

Abstract
Significant prior evidence indicates that centrally acting oxytocin robustly modulates stress responsiveness and anxiety-like behavior, although the neural mechanisms behind these effects are not entirely understood. A plausible neural basis for oxytocin mediated stress reduction is via inhibition of corticotropin-releasing hormone (CRH) neurons in the paraventricular nucleus of the hypothalamus (PVN) that regulate activation of the hypothalamic-pituitary-adrenal (HPA) axis. Previously, we have shown that following subcutaneous injection of 2.0 M NaCl, oxytocin (OT) synthesizing neurons are activated in the rat PVN, an oxytocin receptor (Oxtr) dependent inhibitory tone develops on a subset of parvocellular neurons, and stress-mediated increases in plasma corticosterone levels are blunted. Here, we utilized transgenic male CRH-reporter mice to selectively target PVN CRH neurons for whole-cell recordings. These experiments reveal that acute salt loading produces tonic inhibition of PVN CRH neurons through a mechanism that is largely independent of synaptic activity. Further studies reveal that a subset of CRH neurons within the PVN synthesize mRNA for Oxtr(s). Salt induced Oxtr-dependent inhibitory tone was eliminated in individual PVN CRH neurons filled with GDP-β-S. Additional electrophysiological studies suggest that reduced excitability of PVN CRH neurons in salt loaded animals is associated with increased activation of inwardly rectifying potassium channels. Nevertheless, substantial effort to recapitulate the core effects of salt loading by activating Oxtr(s) with an exogenous agonist produced mixed results. Collectively, these results enhance our understanding of how oxytocin receptor-mediated signaling modulates the function of CRH neurons in the PVN.
Keywords: oxytocin, corticotropin-releasing hormone, paraventricular nucleus, tonic inhibition, oxytocin receptor, stress, hypothalamus-pituitary-adrenal axis
Introduction
The neuroendocrine response to acute stress is largely defined by changes in the state of activation of the sympathetic nervous system and of the hypothalamic-pituitary-adrenal (HPA) axis. Both effects depend in large part on modulation of distinct populations of neurons within the paraventricular nucleus of the hypothalamus (PVN). These include preautonomic parvocellular neurons, which project to preganglionic sympathetic neurons in the hindbrain and spinal cord to regulate sympathetic output, and corticotropin-releasing hormone (CRH) positive parvocellular neurosecretory neurons, which project to the median eminence to regulate activation of the HPA axis. Both populations of neurons are subject to modulatory control by a wide range of endogenous and exogenous compounds.
Oxytocin is one such neuromodulator, an endogenous neuropeptide produced predominantly in the paraventricular and supraoptic nuclei of the hypothalamus. Significant prior evidence suggests that endogenous oxytocin, acting in the PVN, is likely to directly modulate stress-induced activation of the HPA axis. Indeed, a variety of distinct psychological stressors associated with increased HPA activity promote central release of oxytocin [1–5], chronic administration of oxytocin into the PVN blunts stress responsiveness [6,7], and intracerebral infusion of an Oxtr antagonist promotes stress-induced release of both adrenocorticotropic hormone (ACTH) and corticosterone into the blood [8]. Similarly, oxytocin knockout mice produce elevated levels of CRH mRNA in the PVN in response to restraint stress [9], while lactating rodents demonstrate both elevated levels of oxytocin release and reduced levels of CRH mRNA in the PVN [10,11]. Collectively, these data suggest that oxytocin release, produced in response to psychological stressors, initiates a negative feedback mechanism that ultimately dampens stress responsiveness in part through down-regulation of CRH synthesis in the PVN [12,13].
Intriguingly, several findings from our group suggest that oxytocin release, produced by a stressor that is not associated with robust activation of the HPA axis, likely also dampens stress responsiveness, but through a mechanism not likely to involve genomic modulation of CRH synthesis. Specifically, we have reported that acute hypernatremia, a peripheral stressor that promotes vasopressin dependent antidiuresis and oxytocin dependent natriuresis [14–17], also directly reduces HPA axis activation produced by subsequent restraint stress [18]. Notably, additional work in both rats and mice revealed that this effect is associated with increased activation of c-Fos in PVN oxytocinergic neurons, decreased activation of c-Fos in PVN CRH neurons, and development of an Oxtr dependent inhibitory tone on a subset of PVN parvocellular neurons that lack both a prominent A-type potassium current and a robust low threshold spike [19–21].
The intrinsic electrophysiological phenotype of the PVN neurons that were inhibited by oxytocin after acute hypernatremia in Frazier et al (2013) were consistent with the interpretation that they were CRH expressing neurosecretory neurons involved in the regulation of the HPA axis [22], but this phenotype was not directly confirmed. Therefore, in the current study, we used male mice that express tdTomato in CRH neurons to directly test the hypothesis that acute hypernatremia creates an Oxtr-mediated inhibitory tone on PVN CRH neurons, and to further explore the cellular mechanisms and downstream effects of this inhibition. Our results strongly suggest that acute hypernatremia produces an Oxtr-dependent inhibitory tone in PVN CRH neurons, and that this effect depends on Oxtr(s) specifically expressed by these neurons. Further experiments suggest inhibition of PVN CRH neurons as observed after acute hypernatremia likely involves increased activation of a potassium ionophore, and is associated with a clear reduction in neuronal gain. That said, despite substantial effort, we were not able to fully recapitulate all of the effects of acute hypernatremia on PVN CRH neurons in experiments that relied solely on exposure to exogenous Oxtr agonists. That observation may simply highlight technical difficulties with effectively reproducing the kinetic and concentration profiles of endogenous Oxtr activation, but might also suggest that there remain some undiscovered complexities in this system.
Materials and Methods
Animals
CRH -reporter mice
All animal procedures used in this study were in compliance with the United States National Research Council and United States Public Health Service Guides on the Care and Use of Laboratory Animals, and were reviewed and approved by the Institutional Animal Care and Use Committee at the University of Florida. Most studies were performed on CRH-reporter mice (3–6 months of age) that expressed red fluorescent protein (tdTomato) in neurons that produce CRH. These mice were generated by crossing Crh-IRES-Cre mice (B6(Cb)-Crhtm1(cre)Zjf/J mice, Jackson Laboratories Stock #012704) with mice that express tdTomato behind a floxed stop codon in the Rosa26 locus under control of the CAG promoter (B6.Cg-Gt(ROSA)26Sortm14(CAG-TdTomato)Hze/J, Jackson Laboratories Stock #007914). These same CRH-reporter mice have been previously found, by our laboratory and others, to express mRNA for CRH in 80–95% of tdTomato positive neurons in the hypothalamus [20,23].
CRH-Oxtr KO mice
Mice used in these experiments were on a C57BL/6J background. In order to generate mice that lack Oxtr(s) in CRH neurons and their littermate controls, mice homozygous for the Oxtr-flox knock-in gene and also heterozygous for the CRH-Cre knock-in gene were bred with those expressing only the Oxtr-flox gene. Oxtr-flox knock-in mice (Jackson laboratories; Stock # 008471) express loxP sites that flank exons 2–3 of the Oxtr gene (Lee et al., 2008).
Saline injections and slice preparation
On the day of experimentation, male mice were assigned to one of two groups: isotonic or hypertonic. Mice in the isotonic group received subcutaneous injections of 0.1 mL of 0.15 M NaCl, while mice in the hypertonic group received subcutaneous injections of 0.1 mL of 2.0 M NaCl. All animals were euthanatized 1-hour post-injection. Mice in the isotonic group were allowed free access to water in the one hour between injection and euthanasia, while mice in the hypertonic group were denied access to water between the time of injection and euthanasia. To minimize pain and irritation, each injection was preceded by 2% lidocaine (~0.01 mL). Prior work from our group has revealed that this protocol increases plasma sodium concentration without affecting indices of blood volume [20]. Sixty minutes after saline injections, mice were administered ketamine (80–100 mg/kg, IP) and were rapidly decapitated using a rodent guillotine. The brain was quickly removed, and coronal sections (300 μm thick) through the PVN were made using a Leica VT 1000s vibratome. Slices were incubated for 30 minutes in a dissecting solution maintained at 30–35°C that contained in mM: 124 NaCl, 2.5 KCl, 1.23 NaH2PO4, 2.5 MgSO4, 10 D-glucose, 1 CaCl2, and 25.9 NaHCO3, saturated with 95% O2–5% CO2. After equilibrating at room temperature for at least 30 minutes, slices were transferred to a slice chamber for experimental use.
In Vitro Electrophysiology
For whole-cell recording, slices were continuously perfused at a rate of 1.2–1.5 mL/min with aCSF that contained (in mM): 126 NaCl, 3 KCl, 1.2 NaH2PO4, 1.5 MgSO4, 11 D-glucose, 2.4 CaCl2, and 25.9 NaHCO3. This solution was saturated with 95% O2 and 5% CO2, and bath temperature was maintained at 28±2°C. For experiments that required minimal spontaneous synaptic activity in the slice, this ACSF was supplemented with TTX (1μM), to block voltage-gated sodium channels, DNQX (20 μM) and DL-2-amino-5-phosphonopenatanoic acid (APV, 40 μM), to block ionotropic glutamate receptors, picrotoxin (100 μM) to block both synaptic and extrasynaptic GABAA receptors. For most experiments the patch electrode was filled with a K-gluconate based internal solution that contained (in mM): 130 K-gluconate, 10 KCl, 10 NaCl, 2 MgCl2, 1 EGTA, 2 Na2ATP, 0.3 NaGTP, and 10 HEPES, pH adjusted to 7.3 using KOH and volume adjusted to 285–300 mOsm. For experiments that involved intracellular inhibition of G-protein coupled receptors, the GTP in this internal was replaced with GDP-β-S (300 μM), and cells were allowed to equilibrate for 30 minutes after whole-cell configuration was attained to allow intracellular diffusion of GDP-β-S into the cell from the patch pipette. Control cells in Fig. 4 were also allowed the same 30-minute period for whole-cell dialysis before experiments began. For experiments designed to measure spontaneous synaptic currents, a Cs-gluconate based internal solution was used, which contained, in mM: 140 Cs-gluconate, 3 NaCl, 1 MgCl2, 0.2 EGTA, 4 Na2ATP, 0.3 NaGTP, 5 QX-314-Cl, and 10 HEPES, pH adjusted to 7.3 using CsOH and volume adjusted to 285–300 mOsm. In all cases, slices were visualized using an Olympus BX51W1 microscope using a combination infrared differential interference contrast (IR DIC) and epifluorescence microscopy. Whole-cell voltage-clamp recordings were performed using micropipettes pulled from borosilicate glass using a Flaming/Brown electrode puller (Sutter P-97; Sutter Instruments, Novato, California). Electrode open tip resistance was between 4 and 6 MΩ using the K-gluconate internal. Voltage-clamp experiments were performed using an Axon Multiclamp 700A or 700B amplifier (Molecular Devices, Sunnyvale, CA). Data were sampled at 20 kHz, filtered at 2 kHz, and recorded on a computer by a Digidata 1400 A/D converter using Clampex version 10 (Molecular Devices, Sunnyvale, CA). All chemicals used for in vitro electrophysiology were obtained from either Tocris Cookson or Sigma-Aldrich except [d(CH2)51,Tyr(Me)2,Orn8]-oxytocin (Oxtr-A), which was obtained from Bachem.
Immunohistochemistry and two-photon microscopy
Immunohistochemical studies were performed on 300 μm brain slices prepared identically to those used for in-vitro electrophysiology. Slices were preserved in 10% formalin overnight at 4°C. All subsequent steps were performed at 25C on an orbital shaker. Slices were washed with PBST (PBS + 0.1% Triton X-100) 5× 5 min, then incubated in blocking solution (PBST supplemented with 2% bovine serum albumin and 2% normal goat serum) for 48hr. Primary antibodies against neurophysin 1 (1:500 goat anti-mouse neurophysin-1, Millipore MABN844) and tdTomato (1:500 rabbit anti-RFP, Rockland 6004013795) were then applied in blocking solution for 72hr. Slices were washed (5× 5 min with PBST) then incubated in secondary antibodies against mouse (1:500 goat anti-mouse, ThermoFisher A11001) and rabbit (1:500 goat anti-rabbit, ThermoFisher A11012) conjugated with two-photon-excitable fluorophores (488nm and 594nm, respectively) for 24hr. Slices were washed (5× 5 min with PBS), coverslipped with an aqueous mounting medium (Vectashield), and imaged with a two-photon laser scanning microscope (excited by 810 nm femtosecond laser).
