

Abstract
We previously reported a series of p53-elevating anthraquinone compounds with considerable cytotoxicity for acute lymphatic leukemia (ALL) cells. To further develop this class of compounds, we examined the effect of a few key structural features on the anticancer structure-activity relationship in ALL cells. The active analogs showed comparable cytotoxicity and upregulation of p53 but did not induce significant downregulation of MDM2 as seen with the lead compound AQ-101, indicating the importance of the anthraquinone core scaffold for MDM2 regulation. The result from the current study not only contributes to the SAR framework of these anthraquinone derivatives but also opens up new chemical space for further optimization work.
Keywords: Leukemia, Anthraquinone, Structure-activity relationships, p53, covalent inhibitor
Introduction
Leukemia is a group of blood cancers characterized by uncontrolled production of ill-functioning blood cells, particularly the leukocytes (Zou 2007). Due to the central roles played by these cells in the body’s immune system, a common complication of this often fatal disease is recurrent infections, mostly of bacterial and/or fungal origin (Chang et al. 1976; Slats et al. 2005). Based on the origin and duration of occurrence, four broad types of leukemia are presently identified, namely acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), chronic lymphocytic leukemia (CLL), and chronic myeloid leukemia (CML) (Zhao et al. 2018). In 2018, it was estimated that over 400,000 new cases of leukemia were reported, with about 300,000 deaths. It is the most diagnosed cancer in children, about 75% of which are of the ALL type (Bray et al. 2018; Hunger et al. 2020). Despite the availability of a wide range of therapeutic options, the prognosis for leukemia is still poor and treatment is often marred by unbearable side effects (Terwilliger and Abdul-Hay 2017; Watts and Nimer 2018). Therefore, there is a need for new treatments with improved efficacy and a better safety profile. While the exact cause of leukemia is largely unknown, genetic factors are believed to play a part in the etiology of the disease (Hutter 2010; Etzel 2016). The dysregulation of the tumor suppressor p53 protein through mutations, deletions, and inactivation is strongly implicated (Gaidano et al. 1991; Prokocimer et al. 2017). p53 dysregulation is also reported to be responsible for patients’ poor response to conventional therapy (Lozanski et al. 2004). A major pathway for p53 inactivation involves the murine double minute 2 (MDM2) protein, an E3 ligase that mediates the ubiquitination of the p53 protein leading to its proteasomal degradation (Haupt et al. 1997; Momand et al. 1992). Almost half of the ALL cell lines with wild-type p53 (wt-p53) harbors overexpression of MDM2, which leads to inhibition of p53’s tumor-suppressing functions. Downregulation of the MDM2 protein would in turn elevate the levels of the p53 protein and consequently restore its functions.
We had previously reported novel anthraquinone compounds with considerable cytotoxic activity (Anifowose et al. 2020; Draganov et al. 2019; Yang et al. 2011). The prototype is AQ-101 (1, Figure 1), with an IC50 of 0.83 ± 0.18 μM against EU-1 ALL cells (Draganov et al. 2019). AQ-101 was shown to induce MDM2 protein degradation and subsequently an increase in p53 levels in a dose-dependent manner. Further investigations revealed that AQ-101 induced MDM2 self-ubiquitination and reduced p53 polyubiquitination by binding to the MDM2 protein, blocking its dimerization with MDM4 (Gu et al. 2018), which is a crucial step in the proteasomal degradation of p53 (Wade et al. 2013). Many of the tested analogs including AQ-101 showed good selectivity for leukemia cells with wild-type p53 among a panel of cancer cell lines (Draganov et al. 2019). AQ-101 in particular produced complete disease remission in animal models of ALL at a dose of 20 mg/kg (Gu et al. 2018; Draganov et al. 2019). With the aim of further developing these anthraquinone compounds, we sought to achieve further understanding of the structure-activity relationships (SAR) for this class of compounds. We herein report our effort in examining three key structural features of this class of compounds for their in-vitro antileukemic activity (Figure 1).
Figure 1.
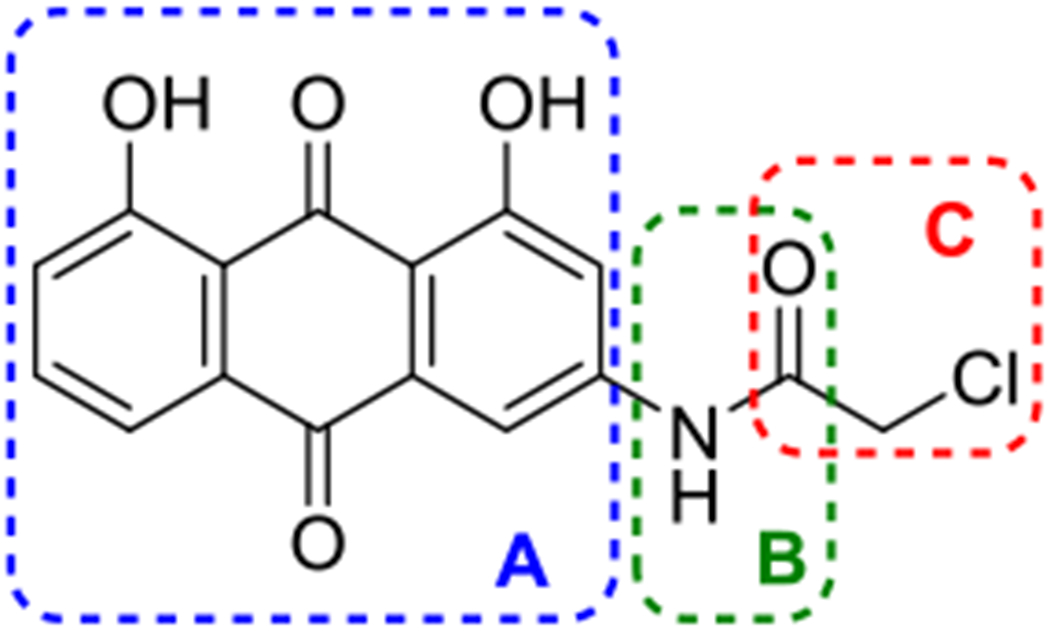
Chemical structure of AQ-101(1) and its three regions (A-C) for modifications.
For further medicinal chemistry work, we first worked on the modification of the anthraquinone core structure (Region A) to determine whether the planar anthraquinone structure and the carbonyl groups are critical to the activity. Secondly, we explored whether the acetamide group (B) poses a hydrolytic liability. If so, whether there are ways to ameliorate this liability. Further, we explored whether the N-H group is essential for activity, possibly as a hydrogen bond donor. Therefore, we were interested in replacing the NH group with a methylene group. Thirdly, based on our previous results of modifications to the alpha-chloroacetamide moiety (C) (Draganov et al. 2019), we explored several other structural variations of this alkylating group.
Results and discussion
Chemistry
Modification to the anthraquinone scaffold (A):
As aforementioned, the dihydroxyanthraquinone scaffold was changed in our previously reported studies (Anifowose et al. 2020; Draganov et al. 2019; Yang et al. 2011). To determine if the anthraquinone structure is essential to the activity, we made three modifications to the dihydroxyanthraquinone core while maintaining the alkylating moiety as the chloroacetamide used in AQ-101: (1) elimination of one keto group to open the anthraquinone central ring to form a diphenyl ketone scaffold, leading to a molecule that is ni longer flat; (2) reduction of the anthraquinone leading to anthracene as the core; (3) elimination of the phenolic hydroxyl groups leading to an anthraquinone core structure.
The diphenyl ketone analogs 3a-c with different substitution poatterns were synthesized by direct acylation of the commercially available amines (2a-c) with chloroacetyl chloride (Scheme 1a). To probe the effect of increasing the electron density of the phenyl ring (ring activation) on activity, diphenyl ketone analogs with a methoxyl or a hydroxyl substituent were also made. To synthesize compound 6, Friedel-Crafts acylation of anisole under neat condition led to intermediate 4. The nitro group was reduced through hydrogenation to give amine 5, which was acylated with chloroacetyl chloride to afford 6 as a pink solid (Scheme 1b). The hydroxyl substituted compound 9 was synthesized similarly by first demethylating compound 4 to give compound 7 (Scheme 1c).
Scheme 1.
