

Abstract
The type I interferon response is an important innate antiviral pathway. Recognition of viral RNA by RIG-I-like receptors (RLRs) activates a signaling cascade that leads to type I interferon (IFN-α/β) gene transcription. Multiple proteins in this signaling pathway (e.g. RIG-I, MDA5, MAVS, TBK1, IRF3) are regulated by (de)ubiquitination events. Most viruses have evolved mechanisms to counter this antiviral response. The leader protease (Lpro) of foot-and-mouth-disease virus (FMDV) has been recognized to reduce IFN-α/β gene transcription; however, the exact mechanism is unknown. The proteolytic activity of Lpro is vital for releasing itself from the viral polyprotein and for cleaving and degrading specific host cell proteins, such as eIF4G and NF-κB. In addition, Lpro has been demonstrated to have deubiquitination/deISGylation activity. Lpro’s deubiquitination/deISGylation activity and the cleavage/degradation of signaling proteins have both been postulated to be important for reduced IFN-α/β gene transcription. Here, we demonstrate that TBK1, the kinase that phosphorylates and activates the transcription factor IRF3, is cleaved by Lpro in FMDV-infected cells as well as in cells infected with a recombinant EMCV expressing Lpro. In vitro cleavage experiments revealed that Lpro cleaves TBK1 at residues 692–694. We also observed cleavage of MAVS in HeLa cells infected with EMCV-Lpro, but only observed decreasing levels of MAVS in FMDV-infected porcine LFPK αVβ6 cells. We set out to dissect Lpro’s ability to cleave RLR signaling proteins from its deubiquitination/deISGylation activity to determine their relative contributions to the reduction of IFN-α/β gene transcription. The introduction of specific mutations, of which several were based on the recently published structure of Lpro in complex with ISG15, allowed us to identify specific amino acid substitutions that separate the different proteolytic activities of Lpro. Characterization of the effects of these mutations revealed that Lpro’s ability to cleave RLR signaling proteins but not its deubiquitination/deISGylation activity correlates with the reduced IFN-β gene transcription.
Author summary
Outbreaks of the picornavirus foot-and-mouth disease virus (FMDV) have significant consequences for animal health and product safety and place a major economic burden on the global livestock industry. Understanding how this notorious animal pathogen suppresses the antiviral type I interferon (IFN-α/β) response may help to develop countermeasures to control FMDV infections. FMDV suppresses the IFN-α/β response through the activity of its Leader protein (Lpro), a protease that can cleave host cell proteins. Lpro was also shown to have deubiquitinase and deISGylase activity, raising the possibility that Lpro suppresses IFN-α/β by removing ubiquitin and/or ISG15, two posttranslational modifications that can regulate the activation, interactions and localization of (signaling) proteins. Here, we show that TBK1 and MAVS, two signaling proteins that are important for activation of IFN-α/β gene transcription, are cleaved by Lpro. By generating Lpro mutants lacking either of these two activities, we demonstrate that Lpro’s ability to cleave signaling proteins, but not its deubiquitination/deISGylase activity, correlates with suppression of IFN-β gene transcription.
Introduction
A virally infected cell activates a plethora of antiviral responses. One of the best-known antiviral responses is the induction of type I interferons (IFN-α/β). Replication of the viral genome generates double-stranded RNA (dsRNA) replication intermediates that can be recognized by cytoplasmic RIG-I like receptors (RLRs). For example, picornaviruses, small (~30 nm) non-enveloped viruses with a positive-sense RNA genome, synthesize replication intermediates that are predominantly recognized by MDA5 [1–4]. Upon recognition of viral dsRNA, MDA5 interacts with MAVS, which subsequently activates TRAF3 and TBK1. TBK1 phosphorylates the transcription factors IRF3 and IRF7, resulting in their activation and dimerization. Simultaneously, TRAF3 interacts with the IKK complex to activate the transcription factor NF-κB. Upon activation, IRF3, IRF7 and NF-κB translocate to the nucleus, where they induce expression of IFN-α/β and other proinflammatory cytokines. Subsequent IFN-α/β signaling via the type I IFN receptor (IFNAR) and the JAK-STAT pathway induces the expression of hundreds of interferon stimulated genes (ISGs) (reviewed in [5,6]).
IFN-α/β gene transcription is extensively regulated by post-translational modification of RLRs and their downstream signaling proteins, including phosphorylation and ubiquitination. Ubiquitin is a 8.5 kDa protein that can be covalently linked through an ε-amino peptide linkage to lysine residues in target proteins. Within the RLR signaling pathway RIG-I, MAVS, TBK1, TRAF3, TRAF6 and IKKγ are ubiquitinated and this affects their molecular interactions, localization, stability, or activity (reviewed in [7,8]). Ubiquitination of RLR signaling proteins can both positively and negatively regulate the signaling pathway, which allows for rapid fine-tuning of the innate immune response against viral infection (reviewed in [7–9]). Consequently, many viruses encode enzymes with deubiquitinating (DUB) activity to manipulate the RLR signaling pathway and thereby suppress expression of IFN-α/β (reviewed in [9]).
In addition to ubiquitin, there are multiple ubiquitin-like modifiers, which can also be attached to target proteins. Of special interest is ISG15, an IFN-induced modifier of 17.5 kDa comprised of two ubiquitin-like domains in tandem. The exact antiviral properties of ISG15 are not yet fully understood (reviewed in [10,11]). Early work on ISG15 depended on mouse models and showed that expression of ISG15 protected mice from viral infection [12–15]. However, important biological and structural differences between ISG15 of murine and human origin have since been reported [16–19]. More recently, a picture is emerging that proteins are ISGylated co-translationally, explaining why predominantly viral proteins and ISGs are ISGylated upon infection in humans [20]. ISGylation of RLR signaling proteins has been reported, but the effect of these modifications on the outcome of the signaling pathway is still unclear ([21–24], reviewed in [11]). In addition, ISG15 has been reported to act as a cytokine [25,26].
IFN-α/β signals in autocrine and paracrine ways to induce a tissue-wide antiviral state, thereby limiting viral spread. To establish infection in their host, it is essential for viruses to suppress both the RLR signaling pathway and the downstream signaling of IFN-α/β. Affecting protein levels of important signaling molecules, either via cleaving them or inducing their degradation, is a strategy commonly used by viruses to suppress antiviral signaling [27–30]. One such example is the picornavirus foot-and-mouth disease virus (FMDV). FMDV is a member of the genus Aphthovirus, which also contains bovine rhinitis A and B viruses, and equine rhinitis A virus (ERAV). The genetic information on the FMDV RNA genome is translated as one polyprotein that is autocatalytically processed into the mature proteins, two of which have been shown to possess proteolytic activity and also been implicated in suppressing IFN-α/β induction (reviewed in [31]). The 3Cpro, the protease that processes the majority of cleavage sites on the polyprotein, cleaves NF-κB essential modulator (NEMO), an adaptor protein that is essential to activate the NFκB and IRF signaling pathways [32].
The second protease on the polyprotein implicated in suppressing IFN-α/β induction is Lpro, a papain-like cysteine protease located at the N-terminus of the polyprotein [33]. Once synthesized, Lpro immediately frees itself from the growing peptide chain by autocleavage at its own C-terminus. Lpro then efficiently cleaves the two isoforms of eIF (eukaryotic initiation factor) 4G to reduce protein synthesis from cellular mRNA [34] and suppresses the induction of IFN-α/β via several mechanisms. Lpro has been shown to induce the degradation of NF-κB subunit p65/RelA [35,36], and decrease the levels of IFN regulatory factor 3 (IRF3) and IRF7 [37]. Further, Lpro can also interact with ADNP, a negative regulator of transcription [38]. In addition to cleaving or degrading important signaling molecules, Lpro possesses deubiquitinase (DUB) activity which has been proposed to modulate RLR signaling [39]. A subsequent study demonstrated that Lpro should be predominantly regarded as a deISGylase rather than a DUB as biochemical evidence showed that Lpro has a 1000-fold higher affinity for ISG15 than for ubiquitin [40]. Structural studies and biochemical studies have shown separate substrate binding sites on Lpro for the viral polyprotein, the isoforms of eIF4G as well as for ubiquitin and ISG15 [40–42], suggesting that it may be possible to uncouple the activities of Lpro by the introduction of specific amino acid substitutions.
We therefore set out to uncouple the different activities of Lpro to discover whether Lpro suppresses RLR signaling through its deISGylase/DUB activity or through its ability to cleave and degrade multiple RLR signaling proteins. In this work, utilizing encephalomyocarditis virus (EMCV) expressing FMDV Lpro (EMCV-Lpro), we identified MAVS and TBK1 as new Lpro substrates and determined the cleavage site in TBK1. By introducing specifically designed mutations into Lpro, we further identified residues that are important for either the cleavage/degradation of RLR signaling proteins or for its deISGylase/DUB activity, thereby uncoupling the two catalytic activities of Lpro. We demonstrate that cleavage/degradation of RLR signaling proteins, but not the deISGylase/DUB activity of Lpro, correlates with suppressing IFN-α/β gene transcription.
Results
FMDV Lpro reduces IFN-β mRNA induction when introduced into a recombinant EMCV containing an inactive stress antagonist
To study the effects of Lpro on the induction of type I IFN in picornavirus-infected cells, we used two previously generated recombinant viruses; EMCV-LZn, which contains inactivating mutations in the zinc-finger domain of the Leader (i.e. EMCV’s RLR signaling antagonist) [1,43,44], and EMCV-Lpro, which was derived from EMCV-LZn and additionally encodes FMDV Lpro at the N-terminus of its polyprotein (Fig 1A) [45]. We also constructed a similar recombinant EMCV carrying a catalytically inactive Lpro (i.e. EMCV-Lpro C51A) [46]. To determine whether EMCV-Lpro can suppress IFN-β induction, we infected HeLa cells with EMCV, EMCV-LZn, EMCV-Lpro or EMCV-Lpro C51A and determined the IFN-β mRNA levels over time via RT-qPCR analysis (Fig 1B). Consistent with previous studies [37,39,47], wt Lpro, but not Lpro C51A, reduced the induction of IFN-β mRNA approximately 10-fold, indicating that the catalytic activity of Lpro is needed to suppress RLR signaling. In conclusion, the viruses that we generated (EMCV-Lpro and EMCV-Lpro C51A) accurately mimic the suppression of RLR signaling by Lpro as previously reported for FMDV-infected cells [47], providing us a model system to determine the mechanism via which Lpro suppresses type I IFN induction.
Fig 1. Lpro reduces IFN-β gene transcription when introduced into a recombinant EMCV containing an inactive stress antagonist.

(A) Schematic representation of the recombinant EMCV-LZn and EMCV-Lpro viruses (the latter was described in detail in [45]). In EMCV-LZn, L contains inactivating mutations in its Zn-finger domain (C19A/C22A) which abolishes its ability to suppress antiviral responses. To generate EMCV-Lpro, the Lbpro gene of FMDV O-strain was introduced at the 5’ end of the EMCV-LZn open reading frame. The Lpro cleavage site at its own C-terminus was mutated to AAA. Instead Lpro is released from this viral polyprotein via an EMCV 3C cleavage site. (B) Hela R19 cells were infected at MOI 10 with the indicated viruses and cells were lysed at 2,4,6,8 h pi. Total RNA was isolated and used for RT-qPCR analysis for IFN-β and actin mRNA, and EMCV vRNA. The IFN-β levels are depicted as a fold induction compared to levels in mock-infected cells, after correction for actin mRNA levels. The EMCV vRNA is depicted as a copy number per cell, calculated from a plasmid standard. Error bars depict the SD. One-way ANOVA with the Dunnet post hoc test was used to determine statistical significance compared to the results for EMCV-LZn-infected cells (*, p<0.05; ***, p<0.001; ****, p<0.0001; ns, no significant difference).
