

Abstract
Gastrointestinal stromal tumor (GIST), generally driven by oncogenic KIT or PDGFRA mutations, is the most common mesenchymal tumor of the gastrointestinal (GI) tract. GIST is most common in the stomach (60%) and small intestine (30%), but can occur anywhere in the GI-tract and the intra-abdominal soft tissues. GIST can show spindle cell or epithelioid morphology, and mitotic count and tumor size are most important prognostic parameters. GISTs in NF1 patients and children are distinctive clinicopathologic groups. Immunohistochemical testing for KIT and sometimes for DOG1/Ano 1 is essential in confirming the diagnosis.
Keywords: gastrointestinal stromal tumor, KIT, PDGFRA, anoctamin 1, succinate dehydrogenase subunit B
INTRODUCTION
Gastrointestinal stromal tumor (GIST) is the designation for the most common mesenchymal tumors of the gastrointestinal (GI) tract. Until some 10 years ago, these tumors were widely considered variants of smooth muscle tumors: leiomyomas if benign and leiomyosarcomas if malignant [1]. The term “gastrointestinal autonomic nerve tumor” (GANT) also refers to GIST, based on identical histologic and immunohisto-chemical features and KIT mutations [2].
The incidence of GIST has been estimated to be 14–20 per million, but minimal incidental GISTs are far more common. Most GISTs occur on a sporadic basis, but some occur in clinical syndromes. The most common of these is neurofibromatosis 1, in which GISTs usually occur in small intestine, often as multiple, clinically indolent tumors. Familial GISTs are based on hereditary KIT/PDGFRA-activating mutations, and pediatric GISTs (almost all gastric) are linked with loss of succinate dehydrogenase subunit B (SDHB) and Carney triad and Carney–Stratakis syndromes (CSS), the latter is autosomal dominant tumor syndrome combining GIST and paraganglioma.
GISTs are now understood as generally KIT-positive, KIT, or PDGFRA mutation driven mesenchymal neoplasms, and this information has been the basis of the new KIT tyrosine kinase inhibitor drugs, imatinib mesylate and second and third generation inhibitors now routinely used in the treatment of metastatic and unresectable GISTs [3–7]. Availability of these new treatments has made specific and accurate identification of GIST all the more important. GIST can be considered as neoplastic derivatives of Cajal cells or their precursors. Cajal cells are a small KIT-positive spindle cell population especially located around the myenteric plexus [8,9].
KIT receptor activating mutations occur in 60–70% of all GISTs, most common of them being exon 11 in-frame deletions or single nucleotide substitutions, exon 9 duplications (intestinal GISTs), and exon 11 internal tandem duplications (gastric GISTs). PDGFRA mutations occur almost exclusively in gastric GISTs, most commonly in exon 18.
GISTs occur throughout the GI tract, usually in persons >50 years of age with a median age of 62–63 years, although a clinicopathologically distinctive pediatric GIST subgroup exists. GISTs have been estimated to occur with a frequency of 14–20 per million inhabitants [10,11]. These tumors manifest variably by GI bleeding associated with tumor ulcer, mass effect, and sometimes by acute abdomen due to tumor perforation. Minimal incidental GISTs may be more common, as found in autopsy studies[12] and thorough examinations of gastroesophageal carcinoma resections; 10% of the latter were found to harbor a minimal incidental GIST in one study [13].
For specific recognition of GIST, one has to consider the following facts: (1) most GI mesenchymal tumors are GISTs, (2) GIST can occur as a metastatic tumor anywhere in the abdominal cavity, as well as in liver, but rarely in lungs or distant peripheral sites, (3) GIST has a rather wide morphologic spectrum including spindle cell and epithelioid variants, and this is especially true for gastric tumors, and (4) histologic differential diagnosis of GIST is relatively broad and requires immunohistochemical testing.
The two most important immunohistochemical markers for the histopathological identification of GIST are KIT and anoctamin 1. KIT is a type III receptor tyrosine kinase transmembrane protein, a growth factor receptor for stem cell factor. Pathologic activation of this receptor is a key element in GIST pathogenesis, and this can now be countered by the new tyrosine kinase inhibitor treatment [3]. KIT expression is fairly specific for GIST among GI mesenchymal tumors, but it is also detected in mast cells, melanomas, and some epithelia, especially in the skin adnexa and the breast [14–16]. KIT is expressed in a great majority of GISTs, but a small percentage of GISTs (3–5%) are only focally positive or negative, and these especially include gastric epithelioid GISTs with PDGFRA mutations [17,18].
Anoctamin 1, also designated as DOG1 (discovered on GIST) and TMEM16A gene product, is a calcium-regulated chloride ion channel protein, an 8-pass transmembrane protein regulating chloride transport [19]. Similar to KIT, anoctamin 1 is also constitutively expressed in Cajal cells and in a vast majority of GISTs, including many KIT-negative GISTs, so that it supplements KIT in the positive identification of GIST. Although anoctamin 1 is relatively specific for GIST among mesenchymal tumors, it is also expressed in some leiomyomas and many GI carcinomas, especially squamous cell ones [20,21].
CD34, a hematopoietic progenitor cell antigen, is commonly present in GISTs but is less specific than KIT and anoctamin 1 [1,22]. Protein kinase C theta has been suggested as a useful GIST marker based on its presence in most GISTs, but our experience as well as a recent report found a relatively low specificity for GIST [23–26]. Smooth muscle markers, alpha smooth muscle actin and heavy caldesmon, and to lesser degree desmin, are also expressed in some GISTs, although by contrast, smooth muscle tumors typically express a full complement of these markers [1].
In this article, we review the pathology of GIST with a special emphasis on specific recognition and prognostication of these tumors. We take a site-specific approach, as GIST presenting at different sites and segments of the GI tract have distinctive clinicopathologic features. The main emphasis is on the most common subsets: gastric and small intestinal GISTs, with special attention to rare clinicopathologic subgroups, such as syndrome-associated GISTs.
GASTRIC GIST
Stomach is the by far most common site for GIST (almost 60%). The clinical as well as histopathological variety is also greatest among this group of GISTs [27]. The prognostication of GISTs in stomach and other sites, based on large series with clinical follow-up, has been summarized in Table I [27–30].
TABLE I.
Prognostication of GIST of Different Sites by Tumor Size and Mitotic Rate Based on Follow-Up Studies of Over >1,700 GISTs Prior to Imatinib
| Tumor parameters | Percentage of patients with progressive disease during long-term follow-up and quantitative characterization of the risk for metastasis | |||||
|---|---|---|---|---|---|---|
| Group | Size | Mitotic rate | Gastric GISTs | Small intestinal GISTs | Duodenal GISTs | Rectal GISTs |
| 1 | ≤2 cm | ≤5/50 HPFs | 0 none | |||
| 2 | >2 ≤ 5 cm | 1.9 very low | 4.3 low | 8.3 low | 8.5 low | |
| 3a | >5 ≤ 10 cm | 3.6 low | 24 moderate | 34 higha | 57 higha | |
| 3b | >10 cm | 12 moderate | 52 high | |||
| 4 | ≤2 cm | >5/50 HPFs | 0a | 50a | b | 54 high |
| 5 | >2 ≤ 5 cm | 16 moderate | 73 high | 50 high | 52 high | |
| 6a | >5 ≤ 10 cm | 55 high | 85 high | 86 higha | 71 higha | |
| 6b | >10 cm | 86 high | 90 high | |||
Table adopted from Miettinen and Lasota. Arch Pathol Lab Med 2006;130:1466–1478.
HPF, high power field. 50 high power fields equal here approximately 5 mm2.
Small number of cases. Groups combined or prognostic prediction less certain.
No tumors encountered with these parameters.
Gastric GISTs vary from small serosal or intramural nodules to large masses that can have variable intraluminal, intramural, and external components. Histologically, these tumors have a wide variation ranging from hypocellular to highly cellular with higher mitotic rates in the latter group. The majority of gastric GISTs are spindle cell tumors, but approximately 20–25% have epithelioid morphology. Clinicopathologic series have established mitotic rate and tumor size as most important histologic prognosticators. In general, gastric GISTs with mitotic rates ≤ 5/50 high power fields (5 mm2 total area) have a favorable prognosis, although tumors >2 cm in size in this group already have some tumor-related mortality. However, gastric GISTs with mitotic rates >5/50 high power fields and tumor size >5 cm have a significant tumor-related mortality. Small gastric GISTs can occur as small serosal or intramural nodules, whereas large tumors have variable intraluminal, intramural, and external components (Fig. 1). Some examples are attached to stomach only with a narrow pedicle. Large tumors are often cystic, in some cases only a narrow rim of viable tumor remaining in the cyst wall. On sectioning, the GIST tissue is typically pinkish-tan, fish flesh-like and can be hemorrhagic.