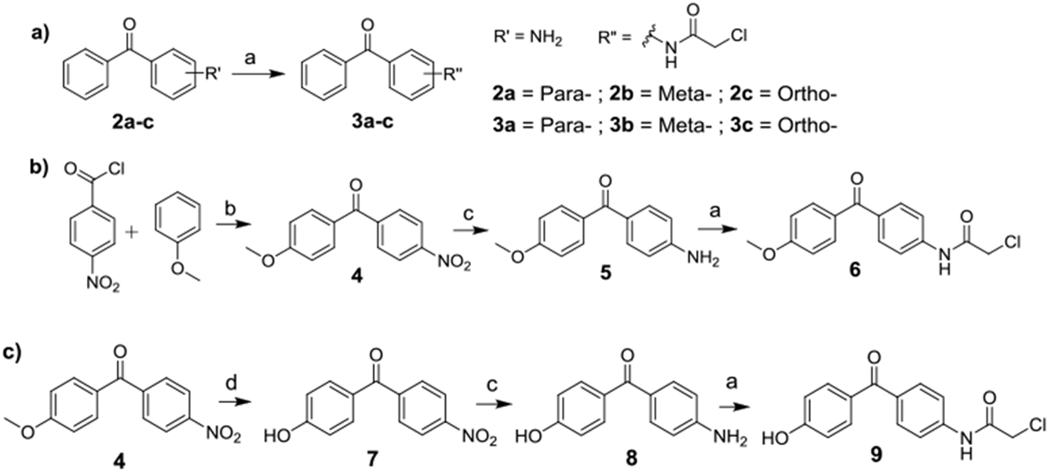
Synthesis of diphenyl ketone analogs.
Reagents and conditions:
(a) chloroacetyl chloride, dioxane, RT, 30 min, 39 - 96%; (b) ZnCl2, neat, 150 °C 30 min, 89%; (c) hydrogen, Pd/C, RT, overnight, 68-72%; (d) BBr3, DCM, 40 °C, overnight, 45%.
Reagents and conditions:
(a) Zn/NaOH, reflux, overnight, 64-75%; (b) chloroacetyl chloride, dioxane, RT, 30 min, 87-91%; (c) chloroacetyl chloride, dioxane, RT, 30 min, 46%.
The two phenolic hydroxyl groups in compound 1 could serve as sites for intermolecular and/or intramolecular hydrogen bond formation, contributing to the observed activity (Malik and Muller 2016). To understand how much these phenolic hydroxy groups and the quinone scaffold contribute anticancer activity, the corresponding anthracene-based analogs 12a,b and 13 were synthesized (Scheme 2). Generally, anthraquinone amine with or without the methoxy group was reduced to form anthracene amine, followed by acylation to form the anthracene analogs 12a and 12b. Alkylation of the 3-amino anthraquinone 10a gave the target anthraquinone analog 13 without the phenolic hydroxyl groups.
Scheme 2.
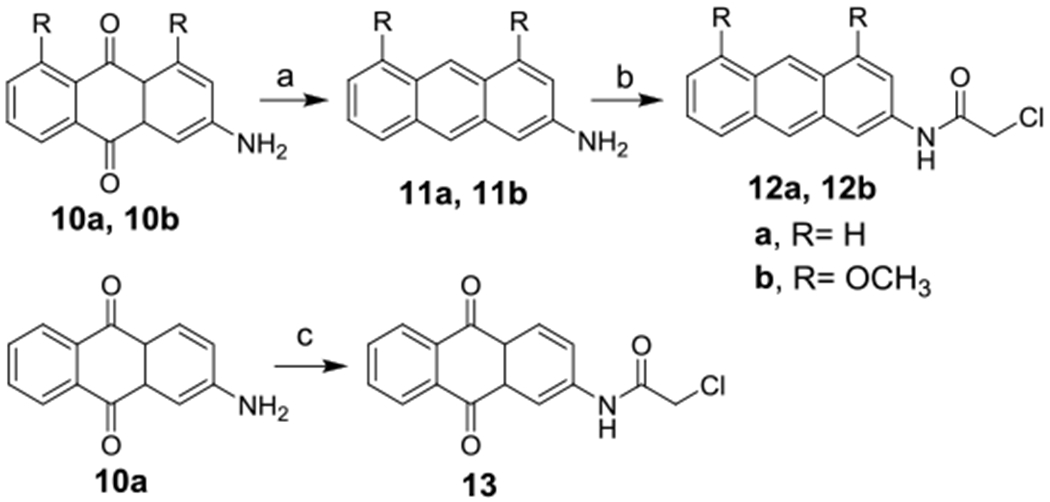
Synthesis of anthracene and anthraquinone analogs.
Modification of the alkylating group (B and C):
Analogs of AQ-101 with the NH replaced by a CH2 moiety were synthesized from the anthraquinone amine 10b (Scheme 3a). By a diazonium-iodination reaction, the key intermediate 14 was formed. Sonogashira coupling with 2-propynol followed by thionylation and hydrolysis afforded 17 with an alpha-hydroxy group. Further chlorination with N-chloro-succinimide (NCS) and triphenylphosphine (TPP) yielded target compound 18. Compounds 19 and 20 were synthesized by acylating 10b with 2-chloropropionyl chloride and chloromethyl chloroformate respectively (Schemes 3b). To synthesize compound 22, amine 10b was converted to A-sulfinyl amine 21 followed by reacting with glycolic acid (Scheme 3c). Compound 25 with the benzylic chloride group as the alkylating moiety was synthesized by chlorination of aloe emodin 24, obtained from reducing the methyl ester of rhein with LiAlH4 (Scheme 3d).
Scheme 3.
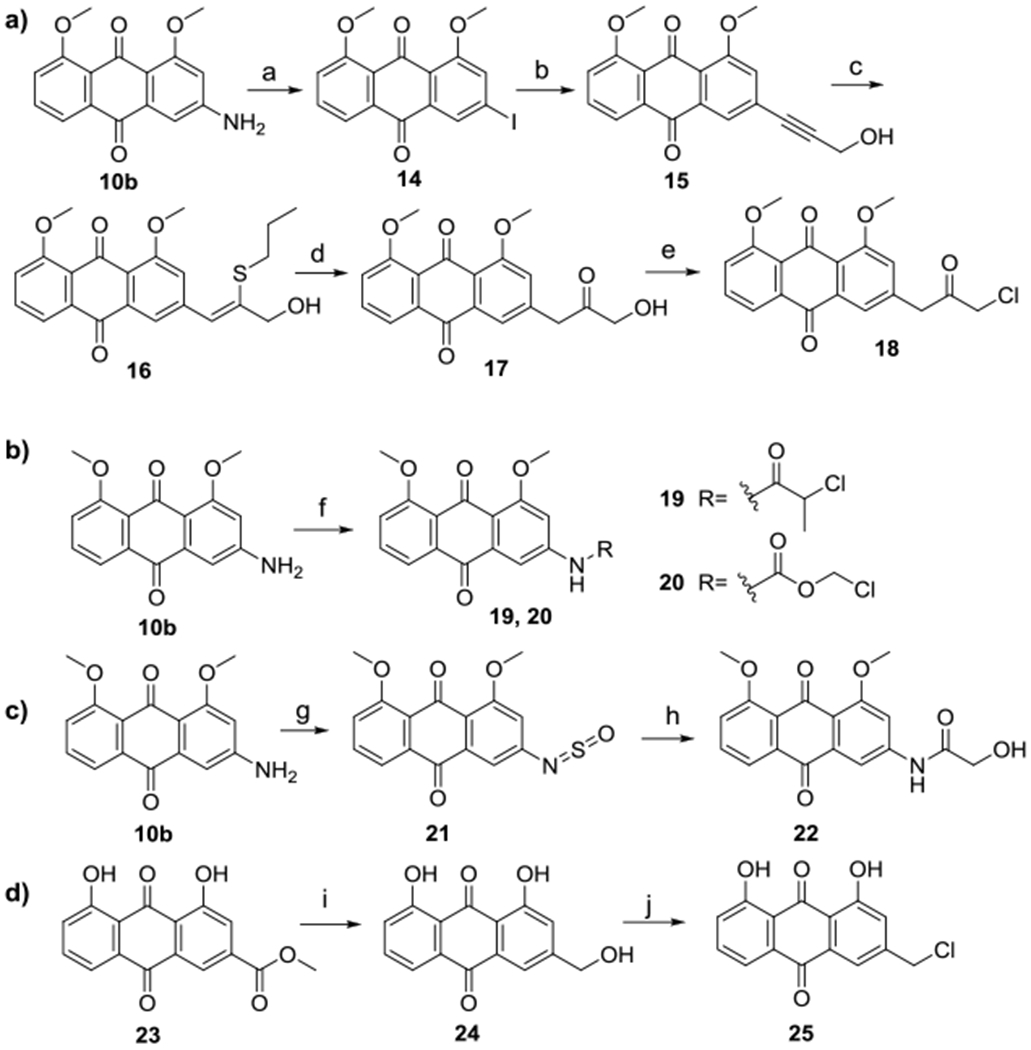
Synthesis of AQ-101 analogs with different alkylating groups.
