

SUMMARY
TRAF-interacting protein with a forkhead-associated domain B (TIFAB) is implicated in myeloid malignancies with deletion of chromosome 5q. Employing a combination of proteomic and genetic approaches, we find that TIFAB regulates ubiquitin-specific peptidase 15 (USP15) ubiquitin hydrolase activity. Expression of TIFAB in hematopoietic stem/progenitor cells (HSPCs) permits USP15 signaling to substrates, including MDM2 and KEAP1, and mitigates p53 expression. Consequently, TIFAB-deficient HSPCs exhibit compromised USP15 signaling and are sensitized to hematopoietic stress by derepression of p53. In MLL-AF9 leukemia, deletion of TIFAB increases p53 signaling and correspondingly decreases leukemic cell function and development of leukemia. Restoring USP15 expression partially rescues the function of TIFAB-deficient MLL-AF9 cells. Conversely, elevated TIFAB represses p53, increases leukemic progenitor function, and correlates with MLL gene expression programs in leukemia patients. Our studies uncover a function of TIFAB as an effector of USP15 activity and rheostat of p53 signaling in stressed and malignant HSPCs.
Graphical Abstract
In Brief
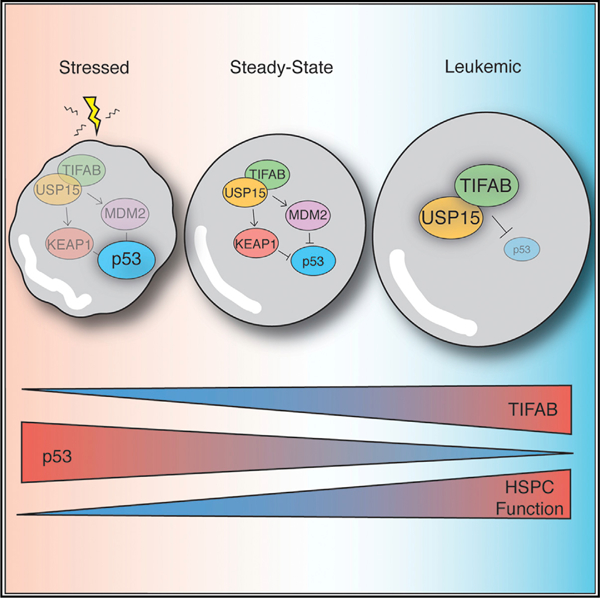
Niederkorn et al. identify TIFAB as a critical node in hematopoietic cells under stressed and oncogenic cell states. Their studies indicate that deregulation of the TIFAB-USP15 complex, as observed in del(5q) myelodysplasia or MLL-rearranged leukemia, modulates p53 activity and has critical functional consequences for stressed and malignant hematopoietic cells.
INTRODUCTION
TRAF-interacting protein with a forkhead-associated domain (TIFA) and its structural homolog, TIFAB, are members of a forkhead-associated (FHA)-domain-containing family of proteins that can recognize and bind phospho-threonine residues on interacting proteins (Matsumura et al., 2004; Takatsuna et al., 2003). Both TIFA and TIFAB harbor a conserved FHA domain, which contains the phospho-threonine recognition sites, and is flanked by structurally distinct N- and C-terminal domains (Matsumura et al., 2004). The conserved FHA domain has been well characterized to mediate protein-protein interactions, thus eliciting effects on cellular signaling.
TIFA and TIFAB have been implicated in various cellular signaling pathways associated with hematopoietic and immune cells. TIFA promotes nuclear factor κB (NF-κB) and c-Jun N-terminal kinase (JNK) signaling in splenic B cells by binding TRAF6 and promoting Toll-like receptor (TLR) signaling following inflammatory stimuli, ultimately leading to cell cycle progression and cell survival under infectious or DNA-damaging conditions (Ea et al., 2004; Fu et al., 2018; Gall et al., 2017; Gaudet et al., 2017; Men et al., 2018; Takatsuna et al., 2003; Zimmermann et al., 2017). Recently, TIFA was implicated as an indispensable cytosolic innate immune sensor for a metabolic intermediate of lipopolysaccharide biosynthesis (Gaudet et al., 2015). TIFAB was identified in an in silico screen for genes containing sequence homology to TIFA and found to interact with TIFA and TRAF6 (Matsumura et al., 2004, 2009). Unlike TIFA, binding of TIFAB to TRAF6 suppresses TRAF6-mediated TLR-NF-κB signaling (Matsumura et al., 2009). One potential explanation for the disparate functions of TIFA and TIFAB is the requirement of an N-terminal threonine (Thr) residue at position 9 in TIFA. Phosphorylation of Thr-9 on TIFA initiates rapid assembly of a large signaling complex including TIFA and TRAF6, which then elicits downstream signaling (Gaudet et al., 2015; Huang et al., 2012). TIFAB lacks a homologous residue at this position. Collectively, these studies identified contrasting roles for TIFA- and TIFAB-dependent signaling. Though relatively small and lacking any intrinsic catalytic functions, TIFA and TIFAB are important mediators of innate immune signaling, with broader implications in immune disorders, cancers, and hematological malignancies.
The TIFAB locus lies within the commonly deleted region on chromosome 5q (del(5q)) in myeloid malignancies, including myelodysplastic syndromes (MDSs) and acute myeloid leukemia (AML) (Varney et al., 2015, 2017; Zhao et al., 1997). MDSs are clonal hematopoietic stem cell disorders characterized by ineffective hematopoiesis and peripheral blood (PB) cytopenias with a propensity to develop AML (Steensma, 2015, 2018; Tefferi and Vardiman, 2009). TIFAB is the only TRAF-interacting protein directly implicated in hematopoietic malignancies, as it resides within a recurring deleted segment of chromosome 5; therefore, the scope of its molecular and cellular functions differs from those relating to TIFA. Mouse genetic studies revealed that TIFAB function is important for maintaining effective hematopoiesis by regulating TRAF6 protein levels (Matsumura et al., 2004; Varney et al., 2015, 2017). Specifically, in the absence of TIFAB, TRAF6 protein levels are elevated, thereby increasing the intensity and duration of TLR inflammatory responses. Moreover, deletion of TIFAB in hematopoietic cells recapitulates features of human MDSs, including progressive cytopenias, altered myeloid differentiation, and propensity to develop bone marrow (BM) failure (Bennett et al., 1982; Starczynowski et al., 2010; Steensma, 2015, 2018; Tefferi and Vardiman, 2009; Weh et al., 1991). Increasing evidence indicates that aberrant innate immune signaling (Barreyro et al., 2018; Fang et al., 2017a, 2017b; Rhyasen et al., 2013; Ribezzo et al., 2019), which we observe in TIFAB-deficient models, is sufficient to induce MDS phenotypes. Although TIFAB is within the deleted region in del(5q) MDSs and AML, it likely has pleotropic functions in hematopoietic cells, yet it remains a poorly characterized member of the TRAF-interacting protein family. By employing a combination of proteomic and genetic approaches, we uncovered a function of TIFAB in stressed and malignant hematopoiesis through regulation of USP15 deubiquitination activity and downstream p53 signaling.
RESULTS
The TIFAB Interactome Implicates Ubiquitin Signaling Networks in Leukemic Cells
To determine the function of TIFAB, we performed tandem-affinity purification and mass spectrometry (MS) analysis of TIFAB-interacting protein complexes in a del(5q) leukemia cell line (HL60). Utilizing HL60 cells that retrovirally express FLAG- and hemagglutinin (HA)-epitope-tagged TIFAB (FLAG/HA-TIFAB), we immunoprecipitated TIFAB-specific binding partners, which were then identified by liquid chromatography (LC)-MS and peptide sequence alignment (Figure 1A). We compared the TIFAB-specific peptides to peptides that precipitated with vector-transduced control HL60 cells (Figure 1B). Any proteins that aligned to sequences in both samples and matched to fewer than three peptides were excluded from further analysis. From the remaining hits, 218 TIFAB-interacting proteins matched to interactions in the STRING database (Jensen et al., 2009). We reasoned that proteins with low specificity and high interconnectedness are more likely to be nonspecific interactions; therefore, we excluded hits below the low-confidence cutoff on the STRING database and excluded highly interconnected proteins via PageRank calculation. From this list, we assembled a TIFAB interactome to visualize the relative interconnectedness of the top 100 TIFAB-interacting proteins ranked by average precursor intensity (Figure 1C; Table S1). We assessed biological pathway enrichment by statistical over-representation using Reactome (Fabregat et al., 2018; Figure 1D). The enriched pathways include innate immune signaling, which we would expect, but also p53-dependent apoptotic pathways and cellular stress response pathways, such as the response to reactive oxygen species and the unfolded protein response. Biological replicates of this experiment were analyzed using two independent MS approaches yielding different sensitivities in spectral resolution. Based on the unique proteins identified, TIFA, ANXA2, and USP15 were reproducible TIFAB-interacting proteins shared between both experiments (Figure 1E). Considering the results from the pathway analyses and ranked TIFAB-interacting peptides, ubiquitin-specific peptidase 15 (USP15) emerged as the lead candidate. USP15 belongs to a family of ubiquitin-specific peptidases that removes ubiquitin moieties from proteins, including MDM2 (Zou et al., 2014), Keap1 (Villeneuve et al., 2013), and Ub-H2B (Long et al., 2014), among others also involved in DNA damage response, cell cycle (Faronato et al., 2013), oxidative stress response (Cornelissen et al., 2014; Villeneuve et al., 2013), response to infection (Zhang et al., 2015), and genome integrity (Fielding et al., 2018). To validate the proteomic screen, HL60 leukemic cells expressing FLAG-TIFAB were immunoprecipitated with anti-FLAG antibodies. Endogenous USP15 immunoprecipitated with FLAG-TIFAB, suggesting that TIFAB and USP15 are bona fide interacting partners (Figure 1F). Our data revealed that TIFAB binds USP15 and may impact cellular stress responses related to p53-dependent signaling pathways.
Figure 1. The TIFAB Interactome Implicates Ubiquitin Signaling Networks in Leukemic Cells.

(A) Schematic overview of workflow for identifying TIFAB-interacting proteins in a human del(5q) AML cell line.
(B) Silver-stained gel for vector-transduced protein eluate and TIFAB-transduced protein eluate from a tandem-affinity purification procedure.
(C) Interaction network for TIFAB-interacting proteins with the top 100 precursor intensity values. STRING interaction data were plotted in Cytoscape (Shannon et al., 2003), which calculates degree, a measure of connectivity (red, low; blue, high).
(D) Dot plot representing p values of statistically over-represented pathways, generated by analyzing TIFAB-interacting proteins in Reactome.
(E) Venn diagram of TIFAB-interacting proteins identified in two independent biological replicates of the proteomics screen (1A) and analyzed using two mass spectrometry (MS) approaches with varying sensitivities.