Fig. 1.
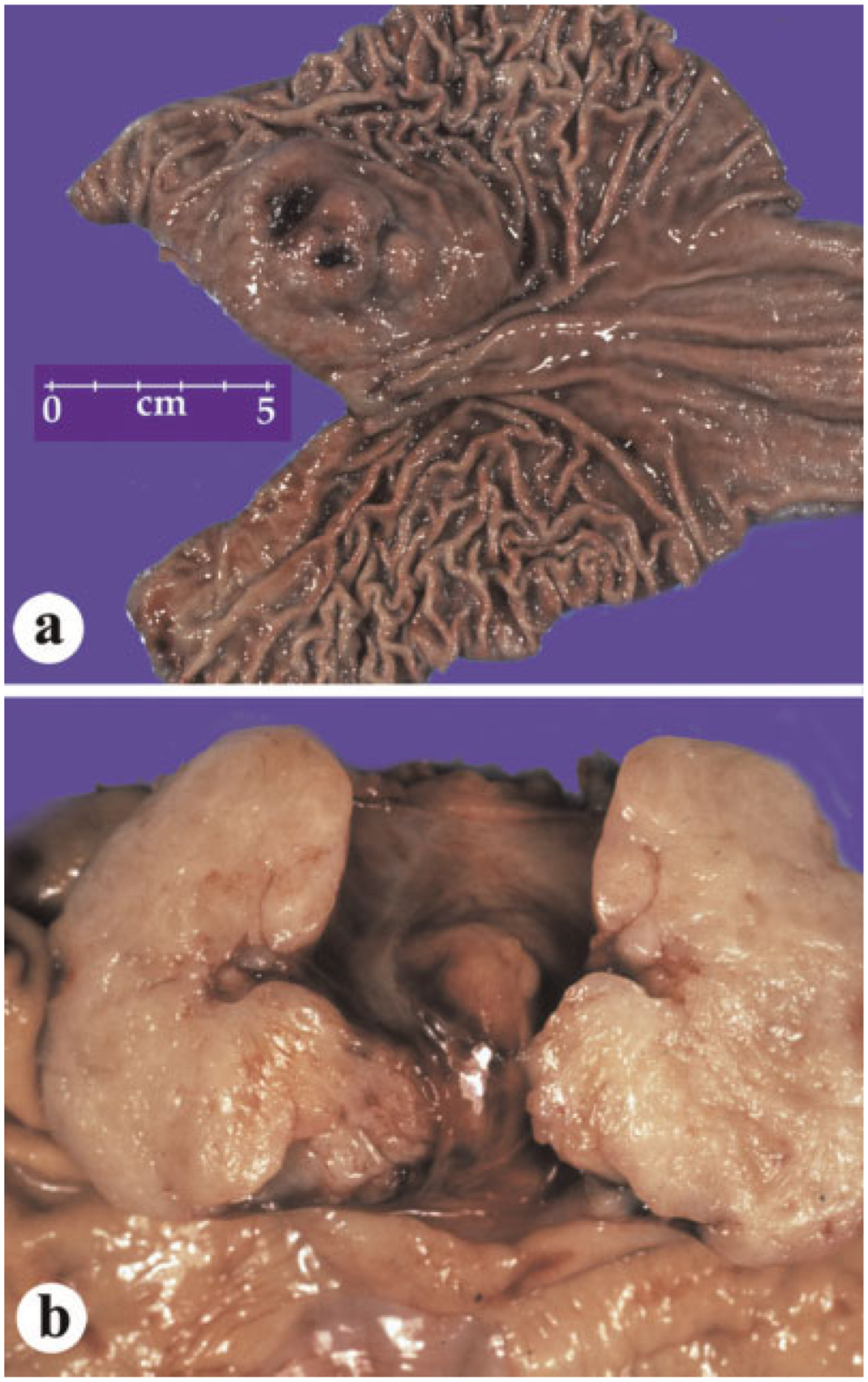
Gross features of gastric GISTs. a: Intraluminal tumor component with ulcer. b: A bivalved GIST shows a typical, pale tan surface on sectioning.
Sclerosing spindle cell morphology is commonly seen among the small, incidental GISTs. These tumors have a low cellularity with a high content of collagenous matrix, sometimes with calcification (Fig. 2a).Palisaded-vacuolated morphology is the most common in gastric GISTs. These tumors have variably prominent perinuclear vacuolization and nuclear palisading (Fig. 2b).
Fig. 2.
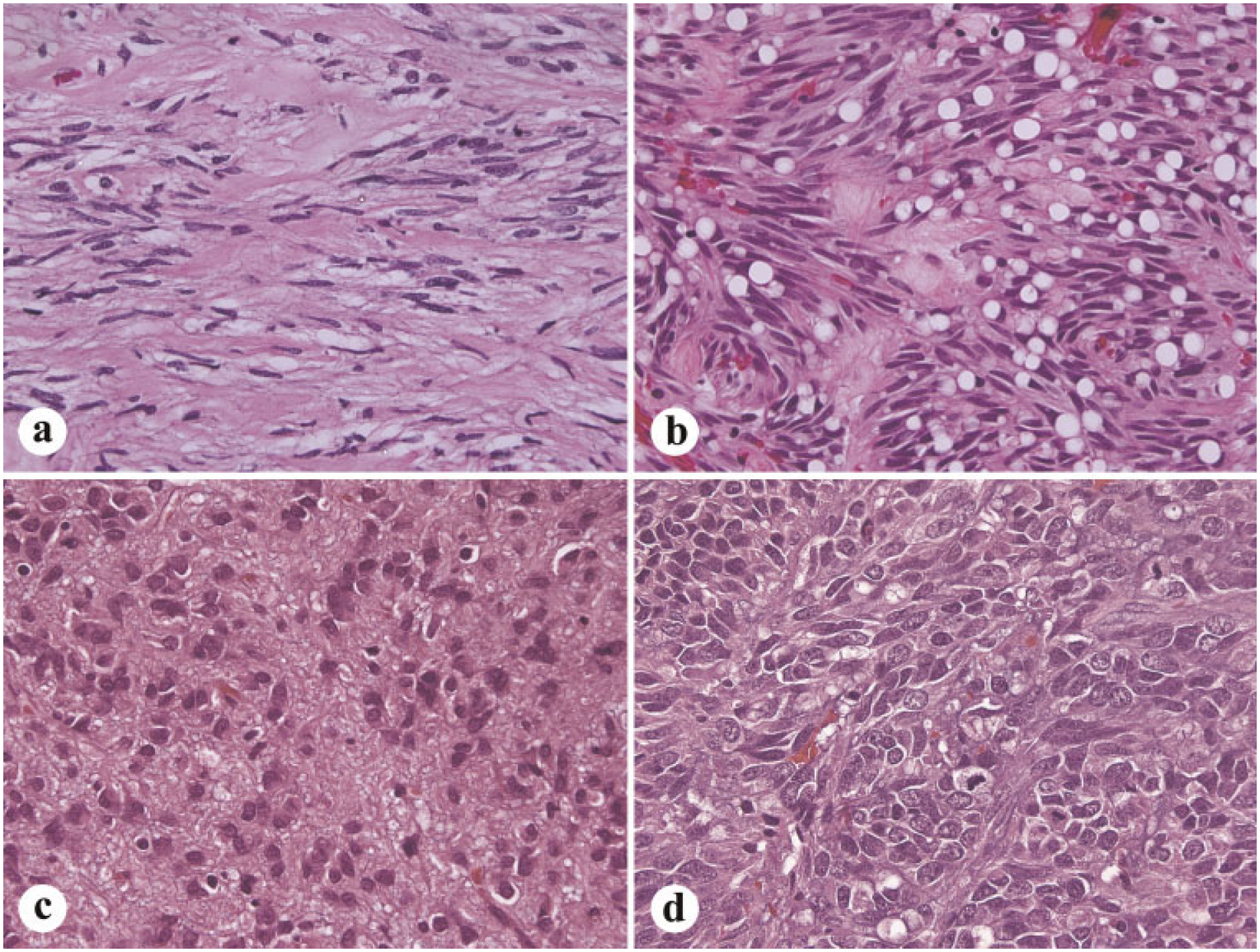
Histologic spectrum of gastric GISTs. a: Sclerosing spindle cell variant is paucicellular and rich in collagenous matrix. b: Palisadedvacuolated type is the most common gastric GIST variant. c: Epithelioid GIST with a sclerosing, collagen-rich matrix. d: Sarcomatous gastric GIST with spindled and epithelioid features.
