

Abstract
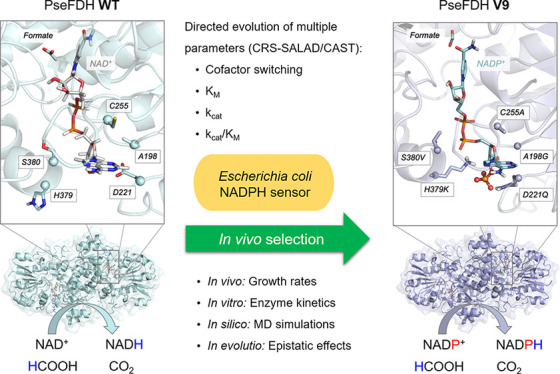
The efficient regeneration of cofactors is vital for the establishment of biocatalytic processes. Formate is an ideal electron donor for cofactor regeneration due to its general availability, low reduction potential, and benign byproduct (CO2). However, formate dehydrogenases (FDHs) are usually specific to NAD+, such that NADPH regeneration with formate is challenging. Previous studies reported naturally occurring FDHs or engineered FDHs that accept NADP+, but these enzymes show low kinetic efficiencies and specificities. Here, we harness the power of natural selection to engineer FDH variants to simultaneously optimize three properties: kinetic efficiency with NADP+, specificity toward NADP+, and affinity toward formate. By simultaneously mutating multiple residues of FDH from Pseudomonas sp. 101, which exhibits practically no activity toward NADP+, we generate a library of >106 variants. We introduce this library into an E. coli strain that cannot produce NADPH. By selecting for growth with formate as the sole NADPH source, we isolate several enzyme variants that support efficient NADPH regeneration. We find that the kinetically superior enzyme variant, harboring five mutations, has 5-fold higher efficiency and 14-fold higher specificity in comparison to the best enzyme previously engineered, while retaining high affinity toward formate. By using molecular dynamics simulations, we reveal the contribution of each mutation to the superior kinetics of this variant. We further determine how nonadditive epistatic effects improve multiple parameters simultaneously. Our work demonstrates the capacity of in vivo selection to identify highly proficient enzyme variants carrying multiple mutations which would be almost impossible to find using conventional screening methods.
Keywords: NADPH biosensor, cofactor switching, formate dehydrogenase, protein engineering, directed evolution, combinatorial mutagenesis, multiparametric optimization, epistasis
Introduction
Cofactor regeneration is vital for the operation of biocatalytic processes taking place either within a living cell or in a cell-free system.1 A considerable amount of research has therefore been invested in developing and optimizing enzymatic systems for the in situ regeneration of key cofactors such as ATP, NADH, and NADPH.2,3 Formate has been long considered as a suitable reducing agent for the regeneration of NADH both in vivo and in vitro.2,4 This is due to several properties: (i) there is an abundance of formate dehydrogenases (FDHs) that can efficiently transfer reducing power from formate to NAD+,5 (ii) formate oxidation is practically irreversible, increasing the efficiency of NADH regeneration, (iii) formate, a small molecule, can easily cross membranes, thus being accessible within cellular compartments, and (iv) the byproduct of formate oxidation, CO2, is nontoxic and can be easily expelled from the system.
Many biocatalytic processes rely on NADPH rather than NADH.6,7 Therefore, in the last 20 years multiple studies have aimed at identifying FDHs that can naturally accept NADP+ or engineering NAD-dependent FDHs to accept the phosphorylated cofactor.8−16 While some of these studies were quite successful, the kinetic efficiencies observed with NADP+ were relatively low, kcat/KM ≤ 30 s–1 mM–1, and the specificities toward this cofactor were also not high, (kcat/KM)NADP/(kcat/KM)NAD ≤ 40. In cell-free systems, the low enzyme efficiency results in the need to add a high amount of FDH to support a sufficient rate of NADPH regeneration, thus increasing the cost of enzyme biosynthesis. In vivo, the rather low specificity of the NADP-dependent FDHs could prevent efficient NADPH regeneration: as the cellular concentration of NAD+ is about 100-fold higher than that of NADP+,17 the reduction of NADP+ is expected to proceed efficiently only if (kcat/KM)NADP/(kcat/KM)NAD approaches or surpasses 100. The affinity toward formate is also an important factor, especially for cellular systems, as formate becomes toxic at high concentrations.18,19 Unfortunately, in previous studies, the affinities toward formate were quite low, with apparent KM values between 50 and 200 mM.
In this study, we aimed to harness the power of natural selection to test a large library of FDH variants and identify those which can support efficient in vivo regeneration of NADPH. By performing a structural analysis of the cofactor binding site, we identified multiple residues that are expected to affect enzyme activity and cofactor specificity. We systematically mutated these residues to generate a library of >106 variants. Screening such a large number of variants would be highly challenging. Instead, we introduced the enzyme library into an E. coli strain which was constructed to be auxotrophic for NADPH.20 By selecting for growth with formate as the NADPH source, we were able to isolate several enzyme variants that support NADPH production at a high rate and specificity, reaching a (kcat/KM)NADP value of 140 s–1 mM–1 (almost 5-fold higher than that of the best variant reported previously11) and a (kcat/KM)NADP/(kcat/KM)NAD value of 510 (more than 14-fold higher than that of the best variant reported previously16). We found that each of these enzyme variants harbor multiple mutations in the cofactor binding pocket. We analyzed the contribution of each mutation using Molecular Dynamics (MD) simulations and steady-state kinetics to determine how nonadditive epistatic effects improve multiple parameters simultaneously. Overall, our work demonstrates the capacity of in vivo selection to identify proficient enzyme variants carrying multiple mutations which would be very difficult to find using conventional screening methods.
Results
Active Site Structure and Dynamics of FDH from Pseudomonas sp. 101
FDH from Pseudomonas sp. 101 (PseFDH) is composed of 400 amino acids and functions as a homodimer in which each subunit has its own active site for binding formate and the cofactor. The C-terminus loop (S382-A393) is close to the active site and may be important for formate and cofactor binding. However, in the available crystal structures of dimeric PseFDH, with or without the NAD+ cofactor and the formate substrate bound, the C-terminus loop is not completely solved,21 suggesting high mobility of this region. We therefore built a computational model based on existing PDB structures (see Figure S1 and computational details in the Supporting Information) to inspect the interactions of specific amino acids with the substrate and the cofactor. The catalytic residues involved in hydride transfer are R284 and H332, while I122 and N146 are important for formate binding (Figure 1).22,23 The active site residues that interact with the NAD+ cofactor bind to the adenosine ribose (D221, H258, E260), phosphodiester (S147, R201, I202, S380), and nicotinamide (T282, D308, S334, G335) groups via hydrogen bonds.
Figure 1.

Active site of a PseFDH monomer. Catalytic residues involved in hydride transfer (green) and formate binding (blue) as well as those interacting with cofactor moieties of adenosine ribose (brown), phosphodiester (black), and nicotinamide (violet) are highlighted. Carbon (yellow), nitrogen (blue), and oxygen (red) atoms of the cofactor are shown in stick format. The picture was created using PyMol and PDB file 2NAD.21
To understand the molecular basis of the specificity of WT PseFDH toward NAD+, we performed MD simulations comparing the conformational dynamics of PseFDH in the presence of either NAD+ or NADP+ bound to the active site (Figure 2). These simulations highlighted the instability of the PseFDH-NADP+ complex. Specifically, the 2′-phosphate group of NADP+ causes a rearrangement of the active site residues, mainly due to the repulsion between the negatively charged carboxylate group of D221 and the 2′-phosphate group (distances explored ca. 4.7 ± 1.0 Å; Figure 2a,b). In contrast, in PseFDH-NAD+, D221 is strongly interacting with the 2′-hydroxyl of NAD+ (2.5 ± 1.2 Å; Figure 2a,b). To enable NADP+ binding to the active site and accommodate the additional negatively charged phosphate, mutagenesis of residue D221 to a neutral or positively charged amino acid seems to be necessary.12
Figure 2.

PseFDH WT NAD+/NADP+ conformational dynamics. (a) Representative structures of a PseFDH wild-type (WT) active site in the presence of NAD+ (gray, left) or NADP+ (cyan, right) and formate extracted from MD simulations (most populated clusters). The presence of the 2′- phosphate group of NADP+ causes a rearrangement of binding pocket residues. In WT-NAD+, the hydrogen bond interaction between D221 and the hydrogen of the 2′-OH group of NAD+ is highlighted in green. In WT-NADP+, the repulsive interaction between D221 and the 2′-phosphate group of NADP+ is shown in red and the salt-bridge interaction between R222 and the 2′-phosphate group of NADP+ in green. Relevant average distances (in Å) obtained from MD simulations are also depicted. (b) Plot of the distance between the carbon of the carboxylate group of D221 and either the 2′-OH group of NAD+ (orange) or the 2′-phosphate group of NADP+ (blue) along representative 500 ns replicas of MD simulations. Average distances (dashed orange line for WT-NAD+ and dashed blue line for WT-NADP+) of 2.5 ± 1.2 and 4.7 ± 1.0 Å are also shown, respectively. (c) Plot of the distance between the carbon of the guanidinium group of R222 and either the oxygen of the 2′-OH group of NAD+ (orange) or the 2′-phosphate group of NADP+ (blue) along representative 500 ns replicas of MD simulations. Average distances (dashed orange line for WT-NAD+ and dashed blue line for WT-NADP+) of 6.2 ± 1.9 and 4.3 ± 0.4 Å are also included, respectively. All distances are represented in Å. The trajectories of the three independent replicates are shown in Figure S2.
The neighboring R222 residue is rather flexible and alternates between two main conformations in apo state MD simulations (Figure S9). On the basis of the PseFDH-NAD+ X-ray structure, it was proposed that the main function of R222 is to maintain the optimal conformation of the active site rather than directly participate in the binding of NAD+.9 Our MD simulations on PseFDH-NAD+ are in line with these observations, as no direct interactions between R222 and NAD+ are observed (Figure 2a,c). Interestingly, MD simulations with NADP+ bound revealed persistent electrostatic and cation-π interactions between R222 and the adenine ring of NADP+ due to R222 intrinsic conformational flexibility (Figure 2a,c). These observations suggest that the mutagenesis of R222 might be detrimental when cofactor specificity is switched from NAD+ to NADP+.