Lpro cleaves the RLR signaling proteins MAVS and TBK1
Lpro has been reported to degrade important signaling proteins such as the p65 subunit of NF-κB, IRF3 and IRF7 [35,37]. To determine whether Lpro targets additional RLR signaling proteins, we subjected cell lysates of HeLa cells infected with EMCV-LZn, EMCV-Lpro or EMCV-Lpro C51A to Western Blot analysis for the signaling proteins MAVS, TBK1 and IRF3, as well as the known Lpro-substrates eIF4G and G3BP1 (Fig 2A). EMCV capsid proteins and tubulin served as infection and loading controls, respectively. Infection with EMCV-Lpro, but not EMCV-Lpro C51A, resulted in the rapid cleavage of eIF4G (from 4 hpi onwards) and the cleavage of G3BP1 (from 6 hpi onwards). We did not observe cleavage or degradation of IRF3 as was suggested by others [37]. In addition to these known cleavages, we observed cleavage of MAVS and TBK1 at 8 hpi. For MAVS, we observed multiple cleavage products ranging in apparent molecular weight from ~45 kDa to ~35 kDa. TBK1 cleavage resulted in a single cleavage product with an apparent molecular weight of ~90–95 kDa. We also attempted to detect MDA5 and investigate whether this dsRNA sensor is targeted by Lpro. Unfortunately, the low levels of MDA5 prevented us from detecting the endogenous protein. MDA5 expression could be boosted by pretreatment with recombinant IFN-α2, but IFN-α2 pretreatment inhibited efficient EMCV infection, thereby interfering with the subsequent analysis.
Fig 2. Lpro cleaves MAVS and TBK1.
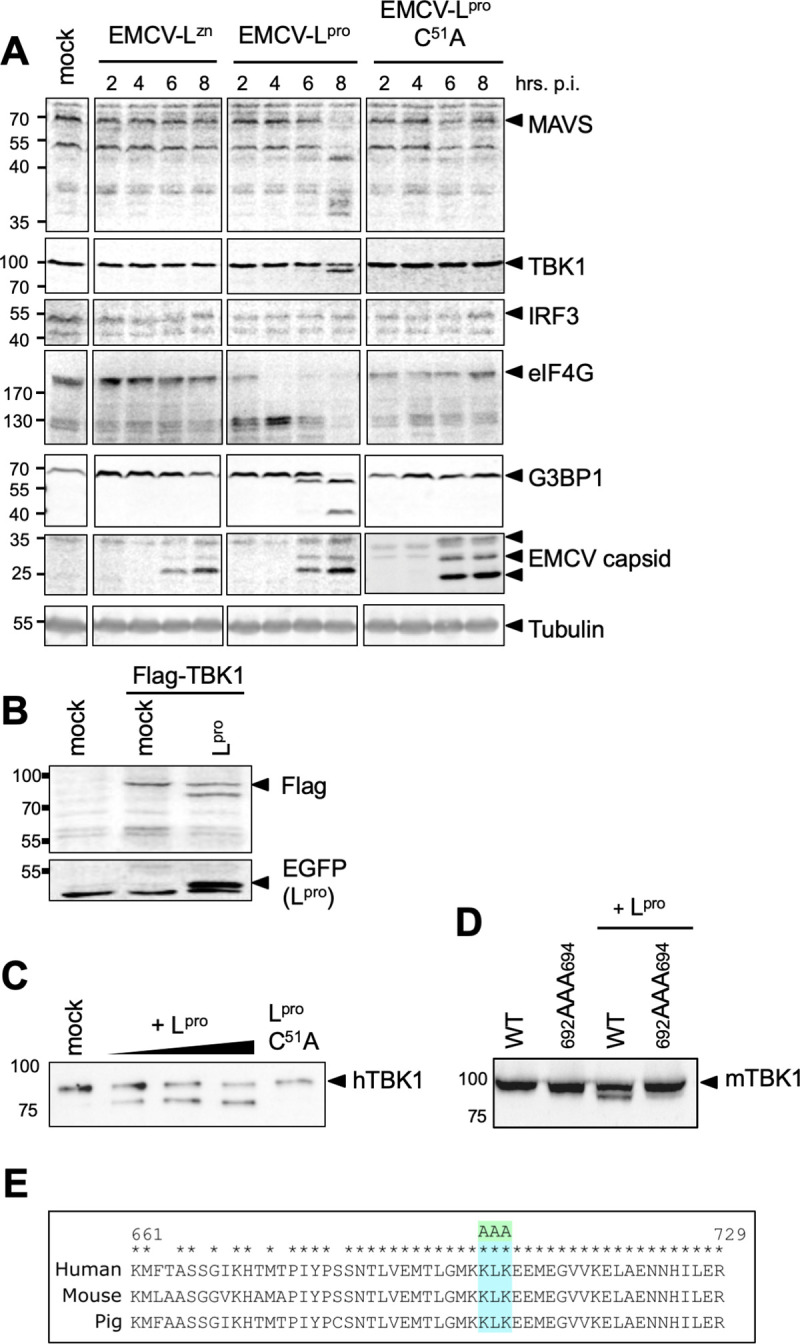
(A) HeLa R19 cells were infected at MOI 10 with the indicated viruses and lysed at 2,4,6,8 h pi. Cell lysates were subjected to Western Blot analysis for MAVS, TBK1, IRF3, eIF4GI, G3BP1, EMCV capsid proteins and tubulin. (B) HEK293T cells were transfected with 1 μg pcDNA-FLAG-TBK1 plasmid and 0.5 μg pIRES-GFP-Lpro plasmid. 16 h post transfection cells were lysed and lysates were subjected to Western Blot analysis for FLAG and GFP. (C) His-hTBK1 was incubated with 0–3 μg sLpro or 3 μg sLpro C51A for 2 h and reaction mixtures were subsequently subjected to Western Blot analysis for His. (D) HeLa OHIO cells were transfected with 7 μg pCS2-6Myc-mTBK1 or pCS2-6Myc-mTBK1692AAA694. 24 h post transfection, cells were lysed and incubated with 2 μg recombinant sLpro for 2 h before being subjected to Western Blot analysis for HA. (E) Comparison of TBK1 amino acids 661–729 of human and mouse origin. Asterisks denote conserved residues. KLK at position 692–694 was mutated to AAA.
We next focused our attention to identifying the cleavage site in TBK1. To this end, we overexpressed N-terminally FLAG-tagged TBK1 together with GFP-tagged Lpro and performed Western Blot analysis. As seen in Fig 2B, GFP-Lpro was able to cleave FLAG-TBK1. We observed an αFLAG-reactive cleavage product migrating at ~90–95 kDa, the same apparent molecular weight as the cleavage product we observed in EMCV-Lpro infected cells (Fig 2A), suggesting that Lpro cleaves TBK1 at its C-terminus. We also co-incubated recombinant His-TBK1 with increasing amounts of recombinantly expressed Lpro and Lpro C51A (Fig 2C). The in vitro incubation of His-TBK1 with wt Lpro also resulted in a ~90–95 kDa αHis-reactive cleavage product, confirming that Lpro cleaves TBK1 at its C-terminus and does not rely on other cellular factors. Incubation of His-TBK1 with catalytically inactive Lpro did not result in the formation of a cleavage product, confirming that the cleavage is dependent on Lpro’s proteolytic activity. Subsequently, we showed that Lpro also cleaves TBK1 of murine origin (Fig 2D), which suggests that Lpro cleaves TBK1 in a conserved region. We identified residues 692KLK694 –which localize at the very C-terminus of TBK1 and are well conserved between human, murine and porcine TBK1 –as a possible cleavage site (Fig 2E). Indeed, mutation of these residues prevented the cleavage of TBK1 by Lpro (Fig 2D), confirming these residues are the cleavage site.
Upon identifying the cleavage site in TBK1, we next investigated whether the cleavage of TBK1 by Lpro inhibits its function in the RLR signaling pathway. To this end, we generated cells in which the endogenous TBK1 gene is replaced with a TBK1 truncation mutant representative of the ~90–95 kDa cleavage product. We first generated HeLa TBK1 k.o. cells using CRISPR/cas9 technology and characterized the remaining RLR signaling capacity of these cells (S1A and S1B Fig). We found that depletion of TBK1 is not sufficient to fully impair RLR signaling, probably because of functional redundancy with IKKε (S1B Fig). TBK1 k.o. cells have a ~10-fold lower IFN-β induction upon infection with EMCV-LZn, transfection of vRNA or upon overexpression of MAVS. As expected, IFN-β induction resulting from transfection of IRF3, which acts downstream of TBK1, was not affected in the TBK1 k.o. cells. Subsequently, we expressed full length TBK1 or TBK1 Δ35aa, which represents the N-terminal cleavage product, in TBK1 k.o. cells (S1C Fig) and determined whether expression of TBK1 and TBK1 Δ35aa restored IFN-β mRNA expression upon transfection of poly (I:C). Expression of full length TBK1 in TBK1 k.o. cells fully restored IFN-β mRNA induction (S1D Fig) and TBK1 Δ35aa was similarly efficient in this (S1E Fig), indicating that the Lpro-generated N-terminal cleavage product is signaling competent.
TBK1 is cleaved during aphthovirus infection
To investigate whether TBK1, MAVS and IRF3 are cleaved during FMDV infection, we infected porcine LFPK αvβ6 cells with wt FMDV-A12 or FMDV-A12 lacking Lpro (leaderless virus, A12-LLV). Western Blot analysis revealed cleavage of TBK1, but not of MAVS or IRF3, during wt FMDV-A12 infection (Fig 3A). TBK1 cleavage was observed from 4 hpi onwards upon infection with wt FMDV, but not upon infection with leaderless FMDV. The cleavage product had an apparent molecular weight of ~90–95 kDa, consistent with our previous observations of the size of this cleavage product. Although a MAVS cleavage product was not detected during FMDV infection, densitometry analysis revealed a strong and progressive decrease in the relative ratio of MAVS/tubulin from 2–6 hpi post infection with wt FMDV compared to mock-infected cells, whereas only a small decrease was detected in leaderless-infected cells (MAVS/tubulin ratio is indicated in Fig 3A). This suggests that expression of Lpro induces degradation of MAVS, also in FMDV-infected cells. Consistent with our observations in EMCV-Lpro infected cells, we did not observe a decrease in IRF3 signal in FMDV-infected cells.
Fig 3. TBK1 is cleaved in aphthovirus infected cells.