RNAscope in situ hybridization
In situ hybridization was performed on brain sections of CRH-tdTomato mice using the RNAScope® Multiplex Fluorescent Assay (n=3), or the RNAScope® Multiplex Fluorescent Assay V2 (n=2; Advanced Cell Diagnostics, Newark, CA, USA) as per the manufacturer’s instructions, and using the same modifications to the pretreatment procedure that we have previously described (Smith et al., 2014; de Kloet et al., 2016). These modifications allow for the visualization of mRNA transcripts (visible as punctate dots) in the presence of preserved tdTomato fluorescence. Briefly, mice were euthanized with an overdose of sodium pentobarbital and transcardially perfused with 4% RNase free paraformaldehyde (PFA). Brains were extracted, post-fixed in 4% PFA for 4 h, and then transferred into RNase free 30% sucrose at 4° C. Brains were then coronally sectioned at 20 μm using a cryostat. Brain sections were rinsed twice in RNase free PBS, mounted onto Superfrost Plus Gold slides, air-dried for 20 min at room temperature and then stored at −80° C. On the day of RNAscope in situ hybridization, brain sections were again air-dried for 30 min at room temperature, incubated in protease IV for 20 min, and then hybridized with specific probes. Probes used in this study were: (1) DapB, negative control, (2) Ubc, positive control, and (3) Oxtr. The probe for Oxtr was used to evaluate the localization of Oxtr mRNAs within CRH-tdTomato neurons in the PVN. The color label for all probes was assigned to FAR RED (Excitation: 647 nm; Emission: 690 ± 10 nm).
Image capture, processing, and analysis.
For in situ hybridization studies, z-stacks throughout the PVN were captured and processed using Axiovision 4.8.2 software and a Zeiss AxioImager fluorescent Apotome microscope (Carl Zeiss Microscopy, Thornwood, NY, USA). In all cases, z-stacks of tdTomato fluorescence and transcripts of interest were captured at 40× magnification. These z-stacks each contained an average of 20 optical sections, with the distance between the optical sections set at 0.5 μm. For each mouse, an average of 4 z-stacks were captured throughout the rostral-caudal extent of the parvocellular neurosecretory region of the PVN. Importantly, when capturing these images, sections hybridized with the positive control probes were used to determine the exposure time and image processing required to provide optimal visualization of RNA signal. Then, as described in detail previously (de Kloet et al., 2016), these same parameters were used for visualization of mRNA transcripts of interest and to assess background fluorescence in sections hybridized with negative control probe (DapB). Importantly, using these exposure times and image processing parameters there was minimal or no fluorescence in sections hybridized with the negative control probe. The projection image shown in the results section was generated from a representative z-stack and then prepared using Adobe Photoshop 7.0 where the brightness and contrast were adjusted to provide optimal visualization. Quantitative cell counts (see results) were performed on individual sections (not z-stacks) and care was taken to not count puncta above or below tdTomato positive neurons.
Data and statistical analysis
The analysis of all electrophysiological data was performed using custom software written in OriginC (OriginLab, Northampton, MA) by CJF. Current density was calculated as the ratio of holding current to whole-cell capacitance (pA/pF) when cells were continuously voltage-clamped at −70 mV. Whole-cell capacitance (Cm) was calculated as τ(1/Ra+1/Rm), as previously described [24]. More specifically, in this equation τ is the monoexponential time constant produced by fitting the whole-cell capacitive transients, as produced by a 50 msec voltage step from −70 mV to −80 mV, between 10% and 90% of their peak amplitude (I0). Ra (access resistance) was calculated as ΔV/I0, and Rm (input resistance) was calculated as (ΔV – RaISS)/ISS, where ISS is the steady-state current observed at the end of the 50 msec voltage step. Spontaneous postsynaptic currents were detected using parameter-based event detection software. Event parameters were chosen to detect the vast majority of distinguishable events while minimizing false positives. Typical threshold amplitude was 6–8 pA and typical threshold area was 20–40 pA*ms. In all cases, an algorithm for detecting complex peaks (peaks that occur during the decay period of a previous event) was employed. This algorithm calculates event amplitude for the second (and subsequent) peaks within the decay period based on extrapolated monoexponential decay from the peak of the initial event. Within groups, a two-tailed, one-sample t-test was used to evaluate changes in current density following the antagonism of oxytocin receptors [null hypothesis, Δ current density = 0]. In experiments that involved comparing a single variable across two groups, a two-tailed, two-sample unpaired t-test was used, and Welch’s correction was applied in cases where the groups had unequal variance. For statistical analyses in Fig. 6C–D and G–I, a two-way repeated-measures ANOVA was employed (salt condition [isotonic or hypertonic] × current or voltage command). When this analysis revealed a main effect of salt loading and/or a significant salt × command interaction, Sidak-Holm post-hoc tests were used to compare individual means. For all electrophysiological experiments testing the effects of bath applied drugs, baseline was considered as 2 −5 mins after initiation of a stable recording, and the drug effect was measured 6–8 minutes after drug application unless otherwise noted (see Figure legends). All data are expressed as mean ± SEM. P-values ≤ 0.05 were considered significant.
Results
Acute salt loading produces an oxytocin receptor-dependent tonic inhibitory current in PVN CRH neurons
Prior work from our group revealed that acute hypernatremia reduces activation of the HPA axis in response to subsequent restraint stress, and produces an inhibitory oxytocinergic tone in a subset of PVN parvocellular neurons [18–20]. In order to determine whether this phenomenon involves direct inhibition of PVN CRH neurons, we crossed Crh-IRES-Cre mice with a tdTomato Cre reporter to create a CRH reporter animal (see methods). Prior work from our group and others has confirmed that tdTomato expression is an excellent marker of PVN CRH neurons in this animal model [20,23,25,26]. Immunohistochemistry for neurophysin 1 (NP1), a carrier protein for oxytocin, in CRH reporter animals revealed that PVN CRH neurons are located in close proximity to NP1 positive and thus presumably oxytocinergic neurons, and also further reinforce that there is minimal overlap between these two populations (Fig. 1A). We performed whole-cell patch clamp recordings from PVN CRH neurons from these animals identified with a combination of epifluorescence and differential interference contrast microscopy (Fig. 1B–D). Intrinsic properties of CRH neurons were calculated from data generated in voltage clamp using brief voltage steps from −70 to −80 mV. Consistent with expectations, we found the vast majority of PVN CRH neurons had high input resistance (typically > 800 MΩ), small whole-cell capacitance (typically < 20 pF). When evaluated in current clamp, these neurons lacked a robust transient outwardly rectifying potassium current (IA), typical of magnocellular neurons, and also lacked a robust low threshold spike, often present in preautonomic neurons [19,22,23].
Figure 1: Acute salt loading produces an oxytocin receptor-dependent tonic inhibitory current in PVN CRH neurons.
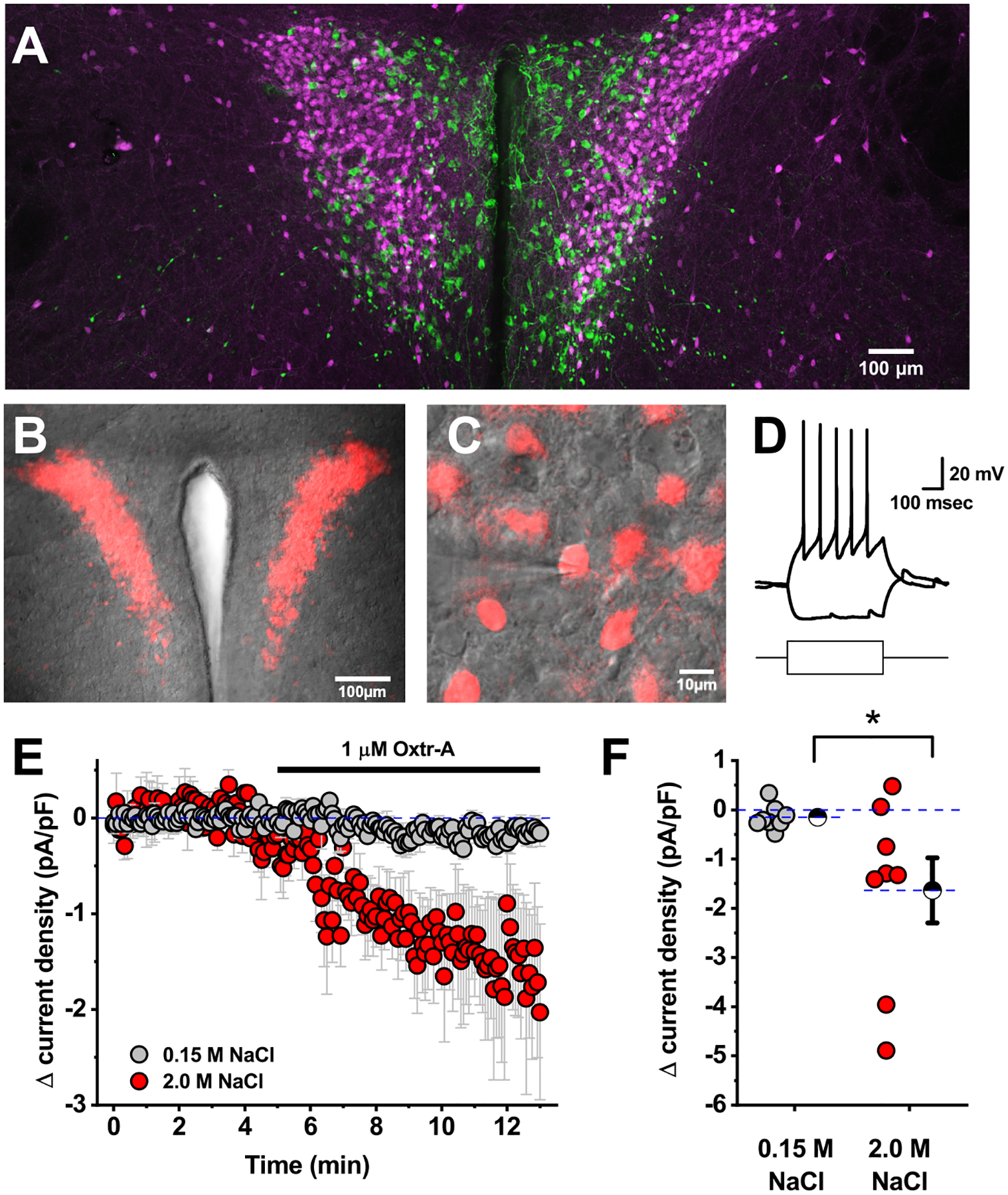
A) Immunohistochemistry for NP1 in CRH reporter animals suggests that PVN oxytocinergic neurons (green) are located in close proximity to PVN CRH neurons (magenta), but that there is minimal if any overlap between these populations. B) tdTomato (red), as a marker for CRH neurons, can also be visualized in combination with DIC (gray) in an in-vitro slice preparation using a combination of epifluorescence and differential interference contrast microscopy. C) CRH neurons can be selectively targeted for patch-clamp analysis by their fluorescence. The patch pipette is visible coming in from the left side of the image. D) Representative response of a CRH neuron to +/−20 pA current steps. The shape of the depolarizing voltage trace is characteristic of CRH neurons (lacking both an IA current and an LTS). A small hyperpolarizing step does not reveal robust hyperpolarization-activated currents. E) Mean change in current density in response to Oxtr-A in CRH neurons from mice that received an isotonic or hypertonic saline injection (grey vs. red, respectively). F) Specific change in current density produced by Oxtr-A for each cell that contributed to D. Hatched symbols with error bars represent mean and SE of each group. Asterisk indicates a significant difference between groups (p=0.04).
In order to evaluate the effect of acute salt loading (see methods) on identified PVN CRH neurons, we voltage-clamped the neurons at −70 mV and bath applied an oxytocin receptor antagonist (Oxtr-A, 1 μM). In animals that received isotonic saline, Oxtr-A had no effect on current density (current per unit capacitance) required to hold the cells at −70 mV. However, in animals that received injections of hypertonic saline, bath application of Oxtr-A produced a significant decrease in current density (−1.6 ± 0.7 pA/pF, n=8, p=0.04 vs. isotonic condition, Fig. 1D–E). This result effectively reveals that salt acute loading produces an oxytocin receptor-dependent tonic inhibitory current in genetically identified PVN CRH neurons.