(F) Immunoblotting of endogenous USP15 on lysates immunoprecipitated for FLAG-TIFAB that were isolated from HL60 cells transduced with retrovirus encoding FLAG-TIFAB and subsequently used for MS analysis.
TIFAB Binds the Catalytic Domain of USP15 and Increases Its Rate of Deubiquitination
To delineate the region of USP15 bound to TIFAB, we utilized a series of N-terminal FLAG-epitope-tagged deletion fragments of USP15 and conducted FLAG immunoprecipitations in HEK293 cells (Figure 2A; Zou et al., 2014). Co-immunoprecipitation of TIFAB with USP15 deletion mutants demonstrated that TI-FAB binds at least two regions of USP15. TIFAB binds USP15 mutants that harbor residues 1–583 and 470–981 (Figure 2A). Both of these USP15 mutants retain at least one ubiquitin-like fold and parts of the ubiquitin C-terminal hydrolase domain (UCH). In contrast, TIFAB bound less efficiently to USP15 mutants 114–981 and 278–981 (Figure 2A). These findings raise the possibility that TIFAB binds more than one domain of USP15 and/or that certain USP15 deletion mutants improperly fold back onto the UCH domain and prevent the binding of TI-FAB. To determine whether TIFAB has a preference for binding catalytically active or inactive USP15, we immunoprecipitated FLAG-TIFAB and then immunoblotted for HA-USP15 or a catalytically inactive mutant, HA-USP15(C269A) (Das et al., 2017). USP15 was readily detected following immunoprecipitation of TIFAB (lane 4) as compared to the nonspecific bands (lanes 2–3) (Figure 2B). In contrast, TIFAB was less efficient at binding to HA-USP15(C269A) (Figure 2B), suggesting that TIFAB has a higher binding affinity for enzymatically active USP15.
Figure 2. TIFAB Binds the Catalytic Domain of USP15 and Increases Its Rate of Deubiquitination.

(A) Top: immunoblotting of HA-TIFAB on lysates immunoprecipitated for FLAG-USP15 deletion mutants that were isolated from HEK293T cells transfected with HA-TIFAB and the indicated FLAG-USP15 mutants. Bottom: schematic of human USP15 with key functional domains. DUSP, domain present in ubiquitin-specific peptidases; UBL, ubiquitin-like domain; UCH, ubiquitin C-terminal hydrolase.
(B) Immunoprecipitation of FLAG-TIFAB and immunoblotting for wild-type or catalytically inactive USP15 (C269A) from HEK293T cells transfected with FLAG-TIFAB alone, HA-USP15 constructs alone, and FLAG-TIFAB combined with HA-USP15 constructs (lanes 4 and 5).
(C) Cell-free deubiquitination assay utilizing internally quenched fluorescent (IQF) diubiquitin as a substrate for USP15. 500 nM K48-linked diubiquitins was incubated with 50 nM USP15 plus mock buffer (black) or USP15 plus 50 nM recombinant TIFAB (red). Relative fluorescent units (RFUs) measured over time by spectrometry are a readout of USP15 activity. Four replicates were analyzed per group for two independent experimental assays.
(D) The reaction rate (RFU/s) of the linear phase of the reaction (~500 s) plotted as a function of diubiquitin substrate concentration using an allosteric sigmoidal model to calculate Vmax. Increasing amounts of diubiquitins were added to the reaction with 50 nM USP15 and 50 nM TIFAB until the rate of ubiquitin hydrolysis by USP15 began to saturate. The USP15 rate of reaction is increased by adding TIFAB to the reaction (red versus black). Four replicates were analyzed per group for two independent experimental assays.
(E) Schematic of the orthogonal deubiquitination assay.
(F) Coomassie-stained gels containing diubiquitin incubated with USP15 alone or USP15 with TIFAB for the indicated times. Diubiquitins for which USP15 has a high affinity (K11, K6, and K48) are in the top row (USP15 dependent). Diubiquitins for which USP15 has low affinity (K29, linear, and K27) are in the bottom row (USP15 independent). Panels were constructed by assembling images from the respective sections of gels containing the proteins indicated.
Since we observed that TIFAB binds the catalytic domain of USP15 and the binding of TIFAB to USP15 is affected by the deubiquitinase activity of USP15, we next sought to determine whether TIFAB binding affects USP15 deubiquitinating activity. To address this, we employed in vitro cell-free deubiquitination assays, which measure cleavage of diubiquitin substrates. Diubiquitins are chemically linked by either lysine 48 (K48) or K63 linkages and harbor a fluorophore that is activated when the covalent bond is cleaved by a deubiquitinase (Figure 2C, top panel; and Figure S1A). First, we incubated recombinant USP15 with quenched-fluorescent K48 diubiquitin substrates in the presence or absence of TIFAB, and then we determined the relative fluorescent units over time as a readout for USP15 deubiquitin enzyme (DUB) activity (Bozza et al., 2012; McGouran et al., 2013). Incubation of USP15 with the diubiquitin substrate resulted in cleavage of the diubiquitin, as indicated by the increase in relative fluorescence (Figure 2C). However, addition of TIFAB resulted in an increase of USP15-mediated cleavage of the diubiquitin as compared to USP15 alone (Figure 2C). As a control, incubation of immunoprecipitated TIFAB or recombinant TIFAB alone with the diubiquitin substrate did not result in diubiquitin cleavage, indicating that TIFAB does not have intrinsic enzymatic activity (Figures S1A and S1B). To evaluate how TIFAB affects the enzymatic properties of USP15, we next determined whether TIFAB binding can accelerate the rate of USP15 deubiquitination. To test this, the in vitro cell-free deubiquitination assay was performed with increasing amounts of diubiquitin substrate, which permits calculating the maximum rate of reaction at saturating concentrations of the substrate. Relative to control samples, which contain only the diubiquitin substrate and enzyme (USP15), addition of TIFAB resulted in faster rates of reaction for USP15 at a given concentration of substrate, indicating that TIFAB increases the velocity of USP15 catalytic activity (Vmax = 13,508 nM/s versus 19,200 nM/s) (Figure 2D).
USP15 has been reported to cleave several types of ubiquitin chains (McGouran et al., 2013), so we next evaluated whether TIFAB affects the ubiquitin linkage preferences of USP15 using an orthogonal assay. We examined the ability of USP15 to cleave diubiquitin substrates containing varying chain types (K6, K11, K27, K29, K48, and linear ubiquitin) incubated alone or with the addition of TIFAB (Figure 2E). Cleavage of diubiquitins to ubiquitin monomers by USP15 is visualized by the disappearance of the larger, diubiquitin substrate and the accumulation of mono-ubiquitins following gel electrophoresis and Coomassie blue staining (Figure S1C). USP15 rapidly hydrolyzed K11, K6, and K48 diubiquitins, which was enhanced upon the addition of TIFAB (Figure 2F, top row). In contrast, USP15 has comparatively weaker specificity for K27, K29, and linear ubiquitin, as the larger diubiquitin substrate remained relatively stable for up to 20 min (Figure 2F, bottom row). Although TIFAB increased the reaction rate of USP15 cleavage of its preferred ubiquitin linked chains, incubation of TIFAB did not result in USP15-mediated cleavage of K27, K29, and linear ubiquitins (Figure 2F), indicating that TIFAB does not change the linkage preferences of USP15. As a negative control, a TIFAB mutant that lacks amino acids in its C-terminal domain is unable to increase USP15 activity (Figure S1D). Importantly, when TIFAB was incubated with another USP enzyme, USP7, we could not detect a discernable increase in the rate of ubiquitin hydrolysis with TIFAB, indicating that the effects of TIFAB are, at least, partly selective to USP15 (Figure S1E). Collectively, our data indicate that TIFAB increases the rate of USP15 deubiquitination without altering the enzyme specificity for ubiquitin chain types.
TIFAB Regulates USP15-Dependent Signaling in Hematopoietic Cells
USP15 has been previously shown to deubiquitinate KEAP1 and MDM2, which share biological functions in cellular stress responses that converge upon p53 (Figure 3A). For this example, USP15 removes K48-linked ubiquitin chains and prevents proteasome-mediated degradation of MDM2 (Zou et al., 2014) and KEAP1 (Villeneuve et al., 2013), which results in reduced p53 expression. To determine whether TIFAB can increase the DUB activity of USP15 by accelerating removal of K48-linked chains from MDM2, we performed a cell-based deubiquitination assay. We co-expressed HA-epitope-tagged ubiquitin and MDM2 along with USP15 and/or TIFAB in HEK293 cells. Transfected cells were also treated with the proteasome inhibitor MG-132 to prevent degradation of MDM2 and allow for accumulation of K48-ubiquitinated MDM2. Immunoprecipitation of MDM2 followed by immunoblotting for ubiquitin revealed that USP15 expression reduces MDM2 K48-linked poly-ubiquitination, as previously described (Zou et al., 2014; Figure 3B, lane 2 versus lane 3). Consistent with our biochemical evidence that TIFAB increases USP15 DUB activity, co-expression of TIFAB and USP15 resulted in further reduction in MDM2 K48-linked ubiquitination (Figure 3B, lane 3 versus lane 5). Interestingly, expression of TIFAB alone was sufficient to moderately reduce MDM2 ubiquitination, potentially by increasing the DUB activity of endogenous USP15 (Figure 3B, lane 2 versus 4). The effects of TIFAB on USP15 function are likely occurring in the cytoplasm, as both proteins are primarily localized to the cytoplasmic fraction (Figure S1F). Our data suggest that TIFAB directly mediates USP15 catalytic function, resulting in deubiquitination of USP15 substrates.
Figure 3. TIFAB Regulates USP15-Dependent Signaling in Hematopoietic Cells.

(A) Schematic overview of TIFAB and USP15 substrates exerting homeostatic control of p53, left, and de-repression of p53 by deletion of TIFAB (middle) or by deletion of USP15 (right).
(B) Immunoblotting of HA-K48-Ub on lysates immunoprecipitated for MDM2 that were isolated from HEK293T cells transfected with TIFAB, USP15, and/or MDM2. Transfected cells were incubated with the proteasome inhibitor (MG-132) to enrich for ubiquitinated MDM2.
(C) Immunoblotting of Usp15, Keap1, and vinculin in Tifab+/+ and Tifab−/− BM mononuclear cells.
(D) Immunoblotting of Mdm2, p53, and vinculin in Tifab+/+ and Tifab−/− Lin− BM cells.
(E) Immunoblotting of USP15, Keap1, p53, and vinculin in Usp15+/+ and Usp15−/− BM mononuclear cells.
(F) Immunoblotting of Mdm2 and vinculin in Usp15+/+ and Usp15−/− BM mononuclear cells.