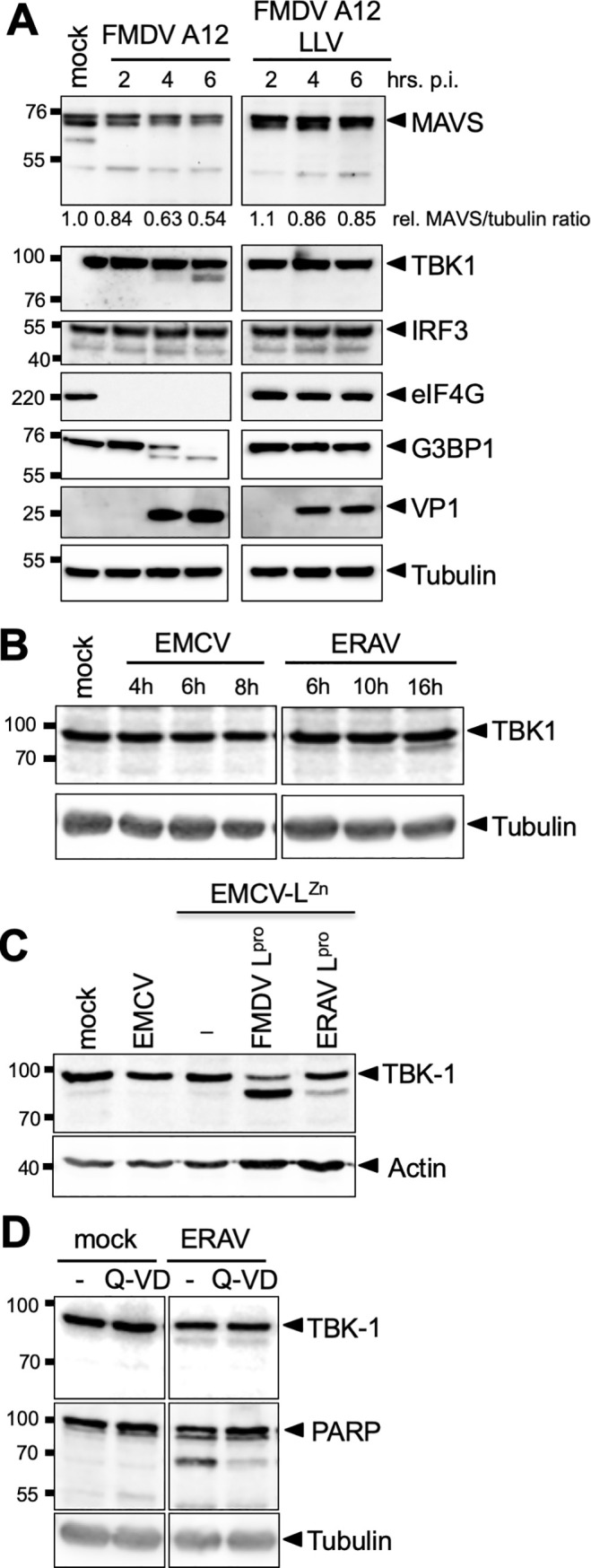
(A) LFPK αvβ6 cells were infected with FMDV-A12 or leaderless FMDV-A12 (LLV) at MOI 10 and lysed at the indicated times postinfection. Lysates were subjected to Western blot analysis for MAVS, TBK1, IRF3, eIF4G, G3BP1, viral protein VP1 and tubulin. (B) HeLa R19 cells were infected with EMCV or ERAV at MOI 10 and lysed at indicated times post infection. Cell lysates were subjected to Western Blot analysis for TBK1 and tubulin. (C) HeLa R19 cells were infected at MOI 10 with the indicated viruses and lysed at 8 hpi. Cell lysates were subjected to Western Blot analysis for TBK1 and actin. (D) HeLa R19 cells were infected with ERAV at MOI 10. Subsequently, the cells were incubated for 16h in the presence or absence of 10 μM Q-VD, a pan-caspase inhibitor. Cell lysates were subjected to Western Blot analysis for TBK1, PARP and tubulin.
To investigate whether the cleavage of TBK1 is conserved amongst aphthoviruses, we infected cells with ERAV, the closest relative of FMDV. Infection with ERAV, but not EMCV, resulted in the cleavage of TBK1 (Fig 3B). However, the cleavage product was less prominent than for EMCV-Lpro, suggesting that cleavage was inefficient or infection was delayed. Notably, in our HeLa cells, ERAV displayed a replicative cycle of ~16 h. This is considerably slower than FMDV, which replicates in 6–8 hours. To study TBK1 cleavage by the two different Lpro’s irrespective of variation in viral replication kinetics, we infected cells with EMCV-Lpro or an EMCV expressing ERAV Lpro (EMCV-ERAV Lpro), for which we previously determined the replication kinetics to be similar [45]. Both viral proteases cleaved TBK1 resulting in a ~90–95 kDa cleavage product (Fig 3C). FMDV Lpro cleaved TBK1 more efficiently than ERAV Lpro, consistent with the results observed during infection with FMDV or ERAV (Fig 3A and 3B). Notwithstanding the differences between ERAV Lpro and FMDV Lpro, our data demonstrate that the ability to cleave TBK1 is conserved amongst these two aphthoviruses.
We also investigated the effect of the pan-caspase inhibitor Q-VD-PH (Q-VD) on TBK1 cleavage in ERAV-infected cells (Fig 3D). While addition of Q-VD decreased the cleavage of known caspase substrate PARP, the cleavage of TBK1 was unaffected. Collectively, these results demonstrate that Lpro directly cleaves TBK1 and that this activity is conserved amongst aphthoviruses.
Construction of Lpro mutants to uncouple cleavage/degradation of RLR signaling proteins from its deISGylase/DUB activity
Lpro also possesses deISGylase and—to a lesser extent—DUB activity [39,40,42], and this latter activity was previously suggested to be important for Lpro’s ability to suppress RLR signaling [39]. To investigate how Lpro reduces the induction of IFN-β gene transcription, we set out to uncouple these two abilities of Lpro. To this end, we introduced previously described mutations in the chimeric EMCV-Lpro and determined whether these mutations affect the deISGylase/DUB activity of Lpro and/or its ability to cleave/degrade RLR signaling proteins. A mutation in the SAP domain (I83A/L86A), was previously reported to abolish the ability of Lpro to suppress type I IFN expression, to degrade signaling proteins (i.e. NF-κB p65, IRF3 and IRF7), and to disrupt its DUB activity [36,37,39], and was therefore included in our screening. Analysis of the crystal structure of Lpro bound to ISG15 suggested that Lpro residues L92, P99 and L102 are important for ISG15 binding [40]. Fig 4A shows the structure of Lpro (grey) in complex with ISG15 (blue) and indicates the residues of ISG15 (W123 and R153, L154, G155) that interact with Lpro. Mutation of L92, P99 or L102 in Lpro reduced its affinity for ISG15 and impaired its deISGylase activity, without affecting eIF4G cleavage [40]. Homology modeling showed that ISG15 and ubiquitin interact with the same surfaces of Lpro [40], suggesting that Lpro’s deISGylase activity also reflects its DUB activity. Two of these mutations (L92A and L102A) were introduced in EMCV-Lpro. In addition, we introduced the mutations L143A and C133S. Mutation C133S was reported to reduce the affinity of Lpro for eIF4G [48] whereas mutation of to L143 to alanine with its shorter side-chain rescued polyprotein processing in the context of the additional mutation L200F [49]. Based on the structure of Lpro, mutation L143A has been predicted to open up the catalytic pocket. Fig 4 shows the locations of the residues that are mutated in this study (Fig 4A) and summarizes the reported effects of these mutations on Lpro’s various proteolytic activities (Fig 4B).
Fig 4. Visualization and summary of the Lpro mutants used in this study.

(A) Standard cartoon view of Lpro (grey) bound to ISG15 (blue) (PDB: 6FFA). Lpro L92 and L102 interact with ISG15 W123, this positions ISG15’s C-terminus (R153, L154 and G155) in the substrate binding cleft of Lpro. *In this structure a C-terminal propargyl warhead replaced G155 [40]. Residues which upon mutation were reported to affect Lpro’s structure or function are shown as colored sticks. Green: C51A inactivates Lpro’s catalytic activity; red: I83A or L86A reduce the DUB activity and IFN induction; pink: L92A and L102A reduce affinity for ISG15; orange: C133S reduces affinity for eIF4G; aquamarine: L143A predicted to open substrate binding pocket. Drawings were generated using PyMOL. (B) Overview of the reported effects of the introduced mutations in Lpro on the various proteolytic activities of Lpro, this includes references and the category of the underlying experimental evidence.
Effect of Lpro mutations on cleavage and/or degradation of RLR signaling proteins
First, we determined the effect of the introduced mutations on Lpro’s ability to cleave or degrade RLR signaling proteins. We infected Hela cells with EMCV, EMCV-LZn or the different EMCV-Lpro carrying the described mutations, lysed the cells at the indicated timepoints and subjected the lysates to Western Blot analysis for MAVS, TBK1, NF-κB subunit p65, IRF3, eIF4G and G3BP1, as well as Lpro and EMCV capsid proteins (Fig 5). Our data show that infection with the different Lpro mutant viruses resulted in Lpro expression and accumulation of EMCV capsid proteins from 6 hpi onwards, which is indicative of efficient infection. Interestingly, the introduced mutations in Lpro had different effects on the cleavage or degradation of RLR signaling proteins. Upon infection with EMCV-Lpro C133S, we observed a ~2 hr delay in eIF4G cleavage, consistent with a previous report [48]. This mutation did not affect the cleavage/degradation of the various RLR signaling proteins. Mutation of L92 or L102 has been reported to reduce the activity of Lpro towards ISG15, without affecting eIF4G cleavage [40]. Indeed, infection with EMCV-Lpro L92A, resulted in cleavage of eIF4G, as well as all other Lpro substrates (i.e. MAVS, TBK1, NF-κB p65 and G3BP1) (Fig 5). Infection with both EMCV-Lpro L92A and EMCV-Lpro L102A resulted in efficient cleavage of RLR signaling proteins, confirming that these two Lpro mutants have the same proteolytic profiles (Fig 6). Of note, the cleavage of eIF4G appears to occur faster by Lpro L92A and L102A compared to wt Lpro, although a more in-depth analysis is necessary to confirm a true difference in eIF4G cleavage kinetics. Importantly, both mutations allow us to separate the deISGylase/DUB activity of Lpro from its ability to cleave RLR signaling proteins. Serendipitously, we observed that Lpro carrying mutation L143A was strongly impaired in degrading NF-κB p65 and cleaving MAVS and TBK1, while cleavage of G3BP1 and eIF4G was delayed but could be observed clearly at later timepoints (eIF4G cleavage is ~2 hr delayed, comparable to the delay observed for Lpro C133S). Mutation of Lpro’s SAP domain (I83A/L86A), which was previously shown to abolish degradation of NF-κB p65 and DUB activity as well as to impair Lpro’s ability to reduce IFN-β mRNA expression [36,39], also affected Lpro’s ability to cleave MAVS and TBK1. Overall, our data demonstrate that the mutations have differential effects on the cleavage/degradation of RLR signaling proteins. Importantly, we demonstrate that Lpro residues L92 and L102, which are essential for its deISGylase/DUB activity, are not essential for is ability to cleave/degrade RLR signaling proteins, indicating that the two different catalytic activities of Lpro can be uncoupled.
Fig 5. Mutation of L143 or SAP domain strongly reduces cleavage and/or degradation of RLR signaling proteins.

HeLa R19 cells were infected at MOI 10 with the indicated viruses and lysed at 2, 4, 6, 8 h pi. Cell lysates were subjected to Western Blot analysis for MAVS, TBK1, NF-κB p65, IRF3, eIF4G, G3BP1, Lpro, EMCV capsid proteins and tubulin.
Fig 6. Neither mutation of L92 or L102 affects the cleavage and/or degradation of RLR signaling proteins.
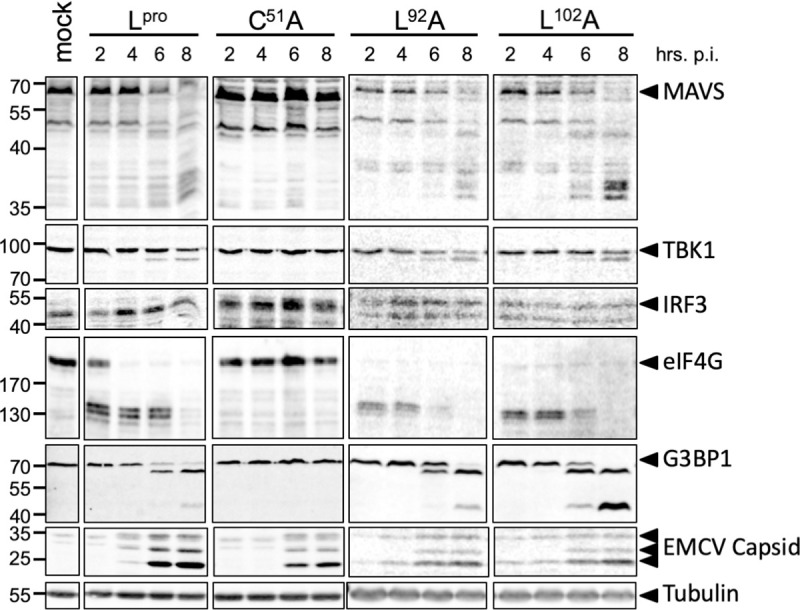
HeLa R19 cells were infected at MOI 10 with the indicated viruses and lysed at 2, 4, 6, 8 hpi. Cell lysates were subjected to Western Blot analysis for MAVS, TBK1, NF-κB p65, IRF3, eIF4G, G3BP1, Lpro, EMCV capsid proteins and tubulin.