Epithelioid gastric GISTs have a spectrum from sclerosing (Fig. 2c) and paucicellular to sarcomatous and mitotically highly active. They have a spectrum from mitotically inactive tumors to sarcomas with high mitotic activity. However, benign atypia, even pleomorphism occurs in epithelioid GISTs. Sarcomatous spindle cell GISTs contain significant mitotic activity (>10 mitoses per 50 high power fields) and nuclear atypia usually manifesting as uniform nuclear enlargement and rarely by pleomorphism (Fig. 2d).
Immunohistochemically, common to all gastric spindle cell GISTs is positivity for KIT (Fig. 3), Anoctamin 1, and CD34, whereas a minority of these tumors is smooth muscle actin positive. Epithelioid tumors are similar, except that some of them are only focally positive for KIT and rare examples entirely KIT-negative. Also, only half of the epithelioid GISTs are CD34-positive.
Fig. 3.
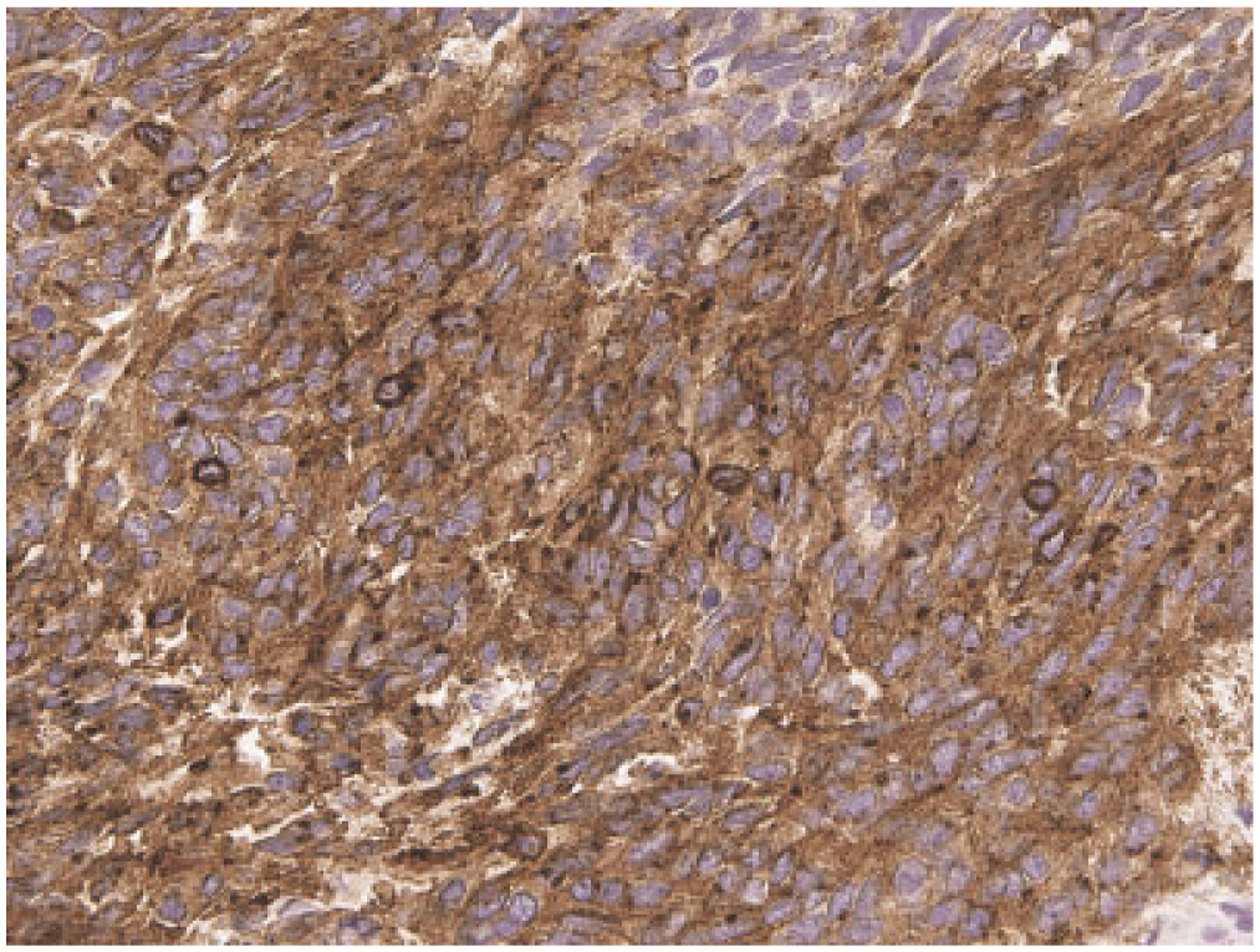
Immunohistochemical positivity for KIT is typical of >95% of GISTs.
Majority of gastric GISTs have KIT mutations, of which in-frame deletions in 5′ end of exon 11 (often involving codons 550–560) are the most common, followed by single nucleotide substitutions essentially restricted to codons 557, 559, 560, and 576 and internal tandem duplications in 3′ end of exon 11. However, approximately 20–25% of gastric GISTs have PDGFRA mutations instead [31,32]. PDGFRA and KIT are homologous genes and might have evolved as duplication of a common ancestral gene. PDGFRA mutations are essentially restricted to gastric GISTs and have predilection to tumors with epithelioid morphology. However, reliable prediction of mutation type requires molecular analysis and sequencing. The most common PDGFRA mutation is a single nucleotide substitution leading into Asp842Val mutation. In contrast to most KIT exon 11 mutant, PDGFRA Asp842Val mutants are primarily resistant to imatinib mesylate, the first generation KIT tyrosine kinase inhibitor drug [33]. KIT exon 13 or exon 17 mutations (single nucleotide substitutions) are rare, and some of these mutant tumors are imatinib resistant [34].
SMALL INTESTINAL GIST
Small intestinal GISTs vary from small intramural or serosal nodules to large masses, which tend to be sessile and externally extending and pedundulated in some cases. Many examples contain intraluminal and external components with an overall dumb bell configuration [28].
Small intestinal GISTs (including the duodenal ones) are a histologically more homogeneous than gastric GISTs. In contrast to gastric GISTs, all small intestinal (and other intestinal) GISTs >5 cm have a significant tumor-related mortality, twice as high as gastric GISTs (around 40–50%). These tumors tend to form predominantly external masses. Histologically they are more homogenous than gastric GISTs, mostly with spindle cell morphology, often with distinctive extracellular collagen globules. Larger tumors typically have internal cystic component that may communicate with the intestinal lumen (Fig. 4). Variants with low mitotic activity often contain distinctive extracellular collagen globules, named as skeinoid fibers by their skein-like ultrastructural appearance [35]. Many examples contain large anucleated pools that consist of complex, entangled cell processes analogous to the Verocay bodies in schwannoma and neuropil material in neuroblastoma (Fig. 5). In fact, this characteristic, as well as common vascular dilatation and hyalinization, are features mimicking schwannoma, with which GISTs often were previously confused prior to modern immunohistochemistry.
Fig. 4.