TIFAB is primarily expressed in hematopoietic cells. Therefore, based on our observations that expression of TIFAB increases USP15 enzyme activity in vitro, we posited that loss of TIFAB would result in impaired USP15 signaling in hematopoietic stem and progenitor cells (HSPCs). Examination of TIFAB-deficient (Tifab−/−) BM cells revealed that the level of the USP15 DUB substrate, KEAP1, was reduced as compared to WT (Tifab+/+) BM cells (Figure 3C). Tifab−/− BM cells also exhibited decreased expression of MDM2 and a corresponding increase in p53 expression (Figure 3D). To determine whether these effects were due to decreased USP15 signaling in hematopoietic cells, we developed a USP15 knockout mouse and examined these USP15 substrates in BM cells (Figure S2). Similar to TI-FAB-deficient BM cells, BM cells from Usp15−/− mice exhibited reduced expression of KEAP1 and MDM2 and a corresponding increase in p53 expression compared to Usp15+/+ BM cells (Figures 3E and 3F). Our data suggest that TIFAB regulates USP15 signaling to substrates in hematopoietic cells (Figure 3A).
Loss of TIFAB Sensitizes HSPCs to p53-Dependent Stress
p53 is critical in hematopoiesis by instructing cell fate under various conditions, including cell cycling and proliferation (Xu et al., 2012), balancing differentiation and self-renewal (Asai et al., 2011; Liu et al., 2009), and responding to DNA damage (Chen et al., 2018). Since TIFAB regulates USP15 DUB activity and substrates implicated in p53 activation, we hypothesized that deregulation of the TIFAB-USP15 complex, such as by genetic deletion of TIFAB, would sensitize hematopoietic cells to a variety of cellular stressors. We assayed the function of TIFAB-deficient BM cells following stress of hematopoietic transplantation, myeloablation, 5-fluorouracil (5-FU) injection, ionizing radiation, and viral infection (Figure 4A). First, we examined myeloablation and hematopoietic recovery of mice reconstituted with TIFAB-deficient or WT BM cells. Although all mice survived immediately following BM transplantation, ~40% of congenic mice transplanted with Tifab−/− BM cells succumbed to hematopoietic defects after 16 months as compared to mice transplanted with Tifab+/+ BM cells, consistent with previous observations (Figures 4B and S3). In contrast, Tifab−/− mice without BM transplantation did not develop overt disease during the same time period (Figure 4B), suggesting that TIFAB-deficient hematopoietic cells exhibit diminished function following myeloablation and transplantation. Gene expression analysis was performed on lineage-negative (Lin−) cKit+Sca1+ (LSK) cells isolated from recipient mice transplanted with Tifab+/+ and Tifab−/− BM cells. Gene set enrichment analysis revealed that TIFAB-deficient HSPCs are associated with altered expression of genes involved in electron transport (orange), ubiquitin-dependent processes (blue), and p53-dependent pathways (green) (Figure 4C). As expected, we also observed changes to immune signaling pathways (red), which are due in part to the role of TIFAB in negatively regulating TRAF6 (Matsumura et al., 2009; Varney et al., 2015, 2017) Following BM transplantation of Tifab+/+ and Tifab−/− BM cells into lethally irradiated congenic recipients, Tifab−/− HSPCs exhibited an increase in a subset of p53 target genes (Figure 4D), consistent with increased p53 activity and with increased p53 protein observed (Figure 3D). Under stress of transplantation, some TIFAB-deficient HSPCs undergo apoptosis, as evidenced by cleavage of caspase-3 (Figure 4E), suggesting that loss of TIFAB primes p53 function. Similarly, examination of p53 target genes in MDS patient samples, which we stratified based on TIFAB expression, revealed an increase in a subset of p53 target genes in TIFAB-”low” patients relative to patients with elevated TIFAB expression (Figure S4).
Figure 4. Loss of TIFAB Sensitizes HSPCs to p53-Dependent Stress.

(A) Overview of experimental scheme.
(B) Kaplan-Meier survival curves for nontransplanted and transplanted mice reconstituted with Tifab+/+ or Tifab−/− BM cells (four groups total). All groups except transplanted Tifab−/− exhibited 100% survival. Summary from two independent transplants analyzed for up to 16 months. A subset of the transplanted mice were previously characterized at early time points (Varney et al., 2015).
(C) Table displaying top 25 enriched pathways in TIFAB-deficient LSK cells, identified by RNA microarray and gene set enrichment analysis, with false discovery rate (FDR) <0.5. Colored pathways denote shared functions.
(D) Heatmap representing Z score values for established p53 target genes in Tifab+/+ and Tifab−/− BM LK cells (Wei et al., 2006).
(E) Immunoblotting of caspase-3 and actin on lysates isolated from BM mononuclear cells of mice reconstituted with Tifab+/+ and Tifab−/− BM cells.
(F) Peripheral blood count recovery after 5-FU injection (150 mg/kg) of mice reconstituted with BM from Tifab+/+ (n = 16), Tifab−/− (n = 16), and Tifab−/−;p53−/− (n = 5) mice. When comparing Tifab−/− and Tifab−/−;p53−/− mice, *p < 0.05.
(G) Immunoblotting of p53 in lineage-negative (Lin−) BM cells isolated from Tifab+/+ and Tifab−/− mice treated with 0, 2, or 6 Gy irradiation in vitro.
(H) Colony formation in methylcellulose of Lin− BM cells isolated from Tifab+/+ or Tifab−/− mice treated in vitro with 0, 2, or 6 Gy irradiation (n = 3). *p < 0.05.
(I) Colony formation in methylcellulose of Lin− BM cells of Tifab+/+ or Tifab−/− after infection with mCherry-expressing lentivirus (LV; pReciever-mCherry) (n = 2). A representative image is shown on the right as an image section of the entire well. *p = 0.09.
(J) Relative number of colonies of Tifab−/− Lin− BM cells transduced with vectors encoding shp53 or control shRNA (shCtl) (n = 3). Data are normalized to Tifab+/+ expressing shControl. A representative image is shown on the right as an image section of the entire well. *p = 0.08.
To apply these functional assays under other forms of hematopoietic stress, we next examined the consequences of TIFAB deletion in response to 5-FU injection. To generate BM chimeric mice, we transplanted Tifab+/+ and Tifab−/− BM cells into lethally irradiated recipients. After confirming that blood counts recovered to normal levels, we treated recipient mice with 150 mg/ kg 5-FU by intraperitoneal injection and tracked PB recovery of white blood cells (WBCs), red blood cells (RBCs), and platelets (PLTs) (Figure 4F). Mice reconstituted with Tifab−/− BM cells exhibited increased sensitivity to 5-FU as compared to mice reconstituted with Tifab+/+ BM cells. This was indicated by lower WBC counts following 5-FU and a compromised recovery of WBCs up to 16 days (Figure 4F). The recovery of RBCs and PLTs following 5-FU was comparable between mice reconstituted with Tifab−/− and Tifab+/+ BM cells. To determine whether the impaired recovery of Tifab-deficient HSPC to 5-FU is due to increased p53 activation, we deleted p53 in TIFAB-deficient mice by crossing Tifab−/− with p53−/− mice (Figures S5A and S5B). Mice reconstituted with Tifab−/−;p53−/− BM cells exhibited improved recovery of WBCs following 5-FU compared to mice reconstituted with Tifab−/− BM cells (Figure 4F), suggesting that p53 signaling is augmented in TIFAB-deficient hematopoietic cells following chemotherapeutic stress.
Based on the well-defined role of p53 in genome integrity, we also subjected TIFAB−/− cells to DNA damage induced by ionizing radiation and viral infection. HSPCs (Lin− BM cells) were isolated from Tifab+/+ and Tifab−/− mice, irradiated with 2 or 6 Gy, and then evaluated for p53 protein and hematopoietic progenitor function by assessing colony formation in methylcellulose. As expected, we observed an increase in p53 expression in Tifab−/− HSPCs as compared to Tifab+/+ HSPC at multiple doses of irradiation (Figure 4G). Moreover, Tifab−/− HSPCs treated with 2 and 6 Gy radiation formed significantly fewer colonies than Tifab+/+ HSPCs (Figure 4H). Finally, Tifab+/+ and Tifab−/− HSPC were infected with lentivirus (pReceiver backbone encoding mCherry for sorting) to induce genotoxic stress. Following infection with lentivirus, Tifab+/+ and Tifab−/− HSPCs were sorted for mCherry-positive cells and plated in methylcellulose. Corroborating our observations with irradiated treatment, virally infected Tifab−/− HPSCs formed fewer colonies as compared to Tifab+/+ HSPCs (Figure 4I). To determine whether the impaired hematopoietic progenitor function of TIFAB-deficient cells was due to elevated p53 activity, we instead infected Tifab+/+ and Tifab−/− HSPCs with lentivirus encoding small hairpin RNAs (shRNAs) targeting p53 (shp53) or non-targeting shRNA (shControl) and evaluated hematopoietic colony formation in methylcellulose. Knockdown of p53 in Tifab−/− HSPCs (shp53-Tifab−/−) restored colony formation following viral infection as compared to Tifab−/− HSPC infected with shControl (Figures 4J and S5C). Based on our observations that loss of TIFAB both compromises USP15 signaling and sensitizes HSPCs to stress, our data support a model where USP15-mediated repression of p53 is ineffective in TIFAB-deficient hematopoietic cells.
TIFAB Is Highly Expressed and Functionally Relevant in Human MLL-Rearranged AML
Given that the TIFAB-USP15 axis suppresses p53-mediated stress in hematopoietic cells, we sought to determine whether this pathway was relevant in AML. Examination of TIFAB RNA expression in The Cancer Genome Atlas (TCGA) dataset revealed that a subset of AML patients expressed relatively higher levels of TIFAB as compared to the entire cohort (Figure 5A). Moreover, AML patients with the highest TIFAB expression levels (top quartile) were significantly enriched in gene expression signatures associated with mixed-lineage leukemia 1 (MLL)-rearranged leukemia (Figure 5B). MLL is a hematopoietic transcription factor involved in chromosomal rearrangements that produce potent fusion oncogenes, such as MLL-AF9, which drive leukemic transcriptional programs and produce an aggressive subtype of leukemias. Although MLL-AF9 is not predicted to directly regulate TIFAB expression (data not shown), when we compared TIFAB mRNA across leukemia cell lines, we observed the highest level of TIFAB expression in MLL-rearranged leukemic cell lines MV4;11 and RS4;11, corroborating our finding in patient samples (Figure 5C). We hypothesized that high expression of TIFAB could indicate a potential functional dependency of leukemic cells on the TIFAB-USP15 complex as a means of inhibiting tumor-suppressive p53 activity. To determine whether TIFAB expression is important for the function of MLL-rearranged AML, we expressed shRNAs targeting TIFAB in MV4;11 cells and evaluated leukemic progenitor function in methylcellulose. Knockdown of TIFAB significantly impaired the leukemic colony-forming potential of MV4;11 cells as compared to MV4;11 cells expressing the scrambled-control shRNA (shControl) (Figures 5D and 5E). However, when we knocked down TIFAB in normal human HSPCs (CD34+), we did not observe a decrease in colony-forming potential (Figure 5D). Likewise, we also knocked down USP15 in MV4;11 leukemic cells and normal human HSPCs. Knockdown of USP15 significantly impaired the colony-forming ability of MV4;11 cells but did not impair normal human HSPCs (Figures 5F and 5G). Our data suggest that the TIFAB-USP15 complex is important for maintaining leukemic progenitor function.