Reagents and conditions:
(a) HNO3/H2SO4, KI, H2O/THF, −5 °C-RT, 30 min, 71%; (b) propagyl alcohol, Pd(PPh3)Cl2, CuI, TEA, dioxane, 80 °C, 6 h, 86%; (c) propanethiol, KOH, ACN, 60 °C , 30 min; (d) H2SO4, methanol, 70 °C, 2 h, 90%; (e) NCS/TPP, DCM, RT, 5 min, 94%; (f) α-methyl chloroacetyl chloride for 19 or chloromethyl chloroformate for 20, dioxane, RT, 30 min, 68-90%; (g) SOCl2, toluene, reflux, 3 h; (h) glycolic acid, ACN, RT, overnight, 45%; (i) LiAlH4, THF, 0 °C - RT, then reflux, 2 h, 65%; (j) SOCl2, DMF, RT, overnight, 83%.
Biological activity
With the structurally modified compounds in hand, the in-vitro activity against the wild type p53-expressing EU-1 ALL cells was tested using the WST-8 assay (Table 1, see Figure 2 for dose-response curves). In order to assess whether these compounds are able to induce MDM2 degradation as the lead compound AQ-101, selected analogs were subjected to Western-blot analysis to probe if they can time-dependently induce MDM2 degradation.
Table 1.
Average IC50 Values of analogs. (Experiment triplicates with 95% confidence intervals of IC50 showing in parentheses)
| Compound NO. | AQ ID | Structure | IC50 (μM) |
|---|---|---|---|
| 1 | AQ-101 | 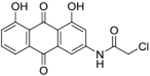 |
1.37 (0.96-1.96) |
| 3a | AQ-270 | 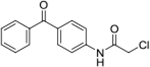 |
2.06 (1.81-2.36) |
| 3b | AQ-271 | 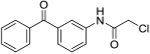 |
> 50 |
| 3c | AQ-272 | 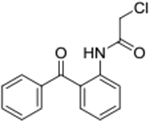 |
> 50 |
| 6 | AQ-285 | 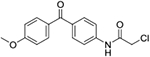 |
2.27 (2.14-2.40) |
| 9 | AQ-273 | 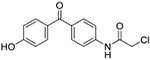 |
3.12 (2.32-4.18) |
| 12a | AQ-332 | 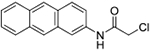 |
>50 |
| 12b | AQ-333 | 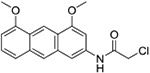 |
>50 |
| 13 | AQ-131 | 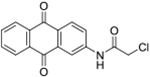 |
0.74 (0.62-0.89) |
| 17 | AQ-283 | 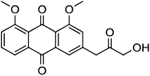 |
>50 |
| 18 | AQ-284 | 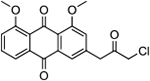 |
1.75 (1.27-2.41) |
| 19 | AQ-317 | 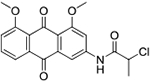 |
17.6 (16.7-18.6) |
| 20 | AQ-340 | 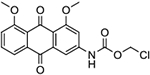 |
>50 |
| 22 | AQ-282 | 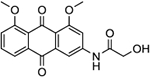 |
≈50 |
| 25 | AQ-300 | 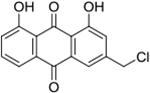 |
25.9 (25.2-26.6) |
Figure 2.
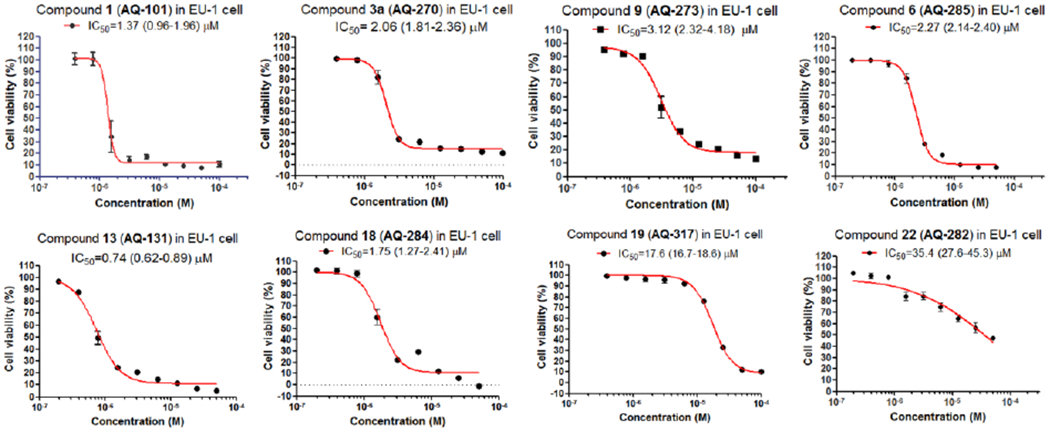
Dose-response curves of the active compounds by WST-8 assay.
Modifying the anthraquinone scaffold resulted in diverse changes in activity. Eliminating the phenolic hydroxyl groups and one ketone group to open the anthraquinone reduced the activity by about 2-fold (3a); however, it did not lead to the abolishment of the activity. The latter part is important because the flat structure of the anthraquinone moiety is a major issue that affects water solubility. Being able to pursue the ring-opened scaffolds suggests ways to further optimize this class of compounds for improved potency and solubility. Changing the substitution position from the para- to the meta- (3b) or ortho- (3c) position completely abolished activity, suggesting the critical nature of the relative positions of these functional groups. Introducing a methoxy or a hydroxy group (6 and 9 respectively) did not enhance activity. It should be noted that a study by Nomura’s group has already revealed that compound 3a was a covalent inhibitor of ubiquitin-like modifier activating enzyme 5 (UBA5), which is a potential target for pancreatic cancer (Roberts et al. 2017). As an analog of 3a, compound 9 was chosen to verify its effect on MDM2 degradation. However, compound 9 at its IC50 concentration did not induce significant changes in MDM2 levels, albeit an increase in p53 level was detected (Figure 3a), suggesting a different mechanism of action for the diphenyl ketone derivatives when compared with the lead compound AQ-101.
Figure 3.
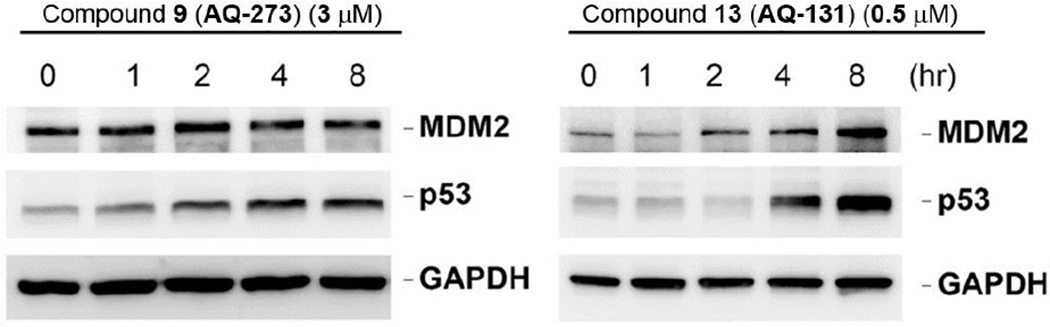
Western blot studies of protein levels in EU-1 ALL cell line showed that (a) compound 9 (AQ-273) and (b) compound 13 (AQ-131) upregulated p53 expression in a time dependent manner but did not significantly downregulate MDM2.
Reducing the anthraquinone core to anthracene (12a and 12b) abolished activity, although the methoxy group in 12b was known to help retain activity in the anthraquinone series as described previously (Draganov et al. 2019). It suggested that the alkylating group alone without attaching to a proper targeting scaffold would not induce significant cytotoxicity, which is an essential premise in designing covalent binding inhibitor (Wang et al. 2017). Our own work using α-chloroacetamide suggests the same. Elimination of the phenolic hydroxyl group in the anthraquinone scaffold (13) allowed for retention of cytotoxicity at a potency level similar with the lead compound. However, Western-blot studies did not show downregulation of MDM2. Interestingly, an increase of MDM2 level was observed along with increased p53 expression (Figure 3b). The reason for the observed phenomenon is not clear. It is possible that this was the result of transactivation of MDM2 by p53 induced by other stress signals. As a feedback loop, p53 can bind to the MDM2 promoter to increase MDM2 expression (Vousden and Prives 2009). However, more work needs to be done in the future to firmly establish the mechanism of action.
In previous studies (Draganov et al. 2019), we have demonstrated that the chloroacetamide was an optimal alkylating moiety among those tested . Preliminary pharmacokinetic studies indicated that AQ-101 “disappeared” very quickly (within minutes) from the blood after i.v. injection in mice (unpublished results). Many reasons could contribute to the short half-life including hydrolytic liability of the acetamide group, covalent binding to albumin cysteine (Turell et al. 2013), and/or fast alkylation of the intended target, thus trapping AQ-101 in the target protein. To address the possible hydrolytic liability issue, we made further modifications to this part by replacing the NH-group in the chloroacetamide group by a methylene moiety. Though the resulting analog 18 showed decreased cytotoxicity (IC50 = 1.75 μM), it is more important to note that this modification did not abolish the activity, suggesting new directions for future optimization work. Preliminary Western-blot studies showed that compound 18 induced p53 expression but did not induce appreciable time-dependent downregulation of the MDM2 in EU-1 cells (Figure 4). Similar to the results with compound 13, more work is needed to affirm the results of the study and to fully elucidate the mechanism of action.