PseFDH Library Design and Construction
Previous studies have highlighted three key residues that should be mutated to change the cofactor specificity of PseFDH. The most important one is D221, as the oxygen atoms of the carboxylate side chain stabilize the NAD+ cofactor via hydrogen bonds. Either D221S24 or D221Q13 has been shown to improve NADP+ binding due to the removal of the repulsive electrostatic interactions with the phosphate group of the cofactor. Second, replacing alanine with glycine at position 198 provides more space for the bulkier cofactor NADP+. This replacement is based on the sequence of the NADP+-dependent FDH from Burkholderia spp. which, instead of the cofactor binding region AXGXXGX17D as in PseFDH, harbors the sequence GXGXXGX17Q (where the underlined glycine and glutamine residues correspond to positions 198 and 221, respectively).24 Moreover, mutation A198G improves the binding of NADP+ when cofactor specificity has already changed: e.g., by the D221Q mutation.12 The third residue is C255,25−27 which interacts with the adenine moiety of NAD+, and its mutation to valine was shown to improve the binding affinity toward NADP+.12 Similarly, the NADP+-dependent FDH from Burkholderia spp. has an isoleucine at this site, highlighting the importance of changing C255 to an aliphatic amino acid.12 However, while A198, D221, and C255 are central for improving the binding of NADP+, they alone are not sufficient for specificity reversal, as reflected by the poor kinetic parameters of the available variants (Table S1).
To identify further residues involved in cofactor specificity, we employed the program CSR-SALAD (Cofactor Specificity Reversal – Structural Analysis and Library Design), which searches for protein structures or homology models containing NAD(H) or NADP(H) ligands.28 As changing the cofactor specificity is usually accompanied by a decrease in the catalytic turnover owing to the existence of activity–selectivity tradeoffs,29 CSR-SALAD further suggests which sites are important to recover catalytic efficiency (i.e., activity–recovery residues). The program suggested several PseFDH residues for mutagenesis, which are divided into two categories: specificity switching (D221, R222, and H223) and activity–recovery (H258, E260, T261, H379, and S380) (Figure S3). However, while CRS-SALAD proposed several specific amino acid substitutions to switch cofactor specificity, it did not predict the exact amino acid substitutions required to recover activity, recommending instead iterative site-saturation mutagenesis, in which each site is individually mutated to the 19 canonical amino acids.
The combination of available mutagenesis data (A198, D221, and C255) and the suggestions from the CSR-SALAD program (D221, R222, H223, H258, E260, T261, H379, and S380) yielded a total of 10 residues for mutagenesis. To reduce the library size, we started with variant A198G and avoided the mutagenesis of R222, as it seemed to be important for NADP+ binding (as shown by the MD simulations above). Therefore, we initially considered eight potential residues for mutagenesis (D221, H223, C255, H258, E260, T261 H379, and S380), the first three of which are directly involved in cofactor specificity and the rest could recover enzyme activity. We further decided to split the activity-recovering residues into two groups according to their distance proximity: group A (H379 and S380) and group B (H258, E260, T261). Since group A residues are closer to D221 and H223 and those of group B are further away (Figure S4), we decided to prioritize group A for mutagenesis with the hope of enabling the emergence of cooperative effects.
We aimed to simultaneously reverse coenzyme specificity (D221, H223, and C255) and recover enzyme activity (H379 and S380) in a single round of mutagenesis using the PseFDH variant A198G as a template. Although all of these five residues could be simultaneously mutated to all 20 amino acids using NNK degeneracy, the library size would be too large (325 = 3.3 × 107), especially considering that a >10-fold oversampling factor is needed for complete library coverage if the expected diversity is not perfect. To reduce the library size, three of these residues were randomized to all 20 amino acids (D221, H223, and H379), while, on the basis of alignments of multiple FDH sequences (using HotSpot Wizard 3.030), C255 was randomized to the 10 aliphatic amino acids and S380 to 6 small side-chain amino acids (Figure 3).
Figure 3.

PseFDH CAST library design. (top) Active site of PseFDH variant A198G (black) highlighting residues selected for mutagenesis for switching cofactor specificity (green and violet), while recovering activity (blue). Residue R222 (black) was not mutated despite being suggested for mutagenesis by CRS-SALAD, as explained above. The carbon (yellow), nitrogen (blue), and oxygen (red) atoms of the cofactor are shown in stick format. The picture was generated with PyMol using PDB file 2NAD.21 (bottom) Amino acid (AA) alphabet, degenerate codon (N = A/T/G/C, K = T/G, D = A/G/T, B = C/G/T, W = A/T, R = A/G), and library size and coverage. The library size was calculated using the program CASTER 2.0.31
This library contained 3.5 × 106 unique variants (1 order of magnitude smaller than the previous library) with a 3-fold oversampling factor being needed to cover 95% of the library (Figure S5). The oversampling factor was calculated with the CASTER 2.0 program, on the basis of the combinatorial active site saturation test (CAST).32 The CAST library was created using the assembly of designed oligos (ADO) method,33 and its quality was checked using a quick quality control (QQC), which consists of sequencing the pooled plasmids in a single reaction. QQC is traditionally assessed via Sanger sequencing due to its practicality and economics.34−38 Using this strategy, we found that the theoretical and observed library diversities are comparable (Figure S6).
In Vivo Selection of PseFDH Variants with Improved NADPH Production
We have recently constructed an E. coli strain deleted in all enzymes that produce NADPH (Δzwf ΔmaeB ΔpntAB ΔsthA Δicd), with the exception of 6-phosphogluconate dehydrogenase.20 This strain is auxotrophic for NADPH and can grow on a minimal medium only when gluconate is added as a source of NADPH (doubling time of 2.1 h; Figure 4a). Without gluconate, this strain can be used as a biosensor for the ability of different enzymes to support the in vivo regeneration of this cofactor.20 We showed that expression of an FDH variant from Mycobacterium vaccae N10 (C145S/A198G/D221Q/C255V, MvaFDH4M)—an engineered enzyme with one of the highest reported activities toward NADPH, kcat/KM = 21 s–1 mM–1 12—can rescue the growth of this strain upon addition of 75 mM formate (red line in Figure 4a, doubling time of 4.1 h).20 As the NAD-dependent malic enzyme, encoded by maeA, is also known to exhibit some NADP+ reduction activity,39 we decided to delete it as well. Indeed, the resulting strain NADPH-Aux (Δzwf ΔmaeB ΔpntAB ΔsthA Δicd ΔmaeA) showed a substantially reduced growth rate upon overexpression of MvaFDH4M (red dotted line in Figure 4a, doubling time >17 h), confirming that maeA has likely contributed to NADPH regeneration in the previous strain. In contrast, expression of PseFDH WT did not complement growth unless gluconate was added (black vs orange lines in Figure 4a).
Figure 4.
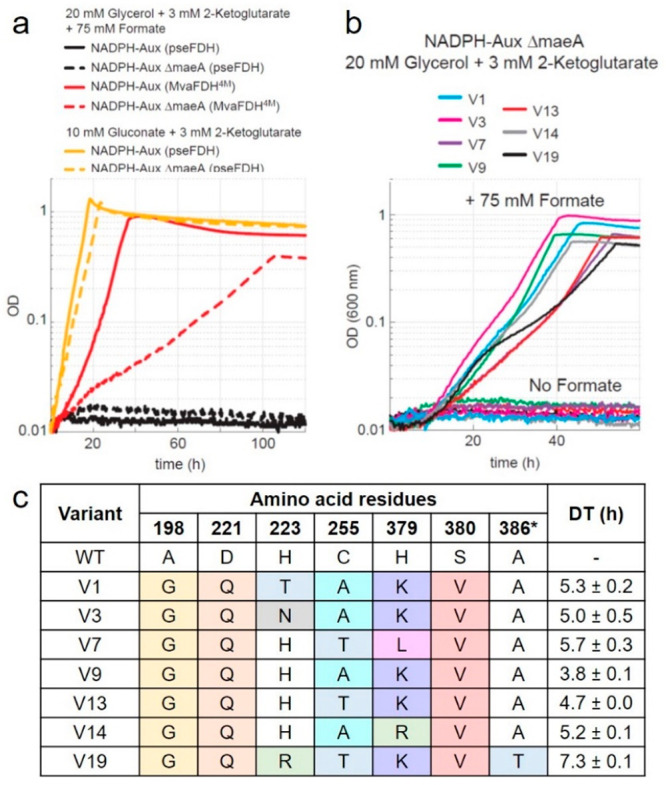
In vivo characterization of FDH variants. (a) Growth profiles of NADPH auxotroph strain with five gene deletions expressing MvaFDH4M (red line) or PseFDH in the presence of formate (black line) or gluconate (yellow line) as a control. Additional deletion of maeA results in a more robust selection strain (NADPH-aux) that displays slower growth with MvaFDH4M (red dotted line) in the presence of formate, no growth with PseFDH in the presence of formate (black dotted lines), and full growth with PseFDH in the presence of gluconate (yellow dotted lines). (b) Growth profiles of NADPH-aux when the seven PseFDH variants isolated from the selection are expressed. Strains were cultured in 20 mM glycerol, 3 mM keto-glutarate, and 75 mM formate. (c) Sequencing results of PseFDH variants with doubling time (DT) ± standard error from triplicates.
We then used the NADPH-Aux strain as a platform to select for variants of the PseFDH library that can efficiently regenerate NADPH under physiological conditions in the presence of formate. We transformed this strain with plasmids carrying the PseFDH variant library with a transformation efficiency of >1 × 107 colony-forming units (CFU)/μg. We streaked the transformed strains on a M9 agar plate containing 30 mM formate. After 5 days, we observed ∼100 colonies on the plate. We chose and cultivated the 21 largest colonies in liquid minimal medium containing 75 mM formate and found that all of them were able to grow under these conditions. We sequenced the 21 plasmids and identified seven unique sequences. The strains carrying these seven different plasmids displayed a doubling time of 4–7 h with formate as the NADPH source (Figure 4b): that is, a growth rate substantially higher than that supported by MvaFDH4M (Figure 4a).
