

Abstract
Cystic fibrosis (CF) is a monogenic autosomal recessive disorder. The clinical manifestations of the disease are caused by ~ 2,000 mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) protein. It is unlikely that any one approach will be efficient in correcting all defects. The recent approvals of ivacaftor, lumacaftor/ivacaftor and elexacaftor/tezacaftor/ivacaftor represent the genesis of a new era of precision combination medicine for the CF patient population. In this review, we discuss targeted translational readthrough approaches as mono and combination therapies for CFTR nonsense mutations. We examine the current status of efficacy of translational readthrough/nonsense suppression therapies and their limitations, including non-native amino acid incorporation at PTCs and nonsense-mediated mRNA decay (NMD), along with approaches to tackle these limitations. We further elaborate on combining various therapies such as readthrough agents, NMD inhibitors, and corrector/potentiators to improve the efficacy and safety of suppression therapy. These mutation specific strategies that are directed towards the basic CF defects should positively impact CF patients bearing nonsense mutations.
Keywords: CFTR, nonsense mutations, translational readthrough, combination therapy
Graphical Abstract
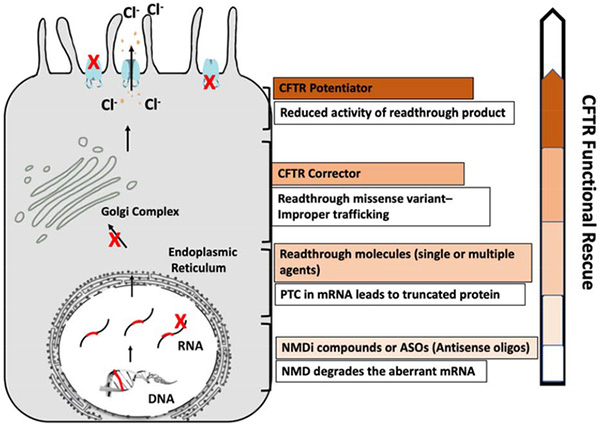
Targeted interventions that can be combined to rescue maximum CFTR function
Cystic Fibrosis
Cystic fibrosis (CF) is an autosomal recessive genetic disease that is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene (1–3). This gene encodes a cAMP-regulated anion channel that mediates Cl− and HCO−3 ion transport in the epithelia of airways, pancreatic ducts, and other tissues (4, 5). The CFTR protein is a member of the ATP-binding cassette class C (ABC-C) family. Consistent with other proteins in this class, it contains two membrane-spanning domains (MSD1 and MSD2) and two nucleotide-binding domains (NBD1 and NBD2) that require ATP binding for channel gating. In addition, CFTR exhibits a unique feature not found in other ABC family members, an intracellular regulatory domain (R domain) that normally requires phosphorylation for channel opening (6, 7). Improper CFTR channel function leads to dysfunction of epithelial tissues in the lung, gastrointestinal tract, pancreas, and reproductive system.
More than 2,000 CFTR gene variants have been identified, many of which are confirmed to be disease-causing (8). Due to the large number of known CFTR mutations, they have been categorized into six different classes based on their effect on CFTR protein expression and function: class I (no protein production), class II (defective protein processing and trafficking), class III (abnormal channel gating), class IV (reduced ion conductance), class V (reduced protein expression through aberrant splicing), and class VI (faulty recycling) (1, 9, 10). While CFTR mutation classification is a useful framework for devising therapeutic approaches to repair molecular defects in CFTR (see Figure 5 in Rowe et al (1)) it is imperfect since many mutations exhibit more than one feature (10) and some molecular approaches extend beyond one specific therapeutic class. The quantity and the function of these disease-causing CFTR variants are the primary determinants of the clinical severity in individual CF patients.
A number of therapeutic compounds called ‘CFTR modulators’ have been identified that restore the expression or function of CFTR (3, 11). Among CFTR modulators, CFTR ‘correctors’ fundamentally target the cellular processing of mutant CFTR protein to ensure its proper folding, maturation, and trafficking to the cellular membrane (12–15). CFTR ‘potentiators’ enhance CFTR channel activity at the cell surface by improving open channel probability, thereby augmenting channel gating (3, 16–18). CFTR modulators are often used in combination to address more than one CFTR defect in a given mutation; the seminal example of this is co-administration of correctors and potentiators to restore activity to F508del CFTR, which exhibits both folding and trafficking abnormalities as well as defective channel gating. Together, these CFTR modulators have demonstrated eminent clinical success in CF patients with mutations that maintain residual CFTR function (class II, III, IV, and others) (19, 20). It is estimated that correctors and potentiators are likely to provide a therapeutic benefit in up to 90% of CF patients, as F508del, gating, and conductance mutations have all exhibited clinical efficacy (21–23).
While current modulator compounds represent a viable treatment option for many CF patients, no pharmacological options are available to individuals who carry nonsense mutations, a subtype of class I mutations that abrogate CFTR protein expression. Nonsense mutations represent single nucleotide point mutations that generate an in-frame premature termination codon (PTC) in the open reading frame (ORF) of CFTR mRNA, causing cessation of translation and preventing the synthesis of full-length protein (24). Additionally, a PTC often triggers nonsense-mediated decay (NMD), a conserved eukaryotic mRNA surveillance pathway that degrades aberrant mRNAs containing a PTC, reducing the pool of PTC-containing transcripts for translation and further limiting the synthesis of truncated or full-length CFTR protein (25–32). These PTC-mediated mechanisms lead to a complete absence of CFTR protein function and thus, with few exceptions, do not respond to the current CFTR modulator compounds that require at least partial CFTR protein expression. Therefore, despite tremendous efforts by the CF research community, there remains an unmet need for the CF population that bears nonsense mutations. In this review, we will discuss the potential of using small-molecule pharmacological agents to suppress PTC mutations in CF patients who carry nonsense mutations.
Potential Treatments for Class I CFTR Mutations
Class I mutations completely abrogate CFTR function and affect more than 11% of the CF population (33). The majority of these Class I lesions are comprised of nonsense mutations (e.g. Y122X, R553X, G542X, and W1282X) (www.cff.org), with the remaining mutations represented by major gene disruptions such as chromosomal deletions, insertions, and other frameshift mutations, including canonical splice site mutations (which overlap with Class V variants) (33). Since 12% of all known disease-causing gene lesions are nonsense mutations according to the Human Gene Mutation Database (34, 35), the prevalence of nonsense mutations in CF patients is not unusual.
Similar to other genetic diseases (24, 36), nonsense mutations are often associated with the most severe form of CF due to a complete absence of CFTR protein. This loss of CFTR can be attributed to premature translation termination as well as nonsense-mediated mRNA decay (NMD), which reduces mRNA stability (37). To address protein deficiencies attributable to in-frame premature termination codons (PTCs) arising from nonsense mutations, a number of different approaches are being explored to suppress translation termination specifically at PTCs, with the aim of restoring sufficient protein expression and function to alleviate genetic diseases. One approach to restore expression of CFTR (or deficient proteins in the setting of other genetic diseases) is translational readthrough, where termination at PTCs is suppressed by the insertion of aminoacyl-tRNAs at a PTC by incorporation into the ribosomal A-site. At a low frequency, near-cognate tRNAs can outcompete the translation termination factor, eRF1 (eukaryotic release factor 1) for binding to A-site bound PTCs (Figure 1). This leads to the incorporation of an amino acid into the polypeptide chain at the site of the PTC, allowing the translation elongation to continue in the correct reading frame until the next in-frame stop codon is encountered (38–40). This readthrough mechanism is being exploited for therapeutic purposes and has shown some success in preclinical studies for CFTR nonsense mutations (38, 41, 42). There are other approaches that target PTCs, including suppressor tRNAs, RNA-directed pseudouridylation, NMD inhibition and gene editing, but these therapeutic strategies are beyond the scope of this review (34, 43, 44).
Figure 1. Translation termination versus readthrough at PTCs:
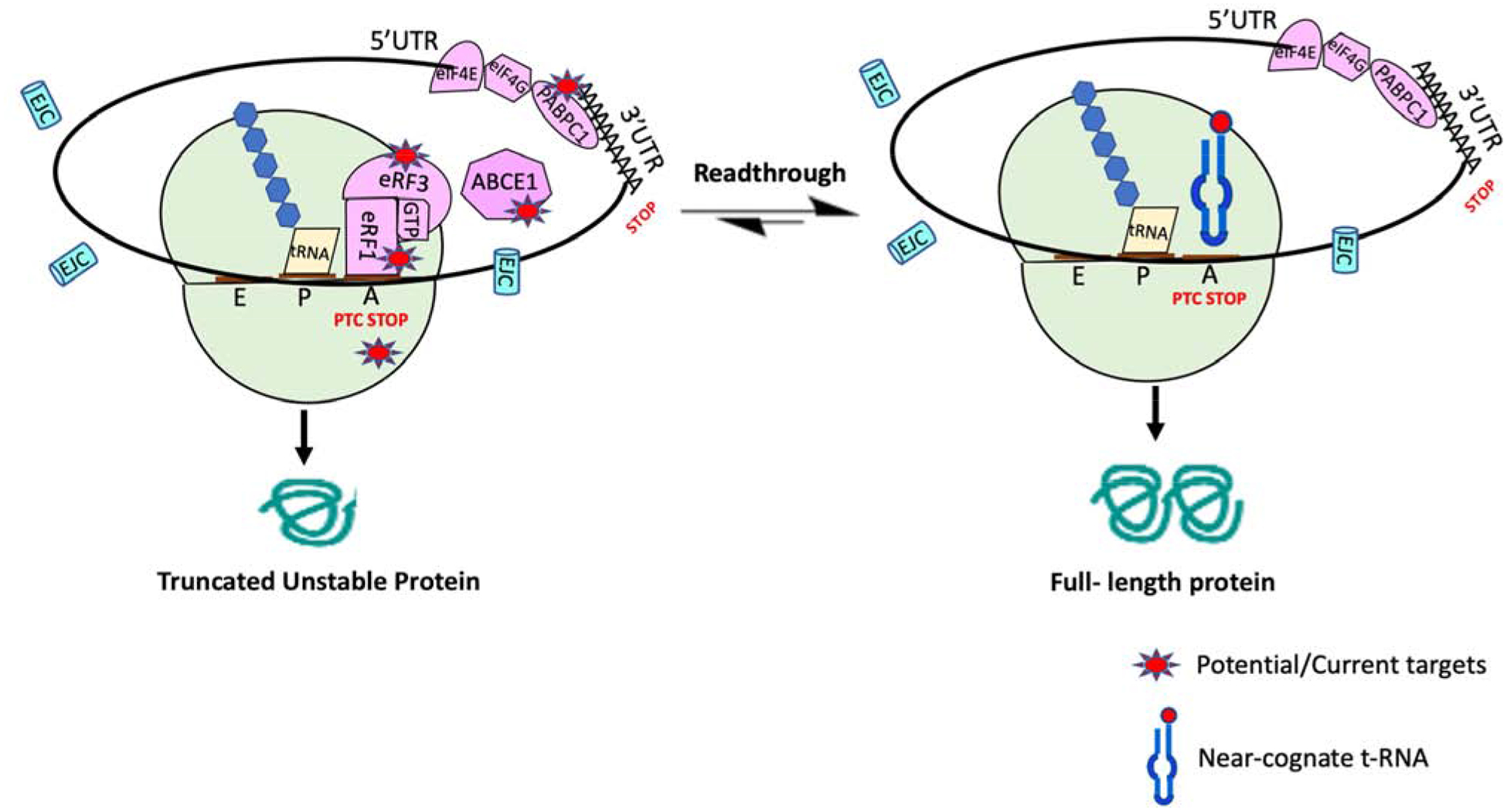
eRF1 and eRF3 bind to a stop codon when it enters the ribosomal A site. PABPC1 binds to eRF3 and promotes efficiency termination. ABCE1 assists in polypeptide release and mediates ribosomal recycling. When a PTC enters the A site, a truncated polypeptide is generated. At a low frequency, near-cognate tRNAs compete with the eRF1/eRF3 complex for binding to A-site bound PTCs, allowing translation elongation to continue in the original ribosomal reading frame to produce a full-length protein. Several therapeutic approaches are attempting to skew this competition more toward elongation in order to generate more full-length protein.