Acute salt loading does not alter spontaneous synaptic neurotransmission to the parvocellular CRH neurons
We next asked whether salt loading might also modulate the excitability of PVN CRH neurons by altering the frequency or amplitude of spontaneous excitatory or inhibitory synaptic currents in the PVN. In order to evaluate those possibilities, we used a Cs-gluconate based internal solution (see methods) to isolate glutamatergic spontaneous excitatory postsynaptic currents (sEPSCs) in PVN CRH neurons voltage clamped at −60 mV, and also to isolate GABAergic spontaneous inhibitory postsynaptic currents (sIPSCs) in the same cells when voltage-clamped at +10 mV (Fig. 2A–B). Overall, these experiments revealed that acute salt loading had no significant effect on either frequency or amplitude of sEPSCs or sIPSCs as observed in PVN CRH neurons (Fig. 2C–D). Specifically, in slices obtained from animals that received isotonic saline, the frequency of sEPSCs was 2.9 ± 0.7 Hz (n=9), while the frequency of sIPSCs was 2.1 ± 0.3 Hz (n=13). In slices extracted from animals that received hypertonic saline theses value were 2.0 ± 0.5 Hz for sEPSC frequency and 2.7 ± 0.7 Hz, for sIPSC frequency (n= 9, 13, and p=0.29 and 0.43, respectively). Mean sEPSC and sIPSC amplitudes were similarly unaffected (see Fig. 2D–E).
Figure 2: Acute salt loading does not alter spontaneous synaptic neurotransmission to PVN CRH neurons.
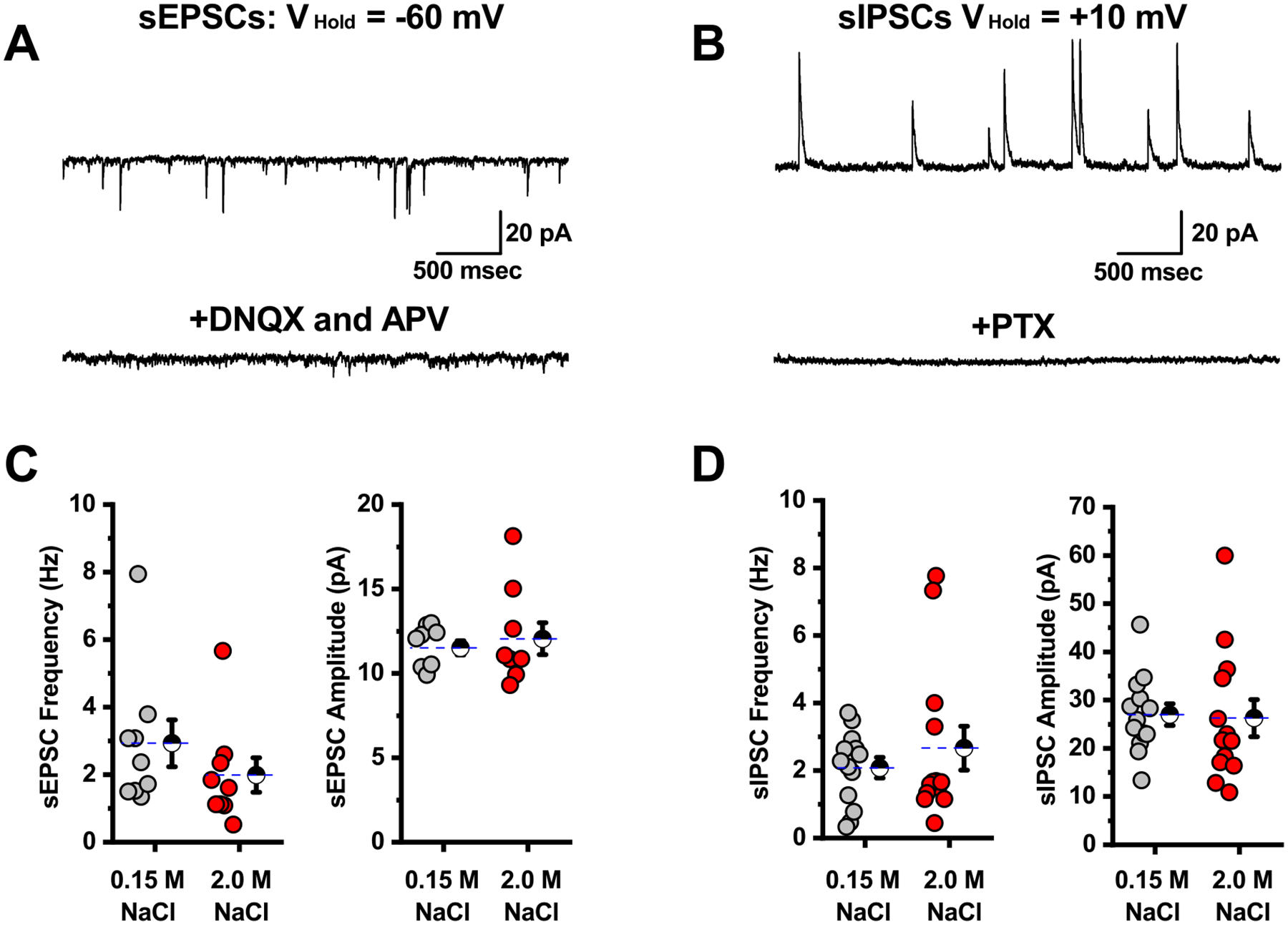
A) Isolated sEPSCs in a representative PVN CRH neuron voltage-clamped at −60 mV (top trace) are blocked by bath application of the ionotropic glutamate receptor antagonists DNQX and APV (bottom trace). B) Isolated sIPSCs in a representative PVN CRH neuron voltage-clamped at +10 mV (top trace) are blocked by bath application of the GABAA receptor antagonist PTX. C) Summary data illustrating sEPSC frequency and amplitude as observed on a cell by cell basis for cells voltage clamped at −60 mV. D) Summary data illustrating sIPSC frequency and amplitude as observed on a cell by cell basis for cells voltage-clamped at +10 mV. Overall, there was no significant difference in the frequency or amplitude of spontaneous glutamatergic or GABAergic currents observed in PVN CRH neurons extracted from slices that received isotonic vs. hypertonic saline (grey vs. red, respectively). For each box plot, large hatched symbols to the right of the data represent the mean of each group, and error bars indicate the SE.
Oxytocinergic receptor-mediated tonic current in CRH neurons is insensitive to glutamate blockers, GABA blockers, and TTX.
Although salt loading did not alter glutamate or GABAA receptor-mediated spontaneous synaptic transmission as observed in PVN CRH neurons, it seemed possible that oxytocin receptor activation could still indirectly modulate CRH neurons by altering transmitter uptake or breakdown in ways that influence activation of extrasynaptic receptors, or by promoting activity-dependent release of a transmitter that does not act through glutamate or GABAA receptors. In order to evaluate those possibilities, we repeated our experiments with Oxtr-A in slices obtained from animals that received hypertonic saline. Experiments were performed exactly as in Fig. 1D (red traces) except that we added DNQX, APV, and PTX, to block not only synaptic, but also extrasynaptic ionotropic glutamate and GABAA receptors, and also TTX (1 μM) to block all activity-dependent synaptic transmission in the slice. Under these conditions, bath application of Oxtr-A still produced a robust decrease in current density in PVN CRH neurons voltage clamped at −70 mV (−1.2 ± 0.3 pA/pF, n=5, p=0.02 on a one-sample t-test vs. null hypothesis of mean = 0, Fig. 3A–B). Notably, this effect is not significantly different than observed in Fig. 1D in the absence of DNQX, APV, PTX, and TTX (p=0.61, two-sample unpaired t-test). Collectively these results suggest that Oxtr-mediated tonic inhibition of PVN CRH neurons as observed in slices from salt loaded animals is independent of synaptic or extrasynaptic activation of ionotropic glutamate or GABAA receptors and does not require ongoing action potential-dependent synaptic transmission.
Figure 3: Oxytocin receptor-mediated tonic current in CRH neurons is insensitive to glutamate blockers, GABA blockers, and TTX.
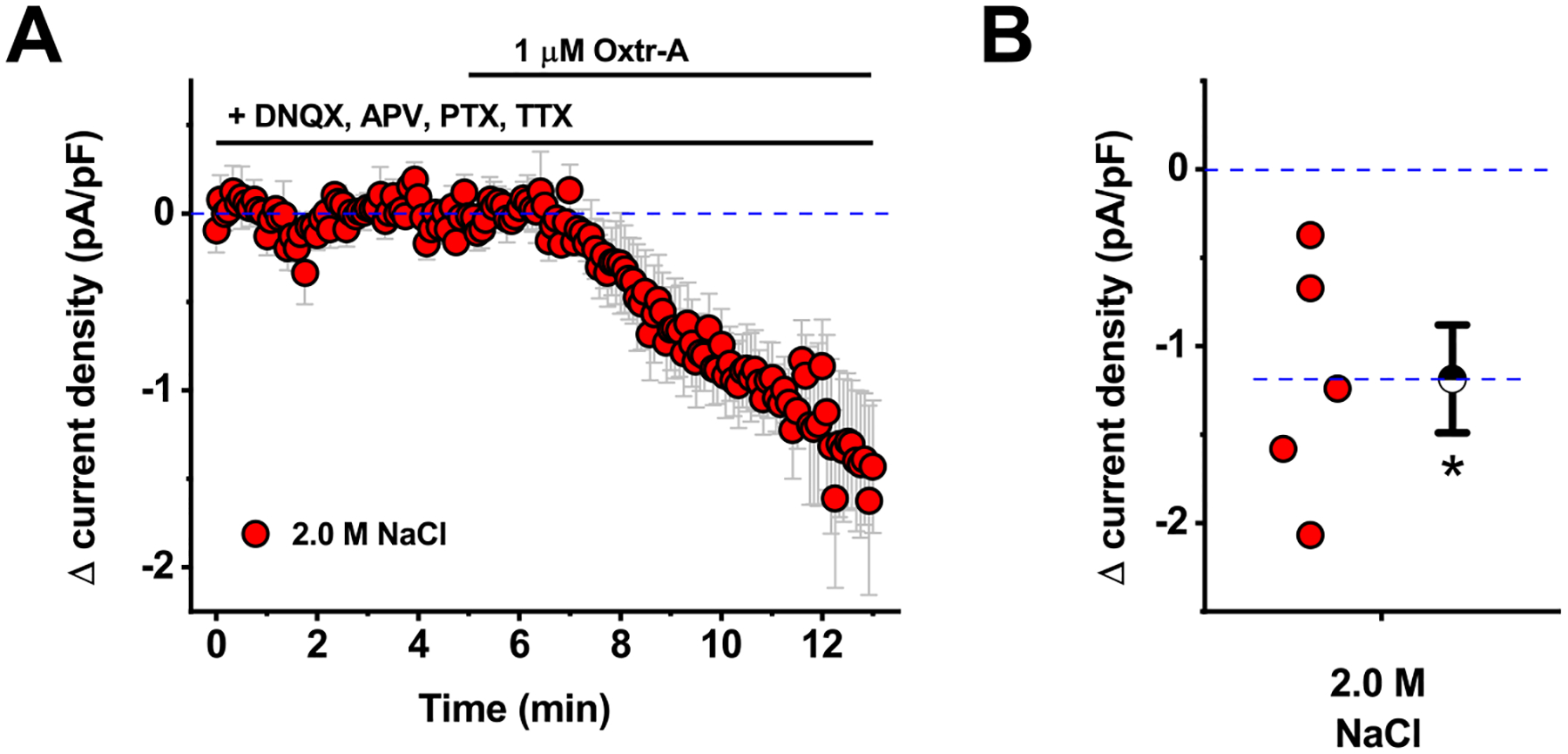
A) Bath application of Oxtr-A reveals an inhibitory tone in PVN CRH neurons obtained from animals that received hypertonic saline, even in the presence of bath applied antagonists for ionotropic glutamate receptors (DNQX and APV), GABAA receptors (PTX), and voltage gated sodium channels (TTX). B) Box plot illustrating specific change in current density produced by Oxtr-A for each cell that contributed to the summary plot in panel A. Hatched symbol to the right of the data represents the population mean, and the error bars indicate the SE. The asterisk indicates mean change is significantly different than 0 (p=0.02).
Inhibitory tone observed in CRH neurons from salt loaded animals is blocked by intracellular inhibition of GPCRs.