Figure 4.
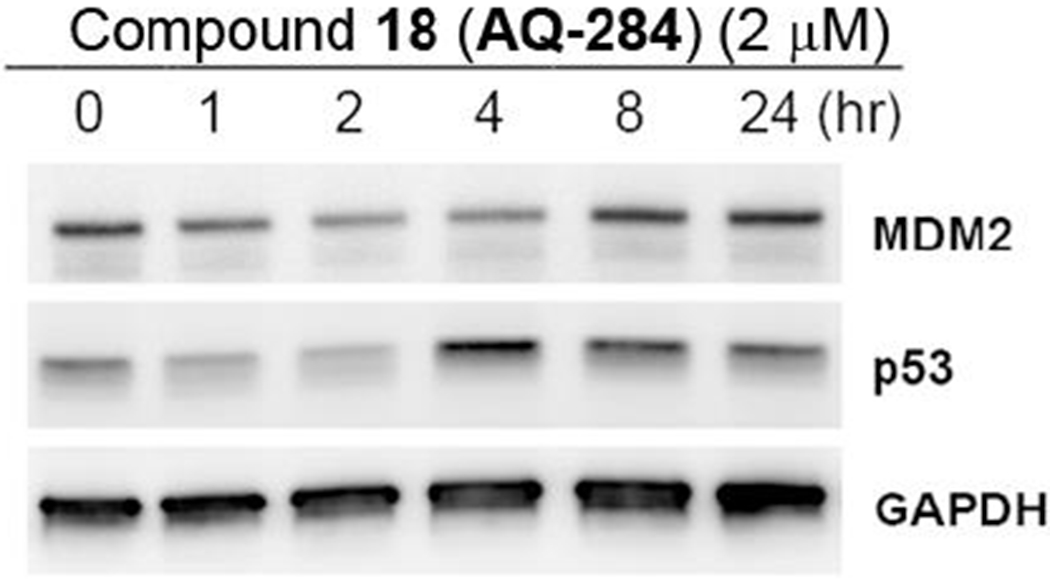
Western blot in EU-1 ALL cell line showed that compound 18 (AQ-284) upregulated p53 expression in a time-dependent manner but did not induce significant downregulation of MDM2.
Modification of the alkylating moiety by increasing steric hindrance to the alkylating moiety through alpha-methylation (19) led to decreased activity. Changing chloroacetamide to chloromethyl carbamate (20) or hydroxyacetamide (22) abolished activity, demonstrating the essential nature of the chloroacetyl group as the electrophilic moiety. However, a more chemically reactive chloromethyl analog 25 was much less active than the lead compound, presumably because of the mispositioned alkylating moiety as a result of the shortened chain.
Conclusion
In conclusion, structural modifications conducted in this study provide valuable SAR information toward the development of such anthraquinone compounds into potential therapy for treating ALL. The results showed that drastic changes such as opening the tricyclic ring or eliminating the phenolic hydroxyl group of anthraquinone core allowed the retention of activity. However, the resulting compounds may function through different mechanism(s) other than downregulating MDM2 in upregulating p53. This aspect needs more work to achieve a better understanding. The α-chloroacetamide remains the most preferable electrophilic moiety, and the N-H group of the acetamide group can be replaced, but with the possibility of modifying the detailed mechanism of action. These findings provide a more detailed picture with regard to the SAR of this class of compounds and will further guide the design of new compounds with improved potency and pharmaceutical property.
Experimental
Biology Experiments
Cytotoxicity Assay
All WST-8 cytotoxicity experiments were conducted in triplicates using Cell Counting Kit-8 (CCK-8, Dojindo, Japan). EU1 cells were cultured in RPMI-1640 medium. The medium was supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Cells were seeded into 96-well plates (3 × 105 in 100 μL volume per well). Compounds to be tested were dissolved in DMSO to make 10 mM stock solutions and then diluted with culture medium to various concentrations; final DMSO concentration was kept at 0.5%. The dissolved compounds were added immediately after seeding. Following incubation for 24 h at 37 °C in humidified atmosphere with 5% CO2; 10 μL of CCK-8 solution was added to each well, and the plate was incubated for an additional 2-4 h at 37 °C before measuring the optical density at 450 nm with a microplate reader (PerkinElmer Victor2, USA). The cell viability of each well was calculated as the percentage of the untreated control according to the manufacturer’s manual. Average IC50 values were computed based on various concentrations by non-linear regression using GraphPad Prism 5.
Western blot
5×106 EU-1 cells in 3 mL RPMI-1640 culture media (supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin) were seeded in a 6 well-plate and incubated for 12 h, then 1 mL of the drug-loaded medium was added. The cells were harvested at a designated time point, washed with cold PBS then centrifuged at 1500 rpm (3 times). The cells were lysed by adding 200 μL of cold NP-40 buffer (supplied with cOmplete™ protease Inhibitor tablet (Roche) and 1mM PMSF). The cell lysates were centrifuged at 12500 rpm at 4 °C for 10 min and total protein concentration in the supernatant was measured with the BCA assay (Thermo-Fisher). 30 μL of the cell lysate was mixed with 10 μL 4x laemmli sample buffer and denatured at 95°C for 5min. The total protein concentration was adjusted with lx laemmli sample buffer. Equal amount of protein sample was loaded onto 4-15% gradient SDS PAGE gel (Bio-rad). After electrophoresis, the protein was transfer to PTFE membrane (Bio-rad) and probed with the corresponding antibody. MDM2 ((SMP14), 1:500 Santa Cruz); p53 ((DO-1), 1:800, Santa Cruz); GAPDH ((0411), 1:2000, Santa Cruz) and HRP conjugated goat anti-mouse secondary antibody (Bio-rad, 1:2000). The chemiluminescent result was detected with Pierce ECL Plus Substrate (Thermo Scientific) and imaged with LSA4000 (GE Healthcare, Fairfield, USA).
Synthesis Experiments
Chemicals were purchased from Sigma-Aldrich, VWR, or Acros. Analytical-grade solvents were used for all reactions except for moisture-sensitive reactions, in which case anhydrous solvents were used. TLC analyses were conducted on silica gel plates (Silica GF254); and column chromatography was carried out on flash silica gel (230–400 mesh). NMR spectra were recorded at 400 MHz for 1H and 100 MHz for 13C on a Bruker Avance NMR spectrometer with TMS (δ = 0.00 ppm) or residual solvent as the internal reference. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, m = multiplet), coupling constant, and integration. High-resolution mass spectrometry (HRMS) was performed using electrospray ionization (ESI) by the GSU Mass Spectrometry Facilities.
General experimental procedure for the synthesis of compounds 3a-c:
Triethylamine (0.03 ml, 0.23 mmol) was added into a solution of commercially available starting material 2a-c (0.15 mmol) in dry dioxane (2 mL). Then, chloroacetyl chloride (0.02 ml, 0.23 mmol) was injected slowly into the mixture and the mixture was stirred at RT for 30 mins. About 10 ml of water was added to quench the reaction. The precipitate formed was filtered to afford 3a (0.035 g), 3b (0.036 g), or 3c (0.037 g) as a white solid, which was further purified by flash chromatography using ethyl acetate/hexane as the eluent.
N-(4-Benzoylphenyl)-2-chloroacetamide (3a).
Yield: 90%. 1H NMR (Chloroform-d) δ 8.56 (s, 1H), 7.90 – 7.82 (m, 2H), 7.79 (dt, J = 8.4, 1.4 Hz, 2H), 7.76 – 7.68 (m, 2H), 7.61 (td, J = 7.4, 1.4 Hz, 1H), 7.50 (td, J = 7.5, 7.1, 1.4 Hz, 2H), 4.23 (d, J = 1.2 Hz, 2H). 13C NMR (Chloroform-d) δ 195.6, 164.2, 140.5, 137.6, 133.9, 132.4, 131.5, 129.91, 128.3, 119.2, 42.9. HRMS (ESI) m/z: Calculated for C15H13O2NCl [M+H]+ 274.0630; Found 274.0629.
N-(3-Benzoylphenyl)-2-chloroacetamide (3b).
Yield: 92%. 1H NMR (Chloroform-d) δ 8.44 (s, 1H), 7.96 (ddd, J = 8.1, 2.3, 1.1 Hz, 1H), 7.89 (t, J = 1.9 Hz, 1H), 7.87 – 7.80 (m, 2H), 7.67 – 7.59 (m, 1H), 7.63 – 7.56 (m, 1H), 7.56 – 7.46 (m, 3H), 4.22 (s, 2H). 13C NMR (Chloroform-d) δ 1960, 164.1, 138.4, 137.1, 136.9, 132.7, 130.1, 129.2, 128.4, 126.8, 124.0, 121.4, 42.8. HRMS (ESI) m/z: Calculated for C15H13O2NCl [M+H]+ 274.0630; Found 274.0629.