We found that the seven unique PseFDH variants contain at least four mutations (besides A198G, which was introduced in the template for mutagenesis) (Figure 4c). D221Q and S380V were present in all variants, suggesting that these mutations are key for switching the enzyme specificity toward NADP+. PseFDH V9—which contained mutations D221Q, C255A, H379K and S380V—displayed the fastest growth, having a doubling time of 3.8 ± 0.1 h. PseFDH V1, V3, V13, and V14 differed from PseFDH V9 in only one residue: that is, H223T, H223N, C255T, and H379R, respectively. These variants exhibit doubling times of 4.7–5.3 h (Figure 4c), suggesting that residues with similar side-chain properties (H, T, N, R) are rather equivalent.
Kinetic Characterization of Improved PseFDH Variants
In order to enable effective in vivo reduction of NADP+, we expected the PseFDH variants within the growing strains to exhibit three key characteristics simultaneously: (i) high efficiency of NADP+ reduction, (ii) high specificity toward NADP+, such that the enzyme would preferably accept this cofactor despite the higher cellular concentration of NAD+, and (iii) sufficiently high affinity toward formate, such that 30 mM would (almost) saturate the enzyme. To characterize the underlying changes in the individual PseFDH enzyme variants, we purified the corresponding proteins (Figure S7) and characterized their kinetic parameters in detail.
Steady-state kinetics using constant concentrations of formate and either NAD+ or NADP+ (Figure S5 and Table S2) demonstrated that all variants displayed higher apparent turnover rates (kcat) for NADP+ (3–6 s–1) than for NAD+ (1–3 s–1), although none reached a kcat value as high as 7.5 s–1 as for PseFDH WT with NAD+ (Figure 5a). Moreover, all PseFDH variants exhibited high affinity toward NADP+, with apparent KM values ranging from 26 to 130 μM (Figure 5b). The variant PseFDH V9 displayed the highest efficiency with NADP+, having (kcat/KM)NADP > 140 s–1 mM–1 (Figure 5c)—almost 5-fold higher than the best NADP-dependent FDH variant previously described.11 The high catalytic efficiency of this enzyme variant can be attributed to a very low KM = 26 μM for NADP+, which is the highest affinity of an FDH toward this cofactor reported thus far (Table S1).
Figure 5.

Enzyme kinetics of FDH variants. Parameters reported for coenzyme turnover (a), affinity (b), catalytic efficiency (c), and specific ratio (d) under saturating amounts of formate as well as for formate turnover (e), affinity (f), catalytic efficiency (g), and specificity ratio (h) under saturating concentrations of coenzymes. The values represent average ± standard error from triplicates. Michaelis–Menten values and curves are found in Table S2 and Figure S16.
We further found that, in the presence of NADP+, all PseFDH variants displayed a higher kcat and a lower apparent KM for formate than observed with NAD+ (Figure 5e,f). This is in line with the hypothesis that the affinity for formate is dependent on the conformation and the stability of the cofactor–enzyme complex, which differ between NAD+ and NADP+.24 For example, PseFDH V9 showed a (kcat/KM)formate value of 0.16 s–1 mM–1 with NADP+, while it was only 0.009 s–1 mM–1 with NAD+ (Figure 5g), resulting in a specificity ratio (kcat/KM)NADP/(kcat/KM)NAD of ∼18 (Figure 5h).
Remarkably, the catalytic efficiency of PseFDH V9 with NADP+ was practically identical to that of PseFDH WT with NAD+ (142 s–1 mM–1), indicating a relative catalytic efficiency (RCE) of 1, which is 4-, 10-, 30-, and 1000-fold higher in comparison to engineered MvaFDH4M,12 PseFDH,9 CboFDH,10 and CmeFDH,8 respectively. Furthermore, all PseFDH variants displayed an increased coenzyme specificity ratio (CSR, defined as (kcat/KM)NADP/(kcat/KM)NAD) in comparison to previously described FDHs15 (Figure 5d). PseFDH V9 showed a CSR of >510, which is 14-fold higher than that of the most specific NADP-dependent FDH variant reported before.16
Nonadditive Epistatic Amino Acid Interactions in PseFDH V9
To gain insight into the contributions of the different mutations to the change in cofactor specificity and catalytic efficiency, we constructed several enzymes carrying subsets of the mutations found in PseFDH V9. We started with the parent template A198G, to which we added D221Q to generate the double variant A198G/D221Q (GQ). Next, we added S380V, generating the triple variant A198G/D221Q/S380V (GQV). Further incorporation of the mutation C255A or H379K resulted in variants A198G/D221Q/C255A (GQA), A198G/D221Q/H379K (GQK), A198G/D221Q/C255A/S380V (GQAV), and A198G/D221Q/H379K/S380V (GQKV). PseFDH V9 contains all five mutations (GQAKV). The “deconvoluted” variants were purified (Figure S7) and characterized in detail (Figure 6 and Table S3).
Figure 6.

Enzyme kinetics of FDH deconvolutants. Parameters for coenzyme turnover (a), affinity (b), catalytic efficiency (c), and specific ratio (d) under saturating amounts of formate as well as for formate turnover (e), affinity (f), catalytic efficiency (g), and specificity ratio (h) under saturating concentrations of coenzymes. The values represent average ± standard error from triplicates. Michaelis–Menten values and curves are found in Table S3 and Figure S17.
Steady-state kinetics of all variants revealed that H379K was essential for sustaining a high kcat value with NADP+: i.e., kcat > 3.5 s–1 in the GQK and GQKV variants (Figure 6a). On the other hand, a high affinity toward NADP+, i.e. KM < 60 μM, was dependent on S380V, as seen in the GQV, GQAV, and GQKV variants (Figure 6b). The combination of S380V and H379K in the GQKV variant resulted in high catalytic efficiency for NADP+ reduction, i.e., a (kcat/KM)NADP value of 100 s–1 mM–1 (Figure 6c). High specificity toward NADP+ could be obtained when S380V was coupled with either C255A (variant GQAV) or H379K (variant GQKV), leading to CSR values of 376 and 249, respectively (Figure 6d).
The H379K mutant also benefited the kcat value with formate in the presence of NADP+ (kcat > 3.5 s–1; Figure 6e). In addition, variants carrying the S380V mutation (variants GQAV and GQKV) had a lower apparent KM value for formate in the presence of NADP+ (KM between 40 and 44 mM, Figure 6f). Together, these two mutations contributed to a higher efficiency with formate and NADP+, i.e. (kcat/KM) = 0.03–0.08 s–1 mM–1 (Figure 6g), and a high specificity ratio of 7 toward formate (Figure 6h). Interestingly, none of the deconvolutants were better than PseFDH V9 at any kinetic parameter, indicating that all mutations were needed to optimize the activity, potentially acting synergistically.
To explore nonadditive (i.e., epistatic) effects of the different mutations, that is, a combination of mutations that results in a value smaller or greater than expected by the contribution of each mutation separately, we used additivity equations.40 The double variant GQ displayed a (kcat/Km)NADP value of 45 s–1 mM–1, whereas the triple variants GQA, GQV, and GQK showed values of 17, 43, and 30 s–1 mM–1, respectively (Figure 6c and Table S3). This means that mutations C255A, S380V, and H379K changed the catalytic efficiency of the GQ variant by −28, −2, and −15 s–1 mM–1, respectively. If we assume additivity, the quadruple variants GQAV and GQVK should have had respective kcat/KM values of 15 and 28 s–1 mM–1 (45 – 28 – 2 = 15 and 45 – 2 – 15 = 28). However, their experimental values are 37 and 100 s–1 mM–1, which are 2.5- and 3.6-fold higher than the expected values. Furthermore, addition of H379K (37 – 15 = 22) or C255A (100 – 28 = 72) into the quadruple variants resulted in PseFDH V9 (GCAQV) with a catalytic efficiency of 142 s–1 mM–1, which is 6.4- and 2-fold higher than the expected value without additivity. These results demonstrate that the interactions between the residues in PseFDH V9 are epistatic (nonadditive) and positive (synergistic).
In order to explore the stepwise evolution of the kinetic parameters in the parent variant A198G toward PseFDH V9 and understand simultaneous enhancement of multiple catalytic parameters, we constructed a multiparametric scheme (Figure 7). We focused on the mutational trajectory A198G → GQ → GQK → GQKV → GQAKV (the respective arrows are highlighted in red in Figure 7). While the parent A198G has no activity toward NADP+, variant GQ showed kcat/KM = 45 s–1 mM–1 for NADP+ and kcat/KM = 0.009 s–1 mM–1 for formate (Figure 7c). Introducing the H379K mutation reduced the catalytic efficiency for NADP+ to 30 s–1 mM–1, while it increased the efficiency for formate to 0.034 s–1 mM–1. This suggests a tradeoff between the catalytic efficiencies with formate and the cofactor. Yet, further introduction of the S380V mutation improved the catalytic efficiency for both NADP+ (100 s–1 mM–1) and formate (0.083 s–1 mM–1). Finally, mutation C255A improved the catalytic efficiency with NADP+ (142 s–1 mM–1) without affecting the catalytic efficiency for formate (0.083 s–1 mM–1). Such simultaneous improvement of multiple catalytic parameters is a major challenge that is difficult to address in traditional protein engineering methods, emphasizing the power of in vivo selection to identify the few superior multimutation variants.
Figure 7.