Effect of Lpro mutations on DUB and deISGylase activities
It has been previously reported that mutations L92A, L102A and I83A/L96A affect Lpro’s deISGylase/DUB activity [39,40]. To determine the effect of mutations L143A or C133S on these activities, mutant Lpro’s were expressed and purified from E. coli and in vitro catalytic activities towards ubiquitin-TAMRA and ISG15-TAMRA were measured (Fig 7A). Lpro C133S and L143A displayed wt-like activity towards ISG15 and ubiquitin. We next determined DUB activity of wt Lpro, Lpro C51A, C133S and L143A in cells. To this end, we transfected HEK-293T cells with a combination of HA-ubiquitin, FLAG-RIG-I and increasing amounts of GFP-Lpro encoding plasmids (0.1, 0.3 and 0.5 μg), and visualized HA-tagged ubiquitinated proteins by Western Blot analysis (Fig 7B). FLAG-RIG-I, which was included to monitor the effects of Lpro-induced translational shut-off on the overexpressed proteins, was clearly detectable even at the highest level of Lpro (i.e. transfection of 0.5 μg of Lpro plasmid), indicating that Lpro-induced translational shut-off did not significantly reduce the protein expression from the transfected plasmids. Both wt Lpro as well as the C133S and L143A mutants displayed DUB activity upon overexpression in cells, as indicated by the reduction of HA-tagged ubiquitinated proteins upon increasing Lpro levels. Notably, we observed ubiquitinated proteins upon transfection of low amount of Lpro C133S plasmid (0.1 μg), although our in vitro data (Fig 7A) indicated that mutation C133S does not reduce the activity for ubiquitin. As the in vitro data are much more quantitative in nature, we consider the relatively decreased DUB activity of Lpro C133S in comparison to Lpro wt or mutant L143A as observed in Fig 7B the result of variations in expression of the transfected plasmids.
Fig 7. Mutation of L143 or C133S does not affect DUB or deISGylase activities.

(A) Ubiquitin-TAMRA or ISG15-TAMRA (substrates) were incubated with 10 μM or 100 nM Lpro (wt or indicated mutant), respectively. The change in fluorescent polarization (mFP) was determined over 60 min, with readings recorded every 60 sec. The average trace of assays performed in technical triplicate is shown. Substrate only and KG-TAMRA peptide served as respective negative and positive controls. (B) HEK293T cells were transfected with 0.5 μg pcDNA-FLAG-RIG-I, 0.5 μg pcDNA-HA-ubiquitin and 0.1–0.5 μg pIRES-GFP-Lpro. 16 h post transfection, cells were lysed and lysates subjected to Western Blot analysis for FLAG, HA, GFP and tubulin.
Thus far, our data showed that mutations C133S and L143A did not affect Lpro’s DUB activity towards a mono-ubiquitin fused to a fluorescent TAMRA molecule. To exclude the possibility that mutations C133S and L143A affect Lpro’s DUB activity towards other substrates, we determined its ability to cleave differently-linked di-ubiquitin molecules. Co-incubation of Lpro with di-ubiquitin molecules of different linkages indicated that Lpro preferentially targets K6- and K48-linked ubiquitin chains, displayed some activity towards K11-, K33-, K63-, and M1-linked chains, but at this enzyme concentration and incubation times has no activity towards K27- or K29-linked ubiquitin chains (Fig 8). Neither mutation L143A nor C133S affected the ability of Lpro to cleave the di-ubiquitin molecules. Overall, our analysis shows that Lpro carrying mutation L143A or C133S have wt-like deISGylase and DUB activity. Importantly, as mutation L143A impairs Lpro’s ability to cleave/degrade RLR signaling proteins (Fig 5), but does not affect its deISGylase/DUB activity, introduction of this mutation also allows us to make a distinction between the two proteolytic activities of Lpro.
Fig 8. Mutation of L143 or C133 does not affect activity towards differently linked ubiquitin.

Wild-type Lpro and the Lpro mutants (C133S, L143A) were incubated with each of the eight di-ubiquitin chain types for the indicated times, separated by SDS-PAGE, and visualized by silver-staining. The concentration of Lpro used in the cleavage assays is indicated.
Lpro’s ability to reduce IFN-β mRNA levels correlates with its ability to cleave/degrade RLR signaling proteins, not with its deISGylase/DUB activity
Having characterized how the different mutations in Lpro affect its deISGylase/DUB activity or the cleavage/degradation of RLR proteins, we next set out to determine which mutants reduce the induction of IFN-β mRNA. We infected HeLa cells with EMCV, EMCV-LZn or the different EMCV-Lpro mutants and determined the IFN-β mRNA and EMCV vRNA levels over time via RT-qPCR analysis (Fig 9). Wildtype Lpro as well as Lpro C133S, L92A and L102A consistently reduced the induction of IFN-β mRNA, while Lpro C51A and L143A were unable to do so (Fig 9A). The SAP domain mutant (EMCV-Lpro I83A/L86A) also failed to suppress IFN-β mRNA levels (Fig 9B), which is in agreement with observations in FMDV-infected cells [36,39]. Notably, infection with EMCV Lpro L143A, which displayed wt deISGylase/DUB activity but is strongly impaired in its ability to cleave/degrade RLR signaling proteins MAVS, TBK1 and NFκB p65, failed to suppress the induction of IFN-β mRNA. In contrast, Lpro L92A and L102A,which are strongly impaired in their deISGylase/DUB activity [40] but not in their ability to cleave/degrade RLR signaling proteins, reduced IFN-β mRNA levels. These combined observations (summarized in Fig 10) demonstrate that cleavage/degradation of RLR signaling proteins, but not the deISGylase/DUB activity of Lpro, correlate with suppressing IFN-α/β gene transcription. Notably, Lpro C133S reduced the induction of IFN-β mRNA despite a 2h delay in eIF4G cleavage, indicating that the rapid eIF4G cleavage and the subsequent translational shut-off is not sufficient to suppress RLR signaling.
Fig 9. Reduction in IFN-β gene transcription correlates with cleavage and/or degradation of RLR signaling proteins.

(A+B) Hela R19 cells were infected at MOI 10 with the indicated viruses and cells were lysed at 8 hpi. Total RNA was isolated and used for RT-qPCR analysis for IFN-β and actin mRNA (upper panels), and EMCV vRNA (lower panels). The IFN-β levels are depicted as a fold induction compared to levels in mock-infected cells, after correction for actin mRNA levels. The EMCV vRNA is depicted as a copy number per cell, calculated from a plasmid standard. Error bars represent the SD. One-way ANOVA with the Dunnet post hoc test was used to determine statistical significance compared to the results for EMCV-LZn-infected cells (**, p<0.01; ***, p<0.001; ****, p<0.0001).
Fig 10. Overview of the effects of Lpro mutations on the different proteolytic activities of Lpro as well as reduction in IFN-β gene transcription.

(A) Standard cartoon view with transparent surface of Lpro bound to E69 inhibitor shown as blue sticks (PDB: 4QBB). Residues which upon mutation were reported to affect Lpro’s structure or function are shown as colored sticks. Green: C51A inactivates Lpro’s catalytic activity; red: I83A or L86A reduce the DUB activity and IFN induction; pink: L92A and L102A reduce affinity for ISG15; orange: C133S reduces affinity for eIF4G; aquamarine: L143A predicted to open substrate binding pocket. Drawings were generated using PyMol. (B) Overview of the effects of introduced mutations on cleavage and/or degradation of host proteins, deISGylase/DUB activity, and their ability to reduce IFN-β gene transcription. Coloring of mutations is consistent with panel A. The activities of the mutations have been scored + +, +, + /–, or–according to the following criteria. + +, activity is similar to wt Lpro; +, moderately reduced activity; + /–, severely impaired activity;–, no activity.
Discussion
FMDV suppresses IFN-α/β both at the mRNA and at the protein level [47], but the molecular mechanism underlying the reduced induction of IFN-α/β gene transcription (RLR signaling) is poorly understood. Both the DUB activity of Lpro as well as its ability to cleave/degrade RLR signaling proteins have been implicated in the suppression of RLR signaling [35,37,39]. In this study, we identified MAVS and TBK1 as novel Lpro substrates and mapped the cleavage site in TBK1. Moreover, by introducing specific mutations we were able to separate Lpro’s deISGylase/DUB activity from its ability to target RLR signaling proteins. Using Lpro carrying either of these uncoupling mutations, we demonstrated that the cleavage/degradation of RLR signaling proteins, not the deISGylase/DUB activity, correlates with the ability to reduce IFN-β gene transcription. Collectively, our data strongly suggest that the ability of Lpro to cleave/degrade RLR signaling proteins is needed to reduce the IFN-β mRNA levels.
We identified TBK1 as a new Lpro substrate and identified the cleavage site. We observed cleavage of TBK1 both in HeLa cells infected with EMCV-Lpro and in FMDV-infected LFPK cells, and we demonstrated that the Lpro cleavage site is located towards the C terminus of TBK1, more specifically in the coiled-coil 2 (CC2) domain. Previous work indicated that TBK1 ΔCC2 was signaling competent upon overexpression of TBK1. However, it was also shown that mutation of residues L693 and K694, which are located in the CC2 domain, abolishes IFN-β mRNA induction upon polyI:C transfection and VSV infection [50]. Lpro cleaves TBK1 at these residues. Yet, we observed that the N-terminal cleavage product restored RLR signaling in TBK1 k.o. cells upon poly(I:C) stimulation. Whether this implies that the Lpro-mediated cleavage of TBK1 does not contribute to the viral strategy to suppress RLR signaling and IFN-β gene transcription remains unknown. Unfortunately, it is very difficult to investigate the effect of TBK1 cleavage by Lpro on IFN-β gene transcription in infected cells, as Lpro also cleaves other RLR signaling proteins, i.e. MAVS (this study) and LGP2 ([51], see below), and the transcription factor NF-κB. Dissection of the effect of TBK1 cleavage on IFN-β gene transcription from the effect of MAVS, LGP2 and NF-κB cleavage would require the identification of all Lpro cleavage sites in these known targets followed by the generation of cells with cleavage-resistant versions of these proteins. It remains a question whether such an approach will yield conclusive answers as other RLR signaling proteins may be targeted by Lpro, which have not yet been identified. Hence, the relative importance of the Lpro-mediated cleavage of TBK1 for the viral suppression of IFN-β gene transcription remains unknown.
Apart from its role in RLR signaling, TBK1 has been suggested to be involved in autophagosome maturation. TBK1 was identified as a factor in the autophagosomal clearance of herpes simplex virus 1 and mycobacteria [52,53]. A recent report showed that TBK1 phosphorylates lipidated LC-3 to prevent premature removal of LC3 from autophagosomal membranes by ATG4, thereby facilitating a unidirectional flow from the autophagosome to the lysosome [54]. Many picornaviruses hijack autophagic pathways to generate sites for viral RNA replication and to facilitate non-lytic release of virions [55–61] Possibly, cleavage of TBK1 by Lpro facilitates the use of autophagy to aid viral infection and propagation.
