

Abstract
Objectives:
Smoking is a common risk factor for chronic obstructive pulmonary disease (COPD) and lung cancer. Although COPD patients have higher risk of lung cancer compared to non-COPD smokers, the molecular links between these diseases are not well-defined. This study aims to identify genes that are downregulated by cigarette smoke and commonly repressed in COPD and lung cancer.
Materials and Methods:
Primary human airway epithelial cells (HAEC) were exposed to cigarette-smoke-extract (CSE) for 10-weeks and significantly suppressed genes were identified by transcriptome array. Epigenetic abnormalities of these genes in lung adenocarcinoma (LUAD) from patients with or without COPD were determined using genome-wide and gene-specific assays and by in vitro treatment of cell lines with trichostatin-A or 5-aza-2-deoxycytidine.
Results:
The ten most commonly downregulated genes following chronic CSE exposure of HAEC and show promoter hypermethylation in LUAD were selected. Among these, expression of CCNA1, SNCA, and ZNF549 was significantly reduced in lung tissues from COPD compared with non-COPD cases while expression of CCNA1 and SNCA was further downregulated in tumors with COPD. The promoter regions of all three genes were hypermethylated in LUAD but not normal or COPD lungs. The reduced expression and aberrant promoter hypermethylation of these genes in LUAD were independently validated using data from the Cancer Genome Atlas project. Importantly, SNCA and ZNF549 methylation detected in sputum DNA from LUAD (52% and 38%) cases were more prevalent compared to cancer-free smokers (26% and 15%), respectively (p<0.02).
Conclusions:
Our data show that suppression of CCNA1, SNCA, and ZNF549 in lung cancer and COPD occurs with or without promoter hypermethylation, respectively. Detecting methylation of these and previously identified genes in sputum of cancer-free smokers may serve as non-invasive biomarkers for early detection of lung cancer among high risk smokers including COPD patients.
Keywords: Methylation, Epigenetic, COPD, NSCLC, Sputum, biomarkers
1. Introduction
Cigarette-smoke (CS) exposure plays a major role in the development of chronic obstructive pulmonary disease (COPD) and lung cancer [1]. Several independent studies have shown that smokers with COPD have a 2–4-fold higher risk of developing lung cancer compared with smokers without COPD [2,3]. Evaluation of the National Lung Screening Trial (NLST) data for patients with spirometry information [4], a meta-analysis of eighteen published prospective cohort studies involving 12,442 lung cancer cases [5], and another meta-analysis involving 31 case-control and 8 cohort studies found that the relative risk for lung cancer is higher for patients with COPD than those without [6]. These findings suggest that smoking-induced molecular alterations in lung cancer may already be present in patients with COPD and may contribute to the increased risk of lung cancer among COPD cases [7].
Epigenetic modifications link the impact of environmental factors such as smoking with inherent or acquired genetic predisposition to various lung diseases and increase susceptibility [8]. Aberrant DNA methylation of CpG islands across the promoter regions of tumor-suppressor genes is one of the most common epigenetic change caused by smoking [9–13]. Our previous epigenome-wide association study compared the epigenetic changes found in lung tumors and cancer-free lung tissue pairs from non–small-cell lung cancer (NSCLC) cases with or without COPD [14]. We found that expression of some genes, such as CCDC37 and MAP1B, is downregulated in the lungs of COPD subjects, further reduced in lung tumors, and even more suppressed in tumors from COPD cases. The suppression of these genes was enhanced in lung tumors by promoter hypermethylation that establishes a long-term loss of promoter activity [15,16]. We have also shown that high methylation index (methylation of ≥ 3 genes in a 12 gene-panel) in sputum predicts decline in forced expiratory volume in 1 second (FEV1), lung cancer incidence, and all-cause mortality in smokers [17].
Based on these findings we hypothesized that CS exposure suppresses expression of specific genes in airway epithelial cells, and these changes become more evident in lung cancer due to clonal expansion of the transformed cells. We and others have previously demonstrated that the promoter regions of genes with aberrantly repressed expression are prone to methylation during transformation [16,18]. The addition of promoter methylation not only reinforces the downregulation of such genes and stabilizes long term repression, but also provides potential DNA biomarkers that aid early detection in noninvasive samples such as sputum from high risk smokers. This study compared genome-wide downregulation of genes in primary human airway epithelial cells following chronic CS-extract (CSE) exposure and promoter CpG island methylation in COPD and lung cancer to identify genes commonly suppressed in the two major lung diseases. Importantly, the identified epigenetic changes were more frequently detected in the sputum of lung cancer cases compared to cancer-free smokers, suggesting that they may be used as part of a biomarker panel. The use of these and our previously validated noninvasive sputum biomarkers [17,19–21] together with low dose computed tomography (LDCT) could improve the specificity of early detection and treatment strategies for lung cancer.
2. Materials and methods
2.1. Study subjects and samples
Frozen lung adenocarcinoma (LUAD) samples from 90 patients (47 with and 43 without COPD) were obtained from tumor banks at the University of New Mexico (UNM) and the Mayo Clinic. Characteristics of these patients are shown in Table S1. All tumors have ≥ 75% tumor purity, and cancer-free lung tissues collected from sites most distant to the tumor in the resected lobe were available for a subset of these cases. COPD status and severity was defined according to the Global Initiative for Chronic Obstructive Lung Disease (GOLD) classification [22]. Sputum samples from the Lovelace Smokers cohort (n=108) and New Mexico Lung Cancer cohort (n=29), whose participants are from the Albuquerque, NM metropolitan area since 2001 [23–26] were used for methylation analysis. Characteristics of these subjects are described under Table S2. Normal human bronchial epithelial cells (NHBEC) collected from 5 cancer-free smokers at UNM through diagnostic bronchoscopy [10] and peripheral blood mononuclear cells (PBMC) obtained from 5 healthy donors were used as normal control. Primary human airway epithelial cells (HAEC) isolated from the lungs of 11 cancer-free never-smokers that were not suited for transplantation under the protocol and consent form approved by the University of North Carolina School of Medicine were used for air-liquid-interface cultures. All samples were obtained with written informed consent from patients, and the study was approved by each institute’s Ethics Committee. In addition, five human bronchial epithelial cell lines (HBEC) immortalized as described [27], and 20 NSCLC cell lines (Table S3), whose sources, authentications, and handling methods are described in the online supplement were also studied.
