaluated using MES screens by Deng et al. [138,139]. SAR study of compounds 152 revealed the role of alkyl groups and their size on anticonvulsant activity. Among them, compound 152g (R = n-heptane) was the most potent (ED50: 6.9 mg/kg and PI: 9.5) and exhibited anticonvulsant activity via GABA-modulating mechanisms in sc-PTZ, isoniazid, 3-MP, thiosemicarbazide and Bicuculline induced seizures tests. Compound 154a exhibited promising anti-MES activity with an ED50 of 26.3 mg/kg and a superior PI value of 12.6.
Deng et al. synthesized a set of 6-(substituted-phenyl)thiazolo[3,2-b][1,2,4]triazole derivatives 155 (Fig. 34) to screen their anticonvulsant activity [140]. Results indicated that compound 155c was found to be more selective in MES screen with an ED50 and PI value of 49.1 and 1.9 respectively, while 155n was found to be active in both MES test and PTZ test. In the PTZ screening, compound 155n exhibited an ED50 value 63.4 mg/kg and a TD50 of 105.6 mg/kg, resulting in a high PI value of 1.7 when compared with standard carbamazepine (PI < 0.44). Further, neurotoxicity of the compounds was measured using rotarod test, which indicated that most of the compounds exhibited high level of neurotoxicity.
Cao et al. synthesized a series of 7-alkoxy-2,4-dihydro-1H-benzo[b][1,2,4]triazolo[4,3-d][1,4]-thiazin-1-ones 156 (Fig. 34) and evaluated for their anticonvulsant activity [141]. Compound 156a exhibited significant anticonvulsant activity in MES test with ED50 value of 9.2 mg/kg and PI value of 15.4 which was superior to standard carbamazepine (ED50 and PI values of 11.8 and 6.4, respectively).
Several triazolo[4,3-a]quinazolin-5(4H)-ones 157 and pyrido[3,2-e][1,2,4]triazolo[4,3-a]pyrimidin-5(6H)-ones 158 (Fig. 34) were synthesized as anticonvulsant agents by Zhang et al. [142,143]. Based on the anticonvulsant and neurotoxicity screening data, compounds 157a and 157b showed wide margins of safety with PI value of >25.5 and > 26.0, and significant oral activity against MES-induced seizures in mice with an ED50 of 88.0 and 94.6 mg/kg, respectively. SAR study of compounds 158 revealed that presence of halogen atom (F and Cl) and the position of the halogen atom on the benzyl group influenced the activity and in N-alkyl derivatives the anticonvulsant activity gradually decreased with increase in the alkyl chain length.
Guan et al. have reported synthesis and anticonvulsant activity of 6-alkoxy- [1,2,4]triazolo[4,3-b]pyridazine derivatives 159 (Fig. 34) in which compound 159i showed anticonvulsant activity with median effective dose (ED50) of 17.3 mg/kg and median toxicity dose (TD50) of 380.3 mg/kg, and PI of 22.0 in the anti-MES test [144].
Phenytoin-1,2,4-triazole hydrids 160 (Fig. 35 ) were synthesized and evaluated for their anticonvulsant activity using MES and scPTZ screens in mice by Botros et al. [145]. Hybrids 160b-e, containing aromatic ring at N-4 position of the triazole, displayed higher protection in MES screen against electrically induced seizures than the ethyl substituted analogue 160a at a dose of 100 mg/kg.
Fig. 35.

Substituted triazolthiones with anticonvulsant activity.
Several 1,2,4-triazole-3-thione derivatives having 4-aryl group 161 and 4-alkyl group 162 (Fig. 35) were evaluated for their anticonvulsant activity by Plech et al. [146,147]. The MES and neurotoxicity tests demonstrated that compound 161a with the ED50 of 35.2 mg/kg, TD50 of 136.7 mg/kg and PI of 3.9 possess the most potent activity. Compounds 162a-g showed better activity as compared to standard drug valproate. Results revealed that elongation of alkyl fragment from –C2H5 to –C4H9 at 4-position of 1,2,4-triazole increased the activity approximately 4-fold (from 152 mg/kg to 38.5 mg/kg), due to increase in the lipophilicity of the molecule. Chromatographic tests showed that analogues 162h and 162i with C10 and C12 alkyl chain, respectively lack anticonvulsant effect due to the inability to cross the blood brain barrier (BBB).
To gain more insights into SARs, Plech’s et al. synthesized several 4-alkyl-1,2,4-triazole-3-thiones by replacing the 5-(3-chlorophenyl) group by 5-(3-chlorobenzyl/2,3-dichlorophenyl). In the analogues containing 5-(3-chlorobenzyl) group 163 (Fig. 35), the presence of –CH2- linker improves the potency, time-course profile and safety due to increase in molecule flexibility [148]. Based on the activity and toxicity profile, compound 163d showed the most promising potential as anticonvulsant agent (ED50: 72.1 mg/kg, TD50: >1000 mg/kg and PI: >13.9 after 15 min). Radioligand binding assay indicated that these compounds excluded the possibility of direct or allosteric modulation of GABAA receptors.
Deng et al. have reported the synthesis of some new triazole-containing quinolinones 164 (Fig. 36 ) and screened for their anticonvulsant and antidepressant activity by using MES and forced swimming test (FST) [149]. Compound 164a exhibited most potent antidepressant activity and higher efficacy than the reference drug fluoxetine. SAR study revealed that compounds 164b and 164c having an n-pentyl and a hexyl chain attached to the core quinolinone fragment, respectively showed the highest anticonvulsant activities and provided 100% protection at the dose of 100 mg/kg.
Fig. 36.

1,2,4-Triazoles derivatives 164–168 with anticonvulsant activity.
A series of purine containing triazoles 165 (Fig. 36) were synthesized and evaluated for anticonvulsant activity using MES and scPTZ models in mice by Wang et al. [150]. Among the tested compounds, 165a was the most active compound with ED50 of 23.4 mg/kg and PI value of >25.6, which is higher than the reference drug, carbamazepine whose PI value was 6.4. Moreover, compound 165a exhibited significant oral activity against MES-induced seizures (ED50: 39.4 mg/kg). SAR study revealed the significance of triazole ring as shown in Fig. 36.
Sari et al. synthesized a series of ester derivatives of 1-(2-naphthyl)-2-(1H-1,2,4-triazol-1-yl)ethanone oxime 166 (Fig. 36) and evaluated them in vivo for anticonvulsant and neurotoxic effects by MES, scMET-induced seizures and rotarod tests [151]. Docking study using homology models of Na+ channel inner pore and GABAAR revealed that the compounds exerted anticonvulsant activity by inhibiting voltage-gated sodium channels (VGSC) and allosterically modulating GABAAR.
Abuelhassan et al. reported the anticonvulsant activity of 1,5-diaryl-1H-1,2,4-triazole-3-carboxamide derivatives 167 (Fig. 36) against MES, scPTZ and Strychnine animal screen methods [152]. Most of the compounds showed parallel activity pattern with standard phenytoin and valparate. Compound 167a and 167b showed 100% of sodium valproate activity and phenytoin activity, respectively after 0.5 and 4 h in scPTZ model. The pharmacophoric results for the selected compounds revealed that the compounds showed good fitting on the pharmacophoric query with good RMSDX results.
Liu et al. have synthesized and evaluated 1,2,4-triazole-3-thiol derivatives 168 (Fig. 36) for their anticonvulsant activity and neurotoxicity by using MES, scPTZ, and rotarod tests. Among them, compounds 168a and 168b exhibited significant anticonvulsant activity with the ED50 value of 50.8 and 54.8 mg/kg in the MES test and 76.0 and 52.8 mg/kg in the scPTZ seizures test, respectively [153].
2.5. Antituberculosis agents
Isoniazid (isonicotinic acid hydrazide) is the most effective antimycotic drug used for treatment of tuberculosis (TB) for more than 5 decades. Unfortunately, side effect of isoniazid and the emergence of drug-resistant tuberculosis provoked medicinal chemists to design novel anti-TB agents. Several 1,2,4-triazole derivatives have been synthesized with the aim to explore new anti-TB agents.
Krishana et al. reported the synthesis of a series of diphenylamine containing 1,2,4-triazoles 169–172 (Fig. 37 ) and screened against Mycobacterium tuberculosis H37RV (Mtb H37Rv) species using standard Microplate Alamar Blue Assay (MABA) and agar dilution method [154]. Among the tested compounds, compounds 169a, 169d and 169e displayed potent antimycobacterial activity with MIC value in the range of 0.2–3.125 μM. Compound 169a showed more significant activity comparable to the standard drug isoniazid. SAR study revealed that mannich base 169 and 170 displayed better activity as compared to triazoloquinazolinones 171 and triazolothiazolidinones 172. The cytotoxicity of the most active compounds were evaluated against Vero (African Green monkey kidney epithelial cells) and HepG2 cell line. It was observed that compounds were not cytotoxic.
Fig. 37.

Substituted triazolthiones 169–177 with anti-TB potential.
Castelino et al. have reported the design and synthesis of Schiff bases of 1,2,4-triazole-bearing haloarene moiety 173 (Fig. 37) and screened for in vitro anti-TB properties using disc diffusion method (ZOI test) and microplate Alamar Blue assay (MABA) method (MIC test) towards Mtb H37Rv strain [155]. Compounds 173a and 173h having two fluorine atoms at positions 2 and 4 were found to exhibit the highest activity for antituberculosis screening as well as for neutrophil function test. Acute oral toxicity studies revealed that some of the compounds were safe even at the dose of 2000 mg/kg body weight.
S-Substituted 4-alkyl-5-(3,5-dinitrophenyl)-4H-1,2,4-triazole-3-thiols and their 3-nitro-5-(trifluoromethyl)phenyl analogues 174 (MIC: 0.03–2 μM) (Fig. 37) were endowed with excellent and selective antimycobacterial activities against Mtb strains, including clinically isolated MDR strains [156]. SAR studies revealed the crucial role of position of 3,5-dinitrophenyl fragment on anti-TB activity.
Several hybrid triazoles 175–177 (Fig. 37) were designed, synthesized and evaluated by Dixit et al. [157] as growth and efflux inhibitor of TB against Mtb H37Rv and M. smegmatis mc2155. Pharmacologically active compounds were further tested for their cytotoxicity against human monocyte to assess their ex-vivo cytotoxicity toward eukaryotic cells. Further, the compounds which exhibited higher inhibition and less toxicity were subjected to secondary evaluation of growth and efflux inhibition on Mtb H37Rv and synergistic action with first line and second line anti-TB drugs. One of the compounds 176 having R = CH3 and R1 = C3H7 exhibited potent inhibitory growth in both M. tuberculosis and M. smegmatis mc2155 as well as efflux (5 fold better than thioridazine (TZ)) have found to show very less toxicity compared to TZ towards human macrophages (16 folds) and proved as a better dual inhibitor which is better than TZ devoid of any CNS related side effects.
Evaluation of a series of novel 3-substituted triazolophthalazines 178 (Fig. 38 ) for anti-TB activity revealed that compounds 178a-d exhibited moderate to excellent in vitro activities (MIC: 0.5–4 μg/mL) against Mtb H37Rv [158]. Furthermore, the most active compounds 178b-d showed activity to a similar extent against various MDR-Mtb strains, thus revealing a distinct mode of action.
Fig. 38.

Fused 1,2,4-triazoles anti-TB potential.
Various triazolopyrimidines 180 (Fig. 38) were designed and synthesized as anti-TB agents by Zuniga et al. [159] via the by modification of compound 179, identified from a whole-cell screen against M. tuberculosis, at the C-5, C-7 and C-2 positions. A number of compounds exhibited sub-micromolar activity against M. tuberculosis with MIC90 value in the range of 0.52–10 μM with no cytotoxicity against HepG2 cells. Three compounds 179, 180a and 180b displayed selectivity with MIC99 values of 3.1, 13 and 1.6 μM, respectively for M. tuberculosis over M. smegmatis, E. coli, P. aeruginosa, B. subtilis and yeast S. cerevisiae.
A series of isopropylthiazole clubbed triazole derivatives 181 and dihydro triazolothiadiazoles 182 were synthesized and screened for their anti-TB activity by Kumar et al. [160,161]. Compounds 181a, 181b, 182a and 182b exhibited potent in vitro activity against Mtb H37Rv strain at MIC 4 μg/mL (Fig. 38). Bonde et al. reported that among the ten screened 1,2,4-triazolo[3,4-b] [1,3,4]thiadiazoles containing pyrazin-2-yloxymethyl moiety 183, two compounds 183a (MIC: 0.4 μg/mL) and 183b (MIC: 1.0 μg/mL) exhibited a promising antimycobacterial activity against H37Rv strain (Fig. 38) [162].
Several triazolothiadiazoles and triazolothiadiazines 184 and 185 (Fig. 38) with structural modifications at C-3 and C-6 positions of fused system were reported to inhibit the growth of Mtb H37Rv, MDR-TB (isoniazid and rifampin resistant strains) and DR-TB (rifampin resistant strains) by Li et al. [163,164]. SAR revealed that the electron-withdrawing groups at para position of the phenyl ring at the 3-position and p-bromophenoxymethyl group at the 6-position on the triazolothiadiazole 184 displayed significant enhancement in potency. Two highly active lead compounds 184a and 184b (MIC for H37Rv: 0.5 μg/mL; MDR-TB: 4.0 μg/mL; DR-TB: 0.5–1.0 μg/mL) also showed potential inhibitory activity on M. tuberculosis shikimate dehydrogenase (Mtb SD) with IC50 value of 6.82 μg/mL and 14.42 μg/mL, respectively. SAR of triazolothiadiazines 185 regarding anti-TB activity led to the identification of two highly potent compounds 185a (MIC for Mtb H37Rv, DR-TB and MDR-TB strains: 0.25, 0.25 and 2.0 μg/mL) and 185b (MIC for Mtb H37Rv, DR-TB and MDR-TB strains: 1.0, 2.0 and 4.0 μg/mL) which were endowed with potent Mtb SD inhibitory properties with IC50 values 86.39 and 73.57 μg/mL, respectively. Triazolothiadiazole IMB-SD62 184c, inhibitor of Mtb SD, showed in vivo anti-TB activity against acute Mtb H37Rv infection in mice with a 1.7 log reduction in the lung CFU counts and 14% oral bioavailability in a preliminary pharmacokinetic study [165].
Papadopoulou et al. screened the anti-TB potential of 3-nitro-1,2,4-triazole-linked sulfonamides 186 and amides 187–188 (Fig. 39 ) against Mtb H37Rv under aerobic or hypoxic conditions [166,167]. Among them, compounds 186a and 186b exhibited excellent activity MIC of 1.56 and 3.13 μg/mL, respectively, superior to ethambutol (MIC: 6.25 μg/mL). The study revealed the reduction in aerobic anti-TB activity with decrease in length of the linker from a 4-methylene to a 3-methylene and 2-methylene linker between the nitrotriazole ring and the sulfamido group. Among amides, compounds 187a and 187b active against aerobic and hypoxic Mtb displayed bactericidal and intracellular antitubercular activities. Moreover, compound 188 was selectively active against aerobic Mtb and exhibited good in vitro ADMET characteristics, representing excellent caco-2 permeability, an efflux ratio of 0.39, good microsomal stability and lack of hepatotoxicity (Fig. 39).
Fig. 39.

Various 1,2,4-triazoles with anti-TB potential.
A set of coumarin-3-yl-methyl-1,2,3-triazolyl-1,2,4-triazol-3(4H)-ones 189 (Fig. 39) were synthesized and screened for their anti-TB activity by using the Microplate Alamar Blue assay by Somagond et al. [168]. The preliminary in vitro results indicated that compounds 189a-g displayed excellent anti-TB activities against Mtb H37Rv with MIC of 1.60 μg/mL and were ∼2 folds more active than standard drug pyrazinamide (MIC: 3.12 μg/mL). Docking studies illustrated that 189d and 189g fitted well into the binding pocket of InhA-D148G (4DQU).
Ozadali et al. synthesized some thiazolylhydrazones 190 (Fig. 39) and reported their anti-TB activity [169]. Compounds 190a-d with MIC value of 3.76–4.33 μM were found to be equally active as ethambutol (MIC: 7.65 μM) and ciprofloxacin (MIC: 4.71 μM). In general, the presence of NO2, Cl and F atoms on phenyl ring is found to increase antimycobacterial activity remarkably.
A series of novel substituted 4H-1,2,4-triazol-3-yl cycloalkanols 191 (Fig. 39) has been designed and screened for anti-TB activity against Mtb H37Rv using resazurin microtiter assay by Desai et al. [170]. SAR revealed that compounds 191a-e with 4-pyridyl substituent on triazole ring exhibited excellent anti-TB activity with MIC value in the range 0.59–0.95 μg/mL and low cytotoxicity against the Vero Cell line C1008 with SI > 28.
Twenty 1,2,4-triazol-1-yl-pyrazole based spirooxindolopyrrolizidines 192 (Fig. 39) were synthesized and evaluated for their anti-TB potential against Mtb H37Rv by Pogaku et al. [171]. Among all, most active compound 192a was 2 folds more potent (MIC: 0.78 μg/mL) than the standard drug ethambutol and compounds 192b-g were equally potent to ethambutol (MIC: 1.56 μg/mL). SAR study revealed the significance of substituents at 5-position of isatin ring and substituents at para- and meta-positions of the aryl ring attached to the pyrrolizidine ring (Fig. 39). Compounds 192a-g exhibited low cytotoxicity against RAW 264.7 cells.
2.6. Antiviral agents
Goma’a et al. designed and synthesized several 1,2,4-triazole derivatives with ethyl 2-((5-amino-1H-1,2,4-triazol-3-yl)thio)acetate as the starting material. Among the compounds studied, compound 193 (Fig. 40 ) was found to be the most potent compound, which could reduce the viral plaques by 50% at a dose of 80 μM against herpes simplex virus-1 (HSV-1), grown on Vero African green monkey kidney cells. Moreover, compound 193 possessed higher selectivity than acyclovir (>200 μM vs 80 μM) [172]. Docking studies revealed that compound 193 interacted into the active site of HSV-1 thymidine kinase mainly by making many hydrogen bonds.
Fig. 40.

1,2,4-Triazole derivatives as antiviral agents.
Henen et al. reported synthesis of number of 1,2,4-triazolo[4,3-a]quinoxaline derivatives as antiviral and antimicrobial agents. Among them, compound 194 (Fig. 40) showed most promising anti-HSV-1 activity with 25% plaque reduction at 20 mg/mL [173]. Pandey et al. synthesized 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines 195 (Fig. 40) and screened them for their antiviral activity against Japanese encephalitis virus (JEV) and HSV-1 [11]. Among them, compound 195c (ED50 7.8 μg/mL) showed moderate anti-JEV activity with 50% inhibition and therapeutic index (TI) value 32.