Translation Termination Overview
Translation termination is a highly efficient process, with >99% efficiency at natural termination codons (NTCs). Translation termination is triggered by the entry of a stop codon (UAA, UAG, or UGA) into the ribosomal acceptor site (A-site). Unlike the decoding of sense codons by tRNAs, stop codons are decoded by a complex of proteins called the termination complex. The termination complex is minimally composed of the eurkaryotic release factors 1 (eRF1) and 3 (eRF3). eRF1 binds directly to stop codons located in the ribosomal A site, akin to tRNA binding of conventional codons. The overall three-dimensional structure of eRF1 is similar to that of a tRNA (45). eRF1 bears several conserved elements, including the Asn–Ile–Lys–Ser (NIKS), Gly–Thr–Ser (GTS), and YxCxxxF (YCF) motifs that participate in stop codon recognition with high specificity (46, 47). eRF3, a GTPase, binds to eRF1 to facilitate stop codon recognition (48). eRF1 also contains a unique Gly–Gly–Gln (GGQ) motif that extends into the peptidyl transferase center and promotes liberation of the polypeptide upon GTP hydrolysis by eRF3 (49–53). These structural motifs represent potential targets for structural-based drug design.
This mechanism of stop codon recognition is similar at both natural and premature stop codons. However, the frequency of readthrough at PTCs is approximately 10-fold greater (0.01–1%) than the readthrough rate at NTCs (0.001–0.1%) (24, 54–56). Various factors likely mediate this difference in termination efficiency at PTCs. For instance, the interactions between the cytoplasmic poly(A) binding protein (PABPC1) and eRF3, which facilitates efficient translation termination, are likely to be less efficient at a PTC due to its increased distance from the 3′-UTR (3’-untranslated region) (40, 57, 58). Furthermore, ABCE1/RLI1, an essential member of the ABC family of proteins, plays an important role in translation initiation, termination and ribosomal recycling. ABCE1/RLI1 stimulates eRF1-mediated peptidyl hydrolysis and the dissociation of the post-translation complex into free 60S subunits, mRNA-, and tRNA-bound 40S subunits for use in subsequent rounds or translation (59). The efficiency of ABCE1/RLI1 binding to eRF1 at PTCs may also be subverted due to the increased distance of PTCs from the 3’ UTR, altering otherwise favorable stoichiometry for this process to occur (60). This distinction in translation termination efficiency at normal termination codons as compared to PTCs lays the foundation for pharmacologic approaches for diseases caused by PTC mutations.
Potential Therapeutic Targets for Readthrough
Recent studies that have examined the molecular interactions occurring during translation termination have identified subtle changes in the spatial orientation of various factors associated with the termination complex (61). While eRF1 is the main catalytic factor for translation termination, other factors have also been found to play important roles in mediating termination efficiency (Figure 1). For example, the cytoplasmic poly(A)-binding protein (PABPC1) binds to the poly(A)-tail of mRNAs and interacts with the translation initiation factors eIF4E and eIF4F to circularize the mRNA during translation, which increases mRNA stability and enhances translation rates (62–64). However, it has been shown recently that PABPC1 enhances translation termination efficiency through its interaction with the termination complex (65–67), specifically via eRF3. Thus interfering with these interactions could represent an approach to augment translational readthough, particularly if it could be designed in such a way as to preferentially block blinding at PTCs as compared to NTCs.
As another example, several translation initiation factors recently have been shown to be important contributors to the translation termination process, elucidating a stringent association between various phases of translation (68–70). Deletion of HCR1 (a subunit of eIF3 that has a principle role in ribosomal recycling) increases readthrough activity by promoting GDP-eRF3 release from the ribosomes (70, 71). In addition, eIF3j is known to bind at the ribosomal decoding center (72), which serves a proofreading function during both elongation and termination (73, 74).
Studies in Saccharomyces cerevisiae have identified additional factors that play significant roles in translation termination, and may also be relevant to PTC readthrough in mammalian cells (75). The ATP-dependent DEAD-box RNA helicase Dbp5 (homologous to DDX19 in mammals) is generally associated with the nuclear pore complex and is involved in transfer of mature mRNA/protein complexes into the cytoplasm. During the formation of the translation termination complex, the mRNA export factor Gle1p, in association with inositol hexakisphosphate (IP6), directs Dbp5 to properly position eRF1 at stop codons in ribosomal A site. Dbp5 also recruits eRF3 to the termination complex (69, 75–77). Discovery of these and other novel aspects of the translation termination machinery suggests the potential for exploring these pathways to identify new ways to stimulate readthrough at PTCs.
Importance of Local PTC Sequence Context for Readthrough Efficiency
Biochemical and genetic studies, and more recently, ribosomal profiling, have shown that the stop codon and the mRNA sequence context surrounding the stop codon significantly contribute to translation termination efficiency (55, 78–80). Differences in readthrough susceptibility have been found based on the identity of the stop codon. In general, UGA is most susceptible to readthrough followed by UAG and UAA. The nucleotide directly following a stop codon (the +4 nucleotide) also has a strong effect on readthrough susceptibility, where the presence of a cytidine downstream of a stop codon generally correlates with more permissive readthrough (55, 56).
Extended mRNA context may also affect readthrough efficiency. At least 6 nucleotides of local upstream and downstream of a stop codon has been shown to affect the efficiency of translation termination (81–84). Cridge et. al recently showed that the +5, +6 and +8 nucleotides downstream of the UGA stop codon has a strong influence on translation efficiency and fidelity (78). In addition, the presence of two adenines at two positions 5’ of stop codon are associated with a high frequency of readthrough, irrespective of the stop codon or downstream mRNA sequences (85). More distant sequence context may also affect readthrough efficiency. For example, approximately 30 nucleotides are present within the ribosomal mRNA channel, which could affect the efficiency of translation termination or the recruitment of near-cognate aminoacyl-tRNAs (78).
The sequence context has been shown to affect the clinical severity of genetic disorders by affecting the frequency of spontaneous readthrough of a PTC and therefore, the amount of residual protein function. In a patient with junctional epidermolysis bullosa due to nonsense mutations in the LAMA3 gene (R943X/R1159X) (41, 86), the R943X allele encoded a hypothetical consensus sequence (AGU-UGA-CUA) that is thought to be amenable to readthrough. In addition, the LAMA3 mRNA levels were relatively preserved in this patient. This resulted in a sufficient amount of spontaneous readthrough to produce full length laminin α3 protein, ameliorating disease severity (86). This important finding shows that the PTC context may help predict phenotypic severity, and is likely to be important in selecting a readthrough treatment for specific PTC mutations to maximize the potential for efficacy, especially if the competition between readthrough and termination is only modestly affected by treatment.
Readthrough as a Therapeutic Approach for CF and Duchenne’s muscular dystrophy
Nearly two decades ago, suppressing translation termination at nonsense mutations was first described as a potential therapeutic approach for genetic diseases caused by nonsense mutations, with CF being the first disease model in which this approach was examined (87). Multiple studies have shown that some aminoglycoside antibiotics promote translational readthrough in cell (56, 57, 87) and animal models (88–90). By binding to the ribosomal decoding site, aminoglycosides can inhibit ribosomal proofreading and enhance the frequency of near-cognate tRNA accommodation at A-site bound PTCs (73, 74). This allows rescue of full-length protein synthesis, with the potential of restoring at least partial function depending on the sensitivity of the protein product to the amino acid inserted at the PTC (73, 90).
In a subset of Duchenne muscular dystrophy (DMD) patients carrying nonsense mutations, the aminoglycoside, gentamicin, increased dystrophin expression and reduced serum creatinine kinase levels (91, 92). Similarly, pilot clinical trials in which gentamicin was administered to CF patients showed restoration of CFTR protein and function in nasal epithelium in approximately half of the patients (93, 94). However, brief proof-of-concept studies in CF patients (93–95) found that neither amikacin nor gentamicin restored enough functional CFTR protein to impart long-term clinical benefit (96). These studies indicate that aminoglycosides can restore full-length functional protein via readthrough of PTCs, but only in a fraction of patients. In addition, administration of aminoglycosides for lifelong genetic diseases is not feasible due to their association with off-target sites such as mitochondrial ribosomes (56, 90, 97) and lysosomal membranes (98, 99), leading to the renal and ototoxicity (99, 100).
To improve the efficiency of aminoglycosides to promote readthrough while also reducing their toxicity, a medicinal chemistry approach was used to synthesize aminoglycoside derivatives that bind more tightly to cytoplasmic ribosomes and less tightly to mitochondrial ribosomes (101, 102). ELX-02 (formerly NB124), was developed by Eloxx Pharmaceuticals (Herzliya, Israel) to bring this approach to fruition (chemical structures of gentamicin, amikacin and ELX-02 in Figure 2). ELX-02 is less toxic than conventional aminoglycosides and more efficiently promotes readthrough than gentamicin in mammalian cells (103, 104). For example, in human bronchial epithelial cells, gentamicin did not restore CFTR function, while NB-124 and similar derivatives restored ~7% of wild-type CFTR activity (103, 105). ELX-02 established a better safety and pharmacokinetics profile than conventional aminoglycosides as observed in two Phase 1a, randomized, double-blind placebo- controlled studies in healthy volunteers (106). ELX-02 is currently advancing to Phase 2 clinical trials for both CF and cystinosis patients who carry the G542X nonsense mutation (101, 106–109). Given gentamicin improved sweat chloride in patients with particularly susceptible nonsense codons (110), and the strong association of sweat chloride response and clinical outcome in other mutations of CF (111), this represents a critical near-term effort for people with CF caused by nonsense mutations. ELX-02 as part of multi-modal CFTR therapy could also be a consideration for future studies.