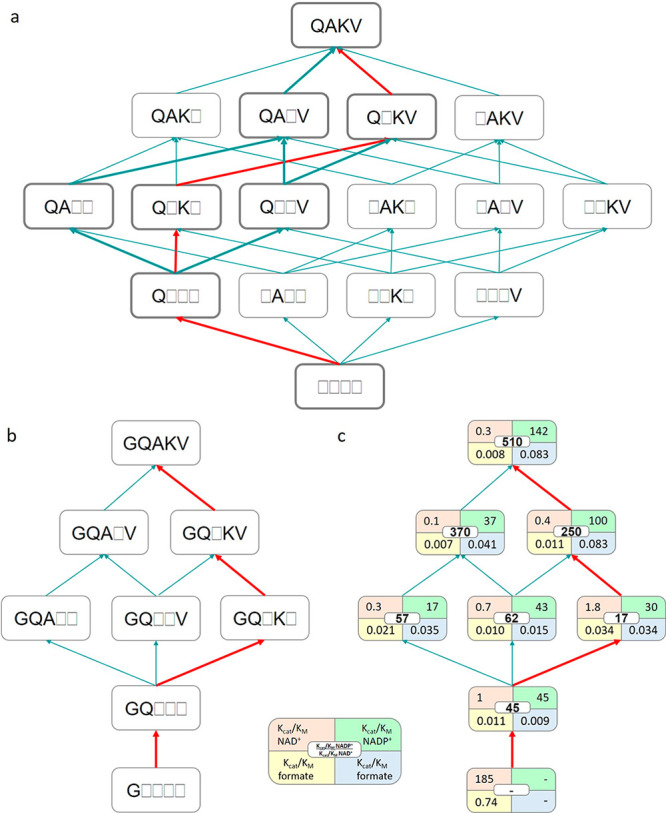
Fitness landscapes in variants derived from PseFDH V9. (a) The introduction of the four additional mutations (D221Q, C255A, H379K, and S380V) into variant A198G yields 4! = 24 possible evolutionary pathways. Thick lines indicate the four possible pathways that can be explored by stepwise evolution with the available double and triple mutants. (b) Variants and the four possible pathways from double mutant A198G/D221Q (GQ) via triple variants A198G/D221Q/C255A (GQA), A198G/D221Q/S380V (GQV), and A198G/D221Q/H379K (GQK) and quadruple mutants A198G/D221Q/C255A/S380V (GQAV) and A198G/D221Q/H379K/S380V (GQKV) toward PseFDH V9 or quintuple mutant A198G/D221Q/C255A/H379K/S380V (GQAKV). (c) Catalytic efficiencies (mM–1 s–1) of the deconvoluted variants (shown in the left) toward either NAD+/formate (orange/yellow) or NADP+/formate (green/blue). The coenzyme specificity ratio (CSR) toward NADP+ ((kcat/KM)NADP+/(kcat/KM)NAD+) is shown in the middle (white). The red lines correspond to the pathway exemplified and described in the main text.
Molecular Basis of PseFDH V9 Specificity toward NADP+
To get a better understanding of the origin of the high affinity of PseFDH V9 toward NADP+, we performed MD simulations to analyze its conformational dynamics and interactions occurring with formate and NADP+, using PseFDH WT as a reference. In PseFDH V9, D221 is mutated to glutamine, eliminating the repulsive electrostatic interactions with the phosphate group of NADP+, as described earlier (Figure 2). The hydrogen bonds between the amide group of the newly introduced glutamine and both the 2′-phosphate and the 3′-OH group of NADP+ are indeed frequently observed along the MD trajectories (5.2 ± 1.7 Å; Figure 8a and Figure S8). This allows the adenine ring of NADP+ to stay in an orientation similar to that observed in PseFDH WT with the natural NAD+ cofactor (Figure S8). As discussed above, R222 stabilizes the NADP+ cofactor through the formation of a salt bridge with the 2′-phosphate group. This stabilization is observed in both PseFDH WT and PseFDH V9 (Figure 8b and Figure S9).
Figure 8.

Conformational dynamics of PseFDH V9. A representative structure of the reshaped active site with NADP+ (cyan) and formate extracted from MD simulations (most populated cluster) is shown in the center with introduced mutations (Cα atoms depicted as spheres). (a) Representative structure of hydrogen bonds between D221Q and 2′-phosphate and the 3′-OH group of NADP+. The average distance between the amide group of D221Q and 3′-OH group of NADP+ (5.2 ± 1.7 Å) is depicted. (b) Representative structure of the salt-bridge interaction between the guanidinium group of R222 and the 2′-phosphate group of NADP+ (mean distance of 4.5 ± 0.9 Å) and the cation−π interaction between the guanidinium group of R222 and the adenine group of NADP+. (c) Representative structure of the salt-bridge interaction between the amino group of H379K and the 2′-phosphate group of NADP+ (4.8 ± 2.0 Å) and the salt-bridge interaction between the amino group of H379K and the linker 4′-phosphate group of NADP+ (6.9 ± 2.7 Å). (d) Representative structure of the CH−π interaction between the adenine ring of NADP+ and the β-carbon of the side chain of C255A. The average distance between the center of mass (COM) of the NADP+ adenine ring and the side chain of C255A (4.7 ± 0.7 Å) is depicted. (e) Representative structure of the interactions between the side chain of S380V and the side chain of P256 (with an average distance of 6.5 ± 1.1 Å) and the interactions between the side chain of S380V and the nicotinamide ribose group of NADP+ (8.3 ± 1.4 Å). All representative structures are extracted from the most populated clusters of three replicas of 500 ns of MD simulations for V9-NADP+. All distances are represented in Å.
In PseFDH V9, H379 is replaced by lysine, which according to our simulations establishes transient and frequent salt-bridge interactions with the 2′-phosphate group of NADP+ (4.8 ± 2.0 Å; Figure 8c and Figure S10). In contrast, the distance between H379 and the 2′-phosphate group in PseFDH WT is 9.2 ± 2.5 Å. In addition, the newly introduced lysine, H379K, establishes salt-bridge interactions with the linker 4′-phosphate group of the NADP+ cofactor (6.9 ± 2.7; Figure 8c). This stabilization does not occur in PseFDH WT, as the distance between H379 and the linker 4′-phosphate group is rather long (11.0 ± 1.2 Å; Figure S10).
The C255A mutation, found in a loop surrounding the cofactor, partially explains PseFDH V9 improved binding of the adenine ring of NADP+. The substitution of the bulkier cysteine with an alanine provides extra space in this region, leaving room for the adenine ring to favor its cation−π interaction with R222. Moreover, a hydrophobic CH−π interaction between the C255A side chain and the adenine ring of the cofactor stabilizes NADP+ in the active site (with a mean distance of 4.7 ± 0.7 Å; Figure 8d and Figure S11). Finally, the S380V mutation introduces an aliphatic side chain that increases the hydrophobic character of this region of the binding pocket. The valine side chain establishes hydrophobic interactions with P256 that is located in the loop surrounding the cofactor next to C255A (Figure 8e). This interaction is responsible for wrapping NADP+ in the binding pocket, and it was not observed in PseFDH WT (Figure S12).
Overall, the mutations found in PseFDH V9 have reshaped the active site to stabilize NADP+ in a conformation similar to that observed in PseFDH WT with the natural NAD+ cofactor. The MD simulations show that, in comparison to the PseFDH WT with NAD+ bound, in PseFDH V9 the nicotinamide ring of the NADP+ cofactor exhibits slightly higher flexibility and explores different orientations in the active site. To evaluate the catalytic efficiency when the cofactor specificity is switched, we analyzed the near attack conformations (NACs) explored by formate with respect to the cofactor nicotinamide ring for productive hydride transfer along the MD trajectories (Figure S13).41 As a reference, we used a DFT-optimized model for the ideal transition state (TS) geometry for the hydride transfer between formate and the nicotinamide ring (see computational details and Figure S13). Formate bound to PseFDH WT with NAD+ and to PseFDH V9 with NADP+ can explore catalytically competent poses in the MD simulations (i.e., C4NAD+/NADP+–H1HCOO– distances below 4 Å; and N1NAD+/NADP+–C4NAD+/NADP+–H1HCOO– attack angles of ca. 100–130°; see Figure S13). However, in comparison to PseFDH WT with NAD+, PseFDH V9 with NADP+ shows a wider dispersion in terms of the proper attack angle for hydride transfer, which can be related to the higher flexibility of the nicotinamide ring. This explains the slightly lower kcat value of PseFDH V9 with NADP+ with respect to that for PseFDH WT with NAD+ (Figure 5a) and the significant increase in apparent KM values toward formate in PseFDH V9 in comparison to PseFDH WT (Figure 5f), despite the fact that the relative catalytic efficiency (RCE) of 1 is unique among all engineered FDHs (Table S1).42
Discussion
Cofactor switching is very important for protein and metabolic engineering efforts as a tool to balance pathway activity and to recycle cofactors for maximizing product yield.28 A comprehensive review identified 103 enzymes engineered toward accepting a different cofactor, 52 of which (50%) had switched specificity from NAD+ to NADP+; however, the catalytic efficiency of the engineered enzymes was reduced in 70% of the cases (RCE value <1).42 The simultaneous optimization of multiple parameters is an especially difficult task due to tradeoffs, where improvement in one parameter usually worsens another.29,43,44
In most previous studies, small libraries were constructed on the basis of rational design and site-directed mutagenesis because the screening step limits the amount of enzyme variants that can be tested (e.g., low-throughput GC or HPLC). This is a general barrier for engineering selective enzymes.45 In contrast, with the use of in vivo screening based on the NADPH auxotroph E. coli strain, we tested a much larger library in a single experiment. This was made possible, as the survival and growth of the bacterium depends on NADPH regeneration via the engineered FDH variants.20
FDHs from Mycobacterium,12Saccharomyces,9 and Candida(8,46) have been previously engineered to produce NADPH. Of these, MvaFDH4M displayed one of the highest catalytic efficiencies (Table S1) and has been used to regenerate NADPH in cellular and cell-free systems.47,48 On the other hand, most mutagenesis data are available for PseFDH,5 including residues involved in cofactor specificity (A198G, D221Q, R222, H223, and C255). We identified the previously underexplored residues H379 and S380 as important targets to improve catalytic efficiency with NADP+. Interestingly, position 379 in PseFDH is equivalent to position 356 in FDH from Candida boidinii and position 380 in FDH from Granulicella mallensis, both of which are NADP+-dependent and have a lysine residue in this position,49,50 similar to the H379K mutation identified in our study. Overall, it seems that the catalytic efficiency and coenzyme specificity are largely dictated by the charge and polarity of key active site residues.51
Only a few studies used MD simulations to understand the interactions occurring between NAD(P)+ and FDHs variants.52 In our work, MD simulations highlighted the important role of the mutations in PseFDH V9 that act together to reshape and modulate the polarity of the binding pocket of the enzyme, allowing the formation of new polar interactions with NADP+. In particular, H379K and R222 were found to be instrumental for stabilizing the additional negatively charged 2′-phosphate group of NADP+, whereas D221Q reduced the electrostatic repulsion generated by the original aspartate residue of the WT enzyme. C255A and S380V decreased the polarity of the active site while simultaneously reshaping the binding pocket.