Cao et al. synthesized forty-four chiral triazole derivatives 196 and screened them for their in vitro antiviral activities against enterovirus 71 (EV71) and coxsackievirus B3 (CVB3) [12]. In this study, compounds 196a and 196b (Fig. 40) showed significant potency against the tested viruses with a SI of 21.7 and 24.7, respectively more active than ribavirin (SI: 15) for EV71. Compound 196a (16 μg/mL) exhibited 88.1% inhibition against EV71. SARs indicated that short alkyl chain (R) and 4-methoxyphenyl or benzyl units (Ar) are favourable for antiviral activities.
Evaluation of a series of synthesized [1,2,4]triazolo[4,3-a]pyrimidin-5(4H)-ones 197 (Fig. 40) for antiviral potential against representative human enteroviruses including Coxsackievirus B1 (Cox B1), Coxsackievirus B3 (Cox B3), Poliovirus 3 (PV3), Human Rhinovirus 14 (HRV14), Human Rhinovirus 21 (HRV 21) and Human Rhinovirus 71 (HRV 71), makes compound 197a (1.6–8.85 μM) promising lead compound for developing broad spectrum anti-enterovirus drugs [174].
Nine quinoxaline derivatives were prepared and evaluated for their antiviral activity against hepatitis C virus (HCV), hepatitis B virus (HBV), HSV-1, and human cytomegalovirus HCMV by El-Zahabi [175]. The in vitro screening data indicated that the pyridinyl triazole derivative 198 (Fig. 40) exhibited highly potent activity against HCMV with IC50 of <0.05 μM than that of reference drug ganciclovir (IC50: 0.59 μM). In addition, it also exhibited eleven times higher SI (>3000 μM) against HCMV than ganciclovir (SI > 256 μM).
Massari et al. via hit-to-lead optimization studies identified two hybrid molecules 199 and 200 (Fig. 40), having triazolopyrimidine and cycloheptathiophene scaffolds, as potent Flu polymerase PA-PB1 subunits inhibitors [176]. Along with PA-PB1 interaction inhibitor, 199 also exhibited broad anti-Flu activity with no cytotoxicity.
Sixteen triazole derivatives 201 (Fig. 40) were evaluated for their anti-MERS-CoV activity through the inhibition of helicase and ATPase activity using the FRET assay by Zaher et al. [177]. Among them, compounds 201a and 201b were the most potent MERS-CoV helicase inhibitors with ATPase IC50 values of 0.47 and 0.51 μmol/L, respectively.
Zhan et al. designed and synthesized a series of 2-(2-(2,4-dichlorophenyl)-2H-1,2,4-triazol-3-ylthio)-N-arylacetamide derivatives 202 as potent HIV-1 inhibitors [178]. Among them, six compounds 202a-f (Fig. 41 ) exhibited better inhibition of wild-type HIV-1 (IIIB) replication with ED50 value ranged from 2.78 to 6.21 μM than reference drug dideoxyinosine. Compound 202c showed most promising potency with EC50 value of 2.78 μM and SI of 67 against HIV-1(IIIB) and EC50 value of 7.42 μM against K103N mutant strain.
Fig. 41.

1,2,4-Triazole derivatives as anti-HIV agents.
In another work, two series of [1,2,4]triazolo[1,5-a]pyrimidine derivatives 203 and 204 (Fig. 41) were rationally designed via structure-based core refining approach, synthesized and evaluated for their anti-HIV activities by same group [179,180]. Among the series of 203, compound 203a was the most active against wild-type and double resistant mutant strain (K103N + Y181C) of HIV-1 with EC50 value of 0.02 μM and 7.61 μM, respectively. Among the series 204, compound 204a with an EC50 value of 8.1 nM against wt HIV-1 exhibited 38–2800 folds more potent activity than references didanosine, lamivudine, nevirapine and delavirdine mesylate.
A set of acetamide derivatives of doravirine were synthesized as potent HIV-1 NNRTIs using the structure-based drug design strategy by Wang et al. [181]. The study indicated that most active compound 205 (Fig. 41) with EC50 of 54.8 nM against wt HIV-1 was more potent than reference lamivudine (EC50: 12.8 μM) and comparable to doravirine (EC50: 13 nM).
2.7. Antiparasitic agents
Bhatt et al. reported in vitro antimalarial efficacy of pyrazole-linked triazolo-pyrimidine hybrids 206 (Fig. 42 ) and active hybrids (IC50: 0.034–0.09 μg/mL) for inhibition of Plasmodium falciparum dihydrofolate reductase (PfDHFR) via docking and in vitro studies [182]. The SAR of hybrids 206 indicated the significance of electron-withdrawing group at R1 position; -Br group at R2 position and substitution with methyl group at R3 position for antimalarial potency. Compound 206a with IC50 value of 0.023 μg/mL and SI of 652 exhibited good inhibitory activity in DHFR inhibition assay than reference pyrimethamine.
Fig. 42.

1,2,4-Triazole derivates as antimalarial agents.
Prasad et al. reported that triazole-pyrazole hybrids 207 (IC50: 0.041–1.50 μg/mL) (Fig. 42) displayed significant antiplasmodial activities. Among them, compounds 207d, 207e and 207g (IC50: 0.041, 0.054 and 0.092 μg/mL) displayed more potent activity against the P. falciparum strain in comparison to reference quinine (IC50: 0.286 μM) [183].
The antiplasmodial activities of triazolium salts 208 (Fig. 42) against chloroquine resistant ItG strain of P. falciparum were assessed by Vlahakis et al. [184]. Among them, compound 208a was found to be highly potent with IC50 value of 100 nM and SI of 1430. They hypothesized that potency of compounds in parasite cultures is due to presence of an electron deficient core attached to hydrophobic side groups which interact with a negatively charged moiety on the parasite merozoite.
McConville et al. used a rational approach to investigate carbamoyl triazoles 209, known serine protease inhibitors, as promising antimalarial agents [185]. Among them, compound 209a exhibited potent in vitro antiplasmodial activity (IC50: 10 nM) against 3D7 strain of P. falciparum with in vivo oral efficacy in a SCID mouse model of P. falciparum infection with an ED50 and ED90 of about 100 and 150 mg/kg, respectively. SAR study revealed the significance of N-methyl and R group on benzyl ring as shown in Fig. 42.
The selection from the Tres Cantos Anti-Malarial Set (TCAMS) and an extensive SAR exploration of three carboxamide series by Rueda et al. [186] resulted in cyclopropyl carboxamides 210 (Fig. 42) with improved profile. Further optimization of substitution pattern (R) on phenyl ring adjacent to cyclopropyl ring identified compound 210a (IC50: 3 nM) as a highly potent in vitro inhibitor of P. falciparum 3D7 strain. Notably, compound 210a exhibited 55% oral bioavailability in CD-1 mice and in vivo activity with the ED50 of 12 mg/kg in nonmyelo-depleted P. falciparum murine model.
Among several triazole sulphonamide derivatives 211 (Fig. 42), compounds 211a and 211b with IC50 of 0.023 and 0.025 μg/mL, respectively against chloroquine resistant strain of P. falciparum, were comparable to chloroquine (IC50: 0.020 μg/mL) and 10-fold more potent than quinine (IC50: 0.268 μg/mL) [187].
Among the synthesized triazole Schiff bases 212 (Fig. 42), compounds 212a-c (IC50: 0.230–0.282 μM) displayed higher antimalarial potency against P. falciparum than pyrimethamine (IC50: 1.005 μM) [188]. Further in vitro enzyme inhibition study indicated that 212b with IC50 of 0.0259 μM was found to be most potent DHFR inhibitor as compared to chloroquine (IC50: 0.0301 μM) and pyrimethamine (IC50: 0.1007 μM).
Dihydroorotate dehydrogenase (DHODH), enzyme in the de novo biosynthetic pathway, has emerged as promising target for development of novel antimalarial agents. Phillips and his co-workers [[189], [190], [191], [192], [193]] synthesized a library of triazolopyrimidine derivatives 213 (Fig. 42) as potent P. falciparum DHODH (PfDHODH) inhibitors. Structure-guided lead optimization of the substitution pattern on [1,2,4]triazolo-[1,5-a]pyrimidine scaffold identified the most promising and selective inhibitor 213a (DSM265, IC50: 33 nM), which exhibited antimalarial activity with EC50 values in a range of 15–57 nM against drug sensitive and resistant strains via DHODH inhibition, a long half-life after oral administration in rodents and in vivo efficacy against P. falciparum in SCID mouse model with ED90 value of 8.1 mg/kg [190]. Subsequent optimization highlighted that compound 213b with both meta-fluorines on the aniline ring and fluoroethyl at C-2 of the triazoloprymidine ring have poor species selectivity toward DHODH [191]. Further exploration of antimalatial drug candidate 213a by replacing SF5-phenyl moiety with a series of CF3-pyridinyls identified a promising compound 213c (DSM421, Fig. 42) with more enhanced drug-like properties which displayed 2-fold higher activity with IC50 of 0.053 μM on PfDHODH than for P. vivax DHODH (IC50: 0.094 μM) [193]. Notably, it also showed equivalent activity against field isolates of P. falciparum and P. vivax.
Antimalarial evaluation of twenty six [1,2,4]triazolo[1,5-a]pyrimidine derivatives 214 (Fig. 42) against chloroquine-resistant W2 strain of P. falciparum revealed that compounds 214a-d displayed most effective antimalarial activity with IC50 in the range of 0.023–0.55 μM [194].
Neglected diseases (NDs) are a diverse group of diseases that affect millions of people. These include Chagas disease, human African trypanosomiasis (HAT), leishmaniasis, soil-transmitted helminthiasis which is caused by Trypanosoma cruzi, Trypanosoma brucei, Leishmania spp., and helminths respectively. Literature analysis also reveals the relevance of 1,2,4-triazole derivatives in neglected diseases [195,196].
Franklim et al. designed and synthesized 1,2,4-triazole derivatives by optimization of previously reported N 4-cyclohexyl-1,2,4-triazole 215 having anti-T. cruzi activity [197]. The study revealed that S-alkylated-triazoles 216a-c (Fig. 43 ) exhibited significant trypanocidal profile with IC50 values 3.18–3.52 and 3.61–4.15 μmol/L against epimastigote and amastigote forms of T. cruzi, respectively, in comparison to lead compound 215 (IC50: 18.30 and 8.87 μmol/L against the epimastigote and amastigote forms, respectively) [198].
Fig. 43.

1,2,4-Triazole derivates as antitrypanosomal agents.
Silva et al. synthesized 1,2,4-triazole derivatives 217 (Fig. 43) employing the bioisosterism and molecular hybridization approaches as nitrofurazone analogues, which have shown selectivity against intracellular amastigotes of T. cruzi Y strain [199]. Compound 217a with IC50 of 5.53 μM and SI > 36 was equipotent to drug benznidazole, but exhibited lower efficacy. Furthermore, 217a was found to be 26 fold more potent than analogue 217b, highlighting the impact of nitro group on antitrypanosomal activity.
Papadopoulou et al. reported antichagasic activity of 3-nitro-1H-1,2,4-triazole-based aliphatic and aromatic amines [[200], [201], [202]]. Compounds 218–227 (Fig. 43) have shown high efficacy against the amastigotes forms of T. cruzi (IC50: 0.04–0.57 μM, SI: 208–1725) and were upto 33.8 fold more potent than the standard drug benznidazole. The SAR revealed that presence of nitro group on the triazole ring is positively correlated with antiparasitic activity. Compounds 218–220 also exhibited significant activity against bloodstream-form (BSF) Trypanosoma brucei rhodesiense trypomastigotes with IC50 ranged from 0.117 to 0.435 μM levels and SI of 220–973. In addition, 3-nitrotriazole based piperazine 225a, benzothiazoles (226a and 226b) and quinoline derivative 227 exhibited significant anti-HAT activity against T. b. rhodesiense trypomastigotes with IC50 of 0.231, 0.204, 0.355 and 0.038 μM, respectively [201,202].
Most of the synthesized 3-nitro-1H-1,2,4-triazole-based amides and sulphonamides (IC50: 28 nM-3.72 μM, SI: 66–2782) were also reported to exhibit significant in vitro activity against T. cruzi intracellular amastigotes by Papadopoulou et al. [203]. Among them, compounds 228 and 229 (Fig. 43) were found to be most active against T. cruzi and 36–58 fold more potent than benznidazole (IC50: 1.562 μM). Further, some nitrotriazole have shown moderate activity profile against the axenic form of L. donovani. The SAR of 3-nitrotriazole-based heteroarylamides/sulfonamides revealed that chlorinated thiophene sulfonamides and benzothiophene amides 230 (Fig. 43) were the most active antichagasic compounds, displaying up to 14 fold higher potency than the standard benznidazole [204].
Papadopoulou et al. designed 3-nitrotriazole based aryloxyphenylamides 231 (Fig. 43) as potent and selective anti-T. cruzi agents [205]. Notably, two most potent compounds 231a and 231b reduced the parasite load after 5 days of treatment at 13 mg/kg/day (ip) in infected mice. Moreover, compounds 231 exhibited selective activity against L. donovani axenic amastigotes. Further optimization of 3-nitrotriazole-based aryloxyphenylacetamides 231 via inserting one additional methylene group between the nitrotriazole ring and the amidic carbonyl resulted in corresponding propanamides 232 (Fig. 43) with a broad spectrum antitrypanosomal activity [206]. In vitro evaluation of compounds 232 revealed excellent and comparable antichagasic activity, 4–214 fold greater anti-HAT activity and smaller antileishmanial activity to that of the corresponding acetamides 231.
Several linear, rigid 3-nitrotriazole-based amides and carbinols (analogues of fluconazole) were synthesized as potent antitrypanosomal agents via their dual functioning as excellent substrates for trypanosomaltype I nitroreductase (NTR) and as inhibitiors of sterol 14α-demethylase (T. cruzi CYP51) enzyme [207]. Carbinols 233 (Fig. 43) displayed excellent in vitro activity with IC50 of 33 nM and SI of 3308 against T. cruzi amastigotes as well as in vivo activity in T. cruzi infected murine model.
Khare et al. using hit-to-lead optimization approach reported a selective kinetoplastid proteasome inhibitor 234 (GNF6702, Fig. 44 ) that does not inhibit the human proteasome [208]. It also exhibited in vivo efficacy which cleared parasites in mouse models of leishmaniasis, Chagas disease and HAT. Inhibition of the proteasome chymotrypsin-like activity is demonstrated as primary mechanism of parasite growth inhibition by GNF6702.
Fig. 44.

Kinetoplastid proteasome inhibitors.
Compounds 234 (GNF6702) and its analogue 235 (NITD689, Fig. 44) showed in vitro concentration-time dependent trypanocidal activity with favourable in vivo pharmacokinetics and significant brain penetration [209]. Importantly, both compounds, act by inhibiting chymotrypsin activity of the 20S proteasome in T. brucei, are efficacious for achieving complete cure in HAT hemolymphatic (1 and 10 mg/kg, respectively, once daily, for four days) and meningoencephalic mouse models (at 30 and 60 mg/kg dose, respectively).
A series of amino acid-coupled 1,2,4-triazoles 236 (Fig. 45 ) were evaluated for their in vitro antileishmanial activity on L. major promastigotes by El-Saghier et al. [9]. Among them, compounds 236a-d (IC50: 0.0312–0.0866 μg/mL) were 36–100 folds more potent than the reference miltefosine (IC50: 3.1924 μg/mL) and comparable to amphotericin B deoxycholate (IC50: 0.0472 μg/mL). Reverse docking approach illustrated mitogen-activated protein kinase (MAPK) as a possible putative antileishmanial target. SAR analysis suggested the hydrophobic moiety with certain topology like isopropyl and indolyl groups is favourable for antileishmanial activity.
Fig. 45.

1,2,4-Triazole derivates as antileishmanial agents.
Evaluation of a series of 5-(pyrazin-2-yl)-4H-1,2,4-triazole-3-thiol derivatives 237 and 238 (IC50: 79.0–382.4 μM, Fig. 45) for in vitro antileishmanial activity against L. donovani promastigotes, makes compounds 237a (IC50: 79.0 μM) and 238a (IC50: 79.0 μM) the most promising antileishmanial agent as compared to standard sodium stibogluconate (IC50: 490.0 μM) [210].
Among coumarin-triazolothiadiazine hybrids, 239 (Fig. 45) demonstrated the highest inhibition (IC50: 0.89 μM) in vitro against the promastigote form of L. major [211]. Süleymanoğlu et al. reported that 4-amino-1,2,4-triazole derivative 240 (Fig. 45) presented antileishmanial activity with MIC of 625 μg/mL in vitro study against L. infantum (MON-183) by microdilution broth assay [212].
2.8. Analgesic and anti-inflammatory agents
A set of hydrazone derivatives of 1,2,4-triazole 241 (Fig. 46 ) was evaluated for radical scavenging and anti-inflammatory activities in vitro and in vivo by Khan et al. [213]. The most potent compound 241a with 64.44% inhibition of edema and a potency of 0.92 at 20 mg/kg body weight after 5 h of inducing inflammation was comparable to reference indomethacin (potency 1.00). Docking study revealed that compound 241a occupied celecoxib binding site in COX with high affinity (Fig. 46) and binding free energy of −10.5 and −11.2 kcal/mol for COX-1 and COX-2, respectively.
Fig. 46.

1,2,4-Triazole derivates as anti-inflammatory agents and illustration of the binding of compound 241a in the COX-2 active site.
Abdel-Aziz et al. reported the synthesis and anti-inflammatory activity of 1,2,4-triazole-3-carboxamides derivatives 242 and 243 (Fig. 46) [214,215]. Compounds 242 and 243 exhibited good anti-inflammatory activity (52–78%) after 3 h with lower ulcerogenic risk compared to indomethacin (78% activity). In vitro COX-1/COX-2 inhibition and docking studies revealed compounds 242f and 242g having less bulky group on amide nitrogen as the most potent COX inhibitors. Compounds 243 demonstrated excellent selectivity towards human COX-2 with selectivity indices (COX-1 IC50/COX-2 IC50) ranged from 62.5 to 2127 [215].
Interestingly, NO-triazole hybrids 244 (Fig. 46) were found to be more potent anti-inflammatory agents than the corresponding ketone intermediates [216]. Among the synthesized oximes, compound 244a exhibited high anti-inflammatory activity (79%) after 4 h and lower ulcerogenicity (ulcer index 0.25). In another study, quinoline incorporating 1,2,4-triazole/oxime hybrids 245a-c (Fig. 46) displayed significant anti-inflammatory activity compared to indomethacin with % edema inhibition of 100%, 101% and 111%, respectively [217].