Figure 2.
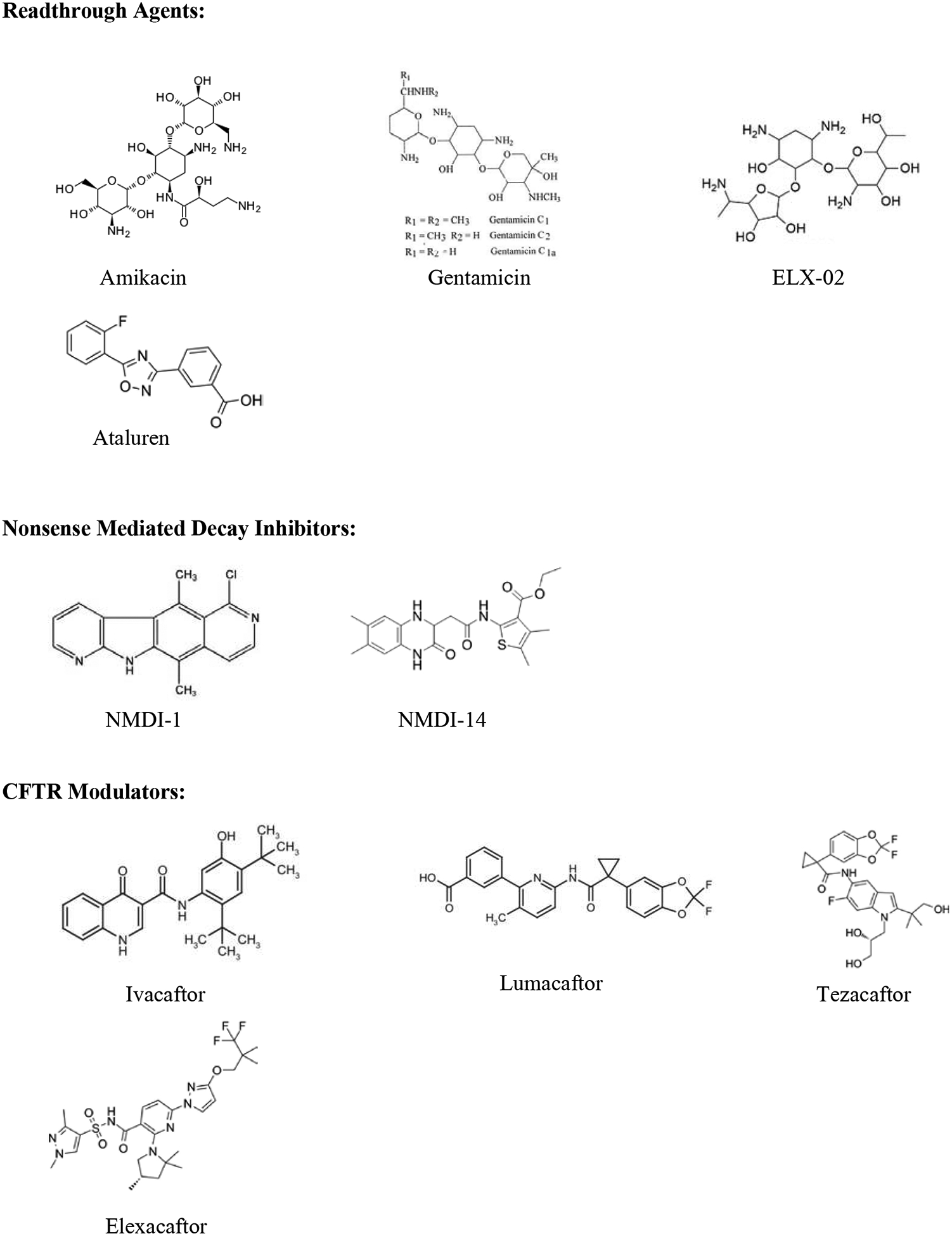
Chemical structures of translational readthrough agents, potential nonsense mediated decay inhibitors, and CFTR modulators.
In addition to the structural modification of aminoglycosides, co-administration of anionic poly amino acids and antioxidants have been shown to reduce aminoglycoside toxicity while maintaining their ability to promote readthrough (112–114). For example, the co-administration of poly-L- aspartic acid with gentamicin was shown to significantly increase the duration of CFTR functional rescue via readthrough compared to gentamicin treatment only (115). Other treatments that reduce the toxicity of aminoglycosides are also in development and could be employed for chronic therapy (116–118).
In an effort to identify more potent, safe, and orally bioavailable readthrough agents for CF and other genetic diseases, PTC Therapeutics, Inc. performed a high throughput screen (HTS) of ~800,000 small molecules using firefly luciferase-based readthrough reporters. From this screen, a hit was identified that was developed into the lead compound, ataluren (also known as PTC124 or translarna, Figure 2) (119). Preclinical studies demonstrated an acceptable safety profile, good oral bioavailability, and an absence of antibacterial activity, distinguishing the agent from aminoglycosides (120, 121). Ataluren was shown to restore dystrophin protein expression in human primary muscle cells and in the mdx mouse model of Duchenne muscular dystrophy (119). Further, ataluren has also been found to restore the CFTR expression and function in a humanized CFTR-G542X transgenic mouse model (122). However, recent randomized, double-blind, placebo-controlled Phase 3 studies showed that ataluren was unable to restore enough CFTR function in CF patients to improve lung function. A confounding factor in these trials is that the readthrough activity of ataluren is significantly inhibited by tobramycin, an aminoglycoside often administered to CF patients for acute and chronic use (39, 123). However, subsequent evaluation of ataluren efficiency in CF patients without tobramycin administration did not lead to significant improvements in lung function or other clinical endpoints, terminating further development of ataluren as a therapeutic for CF (123). The approach to optimize the compound during preclinical development may also have been impacted by stabilization of the luciferase reporter (124). A likely conclusion from these studies is that we are still lacking an effective readthrough therapy for the CF population bearing nonsense mutations and that more effective and safer readthrough agents are needed. Primary human bronchial epithelial (HBE) cells or organoids served as preclinical models to determine the efficacy of potentiators and correctors of CFTR modulator therapies, which advanced in parallel with the development of ataluren. These novel CF models provided an opportunity to retrospectively evaluate ataluren in novel CF model systems. For example, in rectal organoids derived from CF patients harboring nonsense mutations, ataluren was unable to restore CFTR function (125) and our group generated similar unpublished results in primary HBE and HNE cells from CF donors. Future studies may benefit from the use of CF primary cell models and organoids as a benchmark to measure the efficacy of readthrough agents to restore CFTR function to therapeutic levels.
Hurdles to Implementing Readthrough Therapy as a Treatment for CF
Multiple challenges stand in the way of achieving efficient readthrough for treating CF in patients who carry a nonsense mutation. For example, the abundance of mRNA encoding nonsense mutations available for translation and subsequent readthrough is often reduced by the NMD pathway (Figure 3). For the fraction of PTC-containing mRNAs that remain to be translated, the local mRNA context surrounding a PTC influences the pattern of amino acid incorporation at a PTC, affecting the protein stability, folding and/or function of the readthrough product (39, 80). The amino acids incorporated during G418-mediated readthrough of the CFTR G542X mutation (a TGA stop codon) consisted of cysteine, tryptophan, or arginine (39). In contrast, readthrough of the CFTR W1282X mutation (also a TGA) with G418 led to the incorporation of leucine, cysteine or tryptophan (80). Thus, only a fraction of the full-length protein generated by readthrough contained the native amino acid, with the other proteins generated by readthrough contained a non-native amino. Thus, the local sequence surrounding a PTC can alter the amino acids incorporated at a PTC during readthrough. Furthermore, the different variant proteins resulting from readthrough showed differences in CFTR channel maturation, stability and function (80), depending on the amino acid inserted and its physiochemical properties as compared to the native codon (see the graphical abstract Figure). These barriers to readthrough therapy have led to investigations to identify novel approaches to further enhance readthrough efficacy, including the combination of multiple treatments to overcome these challenges, as described below.
Figure 3. Schematic illustration of the aberrant translation termination resulting in the activation of nonsense-mediated mRNA decay (NMD).
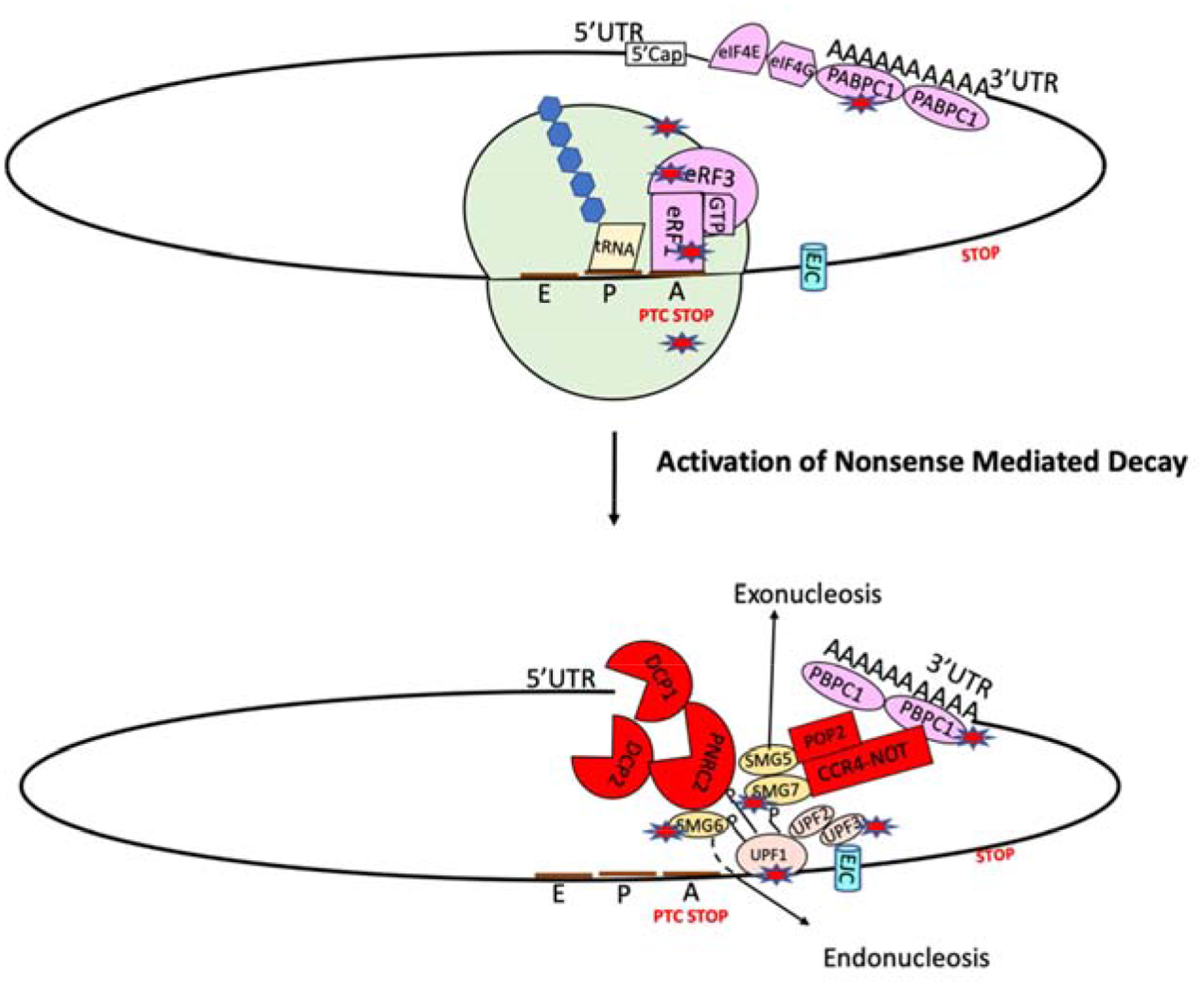
When a ribosome encounters a PTC, the termination factors eRF1 and eRF3 are recruited to the PTC. UPF1 then binds to the termination complex and becomes phosphorylated, allowing an interaction with UPF2 and/or UPF3. This association recruits decay factors, including the SMG6 endonuclease and the SMG5–7 heterodimer, which subsequently recruit the CCR4/NOT complex to promote decay of the mRNA. Star represents the potential therapeutic target.