2.2. Air-liquid-interface (ALI) culture and cigarette-smoke exposure
Primary HAEC isolated from never-smokers (n=11) were driven to differentiation using ALI culture as described [28]. A pair of HAEC cultures from each individual were treated twice-a-week for 10 weeks either with vehicle (culture medium) or medium containing 40 μg/ml cigarette-smoke-extract (CSE). The CSE was prepared using Cigarette smoke (CS) generated from Kentucky Reference cigarettes (Type: 3R4F, Center for Tobacco Reference Products, Kentucky Tobacco Research & Development Center, Lexington, KY). Briefly, a modified AMESA Mark III smoke machine (AMESA Technologies, Switzerland) was used to generate the CS in a 1-m3 chamber using Health Canada Intense puffing regime (puff volume: 55 cc, puff duration 2 seconds, frequency 30 seconds, blocked ventilation holes). The CS was collected by pulling it through pre-weighed glass fiber filters (Type GF/A, GE Whatman), the CS mass was determined from the final weight of the filters at the end of sampling, and the filters were stored at −80°C until use. The filters were incubated for 1 h at 37°C with 5% CO2 in serum-free medium to generate a 100 μg/ml stock solution, fresh CSE solution prepared prior to each treatment, filter-sterilized, and diluted to a final concentration of 40 μg/ml in the culture medium.
2.3. Trichostatin-A or 5-aza-2-deoxycytidine treatments and gene expression analysis
The impact of DNA methylation or histone modification on gene expression were assessed as described [29–31]. Briefly, HAEC and NSCLC cell lines were treated with 500 nM DNA methyltransferases inhibitor 5-aza-2-deoxycytidine (DAC) every 12 h for 96 h or 300 nM histone deacetylase inhibitor Trichostatin-A (TSA) for 18 h prior to harvest in Trizol. For transcriptome analysis, total RNA was isolated, amplified, and biotin labeled using the Illumina TotalPrep RNA Amplification Kit, and 1 μg biotin-labeled cRNA was hybridized, and probes were visualized using Illumina’s iScan and GenomeStudio softwares. All arrays passed quality checks including visual inspection for artifacts and the distribution of signal and background intensity for red and green channels. For gene-specific quantitative PCR, RNA isolated from the various in vitro treatments (CSE, TSA, or DAC) or patient samples was reverse transcribed, expression of target genes quantified as described [29,31,32], and the relative gene expression levels were calculated using ΔΔCT method [33]. RNA-seq data for large number of lung adenocarcinoma (LUAD, n = 517) and normal lung (n = 110) tissue samples was obtained from the Cancer Genome Atlas (TCGA) project https://tcga-data.nci.nih.gov/tcga/ and used for validation of our findings.
2.4. DNA extraction and methylation analysis
DNA extraction, bisulfite modification, and methylation analysis using Combined Bisulfite Modification and Restriction Analysis (CoBRA), Methylation-Specific PCR (MSP), and for sputum samples, nested MSP assays, were conducted as described [19,30,34]. The primer sequences and amplification conditions are shown in Table S4. Genome-wide methylation of lung tumor-normal pairs was quantified using the Infinium HumanMethylation450 BeadChip (HM450K) exactly as described [35]. Briefly, 500 ng bisulfite-modified DNA was whole-genome amplified, enzymatically fragmented, precipitated, re-suspended in hybridization buffer, and hybridized onto the HumanMethylation450 Bead-Chips according to the standard Infinium protocol. The beadchips were then processed through primer extension, immunohistochemistry staining, coated, and imaged on an Illumina iScan. Quantitative methylation data for large number of LUAD (n = 450) and normal lung (n = 74) tissue samples, which like our study generated by TCGA using HM450K, was used for validation of our findings.
2.5. Statistical analysis
Gene methylation and patient characteristics including age, sex, smoking, and COPD status were summarized with mean and standard deviation for continuous variables and proportions for categorical variables. For RNA expression, the statistical platform R and packages from Bioconductor were used for all computation [36,37]. The log ratio of red signal to green signal was calculated after background subtraction and LoEss normalization as implemented in the limma package from Bioconductor [37]. The HM450K data for 485,577 probes across the epigenome of samples evaluated was calculated using GenomeStudio® Methylation module software as methylation beta-value (β=intensity of the Methylated allele (M) / (intensity of the Unmethylated allele (U)+intensity of the Methylated allele (M)+100) as described [35]. HM450K data for lung tissue and tumor samples from COPD and non-COPD cases was compared using β-regression analysis [38]. The association between methylation and COPD or lung cancer was assessed using logistic regression. Area under the curve (AUC) of a receiver operating characteristic (ROC) curve was calculated to assess the prediction performance of logistic regressions for smoking pack years and gene methylation. The methylation status of statistically significant genes as independent variables was included in the basic model to evaluate the delta change in AUC. Gene expression levels were compared using two-tailed T-test for unequal variance (COPD vs. non-COPD cases) and pairwise T-test (tumor vs. normal pairs). Pathway analysis of CSE exposure regulated genes was performed using the Ingenuity® Pathway Analysis (IPA®) program from Qiagen. All analyses were conducted in SAS 9.4 and R 3.4
3. Results
3.1. Discovery of cigarette-smoke-regulated genes linked to COPD and lung cancer
The general strategy to identify genes that are suppressed by chronic CS exposure of airway epithelia and may contribute to COPD and lung cancer is depicted in Figure 1. Primary HAEC derived from 11 never-smokers were differentiated in Air-liquid-interface (ALI) cultures and treated with vehicle or 40 μg/ml CSE twice-a-week for 10 wks. Genome-wide transcriptome analysis identified significant downregulation of 632 genes in the CSE exposed cells (p<0.05, paired t-test). Pathway analysis of these genes revealed that protein kinase A, p53, and interferon signaling pathways are among the top canonical signaling pathways while cellular movement, organization, maintenance, morphology, and cell-to-cell signaling were the top cellular functions affected (Fig. S1A). The genes that are hypermethylated in lung cancer were identified using genome-wide DNA methylation analysis (HM450K) of 20 NSCLC cell lines. About half of the 632 CSE-downregulated genes have a CpG island in their promoter regions and 10 of these, which were significantly downregulated in the majority of HAEC samples (≥ 6/11) and showed promoter hypermethylation in NSCLC cell lines were selected (Table 1). To identify the genes that are reduced in primary lung tumors with possible distinction between cases with or without COPD, we used a list of methylated genes we had previously identified from 35 lung adenocarcinoma (LUAD) patients with (n=18) or without COPD (n=17) [14]. Characteristics of patients whose lung or sputum samples were used in this study are shown under Tables S1 and S2, respectively. Three genes, CCNA1, SNCA, and ZNF549, showed tumor-specific aberrant hypermethylation in multiple NSCLC cell lines and lung tumors but not in any of the distant normal lung tissue (DNLT) samples (Table 1).