Lamie et al. screened a series of triazole Schiff bases containing N-substituted indole 246 (Fig. 46) for their in vitro anti-inflammatory activity [218]. Compound 246a was found to be the most potent inhibitors of cytokine Eselectin and COX-2 enzyme (IC50: 0.98 μM and SI: 8.05).
Biological screening of Schiff and Mannich bases derivatives of 1,2,4-triazoles 247 (Fig. 46) at a dose of 20 mg/kg in rats by Gowda et al. revealed that compounds 247c and 247d showed good anti-inflammatory activity compared to indomethacin whereas compounds 247a, 247b and 247d showed significant analgesic effects [219].
Receptor interacting protein 1 (RIP1) kinase is critical regulator of necroptosis and inflammation. DNA-encoded library (DELs) screening and lead optimization has resulted in identification of clinical candidate GSK2982772 (248, Fig. 46) as first-in-class RIP1 inhibitor which is currently in pre-clinical trials for inflammatory diseases, including psoriasis, rheumatoid arthritis, and ulcerative colitis [220].
Two series of 1,2,4-triazole based benzothiazole derivatives 249 and 250 (Fig. 46) were synthesized and evaluated for their in vitro anti-inflammatory activity and p38α MAP kinase inhibition by Tariq et al. [221,222]. Among the selected compounds for in vivo evaluation, compounds 249a and 250a emerged as the most potent compound with edema inhibition of 84.43% and 85.31%, respectively.
Paprocka et al. reported that 1,2,4-triazole derivatives 251 (Fig. 46) containing methacrylic acid moiety exerted anti-inflammatory activity via modulation of monocytes activation [223]. A novel series of celecoxib derivatives with triazole moiety have been screened for their anti-inflammatory potential by CPE test by Mustafa et al. and most of the them showed higher activity compared to Celecoxib [224].
The pharmacology screening a series of 6-substituted thiazolo[3,2-b]-1,2,4-triazole-5(6H)-one derivatives of ibuprofen revealed that compounds 252a-c (Fig. 47 ) displayed potential in vivo analgesic/anti-inflammatory activity without a gastrointestinal side effect [225].
Fig. 47.

Fused 1,2,4-triazole derivates as anti-inflammatory agents.
Various thieno[3,2-e]triazolo[4,3-a]pyrimidine derivatives were evaluated for anti-inflammatory activity by Rizk et al. [226]. Compounds 253–255 (Fig. 47) showed prominent anti-inflammatory activity comparable with diclofenac Na in the acute and sub-acute inflammatory models. A set of thieno[2,3-d] [1,2,4]triazolo[1,5-a] pyrimidines 256 (Fig. 47) was synthesized and evaluated for their anti-infammatory and analgesic activity by Ashour et al. [227]. Compounds 256a-c (ED50: 23.45–28.15 mg/kg) showed equivalent to moderate anti-infammatory activity at 20 mg/kg oral dose in both acute and sub-acute models compared to reference diclofenac as well as good analgesic profile with a delayed onset of action.
Pan et al. synthesized a series of 4-phenylthieno[2,3-e] [1,2,4]triazolo[4,3-a]pyrimidine-5(4H)-ones 257 (Fig. 47) and screened for their anti-inflammatory activity by xylene-induced ear-edema test [228]. The study indicated that the most potent compound 257a with 50.48% activity at 30 min after intraperitoneal administration was more active than the reference drug indomethacin.
El Shehry et al. synthesized of a series of 3-(2,4-dichlorophenoxy)methyl)-1,2,4-triazolo (thiadiazoles and thiadiazines) and screened for their anti-inflammatory activity [229]. Among them, compounds 258–261 (Fig. 47) showed 36–56% anti-inflammatory activity comparable to the standard indomethacin.
Maddila et al. reported that among the triazolo[3,4-b]thiadiazole derivatives 262 (Fig. 47) screened for their anti-inflammatory potential in the CPE test at 10 mg/kg oral dose, compounds 262a (82.24%) and 262b (83.06%) exhibited potent anti-inflammatory activity than indomethacin (81.55%) [230].
SAR studies on [1,2,4]triazolo[4,3-a] [1,8]naphthyridine scaffold were conducted by Bracci et al. with the objective of improving potency for anti-inflammatory and/or analgesic activities [231]. Results revealed that compound 263b-f (Fig. 47) exhibited good anti-inflammatory activity ranging from 34% to 80% and compound 263c was found to be more potent and effective than the parent compound 263a [232]. Whereas compounds 264a-c (Fig. 47) were endowed with prevalent analgesic activity (74%–96% inhibition in the writhing test in mice, P < 0.01, 50 mg/kg dose) frequently associated with sedative effects.
Guirado et al. synthesized a series of triazolo[4,3-a]quinoxalines 265 (Fig. 47) and evaluated for their anti-inflammatory activity as inhibitors of the pro-inflammatory cytokines TNF-α and IL-6 [233]. Results revealed that compound 265c was found to be the most potent while compounds 265a-d exhibited good levels of inhibition against both cytokines.
Liu et al. synthesized a series of triazolo[3,4-a]phthalazine-3-carboxamide derivatives 266 (Fig. 47) as potent anti-inflammatory agents, which acted on tumor necrosis factor (TNF-α) as inhibitors of NF-κB activation [234]. Moreover, compound 266a exhibited excellent anti-inflammatory activity with 58.19% inhibition at 50 mg/kg (i.p.) against xylene-induced ear edema, with equal efficacy as the standard drug indomethacin (100 mg/kg i.p; 59.21% inhibition).
Several thiazolo[3,2-b]-1,2,4-triazoles derived from naproxen 267 (Fig. 47) exhibited significant analgesic and anti-inflammatory activities with low gastric risk [235]. Moreover, compound 267a was found to be the most selective COX-2 inhibitor with IC50 of 20.5 μM and SI > 4.87.
Some other 1,2,4-triazole derivatives have been synthesized and screened for anti-inflammatory and analgesic activities [[236], [237], [238]].
3. Miscellaneous
Aggarwal et al. reported 1,2,4-triazolo[4,3-a]quinoxaline derivatives 268 and 1,2,4-triazolo[4,3-a]quinoxalin-4(5H)-ones 269 (Fig. 48 ) as effective DNA photocleavers [239,240]. Among all the synthesized molecules, compounds 268c and 269k showed significant photocleavage of supercoiled plasmid ΦX174 and pMaxGFP, respectively under UV irradiation at λ max 312 nm. DNA cleaving efficiency of 269 was found to be dependent on its structure, concentration, and strictly on photoirradiation time. Mechanistic investigations on compound 269k revealed that the DNA photocleavage reaction involves superoxide anion radicals (O2 −∙) (Type-I pathway).
Fig. 48.

1,2,4-Triazolo derivatives with miscellaneous activity.
1,2,4-Triazolo[1,5-a]pyrimidin-7-one 270 (WS-10, Fig. 48) was identified as nontoxic and selective modulator of ABCB1 transporter which plays key roles in the development of multidrug resistance of chemotherapeutic drugs [241]. WS-10 enhanced the intracellular accumulation of paclitaxel in ABCB1 overexpressed SW620/Ad300 cells without affecting the expression or localization of the ABCB1 protein.
A series of triazolopyrimidine hybrids was designed and synthesized as multifunctional anti-Alzheimer agents by Jameel et al. [242,243]. Compounds 271a and 271b (Fig. 48) exhibited more potent acetylcholinesterase (AChE) inhibitory potential with IC50 values 0.065 and 0.092 μM, respectively and high selectivity for AChE over BuChE by ∼28 fold. Notably, these compounds exhibited better Cu2+-induced Aβ1-42 aggregation inhibitory potency.
El-Aleam et al. reported the bronchodilator activity of a set of 1,2,4-triazolo[1,5-a]pyrimidine derivatives 272 (Fig. 48) as phosphodiesterase 4B inhibitors [244]. The study revealed that compounds 272a and 272b with EC50 values of 18.6 and 57.1 μM, respectively, showed better bronchodilator activity than the reference theophylline (EC50: 425 μM).
4. Future perspective
Contemporary medicinal chemistry faces many challenges from several directions, including the need for both potency and specificity of any therapeutic agent. Therefore, in the present perspective, 1,2,4-triazole with broad spectrum biological profile have matured into indispensable heterocyclic scaffold. The work compiled in this review article highlights the findings on 1,2,4-triazoles as a privileged scaffolds endowed with extensive potential therapeutic utility besides applicability in corrosion inhibition, polymers, supramolecular chemistry and material science. Clinical drugs containing triazole nucleus are being used in treating several ailments. Development of resistance in Candida spp against fluconazole, the most efficient anticandiada commercial drug, prompted the pharmacologist to synthesize triazole alcohols as fluconazole analogues to treat fluconazole-resistant fungal strains. 1,2,4-Triazole moiety via hydrogen bonding and dipole interaction can improve the solubility and affinity of the compounds with bimolecular targets. Among the broad spectrum of bioactivities, we comprehensively reviewed the advances in antifungal, antibacterial, anticancer, anticonvulsant, antituberculosis, antiviral, antiparasitic, analgesic and anti-inflammatory activities of 1,2,4-triazole derivatives particularly reported over the past decade.
1,2,4-Triazole derivatives mediate curative effects by acting as promising selective protein/enzyme inhibitors, modulators and receptor antagonists. In this review, we aimed to provide medicinal and pharmaceutical chemists working in area of drug designing and development with a wide data resource about 1,2,4-triazoles derivatives, thus helping them to perform a more organized and fertile drug discovery operation during their experimental studies.
SARs, HTS, hit to lead optimization, molecular hybridization as well as 3D computer modeling would be valuable in structural modifications of 1,2,4-triazole scaffolds for target oriented synthesis, enhancing bioactivities and pharmacokinetic properties and resolving the challenges of multidrug resistance.
5. Conclusion
1,2,4-Triazole is a privileged scaffold in medicinal chemistry having ample potential therapeutic applications continue to expand. This review article is an effort to summarize medicinal chemistry investigations of 1,2,4-triazole derivatives over the last decade, in search for new azaheterocycles which may be a rich source of promising biological activities. It will help the scientific community for rational design and development of novel, target oriented, optimized and varied 1,2,4-triazole based drugs for the treatment of multifactorial diseases. The enriched SAR may pave the way to further explore and develop new 1,2,4-triazole derivatives with improved potency to overcome the resistance.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
Support of this research by D. A. V. College (Lahore), Ambala City is gratefully acknowledged.
References
- 1.Fan Y.L., Ke X., Liu M. Coumarin-triazole hybrids and their biological activities. J. Heterocycl. Chem. 2018;55:791–802. [Google Scholar]
- 2.Cox J.R., Woodcock S., Hillier I.H., Vincent M.A. Tautomerism of 1,2,3- and 1,2,4-triazole in the gas phase and in aqueous solution: a combined ab Initio quantum mechanics and free energy perturbation study. J. Phys. Chem. 1990;94:5499–5501. [Google Scholar]
- 3.Peyton L.R., Gallagher S., Hashemzadeh M. Triazole antifungals: a review. Drugs Today. 2015;51:705–718. doi: 10.1358/dot.2015.51.12.2421058. [DOI] [PubMed] [Google Scholar]
- 4.Zhou C.-H., Wang Y. Recent researches in triazole compounds as medicinal drugs. Curr. Med. Chem. 2012;19:239–280. doi: 10.2174/092986712803414213. [DOI] [PubMed] [Google Scholar]
- 5.Russell P.E. A century of fungicide evolution. J. Agric. Sci. 2005;143:11–25. [Google Scholar]
- 6.Liao L., Jiang C., Chen J., Shi J., Li X., Wang Y., Wen J., Zhou S., Liang J., Lao Y., Zhang J. Synthesis and biological evaluation of 1,2,4-triazole derivatives as potential neuroprotectant against ischemic brain injury. Eur. J. Med. Chem. 2020;190:112114. doi: 10.1016/j.ejmech.2020.112114. [DOI] [PubMed] [Google Scholar]
- 7.Pokuri S., Singla R.K., Bhat V.G., Shenoy G.G. Insights on the antioxidant potential of 1, 2, 4-triazoles: synthesis, screening and QSAR studies. Curr. Drug Metabol. 2014;15:389–397. doi: 10.2174/1389200215666140908101958. [DOI] [PubMed] [Google Scholar]
- 8.Chu X.-M., Wang C., Wang W.-L., Liang L.-L., Liu W., Gong K.-K., Sun K.-L. Triazole derivatives and their antiplasmodial and antimalarial activities. Eur. J. Med. Chem. 2019;166:206–223. doi: 10.1016/j.ejmech.2019.01.047. [DOI] [PubMed] [Google Scholar]
- 9.El-Saghier A.M., Mohamed M.A., Abd-Allah O.A., Kadry A.M., Ibrahim T.M., Bekhit A.A. Green synthesis, antileishmanial activity evaluation, and in silico studies of new amino acid-coupled 1,2,4-triazoles. Med. Chem. Res. 2019;28:169–181. [Google Scholar]
- 10.Bekircan O., Menteşe E., Ülker S., Kucuk C. Synthesis of some new 1,2,4-triazole derivatives starting from 3-(4-chlorophenyl)-5-(4-methoxybenzyl)-4H-1,2,4-triazol with anti-lipase and anti-urease activities. Arch. Pharm. Chem. Life Sci. 2014;347:387–397. doi: 10.1002/ardp.201300344. [DOI] [PubMed] [Google Scholar]
- 11.Pandey V.K., Tusi Z., Tusi S., Joshi M. Synthesis and biological evaluation of some novel 5-[(3-aralkyl amido/imidoalkyl) phenyl]-1,2,4-triazolo[3,4-b]-1,3,4-thiadiazines as antiviral agents. ISRN Org. Chem. 2012:1–7. doi: 10.5402/2012/760517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cao X., Wang W., Wang S., Bao L. Asymmetric synthesis of novel triazole derivatives and their in vitro antiviral activity and mechanism of action. Eur. J. Med. Chem. 2017;139:718–725. doi: 10.1016/j.ejmech.2017.08.057. [DOI] [PubMed] [Google Scholar]
- 13.Kapron B., Czarnomysy R., Wysokinski M., Andrys R., Musilek K., Angeli A., Supuran C.T., Plech T. 1,2,4-Triazole-based anticonvulsant agents with additional ROS scavenging activity are effective in a model of pharmacoresistant epilepsy. J. Enzym. Inhib. Med. Chem. 2020;35:993–1002. doi: 10.1080/14756366.2020.1748026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han S., Zhang F.-F., Xie X., Chen J.-Z. Design, synthesis, biological evaluation, and comparative docking study of 1,2,4-triazolones as CB1 receptor selective antagonists. Eur. J. Med. Chem. 2014;74:73–84. doi: 10.1016/j.ejmech.2013.12.018. [DOI] [PubMed] [Google Scholar]
- 15.Li Y.S., Tian H., Zhao D.S., Hu D.K., Liu X.Y., Jin H.W., Song G.P., Cui Z.N. Synthesis and bioactivity of pyrazole and triazole derivatives as potential PDE4 inhibitors. Bioorg. Med. Chem. Lett. 2016;26:3632–3635. doi: 10.1016/j.bmcl.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 16.Carling R.W., Moore K.W., Street L.J., Wild D., Isted C., Leeson P.D., Thomas S., Connor D., McKernan R.M., Quirk K., Cook S.M., Atack J.R., Wafford K.A., Thompson S.A., Dawson G.R., Ferris P., Castro J.L. 3-Phenyl-6-(2-pyridyl)methyloxy-1,2,4-triazolo[3,4-a]phthalazines and analogues: high-affinity γ-aminobutyric acid-A benzodiazepine receptor ligands with α2, α3, and α5-subtype binding selectivity over α1. J. Med. Chem. 2004;47:1807–1822. doi: 10.1021/jm031020p. [DOI] [PubMed] [Google Scholar]