NMD Inhibition to Augment mRNA Substrate for Readthrough
Decreased abundance of PTC-containing CFTR transcripts due to their sensitivity to NMD limits the efficacy of nonsense suppression therapy (126, 127). Approaches for inhibiting NMD to enhance the availability of mRNA substrates for readthrough therapy include using pharmacological inhibitors (e.g. NMDI-1, NMDI-14, (Figure 2) and SMG1i) (128, 129), small interfering RNAs (siRNA), and antisense oligonucleotides (ASOs) that target NMD factors, attenuating NMD activity (43, 129–131). The potential interconnection between NMD attenuation and readthrough has been shown in vitro and in vivo, including human subjects (24, 89, 127, 129). Patients with higher transcript levels have repeatedly been found to respond better to readthrough therapy because of the availability of more substrate available for translation. This association was also seen in CF patients bearing the W1282X mutation, where improvements in sweat chloride and nasal potential difference measurement were observed following gentamicin treatment in patients with higher levels of CFTR mRNA abundance due to less efficient NMD (127). Similar associations between mRNA levels and readthrough efficacy were also observed in the initial trials of ataluren (132, 133).
Pharmacologic approaches have been attempted to recapitulate the association between increased mRNA abundance and improved readthrough efficacy. Co-administering gentamicin with the NMD inhibitor NMDI-1 generated increased efficacy in an Mucopolysaccharidosis I-Hurler (MPS I-H) mouse model by enhancing the restoration of α-L-iduronidase activity, leading to greater reduction of glycosaminoglycan storage in mouse tissues (129). ASOs that specifically inhibited the Upf3b NMD factor, combined with G418, also increased efficacy in restoring full-length functional hFIX protein in hemophiliac mice bearing the R29X nonsense mutation (130). Furthermore, inhibiting NMD using an ASO directed against SMG1 showed significant rescue of CFTR protein and function in an immortalized primary human bronchial epithelial cell line modified by CRISPR/Cas 9 methodology to express the CFTR W1282X mutation (16HBEge cells) (134, 135). In total, these approaches indicate that inhibition of NMD may improve the therapeutic benefits of readthrough therapy. However, caution must be taken, in addition to degrading PTC-bearing mRNAs, NMD regulates the levels of many physiological transcripts (136–138), plays an important role in embryonic development (137, 139), and appears to influence multiple cellular pathways (140, 141). Thus, additional work is needed to determine whether NMD inhibition can be safely implemented as a therapeutic approach. Even if suitable at low levels in adults, approaching the pathway in children may raise particular safety issues.
CFTR Modulators to Augment Readthrough Therapy for CF
CFTR modulators, which have received US Food and Drug Administration (FDA) approval, primarily consist of two classes of compounds: 1) CFTR correctors, which facilitate protein processing and trafficking, and 2) potentiators, which activate the CFTR channel (142–144). These agents have been approved as a combination therapy to treat CF patients with responsive mutations (2). The CFTR potentiator ivacaftor (formerly VX-770, Figure 2) is effective with multiple CFTR missense mutations where adequate levels of CFTR is present at the cell surface (145). In an single patient study, an individual homozygous for the W1282X mutation showed significant benefits with ivacaftor treatment, where lung function was stabilized and improvements in pulmonary exacerbation frequency, BMI, and insulin requirement were observed (146). The beneficial effects of ivacaftor are largely due to the presence of sufficient levels of truncated W1282X CFTR protein being present at cell surface (146), which can be activated even in the truncated state (147). Haggie et.al has also identified an novel series of small molecules, correctors and potentiators, with potentially greater efficacy for CFTR W1282X mutation (148). These may also be applicable to other terminal (i.e. 3’) mutations when sufficient CFTR mRNA is present (149, 150). However, CFTR correctors and potentiators, alone or in combination, have not exhibited efficacy for CFTR nonsense mutations where cell surface expression of truncated protein is absent, such as G542X, or NMD eliminated protein expression (151).
Beyond the efficacy for CFTR mutations, when combined with readthrough agents, CFTR modulators (correctors and potentiators) have shown improvements in CFTR cell surface expression and function in various in vitro studies (80, 103, 146) by acting on the full-length CFTR protein restored via readthrough; this may be particularly important for missense CFTRs generated upon readthrough (152). We have recently shown that adding CFTR corrector and potentiator treatments doubled the amount of CFTR activity rescued after PTC suppression of the CFTR G542X and W1282X mutations compared to readthrough treatment alone (80). Altogether, these studies suggest the importance of CFTR modulator treatments in combination with PTC suppression therapies is likely applicable for multiple nonsense mutations to augment the function of the readthrough product.
Beyond the CFTR modulators that are already clinically available, alternative CFTR modulators that are specifically designed for PTC readthrough products may have particular advantages, whether as single agents or as part of combination treatments. ASP-11, a novel potentiator for the CFTR W1282X mutation, significantly enhanced CFTR channel activity in combination with ivacaftor in cells derived from a G542X/W1282X patient (153). Additional CFTR modulators are currently being developed by Galapagos/Abbvie, Flately Discovery, and Vertex Pharmaceuticals that may also increase the functionality of proteins generated by readthrough (154). As a prime example, Trikafta, a combination of three CFTR modulators (elexacaftor and tezacaftor are CFTR correctors; ivacaftor, a CFTR potentiator, Figure 2) has been shown to be beneficial in 90% of CF population who carry at least one F508del allele (21–23). However, its efficacy for the CF population bearing a nonsense mutation remains to be determined, and those with a nonsense mutation in trans with an F508del allele did not have an appreciably different response to elexacaftor/tezacaftor/ivacaftor (22).
Dual Readthrough Agent Combination Therapy
Multi-agent corrector therapy has been used to augment F508del processing and trafficking (15, 155, 156). A similar approach could be used to augment readthrough efficacy (157) by combining readthrough agents with distinct mechanisms of action. There are multiple factors involved in translation termination, representing potentially distinct therapeutic targets (Figure 1). Studies in Saccharomyces cerevisiae has shown that translation termination efficiency is dependent on the levels of eRF1 and eRF3 (158). Overexpression of eRF1 increases translation termination efficiency (159); conversely, reduced rates of eRF3 GTP hydrolysis diminishes translation termination efficiency (48). These studies lay the foundation for exploring potential small molecule therapies that can alter the levels of eRF1 or eRF3 to prolong ribosomal pausing at PTCs, providing sufficient time for near-cognate tRNAs to become accommodated at a PTC to promote readthrough. As explained earlier, other factors such as ABCE1 (59), PABPs (65), eIF3 (71) and eIF3j (72) could be exploited for developing novel nonsense suppression therapeutic targets.
Alternatively, phthalimide derivatives are a class of small molecules that have recently been shown to act as enhancers for aminoglycoside-mediated readthrough. The use of enhancer molecules may enable the administration of aminoglycosides at much lower doses in order to minimize their toxicity while also enhancing their readthrough ability for nonsense mutations (160, 161). However, the discovery of how these small molecules function is needed to understand their therapeutic efficacy and to further optimize them. This further raises the hope for combination therapies, where readthrough agents that work by different methods of action can be used to produce the level of translational readthrough that is needed to benefit the CF population bearing nonsense mutations.
Summary
Lessons learned from clinical experiences, significant heterogeneity in response based on characteristics of the nonsense allele, and an improved understanding of challenges of nonsense suppression therapy at the mechanistic level has opened a new horizon for combining therapies to treat CF patients harboring nonsense mutations. The therapeutic paradigm for developing CF modulator combination therapies and the magnificent success of drugs that target mutation-specific molecular defects of CFTR (21, 162, 163) raises the hope for combination treatment therapies that treat CFTR nonsense mutations. Combinations of small molecules that address CFTR protein synthesis, stability, folding and transportation may help enhance the therapeutic levels of CFTR function restored by readthrough. The high therapeutic threshold (estimated as high as 30–35% of normal CFTR function) needed for correction makes it challenging to attain adequate efficacy by nonsense suppression monotherapy, but this will need to be balanced with the risk of toxicity and drug-drug interactions associated with combination therapies (105). Beyond additional treatment options, future research should focus on early identification and selection of appropriate cell or animal models that best recapitulate the disease phenotype to identify the appropriate combination approaches.
Highlights.
Translational readthrough therapy for CFTR nonsense mutations
Translation termination factors are crucial targets for nonsense suppression therapy
NMD inhibition approaches may be effective to enhance the substrates for nonsense suppression therapy
Combine various approaches to enhance the efficacy for nonsense suppression therapy
Acknowledgement
This work was supported by the National Institutes of Health [R35 HL135816, P30 DK072482]; the Cystic Fibrosis Foundation, Bethesda [ROWE19R0]; and Emily’s Entourage grant.