Figure 1.
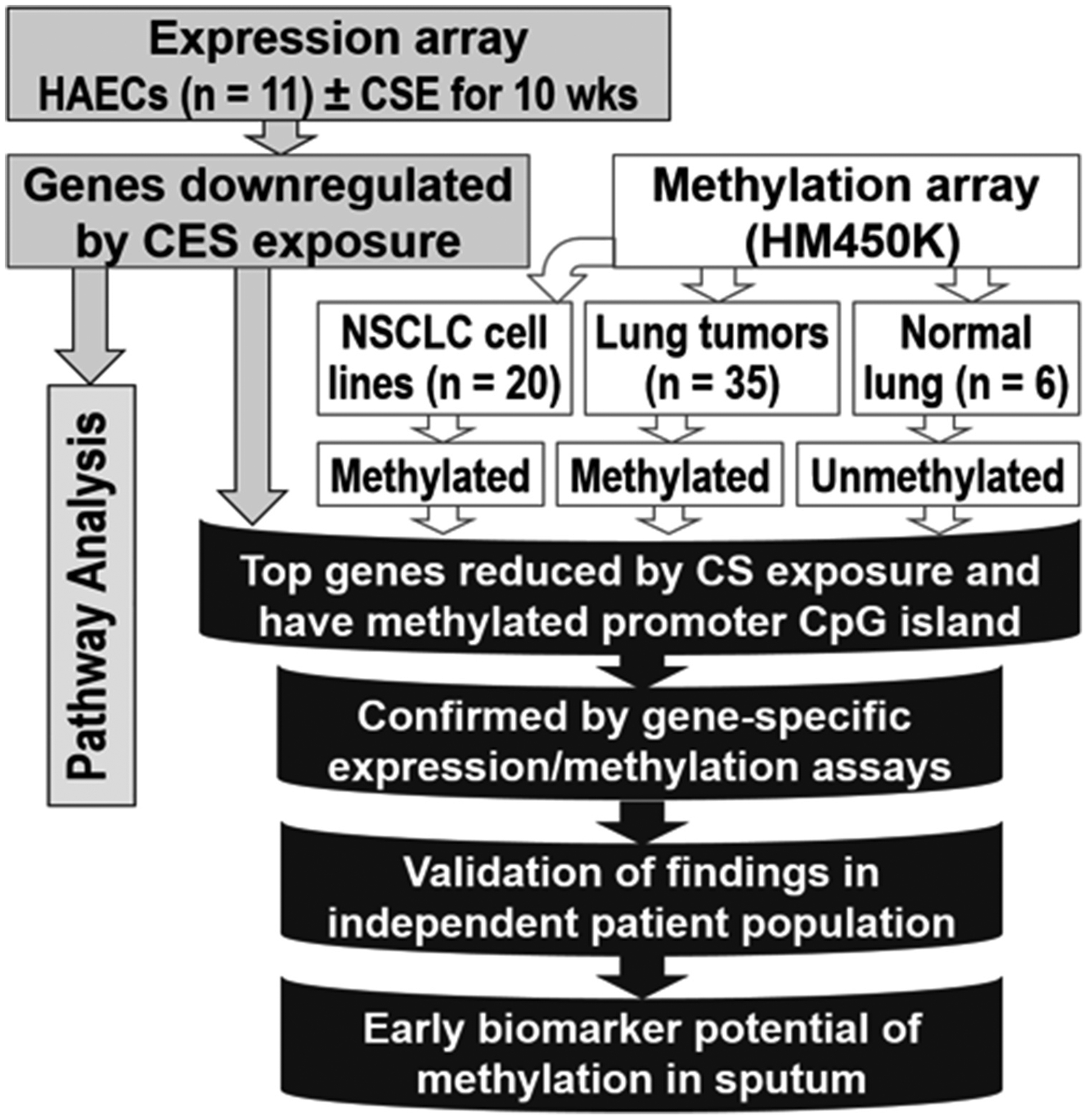
A multifaceted strategy was used to uncover cigarette-smoke regulated genes linking COPD to lung cancer.
Table 1:
Genes suppressed by cigarette smoke (CS) exposure and methylated in lung cancer
| Genes | Down in CS exposed HAECs (n = 11) | Methylated (average promoter CpG island β ≥ 0.2) | ||
|---|---|---|---|---|
| NSCLC cell lines (n = 20) | Adenocarcinoma (n = 35)† | DNLT (n = 6) | ||
| CCNA1 * | 9 (82%) | 4 (20%) | 10 (29%)‡ | 0 (0%) |
| CPA6 | 8 (73%) | 1 (5%) | 25 (71%) | 2 (33) |
| CRISPLD1 | 9 (82%) | 3 (15%) | 31 (89%) | 5 (83) |
| CYYR1 | 7 (64%) | 12 (60%) | 34 (98%) | 6 (100) |
| EFEMP1 | 9 (82%) | 4 (20%) | 35 (100%) | 6 (100) |
| FSD1 | 7 (64%) | 5 (25%) | 34 (97%) | 6 (100) |
| SCARA3 | 6 (55%) | 2 (10%) | 1 (3%) | 0 (0%) |
| SERPINE2 | 11 (100%) | 1 (5%) | 0 (0%) | 0 (0%) |
| SNCA * | 11 (100%) | 9 (45%) | 20 (57%)‡ | 0 (0%) |
| ZNF549 * | 6 (55%) | 4 (20%) | 11 (31%)‡ | 0 (0%) |
Genes that are commonly repressed by CS and methylated in NSCLC cell lines and tumors but not in the distant normal lung tissues (DNLT) were selected for detailed analysis.