Instead of constructing site-saturation mutagenesis (SSM) libraries to recover activity as suggested by the CRS-SALAD program,28 we used combinatorial saturation mutagenesis based on CAST. The advantage of this method is that the simultaneous mutagenesis of (usually close) amino acids can result in strong synergistic (nonadditive) effects that cannot be easily predicted.53 Indeed, we found strong nonadditive effects in the variant GQVK, where a combination of two mutations, which should have lowered a kinetic parameter, actually improved it. This complex type of epistasis would be difficult to identify using SSM libraries. Since H379K in PseFDH V9 stabilizes the 2′-phosphate group of the NADP+ cofactor, the possible mechanism involved in this case is direct interactions between epistatic mutations, one mutation of which directly interacts with the substrate.54 Understanding such an epistatic mechanism is important not only for fundamental science but also for practical applications. For example, the use of epistatic data to train machine-learning algorithms has become a powerful tool to engineer more proficient proteins.55,56
The NADP-dependent FDH variants engineered in this study have various possible uses. In cell-free production systems, our FDH variants can serve to regenerate NADPH more efficiently in comparison to the currently used FDHs. The amount of protein needed to support the required regeneration rate could be reduced by at least 5-fold with our evolved FDH variants, thus saving enzyme synthesis costs. For in vivo NADPH regeneration, the effect of our FDHs might be even higher. Currently, such regeneration is limited by the high concentration of the competing NAD+ substrate. As our best FDH variants show specificity toward NADP+ higher than 500–more than an order of magnitude higher than all previously explored enzymes—they can reduce NADP+ with little competition from NAD+. The high affinity of our enzymes toward formate further makes them especially useful to support in vivo NADPH regeneration, as formate can be added to a microbial culture only at relatively low concentrations due to its toxicity.
With respect to the cofactor substrate (NAD(P)+), the best NADP-dependent FDH variant achieved in this study retained the high efficiency of the WT PseFDH (Figure 5c). However, the catalytic efficiency with formate as a substrate dropped by ∼4-fold (Figure 5g). To further improve the catalytic properties toward formate, a second round of evolution could be performed, where residues involved in formate binding and turnover will be mutated and the growth of the NADPH-Aux strain, transformed with the new library of mutants, will be selected for using low formate concentrations. For example, Jiang et al. recently improved the kcat and KM values of (NADP+-specific) BstFDH toward formate by focusing on binding residues that are conserved across FDHs, including I124 and G146 (I122 and N146 in PseFDH).57
The NADP-dependent FDHs can be especially useful for the ongoing efforts to engineer model microorganisms, such as E. coli, to grow on formate as a sole carbon source.58 Many of the suggested pathways to support formatotrophic growth rely on NADPH as an electron donor. For example, the synthetic reductive glycine pathway59 is dependent on the NADPH-consuming 5,10-methylenetetrahydrofolate dehydrogenase. Growth on formate via the pathway thus requires a high rate of NADPH regeneration. Using an NAD+-dependent FDH to provide reducing power requires high flux via the membrane-associated transhydrogenase to regenerate NAPDH,60,61 at the cost of dissipating the proton motive force and reducing biomass yield. Expressing a NADP+-dependent FDH would enable direct production of NADPH (which can regenerate NADH via the soluble transhydrogenase60), thus saving energy and increasing biomass yield.
In summary, by constructing an E. coli strain auxotrophic to NADPH, we were able to switch, in a single round of mutagenesis, the cofactor specificity of PseFDH, achieving the best kinetic parameters ever reported for FDH with NADP+. Using MD simulations, we were able to uncover the molecular basis of the increased activity and selectivity. We further determined the existence of strong nonadditive epistatic effects, which are difficult to predict via rational design or iterative SSM but are essential to overcome activity and selectivity tradeoffs. The approach we used in this study—especially harnessing the power of natural selection using the NADPH auxotroph strain—can be applied for the engineering and evolution of cofactor specificity of other oxidoreductases, thus expanding the enzymatic toolbox available for biocatalysis.
Experimental Section
Materials and Media
PCR reactions were done using Phusion High-Fidelity polymerase (Thermo Fisher Scientific GmbH, Dreieich, Germany), according to the manufacturer’s instructions. DNA digestions were carried out with FastDigest enzymes from Thermo Fisher Scientific or restriction enzymes from NEB (New England Biolabs, Frankfurt am Main, Germany). All primers were synthesized by Eurofins Genomics GmbH (Ebersberg, Germany), and Sanger sequencing was outsourced to LGC Genomics GmbH (Berlin, Germany). All media and media supplements were ordered from Sigma-Aldrich Chemie GmbH (Munich, Germany) or from Carl Roth GmbH + Co. KG (Karlsruhe, Germany). The cofactors NAD+(Na)2 and NADP+(Na)4 were purchased from Carl Roth GmbH, while chicken egg lysozyme was obtained from Sigma-Aldrich AG and DNase I from Roche Diagnostics. LB medium was used for growth during cloning together with the appropriate antibiotics: streptomycin (100 μg/mL) and chloramphenicol (30 μg/mL). For selection experiments and growth rate experiments, M9 minimal medium was used (47.8 mM Na2HPO4, 22 mM KH2PO4, 8.6 mM NaCl, 18.7 mM NH4Cl, 2 mM MgSO4, 100 μM CaCl2, 134 μM EDTA, 31 μM FeCl3·6H2O, 6.2 μM ZnCl2, 0.76 μM CuCl2·2H2O, 0.42 μM CoCl2·2H2O, 1.62 μM H3BO3, and 0.081 μM MnCl2·4H2O).
Strain Construction
The maeA gene was deleted by recombination of λ-Red in the NADPH auxotroph strain following the procedure described in the previous work.20 For gene deletion, fresh cells were prepared with fresh LB during the morning and recombinase genes were induced by the addition of 15 mM l-arabinose at OD = 0.4–0.5. After incubation of 45 min at 37 °C, cells were harvested and washed three times with ice-cold 10% glycerol (11300g, 30 s, and 4 °C). Approximately 300 ng of Km cassette PCR-product was transformed via electroporation (1 mm cuvette, 1.8 kV, 25 μF, 200 Ω). After selection on Km plates, gene deletions were confirmed via PCR using appropriate oligos. To remove the Km cassette, 50 mM l-rhamnose, which induces flippase gene expression, was added to an exponentially growing 2 mL LB culture at OD 0.5 for ≥3 h at 30 °C. Colonies were screened for Km sensitivity, and antibiotic resistance cassette removal was confirmed by PCR.
Plasmid, Variants, and Library Construction
FDH from Pseudomonas sp. 101 (PseFDH) and a variant version from Mycobacterium vaccae N10 (MycFDH_4M)12 were codon optimized and synthesized with N-terminal 6x His-Tag to ease purification upon expression in E. coli (this codon-optimized enzyme variant, carrying the His-Tag, was shown to be highly active in a previous study61). Gene synthesis was performed by Baseclear (Leiden, The Netherlands). PseFDH was cloned in the expression vector pZ-ASL (p15A origin, streptomycin resistance, strong promoter).62 For library construction, plasmid pZ-ASL-PseFDH was digested with BsmBI and NheI restriction enzymes to open the backbone by cleaving a fragment of 685 bps within the ORF of FDH. The sequence of the resulting fragment was used as a template for building a synthetic gene fragment. The gene fragment was submitted to the online software DNA Works55 to define the optimal overlapping oligos. A total of 36 oligos were ordered with appropriate degenerate codons at the target amino acids (Table S4). As indicated elsewhere33 and in the Supporting Information, the oligos were mixed in equimolar amounts and a first step of polymerase cycling was performed. In the second PCR step, oligos 1 and 36 were added to the sample to amplify the library, which was submitted to Sanger sequencing using oligos 16 and 36 to assess its quality. The fragment and plasmid were digested with restriction enzymes BsmBI and NheI. The plasmid (<20 ng) was also treated with 1 μL of DpnI to eliminate the parent template. Prior to strain transformation, the library was subjected to quick quality control (QQC), in which the pooled plasmids are sequenced in a single reaction. In QQC, the expected and observed variabilities should be similar according to the degenerate codon used. The base distribution was calculated with an automated server: https://pi.matteoferla.com/main/QQC.63 For creation of the PseFDH deconvolutants, target mutations were introduced by PCR on the basis of a QuikChange method using partially overlapping mutagenic primers64 (Table S5) and were confirmed by Sanger sequencing.
Growth and Selection Experiments
Overnight cultures of selected strains were prepared by inoculating these into 4 mL of M9 medium containing 11 mM gluconate and 3 mM 2-ketoglutarate, followed by incubation at 37 °C. Prior to inoculation, cultures were harvested by centrifugation (3960g, 3 min, RT) and washed three times in M9 medium to clean cells from residual carbon sources. Cells were cultivated in M9 medium at 37 °C with a combination of the following carbon sources: 10 mM glucose, 18 mM glycerol, 5 mM 2-ketoglutarate, and 50 or 75 mM formate.
For library screening, the NADPH auxotroph strain was transformed with the library by electroporation and plated on M9 minimal medium plates supplemented with 30 mM formate, 18 mM glycerol, 5 mM 2-ketoglutarate, and streptomycin followed by incubation at 37 °C for selecting variants that can complement growth. The colonies that appeared after several days were transferred to liquid media with 30 mM formate, 18 mM glycerol, and 5 mM α-ketoglutarate, and the growth was monitored for 7 days. The plasmids of the selected strains dubbed as “winners” were extracted and sequenced.