S.M.R reports grants from PTC Therapeutics, grants, personal fees and non-financial support from Vertex Pharmaceuticals Incorporated, grants, personal fees and non-financial support from Novartis, grants from Eloxx, grants from Galapagos/Abbvie, during the conduct of the study; grants from Bayer, grants from Forest Research Institute, grants from AstraZeneca, grants from N30/Nivalis, grants from Galapagos/AbbVie, grants from Proteostasis, grants and personal fees from Celtaxsys, personal fees from Bayer, personal fees from Renovion, grants and personal fees from Synden/Synspira, personal fees from Genentech, personal fees and non-financial support from Boeringher J.S and K.K has no financial interests/personal relationships that could influence the work reported in this paper and has no potential competing interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med 2005; 352: 1992–2001. [DOI] [PubMed] [Google Scholar]
- 2.Quon BS, Rowe SM. New and emerging targeted therapies for cystic fibrosis. Bmj 2016; 352: i859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rowe SM, Verkman AS. Cystic fibrosis transmembrane regulator correctors and potentiators. Cold Spring Harbor perspectives in medicine 2013; 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saint-Criq V, Gray MA. Role of CFTR in epithelial physiology. Cell Mol Life Sci 2017; 74: 93–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mall MA, Hartl D. CFTR: cystic fibrosis and beyond. Eur Respir J 2014; 44: 1042–1054. [DOI] [PubMed] [Google Scholar]
- 6.Jordan IK, Kota KC, Cui G, Thompson CH, McCarty NA. Evolutionary and functional divergence between the cystic fibrosis transmembrane conductance regulator and related ATP-binding cassette transporters. Proc Natl Acad Sci U S A 2008; 105: 18865–18870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gadsby DC, Nairn AC. Regulation of CFTR channel gating. Trends Biochem Sci 1994; 19: 513–518. [DOI] [PubMed] [Google Scholar]
- 8.Ferec C, Cutting GR. Assessing the Disease-Liability of Mutations in CFTR. Cold Spring Harbor perspectives in medicine 2012; 2: a009480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marson FAL, Bertuzzo CS, Ribeiro JD. Classification of CFTR mutation classes. The Lancet Respiratory medicine 2016; 4: e37–e38. [DOI] [PubMed] [Google Scholar]
- 10.Veit G, Avramescu RG, Chiang AN, Houck SA, Cai Z, Peters KW, Hong JS, Pollard HB, Guggino WB, Balch WE, Skach WR, Cutting GR, Frizzell RA, Sheppard DN, Cyr DM, Sorscher EJ, Brodsky JL, Lukacs GL. From CFTR biology toward combinatorial pharmacotherapy: expanded classification of cystic fibrosis mutations. Mol Biol Cell 2016; 27: 424–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kym PR, Wang X, Pizzonero M, Van der Plas SE. Recent Progress in the Discovery and Development of Small-Molecule Modulators of CFTR. Prog Med Chem 2018; 57: 235–276. [DOI] [PubMed] [Google Scholar]
- 12.He L, Aleksandrov LA, Cui L, Jensen TJ, Nesbitt KL, Riordan JR. Restoration of domain folding and interdomain assembly by second-site suppressors of the DeltaF508 mutation in CFTR. FASEB J 2010; 24: 3103–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thibodeau PH, Richardson JM 3rd, Wang W, Millen L, Watson J, Mendoza JL, Du K, Fischman S, Senderowitz H, Lukacs GL, Kirk K, Thomas PJ. The cystic fibrosis-causing mutation deltaF508 affects multiple steps in cystic fibrosis transmembrane conductance regulator biogenesis. J Biol Chem 2010; 285: 35825–35835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mu TW, Ong DS, Wang YJ, Balch WE, Yates JR 3rd, Segatori L, Kelly JW. Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell 2008; 134: 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mijnders M, Kleizen B, Braakman I. Correcting CFTR folding defects by small-molecule correctors to cure cystic fibrosis. Curr Opin Pharmacol 2017; 34: 83–90. [DOI] [PubMed] [Google Scholar]
- 16.Skilton M, Krishan A, Patel S, Sinha IP, Southern KW. Potentiators (specific therapies for class III and IV mutations) for cystic fibrosis. Cochrane Database Syst Rev 2019; 1: CD009841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gees M, Musch S, Van der Plas S, Wesse AS, Vandevelde A, Verdonck K, Mammoliti O, Hwang TC, Sonck K, Stouten P, Swensen AM, Jans M, Van der Schueren J, Nelles L, Andrews M, Conrath K. Identification and Characterization of Novel CFTR Potentiators. Frontiers in pharmacology 2018; 9: 1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Langron E, Prins S, Vergani P. Potentiation of the cystic fibrosis transmembrane conductance regulator by VX-770 involves stabilization of the pre-hydrolytic, O1 state. Br J Pharmacol 2018; 175: 3990–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moss RB, Flume PA, Elborn JS, Cooke J, Rowe SM, McColley SA, Rubenstein RC, Higgins M, Group VXS. Efficacy and safety of ivacaftor in patients with cystic fibrosis who have an Arg117His-CFTR mutation: a double-blind, randomised controlled trial. Lancet Respir Med 2015; 3: 524–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rowe SM, Daines C, Ringshausen FC, Kerem E, Wilson J, Tullis E, Nair N, Simard C, Han L, Ingenito EP, McKee C, Lekstrom-Himes J, Davies JC. Tezacaftor-Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N Engl J Med 2017; 377: 2024–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taylor-Cousar JL, Mall MA, Ramsey BW, McKone EF, Tullis E, Marigowda G, McKee CM, Waltz D, Moskowitz SM, Savage J, Xuan F, Rowe SM. Clinical development of triple-combination CFTR modulators for cystic fibrosis patients with one or two F508del alleles. ERJ Open Res 2019; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Middleton PG, Mall MA, Drevinek P, Lands LC, McKone EF, Polineni D, Ramsey BW, Taylor-Cousar JL, Tullis E, Vermeulen F, Marigowda G, McKee CM, Moskowitz SM, Nair N, Savage J, Simard C, Tian S, Waltz D, Xuan F, Rowe SM, Jain R, Group VXS. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N Engl J Med 2019; 381: 1809–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heijerman HGM, McKone EF, Downey DG, Van Braeckel E, Rowe SM, Tullis E, Mall MA, Welter JJ, Ramsey BW, McKee CM, Marigowda G, Moskowitz SM, Waltz D, Sosnay PR, Simard C, Ahluwalia N, Xuan F, Zhang Y, Taylor-Cousar JL, McCoy KS, Group VXT. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet 2019; 394: 1940–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keeling KM, Wang D, Conard SE, Bedwell DM. Suppression of premature termination codons as a therapeutic approach. Critical reviews in biochemistry and molecular biology 2012; 47: 444–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karousis ED, Nasif S, Muhlemann O. Nonsense-mediated mRNA decay: novel mechanistic insights and biological impact. Wiley interdisciplinary reviews RNA 2016; 7: 661–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hug N, Longman D, Caceres JF. Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic acids research 2016; 44: 1483–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Popp MW, Maquat LE. The dharma of nonsense-mediated mRNA decay in mammalian cells. Molecules and cells 2014; 37: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kervestin S, Jacobson A. NMD: a multifaceted response to premature translational termination. Nature reviews Molecular cell biology 2012; 13: 700–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Silva AL, Romao L. The mammalian nonsense-mediated mRNA decay pathway: to decay or not to decay! Which players make the decision? FEBS Lett 2009; 583: 499–505. [DOI] [PubMed] [Google Scholar]
- 30.Rebbapragada I, Lykke-Andersen J. Execution of nonsense-mediated mRNA decay: what defines a substrate? Curr Opin Cell Biol 2009; 21: 394–402. [DOI] [PubMed] [Google Scholar]
- 31.Fatscher T, Boehm V, Gehring NH. Mechanism, factors, and physiological role of nonsense-mediated mRNA decay. Cell Mol Life Sci 2015; 72: 4523–4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lejeune F. Nonsense-mediated mRNA decay at the crossroads of many cellular pathways. BMB Rep 2017; 50: 175–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilschanski M. Class 1 CF Mutations. Frontiers in pharmacology 2012; 3: 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keeling KM, Xue X, Gunn G, Bedwell DM. Therapeutics Based on Stop Codon Readthrough. Annual review of genomics and human genetics 2014; 15: 371–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mort M, Ivanov D, Cooper DN, Chuzhanova NA. A meta-analysis of nonsense mutations causing human genetic disease. Hum Mutat 2008; 29: 1037–1047. [DOI] [PubMed] [Google Scholar]
- 36.James PD, Raut S, Rivard GE, Poon MC, Warner M, McKenna S, Leggo J, Lillicrap D. Aminoglycoside suppression of nonsense mutations in severe hemophilia. Blood 2005; 106: 3043–3048. [DOI] [PubMed] [Google Scholar]
- 37.Sermet-Gaudelus I, Namy O. New Pharmacological Approaches to Treat Patients with Cystic Fibrosis with Nonsense Mutations. Am J Respir Crit Care Med 2016; 194: 1042–1044. [DOI] [PubMed] [Google Scholar]
- 38.Roy B, Leszyk JD, Mangus DA, Jacobson A. Nonsense suppression by near-cognate tRNAs employs alternative base pairing at codon positions 1 and 3. Proc Natl Acad Sci U S A 2015; 112: 3038–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roy B, Friesen WJ, Tomizawa Y, Leszyk JD, Zhuo J, Johnson B, Dakka J, Trotta CR, Xue X, Mutyam V, Keeling KM, Mobley JA, Rowe SM, Bedwell DM, Welch EM, Jacobson A. Ataluren stimulates ribosomal selection of near-cognate tRNAs to promote nonsense suppression. Proc Natl Acad Sci U S A 2016; 113: 12508–12513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dabrowski M, Bukowy-Bieryllo Z, Zietkiewicz E. Advances in therapeutic use of a drug-stimulated translational readthrough of premature termination codons. Mol Med 2018; 24: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dabrowski M, Bukowy-Bieryllo Z, Zietkiewicz E. Translational readthrough potential of natural termination codons in eucaryotes--The impact of RNA sequence. RNA biology 2015; 12: 950–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mutyam V, Du M, Xue X, Keeling KM, White EL, Bostwick JR, Rasmussen L, Liu B, Mazur M, Hong JS, Falk Libby E, Liang F, Shang H, Mense M, Suto MJ, Bedwell DM, Rowe SM. Discovery of Clinically Approved Agents That Promote Suppression of Cystic Fibrosis Transmembrane Conductance Regulator Nonsense Mutations. American journal of respiratory and critical care medicine 2016; 194: 1092–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Keeling KM. Nonsense Suppression as an Approach to Treat Lysosomal Storage Diseases. Diseases 2016; 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lueck JD, Yoon JS, Perales-Puchalt A, Mackey AL, Infield DT, Behlke MA, Pope MR, Weiner DB, Skach WR, McCray PB Jr., Ahern CA. Engineered transfer RNAs for suppression of premature termination codons. Nat Commun 2019; 10: 822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakamura Y, Ito K. Making sense of mimic in translation termination. Trends Biochem Sci 2003; 28: 99–105. [DOI] [PubMed] [Google Scholar]