- 17.Zhang W., Yuan J. Poly(1-vinyl-1,2,4-triazolium) poly(ionic liquid)s: synthesis and the unique behavior in loading metal ions, Macromol. Rapid Commun. 2016;37:1124–1129. doi: 10.1002/marc.201600001. [DOI] [PubMed] [Google Scholar]
- 18.Swathi N.P., Alva V.D.P., Samshuddin S. A Review on 1,2,4-triazole derivatives as corrosion inhibitors. J. Bio Tribo Corros. 2017;3:42. [Google Scholar]
- 19.Bello-Vieda N.J., Murcia R.A., Muñoz-Castro A., Macías M.A., Hurtado J.J. Coordination polymers containing 1,3-phenylenebis-((1H-1,2,4-triazol-1-yl)methanone) ligand: synthesis and ε-caprolactone polymerization behaviour. Molecules. 2017;22:1860. doi: 10.3390/molecules22111860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cáceres D., Cebrián C., Rodríguez A.M., Carrillo J.R., Díaz-Ortiz Á., Prieto P., Aparicio F., García F., Sánchez L. Optical waveguides from 4-aryl-4H-1,2,4-triazole-based supramolecular structures. Chem. Commun. 2013;49:621–623. doi: 10.1039/c2cc37381e. [DOI] [PubMed] [Google Scholar]
- 21.Rodrigues L.D., Sunil D., Chaithra D., Bhagavath P. 1,2,3/1,2,4-Triazole containing liquid crystalline materials: an up-to-date review of their synthetic design and mesomorphic behaviour. J. Mol. Liq. 2020;297:111909. [Google Scholar]
- 22.Campoy S., Adrio J.L. Antifungals. Biochem. Pharmacol. 2017;133:86–96. doi: 10.1016/j.bcp.2016.11.019. [DOI] [PubMed] [Google Scholar]
- 23.Wang W., Wang S., Liu Y., Dong G., Cao Y., Miao Z., Yao J., Zhang W., Sheng C. Novel conformationally restricted triazole derivatives with potent antifungal activity. Eur. J. Med. Chem. 2010;45:6020–6026. doi: 10.1016/j.ejmech.2010.09.070. [DOI] [PubMed] [Google Scholar]
- 24.Yu S., Chai X., Hu H., Yan Y., Guan Z., Zou Y., Sun Q., Wu Q. Synthesis and antifungal evaluation of novel triazole derivatives as inhibitors of cytochrome P450 14α-demethylase. Eur. J. Med. Chem. 2010;45:4435–4445. doi: 10.1016/j.ejmech.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 25.Xu K., Huang L., Xu Z., Wang Y., Bai G., Wu Q., Wang X., Yu S., Jiang Y. Design, synthesis, and antifungal activities of novel triazole derivatives containing the benzyl group. Drug Des. Dev. Ther. 2015;9:1459–1467. doi: 10.2147/DDDT.S74989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu J., Cao Y., Zhang J., Yu S., Zou Y., Chai X., Wu Q., Zhang D., Jiang Y., Sun Q. Design, synthesis and antifungal activities of novel 1,2,4-triazole derivatives. Eur. J. Med. Chem. 2011;46:3142–3148. doi: 10.1016/j.ejmech.2011.02.042. [DOI] [PubMed] [Google Scholar]
- 27.Jiang Y., Zhang J., Cao Y., Chai X., Zou Y., Wu Q., Zhang D., Jiang Y., Sun Q. Synthesis, in vitro evaluation and molecular docking studies of new triazole derivatives as antifungal agents. Bioorg. Med. Chem. Lett. 2011;21:4471–4475. doi: 10.1016/j.bmcl.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 28.Sheng C., Che X., Wang W., Wang S., Cao Y., Miao Z., Yao J., Zhang W. Design and synthesis of novel triazole antifungal derivatives by structure-based bioisosterism. Eur. J. Med. Chem. 2011;46:5276–5282. doi: 10.1016/j.ejmech.2011.03.019. [DOI] [PubMed] [Google Scholar]
- 29.Wang S., Jin G., Wang W., Zhu L., Zhang Y., Dong G., Liu Y., Zhuang C., Miao Z., Yao J., Zhang W., Sheng C. Design, synthesis and structure-activity relationships of new triazole derivatives containing N-substituted phenoxypropylamino side chains. Eur. J. Med. Chem. 2012;53:292–299. doi: 10.1016/j.ejmech.2012.04.013. [DOI] [PubMed] [Google Scholar]
- 30.Jiang Z., Wang Y., Wang W., Wang S., Xu B., Fan G., Dong G., Liu Y., Yao J., Miao Z., Zhang W., Sheng C. Discovery of highly potent triazole antifungal derivatives by heterocycle-benzene bioisosteric replacement. Eur. J. Med. Chem. 2013;64:16–22. doi: 10.1016/j.ejmech.2013.04.025. [DOI] [PubMed] [Google Scholar]
- 31.Yu S., Chai X., Wang Y., Cao Y., Zhang J., Wu Q., Zhang D., Jiang Y., Yan T., Sun Q. Triazole derivatives with improved in vitro antifungal activity over azole drugs. Drug Des. Dev. Ther. 2014;8:383–390. doi: 10.2147/DDDT.S58680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zou Y., Yu S., Li R., Zhao Q., Li X., Wu M., Huang T., Chai X., Hu H., Wu Q. Synthesis, antifungal activities and molecular docking studies of novel 2-(2,4-difluorophenyl)-2-hydroxy-3-(1H-1,2,4-triazol-1-yl)propyl dithiocarbamates. Eur. J. Med. Chem. 2014;74:366–374. doi: 10.1016/j.ejmech.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 33.Li L., Ding H., Wang B., Yu S., Zou Y., Chai X., Wu Q. Synthesis and evaluation of novel azoles as potent antifungal agents. Bioorg. Med. Chem. Lett. 2014;24:192–194. doi: 10.1016/j.bmcl.2013.11.037. [DOI] [PubMed] [Google Scholar]
- 34.Xie F., Ni T., Zhao J., Pang L., Li R., Cai Z., Ding Z., Wang T., Yu S., Jin Y., Zhang D., Jiang Y. Design, synthesis, and in vitro evaluation of novel antifungal triazoles. Bioorg. Med. Chem. Lett. 2017;27:2171–2173. doi: 10.1016/j.bmcl.2017.03.062. [DOI] [PubMed] [Google Scholar]
- 35.Wu J., Ni T., Chai X., Wang T., Wang H., Chen J., Jin Y., Zhang D., Yu S., Jiang Y. Molecular docking, design, synthesis and antifungal activity study of novel triazole derivatives. Eur. J. Med. Chem. 2018;143:1840–1846. doi: 10.1016/j.ejmech.2017.10.081. [DOI] [PubMed] [Google Scholar]
- 36.Na Y.M. Synthesis and activity of novel indole linked triazole derivatives as antifungal agents. Bull. Kor. Chem. Soc. 2010;31:3467–3470. [Google Scholar]
- 37.Cao X., Sun Z., Cao Y., Wang R., Cai T., Chu W., Hu W., Yang Y. Design, synthesis, and structure-activity relationship studies of novel fused heterocycles-linked triazoles with good activity and water solubility. J. Med. Chem. 2014;57:3687–3706. doi: 10.1021/jm4016284. [DOI] [PubMed] [Google Scholar]
- 38.Shrestha S.K., Garzan A., Garneau-Tsodikova S. Novel alkylated azoles as potent antifungals. Eur. J. Med. Chem. 2017;133:309–318. doi: 10.1016/j.ejmech.2017.03.075. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Y., Tangadanchu V.K.R., Bheemanaboina R.R.Y., Cheng Y., Zhou C.H. Novel carbazole-triazole conjugates as DNA-targeting membrane active potentiators against clinical isolated fungi. Eur. J. Med. Chem. 2018;155:579–589. doi: 10.1016/j.ejmech.2018.06.022. [DOI] [PubMed] [Google Scholar]
- 40.Elias R., Benhamou R.I., Jaber Q.Z., Dorot O., Zada S.L., Oved K., Pichinuk E., Fridman M. Antifungal activity, mode of action variability, and subcellular distribution of coumarin-based antifungal azoles. Eur. J. Med. Chem. 2019;179:779–790. doi: 10.1016/j.ejmech.2019.07.003. [DOI] [PubMed] [Google Scholar]
- 41.Luo Y., Zhang S., Liu Z.J., Chen W., Fu J., Zeng Q.F., Zhu H.L. Synthesis and antimicrobical evaluation of a novel class of 1,3,4-thiadiazole: derivatives bearing 1,2,4-triazolo[1,5-a]pyrimidine moiety. Eur. J. Med. Chem. 2013;64:54–61. doi: 10.1016/j.ejmech.2013.04.014. [DOI] [PubMed] [Google Scholar]
- 42.Çavusoglu B.K., Yurttas L., Cantürk Z. The synthesis, antifungal and apoptotic effects of triazole-oxadiazoles against Candida species. Eur. J. Med. Chem. 2018;144:255–261. doi: 10.1016/j.ejmech.2017.12.020. [DOI] [PubMed] [Google Scholar]
- 43.Patel R.V., Park S.W. Access to a new class of biologically active quinoline based 1,2,4-triazoles. Eur. J. Med. Chem. 2014;71:24–30. doi: 10.1016/j.ejmech.2013.10.059. [DOI] [PubMed] [Google Scholar]
- 44.Lin G.S., Duan W.G., Yang L.X., Huang M., Lei F.H. Synthesis and antifungal activity of novel myrtenal-based 4-methyl-1,2,4-triazole-thioethers. Molecules. 2017;22:1–10. doi: 10.3390/molecules22020193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jin R.-Y., Zeng C.-Y., Liang X.-H., Sund X.-H., Liu Y.-F., Wang Y.-Y., Zhou S. Design, synthesis, biological activities and DFT calculation of novel 1,2,4-triazole Schiff base derivatives. Bioorg. Chem. 2018;80:253–260. doi: 10.1016/j.bioorg.2018.06.030. [DOI] [PubMed] [Google Scholar]
- 46.Zoumpoulakis P., Camoutsis C., Pairas G., Sokovic M., Glamoclija J., Potamitis C., Pitsas A. Synthesis of novel sulfonamide-1,2,4-triazoles, 1,3,4-thiadiazoles and 1,3,4-oxadiazoles, as potential antibacterial and antifungal agents. Biological evaluation and conformational analysis studies. Bioorg. Med. Chem. 2012;20:1569–1583. doi: 10.1016/j.bmc.2011.12.031. [DOI] [PubMed] [Google Scholar]
- 47.Tang R., Jin L., Mou C., Yin J., Bai S., Hu D., Wu J., Yang S., Song B. Synthesis, antifungal and antibacterial activity for novel amide derivatives containing a triazole moiety. Chem. Cent. J. 2013;7:30. doi: 10.1186/1752-153X-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Y., Damu G.L.V., Lv J.S., Geng R.X., Yang D.C., Zhou C.H. Design, synthesis and evaluation of clinafloxacin triazole hybrids as a new type of antibacterial and antifungal agents. Bioorg. Med. Chem. Lett. 2012;22:5363–5366. doi: 10.1016/j.bmcl.2012.07.064. [DOI] [PubMed] [Google Scholar]
- 49.Plech T., Wujec M., Kosikowska U., Malm A., Rajtar B., Polz-Dacewicz M. Synthesis and in vitro activity of 1,2,4-triazole-ciprofloxacin hybrids against drug-susceptible and drug-resistant bacteria. Eur. J. Med. Chem. 2013;60:128–134. doi: 10.1016/j.ejmech.2012.11.040. [DOI] [PubMed] [Google Scholar]
- 50.Plech T., Kaproń B., Paneth A., Kosikowska U., Malm A., Strzelczyk A., Stączek P., Świątek L., Rajtar B., Polz-Dacewicz M. Search for factors affecting antibacterial activity and toxicity of 1,2,4-triazole-ciprofloxacin hybrids. Eur. J. Med. Chem. 2015;97:94–103. doi: 10.1016/j.ejmech.2015.04.058. [DOI] [PubMed] [Google Scholar]
- 51.Mermer A., Faiz O., Demirbas A., Demirbas N., Alagumuthu M., Arumugam S. Piperazine-azole-fluoroquinolone hybrids: conventional and microwave irradiated synthesis, biological activity screening and molecular docking studies. Bioorg. Chem. 2019;85:308–318. doi: 10.1016/j.bioorg.2019.01.009. [DOI] [PubMed] [Google Scholar]
- 52.Cui S.F., Ren Y., Zhang S.L., Peng X.M., Damu G.L.V., Geng R.X., Zhou C.H. Synthesis and biological evaluation of a class of quinolone triazoles as potential antimicrobial agents and their interactions with calf thymus DNA. Bioorg. Med. Chem. Lett. 2013;23:3267–3272. doi: 10.1016/j.bmcl.2013.03.118. [DOI] [PubMed] [Google Scholar]
- 53.Gao L.Z., Xie Y.S., Li T., Huang W.L., Hu G.Q. Synthesis and antibacterial activity of novel [1,2,4]triazolo[3,4-h][1,8]naphthyridine-7-carboxylic acid derivatives. Chin. Chem. Lett. 2015;26:149–151. [Google Scholar]
- 54.Aggarwal N., Kumar R., Dureja P., Khurana J.M. Synthesis, antimicrobial evaluation and QSAR analysis of novel nalidixic acid based 1,2,4-triazole derivatives. Eur. J. Med. Chem. 2011;46:4089–4099. doi: 10.1016/j.ejmech.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 55.Mohamed N.G., Sheha M.M., Hassan H.Y., Avdel-Hafez L.J.M., Omar F.A. Synthesis, antimicrobial activity and molecular modeling study of 3-(5-amino-(2H)-1,2,4-triazol-3-yl]-naphthyridinones as potential DNA-gyrase inhibitors. Bioorg. Chem. 2018;81:599–611. doi: 10.1016/j.bioorg.2018.08.031. [DOI] [PubMed] [Google Scholar]
- 56.Prakash O., Aneja D.K., Hussain K., Lohan P., Ranjan P., Arora S., Sharma C., Aneja K.R. Synthesis and biological evaluation of dihydroindeno and indeno [1,2-e] [1,2,4]triazolo[3,4-b][1,3,4]thiadiazines as antimicrobial agents. Eur. J. Med. Chem. 2011;46:5065–5073. doi: 10.1016/j.ejmech.2011.08.019. [DOI] [PubMed] [Google Scholar]
- 57.Sumangala V., Poojary B., Chidananda N., Arulmoli T., Shenoy S. Facile synthesis, cytotoxic and antimicrobial activity studies of a new group of 6-aryl-3-[4-(methylsulfonyl)benzyl]-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines. Eur. J. Med. Chem. 2012;54:59–64. doi: 10.1016/j.ejmech.2012.04.024. [DOI] [PubMed] [Google Scholar]
- 58.Reddy C.S., Rao L.S., Sunitha B., Nagaraj A. Synthesis and antibacterial activity of N-substituted-[1,2,4]triazoles and 1,2,4-triazole[3,4-b][1,3,4]thiadiazines. Indian J. Chem. 2015;54B:1283–1289. [Google Scholar]
- 59.Abu-Hashem A.A., Hussein H.A.R., Abu-zied K.M. Synthesis of novel 1,2,4-triazolopyrimidines and their evaluation as antimicrobial agents. Med. Chem. Res. 2017;26:120–130. [Google Scholar]
- 60.Fan Z., Shi J., Luo N., Ding M., Bao X. Synthesis, crystal structure, and agricultural antimicrobial evaluation of novel quinazoline thioether derivatives incorporating the 1,2,4-triazolo[4,3-a]pyridine moiety. J. Agric. Food Chem. 2019;67:11598–11606. doi: 10.1021/acs.jafc.9b04733. [DOI] [PubMed] [Google Scholar]
- 61.Plech T., Wujec M., Kosikowska U., Malm A., Kapron B. Studies on the synthesis and antibacterial activity of 3,6-disubstituted 1,2,4-triazolo[3,4-b]1,3,4-thiadiazoles. Eur. J. Med. Chem. 2012;47:580–584. doi: 10.1016/j.ejmech.2011.10.055. [DOI] [PubMed] [Google Scholar]
- 62.Cui P., Li X., Zhu M., Wang B., Liu J., Chen H. Design, synthesis and antimicrobial activities of thiouracil derivatives containing triazolo-thiadiazole as SecA inhibitors. Eur. J. Med. Chem. 2017;127:159–165. doi: 10.1016/j.ejmech.2016.12.053. [DOI] [PubMed] [Google Scholar]
- 63.Barbuceanu S.F., Saramet G., Almajan G.L., Draghici C., Barbuceanu F., Bancescu G. New heterocyclic compounds from 1,2,4-triazole and 1,3,4-thiadiazole class bearing diphenylsulfone moieties. Synthesis, characterization and antimicrobial activity evaluation. Eur. J. Med. Chem. 2012;49:417–423. doi: 10.1016/j.ejmech.2012.01.031. [DOI] [PubMed] [Google Scholar]
- 64.Mange Y.J., Isloor A.M., Malladi S., Isloor S., Fun H.K. Synthesis and antimicrobial activities of some novel 1,2,4-triazole derivatives. Arab. J. Chem. 2013;6:177–181. [Google Scholar]
- 65.Hassan G.S., El-Messery S.M., Al-Omary F.A.M., Al-Rashood S.T., Shabayek M.I., Abulfadl Y.S., Habib E.-S.E., El-Hallouty S.M., Fayad W., Mohamed K.M., El-Menshawi B.S., El-Subbagh H.I. Nonclassical antifolates, Part 4. 5-(2-Aminothiazol-4-yl)-4-phenyl-4H-1,2,4-triazole-3-thiols as a new class of DHFR inhibitors: synthesis, biological evaluation and molecular modeling study. Eur. J. Med. Chem. 2013;66:135–145. doi: 10.1016/j.ejmech.2013.05.039. [DOI] [PubMed] [Google Scholar]
- 66.Yang L., Bao X.P. Synthesis of novel 1,2,4-triazole derivatives containing the quinazolinylpiperidinyl moiety and N-(substituted phenyl)acetamide group as efficient bactericides against the phytopathogenic bacterium Xanthomonas oryzae pv. oryzae. RSC Adv. 2017;7:34005–34011. [Google Scholar]
- 67.Cui J., Jin J., Chaudary A.S., Hsieh Y., Zhang H., Dai C., Damera K., Chen W., Tai P.C., Wang B. Design, synthesis and evaluation of triazole-pyrimidine analogues as SecA inhibitors. ChemMedChem. 2016;11:43–56. doi: 10.1002/cmdc.201500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shi Y., Zhou C.H. Synthesis and evaluation of a class of new coumarin triazole derivatives as potential antimicrobial agents. Bioorg. Med. Chem. Lett. 2011;21:956–960. doi: 10.1016/j.bmcl.2010.12.059. [DOI] [PubMed] [Google Scholar]