Based on the results presented above, we next sought to test the hypothesis that oxytocin receptor-mediated tonic inhibition of PVN CRH neurons, as observed in slices obtained from salt loaded animals, is mediated by direct activation of G-protein coupled Oxtr(s) expressed by the PVN CRH neurons. As a first step towards testing that hypothesis, we again repeated experiments in slices obtained from animals injected with hypertonic saline, as presented in Fig. 1D–E (red data plots), with two important changes. First, we used a more potent and selective oxytocin receptor antagonist, L-368,899-hydrochloride (L-368, 1 μM) in place of Oxtr-A [27]. Second, in a subset of experiments we replaced the GTP in the internal solution with 300 μM GDP-β-S, a non-hydrolysable analog of GDP that competitively inhibits G-protein activation. As such, in the experiments that involved GDP-β-S, we effectively and selectively blocked all G-protein signaling that depends on Gα subunits, but only in the individual neuron that was patched.
The results of these experiments make two important points. First, we found that L-368 reproduced the effect of Oxtr-A when bath applied to PVN CRH neurons voltage clamped at −70 mV in slices obtained from animals that received hypertonic saline. Specifically, we noted a shift in current density of −1.8 ± 0.6 pA/pF (Fig. 4A–B, grey symbols). This effect is significantly bigger than 0 as determined by a one-sample t-test (n=5, p=0.04), and also is not significantly different than the response produced by bath application of Oxtr-A presented in Fig. 1D (−1.6 ±.7 pA/pF, n=8, p=0.85, two-sample unpaired t-test). Second, we found that this effect of L-368 was completely eliminated in cells that were patched with an internal solution that contained GDP-β-S (Δcurrent density = 0.4 ± 0.2 pA/pF, p=0.04, two-sample unpaired t-test vs. control condition, and p=0.08, one-sample t-test vs. null hypothesis of mean = 0). Collectively, these data reinforce the conclusion that tonic inhibition of PVN CRH neurons after acute salt loading depends on activation of Oxtr(s), and further indicate that the mechanism requires G-protein activation specifically in the patched neuron.
Figure 4. Inhibitory tone observed in CRH neurons from salt loaded animals is blocked by intracellular inhibition of GPCRs.
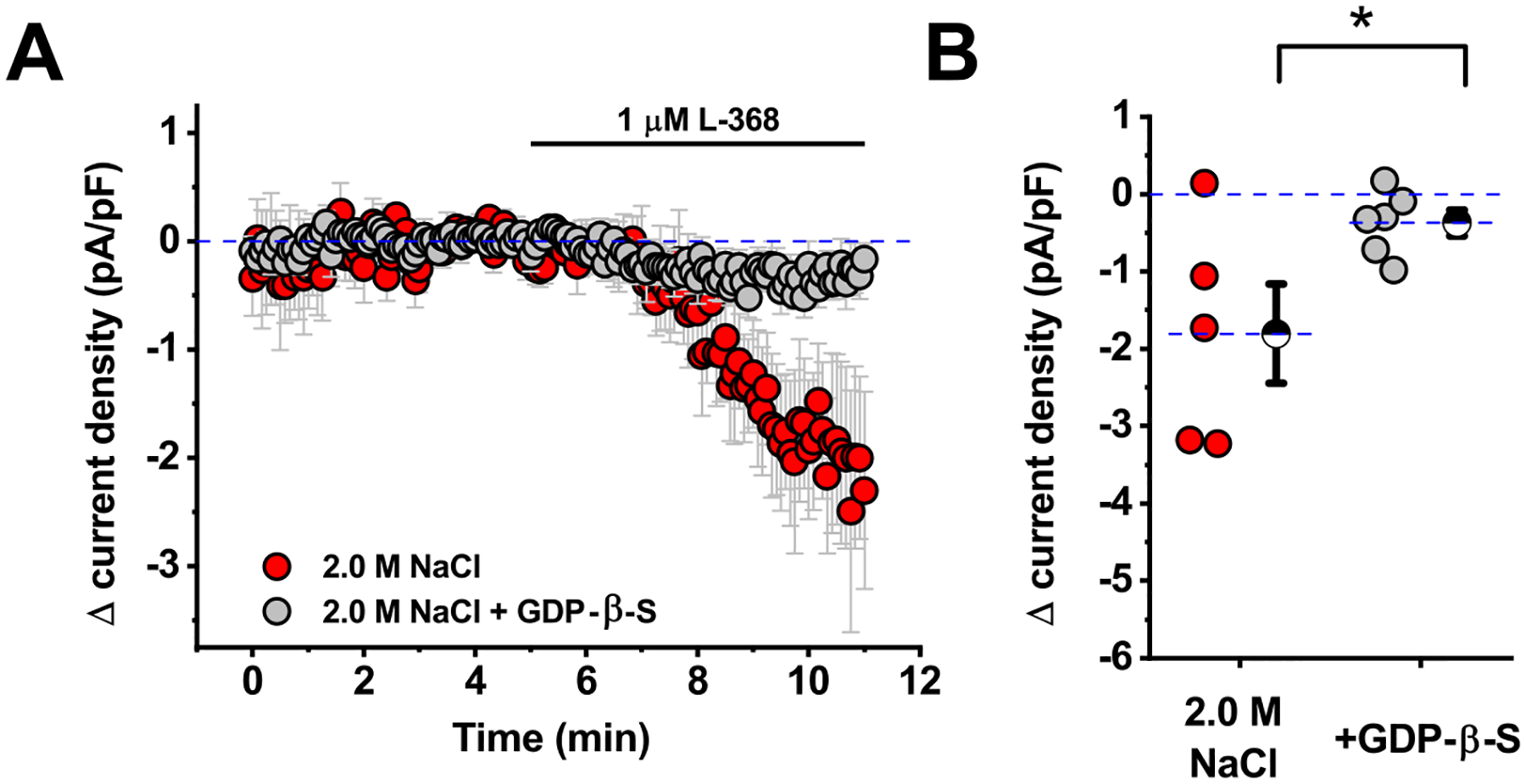
A) Oxytocin receptor-mediated inhibitory tone in PVN CRH neurons obtained from animals that received hypertonic saline (red symbols) is abolished in cells filled via the patch pipette with GDP-β-S (grey symbols). This result suggests that the effect of Oxtr-A depends on GPCRs expressed by the patched neuron. B) Box plot illustrating specific change in current density produced by Oxtr-A (measured 4–6 minutes after drug application) for each cell that contributed to the summary plot in panel A. Large hatched symbols to the right of the data represent the mean of each group, and error bars indicate the SE. The asterisk indicates that GDP-β-S significantly reduced the effect of Oxtr-A (p=0.04). See results for additional analysis.
Oxtr mRNA is expressed in a subset of PVN CRH neurons, and selective deletion of Oxtr(s) from CRH neurons reduces tonic inhibition produced by acute salt loading.
While results presented above provide a compelling argument that the tonic inhibition observed after acute salt loading requires direct activation of Oxtr(s) expressed by PVN CRH neurons, we performed two additional experiments in an attempt to further validate that hypothesis. First, we used in situ hybridization to determine whether PVN CRH neurons synthesize Oxtr mRNA. A total of five brains were used for this analysis, as well as two distinct ISH protocols (see methods). In all cases, Oxtr mRNAs were detected within a clear subset of PVN CRH-tdTomato neurons (Fig. 5A). That said, careful cell counts revealed that co-localization varied considerably, from 82.6 ± 4.5% in 809 PVN CRH neurons examined from three animals using ISH protocol 1, to 29.8% in 571 PVN CRH neurons examined from two animals with ISH protocol 2. Importantly, in both cases, no mRNAs were detected within PVN CRH neurons that were incubated with the negative control probe (Fig. 5B). While it is plausible that protocol differences increase bias towards false positives in protocol 1 and false negatives in protocol 2, it is also plausible that actual cohort variability contributed significantly to these results. Indeed, functional expression of Oxtr(s) is likely to be subject to robust and dynamic regulatory control, e.g. see [28–30]. Overall, we believe these results reinforce the conclusion that a substantial subset of PVN CRH neurons contain detectable levels of Oxtr mRNA. Therefore, we next sought to test the hypothesis that selective genetic deletion of Oxtr(s) from CRH neurons would reduce or eliminate Oxtr mediated inhibition observed after acute salt loading. Towards that end, we crossed mice homozygous for the Oxtr-flox gene with mice heterozygous for the CRH-Cre gene to generate both mice lacking the Oxtr in CRH neurons (CRH-Oxtr KO mice), and the appropriate littermate controls harboring only the Oxtr-flox gene (see Methods for additional details). This breeding scheme created both mice that lack Oxtr in CRH neurons (CRH-Oxtr KO mice) and appropriate littermate controls (see Methods for additional details). Both CRH-Oxtr KO mice and littermate controls were rendered hypertonic following 2.0 M NaCl injection, and putative CRH neurosecretory neurons were identified in vitro and tested with an oxytocin receptor antagonist as in earlier experiments. We found that PVN CRH-like neurons from littermate controls (n=8) did exhibit an Oxtr-dependent tonic inhibition that was blocked by L-368 (Δ current density = −0.91±0.37 pA/pF; one-sample t-test, p=0.04 vs. null hypothesis mean=0; Fig 5C–D). Further, this statistically significant effect was not present in PVN CRH neurons (n=6) from CRH-Oxtr KO mice (Δ current density = −0.23±0.14 pA/pF; one-sample t-test, p=0.16 vs. baseline). These data effectively indicate that selective deletion of Oxtr(s) from PVN CRH neurons prevents the expression of significant Oxtr-mediated tonic inhibition after acute salt loading. Nevertheless, it should also be noted that there was not a significant difference between group means for CRH-Oxtr KO mice and littermates when compared directly to one another using a two-sample t-test (p=0.12).
Figure 5. mRNA for Oxtr(s) is present in the majority of PVN CRH neurons, and selective deletion of Oxtr(s) from CRH neurons reduces tonic inhibition produced by acute salt loading.
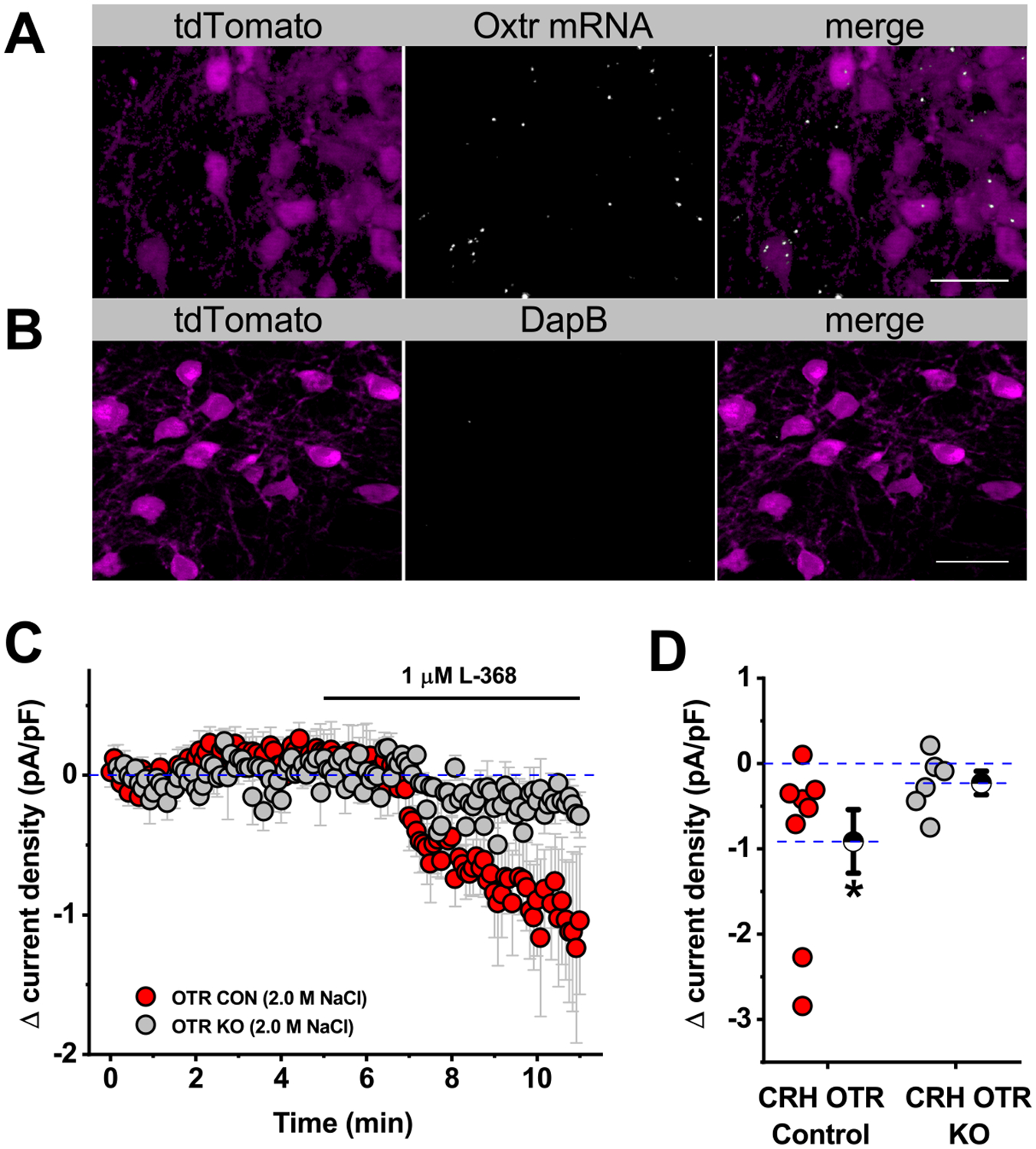
A) Fluorescent in situ hybridization (illustrated here as produced with RNAScope® Multiplex Fluorescent Assay V2) reveals that a subset of CRH-tdTomato neurons express Oxtr mRNA, while B) mRNAs for the DapB negative control are undetectable within CRH-tdTomato neurons localized to adjacent brain sections. The scale bars represent 20 μm. See results section for additional details. C) Conditional knockout of OTR in CRH neurons attenuates oxytocinergic tone in salt-treated animals compared to littermate controls. D) Box plot illustrating specific change in current density produced by Oxtr-A (measured 4–6 minutes after drug application) for each cell that contributed to the summary plot in panel C. Large hatched symbols to the right of the data represent the mean of each group, and error bars indicate the SE. The asterisk indicates that antagonism of Oxtr(s) still causes a significant reduction in current density in littermate controls (p=0.04) that was absent in CRH neurons from Oxtr-CRH KO animals. See results for additional details.