- 46.Chavatte L, Seit-Nebi A, Dubovaya V, Favre A. The invariant uridine of stop codons contacts the conserved NIKSR loop of human eRF1 in the ribosome. The EMBO journal 2002; 21: 5302–5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bulygin KN, Khairulina YS, Kolosov PM, Ven’yaminova AG, Graifer DM, Vorobjev YN, Frolova LY, Kisselev LL, Karpova GG. Three distinct peptides from the N domain of translation termination factor eRF1 surround stop codon in the ribosome. Rna 2010; 16: 1902–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Salas-Marco J, Bedwell DM. GTP hydrolysis by eRF3 facilitates stop codon decoding during eukaryotic translation termination. Molecular and cellular biology 2004; 24: 7769–7778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kisselev L, Ehrenberg M, Frolova L. Termination of translation: interplay of mRNA, rRNAs and release factors? The EMBO journal 2003; 22: 175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Buckingham RH, Grentzmann G, Kisselev L. Polypeptide chain release factors. Mol Microbiol 1997; 24: 449–456. [DOI] [PubMed] [Google Scholar]
- 51.Kisselev LL, Buckingham RH. Translational termination comes of age. Trends Biochem Sci 2000; 25: 561–566. [DOI] [PubMed] [Google Scholar]
- 52.Zavialov AV, Buckingham RH, Ehrenberg M. A posttermination ribosomal complex is the guanine nucleotide exchange factor for peptide release factor RF3. Cell 2001; 107: 115–124. [DOI] [PubMed] [Google Scholar]
- 53.Alkalaeva EZ, Pisarev AV, Frolova LY, Kisselev LL, Pestova TV. In vitro reconstitution of eukaryotic translation reveals cooperativity between release factors eRF1 and eRF3. Cell 2006; 125: 1125–1136. [DOI] [PubMed] [Google Scholar]
- 54.Bonetti B, Fu L, Moon J, Bedwell DM. The efficiency of translation termination is determined by a synergistic interplay between upstream and downstream sequences in Saccharomyces cerevisiae. Journal of molecular biology 1995; 251: 334–345. [DOI] [PubMed] [Google Scholar]
- 55.Cassan M, Rousset JP. UAG readthrough in mammalian cells: effect of upstream and downstream stop codon contexts reveal different signals. BMC Mol Biol 2001; 2: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Manuvakhova M, Keeling K, Bedwell DM. Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. RNA 2000; 6: 1044–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Keeling KM, Bedwell DM. Clinically relevant aminoglycosides can suppress disease-associated premature stop mutations in the IDUA and P53 cDNAs in a mammalian translation system. Journal of molecular medicine 2002; 80: 367–376. [DOI] [PubMed] [Google Scholar]
- 58.Howard MT, Shirts BH, Petros LM, Flanigan KM, Gesteland RF, Atkins JF. Sequence specificity of aminoglycoside-induced stop condon readthrough: potential implications for treatment of Duchenne muscular dystrophy. Annals of neurology 2000; 48: 164–169. [PubMed] [Google Scholar]
- 59.Young DJ, Guydosh NR, Zhang F, Hinnebusch AG, Green R. Rli1/ABCE1 Recycles Terminating Ribosomes and Controls Translation Reinitiation in 3’UTRs In Vivo. Cell 2015; 162: 872–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hellen CUT. Translation Termination and Ribosome Recycling in Eukaryotes. Cold Spring Harb Perspect Biol 2018; 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Poole ES, Askarian-Amiri ME, Major LL, McCaughan KK, Scarlett DJ, Wilson DN, Tate WP. Molecular mimicry in the decoding of translational stop signals. Prog Nucleic Acid Res Mol Biol 2003; 74: 83–121. [DOI] [PubMed] [Google Scholar]
- 62.Eliseeva IA, Lyabin DN, Ovchinnikov LP. Poly(A)-binding proteins: structure, domain organization, and activity regulation. Biochemistry (Mosc) 2013; 78: 1377–1391. [DOI] [PubMed] [Google Scholar]
- 63.Bernstein P, Peltz SW, Ross J. The poly(A)-poly(A)-binding protein complex is a major determinant of mRNA stability in vitro. Molecular and cellular biology 1989; 9: 659–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wells SE, Hillner PE, Vale RD, Sachs AB. Circularization of mRNA by eukaryotic translation initiation factors. Mol Cell 1998; 2: 135–140. [DOI] [PubMed] [Google Scholar]
- 65.Ivanov A, Mikhailova T, Eliseev B, Yeramala L, Sokolova E, Susorov D, Shuvalov A, Schaffitzel C, Alkalaeva E. PABP enhances release factor recruitment and stop codon recognition during translation termination. Nucleic acids research 2016; 44: 7766–7776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hoshino S, Imai M, Kobayashi T, Uchida N, Katada T. The eukaryotic polypeptide chain releasing factor (eRF3/GSPT) carrying the translation termination signal to the 3’-Poly(A) tail of mRNA. Direct association of erf3/GSPT with polyadenylate-binding protein. J Biol Chem 1999; 274: 16677–16680. [DOI] [PubMed] [Google Scholar]
- 67.Kononenko AV, Mitkevich VA, Atkinson GC, Tenson T, Dubovaya VI, Frolova LY, Makarov AA, Hauryliuk V. GTP-dependent structural rearrangement of the eRF1:eRF3 complex and eRF3 sequence motifs essential for PABP binding. Nucleic acids research 2010; 38: 548–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dong J, Lai R, Nielsen K, Fekete CA, Qiu H, Hinnebusch AG. The essential ATP-binding cassette protein RLI1 functions in translation by promoting preinitiation complex assembly. J Biol Chem 2004; 279: 42157–42168. [DOI] [PubMed] [Google Scholar]
- 69.Bolger TA, Folkmann AW, Tran EJ, Wente SR. The mRNA export factor Gle1 and inositol hexakisphosphate regulate distinct stages of translation. Cell 2008; 134: 624–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pisarev AV, Hellen CU, Pestova TV. Recycling of eukaryotic posttermination ribosomal complexes. Cell 2007; 131: 286–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Beznoskova P, Cuchalova L, Wagner S, Shoemaker CJ, Gunisova S, von der Haar T, Valasek LS. Translation initiation factors eIF3 and HCR1 control translation termination and stop codon read-through in yeast cells. PLoS genetics 2013; 9: e1003962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fraser CS, Berry KE, Hershey JW, Doudna JA. eIF3j Is Located in the Decoding Center of the Human 40S Ribosomal Subunit. Mol Cell 2007; 26: 811–819. [DOI] [PubMed] [Google Scholar]
- 73.Prokhorova I, Altman RB, Djumagulov M, Shrestha JP, Urzhumtsev A, Ferguson A, Chang CT, Yusupov M, Blanchard SC, Yusupova G. Aminoglycoside interactions and impacts on the eukaryotic ribosome. Proc Natl Acad Sci U S A 2017; 114: E10899–E10908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Demirci H, Murphy Ft, Murphy E, Gregory ST, Dahlberg AE, Jogl G. A structural basis for streptomycin-induced misreading of the genetic code. Nat Commun 2013; 4: 1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gross T, Siepmann A, Sturm D, Windgassen M, Scarcelli JJ, Seedorf M, Cole CN, Krebber H. The DEAD-box RNA helicase Dbp5 functions in translation termination. Science 2007; 315: 646–649. [DOI] [PubMed] [Google Scholar]
- 76.Khoshnevis S, Gross T, Rotte C, Baierlein C, Ficner R, Krebber H. The iron-sulphur protein RNase L inhibitor functions in translation termination. EMBO Rep 2010; 11: 214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Alcazar-Roman AR, Bolger TA, Wente SR. Control of mRNA export and translation termination by inositol hexakisphosphate requires specific interaction with Gle1. J Biol Chem 2010; 285: 16683–16692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cridge AG, Crowe-McAuliffe C, Mathew SF, Tate WP. Eukaryotic translational termination efficiency is influenced by the 3’ nucleotides within the ribosomal mRNA channel. Nucleic acids research 2018; 46: 1927–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Harrell L, Melcher U, Atkins JF. Predominance of six different hexanucleotide recoding signals 3’ of read-through stop codons. Nucleic acids research 2002; 30: 2011–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Xue X, Mutyam V, Thakerar A, Mobley J, Bridges RJ, Rowe SM, Keeling KM, Bedwell DM. Identification of the amino acids inserted during suppression of CFTR nonsense mutations and determination of their functional consequences. Human molecular genetics 2017; 26: 3116–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Namy O, Hatin I, Rousset JP. Impact of the six nucleotides downstream of the stop codon on translation termination. EMBO Rep 2001; 2: 787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mottagui-Tabar S, Tuite MF, Isaksson LA. The influence of 5’ codon context on translation termination in Saccharomyces cerevisiae. Eur J Biochem 1998; 257: 249–254. [DOI] [PubMed] [Google Scholar]
- 83.Mottagui-Tabar S, Bjornsson A, Isaksson LA. The second to last amino acid in the nascent peptide as a codon context determinant. The EMBO journal 1994; 13: 249–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wei Y, Xia X. The Role of +4U as an Extended Translation Termination Signal in Bacteria. Genetics 2017; 205: 539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tork S, Hatin I, Rousset JP, Fabret C. The major 5’ determinant in stop codon read-through involves two adjacent adenines. Nucleic acids research 2004; 32: 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pacho F, Zambruno G, Calabresi V, Kiritsi D, Schneider H. Efficiency of translation termination in humans is highly dependent upon nucleotides in the neighbourhood of a (premature) termination codon. J Med Genet 2011; 48: 640–644. [DOI] [PubMed] [Google Scholar]
- 87.Bedwell DM, Kaenjak A, Benos DJ, Bebok Z, Bubien JK, Hong J, Tousson A, Clancy JP, Sorscher EJ. Suppression of a CFTR premature stop mutation in a bronchial epithelial cell line. Nature medicine 1997; 3: 1280–1284. [DOI] [PubMed] [Google Scholar]
- 88.Bidou L, Allamand V, Rousset JP, Namy O. Sense from nonsense: therapies for premature stop codon diseases. Trends Mol Med 2012; 18: 679–688. [DOI] [PubMed] [Google Scholar]