Lpro also impacts MAVS integrity. A distinct MAVS cleavage product was observed upon infection with EMCV-Lpro. Remarkably, no MAVS cleavage product was observed in FMDV-infected cells, although MAVS levels progressively declined over time. The reason for this difference is unknown, but may be related to differences in human and porcine cells. Nevertheless, our data indicate that the integrity of MAVS is affected by Lpro in both cell types, suggesting this RLR signaling protein is targeted by FMDV to affect IFN induction. Of note, MAVS is known to localize to the peroxisomes and mitochondrion associated membranes of the ER [62,63], in addition to its default localization on mitochondria. It remains to be established whether Lpro targets all forms of MAVS or specifically targets MAVS at one of these locations. TBK1 and MAVS are not the only RLR signaling proteins that are targeted by Lpro. It was previously reported that overexpression of Lpro induces the degradation of IRF3 and IRF7 [37], and that the p65 subunit of NF-κB is degraded in FMDV-infected cells [35]. We also observed degradation of NF-κB p65 in EMCV-Lpro infected-cells but we did not observe degradation of IRF3. Notably, degradation of IRF3 was also not observed in FMDV-infected cells. Whether IRF3 degradation is restricted to certain cell types or conditions, or merely is an artefact due to overexpression remains unknown.
It is remarkable that Lpro, comprising just 173 amino acids, can carry out several specific proteolytic activities on both the viral and cellular substrates as summarized in Figs 4 and 10. Previous work documented areas of the protease that are required for polyprotein processing [41,49]. These residues included L143 which is part of the P2 pocket that can interact with leucine residues in the substrate at the P2 position. In this work, L143 was identified as being involved in TBK1 and MAVS cleavage; however, its replacement by alanine affected neither the activity on eIF4G nor the deISGylase or DUB activities. Surprisingly, mutation of two residues of the SAP domain, (I83 and L86) also affected TBK1 and MAVS cleavage, even though they are separated by 20 Å (measured between the respective Cα atoms) with helix α4 lying between them. Nevertheless, it cannot be excluded that the SAP mutations cause some destabilization of Lpro, thereby explaining this mutant’s defect in proteolytic activities. Further structural work will be required to understand how Lpro interacts with TBK1 and MAVS.
While this work was in progress, it was reported that LGP2, a factor that is essential for MDA5 activation, is cleaved by Lpro [51]. The mutations in Lpro that impaired the reduction of IFN-β mRNA levels (L143A and mutations in the SAP domain) displayed an overall defect in the cleavage and/or degradation of each of the RLR signaling proteins we studied (i.e. MAVS, TBK1 and NF-κB p65). We anticipate that the cleavage of LGP2 is also likely impaired by introduction of these mutations. Our data suggest that expression of Lpro results in cleavage and/or degradation of multiple RLR signaling proteins (MAVS, TBK1, NF-κB p65, and most likely LGP2). The relative contribution of each cleavage event to the reduction in IFN-β gene transcription remains unknown. A search for other substrates of Lpro is of importance to further our understanding of the role and mechanism of how Lpro reduces IFN-α/β induction. Possibly, such a search may identify RLR signaling proteins that are cleaved earlier than the ones identified so far and may thereby have an influence on the early induction of type I IFN in infected cells.
The Lpro mutants that are defective in either the deISGylase/DUB activity or the cleavage/degradation of RLR signaling proteins allowed us to study which ability is needed to suppress RLR signaling. Mutation L143A, which rendered Lpro unable to reduce IFN-β mRNA levels, impaired the cleavage of RLR signaling molecules, but had no effect on the deISGylase or DUB activity of Lpro. Meanwhile, mutations L92A and L102A resulted in the opposite phenotypic effect; RLR signaling proteins were cleaved with similar efficiency as wt Lpro, but the deISGylase activity was significantly reduced by these mutations [40]. Yet, these Lpro mutants still reduced IFN-β mRNA levels. Collectively, these data indicate that the activity of Lpro to cleave/degrade RLR signaling proteins, not its deISGylase/DUB activity, is important for reduction of IFN-β induction. Medina et. al. have just reported that impairment of the deISGylation activity of Lpro causes viral attenuation in vitro and in vivo [64]. In support of our hypothesis, the mutations introduced by Medina et. al. did not affect IFN or ISG mRNA expression levels [64].
It was previously suggested that the DUB activity of Lpro is important for the suppression of RLR signaling [39], which contrasts our findings. Importantly, most experiments by Wang et. al. relied on overexpression of Lpro, ubiquitin, and several targets proteins (i.e. RIG-I, TBK1, TRAF3 and TRAF6). A recent study showed that Lpro should be predominantly regarded as a deISGylase rather than a DUB, as biochemical evidence showed that Lpro has a 1000-fold higher activity towards ISG15 than ubiquitin [40]. Given the weak DUB activity of Lpro in vitro, it remains to be established whether Lpro genuinely acts as a DUB in FMDV-infected cells under physiological conditions (i.e. without overexpression of components of the ubiquitination system or known ubiquitination target proteins). Previously, we found no differences in the levels of ubiquitinated proteins in cells infected with EMCV expressing wt Lpro or Lpro C51A, whereas the levels of ISGylated protein were decreased in cells infected with EMCV-Lpro [40], suggesting that Lpro predominantly acts as a deISGylase in infected cells.
It is well established that certain viruses (i.a. adenoviruses, herpesviruses and nidoviruses) rely on viral proteases with DUB and deISGylase activity to suppress the induction of IFN-α/β [9]. FMDV Lpro is a papain-like protease and thus Lpro is best compared to other virally encoded papain-like cysteine proteases that suppress IFN-α/β gene transcription. Members of the order Nidovirales (i.e. coronaviruses and arteriviruses) encode one or two papain-like cysteine protease (PLP), referred to as PLpro, or PLP1 and PLP2 when the virus encodes two PLPs. In addition to cleaving the viral polyprotein, PLpro and the equivalent PLP2 have acquired DUB and deISGylase activity [65–73]. Structure-guided mutagenesis of PLpro of MERS-CoV and PLP2 of equine arterivirus (EAV) allowed the DUB activity to be separated from the proteolytic activity portrayed towards the viral polyprotein [72,73]. This uncoupling of these two different proteolytic activities indicated that the DUB activity of PLpro/PLP2 contributes to the suppression of IFN-α/β transcription [72,73]. Unfortunately, it has not been determined whether PLpro/PLP2 cleaves RLR signaling proteins and thus it is unclear what other proteolytic activities could be affected by the mutations that were introduced. Notably, the cleavage site of nidovirus PLpro/PLP2 and FMDV Lpro in ubiquitin and ISG15 is different. While SARS-CoV PLpro breaks the iso-peptide bond between ubiquitin or ISG15 and the target protein [65,66], FMDV Lpro is a non-canonical deISGylase that targets a peptide bond in ISG15 itself, resulting in a diglycylated-lysine in the target protein [40]. In conclusion, nidovirus PLpro/PLP2 and FDMV Lpro are both papain-like proteases, but they likely have evolved different strategies to suppress IFN-α/β gene transcription.
FMDV Lpro and enterovirus 2Apro are structurally different enzymes that share many functions; both cleave translation initiation factor eIF4G [34,74,75], both reduce IFN-α/β gene transcription [27,47,76,77], both have been implicated in the suppression of SG formation [77–79] and both have been suggested to rescue viral translation from the inhibitory effects of p-eIF2 [80,81]. Importantly, Lpro and 2Apro both cleave several RLR signaling proteins, but the only overlapping RLR protein is MAVS [27,76]. Although a causal relationship between cleavage of RLR proteins and suppression of IFN-α/β transcription remains to be established for both proteases, the convergence on the cleavage of MAVS is noteworthy. In the absence of sequence homology, no evolutionary basis for the functional similarities between the two picornavirus proteases can be determined. Possibly, the extensive similarities between FMDV Lpro and enterovirus 2Apro, both picornavirus proteases, is illustrative of the urgency for picornaviruses to suppress these particular antiviral host responses.
Our data suggests that the deISGylase activity of Lpro is not critically needed to suppress RLR signaling, but rather its role should be sought in the broader antiviral activities of ISG15 (reviewed in [10,11]). It should be noted that our experiments were performed in naïve cells at high multiplicity of infection. Expression of the ISGylation machinery as well as ISG15 itself are boosted by IFN-α/β and therefore we cannot formally exclude a role for the DUB and/or deISGylase activity of Lpro in suppressing RLR signaling under different conditions (e.g. in IFN-primed cells). ISG15 has many functions, both intracellular and extracellular. Intracellular ISG15 can act both pro-inflammatory and immunomodulatory, either via ISGylation of target proteins or as free ISG15. Moreover, ISG15 can be secreted to act as a cytokine [25,26]. ISG15 also plays a role in damage repair after clearing viral infection [82] and can regulate cellular processes such as autophagy and metabolism [24,83–85]. How the deISGylase activity of Lpro contributes to efficient in vivo infection, remains to be established.
Materials and methods
Cells and viruses
HeLa R19, HeLa R19 TBK1 k.o. and Hek293T cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% FCS (V/V). HeLa OHIO cells were maintained in DMEM supplemented with 10% FCS (V/V) and 1% penicillin/streptomycin. LFPK αvβ6 cells [86] were obtained from the Foreign Animal Disease Diagnostic Laboratory (FADDL) at the PIADC. These cells were maintained in minimal essential medium (MEM) supplemented with 10% FCS (V/V) and 1% antibiotics and non-essential amino acids. BHK-21 cells used for FMDV propagation were maintained in MEM supplemented with 10% FCS (V/V), 10% tryptose phosphate broth, 1% antibiotics and non-essential amino acids. HeLa R19 TBK1 k.o. cells were generated via CRISPR/cas9 methodology using a pCRISPR plasmid, as described previously [87]. The used gRNA sequences are 5’-GCTACTGCAAATGTCTTTCG-3’ and 5’-GAGGAAAACAGATTGGTT-3’. FMDV A12-WT (wild type) was generated from the full-length serotype A12 infectious clone, pRMC35 [88] and A12-LLV2 (leaderless virus) was derived from the infectious clone lacking the Lb coding region, pRM-LLV2 [89]. Viruses were propagated in BHK-21 and concentrated by polyethylene glycol precipitation, titrated on BHK-21 cells, and stored at -70°C. ERAV (NM-11/67 strain) (gift from D. Rowlands and T. Tuthill [90]) was obtained after passage on HeLa R19 cells and subsequently concentrated by ultracentrifugation through a 30% sucrose cushion at 140,000xg for 16 hours in a SW32Ti rotor and stored at -80°C. Recombinant EMCVs were generated by cloning the genes of interest into the XhoI/NotI restriction sites from the pM16.1-VFETQG-Zn infectious clone that was described previously [45]. EMCV-Lpro viruses were recovered by transfection of run-off RNA transcripts into BHK-21 cells. Upon total CPE, viruses were concentrated by ultracentrifugation (as described for ERAV) and stored at -80°C.
Antibodies
The following antibodies were used for Western blot staining procedures: αFMDV VP1 (rabbit polyclonal Abmade at PIADC), αEMCV capsid (gift from Ann Palmenberg), αLpro (gift from Ewald Beck and Tim Skern), αMAVS (Enzo life science ALX-210-929), αTBK1 (Cell signaling 3504), αIRF3 (Santa cruz sc-9082), αNF-κB-p65/RelA (Santa Cruz Biotechnology SC-8008), αeIF4G (Bethyl laboratories A300-502A), αG3BP1 (BD biosciences clone 23/G3BP), αPARP (Roche Diagnostics #11835238001), αFLAG (Sigma M2), αGFP (Invitrogen OSE00003G), αHis (GE Healthcare, 27-4710-01), αMyc (Clone 4A6, Millipore), αHA (Abcam ab130275) and αtubulin (Sigma DM1A). Respective IRdye680 or IRdye800 conjugated secondary antibodies (LiCOR) or HRP-conjugated secondary antibodies were used for detection.