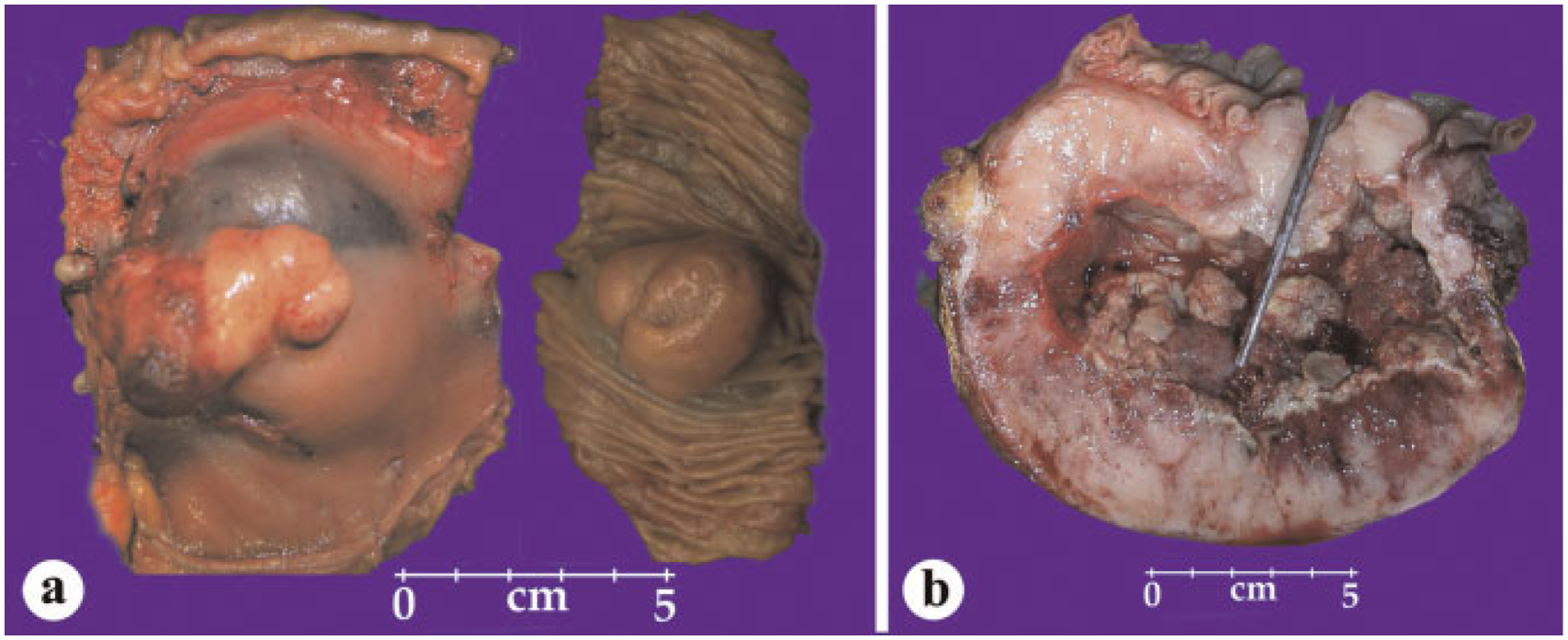
Gross features of small intestinal GISTs. a: A tumor with external and intraluminal components. b: A large tumor contains a central cystic component that communicates with the intestinal lumen, as demonstrated by the probe.
Fig. 5.
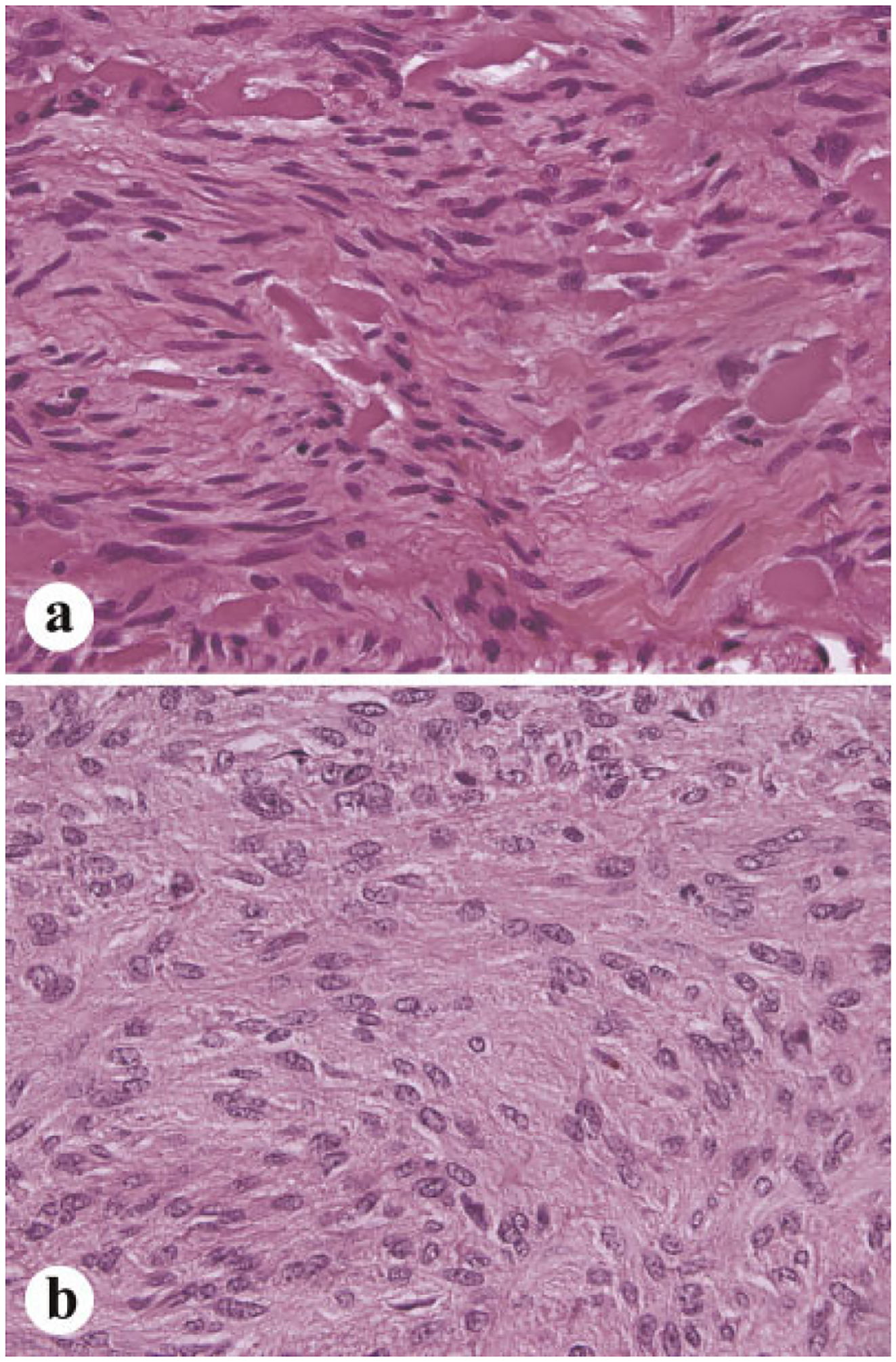
Typical histological examples of small intestinal GISTs. a: Tumor with numerous extracellular collagen globules. b: Spindle cell tumor with anuclear zones resembling verocay bodies or neuropil material.
Nearly, all small intestinal GISTs are immunohistochemically positive for KIT and anoctamin 1 (DOG1). Approximately, 60% of these tumors are positive for CD34 and 30–35% for smooth muscle actin, with almost uniform negativity for desmin. S100 protein is detected in 10–20% of small intestinal GISTs, and this should not lead into misdiagnosis of GIST as a nerve sheath tumor [28].
Small intestinal GISTs typically contain KIT exon 11 mutations including similar deletions and nucleotide substitutions as seen in gastric GISTs. Approximately, 5–10% of small intestinal (and other intestinal) GISTs contain duplication of two codons in KIT exon 9 leading to Ala502 Tyr503dup mutation [31,36]. With rare exceptions, PDGFRA mutations do not occur in small intestinal GISTs.
COLON AND RECTAL GIST
Colonic GISTs are rare (1% of all GISTs) and their histologic and immunohistochemical spectrum closely mirrors that of small intestinal GISTs. Most colonic GISTs have been large tumors when clinically detected, and the prognosis has been poor [37,38]. However, careful studies have detected minute incidental colonic GISTs in surgical colon resections with a frequency of 0.2% in sigmoid colon, the most common colonic segment involved by GIST [39].
GISTs of rectum have a spectrum from very small intramural nodules to large complex masses that extend into pelvis and are often attached to posterior aspect of the prostate potentially clinically and radiologically simulating a prostatic tumor and sometimes seen in prostate biopsies [29,40].