- 69.Fang B., Zhou C.H., Rao X.C. Synthesis and biological activities of novel amine-derived bis-azoles as potential antibacterial and antifungal agents. Eur. J. Med. Chem. 2010;45:4388–4398. doi: 10.1016/j.ejmech.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 70.Zhang Y., Tangadanchu V.K.R., Cheng Y., Yang R.-G., Lin J.-M., Zhou C.-H. Potential antimicrobial isopropanol-conjugated carbazole azoles as dual targeting inhibitors of Enterococcus faecalis. ACS Med. Chem. Lett. 2018;9:244–249. doi: 10.1021/acsmedchemlett.7b00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yin B.T., Yan C.Y., Peng X.M., Zhang S.L., Rasheed S., Geng R.X., Zhou C.H. Synthesis and biological evaluation of α-triazolyl chalcones as a new type of potential antimicrobial agents and their interaction with calf thymus DNA and human serum albumin. Eur. J. Med. Chem. 2014;71:148–159. doi: 10.1016/j.ejmech.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 72.Nam N.H., Parang K. Current targets for anticancer drug discovery. Curr. Drug Targets. 2003;4:159–179. doi: 10.2174/1389450033346966. [DOI] [PubMed] [Google Scholar]
- 73.Martínez-González S., Rodríguez-Arístegui S., de la Oliva C.A.G., Hernández A.I., Cantalapiedra E.G., Varela C., García A.B., Rabal O., Oyarzabal J., Bischoff J.R., Klett J., Albarrán M.I., Cebriá A., Ajenjo N., García-Serelde B., Gomez-Casero E., Cuadrado-Urbano M., Cebrián D., Blanco-Aparicio C., Pastor J. Discovery of novel triazolo[4,3-b]pyridazin-3-yl-quinoline derivatives as PIM inhibitors. Eur. J. Med. Chem. 2019;168:87–109. doi: 10.1016/j.ejmech.2019.02.022. [DOI] [PubMed] [Google Scholar]
- 74.Han M.I., Bekçi H., Uba A.I., Yıldırım Y., Karasulu E., Cumaoğlu A., Karasulu H.Y., Yelekçi K., Yılmaz O., Kuçukguzel S.G. Synthesis, molecular modeling, in vivo study, and anticancer activity of 1,2,4-triazole containing hydrazide-hydrazones derived from (S)-naproxen. Arch. Pharm. Chem. Life Sci. 2019;352 doi: 10.1002/ardp.201800365. [DOI] [PubMed] [Google Scholar]
- 75.Batran R.Z., Dawood D.H., El-Seginy S.A., Ali M.M., Maher T.J., Gugnani K.S., Rondon-Ortiz A.N. New coumarin derivatives as anti-breast and anti-cervical cancer agents targeting VEGFR-2 and p38α MAPK. Arch. Pharm. Chem. Life Sci. 2017;350 doi: 10.1002/ardp.201700064. [DOI] [PubMed] [Google Scholar]
- 76.Qin M., Zhai X., Xie H., Ma J., Lu K., Wang Y., Wang L., Gu Y., Gong P. Design and synthesis of novel 2-(4-(2-(dimethylamino)ethyl)-4H-1,2,4-triazol-3-yl)pyridines as potential antitumor agents. Eur. J. Med. Chem. 2014;81:47-58. doi: 10.1016/j.ejmech.2014.04.059. [DOI] [PubMed] [Google Scholar]
- 77.Qin M., Yan S., Wang L., Zhang H., Zhao Y., Wu S., Wu D., Gong P. Discovery of novel diaryl urea derivatives bearing a triazole moiety as potential antitumor agents. Eur. J. Med. Chem. 2016;115:1–13. doi: 10.1016/j.ejmech.2016.02.071. [DOI] [PubMed] [Google Scholar]
- 78.Liu J., Nie M., Wang Y., Hu J., Zhang F., Gao Y., Liu Y., Gong P. Design, synthesis and structure-activity relationships of novel 4-phenoxyquinoline derivatives containing 1,2,4-triazolone moiety as c-Met kinase inhibitors. Eur. J. Med. Chem. 2016;123:431–446. doi: 10.1016/j.ejmech.2016.07.059. [DOI] [PubMed] [Google Scholar]
- 79.Xu F., Yang Z., Jiang J., Pan W., Yang X., Wu J., Zhu Y., Wang J., Shou Q.-Y., Wu H. Synthesis, antitumor evaluation and molecular docking studies of [1,2,4]triazolo[4,3-b][1,2,4,5]tetrazine derivatives. Bioorg. Med. Chem. Lett. 2016;26:3042–3047. doi: 10.1016/j.bmcl.2016.05.007. [DOI] [PubMed] [Google Scholar]
- 80.Egile C., Kenigsberg M., Delaisi C., Begassat F., Do-Vale V., Mestadier J., Bonche F., Benard T., Nicolas J.P., Valence S., Lefranc C., Francesconi E., Castell C., Lefebvre A.M., Nemecek C., Calvet L., Goulaouic H. The selective intravenous inhibitor of the MET tyrosine kinase SAR125844 inhibits tumor growth in MET-amplified cancer. Mol. Canc. Therapeut. 2015;14:384–394. doi: 10.1158/1535-7163.MCT-14-0428. [DOI] [PubMed] [Google Scholar]
- 81.Zhan Z., Peng X., Liu Q., Chen F., Ji Y., Yao S., Xi Y., Lin Y., Chen T., Xu Y., Ai J., Geng M., Duan W. Discovery of 6-(difluoro(6-(4-fluorophenyl)-[1,2,4]triazolo[4,3-b][1,2,4]triazin-3-yl)methyl)quinoline as a highly potent and selective c-Met inhibitor. Eur. J. Med. Chem. 2016;116:239–251. doi: 10.1016/j.ejmech.2016.03.076. [DOI] [PubMed] [Google Scholar]
- 82.Boezio A.A., Copeland K.W., Rex K., Albrecht B.K., Bauer D., Bellon S.F., Boezio C., Broome M.A., Choquette D., Coxon A., Dussault I., Hirai S., Lewis R., Lin M.-H.J., Lohman J., Liu J., Peterson E.A., Potashman M., Shimanovich R., Teffera Y., Whittington D.A., Vaida K.R., Harmange J.-C. Discovery of (R)-6-(1-(8-fluoro-6-(1-methyl-1H-pyrazol-4-yl)-[1,2,4]triazolo[4,3-a]pyridin-3-yl)ethyl)-3-(2-methoxyethoxy)-1,6-naphthyridin-5(6H)-one (AMG 337), a potent and selective inhibitor of MET with high unbound target coverage and robust in vivo antitumor activity. J. Med. Chem. 2016;59:2328–2342. doi: 10.1021/acs.jmedchem.5b01716. [DOI] [PubMed] [Google Scholar]
- 83.Gu W., Dai Y., Qiang H., Shi W., Liao C., Zhao F., Huang W., Qian H. Discovery of novel 2-substituted-4-(2-fluorophenoxy) pyridine derivatives possessing pyrazolone and triazole moieties as dual c-Met/VEGFR-2 receptor tyrosine kinase inhibitors. Bioorg. Chem. 2017;72:116–122. doi: 10.1016/j.bioorg.2017.04.001. [DOI] [PubMed] [Google Scholar]
- 84.Peterson E.A., Teffera Y., Albrecht B.K., Bauer D., Bellon S.F., Boezio A., Boezio C., Broome M.A., Choquette D., Copeland K.W., Dussault I., Lewis R., Lin M.-H.J., Lohman J., Liu J., Potashman M., Rex K., Shimanovich R., Whittington D.A., Vaida K.R., Harmange J.-C. Discovery of potent and selective 8-fluorotriazolopyridine c-Met inhibitors. J. Med. Chem. 2015;58:2417–2430. doi: 10.1021/jm501913a. [DOI] [PubMed] [Google Scholar]
- 85.Yang Y., Zhang Y., Yang L., Zhao L., Si L., Zhang H., Liu Q., Zhou J. Discovery of imidazopyridine derivatives as novel c-Met kinase inhibitors: synthesis, SAR study, and biological activity. Bioorg. Chem. 2016;70:126–132. doi: 10.1016/j.bioorg.2016.12.002. [DOI] [PubMed] [Google Scholar]
- 86.Shahzad S.A., Yar M., Khan Z.A., Shahzadi L., Naqvi S.A.R., Mahmood A., Ullah S., Shaikh A.J., Sherazi T.A., Bale A.T., Kukułowicz J., Bajda M. Identification of 1,2,4-triazoles as new thymidine phosphorylase inhibitors: future anti-tumor drugs. Bioorg. Chem. 2019;85:209–220. doi: 10.1016/j.bioorg.2019.01.005. [DOI] [PubMed] [Google Scholar]
- 87.Bera H., Tan B.J., Sun L., Dolzhenko A.V., Chui W.-K., Chiu G.N.C. A structure activity relationship study of 1,2,4-triazolo[1,5-a][1,3,5]triazin-5,7-dione and its 5-thioxo analogues on anti-thymidine phosphorylase and associated anti-angiogenic activities. Eur. J. Med. Chem. 2013;67:325–334. doi: 10.1016/j.ejmech.2013.06.051. [DOI] [PubMed] [Google Scholar]
- 88.Eissa I.H., Metwaly A.M., Belal A., Mehany A.B.M., Ayyad R.R., El-Adl K., Mahdy H.A., Taghour M.S., El-Gamal K.M.A., El-Sawah M.E., Elmetwally S.A., Elhendawy M.A., Radwan M.M., ElSohly M.A. Discovery and antiproliferative evaluation of new quinoxalines as potential DNA intercalators and topoisomerase II inhibitors. Arch. Pharm. Chem. Life Sci. 2019;352 doi: 10.1002/ardp.201900123. [DOI] [PubMed] [Google Scholar]
- 89.Ibrahim M.K., Taghour M.S., Metwaly A.M., Belal A., Mehany A.B.M., Elhendawy M.A., Radwan M.M., Yassin A.M., El-Deeb N.M., Hafez E.E., ElSohly M.A., Eissa I.H. Design, synthesis, molecular modeling and anti-proliferative evaluation of novel quinoxaline derivatives as potential DNA intercalators and topoisomerase II inhibitors. Eur. J. Med. Chem. 2018;155:117–134. doi: 10.1016/j.ejmech.2018.06.004. [DOI] [PubMed] [Google Scholar]
- 90.Hou Y.P., Sun J., Pang Z.H., Lv P.C., Li D.D., Yan L., Zhang H.J., Zheng E.X., Zhao J., Zhu H.L. Synthesis and antitumor activity of 1,2,4-triazoles having 1,4-benzodioxan fragment as a novel class of potent methionine aminopeptidase type II inhibitors. Bioorg. Med. Chem. 2011;19:5948–5954. doi: 10.1016/j.bmc.2011.08.063. [DOI] [PubMed] [Google Scholar]
- 91.Cai H., Huang X., Xu S., Shen H., Zhang P., Huang Y., Jiang J., Sun Y., Jiang B., Wu X., Yao H., Xu J. Discovery of novel hybrids of diaryl-1,2,4-triazoles and caffeic acid as dual inhibitors of cyclooxygenase-2 and 5-lipoxygenase for cancer therapy. Eur. J. Med. Chem. 2016;108:89–103. doi: 10.1016/j.ejmech.2015.11.013. [DOI] [PubMed] [Google Scholar]
- 92.El-Husseiny W.M., El-Sayed M.A.-A., Abdel-Aziz N.I., El-Azab A.S., Asirig Y.A., Abdel-Aziz A.A.-M. Structural alterations based on naproxen scaffold: synthesis, evaluation of antitumor activity and COX-2 inhibition, and molecular docking. Eur. J. Med. Chem. 2018;158:134–143. doi: 10.1016/j.ejmech.2018.09.007. [DOI] [PubMed] [Google Scholar]
- 93.Sever B., Altıntop M.D., Kuş G., Özkurt M., Özdemir A., Kaplancıklı Z.A. Indomethacin based new triazolothiadiazine derivatives: synthesis, evaluation of their anticancer effects on T98 human glioma cell line related to COX-2 inhibition and docking studies. Eur. J. Med. Chem. 2016;113:179–186. doi: 10.1016/j.ejmech.2016.02.036. [DOI] [PubMed] [Google Scholar]
- 94.SitaRam G. Celik, Khloya P., Vullo D., Supuran C.T., Sharma P.K. Benzenesulfonamide bearing 1,2,4-triazole scaffolds as potent inhibitors of tumor associated carbonic anhydrase isoforms hCA IX and hCA XII. Bioorg. Med. Chem. 2014;22:1873–1882. doi: 10.1016/j.bmc.2014.01.055. [DOI] [PubMed] [Google Scholar]
- 95.Song Z., Liu Y., Dai Z., Liu W., Zhao K., Zhang T., Hu Y., Zhang X., Dai Y. Synthesis and aromatase inhibitory evaluation of 4-N-nitrophenyl substituted amino-4H-1, 2, 4-triazole derivatives. Bioorg. Med. Chem. 2016;24:4723–4730. doi: 10.1016/j.bmc.2016.08.014. [DOI] [PubMed] [Google Scholar]
- 96.Wang S., Zhao L.-J., Zheng Y.-C., Shen D.-D., Miao E.-F., Qiao X.-P., Zhao L.-J., Liu Y., Huang R., Yu B., Liu H.-M. Design, synthesis and biological evaluation of [1,2,4]triazolo[1,5-a]pyrimidines as potent lysine specific demethylase 1 (LSD1/KDM1A) inhibitors. Eur. J. Med. Chem. 2017;125:940–951. doi: 10.1016/j.ejmech.2016.10.021. [DOI] [PubMed] [Google Scholar]
- 97.Wang S., Li Z.-R., Suo F.-Z., Yuan X.-H., Yu B., Liu H.-M. Synthesis, structure-activity relationship studies and biological characterization of new [1,2,4]triazolo[1,5-a]pyrimidine-based LSD1/KDM1A inhibitors. Eur. J. Med. Chem. 2019;167:388–401. doi: 10.1016/j.ejmech.2019.02.039. [DOI] [PubMed] [Google Scholar]
- 98.Liscio P., Carotti A., Asciutti S., Karlberg T., Bellocchi D., Llacuna L., Macchiarulo A., Aaronson S.A., Schüler H., Pellicciari R., Camaioni E. Design, synthesis, crystallographic studies, and preliminary biological appraisal of new substituted triazolo[4,3-b]pyridazin-8-amine derivatives as tankyrase inhibitors. J. Med. Chem. 2014;57:2807–2812. doi: 10.1021/jm401356t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bi X., Li J., Li J., Shi W., Dai Y., Li Q., Zhang W., Huang W., Qian H., Jiang C. Design, synthesis and biological evaluation of novel 4,5-dihydro-[1,2,4]triazolo[4,3-f]pteridine derivatives as potential BRD4 inhibitors, Bioorg. Med. Chem. 2019;27:2813–2821. doi: 10.1016/j.bmc.2019.05.006. [DOI] [PubMed] [Google Scholar]
- 100.Saez-Calvo G., Sharma A., Balaguer F.A., Barasoain I., Rodriguez-Salarichs J., Olieric N., Munoz-Hernandez H., Berbis M.A., Wendeborn S., Penalva M.A., Matesanz R., Canales A., Prota A.E., Jimenez-Barbero J., Andreu J.M., Lamberth C., Steinmetz M.O., Diaz J.F. Triazolopyrimidines are microtubule-stabilizing agents that bind the vinca inhibitor site of tubulin. Cell Chem. Biol. 2017;24:737–750. doi: 10.1016/j.chembiol.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 101.Alswah M., Bayoumi A.H., Elgamal K., Elmorsy A., Ihmaid S., Ahmed H.E.A. Design, synthesis and cytotoxic evaluation of novel chalcone derivatives bearing triazolo[4,3-a]-quinoxaline moieties as potent anticancer agents with dual EGFR kinase and tubulin polymerization inhibitory effects. Molecules. 2018;23:48. doi: 10.3390/molecules23010048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Driowya M., Leclercq J., Verones V., Barczyk A., Lecoeur M., Renault N., Flouquet N., Ghinet A., Berthelot P., Lebegue N. Synthesis of triazoloquinazolinone based compounds as tubulin polymerization inhibitors and vascular disrupting agents. Eur. J. Med. Chem. 2016;115:393–405. doi: 10.1016/j.ejmech.2016.03.056. [DOI] [PubMed] [Google Scholar]
- 103.El-Sherief H.A.M., Youssif B.G.M., Bukhari S.N.A., Abdel-Aziz M., Abdel-Rahman H.M. Novel 1,2,4-triazole derivatives as potential anticancer agents: design, synthesis, molecular docking and mechanistic studies. Bioorg. Chem. 2018;76:314–325. doi: 10.1016/j.bioorg.2017.12.013. [DOI] [PubMed] [Google Scholar]
- 104.Yang F., He C.-P., Diao P.-C., Hong K.H., Rao J.-J., Zhao P.-L. Discovery and optimization of 3,4,5-trimethoxyphenyl substituted triazolylthioacetamides as potent tubulin polymerization inhibitors. Bioorg. Med. Chem. Lett. 2019;29:22–27. doi: 10.1016/j.bmcl.2018.11.024. [DOI] [PubMed] [Google Scholar]
- 105.Mustafa M., Abdelhamid D., Abdelhafez E.M.N., Ibrahim M.A.A., Gamal-Eldeen A.M., Aly O.M. Synthesis, antiproliferative, anti-tubulin activity, and docking study of new 1,2,4-triazoles as potential combretastatin analogues. Eur. J. Med. Chem. 2017;141:293–305. doi: 10.1016/j.ejmech.2017.09.063. [DOI] [PubMed] [Google Scholar]
- 106.Romagnoli R., Baraldi P.G., Cruz-Lopez O., Cara C.L., Carrion M.D., Brancale A., Hamel E., Chen L., Bortolozzi R., Basso G., Viola G. Synthesis and antitumor activity of 1,5-disubstituted 1,2,4-triazoles as cis-restricted combretastatin analogues. J. Med. Chem. 2010;53:4248–4258. doi: 10.1021/jm100245q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Romagnoli R., Prencipe F., Oliva P., Baraldi S., Baraldi P.G., Brancale A., Ferla S., Hamel E., Bortolozzi R., Viola G. 3-Aryl/heteroaryl-5-amino-1-(3′,4′,5′-trimethoxybenzoyl)-1,2,4-triazoles as antimicrotubule agents. Design, synthesis, antiproliferative activity and inhibition of tubulin polymerization. Bioorg. Chem. 2018;80:361–374. doi: 10.1016/j.bioorg.2018.06.037. [DOI] [PubMed] [Google Scholar]
- 108.Wang X.M., Xu J., Li Y.P., Li H., Jiang C.S., Yang G.D., Lu S.M., Zhang S.Q. Synthesis and anticancer activity evaluation of a series of [1,2,4]triazolo[1,5-a]pyridinylpyridines in vitro and in vivo. Eur. J. Med. Chem. 2013;67:243–251. doi: 10.1016/j.ejmech.2013.06.052. [DOI] [PubMed] [Google Scholar]
- 109.Xu F., Yang Z.-Z., Ke Z.-L., Xi L.-M., Yan Q.-D., Yang W.-Q., Zhu L.-Q., Lin F.-L., Lv W.-K., Wu H.-G., Wang J., Li H.-B. Synthesis, antitumor evaluation and 3D-QSAR studies of [1,2,4]triazolo[4,3-b][1,2,4,5]tetrazine derivatives. Bioorg. Med. Chem. Lett. 2016;26:4580–4586. doi: 10.1016/j.bmcl.2016.08.078. [DOI] [PubMed] [Google Scholar]