- 89.Keeling KM, Xue X, Gunn G, Bedwell DM. Therapeutics based on stop codon readthrough. Annual review of genomics and human genetics 2014; 15: 371–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Malik V, Rodino-Klapac LR, Viollet L, Mendell JR. Aminoglycoside-induced mutation suppression (stop codon readthrough) as a therapeutic strategy for Duchenne muscular dystrophy. Therapeutic advances in neurological disorders 2010; 3: 379–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Malik V, Rodino-Klapac LR, Viollet L, Wall C, King W, Al-Dahhak R, Lewis S, Shilling CJ, Kota J, Serrano-Munuera C, Hayes J, Mahan JD, Campbell KJ, Banwell B, Dasouki M, Watts V, Sivakumar K, Bien-Willner R, Flanigan KM, Sahenk Z, Barohn RJ, Walker CM, Mendell JR. Gentamicin-induced readthrough of stop codons in Duchenne muscular dystrophy. Ann Neurol 2010; 67: 771–780. [DOI] [PubMed] [Google Scholar]
- 92.Politano L, Nigro G, Nigro V, Piluso G, Papparella S, Paciello O, Comi LI. Gentamicin administration in Duchenne patients with premature stop codon. Preliminary results. Acta Myol 2003; 22: 15–21. [PubMed] [Google Scholar]
- 93.Wilschanski M, Yahav Y, Yaacov Y, Blau H, Bentur L, Rivlin J, Aviram M, Bdolah-Abram T, Bebok Z, Shushi L, Kerem B, Kerem E. Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. The New England journal of medicine 2003; 349: 1433–1441. [DOI] [PubMed] [Google Scholar]
- 94.Clancy JP, Bebok Z, Ruiz F, King C, Jones J, Walker L, Greer H, Hong J, Wing L, Macaluso M, Lyrene R, Sorscher EJ, Bedwell DM. Evidence that systemic gentamicin suppresses premature stop mutations in patients with cystic fibrosis. Am J Respir Crit Care Med 2001; 163: 1683–1692. [DOI] [PubMed] [Google Scholar]
- 95.Sermet-Gaudelus I, Renouil M, Fajac A, Bidou L, Parbaille B, Pierrot S, Davy N, Bismuth E, Reinert P, Lenoir G, Lesure JF, Rousset JP, Edelman A. Correction to: In vitro prediction of stop-codon suppression by intravenous gentamicin in patients with cystic fibrosis: a pilot study. BMC medicine 2018; 16: 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Forge A, Schacht J. Aminoglycoside antibiotics. Audiol Neurootol 2000; 5: 3–22. [DOI] [PubMed] [Google Scholar]
- 97.Bottger EC, Schacht J. The mitochondrion: a perpetrator of acquired hearing loss. Hear Res 2013; 303: 12–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mingeot-Leclercq MP, Tulkens PM. Aminoglycosides: nephrotoxicity. Antimicrobial agents and chemotherapy 1999; 43: 1003–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Krause KM, Serio AW, Kane TR, Connolly LE. Aminoglycosides: An Overview. Cold Spring Harbor perspectives in medicine 2016; 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shulman E, Belakhov V, Wei G, Kendall A, Meyron-Holtz EG, Ben-Shachar D, Schacht J, Baasov T. Designer aminoglycosides that selectively inhibit cytoplasmic rather than mitochondrial ribosomes show decreased ototoxicity: a strategy for the treatment of genetic diseases. J Biol Chem 2014; 289: 2318–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nudelman I, Glikin D, Smolkin B, Hainrichson M, Belakhov V, Baasov T. Repairing faulty genes by aminoglycosides: development of new derivatives of geneticin (G418) with enhanced suppression of diseases-causing nonsense mutations. Bioorg Med Chem 2010; 18: 3735–3746. [DOI] [PubMed] [Google Scholar]
- 102.Sabbavarapu NM, Shavit M, Degani Y, Smolkin B, Belakhov V, Baasov T. Design of Novel Aminoglycoside Derivatives with Enhanced Suppression of Diseases-Causing Nonsense Mutations. ACS Med Chem Lett 2016; 7: 418–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Xue X, Mutyam V, Tang L, Biswas S, Du M, Jackson LA, Dai Y, Belakhov V, Shalev M, Chen F, Schacht J, R JB, Baasov T, Hong J, Bedwell DM, Rowe SM. Synthetic aminoglycosides efficiently suppress cystic fibrosis transmembrane conductance regulator nonsense mutations and are enhanced by ivacaftor. Am J Respir Cell Mol Biol 2014; 50: 805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kandasamy J, Atia-Glikin D, Shulman E, Shapira K, Shavit M, Belakhov V, Baasov T. Increased selectivity toward cytoplasmic versus mitochondrial ribosome confers improved efficiency of synthetic aminoglycosides in fixing damaged genes: a strategy for treatment of genetic diseases caused by nonsense mutations. J Med Chem 2012; 55: 10630–10643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Guimbellot J, Sharma J, Rowe SM. Toward inclusive therapy with CFTR modulators: Progress and challenges. Pediatric pulmonology 2017; 52: S4–S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Leubitz A, Frydman-Marom A, Sharpe N, van Duzer J, Campbell KCM, Vanhoutte F. Safety, Tolerability, and Pharmacokinetics of Single Ascending Doses of ELX-02, a Potential Treatment for Genetic Disorders Caused by Nonsense Mutations, in Healthy Volunteers. Clin Pharmacol Drug Dev 2019. [DOI] [PubMed] [Google Scholar]
- 107.Bidou L, Bugaud O, Belakhov V, Baasov T, Namy O. Characterization of new-generation aminoglycoside promoting premature termination codon readthrough in cancer cells. RNA Biol 2017; 14: 378–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nudelman I, Rebibo-Sabbah A, Cherniavsky M, Belakhov V, Hainrichson M, Chen F, Schacht J, Pilch DS, Ben-Yosef T, Baasov T. Development of novel aminoglycoside (NB54) with reduced toxicity and enhanced suppression of disease-causing premature stop mutations. J Med Chem 2009; 52: 2836–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Brasell EJ, Chu LL, Akpa MM, Eshkar-Oren I, Alroy I, Corsini R, Gilfix BM, Yamanaka Y, Huertas P, Goodyer P. The novel aminoglycoside, ELX-02, permits CTNSW138X translational read-through and restores lysosomal cystine efflux in cystinosis. PloS one 2019; 14: e0223954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sermet-Gaudelus I, Renouil M, Fajac A, Bidou L, Parbaille B, Pierrot S, Davy N, Bismuth E, Reinert P, Lenoir G, Lesure JF, Rousset JP, Edelman A. In vitro prediction of stop-codon suppression by intravenous gentamicin in patients with cystic fibrosis: a pilot study. BMC Med 2007; 5: 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fidler MC, Beusmans J, Panorchan P, Van Goor F. Correlation of sweat chloride and percent predicted FEV1 in cystic fibrosis patients treated with ivacaftor. J Cyst Fibros 2017; 16: 41–44. [DOI] [PubMed] [Google Scholar]
- 112.Beauchamp D, Laurent G, Maldague P, Abid S, Kishore BK, Tulkens PM. Protection against gentamicin-induced early renal alterations (phospholipidosis and increased DNA synthesis) by coadministration of poly-L-aspartic acid. J Pharmacol Exp Ther 1990; 255: 858–866. [PubMed] [Google Scholar]
- 113.Williams PD, Hottendorf GH, Bennett DB. Inhibition of renal membrane binding and nephrotoxicity of aminoglycosides. J Pharmacol Exp Ther 1986; 237: 919–925. [PubMed] [Google Scholar]
- 114.Ben Ismail TH, Ali BH, Bashir AA. Influence of iron, deferoxamine and ascorbic acid on gentamicin-induced nephrotoxicity in rats. Gen Pharmacol 1994; 25: 1249–1252. [DOI] [PubMed] [Google Scholar]
- 115.Du M, Keeling KM, Fan L, Liu X, Bedwell DM. Poly-L-aspartic acid enhances and prolongs gentamicin-mediated suppression of the CFTR-G542X mutation in a cystic fibrosis mouse model. J Biol Chem 2009; 284: 6885–6892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mahi-Birjand M, Yaghoubi S, Abdollahpour-Alitappeh M, Keshtkaran Z, Bagheri N, Pirouzi A, Khatami M, Sineh Sepehr K, Peymani P, Karimzadeh I. Protective effects of pharmacological agents against aminoglycoside-induced nephrotoxicity: A systematic review. Expert Opin Drug Saf 2020; 19: 167–186. [DOI] [PubMed] [Google Scholar]
- 117.Yao L, Zhang JW, Chen B, Cai MM, Feng D, Wang QZ, Wang XY, Sun JG, Zheng YW, Wang GJ, Zhou F. Mechanisms and pharmacokinetic/pharmacodynamic profiles underlying the low nephrotoxicity and ototoxicity of etimicin. Acta Pharmacol Sin 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Silan C, Uzun O, Comunoglu NU, Gokcen S, Bedirhan S, Cengiz M. Gentamicin-induced nephrotoxicity in rats ameliorated and healing effects of resveratrol. Biol Pharm Bull 2007; 30: 79–83. [DOI] [PubMed] [Google Scholar]
- 119.Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, Paushkin S, Patel M, Trotta CR, Hwang S, Wilde RG, Karp G, Takasugi J, Chen G, Jones S, Ren H, Moon YC, Corson D, Turpoff AA, Campbell JA, Conn MM, Khan A, Almstead NG, Hedrick J, Mollin A, Risher N, Weetall M, Yeh S, Branstrom AA, Colacino JM, Babiak J, Ju WD, Hirawat S, Northcutt VJ, Miller LL, Spatrick P, He F, Kawana M, Feng H, Jacobson A, Peltz SW, Sweeney HL. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007; 447: 87–91. [DOI] [PubMed] [Google Scholar]
- 120.Wilschanski M, Miller LL, Shoseyov D, Blau H, Rivlin J, Aviram M, Cohen M, Armoni S, Yaakov Y, Pugatsch T, Cohen-Cymberknoh M, Miller NL, Reha A, Northcutt VJ, Hirawat S, Donnelly K, Elfring GL, Ajayi T, Kerem E. Chronic ataluren (PTC124) treatment of nonsense mutation cystic fibrosis. Eur Respir J 2011; 38: 59–69. [DOI] [PubMed] [Google Scholar]
- 121.Hirawat S, Welch EM, Elfring GL, Northcutt VJ, Paushkin S, Hwang S, Leonard EM, Almstead NG, Ju W, Peltz SW, Miller LL. Safety, tolerability, and pharmacokinetics of PTC124, a nonaminoglycoside nonsense mutation suppressor, following single- and multiple-dose administration to healthy male and female adult volunteers. J Clin Pharmacol 2007; 47: 430–444. [DOI] [PubMed] [Google Scholar]
- 122.Du M, Liu X, Welch EM, Hirawat S, Peltz SW, Bedwell DM. PTC124 is an orally bioavailable compound that promotes suppression of the human CFTR-G542X nonsense allele in a CF mouse model. Proc Natl Acad Sci U S A 2008; 105: 2064–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kerem E, Konstan MW, De Boeck K, Accurso FJ, Sermet-Gaudelus I, Wilschanski M, Elborn JS, Melotti P, Bronsveld I, Fajac I, Malfroot A, Rosenbluth DB, Walker PA, McColley SA, Knoop C, Quattrucci S, Rietschel E, Zeitlin PL, Barth J, Elfring GL, Welch EM, Branstrom A, Spiegel RJ, Peltz SW, Ajayi T, Rowe SM, Cystic Fibrosis Ataluren Study G. Ataluren for the treatment of nonsense-mutation cystic fibrosis: a randomised, double-blind, placebo-controlled phase 3 trial. The Lancet Respiratory medicine 2014; 2: 539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Auld DS, Thorne N, Maguire WF, Inglese J. Mechanism of PTC124 activity in cell-based luciferase assays of nonsense codon suppression. Proc Natl Acad Sci U S A 2009; 106: 3585–3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zomer-van Ommen DD, Vijftigschild LA, Kruisselbrink E, Vonk AM, Dekkers JF, Janssens HM, de Winter-de Groot KM, van der Ent CK, Beekman JM. Limited premature termination codon suppression by read-through agents in cystic fibrosis intestinal organoids. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2016; 15: 158–162. [DOI] [PubMed] [Google Scholar]