The histologic features of rectal GISTs are intermediate between small intestinal and gastric GISTs, with most tumors having spindle cell morphology, but occasional epithelioid GISTs similar to those seen in the stomach are also seen in the rectum [29].
ESOPHAGEAL GISTS
This is a very small group of GISTs comprising no more than 1% of all GISTs. These tumors typically occur in the lower esophagus, and majority of reported cases have been large tumors with sarcomatous features. Large esophageal GISTs not infrequently extend externally, potentially creating an impression of a mediastinal tumor. Morphologically most esophageal GISTs are similar to sarcomatous gastric GISTs, and they usually have spindle cell morphology. Prognosis is usually poor due to advanced tumor, but rarely observed small tumors that were incidentally detected were more favorable [41].
EXTRA-GASTROINTESTINAL GIST
Almost 10% of all GISTs seemingly have an origin outside of the tubular GI tract, and sometimes, these are called “extragastrointestinal GISTs” [42].
In fact, this is a heterogeneous group of GISTs that include disseminated abdominal GISTs, for which no specific origin can be determined. On the other hand included are also GISTs that form solitary omental, mesenteric, or retroperitoneal masses and probably have been detached from their proper GI tract origin during tumor growth [43].
Specific studies show that omental solitary GISTs are often of gastric origin showing histological traits typical of gastric GISTs as well as mutation pattern of gastric GISTs, with relatively common occurrence of PDGFRA mutations. Some of these tumors have a prognosis equally good to comparable gastric GISTs, and from a staging perspective, they have to be considered equivalent to primary gastric GISTs [44].
Disseminated omental GISTs are more often from small intestinal origin, as judged by their histologic similarity to intestinal GISTs and similar mutation patterns, including occurrence of KIT exon 9 duplications [44]. Mesenteric GISTs typically show histologic features similar to small intestinal GISTs and they most likely represent external extensions in (small) intestinal GISTs. In fact, many of these tumors are closely associated with or even involve small intestinal wall [45].
Studies are scant on abdominal-peritoneal and retroperitoneal GISTs. In our experience, this group includes a high number of disseminated intestinal GISTs, as judged by histology and KIT mutation patterns. GISTs have been sometimes reported as pancreatic, prostatic or gall bladder tumors. As long as the reported tumors have been true GISTs, their occurrence it better explained as extension of neighboring GISTs from stomach, duodenum, or rectum.
GIST ASSOCIATED WITH NEUROFIBROMATOSIS TYPE 1 (NF)
Patients with NF1 syndrome, the most common autosomal dominant syndrome in humans with a 1:3,000 birth incidence, have been estimated to have at least 200-fold increased risk for GIST, compared with general population. In fact, GISTs may be even more common in these patients, as some autopsy studies have shown as many as one of three NF1 patients to harbor GISTs [46].
NF1 associated GISTs occur at a slightly younger age than GISTs in general. They typically occur in duodenum or small intestine and are often multiple and relatively small being often incidentally detected during other medical surveillance or abdominal surgery. Most of these tumors show favorable histologic parameters (low mitotic rates) and are clinically indolent, but some patients have larger tumors with subsequent metastasis. Hyperplasia of the Cajal cells often occur in association with NF1 syndrome, possibly representing the earliest GIST precursor lesion [46,47].
Histologically, NF1-associated GISTs are similar to other (small) intestinal GISTs with a spindle cell pattern, common content of extracellular collagen globules and generally low mitotic rates. More highly cellular and mitotically active examples occur. Immunohistochemically, these GISTs are positive for KIT and anoctamin 1, and they retain succinate dehydrogenase subunit B expression. In contrast to most sporadic adult GISTs, NF1-associated GISTs lack KIT and PDGFRA mutations [46–48] and are driven by other genetic or epigenetic changes, possibly by the uninhibited RAS-signaling, in connection with the NF1-syndrome [49].
PEDIATRIC GISTS AND CARNEY–STRATAKIS SYNDROME
GISTs are very rare in children, and this group comprises no more than 1% of all GISTs. Notably, many older reports of GISTs in infants and small children deal with other entities, such as inflammatory myofibroblastic tumor.
Pediatric GISTs usually occur in the second decade with a marked female predominance, and most are gastric tumors, with only isolated duodenal and intestinal GISTs reported in children. A majority of pediatric GISTs are clinically indolent, but some patients develop liver and intra-abdominal metastases, with a protracted and unpredictable course of disease. Some patients live many years with liver metastases [50–56].
Pediatric GISTs are linked to two interrelated syndromes: Carney triad (CT) and CSS that can also manifest in (young) adults. CT contains the two of the following: gastric GIST, paraganglioma, and pulmonary chondroma/hamartoma and is not inheritable. The course is usually indolent, and long survival is common even after metastases develop [52,56]. CSS is a combination of gastric GIST and paraganglioma that can occur synchronously or metachronously, even several decades apart. This syndrome is inherited in autosomal dominant pattern and is linked to (and probably caused by) germline loss-of-function alterations in the somatically encoded mitochondrial membrane protein genes for succinate hydrogenase subunits, especially SDHB, previously also known in a subset of paragangliomas [52,53].
The typical histologic features of pediatric, CT, and CS-associated gastric GISTs include multinodularity or apparent tumor multiplicity, often with “plexiform” microscopic growth patterns with tumor nodules spaced between remaining muscularis propria smooth muscle elements. A majority of these GISTs have epithelioid cytology, often with high cellularity, with variable mitotic rates. Some GISTs show a paranganglioma-like pseudo-organoid pattern, whereas a minority of tumors shows a cellular spindle cell pattern. Occurrence of lymph node metastases is unique to pediatric GISTs linked with CT or CS syndromes (Fig. 6a) [53].
Fig. 6.
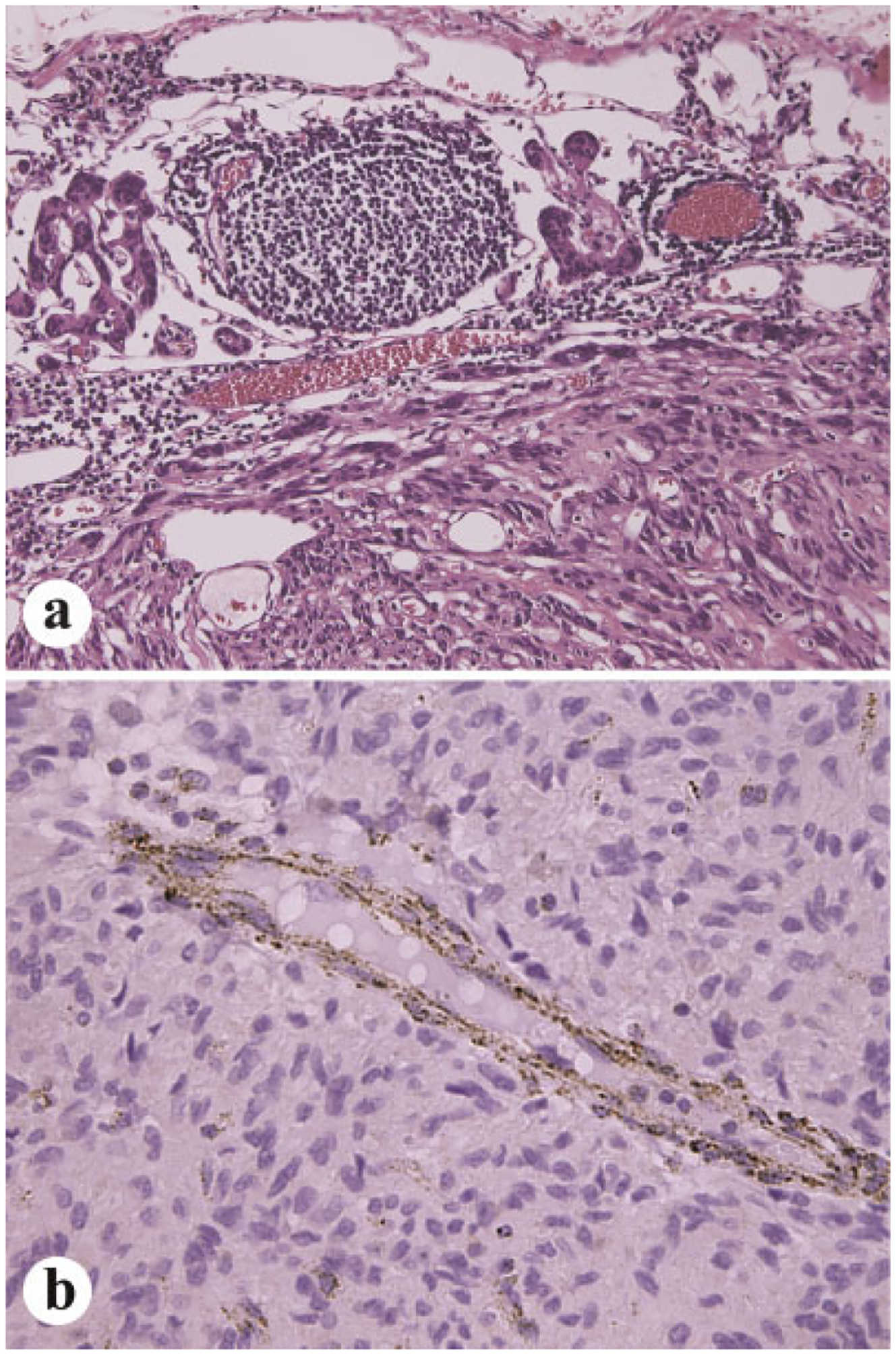
a: Lymph node metastases occur nearly exclusive in pediatric GISTs or those involving related syndromes, Carney triad or Carney–Stratakis syndrome. b: Pediatric GISTs and those in the above mentioned syndromes are typically negative for succinate dehydrogenase subunit B. Note that normal blood vessels are positive.