- 110.Fares M., Abou-Seri S.M., Abdel-Aziz H.A., Abbas S.E.-S., Youssef M.M., Eladwy R.A. Synthesis and antitumor activity of pyrido [2,3-d]pyrimidine and pyrido[2,3-d] [1,2,4]triazolo[4,3-a]pyrimidine derivatives that induce apoptosis through G1 cell-cycle arrest. Eur. J. Med. Chem. 2014;83:155–166. doi: 10.1016/j.ejmech.2014.06.027. [DOI] [PubMed] [Google Scholar]
- 111.Hou Y., Zhu L., Li Z., Shen Q., Xu Q., Li W., Liu Y., Gong P. Design, synthesis and biological evaluation of novel 7-amino-[1,2,4]triazolo[4,3-f]pteridinone, and 7-aminotetrazolo[1,5-f]pteridinone derivative as potent antitumor agents. Eur. J. Med. Chem. 2019;163:690–709. doi: 10.1016/j.ejmech.2018.12.009. [DOI] [PubMed] [Google Scholar]
- 112.Kandeel M.M., Kamal A.M., Abdelall E.K.A., Elshemy H.A.H. Synthesis of novel chromene derivatives of expected antitumor activity. Eur. J. Med. Chem. 2013;59:183–193. doi: 10.1016/j.ejmech.2012.11.011. [DOI] [PubMed] [Google Scholar]
- 113.Kandeel M.M., Refaat H.M., Kassab A.E., Shahin I.G., Abdelghany T.M. Synthesis, anticancer activity and effects on cell cycle profile and apoptosis of novel thieno[2,3-d]pyrimidine and thieno[3,2-e]triazolo[4,3-c]pyrimidine derivatives. Eur. J. Med. Chem. 2015;90:620–632. doi: 10.1016/j.ejmech.2014.12.009. [DOI] [PubMed] [Google Scholar]
- 114.Botros S., Khalil O.M., Kamel M.M., El-Dash Y.S. Synthesis, characterization and cytotoxicity of substituted [1]benzothieno[3,2-e][1,2,4]triazolo[4,3-a]pyrimidines. Acta Chim. Slov. 2017;64:102–116. doi: 10.17344/acsi.2016.2901. [DOI] [PubMed] [Google Scholar]
- 115.Mamta, Aggarwal R., Sadana R., Ilag J., Sumran G. Synthesis and bioevaluation of 6-chloropyridazin-3-yl hydrazones and 6-chloro-3-substituted-[1,2,4]triazolo[4,3-b]pyridazines as cytotoxic agents. Bioorg. Chem. 2019;86:288–295. doi: 10.1016/j.bioorg.2019.01.049. [DOI] [PubMed] [Google Scholar]
- 116.Issa D.A.E., Habiba N.S., Wahab A.E.A. Design, synthesis and biological evaluation of novel 1,2,4-triazolo and 1,2,4-triazino[4,3-a]quinoxalines as potential anticancer and antimicrobial agents. Med. Chem. Commun. 2015;6:202–211. [Google Scholar]
- 117.Wu L., Zhang C., Li W. Regioselective synthesis of 6-aryl-benzo[h][1,2,4]-triazolo[5,1-b]quinazoline-7,8-diones as potent antitumoral agents. Bioorg. Med. Chem. Lett. 2013;23:5002–5005. doi: 10.1016/j.bmcl.2013.06.040. [DOI] [PubMed] [Google Scholar]
- 118.Xue D.-Q., Zhang X.-Y., Wang C.-J., Ma L.-Y., Zhu N., He P., Shao K.-P., Chen P.-J., Gu Y.-F., Zhang X.-S., Wang C.-F., Ji C.-H., Zhang Q.-R., Liu H.-M. Synthesis and anticancer activities of novel 1,2,4-triazolo[3,4-a]phthalazine derivatives. Eur. J. Med. Chem. 2014;85:235–244. doi: 10.1016/j.ejmech.2014.07.031. [DOI] [PubMed] [Google Scholar]
- 119.Husain A., Rashid M., Shaharyar M., Siddiqui A.A., Mishra R. Benzimidazole clubbed with triazolo-thiadiazoles and triazolo-thiadiazines new anticancer agents. Eur. J. Med. Chem. 2013;62:785–798. doi: 10.1016/j.ejmech.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 120.Hu G., Wang C., Xin X., Li S., Li Z., Zhao Y., Gong P. Design, synthesis and biological evaluation of novel 2,4-diaminopyrimidine derivatives as potent antitumor agents. New J. Chem. 2019;43:10190. [Google Scholar]
- 121.Zhang B., Li Y.-H., Liu Y., Chen Y.-R., Pan E.-S., You W.-W., Zhao P.-L. Design, synthesis and biological evaluation of novel 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines bearing furan and thiophene nucleus. Eur. J. Med. Chem. 2015;103:335–342. doi: 10.1016/j.ejmech.2015.08.053. [DOI] [PubMed] [Google Scholar]
- 122.Zhao P.L., Duan A.N., Zou M., Yang H.K., You W.W., Wu S.G. Synthesis and cytotoxicity of 3,4-disubstituted-5-(3,4,5-trimethoxyphenyl)-4H-1,2,4-triazoles and novel 5,6-dihydro-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazole derivatives bearing 3,4,5-trimethoxyphenyl moiety. Bioorg. Med. Chem. Lett. 2012;22:4471–4474. doi: 10.1016/j.bmcl.2012.03.023. [DOI] [PubMed] [Google Scholar]
- 123.Kamel M.M., Abdo N.Y.M. Synthesis of novel 1,2,4-triazoles, triazolothiadiazines and triazolothiadiazoles as potential anticancer agents. Eur. J. Med. Chem. 2014;86:75–80. doi: 10.1016/j.ejmech.2014.08.047. [DOI] [PubMed] [Google Scholar]
- 124.Kulabas N., Tatar E., Ozakpınar O.B., Ozsavcı D., Pannecouque C., Clercq E.D., Küçükgüzel I. Synthesis and antiproliferative evaluation of novel 2-(4H-1,2,4-triazole-3-ylthio)acetamide derivatives as inducers of apoptosis in cancer cells. Eur. J. Med. Chem. 2016;121:58–70. doi: 10.1016/j.ejmech.2016.05.017. [DOI] [PubMed] [Google Scholar]
- 125.Zhao P.L., Mab W.F., Duan A.N., Zou M., Yan Y.C., You W.W., Wu S.G. One-pot synthesis of novel isoindoline-1,3-dione derivatives bearing 1,2,4-triazole moiety and their preliminary biological evaluation. Eur. J. Med. Chem. 2012;54:813–822. doi: 10.1016/j.ejmech.2012.06.041. [DOI] [PubMed] [Google Scholar]
- 126.Zhao P.-L., Chen P., Li Q., Hu M.-J., Diao P.-C., Pan E.-S., You W.-W. Design, synthesis and biological evaluation of novel 3-alkylsulfanyl-4-amino-1,2,4-triazole derivatives. Bioorg. Med. Chem. Lett. 2016;26:3679–3683. doi: 10.1016/j.bmcl.2016.05.086. [DOI] [PubMed] [Google Scholar]
- 127.Wang X.F., Zhang S., Li B.L., Zhao J.J., Liu Y.M., Zhang R.L., Li B., Chen B.Q. Synthesis and biological evaluation of disulfides bearing 1,2,4-triazole moiety as antiproliferative agents. Med. Chem. Res. 2017;26:3367–3374. [Google Scholar]
- 128.Tokala R., Bale S., Janrao I.P., Vennela A., Kumar N.P., Senwar K.R., Godugu C., Shankaraiah N. Synthesis of 1,2,4-triazole-linked urea/thiourea conjugates as cytotoxic and apoptosis inducing agents. Bioorg. Med. Chem. Lett. 2018;28:1919–1924. doi: 10.1016/j.bmcl.2018.03.074. [DOI] [PubMed] [Google Scholar]
- 129.Mavrova A.T., Wesselinova D., Tsenov J.A., Lubenov L.A. Synthesis and antiproliferative activity of some new thieno[2,3-d]pyrimidin-4(3H)-ones containing 1,2,4-triazole and 1,3,4-thiadiazole moiety. Eur. J. Med. Chem. 2014;86:676–683. doi: 10.1016/j.ejmech.2014.09.032. [DOI] [PubMed] [Google Scholar]
- 130.Boraei A.T.A., Gomaa M.S., El Ashry E.S.H., Duerkop A. Design, selective alkylation and X-ray crystal structure determination of dihydro-indolyl-1,2,4-triazole-3-thione and its 3-benzylsulfanyl analogue as potent anticancer agents. Eur. J. Med. Chem. 2017;125:360–371. doi: 10.1016/j.ejmech.2016.09.046. [DOI] [PubMed] [Google Scholar]
- 131.Gomha S.M., Abdel-aziz H.M., Badrey M.G., Abdulla M.M. Efficient synthesis of some new 1,3,4-thiadiazoles and 1,2,4-triazoles linked to pyrazolylcoumarin ring system as potent 5α-reductase inhibitors. J. Heterocycl. Chem. 2019;56:1275–1282. [Google Scholar]
- 132.Kahveci B., Yilmaz F., Menteşe E., Ülker S. Design, synthesis, and biological evaluation of coumarin-triazole hybrid molecules as potential antitumor and pancreatic lipase agents. Arch. Pharm. Chem. Life Sci. 2017;350 doi: 10.1002/ardp.201600369. [DOI] [PubMed] [Google Scholar]
- 133.Shaikh S.K.J., Sannaikar M.S., Kumbar M.N., Bayannavar P.K., Kamble R.R., Inamdar S.R., Joshi S.D. Microwave-expedited green synthesis, photophysical, computational studies of coumarin-3-yl-thiazol-3-yl-1,2,4-triazolin-3-ones and their anticancer activity. Chemistry Select. 2018;3:4448–4462. [Google Scholar]
- 134.Abuelizz H.A., Awad H.M., Marzouk M., Nasr F.A., Alqahtani A.S., Bakheit A.H., Naglahgh A.M., Al-Salahi R. Synthesis and biological evaluation of 4-(1H-1,2,4-triazol-1-yl)benzoic acid hybrids as anticancer agents. RSC Adv. 2019;9:19065. doi: 10.1039/c9ra03151k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Deng X.Q., Quan L.N., Song M.X., Wei C.X., Quan Z.S. Zhe-Shan Quan, Synthesis and anticonvulsant activity of 7-phenyl-6,7-dihydro-[1,2,4]triazolo[1,5-a]pyrimidin-5(4H)-ones and their derivatives. Eur. J. Med. Chem. 2011;46:2955–2963. doi: 10.1016/j.ejmech.2011.04.020. [DOI] [PubMed] [Google Scholar]
- 136.Faizi M., Dabirian S., Tajali H., Ahmadi F., Zavareh E.R., Shahhosseini S., Tabatabai S.A. Novel agonists of benzodiazepine receptors: design, synthesis, binding assay and pharmacological evaluation of 1,2,4-triazolo[1,5-a]pyrimidinone and 3-amino-1,2,4-triazole derivatives. Bioorg. Med. Chem. 2015;23:480–487. doi: 10.1016/j.bmc.2014.12.016. [DOI] [PubMed] [Google Scholar]
- 137.Huang L., Ding J., Li M., Hou Z., Geng Y., Li X., Yu H. Discovery of [1,2,4]-triazolo[1,5-a]pyrimidine-7(4H)-one derivatives as positive modulators of GABAA1 receptor with potent anticonvulsant activity and low toxicity. Eur. J. Med. Chem. 2020;185:111824. doi: 10.1016/j.ejmech.2019.111824. [DOI] [PubMed] [Google Scholar]
- 138.Deng X.Q., Wei C.X., Li F.N., Sun Z.G., Quan Z.S. Design and synthesis of 10-alkoxy-5,6-dihydro-triazolo[4,3-d]benzo[f][1,4]oxazepine derivatives with anticonvulsant activity. Eur. J. Med. Chem. 2010;45:3080–3086. doi: 10.1016/j.ejmech.2010.03.041. [DOI] [PubMed] [Google Scholar]
- 139.Deng X.Q., Song M.X., Wang S.B., Quan Z.S. Synthesis and evaluation of the anticonvulsant activity of 8-alkoxy-4,5-dihydrobenzo[b][1,2,4]triazolo[4,3-d][1,4]thiazepine derivatives. J. Enzym. Inhib. Med. Chem. 2014;29:272–280. doi: 10.3109/14756366.2013.776555. [DOI] [PubMed] [Google Scholar]
- 140.Deng X.Q., Song M.X., Gong G.H., Wang S.B., Quan Z.S. Synthesis and anticonvulsant evaluation of some new 6-(substituted-phenyl)thiazolo[3,2-b][1,2,4]triazole derivatives in mice, Iran. J. Pharm. Res. 2014;13:459–469. [PMC free article] [PubMed] [Google Scholar]
- 141.Cao X., Wang S.B., Deng X.Q., Liu D.C., Quan Z.S. Synthesis and anticonvulsant activity evaluation of 7-alkoxy-2,4-dihydro-1H-benzo[b][1,2,4]triazolo[4,3-d][1,4]thiazin-1-ones in various murine experimental seizure models. Med. Chem. Res. 2014;23:1829–1838. [Google Scholar]
- 142.Zhang H.J., Jin P., Wang S.B., Li F.N., Guan L.P., Quan Z.S. Synthesis and anticonvulsant activity evaluation of 4-phenyl-[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-one and its derivatives. Arch. Pharm. Chem. Life Sci. 2015;348:564–574. doi: 10.1002/ardp.201500115. [DOI] [PubMed] [Google Scholar]
- 143.Zhang H.J., Wang S.B., Wen X., Li J.Z., Quan Z.S. Design, synthesis, and evaluation of the anticonvulsant and antidepressant activities of pyrido[2,3-d]pyrimidine derivatives. Med. Chem. Res. 2016;25:1287–1298. [Google Scholar]
- 144.Guan L.P., Sui X., Deng X.Q., Quan Y.C., Quan Z.S. Synthesis and anticonvulsant activity of a new alkoxy-[1,2,4]triazolo[4,3-b]pyridazine. Eur. J. Med. Chem. 2010;45:1746–1752. doi: 10.1016/j.ejmech.2009.12.077. [DOI] [PubMed] [Google Scholar]
- 145.Botros S., Khalil N.A., Naguib B.H., El-Dash Y. Synthesis and anticonvulsant activity of new phenytoin derivatives. Eur. J. Med. Chem. 2013;60:57–63. doi: 10.1016/j.ejmech.2012.11.025. [DOI] [PubMed] [Google Scholar]
- 146.Plech T., Kaproń B., Łuszczki J.J., Wujec M., Paneth A., Siwek A., Kołaczkowski M., Żołnierek M., Nowak G. Studies on the anticonvulsant activity and influence on GABAergic neurotransmission of 1,2,4-triazole-3-thione-based compounds. Molecules. 2014;19:11279–11299. doi: 10.3390/molecules190811279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Plech T., Luszczki J.J., Wujec M., Flieger J., Pizo M. Synthesis, characterization and preliminary anticonvulsant evaluation of some 4-alkyl-1,2,4-triazoles. Eur. J. Med. Chem. 2013;60:208–215. doi: 10.1016/j.ejmech.2012.11.026. [DOI] [PubMed] [Google Scholar]
- 148.Plech T., Kapron B., Łuszczki J.J., Paneth A., Siwek A., Kołaczkowski M., Zołnierek M., Nowak G. Studies on the anticonvulsant activity of 4-alkyl-1,2,4-triazole-3-thiones and their effect on GABAergic system. Eur. J. Med. Chem. 2014;86:690–699. doi: 10.1016/j.ejmech.2014.09.034. [DOI] [PubMed] [Google Scholar]
- 149.Deng X.Q., Song M.X., Zheng Y., Quan Z.S. Design, synthesis and evaluation of the antidepressant and anticonvulsant activities of triazole-containing quinolinones. Eur. J. Med. Chem. 2014;73:217–224. doi: 10.1016/j.ejmech.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 150.Wang S.B., Jin P., Li F.N., Quan Z.S. Synthesis and anticonvulsant activity of novel purine derivatives. Eur. J. Med. Chem. 2014;84:574–583. doi: 10.1016/j.ejmech.2014.07.074. [DOI] [PubMed] [Google Scholar]
- 151.Sari S., Karakurt A., Uslu H., Kaynak F.B., Calis U., Dalkara S. New (arylalkyl)azole derivatives showing anticonvulsant effects could have VGSC and/or GABAAR affinity according to molecular modeling studies. Eur. J. Med. Chem. 2016;124:407–416. doi: 10.1016/j.ejmech.2016.08.032. [DOI] [PubMed] [Google Scholar]
- 152.Abuelhassan A.H., Badran M.M., Hassan H.A., Abdelhamed D., Elnabtity S., Aly O.M. Design, synthesis, anticonvulsant activity, and pharmacophore study of new 1,5-diaryl-1H-1,2,4-triazole-3-carboxamide derivatives. Med. Chem. Res. 2018;27:928–938. [Google Scholar]
- 153.Liu D.C., Zhang H.J., Jin C.M., Quan Z.S. Synthesis and biological evaluation of novel benzothiazole derivatives as potential anticonvulsant agents. Molecules. 2016;21:e164. doi: 10.3390/molecules21030164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Krishna K.M., Inturi B., Pujar G.V., Purohit M.N., Vijaykumar G.S. Design, synthesis and 3D-QSAR studies of new diphenylamine containing 1,2,4-triazoles as potential antitubercular agents. Eur. J. Med. Chem. 2014;84:516–529. doi: 10.1016/j.ejmech.2014.07.051. [DOI] [PubMed] [Google Scholar]
- 155.Castelino P.A., Dasappa J.P., Bhat K.G., Joshi S.A., Jalalpure S. Some novel Schiff bases of [1,2,4]triazole bearing haloarene moiety synthesis and evaluation of antituberculosis propertiesand neutrophil function test. Med. Chem. Res. 2016;25:83–93. [Google Scholar]
- 156.Karabanovich G., Dusek J., Savkova K., Pavlis O., Pavkova I., Korabecny J., Kucera T., Vlckova H.K., Huszar S., Konyarikova Z., Konecna K., Jand’ourek O., Stolaríkova J., Kordulakova J., Vavrova K., Pavek P., Klimesova V., Hrabalek A., Mikusova K., Roh J. Development of 3,5-dinitrophenyl-containing 1,2,4-triazoles and their trifluoromethyl analogues as highly efficient antitubercular agents inhibiting decaprenylphosphoryl-β-D-ribofuranose 2′-oxidase. J. Med. Chem. 2019;62:8115–8139. doi: 10.1021/acs.jmedchem.9b00912. [DOI] [PubMed] [Google Scholar]
- 157.Dixit P.P., Dixit P.P., Thore S.N. Hybrid triazoles: design and synthesis as potential dual inhibitor of growth and efflux inhibition in tuberculosis. Eur. J. Med. Chem. 2016;107:38–47. doi: 10.1016/j.ejmech.2015.10.054. [DOI] [PubMed] [Google Scholar]
- 158.Veau D., Krykun S., Mori G., Orena B.S., Pasca M.R., Frongia C., Lobjois V., Chassaing S., Lherbet C., Baltas M. Triazolophthalazines: easily accessible compounds with potent antitubercular activity. ChemMedChem. 2016;11:1078–1089. doi: 10.1002/cmdc.201600085. [DOI] [PubMed] [Google Scholar]