Growth experiments of selected strains carrying FDH variants were performed by inoculating M9 medium with the respective strain at a starting OD600 of 0.01 in 96-well microtiter plates (Nunclon Delta Surface, Thermo Scientific) and carried out at 37 °C. Each well contained 150 μL of the culture covered with 50 μL of mineral oil (Sigma-Aldrich) to avoid evaporation. A plate reader (Infinite M200 pro, Tecan) was used for incubation, shaking, and OD600 measurements (controlled by Tecan I-control v1.11.1.0). The cultivation program contained three cycles of four shaking phases, 1 min of each: linear shaking, orbital shaking at an amplitude of 3 mm, linear shaking, and orbital shaking at an amplitude of 2 mm. After each round of shaking (∼12.5 min), the absorbance (OD600 in nm) was measured in each well. Raw data from the plate reader were calibrated to cuvette values according to ODcuvette = ODplate/0.23. Growth curves were plotted in MATLAB (R2017B) and represent averages of triplicate measurements; in all cases, the variability among triplicate measurements was less than 5%.
Protein Expression and Purification
The His-tagged protein was expressed in E. coli BL21 DE3. Cells in terrific broth containing 20 μg/mL of streptomycin were grown at 30 °C for 16 h. Cells were harvested for 15 min at 6000g at 4 °C then resuspended in 2 mL of buffer A (20 mM Tris, 500 mM NaCl, 5 mM Imidazole, pH 7.9) per gram of pellet. The suspension was treated with 10 mg/mL of DNase I, 5 mM MgCl2, and 6 μg/mL of lysozyme on ice for 20 min, upon which cells were lysed by sonication. The lysate was clarified at 45000g at 4 °C for 45 min, and the supernatant was filtered through a 0.4 μm syringe tip filter (Sarstedt, Nümbrecht, Germany). Lysate was loaded onto a pre-equilibrated 1 mL HisTrap FF column and washed with 12% buffer B (20 mM Tris, 500 mM NaCl, 500 mM imidazole, pH 7.9) for 20–30 column volumes until the UV 280 nm band reached the baseline level. The protein was eluted by applying 100% buffer B, collected, and then pooled and desalted into 100 mM Na2HPO4 (pH 7.0). The protein was frozen in N2(l) and stored at −80 °C if not immediately used for assays.
Steady-State Kinetics Analysis
Assays were performed on a Cary-60 UV/vis spectrophotometer (Agilent) at 30 °C using quartz cuvettes (10 mm path length; Hellma). Reactions were performed in 100 mM Na2HPO4 (pH 7.0). Kinetic parameters for one substrate were determined by varying its concentration, while the others were kept constant at 6–10 times their KM value. The reaction procedure was monitored by following the reduction of NADP+ at 340 nm (εNADPH,340 nm = 6.22 mM–1 cm–1). Each concentration was measured in triplicate, and the obtained curves were fit using GraphPad Prism 8. Hyperbolic curves were fit to the Michaelis–Menten equation to obtain apparent kcat and KM values.
Molecular Dynamics Simulations
Protein Preparation
Molecular dynamics simulations (MD) were used to elucidate the molecular basis of cofactor specificity in PseFDH. MD simulations were carried out for the wild-type enzyme (WT-apo, WT-NAD+, and WT-NADP+), for the A198G variant (A198G-apo, A198G-NAD+, and A198G-NADP+) and V9 variant (V9-apo, V9-NAD+, and V9-NADP+). We selected the apo state X-ray crystal structure (Protein Data Bank (PDB) accession number 2GO1) and the holo state X-ray crystal structure (PDB accession number 2GUG crystallized in the presence of formate in the active site) as a starting point to run our MD simulations. In these X-ray structures, the important loop found in a region near cofactor binding (residues 375–400) was unsolved. For this reason, we reconstructed this loop on the basis of FDH X-ray structure PDB 2NAD using Phyre2 (http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index) and Robetta (http://robetta.bakerlab.org/) Web servers. Finally, NAD+ and NADP+ cofactors were placed in the active site of the holo PDB 2GUG by structural alignment with PDB 2NAD (which contains the NAD+ cofactor). See the Supporting Information (protocols and computational model sections) for further descriptions of the models constructed and used in this work. Amino acid protonation states were predicted using the H++ server (http://biophysics.cs.vt.edu/H++). For simulation of the A198G and V9 variants, we used the mutagenesis tool from PyMOL software using the WT crystal structures (PDB 2GO1 and 2GUG) as starting points. From these coordinates, we started the MD simulations.
Substrate Parametrization
The parameters for formate for the MD simulations were generated within the ANTECHAMBER module of AMBER 1665 using the general AMBER force field (GAFF),66 with partial charges set to fit the electrostatic potential generated at the HF/6-31G(d) level by the RESP model.67 The charges were calculated according to the Merz–Singh–Kollman scheme68 using Gaussian 09.69 The parameters for the cofactors NAD+ and NADP+ for the MD simulations were obtained from the Manchester parameter database (http://research.bmh.manchester.ac.uk/bryce/amber/).
Protocol for MD Simulations
To perform the MD simulations, each system was immersed in a pre-equilibrated truncated octahedral box of water molecules with an internal offset distance of 10 Å, using the LEAP module of the AMBER MD package. All systems were neutralized with explicit counterions (Na+ or Cl–). All subsequent calculations were performed using the AMBER force field 14 Stony Brook (ff14SB).76 A two-stage geometry optimization approach was performed. First, a short minimization of the positions of water molecules with positional restraints on the solute by a harmonic potential with a force constant of 500 kcal mol–1 Å–2 was done. The second stage was an unrestrained minimization of all of the atoms in the simulation cell. Then, the systems were gently heated in six 50 ps steps, increasing the temperature by 50 K each step (0–300 K) under constant-volume, periodic-boundary conditions and the particle-mesh Ewald approach70 to introduce long-range electrostatic effects. For these steps, a 10 Å cutoff was applied to Lennard–Jones and electrostatic interactions. Bonds involving hydrogen were constrained with the SHAKE algorithm.71 Harmonic restraints of 10 kcal mol–1 were applied to the solute, and the Langevin equilibration scheme was used to control and equalize the temperature.72 The time step was kept at 2 fs during the heating stages, allowing potential inhomogeneities to self-adjust. Each system was then equilibrated for 2 ns with a 2 fs time step at a constant pressure of 1 atm. Finally, conventional MD trajectories at constant volume and temperature (300 K) were collected. In total, three replicas of 500 ns MD simulations for each system (WT-apo, WT-NAD+, WT-NADP+, A198G-apo, A198G-NAD+, A198G-NADP+, V9-apo, V9-NAD+, and V9-NADP+) in the apo and holo states (with each cofactor and the formate substrate bound) were carried out, collecting a total of 13.5 μs of MD simulations performed at the Galatea cluster (composed of 178 GTX1080 GPUs).
Quantum Mechanics (QM) Calculation Details
DFT calculations were carried out using Gaussian09.69 A truncated computational model was used to model the transition state (TS) structure for the cofactor reduction reaction (H atom transfer). The truncated model consists of a nicotinamide ring and a formate molecule. Geometry optimizations and frequency calculations were performed using the (U)B3LYP functional73−75 with the 6-31+G* basis set. The transition state had one negative force constant corresponding to the desired transformation.
Acknowledgments
This study was funded by the Max Planck Society. This work was partially funded by the German Ministry of Education and Research grant No 031B0194 (project FormatPlant). This study was also supported in part by the European Research Council Horizon 2020 research and innovation program (ERC-2015-StG-679001, S.O.), Spanish MINECO (project PGC2018-102192–B-I00, S.O.; Juan de la Cierva - Incorporación fellowship IJCI-2017-33411, M.G.-B.), and Generalitat de Catalunya AGAUR (SGR-1707, S.O.; Beatriu de Pinós H2020 MSCA-Cofund 2018-BP-00204, M.G.-B.). L.C.R. was funded by the Energy Sustainability Grant 429271 of the National Council of Science and Technology and the Mexican Ministry of Energy (CONACYT-SENER). This work was also funded by the European Research Council (ERC StG 637675 ‘SYBORG’).
Supporting Information Available
This material is available free of charge on the ACS Publications and it contains: The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.0c01487.
Protein library design, quality control, and protocols, structural information on PseFDH as well as descriptions of computational models and data of MD simulations of WT and V9 enzymes, and SDS-PAGE analysis of purified proteins as well as enzyme kinetic data and Michaelis–Menten curves of evolved and deconvoluted enzymes (PDF)
Author Contributions
∇ L.C.-R., C.C.-T., and G.M.M.S. contributed equally to this study.
Author Contributions
L.C.R performed the in vivo experiments. C.C.-T. performed the computational modeling and analysis with support and guidance from M.G.-B. and S.O. L.C.R. and G.M.M.S. performed the kinetic experiments. S.N.L. assisted with the in vivo experiments and constructed the NADPH auxotrophic strain. A.B.-E. and C.G.A.-R. supervised the study and wrote the paper with the input of all authors. All authors revised the work and approved it for publication.
The authors declare the following competing financial interest(s): A.B.-E. is a cofounder of b.fab, exploring the commercialization of microbial bioproduction using formate as a feedstock. The company was not involved in any way in performing or funding this study.