Similar to other GISTs, pediatric gastric GISTs and those in CT and CS syndromes express KIT and anoctamin 1 (DOG1), but lack KIT and PDGFRA mutations (wild type for these genes) [50,51]. In contrast to most adult GISTs, these GISTs are almost invariably immunohistochemically negative for the otherwise ubiquitously expressed mitochondrial complex protein, succinate dehydrogenase subunit B (SDHB, Fig. 6b) [57,58].
FAMILIAL GIST
A very small proportion of GISTs (our estimate: <0.1%) occur in connection with familial GIST syndrome, characterized by activating germline KIT (or in the case of three families, PDGFRA) mutations, similar to the mutations occurring in sporadic GISTs. More than 20 families have been reported worldwide [58–62]. The syndrome has an autosomal dominant pattern of inheritance with a high penetrance, most affected patients developing GISTs by middle age, and rarely manifesting in childhood, in contrast with many other familial syndromes. Typical is the occurrence of multiple, sometimes diffuse GISTs involving large segments of small intestine and colon, or stomach, rectum, and esophagus in some cases. Histologically typical is expansion of the myenteric plexus Cajal cell population, with GISTs showing features otherwise similar to sporadic GISTs.
The clinical course in these cases varies. While some patients enjoy long-lasting periods of indolent tumor growths, others develop liver and intra-abdominal metastases. Some of these patients have other manifestations of KIT overactivation: mastocytosis/urticaria pigmentosa, hyperpigmentation or nevi, and dysphagia related to Cajal cell hyperplasia. Recently, three mouse models with KIT exon 11, 13, and 17 germline mutations have been established that replicate human familial GIST syndrome and also demonstrate the pathogenetic role of KIT mutation in GIST [63–65].
RECOGNITION AND DIFFERENTIAL DIAGNOSIS OF GIST
In view of the now available tyrosine kinase inhibitor treatment, specific identification of GIST is important. Thus, routine immunohistochemical testing for KIT of all GI, intra-abdominal soft tissue, and hepatic mesenchymal tumors not otherwise recognized is recommended in order to comprehensively capture GISTs. Furthermore, GIST can manifest as bone or soft tissue metastasis, so that tumors histologically potentially compatible with GIST and not otherwise recognized should also be tested for GIST markers.
The differential diagnosis of GIST is rather broad and includes a number of entities, most of which are much less common than GIST. These tumors especially include true smooth muscle tumors (leiomyomas and leiomyosarcomas), schwannomas, dedifferentiated liposarcoma, and a number of rare entities. The differential diagnostically most important entities have been summarized in Table II.
TABLE II.
Summary of the Most Important Tumor Entities in the Differential Diagnosis of GIST
| Tumor entity | Preferential site in GI tract | Patient demographics | Key pathologic features contrasting with GIST |
|---|---|---|---|
| Leiomyoma (intramural) | Esophagus, rare elsewhere | Young adults, also older | Eosinophilic spindle cells with immunohistochemical positivity for smooth muscle markers and negativity for KIT and anoctamin 1 |
| Leiomyoma (of muscularis mucosae | Colon and rectum | Old adults | Small mucosal polyp. Tumor cells histologically similar to intramural leiomyoma |
| Leiomyoma, Mullerian type | Colon, also in the abdominal cavity | Middle-aged women | Estrogen and progesterone receptor-positive leiomyomas (low mitotic activity and atypia), comparable to uterine smooth muscle tumors and representing their extrauterine counterparts |
| Leiomyosarcoma | Relatively most common in colon, occurs at all sites | Old adults | Similar to leiomyoma, but contains nuclear atypia and mitotic activity. Negative for KIT and anoctamin 1. Positive for smooth muscle markers, desmin-positivity may vary |
| Glomus tumor | Stomach, nearly exclusively | Adults, marked female predominance | Round tumor cells with variably eosinophilic tumor cells. Positive for smooth muscle actin and negative for KIT |
| Schwannoma | Stomach, colon, rare in small intestine and elsewhere | Older adults | Spindle cells forming microtrabeculae or microfascicles. Tumor cells immunohistochemically positive for S100 protein and GFAP and negative for KIT and anoctamin 1. |
| Inflammatory myofibroblastic tumor | All segments of GI-tract, also other abdominal sites | Infants, children and young adults | Elongated, cytoplasm-rich spindle cells interspersed with lymphocytes and plasma cells. Typically positive for ALK and ALK-gene rearrangement by fluoresce in situ hybridization (FISH). Negative for KIT and anoctamin 1. |
| Inflammatory fibroid polyp | Small intestine, stomach, rare elsewhere | All ages, rare in children | Polypoid intraluminal mass, often in terminal ileum causing intussusception, or a small gastric polyp. Epithelioid to spindled lesional cells in a loose myxofibrous matrix with abundant capillaries. Contains eosinophils and mixed inflammatory cells. KIT negative. |
| Desmoid fibromatosis | Stomach and intestines | From young adulthood on | Moderately cellular, collagenous to myxoid spindle cell tumor. KIT negative. Nuclear positivity for beta-catenin is common |
| Synovial sarcoma | Stomach | Varies | Cellular spindle cell tumor, may involve mucosa or be transmural. Keratin-positive component, negative for KIT |
| Dedifferentiated liposarcoma | Intra-abdominal space, but may also involve intestines | Middle-aged to old adults | Spindle cell tumor with various appearances. Can simulate other tumors such as GIST, smooth muscle tumors, and fibromatosis. Negative for KIT and anoctamin 1. Often shows nuclear positivity for MDM2 |
CONCLUSION
Recognition of GIST in order to enable the patients to receive specific targeted therapy has become important in the past 10 years. This process is aided by understanding the potential occurrence of GIST in almost any segment of the GI tract and intra-abdominal space. It is also helpful to know the morphologic variation of GIST at different sites and application of immunohistochemical markers, especially KIT and anoctamin 1 in the recognition of GIST, as well as KIT/PDGFRA mutation analysis to optimize the clinical management of GIST patients. Pediatric GISTs differ from most adult ones in their lack of KIT and PDGFRA mutations. They are linked to two syndromes, both of which include loss of SDHB expression in tumor.