Acute salt loading increases apparent activation of an inwardly rectifying potassium channel in PVN CRH neurons and reduces neuronal gain.
Prior work from our group has indicated that acute salt loading robustly inhibits activation of the HPA axis, as indicated by lower levels of ACTH and corticosterone subsequent to psychological stress [18–20]. As a next step towards better understanding the mechanism of this inhibition, we carefully evaluated input resistance in PVN CRH neurons from animals that received isotonic vs. hypertonic saline. We found that acute salt loading significantly reduced whole-cell input resistance as measured from a brief voltage step from −70 mV to −80 mV (Isotonic Rm: 1003 ± 128 MOhm, n=7, Hypertonic Rm: 663 ± 70 MOhm, n=9, p=0.03). This finding suggests that salt loading opens a membrane channel, possibly one that reduces cellular excitability. Based on that observation, we next performed an additional series of voltage clamp experiments in order to evaluate the relationship between holding current and command potential between −110 mV and −50 mV (Fig. 6A–D, see legend for details on the protocol). Although a two-way repeated-measures ANOVA on data presented in Fig. 6C reveals no main effect of salt loading (F1,14=3.16, p=0.10), there was a significant salt × voltage interaction (F12,168=9.46, p<0.005). Post hoc tests revealed that PVN CRH neurons from salt loaded animals required significantly more current to hold the membrane potential at all voltages tested negative of −95 mV (p-values < 0.005). Consistent with this observation, the difference between these two I-V plots, illustrated in Fig. 6D, further highlights that salt loading is associated with increased activation of an inwardly rectifying current that reverses near the equilibrium potential for potassium.
Figure 6. Salt loading reduces the excitability of PVN CRH neurons through opening an inwardly rectifying potassium channel.
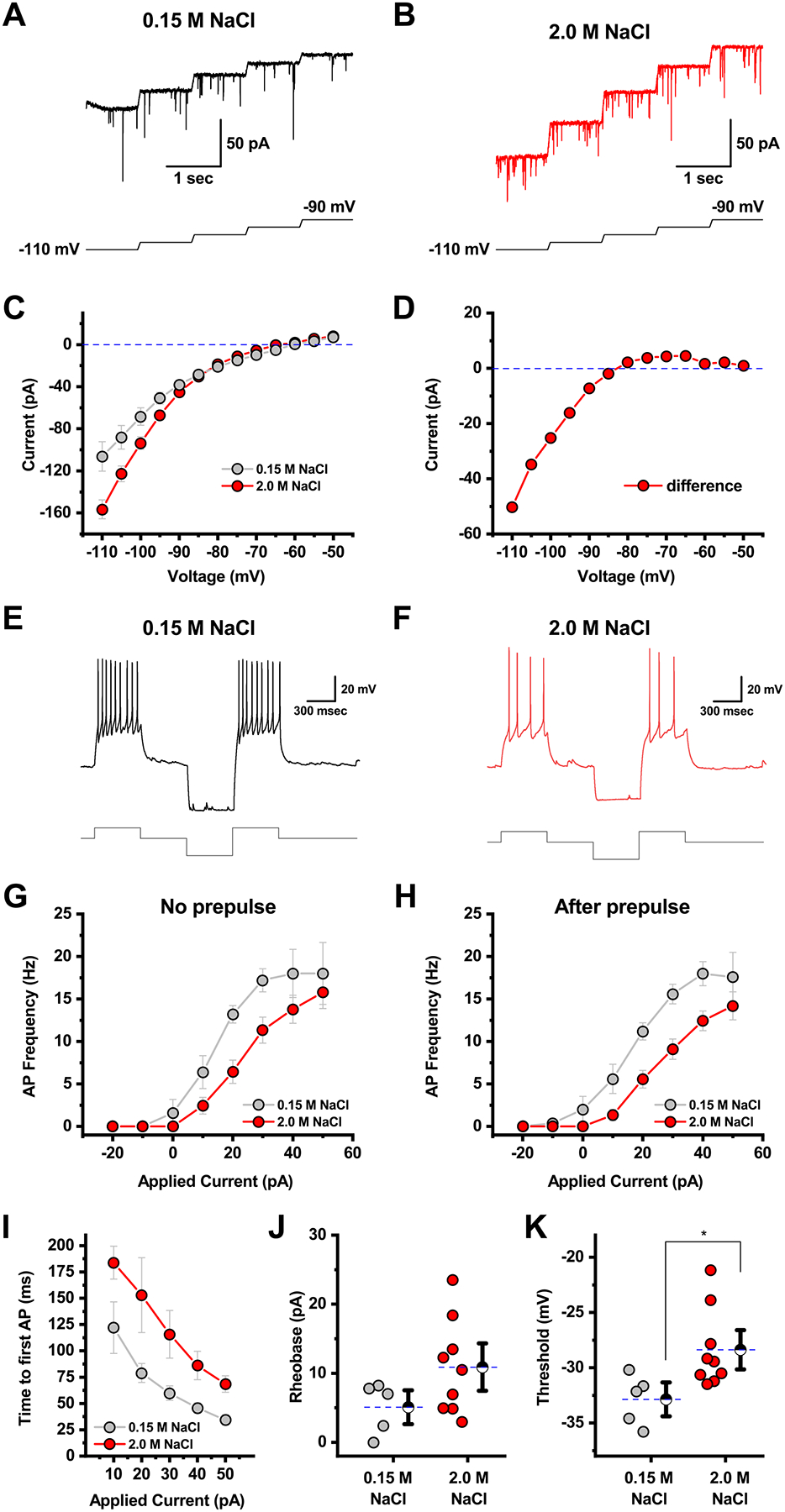
A-B) Top traces illustrate raw data recorded in voltage clamp from a representative cell in the isotonic vs. hypertonic groups, respectively. Data illustrated were obtained between −110 mV and −90 mV. In each panel (A-B),) the lower trace illustrates the voltage protocol used to obtain the illustrated data. Specifically, cells were voltage-clamped at each voltage (in 5 mV increments for 1 second). Transitions between voltages were accomplished with voltage ramps that lasted for 50 msec and moved the membrane potential 5 mV more positive. Although the protocol continued until the voltage was −50 mV, only steps between −110 mV and −90 mV are illustrated in panels A-B. Steady-state current was measured during the last 500 msec of each 1 second voltage step to generate the IV plots in panels C. C) Current/voltage relationship of PVN CRH neurons generated from data generated as in A-B reveals that on average more inhibitory current is required to hold neurons of salt-loaded animals at negative potentials. These I-V plots were generated by measuring the mean current during the last 500 msec recorded at each voltage. In order to eliminate the possibility that variability in spontaneous activity contributed to the observed I-V plots, we also measured tonic current during the last 500 msec of each step using techniques as described in Nahir et al. 2007 [45]. This analysis revealed that core differences in the I-V plots in panel C were not due to differences in spontaneous activity. D) The difference of the current/voltage curves in A reveals salt loading activates an inwardly-rectifying current, which reverses near the equilibrium potential for potassium. E) A representative current-clamp recording showing the response to a PVN CRH neuron obtained from an animal that received isotonic saline to a +30 p current injection delivered either in isolation, or after a −50 pA hyperpolarizing pulse. F) Representative trace from an identical experiment as panel C, in a PVN CRH neuron extracted from a salt loaded animal, highlights decreased responsiveness to a +30 pA current injection, without or with the hyperpolarizing prepulse. G-H) Complete neuronal gain curves constructed from cells recorded as in panels C-D indicate that salt loading produces a statistically significant reduction in neuronal gain. Two cells available for I-V data did not survive through gain experiments and were excluded from this analysis. See results section for additional details. I) Acute salt loading increases time to first action potential in response to injection of 10 – 50 pA current steps. See results section for further details. J-K) Salt loading increases action potential rheobase and threshold, as observed during a slow current ramp, although only the latter effect was statistically significant (n=5, 9, p=0.11 and 0.03, respectively, two-sample unpaired t-test).
Both G-protein coupled inwardly rectifying potassium channels (GIRKs), and potassium permeant hyperpolarization-activated cyclic nucleotide-gated channels (HCNs) can contribute to inward rectification, and their effects on neuronal activity in physiological conditions can be complex. Therefore, in order to more directly evaluate the effect of salt loading on excitability of PVN CRH neurons, we evaluated the response of the neurons to direct current injection in current clamp, with or without a hyperpolarizing prepulse of −50 pA (used to maximize the contribution of any hyperpolarization-activated currents). Fig. 6E illustrates the response of a representative PVN CRH neuron to a +30 pA current injection, both with and without the hyperpolarizing prepulse, in an animal that received isotonic saline. Fig. 6F illustrates reduced firing in response to an identical stimulus in a representative trace recorded from a PVN CRH neuron obtained from a salt loaded animal. Complete neuronal gain curves for isotonic and hypertonic groups with and without a hyperpolarizing prepulse indicate that there was a main effect of salt loading on action potential frequency (salt loading reduced neuronal gain, both without, and with, a hyperpolarizing prepulse, F1,12=7.99, p=0.02, and F1,12=12.86, p<0.005, respectively, Fig. 6G–H). We also noted that salt loading increased the time to first action potential, for current injections between 10 and 50 pA, with the difference being larger in response to smaller current injections (main effect of salt loading, F1,6=9.33, p=0.02, Fig. 6I). Similarly, we measured both the rheobase and the threshold of the first action potential observed during a slow current ramp (10 pA/sec)). There was a trend toward an increase in the rheobase from 5.1 ± 1.6 pA to 10.9 ± 2.3 pA (Fig. J; n=5,9, p=0.11), and an increase in the threshold from −32.9 ± 1.0 mV to −28.4 ± 1.2 mV in salt loaded mice (Fig. 6K; n=5,9, p=0.03). Collectively, these data further reinforce the conclusion that salt loading produces a clear and functional reduction in the excitability of PVN CRH neurons.
Efforts to recapitulate the effects of salt loading in experiments that involve exogenous Oxtr agonists produce mixed results.
We next sought to test the hypothesis that bath application of an Oxtr agonist would increase current density in PVN CRH neurons from control animals that received neither isotonic nor hypertonic saline injection. In contrast to that hypothesis, we found that bath application of oxytocin (600 nM) had no significant effect on current density in PVN CRH neurons voltage clamped at −50 mV (Δcurrent density = −0.17 ± 0.14 pA/pF, n=4, p=0.91, Fig. 7A). Indeed, multiple additional efforts involving bath application of either oxytocin or Thr4,Gly7-OXT (TGOT) at concentrations between 20 nM and 600 nM, in both control and salt loaded animals, failed to produce any reliable effect on holding current or current density in PVN CRH neurons (not illustrated), and similarly did not reduce intrinsic excitability in a manner consistent with the effects of salt loading presented in Figure 6. While there are a number of potential hurdles involved in bath applying exogenous peptide agonists, positive control experiments (using the same solutions and delivery methods that were tested in the PVN) demonstrated clear effects of identical bath application of both oxytocin and TGOT in other brain areas and cell types, consistent with prior results from our laboratory [31].