The 35 lung adenocarcinoma samples were obtained from 18 COPD and 17 non-COPD cases.
CCNA1, SNCA, and ZNF549 promoters showed tumor-specific methylation. All 3 genes were methylated in 4/35 (11%) of the samples, whereas at least 2 or 1 of the three genes were methylated in 14/35 (40%) and 23/35 (66%) of the adenocarcinoma samples, respectively.
3.2. Suppression of CS-regulated genes in COPD and lung cancer
We confirmed the suppression of CCNA1, SNCA, and ZNF549 by chronic CSE exposure of differentiated HAEC using q-PCR (Fig. S1B). Whether the suppression was potentially due to histone modifications was indirectly investigated by treating the CS-treated HAEC with the histone deacetylase inhibitor, Trichostatin-A (TSA). SNCA and ZNF549 expression levels already responded to TSA alone and the suppression by chronic CS was partially restored (Fig. S1B). Whether these genes are progressively suppressed during the development of COPD and lung carcinogenesis was investigated using mRNA from normal lung tissues (NLT) obtained from never-smokers (NLT-NS, n=5) and smokers (NLT-S, n=21), tumors with distant normal lung tissue (DNLT) pairs from LUAD patients (n=37), and NSCLC cell lines (n=20). Since the tumors and DNLT pairs were obtained from smokers with (n=20) or without (n=17) COPD, they were subdivided into four groups. DNLT with (DNLT+COPD, n=20) and without COPD (DNLT-COPD, n=17), and tumors with (Tumor+COPD, n=20) and without COPD (Tumor-COPD, n=17). Expression of CCNA1, SNCA, and ZNF549 in normal lung tissue from smokers (NLT-S and DNLT-COPD) was not significantly different from the expression in never-smokers (NLT-NS, Fig. 2A–C). However, expression of the three genes was significantly suppressed in DNLT+COPD compared to the normal lung tissues (NLT-NS, NLT-S) and/or DNLT-COPD. CCNA1 and SNCA mRNA levels were also significantly reduced in Tumor-COPD and further reduced in Tumor+COPD (Fig. 2A–B). In contrast, the reduced ZNF549 mRNA levels were mainly associated with COPD (DNLT+COPD and Tumor+COPD) and Tumor-COPD did not show significant suppression (Fig. 2C). All three genes were significantly reduced in NSCLC cell lines compared to normal lung (LUAD-COPD). Analysis of RNA-seq data from the Cancer Genome Atlas (TCGA) database independently validated the significant repression (p<0.0001) of CCNA1 and SNCA in LUAD tumors (n=517) compared to normal lung tissue (n=110, Fig. 2D). Our data showed that ZNF549 expression in lung tumors without COPD (Tumor-COPD, Fig. 2C) was not significantly reduced and this was also supported by the similar expression of the gene in normal vs. LUAD samples from TCGA, where the COPD status of cases are unknown.
Figure 2. Gene expression changes in COPD and lung cancer.

The expression levels of (A) CCNA1, (B) SNCA, and (C) ZNF549 in normal lung tissues (NLT) obtained from cancer-free never smokers (NLT-NS) and smokers (NLT-S), tumor-normal pairs from lung cancer patients, and NSCLC cell lines were quantified using gene-specific expression assays. The distant normal lung tissue (DNLT) and tumor pairs were all from smoker LUAD cases with or without COPD. Thus, they were separated into the following 4-groups: DNLT without COPD (DNLT-COPD) or with COPD (DNLT+COPD) and tumors without COPD (Tumor-COPD) or with COPD (Tumor+COPD). Expression is measured relative to the level in NLT-NS and the significant changes from NLT-NS (✭), NLT-S (◆), DNLT-COPD (Δ), DNLT+COPD (☐), or Tumor-COPD (■) are shown. D) RNA-seq data for the expression of CCNA1, SNCA, and ZNF549 in normal lung tissues and LUAD samples from TCGA database was used to independently validate the repression of these genes.
3.3. Tumor-specific methylation of CS-regulated genes in lung cancer
The methylation status of CCNA1, SNCA, and ZNF549 promoter CpG islands were determined using quantitative and qualitative assays. The HumanMethylation450 BeadChip (HM450K) quantifies the methylation levels of 9, 10, and 7 probes across the promoter CpG islands of CCNA1, SNCA, and ZNF549, respectively (Fig. S2A–C). The lowest level of methylation for all three genes was detected in normal bronchial epithelial cells (NHBEC7) and distant normal lung tissue (DNLT), while the highest methylation levels were found in the NSCLC cell lines Calu6 (for CCNA1 and ZNF549) and SKLU1 (for SNCA, Fig S2A–C). The methylation levels of all three genes were significantly higher in the tumors than the DNLT. Similarly, the DNLT-COPD and DNLT+COPD samples, regardless of the COPD status, showed the lowest methylation levels for all three genes compared to lung tumors with or without COPD (Fig. 3A–C). Overall, tumors from COPD patients (LUAD+COPD) showed the highest methylation among primary tissues. The aberrant promoter CpG island hypermethylation of all three genes in lung cancer regardless of COPD status was independently validated using TCGA data for 450 LUAD cases (Fig. 4A–3). Details of the methylation data for the TCGA samples are shown in Tables S5 – S7.
Figure 3. Differentially methylated regions across the promoter regions of (A) CCNA1, (B) SNCA, and (C) ZNF549 in lung cancer.

The human methylation450beadchip (HM450K) was used to quantify the methylation levels across the promoter CpG islands of the three genes in lung tumor and distant normal lung tissue pairs from smoker LUAD cases with or without COPD. The y-axis shows the level of methylation in β–values ranging 0 to 1 (1 indicating complete or 100% methylation). The x-axis labels indicate the shortened HM450K probe ID numbers of targeted CpGs. The full probe ID numbers along with detailed methylation data for TCGA samples are shown in supplementary Tables S3 – S5.