RT-PCR analysis
HeLa R19 cells were seeded in 24-wells plates and the next day infected with the indicated viruses at MOI 10 or transfected with the indicated plasmids or vRNA. Plasmids were transfected using Fugene6 (Promega) and vRNA was transfected using Lipofectamine 2000 (Invitrogen), both according to the manufacturer’s instructions. Preparation of viral dsRNA and the pcDNA-GFP-MAVS construct have been described previously [1,44]. pEGFP-IRF3 [D5] construct was a kind gift from John Hiscott [91]. At the indicated time points cells were lysed and cellular RNA was isolated using total RNA isolation kit (Machery-Nagel) according to manufacturer’s instructions. Reverse transcription was set up using TaqMan Reverse Transcription Reagents (Applied Biosystems) before performing qPCR analysis with SYBR green (Roche) as described previously [92].
Western Blot analysis of transfected cells
The pIRES-EGFP-FMDV Lpro plasmid was described previously [45]. The pcDNA-FLAG-TBK1 plasmid was a gift from John Hiscott [93] and the pEF-FLAG-RIG-I was a gift from Takashi Fujita [94]. HA-ubiquitin was expressed from a pCMV5 plasmid. Hek293T cells were seeded in 6-well plates and the next day transfected with 1.5 μg of total plasmid using Fugene6 (Promega) according to manufacturer’s instructions. 16h posttransfection cells were lysed 100 μl lysisbuffer (100 mM Tris pH 8.0, 1 mM EDTA, 50 mM NaCl, 1% NP40, protease inhibitor mix (Roche)). Post nuclear lysate was obtained by centrifugation at 15000xg at 4°C for 15 min. The amount of total protein in the lysates was determined using BCA assay (ThermoFisher) and 100 μg of protein was resolved using reducing sodiumdodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to 0.2 μm nitrocellulose membranes by wet electrophoretic transfer. Membranes were incubated 1h in blocking buffer (PBS + 0.1% Tween 20 + 2% BSA) and successively incubated overnight with primary antibodies diluted in blocking buffer and then for 30 min with respective secondary antibodies diluted in blocking buffer. Between and after the incubations, the membranes were washed three times with PBS+0.1% Tween-20. Finally, membranes were washed once with PBS and scanned using an Odyssey Imager (Li-COR).
Western Blot analysis of infected cells
HeLa R19 cells were seeded in 10 cm dishes and infected the next day with the indicated viruses at MOI 10. At the indicated time points cells were released using trypsin, washed once in PBS and lysed in 100 μl lysis buffer (100 mM Tris pH 8.0, 1 mM EDTA, 50 mM NaCl, 1% NP40, protease inhibitor mix (Roche)). Subsequent steps are identical as described for transfected cells. For the analysis of FMDV-infected LFBK αvβ6 cultures, cells were lysed in lysis buffer (0.5% NP-40 substitute, 50mM Tris pH 7.5, 150mM NaCl, 1mM EDTA). Lysates were incubated at 4°C for 10 min and cellular debris was collected by centrifugation at 10,000xg for 15 min at 4°C. 40 ng of protein was resolved by SDS-PAGE, transferred by Western blot and secondary antibodies conjugated with horseradish peroxidase (Pierce) were used for detection of proteins. Following incubation with appropriate primary and secondary antibodies, protein bands were visualized using SuperSignal West Dura Extended Duration Substrate (ThermoScientific, Rockford, IL, USA) according to the manufacturer’s directions.
In vitro TBK1 cleavage
sLpro was expressed and purified as reported previously [95]. 275 ng of His-hTBK1 (Millipore) was incubated with 0–3 μg sLpro for 2 h at 30°C in a HEPES buffer (20 mM HEPES pH 7.4, 150 mM KCl, 1 mM EDTA) before the reaction mixture was dissolved on SDS-PAGE. Proteins were transferred to nitrocellulose and Western blot staining for the his-tag was performed. Myc-mTBK1 and Myc-mTBK1692AAA694 were transiently expressed in HeLa OHIO cells from plasmid pCS2-6Myc-mTBK1, a gift from T. Decker. 20 μg myc-tagged mTBK1 containing cell lysate was incubated with 2 μg sLpro for 2 h at 30°C in a HEPES buffer before resolving the reaction mixture on SDS-PAGE, transferring the protein to nitrocellulose membrane and performing Western blot staining for Myc.
In vitro DUB and deISGylase assays
Ubiquitin/ISG15-TAMRA assays were performed according to [96]. Di-ubiquitin in vitro cleavage assays were performed as described previously [97].
Supporting information
(A) HeLa R19 TBK1 k.o. cells were generated using CRISPR/cas9 technology. Wt and TBK1 k.o. cells were lysed and lysates subjected to Western Blot analysis for TBK1 and tubulin. (B) IFN-β induction upon various triggers of RLR signaling was compared in wt and TBK1 k.o. cells. Cells were infected with EMCV-LZn virus at MOI 10, transfected with 20 ng vRNA, or transfected with 1 μg of plasmid expressing MAVS or IRF3. Cells were lysed at 8 h pi or transfection. Total RNA was isolated and used for RT-qPCR analysis for IFN-β and actin mRNA. The IFN-β levels are depicted as a fold induction compared to levels in mock-treated cells, after correction for actin mRNA levels. Error bars depict the SD. (C) HeLa R19 TBK1 k.o. cells were transfected with 2 μg plasmid expressing full-length or truncated TBK1 (TBK1 Δ35aa). TBK1 Δ35aa is representative for the Lpro-generated N-terminal cleavage product. Cells were lysed and lysates subjected to Western Blot analysis for TBK1 and tubulin. (D) TBK1 k.o. cells were reconstituted with full-length TBK1 as described for (C) and subsequently transfected with 100 ng poly(I:C). Cells were lysed at 8 h post transfection of poly(I:C). Total RNA was isolated and used for RT-qPCR analysis for IFN-β and actin mRNA. The IFN-β levels are depicted as a fold induction compared to levels in mock-treated cells, after correction for actin mRNA levels. Error bars depict the SD. (E) TBK1 k.o. cells were reconstituted with full-length or truncated TBK1 (TBK1 Δ35aa) as described for (C). Subsequent steps as described for (D). Error bars depict the SD.
(TIF)
Acknowledgments
We are grateful to John Hiscott and Takashi Fujita for supplying plasmids, Ann Palmenberg and Ewald Beck for providing antibodies, and David Rowlands and Toby Tuthill for supplying ERAV.
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
This work was supported by a Vici grant from the Netherlands Organization for Scientific Research (NWO-918.12.628) and the European Union (Horizon 2020 Marie Skłodowska-Curie ETN “INITIATE”, grant agreement number 813343). LJV was supported by the Graduate Programme Infection and Immunity (NWO-022.004.018) and MAL was supported by a Veni grant (NWO-863.13.008), both from the Netherlands Organization for Scientific Research. GNM and TDLS were supported by USDA-ARS, CRIS project number 1940-32000-061-00D, and USDA-KSU Non-Assistance Cooperative Agreement #58-8064-8-010. KMO and TS were supported by grants P28183 and W1258 from the Austrian Science Foundation (both to TS). KNS and DK were supported by the Medical Research Council (Grant U105192732), the European Research Council (Grants 309756 and 724804), and the Lister Institute for Preventive Medicine (all to DK). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Feng Q, Hato SV V., Langereis MAA, Zoll J, Virgen-Slane R, Peisley A, et al. MDA5 Detects the Double-Stranded RNA Replicative Form in Picornavirus-Infected Cells. Cell Rep. 2012;2: 1187–1196. 10.1016/j.celrep.2012.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang JP, Cerny A, Asher DR, Kurt-Jones EA, Bronson RT, Finberg RW. MDA5 and MAVS mediate type I interferon responses to coxsackie B virus. J Virol. 2010;84: 254–60. 10.1128/JVI.00631-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuo R-L, Kao L-T, Lin S-J, Wang RY-L, Shih S-R. MDA5 Plays a Crucial Role in Enterovirus 71 RNA-Mediated IRF3 Activation. Jin D-Y, editor. PLoS One. 2013;8: e63431 10.1371/journal.pone.0063431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hüsser L, Alves MP, Ruggli N, Summerfield A. Identification of the role of RIG-I, MDA-5 and TLR3 in sensing RNA viruses in porcine epithelial cells using lentivirus-driven RNA interference. Virus Res. 2011;159: 9–16. 10.1016/j.virusres.2011.04.005 [DOI] [PubMed] [Google Scholar]
- 5.Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14: 36–49. 10.1038/nri3581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goubau D, Deddouche S, Reis e Sousa C. Cytosolic Sensing of Viruses. Immunity. 2013;38: 855–869. 10.1016/j.immuni.2013.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiang C, Gack MU. Post-translational Control of Intracellular Pathogen Sensing Pathways. Trends Immunol. 2017;38: 39–52. 10.1016/j.it.2016.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heaton SM, Borg NA, Dixit VM. Ubiquitin in the activation and attenuation of innate antiviral immunity. J Exp Med. 2016;213: 1–13. 10.1084/jem.20151531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bailey-Elkin BA, Knaap RCM, Kikkert M, Mark BL. Structure and Function of Viral Deubiquitinating Enzymes. J Mol Biol. 2017;429: 3441–3470. 10.1016/j.jmb.2017.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perng Y-C, Lenschow DJ. ISG15 in antiviral immunity and beyond. Nat Rev Microbiol. 2018;16: 423–439. 10.1038/s41579-018-0020-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dzimianski J V., Scholte FEM, Bergeron É, Pegan SD. ISG15: it’s Complicated. J Mol Biol. 2019;S0022–2836: 30136–6. 10.1016/j.jmb.2019.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lai C, Struckhoff JJ, Schneider J, Martinez-Sobrido L, Wolff T, García-Sastre A, et al. Mice Lacking the ISG15 E1 Enzyme UbE1L Demonstrate Increased Susceptibility to both Mouse-Adapted and Non-Mouse-Adapted Influenza B Virus Infection. J Virol. 2009;83: 1147–1151. 10.1128/JVI.00105-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ketscher L, Hannß R, Morales DJ, Basters A, Guerra S, Goldmann T, et al. Selective inactivation of USP18 isopeptidase activity in vivo enhances ISG15 conjugation and viral resistance. Proc Natl Acad Sci. 2015;112: 1577–1582. 10.1073/pnas.1412881112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rahnefeld A, Klingel K, Schuermann A, Diny NL, Althof N, Lindner A, et al. Ubiquitin-like protein ISG15 (interferon-stimulated gene of 15 kDa) in host defense against heart failure in a mouse model of virus-induced cardiomyopathy. Circulation. 2014;130: 1589–600. 10.1161/CIRCULATIONAHA.114.009847 [DOI] [PubMed] [Google Scholar]