- 159.Zuniga E.S., Korkegian A., Mullen S., Hembre E.J., Ornstein P.L., Cortez G., Biswas K., Kumar N., Cramer J., Masquelin T., Hipskind P.A., Odingo J., Parish T. The synthesis and evaluation of triazolopyrimidines as anti-tubercular agents. Bioorg. Med. Chem. 2017;25:3922–3946. doi: 10.1016/j.bmc.2017.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kumar G.V.S., Rajendraprasad Y., Mallikarjuna B.P., Chandrashekar S.M., Kistayya C. Synthesis of some novel 2-substituted-5-[isopropylthiazole] clubbed 1, 2, 4-triazole and 1, 3, 4-oxadiazoles as potential antimicrobial and antitubercular agents. Eur. J. Med. Chem. 2010;45:2063–2074. doi: 10.1016/j.ejmech.2010.01.045. [DOI] [PubMed] [Google Scholar]
- 161.Kumar G.V.S., Rajendraprasad Y., Mallikarjuna B.P., Chandrashekar S.M. Synthesis and pharmacological evaluation of clubbed isopropylthiazole derived triazolothiadiazoles, triazolothiadiazines and mannich bases as potential antimicrobial and antitubercular agents. Eur. J. Med. Chem. 2010;45:5120–5129. doi: 10.1016/j.ejmech.2010.08.023. [DOI] [PubMed] [Google Scholar]
- 162.Bonde C.G., Peepliwal A., Gaikwad N.J. Synthesis and antimycobacterial activity of azetidine-, quinazoline-, and triazolo-thiadiazole-containing pyrazines. Arch. Pharm. Chem. Life Sci. 2010;343:228–236. doi: 10.1002/ardp.200900165. [DOI] [PubMed] [Google Scholar]
- 163.Li Z., Liu Y., Bai X., Deng Q., Wang J., Zhang G., Xiao C., Mei Y., Wang Y. SAR studies on 1,2,4-triazolo[3,4-b][1,3,4]thiadiazoles as inhibitors of Mtb shikimate dehydrogenase for the development of novel antitubercular agents. RSC Adv. 2015;5:97089–97101. [Google Scholar]
- 164.Li Z., Bai X., Deng Q., Zhang G., Zhou L., Liu Y., Wang J., Wang Y. Preliminary SAR and biological evaluation of antitubercular triazolothiadiazine derivatives against drug-susceptible and drug-resistant Mtb strains. Bioorg. Med. Chem. 2017;25:213–220. doi: 10.1016/j.bmc.2016.10.027. [DOI] [PubMed] [Google Scholar]
- 165.Deng Q., Meng J., Liu Y., Guan Y., Xiao C. IMB-SD62, a triazolothiadiazoles derivative with promising action against tuberculosis. Tuberculosis. 2018;112:37–44. doi: 10.1016/j.tube.2018.07.006. [DOI] [PubMed] [Google Scholar]
- 166.Papadopoulou M.V., Bloomer W.D., Rosenzweig H.S., Arena A., Arrieta F., Rebolledo J.C.J., Smith D.K. Nitrotriazole and imidazole-based amides and sulfonamides as antitubercular agents. Antimicrob. Agents Chemother. 2014;58:6828–6836. doi: 10.1128/AAC.03644-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Papadopoulou M.V., Bloomer W.D., Rosenzweig H.S. The antitubercular activity of various nitro(triazole/imidazole)-based compounds. Bioorg. Med. Chem. 2017;25:6039–6048. doi: 10.1016/j.bmc.2017.09.037. [DOI] [PubMed] [Google Scholar]
- 168.Somagond S.M., Kamble R.R., Bayannavar P.K., Shaikh S.K.J., Joshi S.D., Kumbar V.M., Nesaragi A.R., Kariduraganavar M.Y. Click chemistry based regioselective one-pot synthesis of coumarin-3-yl-methyl-1,2,3-triazolyl-1,2,4-triazol-3(4H)-ones as newer potent antitubercular agents. Arch. Pharm. Chem. Life Sci. 2019;352 doi: 10.1002/ardp.201900013. [DOI] [PubMed] [Google Scholar]
- 169.Ozadali K., Tan O.U., Yogeeswari P., Dharmarajan S., Balkan A. Synthesis and antimycobacterial activities of some new thiazolylhydrazone derivatives. Bioorg. Med. Chem. Lett. 2014;24:1695–1697. doi: 10.1016/j.bmcl.2014.02.052. [DOI] [PubMed] [Google Scholar]
- 170.Desai N.H.P., Bairwa R., Kakwani M., Tawari N., Ray M.K., Rajan M.G., Degani M. Novel 4H-1,2,4-triazol-3-yl cycloalkanols as potent antitubercular agents. Med. Chem. Res. 2013;22:401–408. [Google Scholar]
- 171.Pogaku V., Krishna V.S., Sriram D., Rangan K., Basavoju S. Ultrasonication-ionic liquid synergy for the synthesis of new potent antituberculosis 1,2,4-triazol-1-yl-pyrazole based spirooxindolopyrrolizidines. Bioorg. Med. Chem. Lett. 2019;29:1682–1687. doi: 10.1016/j.bmcl.2019.04.026. [DOI] [PubMed] [Google Scholar]
- 172.Goma’a H.A.M., Ghaly M.A., Abou-zeid L.A., Badria F.A., Shehata I.A., El-Kerdawy M.M. Synthesis, biological evaluation and in silico studies of 1,2,4-triazole and 1,3,4-thiadiazole derivatives as antiherpetic agents. Chemistry Select. 2019;4:6421–6428. [Google Scholar]
- 173.Henen M.A., El Bialy S.A.A., Goda F.E., Nasr M.N.A., Eisa H.M. [1,2,4]Triazolo[4,3-a]quinoxaline: synthesis, antiviral, and antimicrobial activities. Med. Chem. Res. 2012;21:2368–2378. [Google Scholar]
- 174.Biswas B.K., Malpani Y.R., Ha N., Kwon D.H., Shin J.S., Kim H.S., Kim C., Han S.B., Lee C.K., Jung Y.S. Enterovirus inhibitory activity of C-8-tert-butyl substituted 4-aryl-6,7,8,9-tetrahydrobenzo[4,5]thieno[3,2-e][1,2,4]triazolo[4,3-a]pyrimidin-5(4H)-ones. Bioorg. Med. Chem. Lett. 2017;27:3582–3585. doi: 10.1016/j.bmcl.2017.05.030. [DOI] [PubMed] [Google Scholar]
- 175.El-Zahabi H.S.A. Synthesis, characterization, and biological evaluation of some novel quinoxaline derivatives as antiviral agents. Arch. Pharm. Chem. Life Sci. 2017;350 doi: 10.1002/ardp.201700028. [DOI] [PubMed] [Google Scholar]
- 176.Massari S., Nannetti G., Desantis J., Muratore G., Sabatini S., Manfroni G., Mercorelli B., Cecchetti V., Palu G., Cruciani G., Loregian A., Goracci L., Tabarrini O. A broad anti-influenza hybrid small molecule that potently disrupts the interaction of polymerase acidic protein-basic protein 1 (PAPB1) subunits. J. Med. Chem. 2015;58:3830–3842. doi: 10.1021/acs.jmedchem.5b00012. [DOI] [PubMed] [Google Scholar]
- 177.Zaher N.H., Mostafa M.I., Altaher A.Y. Design, synthesis and molecular docking of novel triazole derivatives as potential CoV helicase inhibitors. Acta Pharm. 2020;70:145–159. doi: 10.2478/acph-2020-0024. [DOI] [PubMed] [Google Scholar]
- 178.Zhan P., Chen X., Li X., Li D., Tian Y., Chen W., Pannecouque C., Clercq E.D., Liu X. Arylazolylthioacetanilide. Part 8: design, synthesis and biological evaluation of novel 2-(2-(2,4-dichlorophenyl)-2H-1,2,4-triazol-3-ylthio)-N-arylacetamides as potent HIV-1 inhibitors. Eur. J. Med. Chem. 2011;46:5039–5045. doi: 10.1016/j.ejmech.2011.08.011. [DOI] [PubMed] [Google Scholar]
- 179.Wang L., Tian Y., Chen W., Liu H., Zhan P., Li D., Liu H., De Clercq E., Pannecouque C., Liu X. Fused heterocycles bearing bridgehead nitrogen as potent HIV-1 NNRTIs. Part 2: discovery of novel [1,2,4]triazolo[1,5-a]pyrimidines using a structure-guided core-refining approach. Eur. J. Med. Chem. 2014;85:293–303. doi: 10.1016/j.ejmech.2014.07.104. [DOI] [PubMed] [Google Scholar]
- 180.Huang B., Li C., Chen W., Liu T., Yu M., Fu L., Sun Y., Liu H., Clercq E.D., Pannecouque C., Balzarini J., Zhan P., Liu X. Fused heterocycles bearing bridgehead nitrogen as potent HIV-1 NNRTIs. Part 3: optimization of [1,2,4]triazolo[1,5-a]pyrimidine core via structure-based and physicochemical property-driven approaches. Eur. J. Med. Chem. 2015;92:754–765. doi: 10.1016/j.ejmech.2015.01.042. [DOI] [PubMed] [Google Scholar]
- 181.Wang Z., Yu Z., Kang D., Zhang J., Tian Y., Daelemans D., Clercq E.D., Pannecouque C., Zhan P., Liu X. Design, synthesis and biological evaluation of novel acetamide-substituted doravirine and its prodrugs as potent HIV-1 NNRTIs. Bioorg. Med. Chem. 2019;27:447–456. doi: 10.1016/j.bmc.2018.12.039. [DOI] [PubMed] [Google Scholar]
- 182.Bhatt J.D., Chudasama C.J., Patel K.D. Microwave assisted synthesis of pyrimidines in ionic liquid and their potency as non-classical malarial antifolates. Arch. Pharm. Chem. Life Sci. 2016;349:791–800. doi: 10.1002/ardp.201600148. [DOI] [PubMed] [Google Scholar]
- 183.Prasad P., Kalola A.G., Patel M.P. Microwave assisted one-pot synthetic route to imidazo[1,2-a]pyrimidine derivatives of imidazo/triazole clubbed pyrazole and their pharmacological screening. New J. Chem. 2018;42:12666–12676. [Google Scholar]
- 184.Vlahakis J.Z., Lazar C.C., Crandall I.E., Szarek W.A. Anti-plasmodium activity of imidazolium and triazolium salts. Bioorg. Med. Chem. 2010;18:6184–6196. doi: 10.1016/j.bmc.2010.05.020. [DOI] [PubMed] [Google Scholar]
- 185.McConville M., Fernandez J., Angulo-Barturen I., Bahamontes-Rosa N., Ballell-Pages L., Castaneda P., Cozar C., Crespo B., Guijarro L., Jimenez-Diaz M.B., Martinez-Martinez M.S., de Mercado J., Santos-Villarejo A., Sanz L.M., Frigerio M., Washbourn G., Ward S.A., Nixon G.L., Biagini G.A., Berry N.G., Blackman M.J., Calderon F., O’Neill P.M. Carbamoyl triazoles, known serine protease inhibitors, are a potent new class of antimalarials. J. Med. Chem. 2015;58:6448–6455. doi: 10.1021/acs.jmedchem.5b00434. [DOI] [PubMed] [Google Scholar]
- 186.Rueda L., Castellote I., Castro-Pichel J., Chaparro M.J., Rosa J.C., Garcia-Perez A., Gordo M., Jimenez-Diaz M.B., Kessler A., Macdonald S.J.F., Martinez M.S., Sanz L.M., Gamo F.J., Fernandez E. Cyclopropyl carboxamides: a new oral antimalarial series derived from the tres cantos anti-malarial set (TCAMS) ACS Med. Chem. Lett. 2011;2:840–844. doi: 10.1021/ml2001517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Mistry B.D., Desai K.R., Intwala S.M. Synthesis of novel sulfonamides as potential antibacterial, antifungal and antimalarial agents. Indian J. Chem. B. 2015;54:128–134. [Google Scholar]
- 188.Thakkar S.S., Thakor P., Doshi H., Ray A. 1,2,4-Triazole and 1,3,4-oxadiazole analogues: synthesis, MO studies, in silico molecular docking studies, antimalarial as DHFR inhibitor and antimicrobial activities. Bioorg. Med. Chem. 2017;25:4064–4075. doi: 10.1016/j.bmc.2017.05.054. [DOI] [PubMed] [Google Scholar]
- 189.Gujjar R., El Mazouni F., White K.L., White J., Creason S., Shackleford D.M., Deng X., Charman W.N., Bathurst I., Burrows J., Floyd D.M., Matthews D., Buckner F.S., Charman S.A., Phillips M.A., Rathod P.K. Lead optimization of aryl and aralkyl amine-based triazolopyrimidine inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase with antimalarial activity in mice. J. Med. Chem. 2011;54:3935–3949. doi: 10.1021/jm200265b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Coteron J.M., Marco M., Esquivias J., Deng X., White K.L., White J., Koltun M., El Mazouni F., Kokkonda S., Katneni K., Bhamidipati R., Shackleford D.M., Angulo-Barturen I., Ferrer S.B., Jimenez-Díaz M.B., Gamo F.-J., Goldsmith E.J., Charman W.N., Bathurst I., Floyd D., Matthews D., Burrows J.N., Rathod P.K., Charman S.A., Phillips M.A. Structure-guided lead optimization of triazolopyrimidine-ring substituents identifies potent Plasmodium falciparum dihydroorotate dehydrogenase inhibitors with clinical candidate potential. J. Med. Chem. 2011;54:5540–5561. doi: 10.1021/jm200592f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Deng X., Kokkonda S., El Mazouni F., White J., Burrows J.N., Kaminsky W., Charman S.A., Matthews D., Rathod P.K., Phillips M.A. Fluorine modulates species selectivity in the triazolopyrimidine class of Plasmodium falciparum dihydroorotate dehydrogenase inhibitors. J. Med. Chem. 2014;57:5381–5394. doi: 10.1021/jm500481t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Kokkonda S., Deng X., White K.L., Coteron J.M., Marco M., de las Heras L., White J., El Mazouni F., Tomchick D.R., Manjalanagara K., Rudra K.R., Chen G., Morizzi J., Ryan E., Kaminsky W., Leroy D., Martinez-Martinez M.S., Jimenez-Diaz M.B., Bazaga S.F., Angulo-Barturen I., Waterson D., Burrows J.N., Matthews D., Charman S.A., Phillips M.A., Rathod P.K. Tetrahydro-2-naphthyl and 2-indanyl triazolopyrimidines targeting Plasmodium falciparum dihydroorotate dehydrogenase display potent and selective antimalarial activity. J. Med. Chem. 2016;59:5416–5431. doi: 10.1021/acs.jmedchem.6b00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Phillips M.A., White K.L., Kokkonda S., Deng X., White J., El Mazouni F., Marsh K., Tomchick D.R., Manjalanagara K., Rudra K.R., Wirjanata G., Noviyanti R., Price R.N., Marfurt J., Shackleford D.M., Chiu F.C., Campbell M., Jimenez-Diaz M.B., Bazaga S.F., Angulo-Barturen I., Martinez M.S., Lafuente-Monasterio M., Kaminsky W., Silue K., Zeeman A.M., Kocken C., Leroy D., Blasco B., Rossignol E., Rueckle T., Matthews D., Burrows J.N., Waterson D., Palmer M.J., Rathod P.K., Charman S.A. A triazolopyrimidine based dihydroorotate dehydrogenase inhibitor with improved drug-like properties for treatment and prevention of malaria. ACS Infect. Dis. 2016;2:945–957. doi: 10.1021/acsinfecdis.6b00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Pinheiro L.C.S., Feitosa L.M., Gandi M.O., Silveira F.F., Boechat N. The development of novel compounds against malaria: quinolines, triazolpyridines, pyrazolopyridines and pyrazolopyrimidines. Molecules. 2019;24:4095. doi: 10.3390/molecules24224095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.de Macedo-Silva S.T., Visbal G., Godinho J.L.P., Urbina J.A., de Souza W., Rodrigues J.C.F. In vitro antileishmanial activity of ravuconazole, a triazole antifungal drug, as a potential treatment for leishmaniasis. J. Antimicrob. Chemother. 2018;73:2360–2373. doi: 10.1093/jac/dky229. [DOI] [PubMed] [Google Scholar]
- 196.de Macedo-Silva S.T., Urbina J.A., de Souza W., Rodrigues J.C.F. In vitro activity of the antifungal azoles itraconazole and posaconazole against Leishmania amazonensis. PloS One. 2013;8 doi: 10.1371/journal.pone.0083247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Franklim T.N., Freire-de-Lima L., Diniz J.N.S., Previato J.O., Castro R.N., Mendonca-Previato L., de Lima M.E.F. Design, synthesis and trypanocidal evaluation of novel 1,2,4-triazoles-3-thiones derived from natural piperine. Molecules. 2013;18:6366–6382. doi: 10.3390/molecules18066366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Franklim T.N., Freire-de-Lima L., Chaves O.A., LaRocque-de-Freitas I.F., da Silva-Trindade J.D., Netto-Ferreira J.C., Freire-de-Lima C.G., Decoté-Ricardo D., Previato J.O., Mendonca-Previatob L., de Lima M.E.F. Design, synthesis, trypanocidal activity, and studies on human albumin interaction of novel S-alkyl-1,2,4-triazoles. J. Braz. Chem. Soc. 2019;30:1378–1394. [Google Scholar]
- 199.Silva F.T., Franco C.H., Favaro D.C., Freitas-Junior L.H., Moraes C.B., Ferreira E.I. Design, synthesis and antitrypanosomal activity of some nitrofurazone 1,2,4-triazolic bioisosteric analogues. Eur. J. Med. Chem. 2016;121:553–560. doi: 10.1016/j.ejmech.2016.04.065. [DOI] [PubMed] [Google Scholar]
- 200.Papadopoulou M.V., Bourdin Trunz B., Bloomer W.D., McKenzie C., Wilkinson S.R., Prasittichai C., Brun R., Kaiser M., Torreele E. Novel 3-nitro-1H-1,2,4-triazole-based aliphatic and aromatic amines as anti-chagasic agents. J. Med. Chem. 2011;54:8214–8223. doi: 10.1021/jm201215n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Papadopoulou M.V., Bloomer W.D., Rosenzweig H.S., Kaiser M., Chatelain E., Ioset J.-R. Novel 3-nitro-1H-1,2,4-triazole-based piperazines and 2-amino-1,3-benzothiazoles as antichagasic agents. Bioorg. Med. Chem. 2013;21:6600–6607. doi: 10.1016/j.bmc.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Papadopoulou M.V., Bloomer W.D., Rosenzweig H.S., Kaiser M. The antitrypanosomal and antitubercular activity of some nitro(triazole/imidazole)-based aromatic amines. Eur. J. Med. Chem. 2017;138:1106–1113. doi: 10.1016/j.ejmech.2017.07.060. [DOI] [PubMed] [Google Scholar]