Figure 4. Independent validation of tumor-specific promoter hypermethylation of (A) CCNA1, (B) SNCA, and (C) ZNF549 in lung cancer.

Quantitative methylation (HM450K) data for the CpG islands across the promoter regions of the three genes in normal lung and LUAD samples were obtained from the publicly available TCGA (the Cancer Genome Atlas) database. The tumor specific hypermethylation shown across the promoter CpG islands of the three genes independently validated our findings. The details of the figures are similar to Figure 3 above.
The tumor-specific methylation of these genes, which is often necessary for simple biomarker screening of large patient samples, was further verified using two independent methylation assays, the semi-quantitative Combined Bisulfite Modification and Restriction Analysis (CoBRA, Fig. S3A–C), and the highly sensitive, qualitative Methylation-Specific PCR (MSP, Table S8) assays. These assays confirmed that all three promoter CpG islands were unmethylated in normal samples including differentiated HAEC cultures with or without CSE exposure (Fig. S3A–C top panels), normal bronchial epithelial cells, and peripheral blood mononuclear cells (Fig. S3A–C middle panels). In contrast, all three promoter CpG islands were hypermethylated in a subset of NSCLC cell lines including complete methylation in some cell lines (shown by the complete digestion in the presence of the BstU1 restriction enzyme, Fig. S3A–C bottom panels). Evaluation of these genes using the highly sensitive MSP assay showed that the CCNA1, SNCA, and ZNF549 promoters were methylated in 60, 65, and 25% of NSCLC cell lines (n=20) and 56, 87, and 40% of LUAD tumors (n=55), respectively, (Table S8).
3.4. Epigenetic changes suppress transcription of these genes
CCNA1 expression is completely silenced in NSCLC cell lines with hypermethylated promoter CpG island (e.g. Calu6, Calu3) but expressed similar to DNLT in those without promoter methylation e.g. SKLU1 (Fig. S2A, S3A, and S4A). Similarly, complete or significant silencing of SNCA and ZNF549 were seen in cell lines where the promoter CpG islands of these genes were hypermethylated (Fig. S2B–C, S3B–C, and S4B–C). The reversibility of epigenetic silencing of these genes in lung cancer was investigated using in vitro treatment of these cell lines with the histone deacetylase inhibitor Trichostatin-A (TSA) or the DNA methyltransferases inhibitor 5-aza-2-deoxycytidine (DAC) as described [30]. Compared to vehicle (culture media), treatment of NSCLC cells with DAC partially or completely restored expression of CCNA1, SNCA, and ZNF549 (Fig S4A–C). In contrast, TSA treatment increased expression only in the cell lines with partial or no methylation such as SNCA and ZNF549 in H358 (Fig. S4B–C) or ZNF549 in H1838 (Fig. S4C). These results indicate that DNA methylation rather than histone deacetylation is primarily responsible for the transcriptional silencing or repression of these genes in lung cancer.
3.5. Methylation of CCNA1, SNCA, and ZNF549 in sputum samples
Aberrant promoter methylation of lung cancer-related genes in sputum samples from high risk smokers can be detected up to two years prior to lung cancer diagnosis [19,20]. Therefore, the potential use of CCNA1, SNCA, and ZNF549 promoter methylation as non-invasive biomarkers in sputum samples from smokers with or without COPD and/or LUAD was evaluated. The prevalence for methylation of all three genes in the sputum samples from cancer-free smokers with COPD (n=32) was similar to smokers without COPD (n=76, Table 2). In a separate set of analyses, the methylation of each gene in sputum samples from cancer-free smokers (with or without COPD, n=108) were compared with sputum samples from smokers with LUAD (n=29). CCNA1 methylation was not detected in any of the sputum samples from LUAD cases. In contrast, methylation of SNCA and ZNF549 was increased from 26% and 15% in the sputum of cancer-free smokers (lung cancer controls) to 52% and 38% in sputum from LUAD cases (Odds ratio = 3.06 and 3.44), respectively (Table 2). The AUC classification accuracy for lung cancer was 59% for pack years, 70% for SNCA and 68% for ZNF549, and improved to 72% when all three variables were included in the model (Supplemental Table S9).
Table 2:
Prevalence for methylation of genes in sputum samples from cancer-free smokers (CFS) with or without COPD and smokers with lung adenocarcinoma (LUAD)
| Genes | Methylated | ||||
|---|---|---|---|---|---|
| COPD status of CFS, n (%) | LUAD, n (%) | OR (95% CI)†† (CFS vs. LUAD) | |||
| Non-COPD (n = 76) | COPD (n = 32) | Total (n = 108) | (n = 29) | ||
| CCNA1 | 5/76 (7%) | 3/32 (9%) | 8/108 (7%) | 0/29 (0%) | ND |
| SNCA | 21/76 (28%) | 7/32 (22%) | 28/108 (26%) | 15/29 (52%) | 3.06 (1.3—7.1) |
| ZNF549† | 9/75 (12%) | 7/31 (23%) | 16/106 (15%) | 11/29 (38%) | 3.44 (1.4 – 8.6) |
Statistically significant differences are shown in bold.
ZNF549 assay failed for 2 samples, one non-COPD and one COPD.
The differences in the methylation of each of the three genes is compared between total CFS (COPD and non-COPD samples combined) versus LUAD. ND (not determined).
4. Discussion
This study combined genome-wide gene expression and DNA methylation analyses to identify genes that are downregulated by cigarette-smoke and may contribute to COPD and lung cancer development. The suppression of CCNA1, SNCA, and ZNF549 expression following chronic CSE-exposure and in the lungs of COPD patients was potentially mediated by histone modifications without abnormal methylation of their promoter CpG islands. However, extensive promoter hypermethylation that is associated with transcriptional repression of these genes was found specifically in the tumors but not normal lung tissue samples from LUAD cases. The aberrant methylation of these genes was also detected in sputum samples from high risk smokers, suggesting that these changes may have originated from early (pre)malignant cells and could serve as novel biomarkers for early detection of lung cancer among heavy smokers. The reduced expression of these genes in CSE-exposed primary HAEC and further suppression in lung tissue from COPD patients and in lung tumors show that epigenetic repression of these genes is a common abnormality in COPD and lung cancer. Taken together, these findings will support future studies to establish a potential mechanistic link between the two major smoking-induced lung diseases.