- 15.Lenschow DJ, Lai C, Frias-Staheli N, Giannakopoulos N V., Lutz A, Wolff T, et al. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc Natl Acad Sci U S A. 2007;104: 371–6. 10.1073/pnas.0609870104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daczkowski CM, Dzimianski J V., Clasman JR, Goodwin O, Mesecar AD, Pegan SD. Structural Insights into the Interaction of Coronavirus Papain-Like Proteases and Interferon-Stimulated Gene Product 15 from Different Species. J Mol Biol. 2017;429 10.1016/j.jmb.2017.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bogunovic D, Byun M, Durfee LA, Abhyankar A, Sanal O, Mansouri D, et al. Mycobacterial disease and impaired IFN-γ immunity in humans with inherited ISG15 deficiency. Science (80-). 2012;337: 1684–8. 10.1126/science.1224026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, Yuan C, et al. Human intracellular ISG15 prevents interferon-α/β over-amplification and auto-inflammation. Nature. 2015;517: 89–93. 10.1038/nature13801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Speer SD, Li Z, Buta S, Payelle-Brogard B, Qian L, Vigant F, et al. ISG15 deficiency and increased viral resistance in humans but not mice. Nat Commun. 2016;7: 11496 10.1038/ncomms11496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Durfee LA, Lyon N, Seo K, Huibregtse JM. The ISG15 Conjugation System Broadly Targets Newly Synthesized Proteins: Implications for the Antiviral Function of ISG15. Mol Cell. 2010;38: 722–32. 10.1016/j.molcel.2010.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shi HX, Yang K, Liu X, Liu XY, Wei B, Shan YF, et al. Positive Regulation of Interferon Regulatory Factor 3 Activation by Herc5 via ISG15 Modification. Mol Cell Biol. 2010;30: 2424–36. 10.1128/MCB.01466-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ganesan M, Poluektova LY, Tuma DJ, Kharbanda KK, Osna NA. Acetaldehyde Disrupts Interferon Alpha Signaling in Hepatitis C Virus-Infected Liver Cells by Up-Regulating USP18. Alcohol Clin Exp Res. 2016;40: 2329–2338. 10.1111/acer.13226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim M-J, Hwang S-Y, Imaizumi T, Yoo J-Y. Negative Feedback Regulation of RIG-I-Mediated Antiviral Signaling by Interferon-Induced ISG15 Conjugation. J Virol. 2008;82: 1474–83. 10.1128/JVI.01650-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du Y, Duan T, Feng Y, Liu Q, Lin M, Cui J, et al. LRRC25 inhibits type I IFN signaling by targeting ISG15‐associated RIG‐I for autophagic degradation. EMBO J. 2018;37: 351–366. 10.15252/embj.201796781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bogunovic D, Boisson-Dupuis S, Casanova JL. ISG15: Leading a double life as a secreted molecule. Experimental and Molecular Medicine. 2013. 10.1038/emm.2013.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Swaim CD, Scott AF, Canadeo LA, Huibregtse JM. Extracellular ISG15 Signals Cytokine Secretion through the LFA-1 Integrin Receptor. Mol Cell. 2017; 10.1016/j.molcel.2017.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feng Q, Langereis MA, Lork M, Nguyen M, Hato S V, Lanke K, et al. Enterovirus 2Apro targets MDA5 and MAVS in infected cells. J Virol. 2014;88: 3369–78. 10.1128/JVI.02712-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qian S, Fan W, Liu T, Wu M, Zhang H, Cui X, et al. Seneca Valley Virus Suppresses Host Type I Interferon Production by Targeting Adaptor Proteins MAVS, TRIF, and TANK for Cleavage. J Virol. 2017;91: e00823–17. 10.1128/JVI.00823-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang H-L, Ye H-Q, Liu S-Q, Deng C-L, Li X-D, Shi P-Y, et al. West Nile Virus NS1 Antagonizes Interferon Beta Production by Targeting RIG-I and MDA5. J Virol. 2017;91: e02396–16. 10.1128/JVI.02396-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qin Y, Xue B, Liu C, Wang X, Tian R, Xie Q, et al. NLRX1 Mediates MAVS Degradation To Attenuate the Hepatitis C Virus-Induced Innate Immune Response through PCBP2. J Virol. 2017;91: e01264–17. 10.1128/JVI.01264-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Medina GN, Segundo FD-S, Stenfeldt C, Arzt J, de los Santos T. The Different Tactics of Foot-and-Mouth Disease Virus to Evade Innate Immunity. Front Microbiol. 2018;12: e2644 10.3389/fmicb.2018.02644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang D, Fang L, Li K, Zhong H, Fan J, Ouyang C, et al. Foot-and-mouth disease virus 3C protease cleaves NEMO to impair innate immune signaling. J Virol. 2012;86: 9311–9322. 10.1128/JVI.00722-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guarné A, Tormo J, Kirchweger R, Pfistermueller D, Fita I, Skern T. Structure of the foot-and-mouth disease virus leader protease: A papain-like fold adapted for self-processing and eIF4G recognition. EMBO J. 1998;17: 7469–7479. 10.1093/emboj/17.24.7469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Devaney MA, Vakharia VN, Lloyd RE, Ehrenfeld E, Grubman MJ. Leader protein of foot-and-mouth disease virus is required for cleavage of the p220 component of the cap-binding protein complex. J Virol. 1988;62: 4407–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Los Santos T, Diaz-San Segundo F, Grubman MJ. Degradation of nuclear factor kappa B during foot-and-mouth disease virus infection. J Virol. 2007;81: 12803–12815. 10.1128/JVI.01467-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de los Santos T, Diaz-San Segundo F, Zhu J, Koster M, Dias CCA, Grubman MJ. A Conserved Domain in the Leader Proteinase of Foot-and-Mouth Disease Virus Is Required for Proper Subcellular Localization and Function. J Virol. 2008;83: 1800–10. 10.1128/JVI.02112-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang D, Fang L, Luo R, Ye R, Fang Y, Xie L, et al. Foot-and-mouth disease virus leader proteinase inhibits dsRNA-induced type I interferon transcription by decreasing interferon regulatory factor 3/7 in protein levels. Biochem Biophys Res Commun. 2010;399: 72–78. 10.1016/j.bbrc.2010.07.044 [DOI] [PubMed] [Google Scholar]
- 38.Medina GN, Knudsen GM, Greninger AL, Kloc A, Díaz-San Segundo F, Rieder E, et al. Interaction between FMDV Lpro and transcription factor ADNP is required for optimal viral replication. Virology. 2017;505: 12–22. 10.1016/j.virol.2017.02.010 [DOI] [PubMed] [Google Scholar]
- 39.Wang D, Fang L, Li P, Sun L, Fan J, Zhang Q, et al. The Leader Proteinase of Foot-and-Mouth Disease Virus Negatively Regulates the Type I Interferon Pathway by Acting as a Viral Deubiquitinase. J Virol. 2011;85: 3758–3766. 10.1128/JVI.02589-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swatek KN, Aumayr M, Pruneda JN, Visser LJ, Berryman S, Kueck AF, et al. Irreversible inactivation of ISG15 by a viral leader protease enables alternative infection detection strategies. Proc Natl Acad Sci U S A. 2018;115: 2371–2376. 10.1073/pnas.1710617115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steinberger J, Skern T. The leader proteinase of foot-and-mouth disease virus: structure-function relationships in a proteolytic virulence factor. Biol Chem. 2014;395: 1179–1185. 10.1515/hsz-2014-0156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Swatek KN, Usher JL, Kueck AF, Gladkova C, Mevissen TET, Pruneda JN, et al. Insights into ubiquitin chain architecture using Ub-clipping. Nature. 2019;572: 533–7. 10.1038/s41586-019-1482-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hato S V, Ricour C, Schulte BM, Lanke KHW, de Bruijn M, Zoll J, et al. The mengovirus leader protein blocks interferon-alpha/beta gene transcription and inhibits activation of interferon regulatory factor 3. Cell Microbiol. 2007;9: 2921–2930. 10.1111/j.1462-5822.2007.01006.x [DOI] [PubMed] [Google Scholar]
- 44.Langereis MA, Feng Q, van Kuppeveld FJ. MDA5 localizes to stress granules, but this localization is not required for the induction of type I interferon. J Virol. 2013;87: 6314–25. 10.1128/JVI.03213-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Visser LJ, Medina GN, Rabouw HH, de Groot RJ, Langereis MA, de Los Santos T, et al. Foot-and-Mouth Disease Virus Leader Protease Cleaves G3BP1 and G3BP2 and Inhibits Stress Granule Formation. J Virol. 2019;93: e00922–18. 10.1128/JVI.00922-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roberts PJ, Belsham GJ. Identification of critical amino acids within the foot-and-mouth disease virus Leader protein, a cysteine protease. Virology. 1995;213: 140–6. 10.1006/viro.1995.1554 [DOI] [PubMed] [Google Scholar]
- 47.de Los Santos T, de Avila Botton S, Weiblen R, Grubman MJ. The leader proteinase of foot-and-mouth disease virus inhibits the induction of beta interferon mRNA and blocks the host innate immune response. J Virol. 2006;80: 1906–14. 10.1128/JVI.80.4.1906-1914.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aumayr M, Fedosyuk S, Ruzicska K, Sousa-Blin C, Kontaxis G, Skern T. NMR analysis of the interaction of picornaviral proteinases Lb and 2A with their substrate eukaryotic initiation factor 4GII. Protein Sci. 2015;24: 1979–1996. 10.1002/pro.2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mayer C, Neubauer D, Nchinda a. T, Cencic R, Trompf K, Skern T. Residue L143 of the Foot-and-Mouth Disease Virus Leader Proteinase Is a Determinant of Cleavage Specificity. J Virol. 2008;82: 4656–4659. 10.1128/JVI.02077-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goncalves A, Bürckstümmer T, Dixit E, Scheicher R, Górna MW, Karayel E, et al. Functional dissection of the TBK1 molecular network. PLoS One. 2011;6: e23971 10.1371/journal.pone.0023971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rodríguez Pulido M, Sánchez-Aparicio MT, Martínez-Salas E, García-Sastre A, Sobrino F, Sáiz M. Innate immune sensor LGP2 is cleaved by the Leader protease of foot-and-mouth disease virus. PLoS Pathog. 2018;14: e1007135 10.1371/journal.ppat.1007135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pilli M, Arko-Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, et al. TBK-1 Promotes Autophagy-Mediated Antimicrobial Defense by Controlling Autophagosome Maturation. Immunity. 2012;37: 223–34. 10.1016/j.immuni.2012.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ahmad L, Mashbat B, Leung C, Brookes C, Hamad S, Krokowski S, et al. Human TANK-binding kinase 1 is required for early autophagy induction upon herpes simplex virus 1 infection. J Allergy Clin Immunol. 2019;143: 765–769.e7. 10.1016/j.jaci.2018.09.013 [DOI] [PubMed] [Google Scholar]
- 54.Herhaus L, Bhaskara RM, Lystad AH, Gestal‐Mato U, Covarrubias‐Pinto A, Bonn F, et al. TBK1‐mediated phosphorylation of LC3C and GABARAP‐L2 controls autophagosome shedding by ATG4 protease. EMBO Rep. 2020;21: e48317 10.15252/embr.201948317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jackson WT, Giddings TH, Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, et al. Subversion of Cellular Autophagosomal Machinery by RNA Viruses. Sugden B, editor. PLoS Biol. 2005;3: e156 10.1371/journal.pbio.0030156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schlegel A, Giddings TH, Ladinsky MS, Kirkegaard K. Cellular origin and ultrastructure of membranes induced during poliovirus infection. J Virol. 1996;70: 6576–88. Available: http://www.ncbi.nlm.nih.gov/pubmed/8794292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hsu N-Y, Ilnytska O, Belov G, Santiana M, Chen Y-H, Takvorian PM, et al. Viral Reorganization of the Secretory Pathway Generates Distinct Organelles for RNA Replication. Cell. 2010;141: 799–811. 10.1016/j.cell.2010.03.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van der Grein SG, Defourny KAY, Rabouw HH, Galiveti CR, Langereis MA, Wauben MHM, et al. Picornavirus infection induces temporal release of multiple extracellular vesicle subsets that differ in molecular composition and infectious potential. PLoS Pathog. 2019;15: e1007594 10.1371/journal.ppat.1007594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bird SW, Maynard ND, Covert MW, Kirkegaard K. Nonlytic viral spread enhanced by autophagy components. Proc Natl Acad Sci U S A. 2014;111: 13081–6. 10.1073/pnas.1401437111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.O’Donnell V, Pacheco JM, LaRocco M, Burrage T, Jackson W, Rodriguez LL, et al. Foot-and-mouth disease virus utilizes an autophagic pathway during viral replication. Virology. 2011;410: 142–50. 10.1016/j.virol.2010.10.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gladue DP, O’Donnell V, Baker-Branstetter R, Holinka LG, Pacheco JM, Fernandez-Sainz I, et al. Foot-and-Mouth Disease Virus Nonstructural Protein 2C Interacts with Beclin1, Modulating Virus Replication. J Virol. 2012;86: 12080–90. 