Figure 5. TIFAB Is Highly Expressed and Functionally Relevant in Human MLL-Rearranged AML.

(A) TIFAB mRNA expression in AML patients. Original TIFAB expression values (from TCGA) are normalized using the RNA-Seq by Expectation Maximization (RSEM) software. The mean values of all samples were subtracted from each sample’s RSEM count, which is depicted as “TIFAB mRNA.”
(B) Toppgene pathway analysis was performed on genes expressed in TIFAB-“high”-expressing AML patients (top quartile).
(C) Relative TIFAB mRNA expression in a panel of human leukemic cell lines normalized to HL60 cells, which are haploinsufficient for TIFAB. Red bars denote cell lines with MLL rearrangements.
(D) Colony formation in methylcellulose of CD34+ cells (left) and MV4;11 cells (right) expressing control shRNA (shControl) or shRNA targeting TIFAB (shTIFAB). *p < 0.005.
(E) Representative images of MV4;11 colonies from (D) (n = 3) as image sections of the entire well.
(F) Colony formation in methylcellulose of CD34+ cells (left) and MV4;11 cells (right) expressing control shRNA (shControl) or shRNA targeting USP15 (shUSP15). *p < 0.05.
(G) Representative images of MV4;11 colonies from (F) (n = 3) as image sections of the entire well.
TIFAB Is Critical for the Function of a Mouse Model of MLL-AF9 AML
To further interrogate the relevance of TIFAB in MLL-rearranged AML, Lin-BM cells isolated from Tifab+/+ and Tifab−/− mice were retrovirally transduced with vectors expressing MLL-AF9 (MSCV-IRES-GFP) (Figure 6A). This retroviral MLL-AF9 model generates a rapid, fully penetrant myeloid leukemia in vivo (Martin et al., 2003). We first assessed leukemic progenitor function in colony-forming-cell assays. Tifab−/−;MLL-AF9 cells formed significantly fewer colonies in methylcellulose than Tifab+/+;MLL-AF9 cells (p < 0.0005) (Figure 6B), suggesting that TIFAB is important for MLL-rearranged AML progenitor function. To determine whether TIFAB is required for leukemia development in vivo, Tifab+/+;MLL-AF9 and Tifab−/−;MLL-AF9 cells were transplanted (along with helper BM mononuclear cells) into lethally irradiated recipient mice. Mice transplanted with Tifab+/+;MLL-AF9 cells developed overt leukemia with a median survival of 140 days (p = 0.0031) (Figures 6C and S6). In contrast, only one mouse transplanted with Tifab−/−;MLL-AF9 cells developed overt leukemia with a detectable GFP+ (MLL-AF9) population in its PB (Figure S6), indicating that TIFAB is important for the function of MLL-rearranged AML development.
Figure 6. TIFAB Is Critical for the Function of a Mouse Model of MLL-AF9 AML.

(A) Generation of Tifab+/+ and Tifab−/− MLL-AF9 leukemia model and schematic overview of the experimental setup.
(B) Colony formation in methylcellulose of Tifab+/+;MLL-AF9 or Tifab−/−;MLL-AF9. Two biological experiments were performed in triplicate. Representative images are shown. ***p < 0.0005.
(C) Kaplan-Meier plot for overall survival of recipient mice transplanted with Tifab+/+;MLL-AF9 or Tifab−/−;MLL-AF9 cells.
(D) Relative mRNA expression of the indicated genes in Tifab+/+;MA9 or Tifab−/−;MA9 cells. Three independent experiments were performed in triplicate. Error bars represent SD. *p < 0.05, ***p < 0.0005.
(E) Cell growth analysis in liquid culture of Tifab+/+;MLL-AF9 or Tifab−/−;MLL-AF9 cells expressed as live cell number per day, determined by trypan blue exclusion. *p < 0.05.
(F) Relative cell growth of Tifab−/−;MLL-AF9 (GFP+) cells transduced with lentivirus encoding empty vector (Vector) or USP15 and mCherry was determined by flow cytometry (GFP+mCherry+) at the indicated time points (n = 6 replicates). *p < 0.05.
(G) Relative colony formation in methylcellulose of Tifab+/+;MLL-AF9 or Tifab−/−;MLL-AF9 cells treated with vehicle (DMSO) or doxorubicin (Dox; 5 nM). The number of colonies was normalized to DMSO controls for each group (n = 2 biological experiments, performed in triplicate). ***p < 0.0005.
(H) Experimental setup for generation of Tifab−/−;MLL-AF9 knockin (KI) mice.
(I) Colony formation in methylcellulose of cKit+ MLL-AF9 KI cells expressing FLAG-TIFAB (MSCV-IRES-GFP) or empty vector. Representative images are shown to the right as image sections of the entire well (n = 3). *p < 0.005.
(J) Immunoblotting of p53 and FLAG-TIFAB on lysate isolated from MLL-AF9 KI cKit+ BM cells transduced with FLAG-TIFAB (MSCV-IRES-GFP) or empty vector, with actin as a loading control.
To evaluate whether the impaired function of TIFAB-deficient AML cells is associated with the USP15-p53 axis, we examined the expression of p53 target genes. Expression of the pro-apoptotic genes p21 and Bax were markedly increased in Tifab−/−;MLL-AF9 cells as compared with Tifab+/+;MLL-AF9, suggesting that there is an increase in p53 transcriptional activity (Figure 6D). We next evaluated the growth and viability of TIFAB-deficient AML cells in vitro. Indeed, the TIFAB−/−;MLL-AF9 cells exhibit significantly reduced proliferation compared to Tifab+/+;MLL-AF9 cells (Figure 6E). To establish whether diminished USP15 signaling is relevant to the phenotype observed in TIFAB-deficient AML, we attempted to rescue the cell proliferation defect by exogenously expressing USP15 in TIFAB−/−;MLL-AF9 cells. TIFAB−/−;MLL-AF9 cells were transduced with vectors encoding USP15 (EF1a-USP15-IRES-mCherry) or empty control (pReciever: EF1a-IRES-mCherry). The cell populations expressing the empty vector or Usp15 were then monitored by flow cytometry for mCherry expression. As compared to control TIFAB−/−;MLL-AF9 cells (vector only), USP15-expressing TIFAB−/−;MLL-AF9 cells exhibited a modest yet significantly increased recovery of leukemic cell proliferation (Figure 6F). These findings suggest that TIFAB is important for MLL-rearranged AML, partly by maintaining USP15 signaling.
Given the increase in p53-dependent signaling in the TIFAB−/−;MLL-AF9 cells, we also explored whether TIFAB-deficient MLL-AF9 AML cells are sensitive to chemotherapy. We examined the leukemic progenitor function of Tifab+/+;MLL-AF9 and Tifab−/−;MLL-AF9 cells in methylcellulose treated with the chemotherapeutic agent doxorubicin (5 nM) or a vehicle (DMSO). Treatment of TIFAB−/−;MLL-AF9 cells with doxorubicin resulted in reduced leukemic colony formation as compared to Tifab+/+:MLL-AF9 cells (Figure 6G), suggesting that reduced TIFAB expression sensitizes leukemic cells to chemotherapy.
Lastly, we investigated whether overexpression of TIFAB in MLL-rearranged AML suppresses p53 signaling and can increase the leukemic progenitor function. Given that the retroviral model of MLL-AF9 yields highly aggressive leukemic cells, we instead utilized the MLL-AF9 knockin (KI) model, which exhibits predominantly myeloid leukemia with delayed latency as compared to the MLL-AF9 retroviral AML model (Dobson et al., 1999). HSPCs (cKit+) BM cells isolated from MLL-AF9 knockin mice were transduced with retroviral vectors encoding TIFAB (MSCV-IRES-GFP) or an empty vector (Figure 6H). Transduced GFP+ MLL-AF9 KI cells were isolated by flow cytometric sorting and then examined for leukemic progenitor function in vitro. MLL-AF9 KI cells overexpressing TIFAB formed significantly more colonies as compared to control cells after the first and secondary re-plating (p < 0.005) (Figure 6I), indicating that the overexpression of TIFAB increases MLL-AF9 leukemic progenitor function. The increase in leukemic progenitor function observed in methylcellulose corresponded with reduced expression of p53 (Figure 6J), further implicating TIFAB as an important negative regulator of p53 in AML.
DISCUSSION
In our present study, we characterized the molecular and biochemical functions of TIFAB, a member of the FHA-domain family. Using a combination of proteomics screens, RNA sequencing, and genetic studies in human and murine hematopoietic cells, we identified unexpected roles of TIFAB in stress response associated with p53 signaling, ubiquitin-mediated proteasome regulation, and leukemia. Specifically, we identified that TIFAB binds and regulates the activity of the deubiquitinase, USP15. Employing cell-based and cell-free approaches, we found that TIFAB promotes the deubiquitinating activity of USP15, thus identifying a function for an FHA-domain-containing protein as a modifier of ubiquitin signaling. USP15 contains several binding and regulatory domains, which may directly regulate its activity or specificity by mediating interactions with other proteins or by post-translational modification. Recently, two phospho-threonine residues (Thr149 and Thr219) were identified on USP15 (Das et al., 2019). In that study, phosphorylation of USP15 at Thr149 or Thr219 affected USP15 interaction with its substrates; however, they did not affect intrinsic USP15 catalytic activity (Das et al., 2019). Moreover, the structure of the USP15 catalytic core domain revealed that USP15 displays a catalytically incompetent open configuration of the ubiquitin C-terminal tail with active site loops, similar to the flexibility of the USP7 catalytic domain (Ward et al., 2018). Thus, USP15 must undergo conformational changes upon substrate binding to initiate ubiquitin linkage hydrolysis. Although the precise mechanism by which TIFAB increases the catalytic function of USP15 is not resolved in our studies, we propose several possibilities. TIFAB may increase the efficiency of substrate capture by USP15 and induce an allosteric conformational change leading to increased catalytic rates for ubiquitin chain processing, or it may preferentially bind to active USP15 and stabilize this conformation of the enzyme. Future structural and biochemical studies will help to inform the precise mechanisms that coordinate the phosphorylation of USP15, the binding of TIFAB, and the conformation of USP15.