Epigenetic modifications are key mechanisms that allow cells to regulate gene expression networks in response to environmental stimuli [39]. Cigarette-smoke exposure modulates expression of many genes and induces genetic and epigenetic lesions across the genome [40]. Our group has identified expression changes of many genes in airway epithelial cells following exposure to cigarette-smoke or CSE [41,42]. We have previously demonstrated that in vitro exposure of bronchial epithelial cells to tobacco carcinogens initially suppresses expression of genes and microRNAs through histone modifications (e.g. acetylation, methylation) [13,18]. These early changes create a condensed chromatin structure that leads to a reversible and transient transcriptional repression. In addition, the transient downregulation or silencing of these genes and the associated reduction in promoter activity make their promoter CpG islands prone to aberrant methylation, which then leads to the establishment of a long-term repressive state. These early changes become more evident during malignant transformation and clonal expansion make these abnormalities easily detectable, and potential biomarkers for early detection of lung cancer. Our findings that show initial suppression of CCNA1, SNCA, and ZNF549 following chronic CSE exposure with no apparent changes in promoter methylation and the detection of hypermethylation in the tumor samples support this pattern. The role of cigarette-smoke exposure in creating DNA adducts and impacting DNA repair pathways within lung epithelia and how these genetic lesions contribute to transcriptional repression and epigenetic changes in chronic respiratory diseases such as COPD and lung cancer have also been previously demonstrated [21,43].
Early detection is the most critical step for improving cancer survival. Unfortunately, unlike for breast and colorectal cancers, highly effective and specific screening method for lung cancer is not yet available. As a result, many lung cancer patients are diagnosed (often with advanced disease) following x-rays or scans for unrelated reasons, and lung cancer remains the leading cause of cancer-related mortality worldwide. Since 2015, the Centers for Medicare & Medicaid Services (CMS) approved the use of low dose computed tomography (LDCT) for lung cancer screening if certain eligibility requirements are met. However, the promise of LDCT in improving survival comes with an increased treatment-related risk and cost due to high false-positive rates [37]. Thus, the use of non-invasive methods to supplement lung cancer screening approaches for high risk individuals including COPD patents is urgently needed. The discovery of novel sputum biomarkers in this and previous studies will contribute towards achieving this goal. The role of CCNA1, SNCA, and ZNF549 in linking COPD to lung cancer needs independent validation. Although we used TCGA data to validate these genes as lung cancer biomarkers, their suppression in COPD could not be independently verified due to absence of pulmonary function test data for an adequate number of TCGA samples.
The concurrent suppression of CCNA1 and SNCA with the development of lung adenocarcinoma supports the hypothesis that these genes may have a tumor-suppressor role in lung cancer. CCNA1 is a member of the cyclin family whose protein levels dramatically fluctuate during cell cycle and function as regulators of cyclin-dependent kinases (CDKs). CCNA1 binds to CDK2 and CDC2 kinases to modulate two distinct kinase activities in the S and G2-M phases of the cell cycle, respectively. It also regulates various cellular functions by binding to key cell cycle regulators such as E2F-1, Rb, and p21 proteins [44,45]. CCNA1 knockdown in lung cancer cells increases cancer properties such as epithelial-to-mesenchymal transition (EMT), cell migration, and invasion [46] suggesting that its repression in COPD may similarly contribute to lung cancer development. In agreement with this premise, the detection of CCNA1 methylation and its prognostic values have been previously reported in various malignancies including lung cancer [47–49]. SNCA encodes for α-synuclein that is abundantly expressed in the brain and helps to integrate presynaptic signaling and membrane trafficking. Thus, SNCA abnormalities including its promoter methylation are primarily associated with neurological disorders such as Parkinson and Alzheimer’s diseases [50–52]. The downregulation of SNCA expression in LUAD is also correlated with shorter survival time of patients [53]. Our findings confirm SNCA downregulation in LUAD and also revealed its repression in cancer-free COPD lung. ZNF549 encodes for a zinc-finger protein that is ubiquitously expressed including in the lungs. Yet, it is also one of the genes that show significantly higher methylation in nasopharyngeal carcinoma [54], suggesting aberrant methylation of its gene promoter occurs in various tumors. However, the normal function of ZNF549 and its potential role in COPD or lung cancer are unclear.
Supplementary Material
Highlights.
COPD increases lung cancer risk while cigarette smoke (CS) leads to both diseases.
Genome-wide assays were used to find CS suppressed genes in lung epithelial cells.
CCNA1, SNCA, and ZNF549 were epigenetically repressed in COPD and lung cancer.
Aberrant methylation of these genes is frequent in the sputum of lung cancer cases.
These sputum biomarkers may aid early detection of lung cancer in smokers with COPD.
Acknowledgements
We thank The Cancer Genome Atlas (TCGA) project that was established by the NCI and NHGRI. Publicly available data generated by TCGA, dbGaP accession number phs000178.v8.p7, was used to validate part of our findings. Information about TCGA and the investigators and institutions that constitute the TCGA research network can be found at http://cancergenome.nih.gov/.
Funding
This study was supported by R01 CA164782 (Drs. Belinsky and Tesfaigzi) and in part by R21 CA161561 and R01CA193532 (Dr. Tessema), RO1 HL068111 (Dr. Tesfaigzi), and P30CA11800.
Abbreviations
- ALI
Air-liquid-interface
- COPD
Chronic obstructive pulmonary disease
- CoBRA
Combined bisulfite modification and restriction analysis
- CS
Cigarette smoke
- CSE
Cigarette smoke extracts
- DAC
5-aza-2-deoxycytidin
- DNLT
Distant normal lung tissue
- HAEC
Human airway epithelial cells
- HM450K
HumanMethylation450 beadchip
- LUAD
Lung adenocarcinoma
- MSP
Methylation specific PCR
- NHBEC
Normal human bronchial epithelial cells
- NS
Never-smokers
- TCGA
The cancer genome atlas
- TSA
Trichostatin-A
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Availability of data and materials
All HumanMethylation450 beadchip and transcriptome array data are available on request.