N-(2-Benzoylphenyl)-2-chloroacetamide (3c).
Yield: 96%. 1H NMR (Chloroform-d) δ 11.64 (s, 1H), 8.63 (dd, J = 8.9, 1.2 Hz, 1H), 7.77 – 7.70 (m, 2H), 7.61 (tt, J = 7.6, 1.2 Hz, 3H), 7.54 – 7.45 (m, 2H), 7.17 (td, J = 7.6, 1.2 Hz, 1H), 4.21 (s, 2H). 13C NMR (Chloroform-d) δ 199.1, 165.4, 139.2, 138.3, 134.1, 133.5, 132.6, 130.0, 128.3, 124.2, 123.1, 121.5, 43.1. HRMS (ESI) m/z: Calculated for C15H13O2NCl [M+H]+ 274.0631; Found 274.0629.
Synthesis of (4-methoxyphenyl)(4-nitrophenyl)methanone (4):
Into a 50-ml round-bottom flask, para-nitrobenzoyl chloride (0.50 g 2.72 mmol) was added successively together with ZnCl2 (0.073 g, 0.54 mmol) and methoxybenzene (0.87 ml, 8 mmol). The neat mixture was heated to about 150 °C for about 30 mins. At the consumption of the starting material, the mixture was diluted with ethyl acetate and washed successively with HCl (100 ml, 1 M), NaHCO3 (100 ml, Saturated). The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The pure product (0.12 g) was crystallized from DCM/Hexane. Yield: 89%. 1H NMR (Chloroform-d) δ 8.35 – 8.29 (m, 2H), 7.92 – 7.85 (m, 2H), 7.85 – 7.77 (m, 2H), 7.03 – 6.95 (m, 2H), 3.91 (s, 3H). 13C NMR (Chloroform-d) δ 193.4, 164.0, 149.4, 143.7, 132.6, 130.3, 128.9, 123.4, 113.9, 55.6. HRMS (ESI) m/z: Calculated for C14H14O4NNa [M+Na]+ 280.0583; Found 280.0586.
Synthesis of compound (4-Aminophenyl)(4-methoxyphenyl)methanone (5):
Into a 100-ml round-bottomed flask, compound 4 (1.00 g, 3.89 mmol) was added and dissolved in 10 ml of methanol followed by addition of 50 mg of Pd/C. Hydrogen gas was introduced via a balloon and the reaction was left to stir overnight at room temperature. At the consumption of the starting material, the reaction mixture was filtered with a 0.45 μm syringe filter to obtain the filtrate. Methanol was removed by vacuo to give the product as a white solid (0.64 g). Yield: 72%. 1H NMR (Chloroform-d) δ 7.80 – 7.73 (m, 2H), 7.73 – 7.64 (m, 2H), 6.99 – 6.91 (m, 2H), 6.71 – 6.62 (m, 2H), 4.32 (s, 2H), 3.86 (s, 3H). 13C NMR (Chloroform-d) δ 194.4, 162.4, 151.0, 132.6, 132.0, 131.3, 127.6, 113.6, 113.3, 55.4. HRMS (ESI) m/z: Calculated for C14H13O2NNa [M+Na]+ 250.0832; Found 250.0832.
Synthesis of 2-Chloro-N-(4-(4-methoxybenzoyl)phenyl)acetamide (6):
Triethylamine (0.66 mmol) was added into a solution of 5 (0.10 g, 0.44 mmol) in dry dioxane (2 mL). Then, chloroacetyl chloride (0.05 ml, 0.66 mmol) was slowly injected into the mixture, and the mixture was stirred at RT for 30 mins. Then, about 10 ml of water was added. The precipitate formed was filtered to afford the desired products as a pink solid (0.12 g). Yield: 93%. 1H NMR (Chloroform-d) δ 8.53 (s, 1H), 7.86 – 7.78 (m, 4H), 7.74 – 7.67 (m, 2H), 7.02 – 6.94 (m, 2H), 4.24 (s, 2H), 3.91 (s, 3H). 13C NMR (Chloroform-d) δ 194.4, 164.1, 163.2, 140.1, 134.6, 132.4, 131.2, 130.1, 119.1, 113.6, 55.5, 42.9. HRMS (ESI) m/z: Calculated for C16H14O3NClNa [M+Na]+ 326.0573; Found 326.0560.
Synthesis of (4-Hydroxyphenyl)(4-nitrophenyl)methanone (7):
Into a 20ml sealed tube, compound 4 (0.25 g, 0.97 mmol) was added and dissolved in 5 ml of dry DCM followed by slowly injection of BBr3 (1M in DCM, 3.9 ml, 3.9 mmol). The reaction was then stirred at 40 °Cin a sealed tube overnight. At the consumption of the starting material, water (10 ml) was added, and the mixture stirred for another 1 hour. At the consumption of the starting material, the mixture was extracted with ethyl acetate, washed with brine, and purified by silica gel column chromatography (Hex/EA: 9/1) to yield 0.11 g of 7. Yield: 45%. 1H NMR (DMSO-d6) δ 10.63 (s, 1H), 8.40 – 8.31 (m, 2H), 7.95 – 7.84 (m, 2H), 7.76 – 7.62 (m, 2H), 6.96 – 6.88 (m, 2H). 13C NMR (DMSO-d6) δ 193.3, 163.2, 149.4, 144.3, 133.2, 130.7, 130.6, 127.4, 124.1, 124.0, 116.0. HRMS (ESI) m/z: Calculated for C13H9O4NNa [M+Na]+ 266.0428; Found 266.0429.
Synthesis of (4-Aminophenyl)(4-hydroxyphenyl)methanone (8):
Into a 25 ml vial, 7 (0.05 g, 0.20 mmol) was added and dissolved in 5 ml of methanol followed by addition of 5 mg of Pd/C. Hydrogen gas was introduced via a balloon and the reaction was left to stir overnight at room temperature. At the consumption of the starting material, the reaction mixture was filtered with a 0.45 μm syringe filter to obtain the filtrate. Then, methanol was removed by vacuo to give the product as a white solid (0.027 g). Yield: 68%. 1H NMR (DMSO-d6) δ 10.15 (s, 1H), 7.54 (dd, J = 8.6, 1.7 Hz, 4H), 6.86 (dd, J = 8.6, 1.7 Hz, 2H), 6.63 – 6.55 (m, 2H), 6.02 (s, 2H). 13C NMR (DMSO-d6) δ 192.8, 161.0, 153.5, 132.6, 132.0, 130.1, 125.0, 115.2, 112.9. HRMS (ESI) m/z: Calculated for C13H11O2NNa [M+Na]+ 236.0676; Found 236.0687.
Synthesis of 2-Chloro-N-(4-(4-hydroxybenzoyl)phenyl)acetamide (9):
Triethylamine (0.02 ml, 0.10 mmol) was added into a solution of 8 (0.015 g, 0.07 mmol) in dry dioxane (2 mL). Then, chloroacetyl chloride (0.01 ml, 0.10 mmol) was injected slowly into the mixture, and the mixture was stirred at RT for 30 mins. After completion of the reaction monitored by TLC, about 10 ml of water was added. The precipitate formed was filtered to afford the desired product as a white solid (0.008 g). Yield: 39%. 1H NMR (DMSO-d6) δ 10.63 (s, 1H), 10.38 (s, 1H), 7.79 – 7.62 (m, 6H), 6.94 – 6.86 (m, 2H), 4.32 (s, 2H). 13C NMR (DMSO-d6) δ 193.6, 165.6, 162.1, 142.2, 133.4, 132.7, 131.1, 128.6, 119.0, 115.6. HRMS (ESI) m/z: Calculated for C15H12O3NClNa [M+Na]+ 312.0417; Found 312.0403.
Synthesis of Anthracen-2-amine compound (11a):
Into a 100-ml round-bottom flask, 10a (0.5 g, 2.24 mmol) was added followed by addition of 20 ml of NaOH (8%, aqueous solution). Then, zinc dust (2.00 g, 30.00 mmol) was added, and the mixture was stirred at reflux overnight. At the consumption of the starting material, the mixture was neutralized with 30 ml of HC1 (10%). The mixture was extracted with dichloromethane (30 ml), and the organic layer was washed with water (200 ml) and brine (200 ml). The resultant organic portion was dried over anhydrous sodium sulfate and purified by silica-gel column chromatography to obtain 11a (0.33 g). Yield: 75%. 1H NMR (DMSO-d6) δ 8.28 (s, 1H), 8.03 (s, 1H), 7.85 – 7.93 (m, 2H), 7.81 (d, J = 9.0 Hz, 1H), 7.41 – 7.33 (m, 1H), 7.29 (dd, J = 8.4, 6.5 Hz, 1H), 7.05 (dd, J = 9.0, 2.2 Hz, 1H), 6.89 (d, J = 2.1 Hz, 1H), 5.56 (s, 2H). 13C NMR (DMSO-d6) δ 146.5, 134.1, 132.4, 129.4, 128.9, 128.5, 127.5, 127.0, 126.2, 125.6, 123.5, 121.4, 121.3, 103.2. HRMS (ESI) m/z: Calculated for C14H12N [M+H]+ 194.0960; Found 194.0964.