- 203.Papadopoulou M.V., Bloomer W.D., Rosenzweig H.S., Chatelain E., Kaiser M., Wilkinson S.R., McKenzie C., Ioset J.R. Novel 3-nitro-1H-1,2,4-triazole-based amides and sulfonamides as potential antitrypanosomal agents. J. Med. Chem. 2012;55:5554–5565. doi: 10.1021/jm300508n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Papadopoulou M.V., Bloomer W.D., Rosenzweig H.S., Wilkinson S.R., Kaiser M. Novel nitro(triazole/imidazole)-based heteroarylamides/sulphonamides as potential antitrypanosomal agents. Eur. J. Med. Chem. 2014;87:79–88. doi: 10.1016/j.ejmech.2014.09.045. [DOI] [PubMed] [Google Scholar]
- 205.Papadopoulou M.V., Bloomer W.D., Rosenzweig H.S., O’Shea I.P., Wilkinson S.R., Kaiser M., Chatelain E., Ioset J.R. Discovery of potent nitrotriazole-based antitrypanosomal agents: in vitro and in vivo evaluation. Bioorg. Med. Chem. 2015;23:6467–6476. doi: 10.1016/j.bmc.2015.08.014. [DOI] [PubMed] [Google Scholar]
- 206.Papadopoulou M.V., Bloomer W.D., Rosenzweig H.S., Wilkinson S.R., Szular J., Kaiser M. Nitrotriazole-based acetamides and propanamides with broad spectrum antitrypanosomal activity. Eur. J. Med. Chem. 2016;123:895–904. doi: 10.1016/j.ejmech.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Papadopoulou M.V., Bloomer W.D., Lepesheva G.I., Rosenzweig H.S., Kaiser M., Aguilera-Venegas B., Wilkinson S.R., Chatelain E., Ioset J.-R. Novel 3-nitrotriazole-based amides and carbinols as bifunctional antichagasic agents. J. Med. Chem. 2015;58:1307–1319. doi: 10.1021/jm5015742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Khare S., Nagle A.S., Biggart A., Lai Y.H., Liang F., Davis L.C., Barnes S.W., Mathison C.J.N., Myburgh E., Gao M.-Y., Gillespie J.R., Liu X., Tan J.L., Stinson M., Rivera I.C., Ballard J., Yeh V., Groessl T., Federe G., Koh H.X.Y., Venable J.D., Bursulaya B., Shapiro M., Mishra P.K., Spraggon G., Brock A., Mottram J.C., Buckner F.S., Rao S.P.S., Wen B.G., Walker J.R., Tuntland T., Molteni V., Glynne R.J., Supek F. Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature. 2016;537:229–233. doi: 10.1038/nature19339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.S Rao S.P., Lakshminarayana S.B., Jiricek J., Kaiser M., Ritchie R., Myburgh E., Supek F., Tuntland T., Nagle A., Molteni V., Mäser P., Mottram J.C., Barrett M.P., Diagana T.T. Anti-trypanosomal proteasome inhibitors cure hemolymphatic and meningoencephalic murine infection models of african trypanosomiasis. Trav. Med. Infect. Dis. 2020;5:28. doi: 10.3390/tropicalmed5010028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Patil S.R., Asrondkar A., Patil V., Sangshetti J.N., Khan F.A.K., Damale M.G., Patil R.H., Bobade A.S., Shinde D.B. Antileishmanial potential of fused 5-(pyrazin-2-yl)-4H-1,2,4-triazole-3-thiols: synthesis, biological evaluations and computational studies. Bioorg. Med. Chem. Lett. 2017;27:3845–3850. doi: 10.1016/j.bmcl.2017.06.053. [DOI] [PubMed] [Google Scholar]
- 211.Ibrar A., Zaib S., Jabeen F., Iqbal J., Saeed A. Unraveling the alkaline phosphatase inhibition, anticancer, and antileishmanial potential of coumarin-triazolothiadiazine hybrids: design, synthesis, and molecular docking analysis. Arch. Pharm. Chem. Life Sci. 2016;349:553–565. doi: 10.1002/ardp.201500392. [DOI] [PubMed] [Google Scholar]
- 212.Süleymanoglu N., Ünver Y., Ustabas Resat, Direkel S., Alpaslan G. Antileishmanial activity study and theoretical calculations for 4-amino-1,2,4-triazole derivatives. J. Mol. Struct. 2017;1144:80–86. [Google Scholar]
- 213.Khan B., Naiyer A., Athar F., Ali S., Thakur S.C. Synthesis, characterization and anti-inflammatory activity evaluation of 1,2,4-triazole and its derivatives as a potential scaffold for the synthesis of drugs against prostaglandin-endoperoxide synthase. J. Biomol. Struct. Dyn. 2020:1–19. doi: 10.1080/07391102.2019.1711193. [DOI] [PubMed] [Google Scholar]
- 214.Abdel-Aziz M., Beshr E.A., Abdel-Rahman I.M., Ozadali K., Tan O.U., Aly O.M. 1-(4-Methoxyphenyl)-5-(3,4,5-trimethoxyphenyl)-1H-1,2,4-triazole-3-carboxamides: synthesis, molecular modeling, evaluation of their anti-inflammatory activity and ulcerogenicity. Eur. J. Med. Chem. 2014;77:155–165. doi: 10.1016/j.ejmech.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 215.Abuo-Rahma G.E.A.A., Abdel-Aziz M., Farag N.A., Kaoud T.S. Novel 1-[4-(aminosulfonyl)phenyl]-1H-1,2,4-triazole derivatives with remarkable selective COX-2 inhibition: design, synthesis, molecular docking, anti-inflammatory and ulcerogenicity studies. Eur. J. Med. Chem. 2014;83:398–408. doi: 10.1016/j.ejmech.2014.06.049. [DOI] [PubMed] [Google Scholar]
- 216.Abuo-Rahma G.E.A.A., Abdel-Aziz M., Beshr E.A.M., Ali T.F.S. 1,2,4-Triazole/oxime hybrids as new strategy for nitric oxide donors: synthesis, anti-inflammatory, ulceroginicity and antiproliferative activities. Eur. J. Med. Chem. 2014;71:185–198. doi: 10.1016/j.ejmech.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 217.Mohassab A.M., Hassan H.A., Abdelhamid D., Abdel-Aziz M., Dalby K.N., Kaoud T.S. Novel quinoline incorporating 1,2,4-triazole/oxime hybrids: synthesis, molecular docking, anti-inflammatory, COX inhibition, ulceroginicity and histopathological investigations. Bioorg. Chem. 2017;75:242–259. doi: 10.1016/j.bioorg.2017.09.018. [DOI] [PubMed] [Google Scholar]
- 218.Lamie P.F., Ali W.A.M., Bazgier V., Rárová L. Novel N-substituted indole Schiff bases as dual inhibitors of cyclooxygenase-2 and 5-lipoxygenase enzymes: synthesis, biological activities in vitro and docking study. Eur. J. Med. Chem. 2016;123:803–813. doi: 10.1016/j.ejmech.2016.08.013. [DOI] [PubMed] [Google Scholar]
- 219.Gowda J., Khader A.M.A., Kalluraya B., Shree Padma, Shabaraya A.R. Synthesis, characterization and pharmacological activity of 4-{[1-substituted aminomethyl-4-arylideneamino-5-sulfanyl-4,5-dihydro-1H-1,2,4-triazol-3-yl]methyl}-2H-1,4-benzothiazin-3(4H)-ones. Eur. J. Med. Chem. 2011;46:4100–4106. doi: 10.1016/j.ejmech.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 220.Harris P.A., Berger S.B., Jeong J.U., Nagilla R., Bandyopadhyay D., Campobasso N., Capriotti C.A., Cox J.A., Dare L., Dong X., Eidam P.M., Finger J.N., Hoffman S.J., Kang J., Kasparcova V., King B.W., Lehr R., Lan Y., Leister L.K., Lich J.D., MacDonald T.T., Miller N.A., Ouellette M.T., Pao C.S., Rahman A., Reilly M.A., Rendina A.R., Rivera E.J., Schaeffer M.C., Sehon C.A., Singhaus R.R., Sun H.H., Swift B.A., Totoritis R.D., Vossenkämper A., Ward P., Wisnoski D.D., Zhang D., Marquis R.W., Gough P.J., Bertin J. Discovery of a first-in-class receptor interacting protein 1 (RIP1) kinase specific clinical candidate (GSK2982772) for the treatment of inflammatory diseases. J. Med. Chem. 2017;60:1247–1261. doi: 10.1021/acs.jmedchem.6b01751. [DOI] [PubMed] [Google Scholar]
- 221.Tariq S., Kamboj P., Alam O., Amir Mohd. 1,2,4-Triazole-based benzothiazole/benzoxazole derivatives: design, synthesis, p38α MAP kinase inhibition, anti-inflammatory activity and molecular docking studies. Bioorg. Chem. 2018;81:630–641. doi: 10.1016/j.bioorg.2018.09.015. [DOI] [PubMed] [Google Scholar]
- 222.Tariq S., Alam O., Amir Mohd. Synthesis, p38α MAP kinase inhibition, anti-inflammatory activity, and molecular docking studies of 1,2,4-triazole-based benzothiazole-2-amines. Arch. Pharm. Chem. Life Sci. 2018;351 doi: 10.1002/ardp.201700304. [DOI] [PubMed] [Google Scholar]
- 223.Paprocka R., Wiese M., Eljaszewicz A., Helmin-Basa A., Gzella A., Modzelewska-Banachiewicz B., Michalkiewicz J. Synthesis and anti-inflammatory activity of new 1,2,4-triazole derivatives. Bioorg. Med. Chem. Lett. 2015;25:2664–2667. doi: 10.1016/j.bmcl.2015.04.079. [DOI] [PubMed] [Google Scholar]
- 224.Mustafa G., Angeli A., Zia-ur-Rehman M., Akbar N., Ishtiaq S., Supuran C.T. An efficient method for the synthesis of novel derivatives 4-{5-[4-(4-amino-5-mercapto-4H-[1,2,4]triazol-3-yl)-phenyl]-3-trifluoromethyl-pyrazol-1-yl}-benzenesulfonamide and their anti-inflammatory potential. Bioorg. Chem. 2019;91:103110. doi: 10.1016/j.bioorg.2019.103110. [DOI] [PubMed] [Google Scholar]
- 225.Uzgören-Baran A., Tel B.C., Sarıgöl D., Öztürk E.I., Kazkayası I., Okay G., Ertan M., Tozkoparan B. Thiazolo[3,2-b]-1,2,4-triazole-5(6H)-one substituted with ibuprofen: novel non-steroidal anti-inflammatory agents with favorable gastrointestinal tolerance. Eur. J. Med. Chem. 2012;45:1906–1911. doi: 10.1016/j.ejmech.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 226.Rizk O.H., Shaaban O.G., El-Ashmawy I.M. Design, synthesis and biological evaluation of some novel thienopyrimidines and fused thienopyrimidines as anti-inflammatory agents. Eur. J. Med. Chem. 2012;55:85–93. doi: 10.1016/j.ejmech.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 227.Ashour H.M., Shaaban O.G., Rizk O.H., El-Ashmawy I.M. Synthesis and biological evaluation of thieno [2’,3’:4,5]pyrimido[1,2-b][1,2,4]triazines and thieno[2,3-d][1,2,4]triazolo[1,5-a]pyrimidines as anti-inflammatory and analgesic agents. Eur. J. Med. Chem. 2013;62:341–351. doi: 10.1016/j.ejmech.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 228.Pan F.-J., Wang S.-B., Liu D.-C., Gong G.-H., Quan Z.-S. Synthesis of 4-phenylthieno[2,3-e][1,2,4]triazolo[4,3-a]pyrimidine-5(4H)-one derivatives and evaluation of their anti-inflammatory activity. Lett. Drug Des. Discov. 2016;13:141–148. [Google Scholar]
- 229.El Shehry M.F., Abu-Hashem A.A., El-Telbani E.M. Synthesis of 3-((2,4-dichlorophenoxy)methyl)-1, 2, 4-triazolo(thiadiazoles and thiadiazines) as anti-inflammatory and molluscicidal agents. Eur. J. Med. Chem. 2010;45:1906–1911. doi: 10.1016/j.ejmech.2010.01.030. [DOI] [PubMed] [Google Scholar]
- 230.Maddila S., Gorle S., Singh M., Lavanya P., Jonnalagadda S.B. Synthesis and anti-inflammatory activity of fused 1,2,4-triazolo-[3,4-b][1,3,4]thiadiazole derivatives of phenothiazine. Lett. Drug Des. Discov. 2013;10:977–983. [Google Scholar]
- 231.Braccio M.D., Grossi G., Alfei S., Ballabeni V., Tognolini M., Flammini L., Giorgio C., Bertoni S., Barocelli E. 1,8-Naphthyridines IX. Potent anti-inflammatory and/or analgesic activity of a new group of substituted 5-amino[1,2,4]triazolo[4,3-a][1,8]naphthyridine-6-carboxamides, of some their Mannich base derivatives and of one novel substituted 5-amino-10-oxo-10H-pyrimido[1,2-a][1,8]naphthyridine-6-carboxamide derivative. Eur. J. Med. Chem. 2014;86:394–405. doi: 10.1016/j.ejmech.2014.08.069. [DOI] [PubMed] [Google Scholar]
- 232.Roma G., Braccio M.D., Grossi G.C., Piras D., Ballabeni V., Tognolini M., Bertoni S., Barocelli E. 1,8-Naphthyridines VIII. Novel 5-aminoimidazo[1,2-a] [1,8]naphthyridine-6-carboxamide and 5-amino[1,2,4]triazolo[4,3-a] [1,8]naphthyridine-6-carboxamide derivatives showing potent analgesic or anti-inflammatory activity, respectively, and completely devoid of acute gastrolesivity. Eur. J. Med. Chem. 2010;45:352–366. doi: 10.1016/j.ejmech.2009.10.020. [DOI] [PubMed] [Google Scholar]
- 233.Guirado A., Lopez Sanchez J.I., Ruiz-Alcaraz A.J., Bautista D., Galvez J. Synthesis and biological evaluation of 4-alkoxy-6,9-dichloro[1,2,4]triazolo[4,3-a]quinoxalines as inhibitors of TNF-α and IL-6 Eur. J. Med. Chem. 2012;54:87–94. doi: 10.1016/j.ejmech.2012.04.035. [DOI] [PubMed] [Google Scholar]
- 234.Liu D.-C., Gong G.-H., Wei C.-X., Jin X.-J., Quan Z.-S. Synthesis and anti-inflammatory activity evaluation of a novel series of 6-phenoxy-[1,2,4]triazolo[3,4-a]phthalazine-3-carboxamide derivatives. Bioorg. Med. Chem. Lett. 2016;26:1576–1579. doi: 10.1016/j.bmcl.2016.02.008. [DOI] [PubMed] [Google Scholar]
- 235.Sarigol D., Uzgoren-Baran A., Tel B.C., Somuncuoglu E.I., Kazkayasi I., Ozadali-Sari K., Unsal-Tan O., Okay G., Ertan M., Tozkoparan B. Novel thiazolo[3,2-b]-1,2,4-triazoles derived from naproxen with analgesic/anti-inflammatory properties: synthesis, biological evaluation and molecular modeling studies. Bioorg. Med. Chem. 2015;23:2518–2528. doi: 10.1016/j.bmc.2015.03.049. [DOI] [PubMed] [Google Scholar]
- 236.Ahirwar J., Ahirwar D., Lanjhiyana S., Jha A.K., Dewangan D., Badwaik H. Analgesic and anti-inflammatory potential of merged pharmacophore containing 1,2,4-triazoles and substituted benzyl groups via thio linkage. J. Heterocycl. Chem. 2018;55:2130–2141. [Google Scholar]
- 237.Sert-Ozgur S., Tel B.C., Somuncuoglu E.I., Kazkayasi I., Ertan M., Tozkoparan B. Design and synthesis of 1,2,4-triazolo[3,2-b]-1,3,5-thiadiazine derivatives as a novel template for analgesic/anti-inflammatory activity. Arch. Pharm. Chem. Life Sci. 2017;350 doi: 10.1002/ardp.201700052. [DOI] [PubMed] [Google Scholar]
- 238.Chidananda N., Poojary B., Sumangala V., Kumari N.S., Shetty P., Arulmoli T. Facile synthesis, characterization and pharmacological activities of 3,6-disubstituted 1,2,4-triazolo[3,4-b][1,3,4]thiadiazoles and 5,6-dihydro-3,6-disubstituted-1,2,4-triazolo[3,4-b][1,3,4]thiadiazoles. Eur. J. Med. Chem. 2012;51:124–136. doi: 10.1016/j.ejmech.2012.02.030. [DOI] [PubMed] [Google Scholar]
- 239.Aggarwal R., Sumran G., Kumar V., Mittal A. Copper(II) chloride mediated synthesis and DNA photocleavage activity of 1-aryl/heteroaryl-4-substituted-1,2,4-triazolo[4,3-a]quinoxalines. Eur. J. Med. Chem. 2011;46:6083–6088. doi: 10.1016/j.ejmech.2011.10.032. [DOI] [PubMed] [Google Scholar]
- 240.Sumran G., Aggarwal R., Mittal A., Aggarwal A., Gupta A. Design, synthesis and photoinduced DNA cleavage studies of [1,2,4]-triazolo[4,3-a]quinoxalin-4(5H)-ones. Bioorg. Chem. 2019;88:102932. doi: 10.1016/j.bioorg.2019.102932. [DOI] [PubMed] [Google Scholar]
- 241.Chang L., Xiao M., Yang L., Wang S., Wang S.-Q., Bender A., Hu A., Chen Z.-S., Yu B., Liu H.-M. Discovery of a non-toxic [1,2,4]triazolo[1,5-a]pyrimidin-7-one (WS-10) that modulates ABCB1-mediated multidrug resistance (MDR) Bioorg. Med. Chem. 2018;26:5006–5017. doi: 10.1016/j.bmc.2018.08.021. [DOI] [PubMed] [Google Scholar]
- 242.Jameel E., Meena P., Maqbool M., Kumar J., Ahmed W., Mumtazuddin S., Tiwari M., Hoda N., Jayaram B. Rational design, synthesis and biological screening of triazine-triazolopyrimidine hybrids as multitarget anti-Alzheimer agents. Eur. J. Med. Chem. 2017;136:36–51. doi: 10.1016/j.ejmech.2017.04.064. [DOI] [PubMed] [Google Scholar]
- 243.Kumar J., Meena P., Singh A., Jameel E., Maqbool M., Mobashir M., Shandilya A., Tiwari M., Hoda N., Jayaram B. Synthesis and screening of triazolopyrimidine scaffold as multi-functional agents for Alzheimer’s disease therapies. Eur. J. Med. Chem. 2016;119:260–277. doi: 10.1016/j.ejmech.2016.04.053. [DOI] [PubMed] [Google Scholar]
- 244.El-Aleam R.H.A., George R.F., Lee K.J., Keeton A.B., Piazza G.A., Kamel A.A., El-Daly M.E., Hassan G.S., Abdel-Rahman H.M. Design and synthesis of 1,2,4-triazolo[1,5-a]pyrimidine derivatives as PDE 4B inhibitors endowed with bronchodilator activity. Arch. Pharm. Chem. Life Sci. 2019;352 doi: 10.1002/ardp.201900002. [DOI] [PubMed] [Google Scholar]