DUB enzymes are becoming attractive therapeutic targets due to their ability to regulate the stability of many important oncogenes, oncogene-dependent genes, and tumor suppressors. For example, chemical inhibition of USP7 and USP10 with small molecules resulted in degradation of MDM2 in multiple myeloma (Chauhan et al., 2012) and FLT3-ITD in AML (Weisberg et al., 2017), respectively. In solid tumors, RNA-mediated inhibition of USP15 in a patient-derived glioblastoma model destabilized SMAD2 and the oncogenic capacity of glioma-initiating cells (Eichhorn et al., 2012). Modulating either the level or the activity of USP enzymes is an effective approach to simultaneously alter the stability of downstream substrates. Therefore, understanding the regulation of both catalytically dependent and independent functions of USP15 will inform future studies on targeting USP15 with small-molecule inhibitors (Zhang et al., 2015; Padmanabhan et al., 2018).
USP15 has previously been implicated in cellular stress responses, including antioxidant response, cell-cycle exit, response to infection, and p53-mediated apoptosis. In a recent finding, USP15 was shown to deubiquitinate and stabilize the E3 ubiquitin ligase MDM2, resulting in diminished p53 expression and function (Zou et al., 2014). Knockdown of USP15 in human melanoma and colon cancer cell lines sensitized cells to apoptosis by destabilization of MDM2 and increased expression of p53. Consistent with the described function of USP15-dependent regulation of p53, TIFAB-deficient cells were sensitive to genotoxic stress, including 5-FU treatment, DNA damage, and viral infection. Importantly, loss of TIFAB coincided with reduced USP15 signaling in vitro and in vivo. In contrast, exogenous TIFAB, which increases USP15 deubiquitinase activity, suppressed p53 levels and increased leukemic progenitor function in an MLL-AF9 leukemia murine model. These observations suggest that TIFAB is an important regulator of HSPC response to cellular stress, as p53 function is exacerbated in TIFAB-deficient HSPCs, while overexpression of TIFAB dampens p53 in pre-leukemic and leukemic murine models. In del(5q) MDS, a subtype of MDS characterized by an interstitial deletion of chromosome 5q, patients exhibit increased p53 activation in their hematopoietic cells (Liu et al., 2017; Saft et al., 2014; Sallman et al., 2014; Schneider et al., 2016; Wei et al., 2013). Since TIFAB is deleted in nearly all cases of del(5q) MDS, our data support a model wherein the loss of TIFAB sensitizes HSPCs to p53-dependent stress via a USP15 axis.
Several studies have uncovered intriguing relationships between TIFAB expression and function of myeloid leukemia cells. In a retroviral insertional mutagenesis screen for genes that cooperate with haploinsufficiency of another del(5q) MDS/AML gene, Egr1, mice that developed a myeloid neoplasm consistently harbored insertional sites that disrupted genomic regions proximal to the TIFAB locus, suggesting that loss of TIFAB is important for the biology of pre-leukemic myeloid neoplasms (Stoddart et al., 2016). In two independent leukemia studies, expression of TIFAB was reduced upon treatment with the bromo-domain inhibitor JQ1 in acquired-resistant MLL-AF9; KRas-G12D leukemia cells (Rathert et al., 2015) and following suppression of MLL-AF9 leukemic stem cells by the NF-κB-inducing kinase (NIK) (Xiu et al., 2018). These later studies support a model wherein the maintenance of TIFAB expression is important for myeloid leukemia pathogenesis. By generating TIFAB-deficient MLL-AF9 leukemia models and evaluating the requirement of TIFAB in leukemic cell function, we reveal an oncogenic role of TIFAB. Expression of TIFAB and USP15 is required in MLL-AF9 leukemia for maintenance of leukemic cell function, such that deletion of either TIFAB or USP15 drastically impairs leukemic cell function in vitro and in vivo.
The bimodal regulation of TIFAB, through its haploinsufficiency in del(5q) MDS or its elevated mRNA expression in a subset of AML patients, reveals an intriguing paradigm for the function of TIFAB in pre-malignant versus overtly malignant cell states. Tifab deficiency in a murine model results in progressive cytopenias via TRAF6 overexpression in HSPCs (Varney et al., 2015). Chronic innate immune signaling in HSPCs via TRAF6 is likely a determinant of refractory cytopenias and progression of MDSs in patients, as mouse models exhibiting cell-intrinsic activation of the innate immune pathway acquire hematopoietic defects resembling human MDSs (Chen et al., 2013; Fang et al., 2014, 2017b; Keerthivasan et al., 2014; Starczynowski et al., 2010, 2011; Taganov et al., 2006; Varney et al., 2017). Moreover, miR-146a, a TIFAB neighboring gene on chromosome 5q, is a direct negative regulator of TRAF6 and is co-deleted with TIFAB in del(5q) MDS (Starczynowski et al., 2010; Varney et al., 2017). As described herein, Tifab deficiency also sensitizes HSPCs to p53-dependent cellular stress. The combination of chronic innate immune signaling, via loss of miR-146a and TIFAB, and increased sensitivity to p53-dependent stress via loss of TIFAB are likely contributing factors to the development of BM failure in del(5q) MDS patients (Saft et al., 2014; Sallman et al., 2014; Schneider et al., 2016; Wei et al., 2013; Xu et al., 2012). Interestingly, deletion of RPS14, another del(5q) MDS gene, results in p53 activation and chronic innate immune signaling via expression of S100A8/A9 (Ribezzo et al., 2019; Schneider et al., 2016). Collectively, we posit that these combined genetic alterations on chromosome 5q could pose a constraint on malignant transformation and induce a selective pressure of clonally derived mutant del(5q) HSPCs. In fact, patients with isolated del(5q) lesions have more favorable prognoses than patients with del(5q) plus additional aberrations (Germing et al., 2012; Jädersten et al., 2011; Mallo et al., 2011); the latter is associated with an increased risk of transformation to AML. It is possible that HSPCs deficient for TIFAB must overcome this barrier with additional oncogenic lesions to initiate leukemogenesis. Interestingly, in a subset of AML patients, we observe relatively high expression of TIFAB and our functional studies indicate that TIFAB is partly required for MLL-AF9 leukemic progenitor function. We also observed that overexpression of TIFAB reduced p53 protein levels and increased leukemic progenitor function in vitro. Data from our studies and others support a paradigm in myeloid malignancies that the loss of TIFAB via del(5q) impairs the function of pre-malignant HSPCs by activating p53 and inducing chronic innate immune signaling via TRAF6 (Fang et al., 2014; Ribezzo et al., 2019; Starczynowski et al., 2010, 2011; Varney et al., 2015). However, overexpression of TIFAB, as observed in subsets of AML patients, may alleviate this cellular stress, suppress p53, and therefore promote leukemic transformation. Future studies are required to carefully track and dissect the potential factors regulating TIFAB expression in individual patients as they progress from del(5q) MDS to overt AML.
Collectively, our studies suggest that TIFAB is a critical node in hematopoietic cells under both stressed and oncogenic cell states. In summary, our data support a model in which deregulation of the TIFAB-USP15 complex, such as in del(5q) MDS or MLL-rearranged leukemia, modulates p53 activity and has critical functional consequences for stressed and malignant hematopoietic cells.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Daniel Starczynowski (daniel.starczynowski@cchmc.org). Plasmids used in this study were either purchased from or are publicly available through several online repositories including Addgene, Genecopoiea, and Sigma-Aldrich, or have been gifted by colleagues. Sources and identifiers of these constructs are provided in the Key Resources Table. All unique resources generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| FLAG® M2 antibody - clone M2 | Sigma-Aldrich | Cat# F3165-.2MG |
| HA antibody | Sigma-Aldrich | Cat# H6908-.2ML |
| His-tag antibody | Cell Signaling | Cat# 2366 |
| KEAP1 (P586) antibody | Cell Signaling | Cat# 4678 |
| MDM2 antibody | Abcam | Cat# ab16895 |
| MDM2 antibody | Santa Cruz | Cat# sc-965 |
| p53 antibody | Cell Signaling | Cat# 2524S |
| Ubiquitin-Histone 2B (Lys 120) antibody | Cell Signaling | Cat# 5546P |
| USP15 antibody | Abcam | Cat# ab71713 |
| Usp15 antibody | Proteintech | Cat# 14354–1-AP |
| USP15 antibody (2D5) | Santa Cruz | Cat# sc-100629 |
| Vinculin | Cell Signaling | Cat# 13901S |
| Pan-Actin | Cell Signaling | Cat# 4968 |
| GAPDH | Cell Signaling | Cat# 5174 |
| Bacterial and Virus Strains | ||
| VSVG lenti- and retrovirus | Cincinnati Children’s Viral Vector Core: https://www.cincinnatichildrens.org/research/cores/translational-core-laboratory/viral-vector-core | N/A |
| Biological Samples | ||
| Normal human hematopoietic stem and progenitor cells (CD34+) | Cincinnati Children’s Cell Processing Core: https://www.cincinnatichildrens.org/research/cores/translational-core-laboratory/cell-processing-core | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| SilverQuest Silver Staining Kit | Life Technologies | Cat# LC6070 |
| Methocult GF M3434 | Stem Cell Technologies | Cat# 0.03434 |
| Methocult H4434 Classic | Stem Cell Technologies | Cat# 0.04434 |
| RPMI | Fisher | Cat# SH30027.01 |
| IMDM | Corning | Cat# 10–016-CV |
| DMEM | Fisher | Cat# SH30022FS |
| TransIT-LT1 transfection reagent | Mirus | Cat# MIR2306 |
| Proeastome inhibitor MG-132 | Sigma-Aldrich | Cat# C2211–5MG |
| Penicillin/Steptomycin | ThermoFisher | Cat# SV30010 |
| mouse IL-3 cytokine | PeproTech | Cat# 213–13 |
| human IL-6 cytokine | PeproTech | Cat# 200–06 |
| mouse SCF cytokine | PeproTech | Cat# 250–03 |