Ethics approval and consent to participate
All samples were obtained with written informed consent from patients, and the study was approved by each participating institute’s Ethics Committee.
Competing interests
The authors declare that they have no competing interests.
Disclosure: This study was supported by R01 CA164782 and in part by R21 CA161561, R01CA193532, RO1 HL068111, and P30CA11800 from the National Institutes of Health.
Appendix A. Supplementary data
Supplementary material related to this article can be found, in the online version.
REFERENCES
- [1].Celli BR, Chronic obstructive pulmonary disease and lung cancer: common pathogenesis, shared clinical challenges, Proc Am Thorac Soc. 9 (2012) 74–79. [DOI] [PubMed] [Google Scholar]
- [2].Nakayama M, Satoh H, Sekizawa K, Risk of cancers in COPD patients, Chest. 123 (2003) 1775–1776. [DOI] [PubMed] [Google Scholar]
- [3].Papi A, Casoni G, Caramori G, et al. , COPD increases the risk of squamous histological subtype in smokers who develop non-small cell lung carcinoma, Thorax. 59 (2004) 679–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Young RP, Duan F, Chiles C, et al. , Airflow Limitation and Histology Shift in the National Lung Screening Trial. The NLST-ACRIN Cohort Substudy, Am J Respir Crit Care Med. 192 (2015) 1060–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zhang X, Jiang N, Wang L, et al. , Chronic obstructive pulmonary disease and risk of lung cancer: a meta-analysis of prospective cohort studies, Oncotarget. 8 (2017) 78044–78056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Brenner DR, McLaughlin JR, Hung RJ, Previous lung diseases and lung cancer risk: a systematic review and meta-analysis, PLoS One. 6 (2011) e17479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Houghton AM, Mechanistic links between COPD and lung cancer, Nat Rev Cancer. 13 (2013) 233–245. [DOI] [PubMed] [Google Scholar]
- [8].Wu DD, Song J, Bartel S, et al. , The potential for targeted rewriting of epigenetic marks in COPD as a new therapeutic approach, Pharmacol Ther. 182 (2018) 1–14. [DOI] [PubMed] [Google Scholar]
- [9].Belinsky SA, Nikula KJ, Palmisano WA, et al. , Aberrant methylation of p16(INK4a) is an early event in lung cancer and a potential biomarker for early diagnosis, Proc Natl Acad Sci U S A. 95 (1998) 11891–11896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Belinsky SA, Palmisano WA, Gilliland FD, et al. , Aberrant promoter methylation in bronchial epithelium and sputum from current and former smokers, Cancer Res. 62 (2002) 2370–2377. [PubMed] [Google Scholar]
- [11].Sood A, Petersen H, Blanchette CM, et al. , Wood smoke exposure and gene promoter methylation are associated with increased risk for COPD in smokers, Am J Respir Crit Care Med. 182 (2010) 1098–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Suzuki M, Wada H, Yoshino M, et al. , Molecular characterization of chronic obstructive pulmonary disease-related non-small cell lung cancer through aberrant methylation and alterations of EGFR signaling, Ann Surg Oncol. 17 (2010) 878–888. [DOI] [PubMed] [Google Scholar]
- [13].Tellez CS, Juri DE, Do K, et al. , EMT and stem cell-like properties associated with miR-205 and miR-200 epigenetic silencing are early manifestations during carcinogen-induced transformation of human lung epithelial cells, Cancer Res. 71 (2011) 3087–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tessema M, Yingling CM, Picchi MA, et al. , Epigenetic Repression of CCDC37 and MAP1B Links Chronic Obstructive Pulmonary Disease to Lung Cancer, J Thorac Oncol. 10 (2015) 1181–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Clark SJ, Melki J, DNA methylation and gene silencing in cancer: which is the guilty party?, Oncogene. 21 (2002) 5380–5387. [DOI] [PubMed] [Google Scholar]
- [16].Tessema M, Yingling CM, Grimes MJ, et al. , Differential epigenetic regulation of TOX subfamily high mobility group box genes in lung and breast cancers, PLoS One. 7 (2012) e34850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Leng S, Diergaarde B, Picchi MA, et al. , Gene Promoter Hypermethylation Detected in Sputum Predicts FEV1 Decline and All-Cause Mortality in Smokers, Am J Respir Crit Care Med. 198 (2018) 187–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Damiani LA, Yingling CM, Leng S, et al. , Carcinogen-induced gene promoter hypermethylation is mediated by DNMT1 and causal for transformation of immortalized bronchial epithelial cells, Cancer Res. 68 (2008) 9005–9014. [DOI] [PubMed] [Google Scholar]
- [19].Belinsky SA, Liechty KC, Gentry FD, et al. , Promoter hypermethylation of multiple genes in sputum precedes lung cancer incidence in a high-risk cohort, Cancer Res. 66 (2006) 3338–3344. [DOI] [PubMed] [Google Scholar]
- [20].Leng S, Do K, Yingling CM, et al. , Defining a gene promoter methylation signature in sputum for lung cancer risk assessment, Clin Cancer Res. 18 (2012) 3387–3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Leng S, Liu Y, Weissfeld JL, et al. , 15q12 Variants, Sputum Gene Promoter Hypermethylation, and Lung Cancer Risk: A GWAS in Smokers, J Natl Cancer Inst. 107 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Pauwels RA, Buist AS, Calverley PM, et al. , Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary, Am J Respir Crit Care Med. 163 (2001) 1256–1276. [DOI] [PubMed] [Google Scholar]
- [23].Hunninghake GM, Cho MH, Tesfaigzi Y, et al. , MMP12, lung function, and COPD in high-risk populations, N Engl J Med. 361 (2009) 2599–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Leng S, Liu Y, Thomas CL, et al. , Native American ancestry affects the risk for gene methylation in the lungs of Hispanic smokers from New Mexico, Am J Respir Crit Care Med. 188 (2013) 1110–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Leng S, Stidley CA, Liu Y, et al. , Genetic determinants for promoter hypermethylation in the lungs of smokers: a candidate gene-based study, Cancer Res. 72 (2012) 707–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Leng S, Stidley CA, Willink R, et al. , Double-strand break damage and associated DNA repair genes predispose smokers to gene methylation, Cancer Res. 68 (2008) 3049–3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ramirez RD, Sheridan S, Girard L, et al. , Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins, Cancer Res. 64 (2004) 9027–9034. [DOI] [PubMed] [Google Scholar]