Figure 7: Acute bath and local application of oxytocin to PVN CRH neurons.
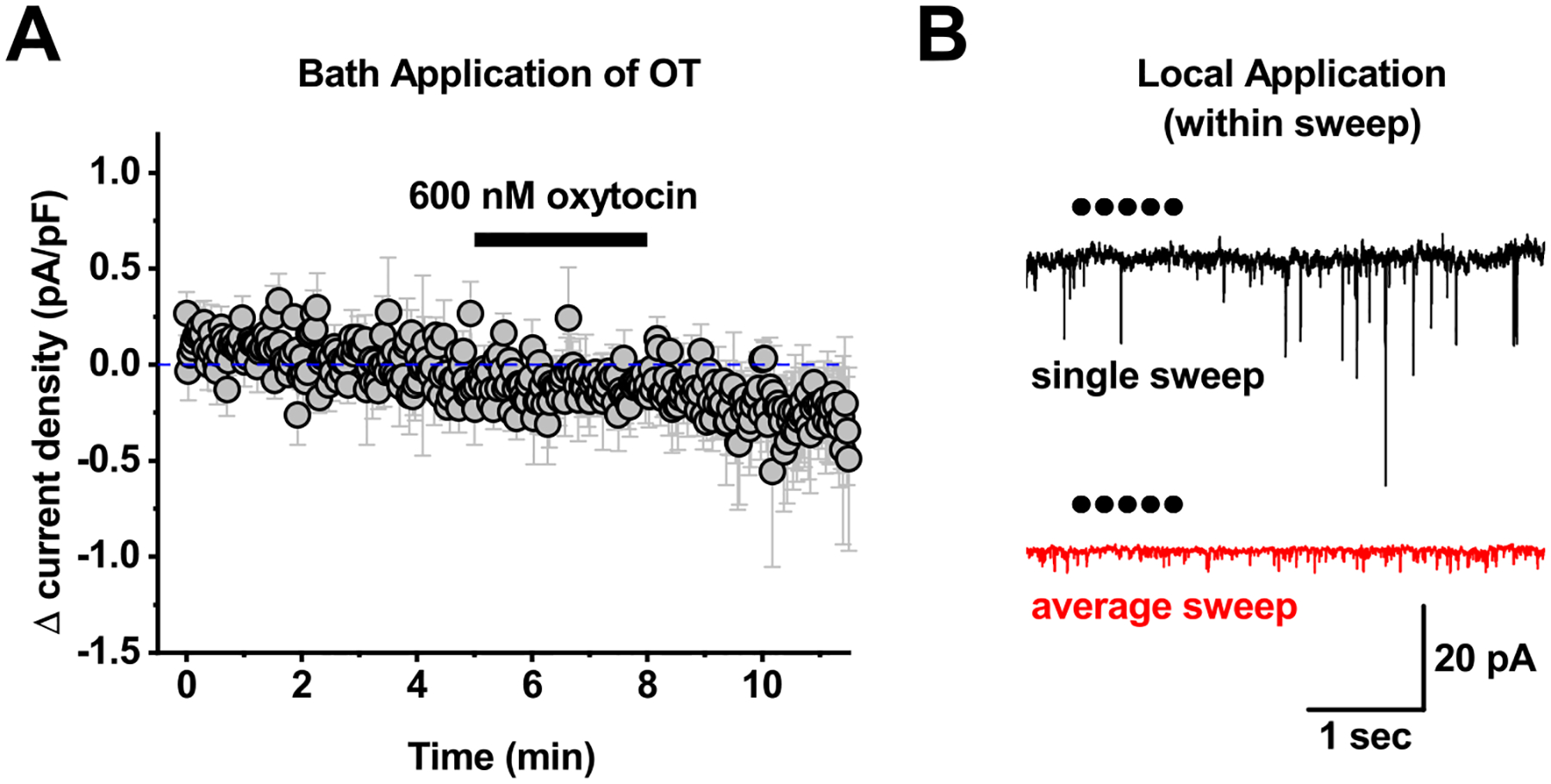
A) Bath application of 600 nM oxytocin failed to produce any clear response in PVN CRH neurons voltage clamped at −50 mV. B) For local application experiments, 1 μM oxytocin was loaded into a glass pipette, which was connected to a picospritzer. The tip of the pipette was then placed within ~20 um of the cell soma, and 5–10 msec puffs of oxytocin were delivered at 5 Hz every 30 seconds. The top trace in panel B illustrates a single sweep recorded during one such application. Each dot represents a single 10 msec puff. The bottom trace is an average of 12 sweeps delivered at 30 second intervals from the same cell as illustrated in the top trace. This experiment was repeated 6 times, and none of the 6 PVN CRH neurons tested showed any rapid response to local application of OT. Specifically, when data in each sweep was baseline subtracted to a 1.9 second period immediately before local application, mean current observed in the 6 seconds immediately after local application was identical to baseline (0.08 ± 1.19 pA, n-6, p=0.7).
Based on these results (in combination with those presented earlier in the study), it seemed plausible that complications related to receptor desensitization and/or internalization might be uniquely problematic for experiments involving bath application of oxytocin or TGOT in the PVN. Therefore, we used local application techniques to rapidly apply oxytocin (10 msec puffs of 1 μM oxytocin at 5 Hz for 1 second, repeated every 30 seconds) directly to individual PVN CRH neurons during whole-cell recording. There was no significant effect of local application of oxytocin on holding current in PVN CRH neurons during or in the seconds immediately after each application (Fig. 7B, see legend for additional details). However, across six cells tested, we did note a small but consistent increase in current density over the first several minutes of local application (Δcurrent density was 0.20 ± 0.028 pA/pF, measured from 3–4 minutes after the first application, n=6, p<0.001). This effect should be interpreted cautiously, as the effect is quite small, and the experimental design (being focused on expected faster effects of local application) did not provide for a long baseline recording period. Nevertheless, these results suggested to us that both concentration of agonist and duration of exposure might be important variables in evaluating the effects of Oxtr agonists on PVN CRH neurons. Therefore in a final effort to reveal clear effects of exogenous Oxtr agonists on PVN CRH neurons we performed a series of experiments that involved preincubation of PVN slices (for a minimum of 1 hour) in control conditions, in 200 nM oxytocin, or in 200 nM oxytocin + 1 μM L-368. In each group, we measured holding current and current density at −70 mV from multiple PVN CRH neurons. Results obtained from slices incubated in either 200 nM oxytocin or in oxytocin plus L-368 were compared to control slices obtained from the same preparations. Intriguingly, and more consistent with the initial hypothesis, results from these experiments indicated that preincubation in 200 nM oxytocin increased both holding current and current density in PVN CRH neurons. Specifically, holding current at −70 mV increased from −12.9 ± 1.94 pA in control conditions to −7.07 ± 1.05 pA in neurons preincubated in 200 nM oxytocin (n=12 in both groups, p=0.016, Fig. 8A, left panel), while current density similarly increased from −1.83 ± 0.31 pA to −0.95 ± 0.27 pA (n=12 in both groups, p=0.045, Fig. 8A, right panel). Importantly, neither of these effects was observed in slices preincubated in both 200 nM oxytocin and 1 μM L-368 (Fig. 8B), indicating Oxtr activation is responsible for these effects. While it would be of interest to know if these changes are also reflected in the resting membrane potential, direct measurement of the resting potential in these cells can be complicated by the fact that a subset of them fire spontaneously when current clamped at I=0 (5 of 12 in the control group vs. 2 of 12 in the group preincubated in 200 nM OT). Notably, although preincubation in 200 nM oxytocin produced clear inhibitory effects as reported by changes in holding current and current density, it did not effectively recapitulate core effects of salt loading on the input resistance, the current-voltage relationship, or the neuronal gain (Fig. 9). Overall the results of these experiments suggest either that there remain some undiscovered difficulties with using exogenous agonists to model the concentration and kinetic profiles of endogenous Oxtr activation during salt loading, or alternatively, that there remain some undiscovered (and potentially Oxtr independent) complexities contributing to the overall effects of salt loading on PVN CRH neurons.
Figure 8. Preincubation of PVN slices in 200 nM oxytocin produces an Oxtr dependent increase in holding current and current density.
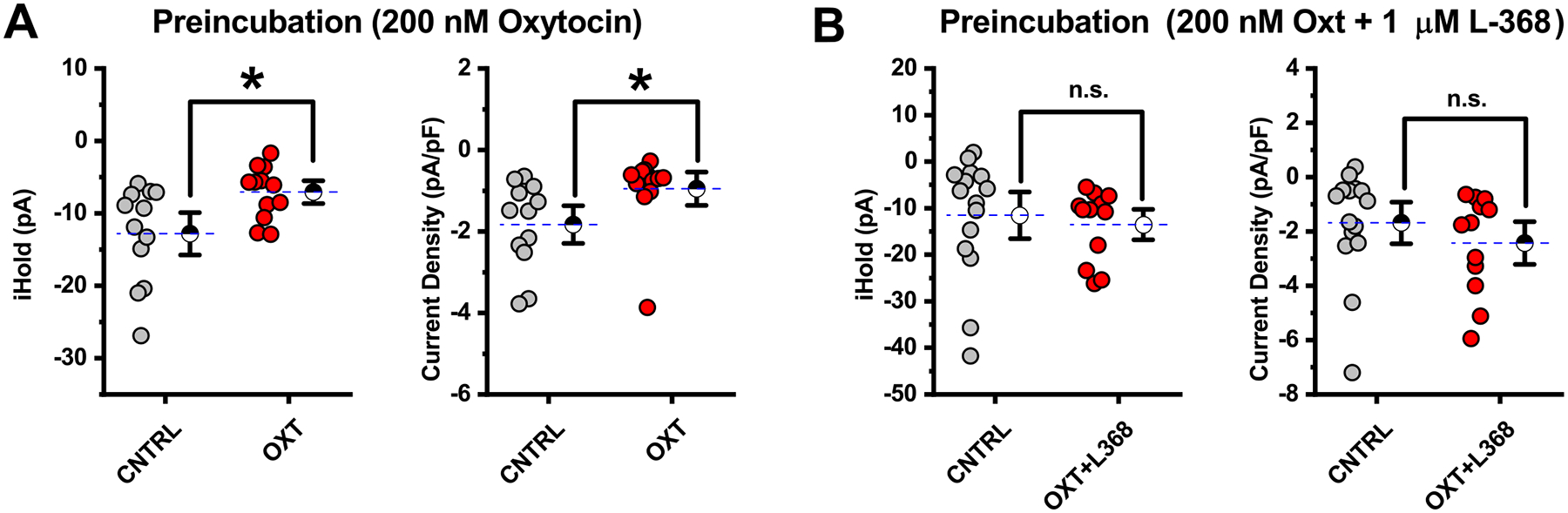
A) Slices incubated 200 nM oxytocin for a minimum of one hour prior to whole-cell recording were compared to control slices from the same animals incubated in normal ACSF. Results indicated that preincubation in 200 nM oxytocin produced a clear increase in holding current and current density relative to preparation matched controls. Asterisks indicated P<0.05, see text in the results section for further details. B) The effects produced by preincubation of slices in 200 nM oxytocin presented in panel A were not observed in slices that were incubated in 200 nM oxytocin + 1 μM L-368. This result highlights a clear role for Oxtr(s) in producing the inhibitory effect of preincubation highlighted in panel A. See text of the results for further details.
Figure 9. Preincubation of PVN slices in 200 nM oxytocin failed to produce a shift in neuronal gain comparable to that observed in salt loaded animals.