Synthesis of compound N-(Anthracen-2-yl)-2-chloroacetamide (12a):
Triethylamine (0.03 ml, 0.23 mmol) was added into a solution of 11a (0.03 g, 0.16 mmol) in dry dioxane (2 mL). Then, chloroacetyl chloride (0.02 ml, 0.23 mmol) was injected slowly into the mixture and the mixture was stirred at RT for 30 mins. Then, about 10 ml of water was added. The precipitate formed was filtered to afford the desired product as a white solid (0.04 g). Yield: 91%. 1H NMR (DMSOd6) δ 10.58 (s, 1H), 8.47 – 8.55 (m, 3H), 8.16 – 7.94 (m, 3H), 7.64 – 7.38 (m, 3H), 4.35 (s, 2H). 13C NMR (DMSO-d6) δ 165.5, 135.8, 132.2, 131.8, 131.0, 129.5, 129.1, 128.5, 128.2, 126.4, 126.2, 125.7, 125.6, 121.1, 115.0, 44.1. HRMS (ESI) m/z: Calculated for C16H12ClNO [M+H]+ 270.0678; Found 270.0680.
Synthesis of compound 4,5-Dimethoxyanthracen-2-amine (11b):
In a 100-ml round-bottom flask was added compound 10b (0.07 g, 0.25 mmol) followed by the addition of 10 ml of NaOH (8%, aqueous solution). Then, zinc dust (229 mg, 3.52 mmol) was added, and the mixture was stirred at reflux overnight. At the consumption of the starting material, the mixture was neutralized with 12 ml of HCl (10%). The mixture was then extracted with dichloromethane (10 ml) and the organic layer was washed with water (100 ml) and brine (100 ml). The resultant organic portion was dried over anhydrous sodium sulfate and purified by silica-gel column chromatography to obtain a grey solid (0.04 g). Yield: 64%. 1H NMR (Chloroform-d) δ 9.08 (s, 1H), 8.00 (s, 1H), 7.46 (d, J = 8.5 Hz, 1H), 7.32 (dd, J = 8.5, 7.4 Hz, 1H), 6.70 (t, J = 1.3 Hz, 1H), 6.64 (d, J = 7.3 Hz, 1H), 6.28 (d, J = 2.0 Hz, 1H), 4.07 (d, J = 6.0 Hz, 6H). 13C NMR (DMSO-d6) δ 156.2, 155.8, 135.0, 133.6, 126.1, 121.0, 120.7, 119.7, 119.6, 115.0, 100.7, 98.2, 55.8, 55.7. HRMS (ESI) m/z: Calculated for C16H16NO2 [M+H]+ 254.1172; Found 254.1176.
Synthesis of 2-Chloro-N-(4,5-dimethoxyanthracen-2-yl)acetamide (12b):
Triethylamine (0.02 ml, 0.18 mmol) was added into a solution of 11b (0.03 g, 0.12 mmol) in dry dioxane (2 mL). Then, chloroacetyl chloride (0.01 ml, 0.18 mmol) was injected slowly into the mixture and the mixture was stirred at RT for 30 mins. At the consumption of the starting material, about 10 ml of water was added. The precipitate formed was filtered to afford the desired product as a white solid (0.034 g). Yield: 87%. 1H NMR (DMSO-d6) δ 10.52 (s, 1H), 8.97 (s, 1H), 8.36 (s, 1H), 8.06 (s, 1H), 7.58 (d, J = 8.5 Hz, 1H), 7.42 (t, J = 8.0 Hz, 1H), 7.01 (d, J = 1.7 Hz, 1H), 6.86 (d, J = 7.4 Hz, 1H), 4.34 (s, 2H), 4.04 (d, J = 3.7 Hz, 6H). 13C NMR (Chloroform-d) δ 157.1, 156.1, 144.0, 134.2, 133.7, 125.6, 122.3, 121.7, 120.7, 119.9, 119.7, 115.7, 100.3, 99.0, 96.7, 55.5, 55.4. HRMS (ESI) m/z: Calculated for C18H17NClO3 [M+H]+ 330.0889; Found 330.0891.
Synthesis of 2-Chloro-N-(9,10-dioxo-9,10-dihydroanthracen-2-yl)acetamide (13):
Triethylamine (0.03 ml, 0.20 mmol) was added into a solution of 10a (0.03 g, 0.13 mmol) in dry dioxane (2 mL). Then, chloroacetyl chloride (0.02 ml, 0.20 mmol) was injected slowly into the mixture and the mixture was stirred at RT for 30 mins. Then, about 10 ml of water was added. The precipitate formed was filtered to afford the desired products as a white solid. Isolated yield: 46%. 1H NMR (Chloroform-d) δ 8.62 (s, 1H), 8.41 – 8.27 (m, 4H), 8.25 (d, J = 2.3 Hz, 1H), 7.89 – 7.78 (m, 2H), 4.29 (s, 2H). 13C NMR (DMSO-d6) δ 182.8, 181.8, 166.0, 144.3, 135.0, 134.7, 134.6, 133.5, 133.5, 129.0, 128.8, 127.2, 127.1, 124.5, 116.5, 44.0. HRMS (ESI) m/z: Calculated for C16H11NC103 [M+H]+ 300.0423; Found 300.0422.
Synthesis of 3-Iodo-1,8-dimethoxyanthracene-9,10-dione (14):
Compounds 10b (0.55 mmol) was dissolved in 5 ml of THF and 5 ml of water. Then, 2 M H2SO4 (1 ml) was added and the reaction mixtures; the mixture was stirred at −5 °C for about 10 mins. Sodium nitrite (0.045 g, 0.66 mmol) in 0.5 ml of water was then added and the mixture was stirred at −5 °C for 5 mins. KI (0.18 g, 1.10 mmol) was then added gradually while gas evolution was observed. When the reaction was completed, the mixture was diluted with water and the resulting black precipitate was filtered. The crude product was purified by silica-gel column chromatography to obtain compounds as a bright Yellow solid. Yield: 0.13g (71%). 1H NMR (Chloroform-d) δ 8.13 (s, 1H), 7.79 (d, J = 7.7 Hz, 1H), 7.67 – 7.57 (m, 2H), 7.31 (s, 1H), 3.99 (d, J = 2.9 Hz, 6H). 13C NMR (Chloroform-d) δ 182.7, 182.1, 159.5, 159.4, 134.8, 134.1, 128.0, 127.0, 123.6, 123.1, 119.0, 118.3, 100.5, 56.8, 56.5. HRMS (ESI) m/z: Calculated for C16H12O4I [M+H]+ 394.9773; Found 394.9775.
Synthesis of 3-(3-hydroxyprop-1-yn-1-yl)-1,8-dimethoxyanthracene-9,10-dione (15):
Compounds 14 (0.50 mmol) was dissolved in 10 ml of dioxane. Propargyl alcohol (0.04 ml, 0.61 mmol) was then added, followed by addition of 5 ml of TEA as a co-solvent. Under argon, Pd(PPh3)2Cl2 (0.018 g, 0.03 mmol) and Cul (0.01 g, 0.05 mmol) was quickly added. The mixture was then stirred at 80 °C for about 5 hours. At the consumption of the starting material, the solvent was removed in vacuo; the residue was dissolved with DCM and washed with water (150 ml) and brine (150 ml) successively and dried over anhydrous sodium sulphate. The solvent was removed, and the crude product was purified by silica-gel column chromatography (DCM/MeOH, 200/1). The product was isolated as a yellow solid. Yield: 0.14 g (86%). 1H NMR (DMSO-d6) δ 7.72 (t, J = 8.0 Hz, 1H), 7.63 (dd, J = 7.7, 1.1 Hz, 1H), 7.56 (d, J = 1.5 Hz, 1H), 7.51 (dd, J = 8.5, 1.2 Hz, 1H), 7.45 (d, J = 1.5 Hz, 1H), 5.48 (t, J = 6.0 Hz, 1H), 4.37 (d, J = 6.0 Hz, 2H), 3.91 (d, J = 6.2 Hz, 6H). 13C NMR (DMSO-d6) δ 183.0, 180.9, 159.2, 159.2, 134.7, 134.6, 134.2, 128.3, 123.7, 123.4, 120.8, 120.8, 119.5, 118.6, 94.2, 82.8, 56.7, 56.7, 49.9. HRMS (ESI) m/z: Calculated for C19H14O5Na [M+Na]+ 345.0727; Found 345.0739.