REFERENCES
- 1.Miettinen M, Lasota J: Gastrointestinal stromal tumors. Review of morphology, molecular pathology, prognosis, and differential diagnosis. Arch Pathol Lab Med 2006;130:1466–1478. [DOI] [PubMed] [Google Scholar]
- 2.Lee JR, Joshi V, Griffin JW Jr, et al. : Gastrointestinal autonomic nerve tumor: Immunohistochemical and molecular identity with gastrointestinal stromal tumor. Am J Surg Pathol 2001;25:979–987. [DOI] [PubMed] [Google Scholar]
- 3.Kitamura Y, Hirota S: Kit as a human oncogenic tyrosine kinase. Review. Cell Mol Life Sci 2004;61:2924–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hirota S, Isozaki K, Moriyama Y, et al. : Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998;279:577–580. [DOI] [PubMed] [Google Scholar]
- 5.Heinrich MC, Corless CL, Duensing A, et al. : PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003;299:708–710. [DOI] [PubMed] [Google Scholar]
- 6.Demetri G: Targeting the molecular pathophysiology of gastrointestinal stromal tumors with imatinib. Mechanisms, successes, and challenges to rational drug development. Hematol Oncol Clin North Am 2002;16:1115–1124. [DOI] [PubMed] [Google Scholar]
- 7.Blay JY, von Mehren M, Blackstein ME: Perspective on updated treatment guidelines for patients with gastrointestinal stromal tumors. Cancer 2010;116:5126–5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kindblom LG, Remotti HE, Aldenborg F, et al. : Gastrointestinal pacemaker cell tumor (GIPACT). Gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol 1998;153:1259–1269. [PMC free article] [PubMed] [Google Scholar]
- 9.Huizinga JD, Thuneberg L, Kluppel M, et al. : W/kit gene required for interstitial cells of Cajal and for intestinal pacemaker activity. Nature 1993;373:347–349. [DOI] [PubMed] [Google Scholar]
- 10.Nilsson B, Bumming P, Medis-Kindblom JM, et al. : Gastrointestinal stromal tumors: The incidence, prevalence, clinical course, and prognostication in the preimatinib mesylate era—A population-based study in western Sweden. Cancer 2005;103:821–829. [DOI] [PubMed] [Google Scholar]
- 11.Tryggvason G, Gislason HG, Magnusson MK, et al. : Gastrointestinal stromal tumors in Iceland, 1990–2003: The Icelandic GIST study, a population-based incidence and pathologic risk stratification study. Int J Cancer 2005;117:289–293. [DOI] [PubMed] [Google Scholar]
- 12.Agaimy A, Wunsch PH, Hofstaedter F, et al. : Minute sclerosing stromal tumors (GIST tumorlets) are common in adults and frequently show KIT mutations. Am J Surg Pathol 2007;31:113–120. [DOI] [PubMed] [Google Scholar]
- 13.Abraham SC, Krasinskas AM, Hofstetter WL, et al. : Seedling mesenchymal tumors (gastrointestinal stromal tumors and leiomyomas) are common in cidental tumors of thevesophagogastric junction. Am J Surg Pathol 2007;31:1629–1635. [DOI] [PubMed] [Google Scholar]
- 14.Tsuura Y, Hiraki H, Watanabe K, et al. : Preferential localization of c-kit product in tissue mast cells, basal cells of skin, epithelial cells of breast, small cell lung carcinoma and seminoma/dysgerminoma in human: Immunohistochemical study on formalin-fixed, paraffin-embedded tissues. Virchows Arch 1994;424:135–141. [DOI] [PubMed] [Google Scholar]
- 15.Sarlomo-Rikala M, Kovatich A, Barusevicius A, et al. : CD117: A sensitive marker for gastrointestinal stromal tumors that is more specific than CD34. Mod Pathol 1998;11:728–734. [PubMed] [Google Scholar]
- 16.Hornick JL, Fletcher CD: Immunohistochemical staining of KIT (CD117) in soft tissue sarcomas is very limited in distribution. Am J Clin Pathol 2002;117:188–193. [DOI] [PubMed] [Google Scholar]
- 17.Medeiros F, Corless CL, Duensing A, et al. : KIT-negative gastrointestinal stromal tumors: Proof of concept and therapeutic implications. Am J Surg Pathol 2004;28:889–894. [DOI] [PubMed] [Google Scholar]
- 18.Lasota J, Stachura J, Miettinen M: GISTs with PDGFRA exon 14 mutations represent subset of clinically favorable gastric tumors with epithelioid morphology. Lab Invest 2006;86:94–100. [DOI] [PubMed] [Google Scholar]
- 19.Yang YD, Cho H, Koo JY, et al. : TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 2008;455:1210–1215. [DOI] [PubMed] [Google Scholar]
- 20.Miettinen M, Wang ZF, Lasota J, et al. : Familial gastrointestinal stromal tumours with germline mutation of the KIT gene. Nat Genet 1998;19:323–324. [DOI] [PubMed] [Google Scholar]
- 21.Lee CH, Liang CW, Espinosa I: The utility of discovered on gastrointestinal stromal tumor 1 (DOG1) antibody in surgical pathology—The GIST of it. Adv Anat Pathol 2010;17:222–232. [DOI] [PubMed] [Google Scholar]
- 22.van de Rijn M, Rouse RV: CD34—A review. Appl Immunohistochem 1994;2:71–80. [Google Scholar]
- 23.Duensing A, Joseph NE, Medeiros F, et al. : Protein kinase C theta (PKCtheta) expression and constitutive activation in gastrointestinal stromal tumors (GISTs). Cancer Res 2004;64:5127–5131. [DOI] [PubMed] [Google Scholar]
- 24.Blay P, Astudillo A, Buesa JM, et al. : Protein kinase C theta is highly expressed in gastrointestinal stromal tumors but not in other mesenchymal neoplasias. Clin Cancer Res 2004;10:4089–4095. [DOI] [PubMed] [Google Scholar]
- 25.Motegi A, Sakurai S, Nakayama H, et al. : PKC theta, a novel immunohistochemical marker for gastrointestinal stromal tumors (GIST), especially useful for identifying KIT-negative tumors. Pathol Int 2005;55:106–112. [DOI] [PubMed] [Google Scholar]
- 26.Novelli M, Rossi S, Rodriquez-Justo M, et al. : DOG1 and CD117 are the antibodies of choice in the diagnosis of gastrointestinal stromal tumours. Histopathology 2010;57:259–270. [DOI] [PubMed] [Google Scholar]
- 27.Miettinen M, Sobin LH, Lasota J: Gastrointestinal stromal tumors of the stomach: A clinicopathologic, immunohistochemical, and molecular genetic studies of 1765 cases with long-term follow-up. Am J Surg Pathol 2005;29:52–68. [DOI] [PubMed] [Google Scholar]
- 28.Miettinen M, Makhlouf HR, Sobin LH, et al. : Gastrointestinal stromal tumors (GISTs) of the jejunum and ileum—A clinicopathologic, immunohistochemical and molecular genetic study of 906 cases prior to imatinib with long-term follow-up. Am J Surg Pathol 2006;31:477–489. [DOI] [PubMed] [Google Scholar]
- 29.Miettinen M, Furlong M, Sarlomo-Rikala M, et al. : Gastrointestinal stromal tumors, intramural leiomyomas, and leiomyosarcomas in the rectum and anus. A clinicopathologic, immunohistochemical, and molecular genetic study of 144 cases. Am J Surg Pathol 2001;25:1121–1133. [DOI] [PubMed] [Google Scholar]
- 30.Miettinen M, Kopczynski J, Maklouf HR, et al. : Gastrointestinal stromal tumors, intramural leiomyomas and leiomyosarcomas in the duodenum—A clinicopathologic, immunohisto-chemical and molecular genetic study of 167 cases. Am J Surg Pathol 2003;27:625–641. [DOI] [PubMed] [Google Scholar]
- 31.Lasota J, Miettinen M: Clinical significance of oncogenic KIT and PDGFRA mutations in gastrointestinal stromal tumors. Histopathology 2008;53:245–266. [DOI] [PubMed] [Google Scholar]
- 32.Lasota J, Miettinen M, Lasota J, et al. : A great majority of GISTs with PDGFRA mutations represent gastric tumors with low or no malignant potential. Lab Invest 2004;84:874–883. [DOI] [PubMed] [Google Scholar]
- 33.Corless CL, Schroeder A, Griffith D, et al. : PDGFRA mutations in gastrointestinal stromal tumors: Frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol 2005;23:5357–5364. [DOI] [PubMed] [Google Scholar]
- 34.Lasota J, Corless CL, Heinrich MC, et al. : Clinicopathologic profile of gastrointestinal stromal tumors (GISTs) with primary KIT exon 13 or exon 17 mutations: A multicenter study of 54 cases. Mod Pathol 2008;21:476–484. [DOI] [PubMed] [Google Scholar]