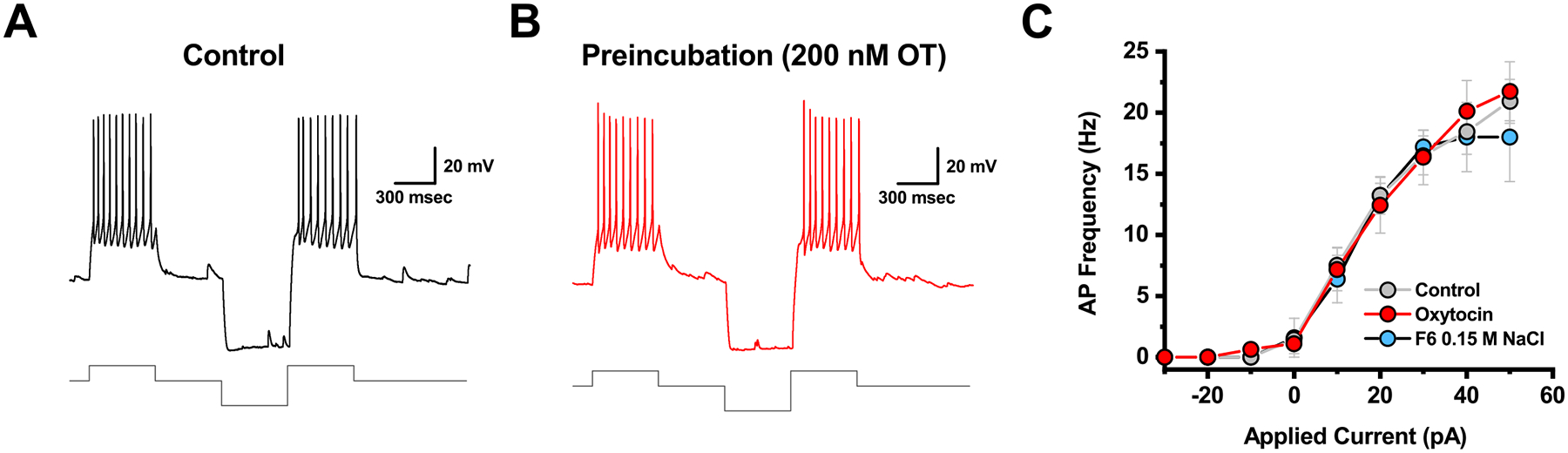
A-B) Illustrate representative data obtained from a cell incubated in control conditions (CNTRL) vs. in 200 nM oxytocin (OXT), respectively. Top trace illustrates firing observed in response to a +30 pA current injection delivered either in isolation, or after a −50 pA hyperpolarizing pulse, exactly as in Fig. 6. The lower trace illustrates the current injected. C) Complete neuronal gain curves constructed from cells recorded as in panels A-B indicate that preincubation in 200 nM oxytocin failed to alter neuronal gain. The gain curve observed in PVN CRH neurons from animals treated with isotonic saline from Fig. 6, is plotted again in this panel (light blue) for comparison.
Discussion
In this study, we used a previously characterized CRH reporter animal [20,23,25,26] to provide the first direct demonstration that acute hypernatremia produces an Oxtr dependent inhibitory tone on identified parvocellular PVN CRH neurons. A first basic question to ask in considering this study is how fundamental information on the identity and physiological properties of PVN CRH neurons observed here aligns with prior data and general expectations. In that regard, it is worth highlighting that we found the vast majority of genetically identified PVN CRH neurons to be parvocellular neurons, with low whole-cell capacitance and high input resistance, that lacked both a clear A-type potassium current and a robust low threshold spike. In those respects, our findings are consistent with early characterizations of ‘Type II’ parvocellular neurosecretory neurons postulated to regulate HPA axis activation [22]. Importantly, these results also align quite well with initial physiological characterizations of PVN CRH neurons in the identical CRH-ires-Cre mouse model as reported by other investigators [23]. Notably, we also report that PVN CRH neurons rarely express NP1, while Wamsteeker Cusulin et al (2013) reports PVN CRH neurons are robustly labeled by peripherally delivered fluorogold, and that they are rarely immunoreactive for either vasopressin or oxytocin. While it is possible that a small subset of PVN magnocellular neurons could express CRH, collectively, we believe the data described above make a compelling case that PVN CRH neurons, as identified in this study are overwhelmingly parvocellular neurosecretory neurons. As such, we conclude that the mechanism revealed here very likely underlies the previously reported ability of acute hypernatremia to dampen activation of the HPA axis as produced by subsequent restraint stress [18–20].
An important aspect of that mechanism, as described in the results section, is the idea that it depends on direct activation of Oxtr(s) that are expressed on the PVN CRH neurons. Several lines of evidence presented here support that conclusion. Most notably, whole-cell recordings reveal an effect of a bath applied Oxtr antagonist on identified PVN CRH neurons obtained from salt loaded animals. This effect is largely insensitive to bath applied antagonists for glutamate and GABA receptors, does not require action potential-dependent transmission in the slice, and is blocked by GDP-β-S delivered intracellularly to the patched neuron. Further, in situ hybridization studies consistently reveal clear expression of Oxtr mRNA in a subset of PVN CRH neurons examined, and conditional knockout of Oxtr from CRH neurons eliminates the effects of hypernatremia observed in vitro. That said, it is worth noting that we observed significant variability in the response to Oxtr antagonists among PVN CRH neurons from salt loaded animals in datasets where Oxtr function was expected to be intact. In our view, this variance is likely produced in part by cell-to-cell differences in levels of functional Oxtr expression in PVN CRH neurons, and may also be due to animal-to-animal variability in the effects of acute hypernatremia on Oxt release. One possibility not directly evaluated in the current study is that Oxtr expression in PVN CRH neurons may be both dynamically regulated and increased in response to acute hypernatremia. It is also worth noting that earlier work by other investigators that relied on single-cell RT-PCR in rat rather than mouse PVN neurons has similarly concluded that a subset of PVN CRH neurons are Oxtr positive [32,33]. However, it should be further noted that on balance those studies also suggest that Oxtr positive CRH neurons are not parvocellular.
In an effort to develop a better understanding of exactly how Oxtr activation confers an inhibitory effect on PVN CRH neurons, we also performed a series of studies that revealed decreased input resistance and decreased neuronal gain in PVN CRH neurons from salt loaded animals compared to controls that received isotonic saline. A careful evaluation of the current/voltage relationship in these two populations suggested that salt loading was associated with increased activation of an inwardly rectifying potassium channel. This result complements one prior study that reveals a clear role for GIRKs in the modulation of neuronal excitability in both parvocellular and magnocellular neurons of the PVN [34]. However, rather than attempting to further explore detailed effects underlying altered excitability observed in PVN CRH neurons from salt loaded animals, we thought it would be desirable to better isolate Oxtr mediated effects (from plausible additional and more complex effects of salt loading) by directly applying Oxtr agonists to PVN CRH neurons in control conditions. However, it is this aspect of the study that has ultimately proven to be the most confounding. Contrary to our expectations, we were unable to produce reliable tonic inhibition of PVN CRH neurons using bath application of either oxytocin or TGOT at concentrations between 20 and 600 nM in either control or salt loaded animals. In that respect, our results are consistent with another recent study that reported bath application of 1 μM oxytocin had no direct effect on PVN CRH neurons [35]. In order to reduce the possibility that bath application was leading to rapid receptor desensitization and/or internalization (e.g. see) [36–39], we attempted to directly activate PVN CRH neurons by locally applying OT via a picospritzer. These experiments also failed to produce a clear or reliable immediate response to oxytocin application, however we did note a small yet statistically significant increase in current density over time when oxytocin was applied in this manner (1 sec 5 Hz train of 10 msec puffs delivered every 30 seconds, with 1 μM oxytocin in the pipette). This result suggested that longer exposure to low concentrations of agonist may be necessary to produce the type of Oxtr mediated inhibition of PVN CRH neurons observed in salt loaded animals. Indeed, a novel experiment involving preincubation of slices in 200 nM oxytocin did produce a clear Oxtr mediated increase in both holding current and current density, which is quite consistent with that hypothesis. Nevertheless, this preincubation protocol still failed to fully reproduce the effects of salt loading on the input resistance, the current-voltage relationship, and the neuronal gain. In summary, the most parsimonious explanation for all of our data is that the primary inhibitory effects of salt loading on PVN CRH neurons are indeed produced at least in significant part by Oxtr dependent activation of a potassium permeant channel, but that detailed pharmacological analysis of this phenomenon is hampered by difficulty effectively mimicking endogenous receptor activation in experiments that depend on exogenous agonists. That said, we cannot definitively rule out the possibility that there may be additional Oxtr independent effects of salt loading that are contributing to some aspects of the inhibition observed in PVN CRH neurons.
In that regard, it is useful to further consider how the data presented here aligns with prior studies that have directly implicated central Oxtr(s) in the modulation of stress responsiveness. As noted in the introduction, significant prior evidence indicates that a variety of acute psychological stressors promote central oxytocin release [1–5], and substantial additional data indicates that centrally acting oxytocin can modulate stress-induced activation of the HPA axis [6–8]. In most prior studies, a specific mechanism for Oxtr dependent modulation of stress-induced activation of the HPA axis is not directly revealed, although several lines of evidence have promoted the idea that, either directly or indirectly, decreased synthesis of CRH in PVN CRH neurons is involved [9,12,13]. In considering our data in light of these studies it is important to emphasize that acute hypernatremia is a peripheral non-psychological stressor that is likely to have very different effects on PVN activity than the psychological stressors previously used to promote central oxytocin release. Indeed, we would hypothesize that hypernatremia is likely to evoke central oxytocin release either via direct osmotic regulation of PVN magnocellular oxytocinergic neurons, or via excitatory input from osmosensitive neurons in circumventricular organs. By contrast, we expect oxytocin release in the PVN following psychological stressors is likely to involve activation of substantially more descending central inputs into the PVN, and is also likely to be confounded with slower genomic effects ultimately produced by circulating glucocorticoids. That said, it seems possible that oxytocin acting locally in the PVN could promote Oxtr-mediated genomic modulation of CRH synthesis, and that this would ultimately act synergistically with mechanisms revealed here to reduce the HPA axis response to additional stressors. Nevertheless, in a broader sense, we believe the results presented here emphasize a new context for Oxtr mediated modulation of the HPA response to stress; one that is not initiated by prior psychological stressors, one that has potential to work on shorter time scales, and one that has some interesting potential social relevance [21].
Finally, it is worth considering what this study helps reveal about differences between acute and chronic hypernatremia with respect to modulation of the HPA axis. Prior studies indicate that chronic salt loading alters CRH expression patterns in hypothalamus [40], is associated with reduced ACTH release from the anterior pituitary [41,42], does not alter stress-induced release of oxytocin despite increasing basal levels of oxytocin in plasma [42], and yet does promote sensitization of the HPA axis during rehydration [43]. Recent work from our group has revealed that 5 days of chronic salt loading produces a moderate reduction in CRH synthesis in neurosecretory regions of the PVN, a clear reduction in sEPSC frequency recorded from PVN CRH neurons, and an attenuation in the HPA axis response to restraint stress that was apparent during recovery rather than immediately after restraint [44]. Broadly speaking, these results reinforce the idea that acute Oxtr mediated modulation of PVN CRH neurons as detailed here is a mechanism likely associated with acute paracrine release of oxytocin in the PVN, and that distinct mechanisms underly long-term adaptation to chronic salt and/or subsequent rehydration.
Acknowledgements
This study was supported by AHA predoctoral fellowship 13PRE17100047 (DP), NIH grants MH104641 (CJF), HL096830 (EGK), HL122494 (EGK), and K99/R00-HL-125805 (ADdK).
Footnotes
Conflict of interest
The authors declare no conflicts of interest.