Synthesis of 3-(3-Hydroxy-2-oxopropyl)-1,8-dimethoxyanthracene-9,10-dione (17):
Into a 20-ml vial, compound 15 (0.16 mmol) was dissolved in acetonitrile (4 ml). Then, propanethiol (0.02 ml, 0.18 mmol) was slowly added at room temperature and catalytic amount of potassium hydroxide (0.03 g, 0.0 5 mmol) in 0.5 ml of water was added and the mixture stirred to 80 °C The starting material was consumed after about 30 mins. Then the solvent was removed, and the crude product 16 was used without purification for the next hydrolysis step. Into 25-ml vial, 16 (0.15 mmol) was dissolved in 5 ml of methanol and 2 M H2SO4 (5 ml) and the mixture was allowed to stir at 80 °C for about 4 hours. At the consumption of the starting material, the mixture was extracted with DCM (5 ml) and dried over anhydrous sodium sulphate. The crude product was purified by silica-gel column chromatography (DCM/MeOH, 200/2) to obtain a yellow solid. Yield: 0.046 g (90%). 1HNMR (Chloroform-d) δ 7.86 (dd, J = 7.7, 1.1 Hz, 1H), 7.72 – 7.63 (m, 2H), 7.34 (s, 1H), 7.34 (d, J = 8.4 Hz, 1H), 7.19 (d, 7= 1.6 Hz, 1H), 4.41 (s, 2H), 4.03 (s, 6H), 3.88 (s, 2H). 13C NMR (Chloroform-d) δ 205.7, 183.8, 182.1, 159.9, 159.5, 139.0, 135.0, 134.6, 134.0, 123.8, 123.2, 119.6, 119.0, 118.8, 118.2, 68.1, 56.6, 56.5, 45.5. HRMS (ESI) m/z: Calculated for C19H16O6Na [M+Na]+ 363.0830; Found 363.0845.
Synthesis of 3-(3-Chloro-2-oxopropyl)-1,8-dimethoxyanthracene-9,10-dione (18):
Into a 20-ml vial, N-chloro-succinimide (0.023 g, 0.17 mmol) and triphenyl phosphene (0.046 g, 0.17 mmol) were dissolved in 2 ml of DCM, and the mixture stirred under argon for about 2 min. Then, 17 (0.15 mmol) in 1 ml of DCM was gradually added and the mixture stirred at room temperature for about 5 min. TLC showed consumption of the starting material and the formation of the product. The mixture was washed with water (50 ml) and brine (50 ml) successively and dried over anhydrous sodium sulphate. The crude product was purified by silica-gel column chromatography (DCM only) to obtain a yellow solid. Yield: 94%. 1H NMR (Chloroform-d) δ 7.83 (dd, J = 7.7, 1.1 Hz, 1H), 7.67 (d, J = 1.6 Hz, 1H), 7.64 (t, J = 8.0 Hz, 1H), 7.31 (dd, J = 8.4, 1.1 Hz, 1H), 7.16 (d,J = 1.6 Hz, 1H), 4.18 (s, 2H), 4.04 (s, 2H), 4.01 (d, J = 1.7 Hz, 6H). 13C NMR (Chloroform-d) δ 198.8, 183.8, 182.4, 159.8, 159.5, 139.2, 135.0, 134.6, 133.9, 123.8, 123.1, 119.8, 119.0, 118.2, 56.6, 56.5, 47.8, 46.5, 29.7. HRMS (ESI) m/z: Calculated for C19H15O5ClNa [M+Na]+ 381.0514; Found 381.0506.
Synthesis of 2-Chloro-N-(4,5-dimethoxy-9,10-dioxo-9,10-dihydroanthracen-2-yl)propenamide (19):
Triethylamine (0.01 ml, 0.08 mmol) was added into a solution of 10b (0.020 g, 0.08 mmol) in dry dioxane (2 mL). Then, 2-chloropropionyl chloride (0.01 ml, 0.08 mmol) was injected slowly into the mixture and the mixture was stirred at room temperature for 10 mins. Next, about 10 ml of water was added and the mixture was extracted with DCM, washed with brine, and dried over anhydrous sodium sulphate. The solvent was removed, and the crude product was purified by silica-gel column chromatography (DCM/MeOH, 150/1). Compound 19 was isolated as a yellow solid (0.027 g). Yield: 90%. 1H NMR (DMSO-d6) δ 10.89 (s, 1H), 7.94 (d, J = 1.9 Hz, 1H), 7.84 (d, J = 2.0 Hz, 1H), 7.75 (t, J = 7.9 Hz, 1H), 7.69 (dd, J = 7.7, 1.2 Hz, 1H), 7.54 (d, J = 8.1 Hz, 1H), 4.71 (q, J = 6.6 Hz, 1H), 3.90 (d, J = 5.3 Hz, 6H), 1.65 (d, J = 6.6 Hz, 3H). 13C NMR (DMSO-d6)d 183.6, 181.7, 168.0, 161.2, 159.7, 142.1, 135.4, 134.6, 133.8, 123.9, 120.3, 119.0, 118.4, 108.9, 108.4, 56.6, 56.5, 55.9, 53.4, 22.4. HRMS (ESI) m/z: Calculated for C19H17NClO5 [M+H]+ 374.0788; Found 374.0790.
Synthesis of Chloromethyl-N-(4,5-dimethoxy-9,10-dioxo-9,10-dihydroanthracen-2-yl)carbamate (20):
Triethylamine (0.01 ml, 0.08 mmol) was added into a solution of 10b (0.020 g, 0.08 mmol) in dry dioxane (2 mL). Then, chloromethyl chloroformate (0.01 ml, 0.10 mmol) was injected slowly into the mixture and the mixture was stirred at room temperature for 10 mins. At the complete consumption of the starting material, the solvent was removed in vacuo, and the crude product was purified by silica-gel column chromatography (Hexane: Ethyl acetate, 1/1). Compound 27 was isolated as a yellow solid (0.018 g). Yield: 68%. 1H NMR (CD3CN) δ 8.52 (s, 1H), 7.78 – 7.61 (m, 4H), 7.44 (d, J = 7.8 Hz, 1H), 5.89 (s, 2H), 3.94 (s, 6H). 13C NMR (DMSO-d6) δ 183.7, 181.1, 160.4, 159.2, 151.4, 143.8, 135.4, 134.8, 134.3, 123.4, 119.6, 119.1, 118.7, 107.8, 82.2, 71.4, 56.7, 56.5. HRMS (ESI) m/z: Calculated for C18H14ClNO6 [M+Na]+ 398.0407; Found 398.2289.
Synthesis of N-(4,5-Dimethoxy-9,10-dioxo-9,10-dihydroanthracen-2-yl)-2-hydroxyacetamide (22)
Compound 10b (0.50 g, 1.76 mmol) was added into a 50 ml round-bottom flask followed by 10 ml of toluene. Then, 5ml of thionyl chloride was added and the mixture was stirred at reflux for about 3 hours. At the consumption of the starting material, the solvent was removed in vacuo to get a the intermediate 21 which was used directly next step without purification. Compound 21 was then transferred into a 20-ml vial containing 5 ml of acetonitrile. Glycolic acid (3.52 mmol) was added and the mixture was stirred at room temperature overnight. At the consumption of 21, acetonitrile was removed by vacuo, and the residue was diluted with water and extracted with DCM (5 ml, x 3). The combined organic layer was washed with water (50 ml) and brine (50 ml) successively and dried over anhydrous sodium sulphate. The solvent was removed, and the crude product was purified by silica-gel column chromatography (DCM/MeOH, 200/1). The product was isolated as a yellow solid (0.26 g). Yield: 45%. 1H NMR (Chloroform-d) δ 8.74 (s, 1H), 8.23 (s, 1H), 7.85 (d, J = 7.6 Hz, 1H), 7.66 (t, J = 8.0 Hz, 1H), 7.49 (d, J = 2.0 Hz, 1H), 7.34 (d, J = 8.4 Hz, 1H), 5.32 (s, 2H), 4.34 (d, J = 5.2 Hz, 2H), 4.03 (d, J = 2.2 Hz, 6H), 2.92 (s, 1H). 13C NMR (DMSO-d6) δ 183.7, 180.7, 172.4, 160.2, 159.2, 144.1, 135.3, 134.5, 134.4, 123.8, 119.5, 119.4, 118.6, 109.0, 109.0, 62.4, 56.7, 56.6. HRMS (ESI) m/z: Calculated for C18H16O6N [M+H]+ 342.0972; Found 342.0972.