10.1128/JVI.01610-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dixit E, Boulant S, Zhang Y, Lee ASY, Odendall C, Shum B, et al. Peroxisomes Are Signaling Platforms for Antiviral Innate Immunity. Cell. 2010;141: 668–81. 10.1016/j.cell.2010.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Horner SM, Liu HM, Park HS, Briley J, Gale M. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc Natl Acad Sci U S A. 2011;108: 14590–5. 10.1073/pnas.1110133108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Medina GN, Azzinaro P, Ramirez-Medina E, Gutkoska J, Fang Y, Diaz-San Segundo F, et al. Impairment of the deISGylation activity of FMDV Lpro causes attenuation in vitro and in vivo. J Virol. 2020; ePub ahead of print. 10.1128/JVI.00341-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lindner HA, Fotouhi-Ardakani N, Lytvyn V, Lachance P, Sulea T, Ménard R. The Papain-Like Protease from the Severe Acute Respiratory Syndrome Coronavirus Is a Deubiquitinating Enzyme. J Virol. 2005;79: 15199–15208. 10.1128/JVI.79.24.15199-15208.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ratia K, Saikatendu KS, Santarsiero BD, Barreto N, Baker SC, Stevens RC, et al. Severe acute respiratory syndrome coronavirus papain-like-protease: Structure of a viral deubiquitinating enzyme. Proc Natl Acad Sci U S A. 2006;103: 5717–22. 10.1073/pnas.0510851103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lindner HA, Lytvyn V, Qi H, Lachance P, Ziomek E, Ménard R. Selectivity in ISG15 and ubiquitin recognition by the SARS coronavirus papain-like protease. Arch Biochem Biophys. 2007;466: 8–14. 10.1016/j.abb.2007.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wojdyla JA, Manolaridis I, van Kasteren PB, Kikkert M, Snijder EJ, Gorbalenya AE, et al. Papain-Like Protease 1 from Transmissible Gastroenteritis Virus: Crystal Structure and Enzymatic Activity toward Viral and Cellular Substrates. J Virol. 2010;84: 10063–73. 10.1128/JVI.00898-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen Z, Wang Y, Ratia K, Mesecar AD, Wilkinson KD, Baker SC. Proteolytic Processing and Deubiquitinating Activity of Papain-Like Proteases of Human Coronavirus NL63. J Virol. 2007;81: 6007–6018. 10.1128/JVI.02747-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mielech AM, Kilianski A, Baez-Santos YM, Mesecar AD, Baker SC. MERS-CoV papain-like protease has deISGylating and deubiquitinating activities. Virology. 2014;450–451: 64–70. 10.1016/j.virol.2014.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Van Kasteren PB, Bailey-Elkin BA, James TW, Ninaber DK, Beugeling C, Khajehpour M, et al. Deubiquitinase function of arterivirus papain-like protease 2 suppresses the innate immune response in infected host cells. Proc Natl Acad Sci U S A. 2013;110: E838–47. 10.1073/pnas.1218464110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.van Kasteren PB, Knaap RCM, van den Elzen P, Snijder EJ, Balasuriya UBR, van den Born E, et al. In vivo assessment of equine arteritis virus vaccine improvement by disabling the deubiquitinase activity of papain-like protease 2. Vet Microbiol. 2015;178: 132–7. 10.1016/j.vetmic.2015.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bailey-Elkin BA, Knaap RCM, Johnson GG, Dalebout TJ, Ninaber DK, van Kasteren PB, et al. Crystal structure of the Middle East respiratory syndrome coronavirus (MERS-CoV) papain-like protease bound to ubiquitin facilitates targeted disruption of deubiquitinating activity to demonstrate its role in innate immune suppression. J Biol Chem. 2014;289: 34667–82. 10.1074/jbc.M114.609644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Haghighat A, Svitkin Y, Novoa I, Kuechler E, Skern T, Sonenberg N. The eIF4G-eIF4E complex is the target for direct cleavage by the rhinovirus 2A proteinase. J Virol. 1996; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ventoso I, MacMillan SE, Hershey JW b., Carrasco L. Poliovirus 2A proteinase cleaves directly the eIF-4G subunit of eIF-4F complex. FEBS Lett. 1998;435: 79–83. 10.1016/s0014-5793(98)01027-8 [DOI] [PubMed] [Google Scholar]
- 76.Wang B, Xi X, Lei X, Zhang X, Cui S, Wang J, et al. Enterovirus 71 Protease 2Apro Targets MAVS to Inhibit Anti-Viral Type I Interferon Responses. PLoS Pathog. 2013;9: e1003231 10.1371/journal.ppat.1003231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Visser LJ, Langereis MA, Rabouw HH, Wahedi M, Muntjewerff EM, de Groot RJ, et al. Essential Role of Enterovirus 2A Protease in Counteracting Stress Granule Formation and the Induction of Type I Interferon. J Virol. 2019;93 10.1128/JVI.00222-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang X, Hu Z, Fan S, Zhang Q, Zhong Y, Guo D, et al. Picornavirus 2A protease regulates stress granule formation to facilitate viral translation. Sarnow P, editor. PLOS Pathog. 2018;14: e1006901 10.1371/journal.ppat.1006901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Visser LJ, Medina GN, Rabouw HH, de Groot RJ, Langereis MA, de Los Santos T, et al. FMDV leader protease cleaves G3BP1 and G3BP2 and inhibits stress granule formation. J Virol. 2018; 10.1128/JVI.00922-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Redondo N, Sanz MA, Welnowska E, Carrasco L. Translation without eIF2 Promoted by Poliovirus 2A Protease. Geraghty RJ, editor. PLoS One. 2011;6: e25699 10.1371/journal.pone.0025699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Moral-López P, Alvarez E, Redondo N, Skern T, Carrasco L. L protease from foot and mouth disease virus confers eIF2-independent translation for mRNAs bearing picornavirus IRES. FEBS Lett. 2014;588: 4053–4059. 10.1016/j.febslet.2014.09.030 [DOI] [PubMed] [Google Scholar]
- 82.Morales DJ, Monte K, Sun L, Struckhoff JJ, Agapov E, Holtzman MJ, et al. Novel Mode of ISG15-Mediated Protection against Influenza A Virus and Sendai Virus in Mice. J Virol. 2015;89: 337–349. 10.1128/JVI.02110-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nakashima H, Nguyen T, Goins WF, Chiocca EA. Interferon-stimulated gene 15 (ISG15) and ISG15-linked proteins can associate with members of the selective autophagic process, histone deacetylase 6 (HDAC6) and SQSTM1/p62. J Biol Chem. 2015;290: 1485–95. 10.1074/jbc.M114.593871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xu D, Zhang T, Xiao J, Zhu K, Wei R, Wu Z, et al. Modification of BECN1 by ISG15 plays a crucial role in autophagy regulation by type I IFN/interferon. Autophagy. 2015;11: 617–628. 10.1080/15548627.2015.1023982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Baldanta S, Fernández-Escobar M, Acín-Perez R, Albert M, Camafeita E, Jorge I, et al. ISG15 governs mitochondrial function in macrophages following vaccinia virus infection. PLOS Pathog. 2017;13: e1006651 10.1371/journal.ppat.1006651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.LaRocco M, Krug PW, Kramer E, Ahmed Z, Pacheco JM, Duque H, et al. A continuous bovine kidney cell line constitutively expressing bovine αVβ6 integrin has increased susceptibility to foot-and-mouth disease virus. J Clin Microbiol. 2013;51: 1714–1720. 10.1128/JCM.03370-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Langereis MA, Rabouw HH, Holwerda M, Visser LJ, van Kuppeveld FJM. Knockout of cGAS and STING Rescues Virus Infection of Plasmid DNA-Transfected Cells. J Virol. 2015;89: 11169–73. 10.1128/JVI.01781-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rieder E, Bunch T, Brown F, Mason PW. Genetically Engineered Foot-and-Mouth Disease Viruses with Poly(C) Tracts of Two Nucleotides Are Virulent in Mice. J Virol. 1993;67: 5139–5145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Piccone ME, Rieder E, Mason PW, Grubman MJ. The foot-and-mouth disease virus leader proteinase gene is not required for viral replication. J Virol. 1995;69: 5376–5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Van Der Schaar HM, Leyssen P, Thibaut HJ, De Palma A, Van Der Linden L, Lanke KHW, et al. A Novel, Broad-Spectrum Inhibitor of Enterovirus Replication That Targets Host Cell Factor Phosphatidylinositol 4-Kinase III-beta. Antimicrob Agents Chemother. 2013;57: 4971–4981. 10.1128/AAC.01175-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lin R, Heylbroeck C, Pitha PM, Hiscott J. Virus-Dependent Phosphorylation of the IRF-3 Transcription Factor Regulates Nuclear Translocation, Transactivation Potential, and Proteasome-Mediated Degradation. Mol Cell Biol. 1998;18: 2986–96. 10.1128/mcb.18.5.2986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rabouw HH, Langereis MA, Knaap RCM, Dalebout TJ, Canton J, Sola I, et al. Middle East Respiratory Coronavirus Accessory Protein 4a Inhibits PKR-Mediated Antiviral Stress Responses. PLoS Pathog. 2016;12: e1005982 10.1371/journal.ppat.1005982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sharma S, TenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science (80-). 2003;300: 1148–51. 10.1126/science.1081315 [DOI] [PubMed] [Google Scholar]
- 94.Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol. 2005;175: 2851–8. 10.4049/jimmunol.175.5.2851 [DOI] [PubMed] [Google Scholar]
- 95.Kirchweger R, Ziegler E, Lamphear BJ, Waters D, Liebig— HD, Sommergruber W, et al. Foot-and-mouth disease virus leader proteinase: Purification of the Lb form and determination of its cleavage site on eIF-4γ. J Virol. 1994;68: 5677–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Geurink PP, El Oualid F, Jonker A, Hameed DS, Ovaa H. A General Chemical Ligation Approach Towards Isopeptide-Linked Ubiquitin and Ubiquitin-Like Assay Reagents. ChemBioChem. 2012;13: 293–7. 10.1002/cbic.201100706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mevissen TET, Kulathu Y, Mulder MPC, Geurink PP, Maslen SL, Gersch M, et al. Molecular basis of Lys11-polyubiquitin specificity in the deubiquitinase Cezanne. Nature. 2016;538: 402–405. 10.1038/nature19836 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) HeLa R19 TBK1 k.o. cells were generated using CRISPR/cas9 technology. Wt and TBK1 k.o. cells were lysed and lysates subjected to Western Blot analysis for TBK1 and tubulin. (B) IFN-β induction upon various triggers of RLR signaling was compared in wt and TBK1 k.o. cells. Cells were infected with EMCV-LZn virus at MOI 10, transfected with 20 ng vRNA, or transfected with 1 μg of plasmid expressing MAVS or IRF3. Cells were lysed at 8 h pi or transfection. Total RNA was isolated and used for RT-qPCR analysis for IFN-β and actin mRNA. The IFN-β levels are depicted as a fold induction compared to levels in mock-treated cells, after correction for actin mRNA levels. Error bars depict the SD. (C) HeLa R19 TBK1 k.o. cells were transfected with 2 μg plasmid expressing full-length or truncated TBK1 (TBK1 Δ35aa). TBK1 Δ35aa is representative for the Lpro-generated N-terminal cleavage product. Cells were lysed and lysates subjected to Western Blot analysis for TBK1 and tubulin. (D) TBK1 k.o. cells were reconstituted with full-length TBK1 as described for (C) and subsequently transfected with 100 ng poly(I:C). Cells were lysed at 8 h post transfection of poly(I:C). Total RNA was isolated and used for RT-qPCR analysis for IFN-β and actin mRNA. The IFN-β levels are depicted as a fold induction compared to levels in mock-treated cells, after correction for actin mRNA levels. Error bars depict the SD. (E) TBK1 k.o. cells were reconstituted with full-length or truncated TBK1 (TBK1 Δ35aa) as described for (C). Subsequent steps as described for (D). Error bars depict the SD.
(TIF)
Data Availability Statement
All relevant data are within the manuscript and its Supporting Information files.