- 35.Min K-W: Small intestinal stromal tumors with skeinoid fibers. Clinicopathological, immunohistochemical, and ultrastructural investigations. Am J Surg Pathol 1992;16:145–155. [DOI] [PubMed] [Google Scholar]
- 36.Antonescu CR, Sommer G, Sarran L, et al. : Association of KIT exon 9 mutations with nongastric primary site and aggressive behavior: KIT mutation analysis and clinical correlates of 120 gastrointestinal stromal tumors. Clin Cancer Res 2003;9:3329–3337. [PubMed] [Google Scholar]
- 37.Miettinen M, Sarlomo-Rikala M, Sobin LH, et al. : Gastrointestinal stromal tumors and leiomyosarcomas in the colon. A clinicopathologic, immunohistochemical, and molecular genetic study of 44 cases. Am J Surg Pathol 2000;24:1339–1352. [DOI] [PubMed] [Google Scholar]
- 38.Hassan I, You N, Dozois EJ, et al. : Clinical, pathologic, and immunohistochemical characteristics of gastrointestinal stromal tumors of the colon and rectum: Implications for surgical management and adjuvant therapies. Dis Col Rectum 2006;49:609–615. [DOI] [PubMed] [Google Scholar]
- 39.Agaimy A, Wunsch PH, Dirnhofer S, et al. : Microscopic gastrointestinal stromal tumors in esophageal and intestinal surgical resection specimens: A clinicopathologic, immunohistochemical and molecular study of 19 lesions. Am J Surg Pathol 2008;32:867–873. [DOI] [PubMed] [Google Scholar]
- 40.Herawi M, Montgomery EA, Epstein JI: Gastrointestinal stromal tumors (GISTs) on prostate needle biopsy: A clinicopathologic study of 8 cases. Am J Surg Pathol 2006;30:1389–1395. [DOI] [PubMed] [Google Scholar]
- 41.Miettinen M, Sarlomo-Rikala M, Sobin LH, et al. : Esophageal stromal tumors: A clinicopathologic, immunohistochemical, and molecular genetic study of 17 cases and comparison with esophageal leiomyomas and leiomyosarcomas. Am J Surg Pathol 2000;24:211–222. [DOI] [PubMed] [Google Scholar]
- 42.Reith JD, Goldblum JR, Lyles RH, et al. : Extragastrointestinal (soft tissue) stromal tumors. An analysis of 48 cases with emphasis on histological predictors of outcome. Mod Pathol 2000;13:577–585. [DOI] [PubMed] [Google Scholar]
- 43.Agaimy A, Wunsch PH: Gastrointestinal stromal tumours: A regular origin in the muscularis propria, but an extremely diverse gross presentation. A review of 200 to critically re-evaluate the concept of so-called extragastrointestinal stromal tumors. Langenbecks Arch Surg 2006;391:322–329. [DOI] [PubMed] [Google Scholar]
- 44.Miettinen M, Sobin LH, Lasota J: Gastrointestinal stromal tumors presenting as omental masses—A clinicopathologic analysis of 95 cases. Am J Surg Pathol 2009;33:1267–1275. [DOI] [PubMed] [Google Scholar]
- 45.Miettinen M, Monihan JM, Sarlomo-Rikala M, et al. : Gastrointestinal stromal tumors/smooth muscle tumors/GISTs in the omentum and mesentery—Clinicopathologic and immunohistochemical study of 26 cases. Am J Surg Pathol 1999;23:1109–1118. [DOI] [PubMed] [Google Scholar]
- 46.Andersson J, Sihto H, Meis-Kindblom JM, et al. : NF1-associated gastrointestinal stromal tumors have unique clinical, phenotypic, and genotypic characteristics. Am J Surg Pathol 2005;29:1170–1176. [DOI] [PubMed] [Google Scholar]
- 47.Miettinen M, Fetsch JF, Sobin LH, et al. : Gastrointestinal stromal tumors in patients with neurofibromatosis 1. A clinicopathologic study of 45 patients with long-term follow-up. Am J Surg Pathol 2006;30:90–96. [DOI] [PubMed] [Google Scholar]
- 48.Kinoshita K, Hirota S, Isozaki K, et al. : Absence of c-kit gene mutations in gastrointestinal stromal tumours from neurofibromatosis type 1 patients. J Pathol 2004;202:80–85. [DOI] [PubMed] [Google Scholar]
- 49.Maertens O, Prenen H, Debiec-Rychter M, et al. : Molecular pathogenesis of multiple gastrointestinal stromal tumors in NF1 patients. Hum Mol Genet 2006;15:1015–1023. [DOI] [PubMed] [Google Scholar]
- 50.Carney JA: Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (Carney triad): Natural history, adrenocortical component, and possible familial occurrence. Mayo Clin Proc 1999;74:543–552. [DOI] [PubMed] [Google Scholar]
- 51.Carney JA, Stratakis CA: Familial paraganglioma and gastric stromal sarcoma: A new syndrome distinct from the Carney triad. Am J Med Genet 2002;108:132–139. [DOI] [PubMed] [Google Scholar]
- 52.Stratakis CA, Carney JA: The triad of paragangliomas, gastric stromal tumours and pulmonary chondromas (Carney triad) and the dyad of paragangliomas and gastric stromal sarcomas (Carney-Stratakis syndrome): Molecular genetics and clinical implications. J Intern Med 2009;266:43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pasini B, McWhinney SR, Bei T, et al. : Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes encoding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. [DOI] [PubMed]
- 54.Prakash S, Sarran L, Socci N, et al. : Gastrointestinal stromal tumors in children and young adults: A clinicopathologic, molecular, and genomic study of 15 cases and review of the literature. J Pediatr Hematol Oncol 2005;27:179–187. [DOI] [PubMed] [Google Scholar]
- 55.Miettinen M, Lasota J, Sobin LH: Gastrointestinal stromal tumors of the stomach in children and young adults: A clinicopathologic, immunohistochemical, and molecular genetic study of 44 cases with long-term follow-up and review of the literature. Am J Surg Pathol 2005;29:1373–1381. [DOI] [PubMed] [Google Scholar]
- 56.Zhang L, Smyrk TC, Young WF, et al. : Gastric stromal tumors in Carney triad are different clinically, patholoigically, and behaviorally from sporadic gastric gastrointestinal stromal tumors: Findings in 104 cases. Am J Surg Pathol 2010;34:53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gill AJ, Chou A, Vilain R, et al. : Immunohistochemistry for SDHB divides gastrointestinal stromal tumors (GISTs) into 2 distinct types. Am J Surg Pathol 2010;34:636–644. [DOI] [PubMed] [Google Scholar]
- 58.Agarwal R, Robson M: Inherited predisposition to gastrointestinal stromal tumor. Hematol Oncol Clin N Am 2009;23:1–13. [DOI] [PubMed] [Google Scholar]
- 59.Carballo M, Roig I, Aguilar F, et al. : Novel c-KIT germline mutation in a family with gastrointestinal stromal tumors and cutaneous hyperpitgmentation. Am J Med Genet 2005;132:361–364. [DOI] [PubMed] [Google Scholar]
- 60.Li FP, Fletcher JA, Heinrich MC, et al. : Familial gastrointestinal stromal tumor syndrome: Phenotypic and molecular features in a kindred. J Clin Oncol 2005;23:2735–2743. [DOI] [PubMed] [Google Scholar]
- 61.Chompret A, Kannengiesser C, Barrois M, et al. : PDGFRA germline mutation in a family with multiple cases of gastrointestinal stromal tumor. Gastroenterology 2004;65:187–197. [DOI] [PubMed] [Google Scholar]
- 62.Chen H, Hirota S, Isozaki K, et al. : Polyclonal nature of diffuse proliferation of interstitial cells of Cajal in patients with familial gastrointestinal stromal tumours. Gut 2002;51:793–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sommer G, Agosti V, Ehlers I, et al. : Gastrointestinal stromal tumors in a mouse model by targeted mutation of the kit receptor tyrosine kinase. Proc Natl Acad Sci USA 2003;100:6706–6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rubin BP, Antonescu CR, Scott-Browne JP, et al. : A knock-in mouse model of gastrointestinal stromal tumor harboring kit K641E. Cancer Res 2005;65:6631–6639. [DOI] [PubMed] [Google Scholar]
- 65.Nakai N, Ishikawa T, Nishitani A, et al. : A mouse model of a human multiple GIST family with KIT-Asp820Tyr mutation generated by a knock-in strategy. J Pathol 2008;214:302–311. [DOI] [PubMed] [Google Scholar]