Synthesis of 1,8-Dihydroxy-3-(hydroxymethyl)anthracene-9,10-dione (24):
Into a 100 ml round-bottom flask, 0.1 g of 23 (0.34 mmol) was dissolved in 5 ml of THF. Then, 0.026 g of LiAlH4 was added at 0 °C and the mixture was stirred at reflux and stirring continued for about 2 hours. At the consumption of the starting material, the mixture was cooled to 0 °C and acidified with dilute H2SO4 (1 ml). Then, the reaction mixture was extracted with DCM (50 ml) and washed with brine (100 ml) and dried over anhydrous sodium sulphate. The solvent was removed, and the crude product was purified by silica-gel column chromatography (DCM /MeOH, 150/1). The desired product was isolated as a yellow solid (0.066 g). Yield: 65%. 1H NMR (DMSO-d6) δ 11.93 (s, 1H), 11.87 (s, 1H), 7.78 (t, J = 7.9 Hz, 1H), 7.72 – 7.62 (m, 2H), 7.55 (s, 1H), 7.35 (dd, J = 8.4, 1.2 Hz, 1H), 7.25 (d, J = 1.5 Hz, 1H), 5.60 (s, 1H), 4.62 (s, 2H). 13C NMR (DMS0-d6) δ 192.0, 181.8, 162.0, 161.7, 154.1, 137.7, 133.7, 133.4, 124.8, 121.1, 119.7, 117.5, 116.2, 114.8, 62.5. HRMS (ESI) m/z: Calculated for C15H15O5 [M+H]+ 271.0599; Found 271.0601.
Synthesis of 3-(Chloromethyl)-1,8-dihydroxyanthracene-9,10-dione (25):
Compound 24 (0.5 g, 1.9 mmol) was added into a 20 ml vial followed by anhydrous DMF (20 mL). Then, 2 ml of thionyl chloride was slowly added under stirring at room temperature and the reaction was stirred overnight. At the consumption of the starting material, the solvent was removed in vacuo and the mixture was diluted with water and extracted with DCM (5 ml, x3). The combined organic layer was washed with water (50 ml) and brine (50 ml) successively and dried over anhydrous sodium sulphate. The solvent was removed, and the crude product was purified by silica-gel column chromatography (DCM only). The desired product was isolated as a yellow solid (0.44 g). Yield: 83%. 1H NMR (Chloroform-d) δ 12.10 (s, 1H), 12.04 (s, 1H), 7.90 – 7.84 (m, 1H), 7.86 (s, 1H), 7.73 (t, J = 8.0 Hz, 1H), 7.40 – 7.31 (m, 2H), 4.64 (s, 2H). 13C NMR (Chloroform-d) δ 192.6, 181.3, 162.8, 162.6, 147.1, 137.4, 134.0, 133.4, 124.8, 123.7, 120.2, 119.8, 115.7, 115.4, 44.6. HRMS (ESI) m/z: Calculated for C15H10O4Cl [M+H]+ 289.0262; Found 289.0262.
Supplementary Material
Acknowledgments
Financial support from the National Institutes of Health (CA180519) is gratefully acknowledged. We also thank the Center for Diagnostics and Therapeutics for a University fellowship to AA, the Georgia Research Alliance for an Eminent Scholar endowment, and GSU internal support.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Supporting Information
The Supporting Information for the NMR spectrum is available on line.
Conflict of interest
The authors declare that they have no conflict of interest.
References
- Anifowose A, Yuan Z, Yang X, Pan Z, Zheng Y, Zhang Z, Wang B (2020) Upregulation of p53 through induction of MDM2 degradation: Amino acid prodrugs of anthraquinone analogs. Bioorg Med Chem Lett 30:126786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68:394–424 [DOI] [PubMed] [Google Scholar]
- Chang HY, Rodriguez V, Narboni G, Bodey GP, Luna MA, Freireich EJ (1976) Causes of death in adults with acute leukemia. Medicine (Baltimore: ) 55:259–268 [DOI] [PubMed] [Google Scholar]
- Draganov AB, Yang X, Anifowose A, De La Cruz LKC, Dai C, Ni N, Chen W, De Los Santos Z, Gu L, Zhou M, Wang B (2019) Upregulation of p53 through induction of MDM2 degradation: Anthraquinone analogs. Bioorg Med Chem 27:3860–3865 [DOI] [PubMed] [Google Scholar]
- Etzel RA (2016) Foreword: Childhood Leukemia and Primary Prevention. Curr Probl Pediatr Adolesc Health Care 46:315–316 [DOI] [PubMed] [Google Scholar]
- Gaidano G, Ballerini P, Gong JZ, Inghirami G, Neri A, Newcomb EW, Magrath IT, Knowles DM, Dalla-Favera R (1991) p53 mutations in human lymphoid malignancies: association with Burkitt lymphoma and chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 88:5413–5417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L, Zhang H, Liu T, Draganov A, Yi S, Wang B, Zhou M (2018) Inhibition of MDM2 by a Rhein-Derived Compound AQ-101 Suppresses Cancer Development in SCID Mice. Mol Cancer Ther 17:497–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, Oren M (1997) Mdm2 promotes the rapid degradation of p53. Nature 387:296–299 [DOI] [PubMed] [Google Scholar]
- Hunger SP, Teachey DT, Grupp S, Aplenc R (2020) 93 - Childhood Leukemia In: Niederhuber JE, Armitage JO, Kastan MB, Doroshow JH, Tepper JE (eds) Abeloff’s Clinical Oncology (Sixth Edition). Content Repository Only!, Philadelphia, pp 1748–1764.e1744 [Google Scholar]
- Hutter JJ (2010) Childhood leukemia. Pediatr Rev 31:234–241 [DOI] [PubMed] [Google Scholar]
- Lozanski G, Heerema NA, Flinn IW, Smith L, Harbison J, Webb J, Moran M, Lucas M, Lin T, Hackbarth ML, Proffitt JH, Lucas D, Grever MR, Byrd JC (2004) Alemtuzumab is an effective therapy for chronic lymphocytic leukemia with p53 mutations and deletions. Blood 103:3278–3281 [DOI] [PubMed] [Google Scholar]
- Malik EM, Muller CE (2016) Anthraquinones As Pharmacological Tools and Drugs. Med Res Rev 36:705–748 [DOI] [PubMed] [Google Scholar]
- Momand J, Zambetti GP, Olson DC, George D, Levine AJ (1992) The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69:1237–1245 [DOI] [PubMed] [Google Scholar]
- Prokocimer M, Molchadsky A, Rotter V (2017) Dysfunctional diversity of p53 proteins in adult acute myeloid leukemia: projections on diagnostic workup and therapy. Blood 130:699–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AM, Miyamoto DK, Huffman TR, Bateman LA, Ives AN, Akopian D, Heslin MJ, Contreras CM, Rape M, Skibola CF, Nomura DK (2017) Chemoproteomic Screening of Covalent Ligands Reveals UBA5 As a Novel Pancreatic Cancer Target. ACS Chem Biol 12:899–904 [DOI] [PubMed] [Google Scholar]
- Slats AM, Egeler RM, van der Does-van den Berg A, Korbijn C, Hahlen K, Kamps WA, Veerman AJ, Zwaan CM (2005) Causes of death--other than progressive leukemia--in childhood acute lymphoblastic (ALL) and myeloid leukemia (AML): the Dutch Childhood Oncology Group experience. Leukemia 19:537–544 [DOI] [PubMed] [Google Scholar]
- Terwilliger T, Abdul-Hay M (2017) Acute lymphoblastic leukemia: a comprehensive review and 2017 update. Blood Cancer J 7:e577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turell L, Radi R, Alvarez B (2013) The thiol pool in human plasma: the central contribution of albumin to redox processes. Free Radic Biol Med 65:244–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden KH, Prives C (2009) Blinded by the Light: The Growing Complexity of p53. Cell 137:413–431 [DOI] [PubMed] [Google Scholar]
- Wade M, Li YC, Wahl GM (2013) MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer 13:83–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zhao J, Yao Y, Wang C, Zhang J, Shu X, Sun X, Li Y, Liu K, Yuan H, Ma X (2017) Covalent binding design strategy: A prospective method for discovery of potent targeted anticancer agents. Eur J Med Chem 142:493–505 [DOI] [PubMed] [Google Scholar]
- Watts J, Nimer S (2018) Recent advances in the understanding and treatment of acute myeloid leukemia. F1000Res 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Sun G, Yang C, Wang B (2011) Novel rhein analogues as potential anticancer agents. ChemMedChem 6:2294–2301 [DOI] [PubMed] [Google Scholar]
- Zhao Y, Wang Y, Ma S (2018) Racial Differences in Four Leukemia Subtypes: Comprehensive Descriptive Epidemiology. Sci Rep 8:548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou GM (2007) Cancer stem cells in leukemia, recent advances. J Cell Physiol 213:440–444. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.