Supplementary Material
References
- Claassens N. J.; Burgener S.; Vogeli B.; Erb T. J.; Bar-Even A. A critical comparison of cellular and cell-free bioproduction systems. Curr. Opin. Biotechnol. 2019, 60, 221–229. 10.1016/j.copbio.2019.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummel W.; Groger H. Strategies for regeneration of nicotinamide coenzymes emphasizing self-sufficient closed-loop recycling systems. J. Biotechnol. 2014, 191, 22–31. 10.1016/j.jbiotec.2014.07.449. [DOI] [PubMed] [Google Scholar]
- Andexer J. N.; Richter M. Emerging enzymes for ATP regeneration in biocatalytic processes. ChemBioChem 2015, 16, 380–386. 10.1002/cbic.201402550. [DOI] [PubMed] [Google Scholar]
- Babel W. The Auxiliary Substrate Concept: From simple considerations to heuristically valuable knowledge. Eng. Life Sci. 2009, 9, 285–290. 10.1002/elsc.200900027. [DOI] [Google Scholar]
- Tishkov V. I.; Popov V. O. Protein engineering of formate dehydrogenase. Biomol. Eng. 2006, 23, 89–110. 10.1016/j.bioeng.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Spaans S. K.; Weusthuis R. A.; van der Oost J.; Kengen S. W. NADPH-generating systems in bacteria and archaea. Front. Microbiol. 2015, 6, 742. 10.3389/fmicb.2015.00742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslan S.; Noor E.; Bar-Even A. Holistic bioengineering: rewiring central metabolism for enhanced bioproduction. Biochem. J. 2017, 474, 3935–3950. 10.1042/BCJ20170377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gul-Karaguler N.; Sessions R. B.; Clarke A. R.; Holbrook J. J. A single mutation in the NAD-specific formate dehydrogenase from Candida methylica allows the enzyme to use NADP. Biotechnol. Lett. 2001, 23, 283–287. 10.1023/A:1005610414179. [DOI] [Google Scholar]
- Serov A. E.; Popova A. S.; Fedorchuk V. V.; Tishkov V. I. Engineering of coenzyme specificity of formate dehydrogenase from Saccharomyces cerevisiae. Biochem. J. 2002, 367, 841–847. 10.1042/bj20020379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreadeli A.; Platis D.; Tishkov V.; Popov V.; Labrou N. E. Structure-guided alteration of coenzyme specificity of formate dehydrogenase by saturation mutagenesis to enable efficient utilization of NADP+. FEBS J. 2008, 275, 3859–3869. 10.1111/j.1742-4658.2008.06533.x. [DOI] [PubMed] [Google Scholar]
- Hatrongjit R.; Packdibamrung K. A novel NADP+-dependent formate dehydrogenase from Burkholderia stabilis 15516: screening, purification and characterization. Enzyme Microb. Technol. 2010, 46, 557–561. 10.1016/j.enzmictec.2010.03.002. [DOI] [Google Scholar]
- Hoelsch K.; Suhrer I.; Heusel M.; Weuster-Botz D. Engineering of formate dehydrogenase: synergistic effect of mutations affecting cofactor specificity and chemical stability. Appl. Microbiol. Biotechnol. 2013, 97, 2473–2481. 10.1007/s00253-012-4142-9. [DOI] [PubMed] [Google Scholar]
- Ihara M.; Kawano Y.; Urano M.; Okabe A. Light driven CO2 fixation by using cyanobacterial photosystem I and NADPH-dependent formate dehydrogenase. PLoS One 2013, 8, e71581 10.1371/journal.pone.0071581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogal S.; Beneventi E.; Cendron L.; Bergantino E. Structural basis for double cofactor specificity in a new formate dehydrogenase from the acidobacterium Granulicella mallensis MP5ACTX8. Appl. Microbiol. Biotechnol. 2015, 99, 9541–9554. 10.1007/s00253-015-6695-x. [DOI] [PubMed] [Google Scholar]
- Alpdagtas S.; Yucel S.; Kapkac H. A.; Liu S.; Binay B. Discovery of an acidic, thermostable and highly NADP(+) dependent formate dehydrogenase from Lactobacillus buchneri NRRL B-30929. Biotechnol. Lett. 2018, 40, 1135–1147. 10.1007/s10529-018-2568-6. [DOI] [PubMed] [Google Scholar]
- Davis B. G.; Celik A.; Davies G. J.; Ruane K. M.. Novel enzyme; Oxford University Innovation Ltd.: 2009. [Google Scholar]
- Bennett B. D.; Kimball E. H.; Gao M.; Osterhout R.; Van Dien S. J.; Rabinowitz J. D. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol. 2009, 5, 593–599. 10.1038/nchembio.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls P. Formate as an inhibitor of cytochrome c oxidase. Biochem. Biophys. Res. Commun. 1975, 67, 610–616. 10.1016/0006-291X(75)90856-6. [DOI] [PubMed] [Google Scholar]
- Warnecke T.; Gill R. T. Organic acid toxicity, tolerance, and production in Escherichia coli biorefining applications. Microb. Cell Fact. 2005, 4, 25. 10.1186/1475-2859-4-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner S. N.; Ramirez L. C.; Krusemann J. L.; Yishai O.; Belkhelfa S.; He H.; Bouzon M.; Doring V.; Bar-Even A. NADPH-Auxotrophic E. coli: A Sensor Strain for Testing in Vivo Regeneration of NADPH. ACS Synth. Biol. 2018, 7, 2742–2749. 10.1021/acssynbio.8b00313. [DOI] [PubMed] [Google Scholar]
- Lamzin V. S.; Dauter Z.; Popov V. O.; Harutyunyan E. H.; Wilson K. S. High resolution structures of holo and apo formate dehydrogenase. J. Mol. Biol. 1994, 236, 759–785. 10.1006/jmbi.1994.1188. [DOI] [PubMed] [Google Scholar]
- Tishkov V. I.; Matorin A. D.; Rojkova A. M.; Fedorchuk V. V.; Savitsky P. A.; Dementieva L. A.; Lamzin V. S.; Mezentzev A. V.; Popov V. O. Site-directed mutagenesis of the formate dehydrogenase active centre: role of the His332-Gln313 pair in enzyme catalysis. FEBS Lett. 1996, 390, 104–108. 10.1016/0014-5793(96)00641-2. [DOI] [PubMed] [Google Scholar]
- Galkin A. G.; Kutsenko A. S.; Bajulina N. P.; Esipova N. G.; Lamzin V. S.; Mesentsev A. V.; Shelukho D. V.; Tikhonova T. V.; Tishkov V. I.; Ustinnikova T. B.; Popov V. O. Site-directed mutagenesis of the essential arginine of the formate dehydrogenase active centre. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 2002, 1594, 136–149. 10.1016/S0167-4838(01)00297-7. [DOI] [PubMed] [Google Scholar]
- Alekseeva A. A.; Fedorchuk V. V.; Zarubina S. A.; Sadykhov E. G.; Matorin A. D.; Savin S. S.; Tishkov V. I. The role of ala198 in the stability and coenzyme specificity of bacterial formate dehydrogenases. Acta Naturae 2015, 7, 60–69. 10.32607/20758251-2015-7-1-60-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkov V. I.; Galkin A. G.; Marchenko G. N.; Egorova O. A.; Sheluho D. V.; Kulakova L. B.; Dementieva L. A.; Egorov A. M. Catalytic properties and stability of a Pseudomonas sp.101 formate dehydrogenase mutants containing Cys-255-Ser and Cys-255-Met replacements. Biochem. Biophys. Res. Commun. 1993, 192, 976–981. 10.1006/bbrc.1993.1511. [DOI] [PubMed] [Google Scholar]
- Yamamoto H.; Mitsuhashi K.; Kimoto N.; Kobayashi Y.; Esaki N. Robust NADH-regenerator: improved alpha-haloketone-resistant formate dehydrogenase. Appl. Microbiol. Biotechnol. 2005, 67, 33–39. 10.1007/s00253-004-1728-x. [DOI] [PubMed] [Google Scholar]
- Savin S. S.; Tishkov V. I. Assessment of Formate Dehydrogenase Stress Stability In vivo using Inactivation by Hydrogen Peroxide. Acta Naturae 2010, 2, 97–102. 10.32607/20758251-2010-2-1-97-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahn J. K.; Werlang C. A.; Baumschlager A.; Brinkmann-Chen S.; Mayo S. L.; Arnold F. H. A General Tool for Engineering the NAD/NADP Cofactor Preference of Oxidoreductases. ACS Synth. Biol. 2017, 6, 326–333. 10.1021/acssynbio.6b00188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawfik D. S. Accuracy-rate tradeoffs: how do enzymes meet demands of selectivity and catalytic efficiency?. Curr. Opin. Chem. Biol. 2014, 21, 73–80. 10.1016/j.cbpa.2014.05.008. [DOI] [PubMed] [Google Scholar]
- Sumbalova L.; Stourac J.; Martinek T.; Bednar D.; Damborsky J. Hot Spot Wizard 3.0: web server for automated design of mutations and smart libraries based on sequence input information. Nucleic Acids Res. 2018, 46, W356–W362. 10.1093/nar/gky417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acevedo-Rocha C. G.; Hoebenreich S.; Reetz M. T. Iterative saturation mutagenesis: a powerful approach to engineer proteins by systematically simulating Darwinian evolution. Methods Mol. Biol. 2014, 1179, 103–128. 10.1007/978-1-4939-1053-3_7. [DOI] [PubMed] [Google Scholar]
- Qu G.; Li A.; Sun Z.; Acevedo-Rocha C. G.; Reetz M. T. The Crucial Role of Methodology Development in Directed Evolution of Selective Enzymes. Angew. Chem., Int. Ed. 2020, 10.1002/anie.201901491. [DOI] [PubMed] [Google Scholar]
- Acevedo-Rocha C. G.; Reetz M. T. Assembly of Designed Oligonucleotides: a useful tool in synthetic biology for creating high-quality combinatorial DNA libraries. Methods Mol. Biol. 2014, 1179, 189–206. 10.1007/978-1-4939-1053-3_13. [DOI] [PubMed] [Google Scholar]
- Acevedo-Rocha C. G.; Reetz M. T.; Nov Y. Economical analysis of saturation mutagenesis experiments. Sci. Rep. 2015, 5, 10654. 10.1038/srep10654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kille S.; Zilly F. E.; Acevedo J. P.; Reetz M. T. Regio- and stereoselectivity of P450-catalysed hydroxylation of steroids controlled by laboratory evolution. Nat. Chem. 2011, 3, 738–743. 10.1038/nchem.1113. [DOI] [PubMed] [Google Scholar]