| CD117 MicoBeads | Miltenyi Biotech | Cat# 130–091-224 |
| CLA1 restriction enzyme | NEB | Cat# R0197S |
| A/G Protein PLUS-Agarose | Santa Cruz | Cat# sc-2003 |
| FLAG® M2 Affinity Gel | Sigma-Aldrich | Cat# A2220–1ML |
| 3X FLAG(R) Peptide | Sigma-Aldrich | Cat# F4799–4MG |
| HA Affinity Matrix from rat IgG1 | Roche | Cat# 11 815 016 001 |
| TIFAB purified recombinant protein (Full-length) | Genscript, for this study | N/A |
| TIFAB purified recombinant protein (deletions) | Genscript, for this study | N/A |
| USP15 purified recombinant protein | Life Sensors | Cat# DB513 |
| USP7 purified recombinant protein | Life Sensors | Cat# DB502 |
| IQF di-Ubiquitin substrate (K48) | Life Sensors | Cat# DU4802 |
| IQF di-ubiquitin substrate (K63) | Life Sensors | Cat# DU6302 |
| di-Ubiquitin purified protein panel (K6, K11, K27, K29, K33, K48, K63, M0) | Life Sensors | Cat# SI200 |
| Recombinant buffer – “Mock” | Genscript, This study | N/A |
| DUB assay buffer | This study | N/A |
| Tandem-affinity purification trition lysis buffer | Singh et al., 2008 | N/A |
| Tandem-affinity purification Buffer C | Singh et al., 2008 | N/a |
| Critical Commercial Assays | ||
| Mouse hematopoietic progenitor cell enrichment kit | Stem Cell Technologies | Cat# 19856 |
| Taqman Gene-expression probe, human beta-Actin | Life Technologies | Hs01060665_g1 |
| Taqman Gene-expression probe, human TIFAB | Applied Biosystems | Hs04185733_m1 |
| Taqman Gene-expression probe, mouse Tifab | Applied Biosystems | Mm04210261_m1 |
| Taqman Gene-expression probe, mouse p21 | Applied Biosystems | Mm00432448_m1 |
| Taqman Gene-expression probe, mouse beta-Actin | ThermoFisher | Mm02619580_g1 |
| Taqman Gene-expression probe, mouse Usp15 | ThermoFisher | Mm00452856_m1 |
| Taqman Gene-expression probe, mouse Trp53 | Life Technoliges | Mm01731290_g1 |
| Taqman Universal Master-Mix | Life Technologies | Cat# 4324020 |
| Deposited Data | ||
| RNA-sequencing of TIFAB-deficient LK cells | Varney et al., 2017 | GEO: GSE87453 |
| RNA Microarray of TIFAB-deficient LSK cells | Varney et al., 2015 | GEO: GSE72936 |
| Experimental Models: Cell Lines | ||
| HL60 | From Aly Karsan Laboratory, ATCC authenticated | ATC CCL-240 |
| MV4;11 | From Lee Grimes Laboratory, ATCC authenticated | ATC CRL-9591 |
| HEK293T | From Susanne Wells Laboratory, ATCC authenticated | ATC CRL-3216 |
| MLL-AF9 immortalized murine lineage negative cells | This study | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Tifab−/−.C57/B6 | This study & Jun-Ichiro Inoue Laboratory, Matsumura et al., 2009 | N/A |
| Mouse: Trp53−/−.B6129S2. | Jackson Laboratories | 002101 |
| Mouse: Tifab−/−;Trp53−/−.C57/B6 | This study | N/A |
| Mouse: Usp15−/−. C57/B6. | This study | N/A |
| Oligonucleotides | ||
| Murine Usp15 Exon 2 Forward Primer (5′−3′): CGAATTGCTAACTACCAGCTTATC | IDT | N/A |
| Murine Usp15 Exon 2 Reverse Primer (5′−3′): GCTCTCCTTGTACTTGCAGG | IDT | N/A |
| Recombinant DNA | ||
| pReceiver-mCherry (negative control) | GeneCopoeia | Cat# EX-NEG-LV214 |
| pReceiver-mCherry-USP15 | GeneCopoeia | Cat# EX-Mm02672-Lv214 |
| USP15 domain-deletion plasmids | Shao-Cong Sun Laboratory, Zou et al., 2014 | N/A |
| pLKO.1.-shScramble-GFP | Daniel Starczynowski Laboratory | N/A |
| pLKO.1-shTIFAB-GFP | Daniel Starczynowski Laboratory | TRCN: 0000182133 |
| pLKO.1-shScramble-mCherry | This study | N/A |
| pLKO.1-shUSP15-mCherry | This study | TRCN: 0000007568 |
| MSCV-pGK-FLAG-HA-TIFAB-GFP | Daniel Starczynowski Laboratory, Varney et al., 2015 | |
| MSCV-pGK-GFP | Daniel Starczynowski Laboratory, Varney et al., 2015 | |
| pLen-Lox_U6_shTrp53 | Addgene | Cat# 59360 |
| pLen-Lox_U6_shNlgn2 | Addgene | Cat# 59358 |
| MSCV-MLL-AF9-GFP | Ahish Kumar Laboratory | N/A |
| pCS2-HA-USP15 | Eun Joo Song Laboratory, Das et al., 2017 | N/A |
| pCS2-HA-USP15-C269A | Eun Joo Song Laboratory, Das et al., 2017 | N/A |
| MDM2 plasmid | Addgene | Cat# 16233 |
| His-Xpress-USP15 | Addgene | Cat# 23216 |
| pcDNA3.0-FLAG-TIFAB | Varney et al., 2015 | N/A |
| HA-Ubiquitin plasmid | Addgene | Cat# 17608 |
| Software and Algorithms | ||
| Statistical over-representation test (pathway enrichment) | Reactome Pathway Database | N/A |
| Prism 7 | GraphPad | N/A |
| Prism 8 | GraphPad | N/A |
| “GetPageRank”: Perl module (Graph::Centrality::Pagerank) | CPAN respository, Cao et al., 2016 | N/A |
| Degree calculation for TIFAB-interaction network | Cytoscape | N/A |
| Protein-interaction Confidence cutoffs | STRING | N/A |
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Animals
This study utilized murine animal models, consisting of adult mice aged 1–6 months. Genetic-deletion models, including Tifab, Tifab/ p53, and Usp15, were bred on a C57/BL/6 background. For bone-marrow transplantation studies, a mix of male and female wild-type BoyJ mice were used as recipients, ensuring comparable proportions between experimental groups. Mice were randomly assigned to experimental groups. Congenic BoyJ recipients were conditioned with lethal, 10Gy total body irradiation prior to all BM transplantations. All transplantations were performed using bone marrow mononuclear cells from C57/Bl6 donors, unless specified otherwise. For 5-FU experiments, mice were injected intraperitoneally with 150mg/kg of 5-FU. Animals were bred and housed in the Association for Assessment and Accreditation of Laboratory Animal Care-accredited animal facility of Cincinnati Children’s Hospital Medical Center. All experiments conform to the regulatory standards of the Institutional Animal Care and Use Committee (IACUC) and adhere to IACUC-approved protocols.
Human Samples
Human hematopoietic stem/progenitor cells from normal donor populations were enriched for CD34+ by the Cell Processing Core at Cincinnati Children’s Hospital Medical Center. Cryopreserved CD34+ samples were provided for experimental use.
Cell Lines
All cells were cultured at 37°C with 5% carbon dioxide. MV4;11, HEK293T, and HL60 cell lines have been authenticated by STR Profiling Services from ATCC (ATCC 135-XV-10). MV4;11 and HL60 cells are cultured in RPMI supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. HEK293T cells were cultured in DMEM supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin.
METHOD DETAILS
Cell lines and cell culture
HL60, THP1, and HEK293T cells were gifted from the laboratories indicated and subsequently authenticated by ATCC. MV4;11 cells were cultured in RPMI with 10% FBS and 1% penicillin/streptomycin. During viral infection or irradiation prior to plating CFCs, murine HSPCs were cultured in IMDM with 10% FBS and 1% penicillin/streptomycin and 10ng/mL of each murine IL-3, human IL-6, and murine SCF. HL60 were previously confirmed to have a deletion of chromosome 5q (Starczynowski et al., 2011).
Viral constructs, transductions, and cell sorting
For tandem affinity purification of TIFAB, HL60 cells were transduced with retrovirus encoding a double-tagged, full length FLAG-HA-TIFAB and green fluorescent reporter under PGK promoter or empty vector (Varney et al., 2015). Cells were sorted for GFP positivity and expanded in liquid culture before being pelleted for lysis. To generate a TIFAB-deficient MLL-AF9 model, Tifab−/− Lineage negative cells were transduced with retrovirus encoding MLL-AF9 and a GFP reporter (Krivtsov et al., 2006). To generate a TIFAB-overexpressing MLL-AF9 model, MLL-AF9 knock-in ckit+ cells were transduced with retrovirus encoding empty vector (pGK-GFP), or encoding a flag-tagged TIFAB construct, pGK-FLAG-HA-TIFAB. Viral infection of Tifab+/+ and Tifab−/− ckit+ cells was performed with an empty lentiviral vector encoding mCherry (pReceiver-mCherry; Genecopoiea, EX-NEG-Lv214). Knockdown of p53 was performed with lentivirus encoding shControl-pLenLox_U6 Nlgn2 (Addgene #59358) or shp53-pLenLox_U6 Trp53 (Addgene #59360). Knockdown of Tifab or Usp15 in MV4;11 cells and CD34+ cells was performed using lentivirus generated from pLKO.1-shTIFAB-GFP or pLKO.1-shUSP15-mCherry, with scrambled-shRNA controls for each construct. All experiments where CFCs were conducted were performed with FACS-sorted cells for GFP+ or mCherry+. All cells were sorted using FACS Aria equipment or Sony SH800S under BSL2 protocol.
Tandem-affinity purification and silver-staining
To isolate TIFAB protein complexes, we performed tandem-affinity purification of FLAG-HA-TIFAB (Singh et al., 2008). Cell pellets of roughly 20 million cells were frozen at −80 degrees until a total cell-pellet size of approximately 1 mL was achieved. Cell pellets were chemically lysed using 300mM salt buffer (LB300) and mechanically lysed. Lysate was spun in an ultracentrifuge and passed through BioRad spin column filters. 25 uL of FLAG-M2-Agarose beads were added to 1mL of lysate and incubated overnight, rocking at 4 degrees. Bead-lysate mixtures were centrifuged and supernatant was aspirated. Beads were combined in 400 ul 150 ug/mL of 3X FLAG peptide and were incubated with intermittent shaking every 15 minutes, on ice, for 2 hours. The 3X FLAG peptide-conjugate supernatant was added to 25 ul of anti-HA Affinity Matrix (Roche 11 815 016 001) per sample and was prepared in Buffer C. 3X FLAG peptide and HA affinity matrix were incubated at 4 degrees overnight with rocking. HA-resin and 3X-FLAG-Peptide mixture were separated by centrifugation and supernatant was aspirated. 40ul of 2X SDS buffer was added to HA resin and samples were boiled at 95 degrees for 5 minutes. Eluate from HA-resin was collected by centrifugation through a filtered spin column (BioRad) and 3ul of sample was loaded for silver-stain gel analysis using SilverQuest Silver Staining Kit (Life Technologies, LC6070). An aliquot of input lysate and final eluate were used to probe for endogenous USP15 before and after FLAG-TIFAB immunoprecipitation (Figure 1).