- [28].Harris JF, Fischer MJ, Hotchkiss JR, et al. , Bcl-2 sustains increased mucous and epithelial cell numbers in metaplastic airway epithelium, Am J Respir Crit Care Med. 171 (2005) 764–772. [DOI] [PubMed] [Google Scholar]
- [29].Tessema M, Klinge DM, Yingling CM, et al. , Re-expression of CXCL14, a common target for epigenetic silencing in lung cancer, induces tumor necrosis, Oncogene. 29 (2010) 5159–5170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Tessema M, Willink R, Do K, et al. , Promoter methylation of genes in and around the candidate lung cancer susceptibility locus 6q23–25, Cancer Res. 68 (2008) 1707–1714. [DOI] [PubMed] [Google Scholar]
- [31].Tessema M, Yingling CM, Liu Y, et al. , Genome-wide unmasking of epigenetically silenced genes in lung adenocarcinoma from smokers and never smokers, Carcinogenesis. 35 (2014) 1248–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Tessema M, Yingling CM, Thomas CL, et al. , SULF2 methylation is prognostic for lung cancer survival and increases sensitivity to topoisomerase-I inhibitors via induction of ISG15, Oncogene. 31 (2012) 4107–4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Livak KJ, Schmittgen TD, Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method, Methods. 25 (2001) 402–408. [DOI] [PubMed] [Google Scholar]
- [34].Tessema M, Yu YY, Stidley CA, et al. , Concomitant promoter methylation of multiple genes in lung adenocarcinomas from current, former and never smokers, Carcinogenesis. 30 (2009) 1132–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bibikova M, Barnes B, Tsan C, et al. , High density DNA methylation array with single CpG site resolution, Genomics. 98 (2011) 288–295. [DOI] [PubMed] [Google Scholar]
- [36].RDc t, R: A language and environment for statistical computing R foundation for statistical computing. Vienna, Austria., (2004.). [Google Scholar]
- [37].Smyth GK, Speed T, Normalization of cDNA microarray data, Methods. 31 (2003) 265–273. [DOI] [PubMed] [Google Scholar]
- [38].Siegmund KD, Statistical approaches for the analysis of DNA methylation microarray data, Hum Genet. 129 (2011) 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Petronis A, Epigenetics as a unifying principle in the aetiology of complex traits and diseases, Nature. 465 (2010) 721–727. [DOI] [PubMed] [Google Scholar]
- [40].Brody JS, Transcriptome alterations induced by cigarette smoke, Int J Cancer. 131 (2012) 2754–2762. [DOI] [PubMed] [Google Scholar]
- [41].Pickett G, Seagrave J, Boggs S, et al. , Effects of 10 cigarette smoke condensates on primary human airway epithelial cells by comparative gene and cytokine expression studies, Toxicol Sci. 114 (2010) 79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Mebratu YA, Schwalm K, Smith KR, et al. , Cigarette smoke suppresses Bik to cause epithelial cell hyperplasia and mucous cell metaplasia, Am J Respir Crit Care Med. 183 (2011) 1531–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Morrow JD, Glass K, Cho MH, et al. , Human Lung DNA Methylation Quantitative Trait Loci Colocalize with Chronic Obstructive Pulmonary Disease Genome-Wide Association Loci, Am J Respir Crit Care Med. 197 (2018) 1275–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Liu D, Liao C, Wolgemuth DJ, A role for cyclin A1 in the activation of MPF and G2-M transition during meiosis of male germ cells in mice, Dev Biol. 224 (2000) 388–400. [DOI] [PubMed] [Google Scholar]
- [45].Yang R, Muller C, Huynh V, et al. , Functions of cyclin A1 in the cell cycle and its interactions with transcription factor E2F-1 and the Rb family of proteins, Mol Cell Biol. 19 (1999) 2400–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wang D, Shi W, Tang Y, et al. , Prefoldin 1 promotes EMT and lung cancer progression by suppressing cyclin A expression, Oncogene. 36 (2017) 885–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Klajic J, Busato F, Edvardsen H, et al. , DNA methylation status of key cell-cycle regulators such as CDKNA2/p16 and CCNA1 correlates with treatment response to doxorubicin and 5-fluorouracil in locally advanced breast tumors, Clin Cancer Res. 20 (2014) 6357–6366. [DOI] [PubMed] [Google Scholar]
- [48].Shames DS, Girard L, Gao B, et al. , A genome-wide screen for promoter methylation in lung cancer identifies novel methylation markers for multiple malignancies, PLoS Med. 3 (2006) e486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Weiss D, Basel T, Sachse F, et al. , Promoter methylation of cyclin A1 is associated with human papillomavirus 16 induced head and neck squamous cell carcinoma independently of p53 mutation, Mol Carcinog. 50 (2011) 680–688. [DOI] [PubMed] [Google Scholar]
- [50].Jowaed A, Schmitt I, Kaut O, et al. , Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’ brains, J Neurosci. 30 (2010) 6355–6359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Nalls MA, Pankratz N, Lill CM, et al. , Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease, Nat Genet. 46 (2014) 989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Yoshino Y, Mori T, Yoshida T, et al. , Elevated mRNA Expression and Low Methylation of SNCA in Japanese Alzheimer’s Disease Subjects, J Alzheimers Dis. 54 (2016) 1349–1357. [DOI] [PubMed] [Google Scholar]
- [53].Yan Y, Xu Z, Hu X, et al. , SNCA Is a Functionally Low-Expressed Gene in Lung Adenocarcinoma, Genes (Basel). 9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zhao W, Mo Y, Wang S, et al. , Quantitation of DNA methylation in Epstein-Barr virus-associated nasopharyngeal carcinoma by bisulfite amplicon sequencing, BMC Cancer. 17 (2017) 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.