- Kille S.; Acevedo-Rocha C. G.; Parra L. P.; Zhang Z. G.; Opperman D. J.; Reetz M. T.; Acevedo J. P. Reducing codon redundancy and screening effort of combinatorial protein libraries created by saturation mutagenesis. ACS Synth. Biol. 2013, 2, 83–92. 10.1021/sb300037w. [DOI] [PubMed] [Google Scholar]
- Hoebenreich S.; Zilly F. E.; Acevedo-Rocha C. G.; Zilly M.; Reetz M. T. Speeding up directed evolution: Combining the advantages of solid-phase combinatorial gene synthesis with statistically guided reduction of screening effort. ACS Synth. Biol. 2015, 4, 317–331. 10.1021/sb5002399. [DOI] [PubMed] [Google Scholar]
- Pullmann P.; Ulpinnis C.; Marillonnet S.; Gruetzner R.; Neumann S.; Weissenborn M. J. Golden Mutagenesis: An efficient multi-site-saturation mutagenesis approach by Golden Gate cloning with automated primer design. Sci. Rep. 2019, 9, 10932. 10.1038/s41598-019-47376-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M.; Tokushige M.; Katsuki H. Studies on regulatory functions of malic enzymes. II. Purification and molecular properties of nicotinamide adenine dinucleotide-linked malic enzyme from Eschericha coli. J. Biochem. 1973, 73, 169–180. [PubMed] [Google Scholar]
- Reetz M. T.; Sanchis J. Constructing and analyzing the fitness landscape of an experimental evolutionary process. ChemBioChem 2008, 9, 2260–2267. 10.1002/cbic.200800371. [DOI] [PubMed] [Google Scholar]
- Hur S.; Bruice T. C. The near attack conformation approach to the study of the chorismate to prephenate reaction. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 12015–12020. 10.1073/pnas.1534873100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanique A. M.; Parra L. P. Protein Engineering for Nicotinamide Coenzyme Specificity in Oxidoreductases: Attempts and Challenges. Front. Microbiol. 2018, 9, 194. 10.3389/fmicb.2018.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acevedo-Rocha C. G.; Gamble C. G.; Lonsdale R.; Li A.; Nett N.; Hoebenreich S.; Lingnau J. B.; Wirtz C.; Fares C.; Hinrichs H.; Deege A. P450-Catalyzed Regio- and Diastereoselective Steroid Hydroxylation: Efficient Directed Evolution Enabled by Mutability Landscaping. ACS Catal. 2018, 8, 3395–3410. 10.1021/acscatal.8b00389. [DOI] [Google Scholar]
- Li G.; Zhang H.; Sun Z.; Liu X.; Reetz M. T. Multiparameter Optimization in Directed Evolution: Engineering Thermostability, Enantioselectivity, and Activity of an Epoxide Hydrolase. ACS Catal. 2016, 6, 3679–3687. 10.1021/acscatal.6b01113. [DOI] [Google Scholar]
- Acevedo-Rocha C. G.; Agudo R.; Reetz M. T. Directed evolution of stereoselective enzymes based on genetic selection as opposed to screening systems. J. Biotechnol. 2014, 191, 3–10. 10.1016/j.jbiotec.2014.04.009. [DOI] [PubMed] [Google Scholar]
- Wu W.; Zhu D.; Hua L. Site-saturation mutagenesis of formate dehydrogenase from Candida bodinii creating effective NADP+-dependent FDH enzymes. J. Mol. Catal. B: Enzym. 2009, 61, 157–161. 10.1016/j.molcatb.2009.06.005. [DOI] [Google Scholar]
- Schwander T.; Schada von Borzyskowski L.; Burgener S.; Cortina N. S.; Erb T. J. A synthetic pathway for the fixation of carbon dioxide in vitro. Science 2016, 354, 900–904. 10.1126/science.aah5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castiglione K.; Fu Y.; Polte I.; Leupold S.; Meo A. Asymmetric whole-cell bioreduction of (R)-carvone by recombinant Escherichia coli with in situ substrate supply and product removal. Biochem. Eng. J. 2017, 117, 102–111. 10.1016/j.bej.2016.10.002. [DOI] [Google Scholar]
- Rozzell J., Hua L., Mayhew M.; Novick S.. Mutants of enzymes and methods for their use; Biocatalytics Inc.: 2003. [Google Scholar]
- Robescu M. S.; Rubini R.; Beneventi E.; Tavanti M.; Lonigro C.; Zito F.; Filippini F.; Cendron L.; Bergantino E. From the amelioration of a NADP+-dependent formate dehydrogenase to the discovery of a new enzyme: round trip from theory to practice. ChemCatChem 2020, 12, 2478. 10.1002/cctc.201902089. [DOI] [Google Scholar]
- Castillo R.; Oliva M.; Marti S.; Moliner V. A theoretical study of the catalytic mechanism of formate dehydrogenase. J. Phys. Chem. B 2008, 112, 10012–10022. 10.1021/jp8025896. [DOI] [PubMed] [Google Scholar]
- Cui D.; Zhang L.; Jiang S.; Yao Z.; Gao B.; Lin J.; Yuan Y. A.; Wei D. A computational strategy for altering an enzyme in its cofactor preference to NAD(H) and/or NADP(H). FEBS J. 2015, 282, 2339–2351. 10.1111/febs.13282. [DOI] [PubMed] [Google Scholar]
- Reetz M. T. The importance of additive and non-additive mutational effects in protein engineering. Angew. Chem., Int. Ed. 2013, 52, 2658–2666. 10.1002/anie.201207842. [DOI] [PubMed] [Google Scholar]
- Miton C. M.; Tokuriki N. How mutational epistasis impairs predictability in protein evolution and design. Protein Sci. 2016, 25, 1260–1272. 10.1002/pro.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadet F.; Fontaine N.; Li G.; Sanchis J.; Ng Fuk Chong M.; Pandjaitan R.; Vetrivel I.; Offmann B.; Reetz M. T. A machine learning approach for reliable prediction of amino acid interactions and its application in the directed evolution of enantioselective enzymes. Sci. Rep. 2018, 8, 16757. 10.1038/s41598-018-35033-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z.; Kan S. B. J.; Lewis R. D.; Wittmann B. J.; Arnold F. H. Machine learning-assisted directed protein evolution with combinatorial libraries. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 8852–8858. 10.1073/pnas.1901979116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H. W.; Chen Q.; Pan J.; Zheng G. W.; Xu J. H. Rational engineering of formate dehydrogenase substrate/cofactor affinity for better performance in NADPH regeneration. Appl. Biochem. Biotechnol. 2020, 10.1007/s12010-020-03317-7. [DOI] [PubMed] [Google Scholar]
- Cotton C. A.; Claassens N. J.; Benito-Vaquerizo S.; Bar-Even A. Renewable methanol and formate as microbial feedstocks. Curr. Opin. Biotechnol. 2020, 62, 168–180. 10.1016/j.copbio.2019.10.002. [DOI] [PubMed] [Google Scholar]
- Yishai O.; Bouzon M.; Doring V.; Bar-Even A. In Vivo Assimilation of One-Carbon via a Synthetic Reductive Glycine Pathway in Escherichia coli. ACS Synth. Biol. 2018, 7, 2023–2028. 10.1021/acssynbio.8b00131. [DOI] [PubMed] [Google Scholar]
- Sauer U.; Canonaco F.; Heri S.; Perrenoud A.; Fischer E. The soluble and membrane-bound transhydrogenases UdhA and PntAB have divergent functions in NADPH metabolism of Escherichia coli. J. Biol. Chem. 2004, 279, 6613–6619. 10.1074/jbc.M311657200. [DOI] [PubMed] [Google Scholar]
- Kim S.; Lindner S. N.; Aslan S.; Yishai O.; Wenk S.; Schann K.; Bar-Even A. Growth of E. coli on formate and methanol via the reductive glycine pathway. Nat. Chem. Biol. 2020, 16, 538. 10.1038/s41589-020-0473-5. [DOI] [PubMed] [Google Scholar]
- Wenk S.; Yishai O.; Lindner S. N.; Bar-Even A. An Engineering Approach for Rewiring Microbial Metabolism. Methods Enzymol. 2018, 608, 329–367. 10.1016/bs.mie.2018.04.026. [DOI] [PubMed] [Google Scholar]
- Acevedo-Rocha C. G.; Ferla M.; Reetz M. T. Directed Evolution of Proteins Based on Mutational Scanning. Methods Mol. Biol. 2018, 1685, 87–128. 10.1007/978-1-4939-7366-8_6. [DOI] [PubMed] [Google Scholar]
- Xia Y.; Chu W.; Qi Q.; Xun L. New insights into the Quik Change process guide the use of Phusion DNA polymerase for site-directed mutagenesis. Nucleic Acids Res. 2015, 43, e12 10.1093/nar/gku1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A.; Betz R. M.; Cerutti D. S.; Cheatham T. E. III; Darden T. A.; Duke R. E.; Giese T. J.; Gohlke H.; Goetz A. W.; Homeyer N.; Izadi S.. AMBER 2016; University of California: San Francisco, 2016.
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Bayly C. I.; Cieplak P.; Cornell W.; Kollman P. A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 1993, 97, 10269–10280. 10.1021/j100142a004. [DOI] [Google Scholar]
- Besler B. H.; Merz K. M. Jr.; Kollman P. A. Atomic charges derived from semiempirical methods. J. Comput. Chem. 1990, 11, 431–439. 10.1002/jcc.540110404. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A., et al. Gaussian09; Gaussian, Inc.: 2009.
- Maier J. A.; Martinez C.; Kasavajhala K.; Wickstrom L.; Hauser K. E.; Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagui C.; Darden T. A. Molecular dynamics simulations of biomolecules: long-range electrostatic effects. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 155–179. 10.1146/annurev.biophys.28.1.155. [DOI] [PubMed] [Google Scholar]
- Ryckaert J. P.; Ciccotti G.; Berendsen H. J. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- Wu X.; Brooks B. R. Self-guided Langevin dynamics simulation method. Chem. Phys. Lett. 2003, 381, 512–518. 10.1016/j.cplett.2003.10.013. [DOI] [Google Scholar]
- Becke A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A: At., Mol., Opt. Phys. 1988, 38, 3098–3100. 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- Jain R.; Ahuja B. L.; Sharma B. K. Density-Functional Thermochemistry. III. The Role of Exact Exchange. Indian J. Pure Ap Phy 2004, 42, 43–48. [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.