Mass-spectrometry and bioinformatic analysis
Tandem-affinity purification samples were generated twice; one replicate was analyzed by the UC Proteomics Laboratory and by the Taplin Mass-Spectrometry Facility. At the UC laboratory, 1D mini gel containing lanes of control (pGK-GFP) and TIFAB (pGK-FLAG-HA-TIFAB-GFP) for in-gel digestions and nLC-MSMS protein ID. The gel was fixed for 2 hr in 10% Acetic acid/50% ethanol and the image captured. The samples were excised from the gel, reduced, alkylated, and digested with trypsin. The peptides were recovered and analyzed by LC-MS/MS on the 5600 TripleTOF. A Protein Pilot search of Homo sapiens was used to identify the proteins. Contaminant proteins were removed from the list, leaving 14 proteins that were uniquely identified with at least 3 distinct peptides at > 99% confidence. At the Taplin facilities, gel sections were digested with trypsin, washed, vacuum-dehydrated and stored until analysis. Prior to analysis, the samples were reconstituted in HPLC solvent, 2.5% acetonitrile, 0.1% formic acid. Peptides were eluted on a reverse-phase capillary column with increasing concentrations of solvent, 97.5% acetonitrile, 0.1% formic acid. Peptides were eluted, ionized, and then entered into an LTQ Orbitrap Velos Pro ion-trap mass spectrometer (ThermoFisher Scientific). Peptide sequences were determined by matching protein databases using Sequest. To process the mass-spec hits similarly to what has been previously described (Cao et al., 2016), we excluded from the TIFAB-interacting proteins those that are found in the vector control samples, keeping only those with more or equal than 3 peptides. We then uploaded the list of proteins to the STRING database and downloaded the interaction list, of which 218 molecules match in the STRING Database (out of the 228). We downloaded 5,006 interactions using STRING’s “low confidence” score cutoff of 0.150 and subsequently kept only the STRING interactions that have 0.900 confidence. Plots of the STRING network were generated using R, and then PageRank of each node in the string network was calculated using “GetPageRank.” PageRank is a measure of “connectedness” of a node within a network (all nodes must be connected by at least one edge). High PageRank fractions refer to highly connected nodes and therefore more likely to be pulled down by chance (proteins with low specificity). We identified overlapping protein names between the files “page-rank_0.700.dat” and “page-rank_0.900.dat,” only considering proteins with PageRank ≤ 0.005. This resulted in a list of 151 protein names. Using the complete list of 151 TIFAB-interacting proteins, we identified statistically over-represented pathways using Reactome.
Plasmids and transfections
USP15 deletion mutant constructs were provided by the Shao-Cong Sun laboratory (Zou et al., 2014). The TIFAB domain-deletion mutant was generated by cloning FLAG-tagged mutant DNA from IDT into pCDNA3.0 vector (Varney et al., 2015). For transient transfection of USP15 and MDM2, pcDNA Xpress-His-USP15 WT (Addgene #23216) and pcDNA MDM2 (Addgene #16233) were used. All transfections were prepared in 1 mL serum free, antibiotic free media with 45 ul of TransIT-LT1 transfection reagent (Mirus MIR2306). Plasmids were added and incubated for 20 minutes before transfection mixture was added to 30% confluent HEK293T cells. Cells were transfected for 24–48 hours. For cell-based deubiquitination of MDM2, HEK293 cells were treated with 25 uM of proteasome inhibitor MG-132 (Sigma) for 5 hours prior to collection. Plasmids encoding HA-tagged USP15 WT and USP15 mutant (C269A) in the pCS2 vector were previously generated and provided by Eun Joo Song (Das et al., 2017).
Immunoprecipitation
A table of reagents used for immunoprecipitation assays is provided in the Key Resources Table. Cell lysates from HEK293T cells were brought to a total of 1 mL using PBS and incubated with FLAG-M2 antibodies at 4 degrees overnight to bind FLAG-USP15 mutant proteins. 50 uL of A/G protein PLUS resin was added for 2 hours at 4 degrees. Resin-protein-complexes were separated by centrifugation and washed 3X for 15 minutes using high salt buffer (Singh et al., 2008). Protein samples were reconstituted with 2X Laemmli sample buffer with beta-mercaptoethanol prior to boiling. Resin was filtered from sample using centrifugation and a BioRad Spin column and then loaded for gel electrophoresis. Cell lysates from HEK293T cells expressing FLAG-TIFAB with HA-USP15-WT and HA-USP15-C269A were immunoprecipitated using FLAG-M2-resin as described above.
Western blotting
A table of all antibodies is provided in the Key Resources Table. Antibody dilutions were prepared at 1:1000 to 1:500 in 5% BSA or dry milk in TBST. Protein samples prepared directly from lysate were prepped in 5X SDS sample buffer. Protein samples used in IPs were prepped in 2X Laemmli sample buffer reconstituted with BME. Immunoblotting was performed in SDS buffer, using BioRad gradient gels, at 175 V for 42 minutes. Precision plus dual color protein marker (BioRad) was used in parallel to samples. Proteins were wet-transferred onto nitrocellulose membranes (0.2 uM, BioRad) at 100V for 1 hr. Membranes were blocked in 5% BSA or milk for 1 hr RT depending on antibodies to be used. Membranes were incubated with primary antibodies overnight at 4 degrees and washed for a total of 30 minutes in TBST before adding secondary diluted to 1:10,000 in 5% milk.
Recombinant TIFAB proteins
Full-length recombinant TIFAB and domain-deletion mutant DNA sequences were generated by Genscript. Codon optimization was performed and TIFAB and mutants were cloned into E.coli Expression Vector pET30a. E. Coli were transformed and grown in 1 L of TB culture. Full-length TIFAB protein was obtained from inclusion bodies by one-step purification on a Ni column. Protein was prepared in several aliquots of storage buffer (50 mM Tris-HCl, 150 mM NaCl, 10% Glycerol, 0.5 M L-Arginine, pH 8.5) and stored at −80 degrees.
Cell-free deubiquitination assays
For the internally-quenched fluorescence assays (IQF), USP15 (LifeSensors, DB513) was used at a final concentration of 200 nM per well in a black 96-well plate. K48 or K63 linked di-ubiquitin molecules (LifeSensors, DU4802 and DU6302) were reconstituted in DUB assay buffer (5% 1M Tris-HCl pH8, 0.005% Tween-20, 1% 1M DTT diluted in H20) to be used at final concentrations ranging from 1.25 nM to 1uM. If needed, individual wells were brought to 150 uL reaction volume total using assay buffer. 50 nM USP15 was incubated alone with K48-linked di-ubiquitin, or with the addition of 50 nM TIFAB. To control for volume and for pH buffering effects, reactions with USP15 alone included buffer generated according to the solution in which recombinant TIFAB was purified (50 mM Tris-HCl, 150 mM NaCl, 10% Glycerol, 0.5 M L-Arginine). Kinetic fluorescence was immediately measured using a SpectraMax microplate reader over time for 30 minutes until the reaction neared saturation (ex. 540, em. 580). For non-fluorescent ubiquitin hydrolysis assays, 2.5 ug of USP15 or USP7 (LifeSensors, DB502) was pre-incubated with Mock buffer or with 3.5 ug recombinant TIFAB brought to 25 uL total volume using assay buffer, and incubated for 30 minutes at 37°C. 2.5 ug of a panel of purified di-ubiquitin chains, purchased from LifeSensors, were added to the mixture, and 5uL aliquots were taken from each reaction over several minutes, quenched in 2X SDS loading buffer, and boiled at 95 degrees for 5 minutes. Each 5 uL aliquot contained 0.5 ug USP15, 0.7 ug TIFAB, and 0.5 ug ubiquitin, which were separated by gel electrophoresis at 175V for 40 minutes and visualized by Coomassie blue staining.
Mice and bone marrow transplantations
Tifab−/− mice were generated on a C57/Bl6 backgrounded and previously characterized (Matsumura et al., 2004; Varney et al., 2015). Usp15−/− mice were generated by CRISPR/Cas9-mediated targeting of conserved USP15 exon 2 using embryonic microinjection of C57/Bl6 wild-type mice. Mosaic gene-edited mice were crossed to C57/Bl6 breeders to generate heterozygotes. Heterozygotes with matching edited alleles were crossed to each other and Usp15−/− pups were validated by sequencing of USP15 exon 2 and by immunoblotting. Targeting vectors and guide RNA sequences will be provided upon request. Tifab−/−;Trp53−/− mice were generated by crossing Tifab−/− mice to Trp53−/− mice, and subsequently crossing Tifab+/−;Trp53+/− mice. Congenic BoyJ recipients were conditioned with lethal, 10 Gy total body irradiation prior to all bone marrow transplantations. All transplantations were performed using total bone marrow from C57/Bl6 donors, unless specified otherwise. For experiments with 5-FU, animals received 150 mg/kg of 5-FU by intraperitoneal injection. Animals were bred and housed in the Association for Assessment and Accreditation of Laboratory Animal Care-accredited animal facility of Cincinnati Children’s Hospital Medical Center.
Generation of Tifab+/+ and Tifab−/− MLL-AF9-expressing cells
To generate a Tifab-deficient MLL-AF9 leukemia model, we isolated lineage negative bone marrow cells from Tifab+/+ and Tifab−/− mice using the EasyStep Mouse Hematopoietic Progenitor Cell Enrichment Kit (Stem Cell Technologies 19856). Lineage negative cells were then transduced with retrovirus encoding the MLL-AF9 fusion gene (MSCV-MLL-AF9-IRES-GFP). Transduced cells were serially re-plated in methylcellulose to enrich for the MLL-AF9-expressing cells (> 95% GFP+ cells remained after 3 platings). At this time, Tifab+/+;MLL-AF9 (GFP+) and Tifab−/−;MLL-AF9 (GFP+) cells were transferred to liquid culture using IMDM with 10% FBS, 1% penicillin-streptomycin, and 10 ng/mL of mouse IL-3, human IL-6, and mouse SCF to be used for in vitro experiments and bone marrow transplantations. For transplantation of these cells into recipients, each BoyJ mouse was lethally irradiated and received 500,000 MLL-AF9 cells with 500,000 bone marrow mononuclear cells from a BoyJ donor. To generate TIFAB overexpressing MLL-AF9 cells, ckit+ cells from MLL-AF9-genetic knock-in donors were enriched using AutoMACS positive selection using CD117 murine microbeads. We transduced ckit+ cells with retrovirus encoding pGK-GFP or pGK-FLAG-HA-TIFAB-GFP. The following day 100,000 sorted GFP+ cells plus 100,000 bone marrow helper cells were transplanted per mouse. Tifab−/−;MLL-AF9 (GFP+) cells were used to generate USP15-overexpressing MLL-AF9 cells by viral transduction with lentivirus encoding USP15 (pReciever: EF1a-USP15-IRES-mCherry) or an empty control vector (pReceiver: EF1a-IRES-mCherry). For doxorubicin treatments, 1,000 cells of Tifab+/+;MLL-AF9 (GFP+) and Tifab−/−;MLL-AF9 (GFP+) were plated per mL of m3434 methylcellulose. The methylcellulose was prepared with a final concentration of 5 nM doxorubicin reconstituted in DMSO or with an equal volume of DMSO for control groups. Colonies were counted manually between 7–10 days when detectable size and confluency were reached.