- 126.Linde L, Kerem B. Nonsense-mediated mRNA decay and cystic fibrosis. Methods Mol Biol 2011; 741: 137–154. [DOI] [PubMed] [Google Scholar]
- 127.Linde L, Boelz S, Nissim-Rafinia M, Oren YS, Wilschanski M, Yaacov Y, Virgilis D, Neu-Yilik G, Kulozik AE, Kerem E, Kerem B. Nonsense-mediated mRNA decay affects nonsense transcript levels and governs response of cystic fibrosis patients to gentamicin. The Journal of clinical investigation 2007; 117: 683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Durand S, Cougot N, Mahuteau-Betzer F, Nguyen CH, Grierson DS, Bertrand E, Tazi J, Lejeune F. Inhibition of nonsense-mediated mRNA decay (NMD) by a new chemical molecule reveals the dynamic of NMD factors in P-bodies. J Cell Biol 2007; 178: 1145–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Keeling KM, Wang D, Dai Y, Murugesan S, Chenna B, Clark J, Belakhov V, Kandasamy J, Velu SE, Baasov T, Bedwell DM. Attenuation of nonsense-mediated mRNA decay enhances in vivo nonsense suppression. PloS one 2013; 8: e60478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Huang L, Low A, Damle SS, Keenan MM, Kuntz S, Murray SF, Monia BP, Guo S. Antisense suppression of the nonsense mediated decay factor Upf3b as a potential treatment for diseases caused by nonsense mutations. Genome Biol 2018; 19: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Nomakuchi TT, Rigo F, Aznarez I, Krainer AR. Antisense oligonucleotide-directed inhibition of nonsense-mediated mRNA decay. Nat Biotechnol 2016; 34: 164–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Thada V, Miller JN, Kovacs AD, Pearce DA. Tissue-specific variation in nonsense mutant transcript level and drug-induced read-through efficiency in the Cln1(R151X) mouse model of INCL. J Cell Mol Med 2016; 20: 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Krall M, Htun S, Slavotinek A. Use of PTC124 for nonsense suppression therapy targeting BMP4 nonsense variants in vitro and the bmp4st72 allele in zebrafish. PLoS One 2019; 14: e0212121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Keenan MM, Huang L, Jordan NJ, Wong E, Cheng Y, Valley HC, Mahiou J, Liang F, Bihler H, Mense M, Guo S, Monia BP. Nonsense Mediated RNA Decay Pathway Inhibition Restores Expression and Function of W1282X CFTR. American journal of respiratory cell and molecular biology 2019. [DOI] [PubMed] [Google Scholar]
- 135.Valley HC, Bukis KM, Bell A, Cheng Y, Wong E, Jordan NJ, Allaire NE, Sivachenko A, Liang F, Bihler H, Thomas PJ, Mahiou J, Mense M. Isogenic cell models of cystic fibrosis-causing variants in natively expressing pulmonary epithelial cells. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2018. [DOI] [PubMed] [Google Scholar]
- 136.Isken O, Maquat LE. The multiple lives of NMD factors: balancing roles in gene and genome regulation. Nat Rev Genet 2008; 9: 699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hwang J, Maquat LE. Nonsense-mediated mRNA decay (NMD) in animal embryogenesis: to die or not to die, that is the question. Curr Opin Genet Dev 2011; 21: 422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lykke-Andersen S, Jensen TH. Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nature reviews Molecular cell biology 2015; 16: 665–677. [DOI] [PubMed] [Google Scholar]
- 139.Li T, Shi Y, Wang P, Guachalla LM, Sun B, Joerss T, Chen YS, Groth M, Krueger A, Platzer M, Yang YG, Rudolph KL, Wang ZQ. Smg6/Est1 licenses embryonic stem cell differentiation via nonsense-mediated mRNA decay. The EMBO journal 2015; 34: 1630–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Nickless A, Bailis JM, You Z. Control of gene expression through the nonsense-mediated RNA decay pathway. Cell & bioscience 2017; 7: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Weischenfeldt J, Damgaard I, Bryder D, Theilgaard-Monch K, Thoren LA, Nielsen FC, Jacobsen SE, Nerlov C, Porse BT. NMD is essential for hematopoietic stem and progenitor cells and for eliminating by-products of programmed DNA rearrangements. Genes Dev 2008; 22: 1381–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Deeks ED. Lumacaftor/Ivacaftor: A Review in Cystic Fibrosis. Drugs 2016; 76: 1191–1201. [DOI] [PubMed] [Google Scholar]
- 143.Bulloch MN, Hanna C, Giovane R. Lumacaftor/ivacaftor, a novel agent for the treatment of cystic fibrosis patients who are homozygous for the F580del CFTR mutation. Expert Rev Clin Pharmacol 2017; 10: 1055–1072. [DOI] [PubMed] [Google Scholar]
- 144.Condren ME, Bradshaw MD. Ivacaftor: a novel gene-based therapeutic approach for cystic fibrosis. J Pediatr Pharmacol Ther 2013; 18: 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Van Goor F, Yu H, Burton B, Hoffman BJ. Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2014; 13: 29–36. [DOI] [PubMed] [Google Scholar]
- 146.Mutyam V, Libby EF, Peng N, Hadjiliadis D, Bonk M, Solomon GM, Rowe SM. Therapeutic benefit observed with the CFTR potentiator, ivacaftor, in a CF patient homozygous for the W1282X CFTR nonsense mutation. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2017; 16: 24–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Rowe SM, Varga K, Rab A, Bebok Z, Byram K, Li Y, Sorscher EJ, Clancy JP. Restoration of W1282X CFTR Activity by Enhanced Expression. Am J Respir Cell Mol Biol 2007; 37: 347–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Haggie PM, Phuan PW, Tan JA, Xu H, Avramescu RG, Perdomo D, Zlock L, Nielson DW, Finkbeiner WE, Lukacs GL, Verkman AS. Correctors and Potentiators Rescue Function of the Truncated W1282X-Cystic Fibrosis Transmembrane Regulator (CFTR) Translation Product. J Biol Chem 2017; 292: 771–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sharma N, Evans TA, Pellicore MJ, Davis E, Aksit MA, McCague AF, Joynt AT, Lu Z, Han ST, Anzmann AF, Lam AN, Thaxton A, West N, Merlo C, Gottschalk LB, Raraigh KS, Sosnay PR, Cotton CU, Cutting GR. Capitalizing on the heterogeneous effects of CFTR nonsense and frameshift variants to inform therapeutic strategy for cystic fibrosis. PLoS genetics 2018; 14: e1007723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gentzsch M, Riordan JR. Localization of sequences within the C-terminal domain of the cystic fibrosis transmembrane conductance regulator which impact maturation and stability. J Biol Chem 2001; 276: 1291–1298. [DOI] [PubMed] [Google Scholar]
- 151.Clancy JP, Cotton CU, Donaldson SH, Solomon GM, VanDevanter DR, Boyle MP, Gentzsch M, Nick JA, Illek B, Wallenburg JC, Sorscher EJ, Amaral MD, Beekman JM, Naren AP, Bridges RJ, Thomas PJ, Cutting G, Rowe S, Durmowicz AG, Mense M, Boeck KD, Skach W, Penland C, Joseloff E, Bihler H, Mahoney J, Borowitz D, Tuggle KL. CFTR modulator theratyping: Current status, gaps and future directions. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2019; 18: 22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Xue X, Mutyam V, Thakerar A, Mobley J, Bridges RJ, Rowe SM, Keeling KM, Bedwell DM. Identification of the Amino Acids Inserted During Suppression of CFTR Nonsense Mutations and Determination of Their Functional Consequences. Hum Mol Genet 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Phuan PW, Son JH, Tan JA, Li C, Musante I, Zlock L, Nielson DW, Finkbeiner WE, Kurth MJ, Galietta LJ, Haggie PM, Verkman AS. Combination potentiator (‘co-potentiator’) therapy for CF caused by CFTR mutants, including N1303K, that are poorly responsive to single potentiators. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2018; 17: 595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Rafeeq MM, Murad HAS. Cystic fibrosis: current therapeutic targets and future approaches. J Transl Med 2017; 15: 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Veit G, Xu H, Dreano E, Avramescu RG, Bagdany M, Beitel LK, Roldan A, Hancock MA, Lay C, Li W, Morin K, Gao S, Mak PA, Ainscow E, Orth AP, McNamara P, Edelman A, Frenkiel S, Matouk E, Sermet-Gaudelus I, Barnes WG, Lukacs GL. Structure-guided combination therapy to potently improve the function of mutant CFTRs. Nature medicine 2018; 24: 1732–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kleizen B, Hunt JF, Callebaut I, Hwang TC, Sermet-Gaudelus I, Hafkemeyer S, Sheppard DN. CFTR: New insights into structure and function and implications for modulation by small molecules. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2019. [DOI] [PubMed] [Google Scholar]
- 157.Keating D, Marigowda G, Burr L, Daines C, Mall MA, McKone EF, Ramsey BW, Rowe SM, Sass LA, Tullis E, McKee CM, Moskowitz SM, Robertson S, Savage J, Simard C, Van Goor F, Waltz D, Xuan F, Young T, Taylor-Cousar JL, Group VXS. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N Engl J Med 2018; 379: 1612–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Carnes J, Jacobson M, Leinwand L, Yarus M. Stop codon suppression via inhibition of eRF1 expression. RNA 2003; 9: 648–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Le Goff X, Philippe M, Jean-Jean O. Overexpression of human release factor 1 alone has an antisuppressor effect in human cells. Molecular and cellular biology 1997; 17: 3164–3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Baradaran-Heravi A, Balgi AD, Zimmerman C, Choi K, Shidmoossavee FS, Tan JS, Bergeaud C, Krause A, Flibotte S, Shimizu Y, Anderson HJ, Mouly V, Jan E, Pfeifer T, Jaquith JB, Roberge M. Novel small molecules potentiate premature termination codon readthrough by aminoglycosides. Nucleic Acids Res 2016; 44: 6583–6598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Rabea SM, Baradaran-Heravi A, Balgi AD, Krause A, Hosseini Farahabadi S, Roberge M, Grierson DS. 2-Aminothiazole-4-carboxamides Enhance Readthrough of Premature Termination Codons by Aminoglycosides. ACS Med Chem Lett 2019; 10: 726–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Habib AR, Kajbafzadeh M, Desai S, Yang CL, Skolnik K, Quon BS. A Systematic Review of the Clinical Efficacy and Safety of CFTR Modulators in Cystic Fibrosis. Sci Rep 2019; 9: 7234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Walker S, Flume P, McNamara J, Solomon M, Chilvers M, Chmiel J, Harris RS, Haseltine E, Stiles D, Li C, Ahluwalia N, Zhou H, Owen CA, Sawicki G, Group VXI. A phase 3 study of tezacaftor in combination with ivacaftor in children aged 6 through 11years with cystic fibrosis. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society 2019. [DOI] [PubMed] [Google Scholar]