References
- 1.Nishioka T, Anselmo-Franci JA, Li P, Callahan MF, Morris M. Stress increases oxytocin release within the hypothalamic paraventricular nucleus. Brain Res. 1998;781: 57–61. doi: 10.1016/s0006-8993(97)01159-1 [DOI] [PubMed] [Google Scholar]
- 2.Torner L, Plotsky PM, Neumann ID, de Jong TR. Forced swimming-induced oxytocin release into blood and brain: Effects of adrenalectomy and corticosterone treatment. Psychoneuroendocrino. 2017;77: 165–174. doi: 10.1016/j.psyneuen.2016.12.006 [DOI] [PubMed] [Google Scholar]
- 3.Wotjak CT, Ganster J, Kohl G, Holsboer F, Landgraf R, Engelmann M. Dissociated central and peripheral release of vasopressin, but not oxytocin, in response to repeated swim stress: New insights into the secretory capacities of peptidergic neurons. Neuroscience. 1998;85: 1209–1222. doi: 10.1016/s0306-4522(97)00683-0 [DOI] [PubMed] [Google Scholar]
- 4.Engelmann Ebner, Landgraf Holsboer, Wotjak1. Emotional Stress Triggers Intrahypothalamic But Not Peripheral Release of Oxytocin in Male Rats. J Neuroendocrinol. 1999;11: 867–872. doi: 10.1046/j.1365-2826.1999.00403.x [DOI] [PubMed] [Google Scholar]
- 5.Ježová D, Michajlovskij N, Kvetńanský R, Makara GB. Paraventricular and Supraoptic Nuclei of the Hypothalamus Are Not Equally Important for Oxytocin Release during Stress. Neuroendocrinology. 1993;57: 776–781. doi: 10.1159/000126436 [DOI] [PubMed] [Google Scholar]
- 6.Windle RJ, Kershaw YM, Shanks N, Wood SA, Lightman SL, Ingram CD. Oxytocin Attenuates Stress-Induced c-fos mRNA Expression in Specific Forebrain Regions Associated with Modulation of Hypothalamo–Pituitary–Adrenal Activity. J Neurosci. 2004;24: 2974–2982. doi: 10.1523/jneurosci.3432-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Windle R, Shanks N, Lightman S, Ingram C. Central Oxytocin Administration Reduces Stress-Induced Corticosterone Release and Anxiety Behavior in Rats1. Endocrinology. 1997;138: 2829–2834. doi: 10.1210/endo.138.7.5255 [DOI] [PubMed] [Google Scholar]
- 8.Neumann Wigger, Torner Holsboer, Landgraf. Brain Oxytocin Inhibits Basal and Stress‐Induced Activity of the Hypothalamo‐Pituitary‐Adrenal Axis in Male and Female Rats: Partial Action Within the Paraventricular Nucleus. J Neuroendocrinol. 2000;12: 235–243. doi: 10.1046/j.1365-2826.2000.00442.x [DOI] [PubMed] [Google Scholar]
- 9.Nomura M, Saito J, Ueta Y, Muglia L, Pfaff D, Ogawa S. Enhanced Up‐Regulation of Corticotropin‐Releasing Hormone Gene Expression in Response to Restraint Stress in the Hypothalamic Paraventricular Nucleus of Oxytocin Gene‐Deficient Male Mice. J Neuroendocrinol. 2003;15: 1054–1061. doi: 10.1046/j.1365-2826.2003.01095.x [DOI] [PubMed] [Google Scholar]
- 10.Walker C-D, Toufexis DJ, Burlet A. Progress in Brain Research. 2001;133: 99–110. doi: 10.1016/s0079-6123(01)33008-x [DOI] [PubMed] [Google Scholar]
- 11.Bealer S, Crowley W. Noradrenergic control of central oxytocin release during lactation in rats. Am J Physiology. 1998;274: E453–8. doi: 10.1152/ajpendo.1998.274.3.e453 [DOI] [PubMed] [Google Scholar]
- 12.Winter J, Jurek B. The interplay between oxytocin and the CRF system: regulation of the stress response. Cell Tissue Res. 2018; 1–7. doi: 10.1007/s00441-018-2866-2 [DOI] [PubMed] [Google Scholar]
- 13.Jurek B, Slattery DA, Hiraoka Y, Liu Y, Nishimori K, Aguilera G, et al. Oxytocin Regulates Stress-Induced Crf Gene Transcription through CREB-Regulated Transcription Coactivator 3. J Neurosci. 2015;35: 12248–12260. doi: 10.1523/jneurosci.1345-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang W, Lee S, Arnason S, öquist. Dehydration natriuresis in male rats is mediated by oxytocin. Am J Physiology. 1996;270: R427–33. doi: 10.1152/ajpregu.1996.270.2.r427 [DOI] [PubMed] [Google Scholar]
- 15.öquist, Huang W, Jacobsson E, Skøtt O, ricker E, Sved A. Sodium excretion and renin secretion after continuous versus pulsatile infusion of oxytocin in rats. Endocrinology. 1999;140: 2814–8. doi: 10.1210/endo.140.6.6831 [DOI] [PubMed] [Google Scholar]
- 16.Tucker A, Stocker SD. Hypernatremia-induced vasopressin secretion is not altered in TRPV1−/− rats. Am J Physiology-regulatory Integr Comp Physiology. 2016;311: R451–R456. doi: 10.1152/ajpregu.00483.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKinley M, Mathai M, McAllen R, McClear R, Miselis R, Pennington G, et al. Vasopressin Secretion: Osmotic and Hormonal Regulation by the Lamina Terminalis. J Neuroendocrinol. 2004;16: 340–347. doi: 10.1111/j.0953-8194.2004.01184.x [DOI] [PubMed] [Google Scholar]
- 18.Krause EG, de Kloet AD, Flak JN, Smeltzer MD, Solomon MB, Evanson NK, et al. Hydration State Controls Stress Responsiveness and Social Behavior. J Neurosci. 2011;31: 5470–5476. doi: 10.1523/jneurosci.6078-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frazier CJ, Pati D, Hiller H, Nguyen D, Wang L, Smith JA, et al. Acute hypernatremia exerts an inhibitory oxytocinergic tone that is associated with anxiolytic mood in male rats. Endocrinology. 2013;154: 2457–67. doi: 10.1210/en.2013-1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith JA, Wang L, Hiller H, Taylor CT, Kloet A, Krause EG. Acute hypernatremia promotes anxiolysis and attenuates stress-induced activation of the hypothalamic–pituitary–adrenal axis in male mice. 2014;136: 91–96. doi: 10.1016/j.physbeh.2014.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith JA, Pati D, Wang L, de Kloet AD, Frazier CJ, Krause EG. Hydration and beyond: neuropeptides as mediators of hydromineral balance, anxiety and stress-responsiveness. Frontiers in systems neuroscience. 2015;9: 46. doi: 10.3389/fnsys.2015.00046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luther J, Daftary S, Boudaba C, Gould G, Halmos CK, Tasker J. Neurosecretory and Non‐Neurosecretory Parvocellular Neurones of the Hypothalamic Paraventricular Nucleus Express Distinct Electrophysiological Properties. J Neuroendocrinol. 2002;14: 929–932. doi: 10.1046/j.1365-2826.2002.00867.x [DOI] [PubMed] [Google Scholar]
- 23.Wamsteeker Cusulin JI, Füzesi T, Watts AG, Bains JS. Characterization of Corticotropin-Releasing Hormone neurons in the Paraventricular Nucleus of the Hypothalamus of Crh-IRES-Cre Mutant Mice. PLoS ONE. 2013;8: e64943. doi: 10.1371/journal.pone.0064943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gillis KD. Techniques for Membrane Capacitance Measurements in Single Channel Recording (2nd Edition), Edited by Sakmann Bert and Neher Erwin. 1995; 155–198. [Google Scholar]
- 25.Walker LC, Cornish LC, Lawrence AJ, Campbell EJ. The Effect Of Acute or Repeated Stress On The Corticotropin Releasing Factor System In The CRH-IRES-Cre Mouse: A Validation Study. Neuropharmacology. 2018; doi: 10.1016/j.neuropharm.2018.09.037 [DOI] [PubMed] [Google Scholar]
- 26.Chen Y, Molet J, Gunn BG, Ressler K, Baram TZ. Diversity of Reporter Expression Patterns in Transgenic Mouse Lines Targeting Corticotropin-Releasing Hormone-Expressing Neurons. Endocrinology. 2015;156: 4769–4780. doi: 10.1210/en.2015-1673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Busnelli M, Bulgheroni E, Manning M, Kleinau G, Chini B. Selective and Potent Agonists and Antagonists for Investigating the Role of Mouse Oxytocin Receptors. Journal of Pharmacology and Experimental Therapeutics. 2013;346: 318–27. doi: 10.1124/jpet.113.202994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perkeybile AM, Carter SC, Wroblewski KL, Puglia MH, Kenkel WM, Lillard TS, et al. Early nurture epigenetically tunes the oxytocin receptor. Psychoneuroendocrino. 2019;99: 128–136. doi: 10.1016/j.psyneuen.2018.08.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Puglia MH, Connelly JJ, Morris JP. Epigenetic regulation of the oxytocin receptor is associated with neural response during selective social attention. Transl Psychiat. 2018;8: 116. doi: 10.1038/s41398-018-0159-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Unternaehrer E, Luers P, Mill J, Dempster E, Meyer A, Staehli S, et al. Dynamic changes in DNA methylation of stress-associated genes (OXTR, BDNF ) after acute psychosocial stress. Transl Psychiat. 2012;2: e150–e150. doi: 10.1038/tp.2012.77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harden SW, Frazier CJ. Oxytocin depolarizes fast‐spiking hilar interneurons and induces GABA release onto mossy cells of the rat dentate gyrus. Hippocampus. 2016;26: 1124–1139. doi: 10.1002/hipo.22595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dabrowska J, Hazra R, Ahern TH, Guo J- D, nald AJ, Mascagni F, et al. Neuroanatomical evidence for reciprocal regulation of the corticotrophin-releasing factor and oxytocin systems in the hypothalamus and the bed nucleus of the stria terminalis of the rat: Implications for balancing stress and affect. Psychoneuroendocrino. 2011;36: 1312–1326. doi: 10.1016/j.psyneuen.2011.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dabrowska J, Hazra R, Guo J- D, DeWitt S, Rainnie DG. Central CRF neurons are not created equal: phenotypic differences in CRF-containing neurons of the rat paraventricular hypothalamus and the bed nucleus of the stria terminalis. Frontiers in Neuroscience. 2013;7: 156. doi: 10.3389/fnins.2013.00156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chu C- P, Jin W- Z, Bing Y- H, Jin Q- H, Kannan H, Qiu D- L. Effects of Stresscopin on Rat Hypothalamic Paraventricular Nucleus Neurons In Vitro. PLoS ONE. 2013;8: e53863. doi: 10.1371/journal.pone.0053863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jamieson B, Nair B, Iremonger K. Regulation of hypothalamic corticotropin‐releasing hormone neurone excitability by oxytocin. Journal of Neuroendocrinology. 2017;29. doi: 10.1111/jne.12532 [DOI] [PubMed] [Google Scholar]
- 36.Raggenbass M, Alberi S, Zaninetti M, Pierson P, Dreifuss J. Vasopressin and oxytocin action in the brain: cellular neurophysiological studies. Progress in brain research. 1998;119: 263–73. [DOI] [PubMed] [Google Scholar]
- 37.Smith M, Ayad V, Mundell S, McArdle C, Kelly E, Bernal LA. Internalization and desensitization of the oxytocin receptor is inhibited by Dynamin and clathrin mutants in human embryonic kidney 293 cells. Mol Endocrinol Baltim Md. 2005;20: 379–88. doi: 10.1210/me.2005-0031 [DOI] [PubMed] [Google Scholar]
- 38.Terenzi MG, Ingram CD. Oxytocin-induced excitation of neurones in the rat central and medial amygdaloid nuclei. Neuroscience. 2005;134: 345–354. doi: 10.1016/j.neuroscience.2005.04.004 [DOI] [PubMed] [Google Scholar]
- 39.Robinson C, Schumann R, Zhang P, Young RC. Oxytocin-induced desensitization of the oxytocin receptor. Am J Obstet Gynecol. 2003;188: 497–502. doi: 10.1067/mob.2003.22 [DOI] [PubMed] [Google Scholar]
- 40.Young SW. Corticotropin‐releasing factor mRNA in the hypothalamus is affected differently by drinking saline and by dehydration. Febs Lett. 1986;208: 158–162. doi: 10.1016/0014-5793(86)81553-8 [DOI] [PubMed] [Google Scholar]
- 41.Dohanics J, Kovacs KJ, Folly G, Makara GB. Long-term salt loading impairs pituitary responsiveness to ACTH secretagogues and stress in rats. Peptides. 1990;11: 59–63. doi: 10.1016/0196-9781(90)90110-q [DOI] [PubMed] [Google Scholar]
- 42.Chowdrey HS, Jessop DS, Patel H, Lightman SL. Altered Adrenocorticotropin, Corticosterone and Oxytocin Responses to Stress during Chronic Salt Load. Neuroendocrinology. 1991;54: 635–638. doi: 10.1159/000125971 [DOI] [PubMed] [Google Scholar]
- 43.Amaya F, Tanaka M, Hayashi S, Tanaka Y, Ibata Y. Hypothalamo-Pituitary-Adrenal Axis Sensitization after Chronic Salt Loading. Neuroendocrinology. 2001;73: 185–193. doi: 10.1159/000054635 [DOI] [PubMed] [Google Scholar]
- 44.Krause EG, Pati D, Frazier CJ. Chronic salt-loading reduces basal excitatory input to CRH neurons in the paraventricular nucleus and accelerates recovery from restraint stress in male mice. Physiology & behavior. 2017;176: 189–194. doi: 10.1016/j.physbeh.2017.03.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nahir B, Bhatia C, Frazier CJ. Presynaptic inhibition of excitatory afferents to hilar mossy cells. Journal of neurophysiology. 2007;97: 4036–47. doi: 10.1152/jn.00069.2007 [DOI] [PubMed] [Google Scholar]