Bone marrow extraction, HSPC enrichment, and colony assays
To generate bone marrow (BM) mononuclear cells for immunoblots, six murine hind-limb bones were collected, crushed, and filtered in PBS (Fang et al., 2018). BM cells were pelleted and subjected to red-blood cell lysis buffer to isolate BM mononuclear cells. For colony forming assays in methylcellulose, isolation and enrichment of hematopoietic stem/progenitor cells (HSPCs) from Tifab+/+ and Tifab−/− mice was performed using the EasyStep Mouse Hematopoietic Progenitor Cell Enrichment Kit (Stem Cell Technologies 19856) for lineage depletion, or by AutoMACS positive selection using CD117 murine microbeads for ckit+ enrichment. Murine HSPCs were plated in 1 mL of MethoCult® GF M3434 (Stem Cell Technologies) per well of an untreated 6-well culture plate. Total colonies were counted manually at 5–10-day intervals, depending on the growth rate of the cells. Where colony images are provided, images were captured using the StemVision plate reader (Stem Cell Technologies).
Quantification and Statistical Analysis
Statistical analyses were performed using GraphPad Prism 7 and Prism 8 software. Unpaired, non-parametric t Tests were used to calculate P values. For experiments with more than two conditions, multiple t tests were performed per group. Statistical significance was determined if p < 0.05. Information on biological and technical replicates, “n,” for experiments can be found in the corresponding figure legend. For pathway enrichment analysis of TIFAB-interacting proteins, P values for each pathway were determined by a statistical over-representation analysis within Reactome.
DATA AND CODE AVAILABILITY
The gene-set enrichment analyses and RNA-sequencing of Tifab+/+ and Tifab−/− HSPCs analyzed during this study have been deposited in NCBI’s Gene Expression Omnibus and are accessible by the following GEO Series accession numbers: GSE72936 (Varney et al., 2015) and GSE87453 (Varney et al., 2017). Mass-spectrometry data for TIFAB-interacting proteins is available in Table S1.
Supplementary Material
Highlights.
TIFAB binds the catalytic domain of USP15 and increases its rate of deubiquitination
TIFAB regulates USP15-dependent signaling in hematopoietic cells
Loss of TIFAB sensitizes HSPCs to p53-dependent stress
TIFAB is overexpressed and functionally relevant in MLL-rearranged AML
ACKNOWLEDGMENTS
This work was supported by grants from the Cincinnati Children’s Hospital Research Foundation, Leukemia Lymphoma Society (Scholar Award), and National Institutes of Health (R35HL135787) to D.T.S. M.N. was supported by a National Institute of Health training and career development grant (F31HL137310), a National Cancer Institute predoctoral to postdoctoral transition award (F99CA234924), and a Pelotonia fellowship. K.D.G. and the UC Mass-Spectrometry Laboratory are supported by the National Institutes of Health shared instrumentation program (S10RR027015). The results shown here are based in part upon data generated by the TCGA Research Network. We thank Jeff Bailey and Victoria Summey for assistance with transplantations (Comprehensive Mouse and Cancer Core at CCHMC). We thank the Viral Vector, DNA Sequencing, and Genotyping Cores at CCHMC for their assistance. We thank Sean Post (MD Anderson Cancer Center), Yi Zheng (CCHMC), Nicolas Nassar (CCHMC), and Gabriel Gracia-Maldonado (CCHMC) for helpful discussion and suggestions. We thank Ben Keener for technical assistance in designing the graphical abstract.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.01.093.
DECLARATION OF INTERESTS
D.T.S. is a consultant for Kurome Therapeutics.
REFERENCES
- Asai T, Liu Y, Bae N, and Nimer SD (2011). The p53 tumor suppressor protein regulates hematopoietic stem cell fate. J. Cell. Physiol 226, 2215–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreyro L, Chlon TM, and Starczynowski DT (2018). Chronic immune response dysregulation in MDS pathogenesis. Blood 132, 1553–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, and Sultan C (1982). Proposals for the classification of the myelodysplastic syndromes. Br. J. Haematol 51, 189–199. [PubMed] [Google Scholar]
- Bozza WP, Liang Q, Gong P, and Zhuang Z (2012). Transient kinetic analysis of USP2-catalyzed deubiquitination reveals a conformational rearrangement in the K48-linked diubiquitin substrate. Biochemistry 51, 10075–10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao LL, Riascos-Bernal DF, Chinnasamy P, Dunaway CM, Hou R, Pujato MA, O’Rourke BP, Miskolci V, Guo L, Hodgson L, et al. (2016). Control of mitochondrial function and cell growth by the atypical cadherin Fat1. Nature 539, 575–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan D, Tian Z, Nicholson B, Kumar KG, Zhou B, Carrasco R, McDermott JL, Leach CA, Fulcinniti M, Kodrasov MP, et al. (2012). A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell 22, 345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Eksioglu EA, Zhou J, Zhang L, Djeu J, Fortenbery N, Epling-Burnette P, Van Bijnen S, Dolstra H, Cannon J, et al. (2013). Induction of myelodysplasia by myeloid-derived suppressor cells. J. Clin. Invest 123, 4595–4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Gao R, Yao C, Kobayashi M, Liu SZ, Yoder MC, Broxmeyer H, Kapur R, Boswell HS, Mayo LD, and Liu Y (2018). Genotoxic stresses promote clonal expansion of hematopoietic stem cells expressing mutant p53. Leukemia 32, 850–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelissen T, Haddad D, Wauters F, Van Humbeeck C, Mandemakers W, Koentjoro B, Sue C, Gevaert K, De Strooper B, Verstreken P, and Vandenberghe W (2014). The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum. Mol. Genet 23, 5227–5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das T, Park JK, Park J, Kim E, Rape M, Kim EE, and Song EJ (2017). USP15 regulates dynamic protein-protein interactions of the spliceosome through deubiquitination of PRP31. Nucleic Acids Res. 45, 4866–4880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das T, Kim EE, and Song EJ (2019). Phosphorylation of USP15 and USP4 regulates localization and spliceosomal deubiquitination. J. Mol. Biol 431, 3900–3912. [DOI] [PubMed] [Google Scholar]
- Dobson CL, Warren AJ, Pannell R, Forster A, Lavenir I, Corral J, Smith AJ, and Rabbitts TH (1999). The mll-AF9 gene fusion in mice controls myeloproliferation and specifies acute myeloid leukaemogenesis. EMBO J 18, 3564–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ea CK, Sun L, Inoue J, and Chen ZJ (2004). TIFA activates IkappaB kinase (IKK) by promoting oligomerization and ubiquitination of TRAF6. Proc. Natl. Acad. Sci. USA 101, 15318–15323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichhorn PJ, Rodón L, Gonzàlez-Juncà, Dirac A, Gili M, Martıńez-Sáez E, Aura C, Barba I, Peg V, Prat A, et al. (2012). USP15 stabilizes TGF-b receptor I and promotes oncogenesis through the activation of TGF-b signaling in glioblastoma. Nat. Med 18, 429–435. [DOI] [PubMed] [Google Scholar]
- Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, Haw R, Jassal B, Korninger F, May B, et al. (2018). The reactome pathway knowledgebase. Nucleic Acids Res 46 (D1), D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J, Barker B, Bolanos L, Liu X, Jerez A, Makishima H, Christie S, Chen X, Rao DS, Grimes HL, et al. (2014). Myeloid malignancies with chromosome 5q deletions acquire a dependency on an intrachromosomal NF-κB gene network. Cell Rep 8, 1328–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J, Bolanos LC, Choi K, Liu X, Christie S, Akunuru S, Kumar R, Wang D, Chen X, Greis KD, et al. (2017a). Corrigendum: Ubiquitination of hnRNPA1 by TRAF6 links chronic innate immune signaling with myelodysplasia. Nat. Immunol 18, 474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J, Bolanos LC, Choi K, Liu X, Christie S, Akunuru S, Kumar R, Wang D, Chen X, Greis KD, et al. (2017b). Ubiquitination of hnRNPA1 by TRAF6 links chronic innate immune signaling with myelodysplasia. Nat. Immunol 18, 236–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J, Muto T, Kleppe M, Bolanos LC, Hueneman KM, Walker CS, Sampson L, Wellendorf AM, Chetal K, Choi K, et al. (2018). TRAF6 mediates basal activation of NF-κB necessary for hematopoietic stem cell homeostasis. Cell Rep 22, 1250–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faronato M, Patel V, Darling S, Dearden L, Clague MJ, Urbé S, and Coulson JM (2013). The deubiquitylase USP15 stabilizes newly synthesized REST and rescues its expression at mitotic exit. Cell Cycle 12, 1964–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielding AB, Concannon M, Darling S, Rusilowicz-Jones EV, Sacco JJ, Prior IA, Clague MJ, Urbé S, and Coulson JM (2018). The deubiquitylase USP15 regulates topoisomerase II alpha to maintain genome integrity. Oncogene 37, 2326–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Huang D, Yuan F, Xie N, Li Q, Sun X, Zhou X, Li G, Tong T, and Zhang Y (2018). TRAF-interacting protein with forkhead-associated domain (TIFA) transduces DNA damage-induced activation of NF-κB. J. Biol. Chem 293, 7268–7280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall A, Gaudet RG, Gray-Owen SD, and Salama NR (2017). TIFA signaling in gastric epithelial cells initiates the cag type 4 secretion system-dependent innate immune response to Helicobacter pylori infection. MBio 8, e01168–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudet RG, Sintsova A, Buckwalter CM, Leung N, Cochrane A, Li J, Cox AD, Moffat J, and Gray-Owen SD (2015). INNATE IMMUNITY. Cytosolic detection of the bacterial metabolite HBP activates TIFA-dependent innate immunity. Science 348, 1251–1255. [DOI] [PubMed] [Google Scholar]
- Gaudet RG, Guo CX, Molinaro R, Kottwitz H, Rohde JR, Dangeard AS, Arrieumerlou C, Girardin SE, and Gray-Owen SD (2017). Innate recognition of intracellular bacterial growth is driven by the TIFA-dependent cytosolic surveillance pathway. Cell Rep 19, 1418–1430. [DOI] [PubMed] [Google Scholar]
- Germing U, Lauseker M, Hildebrandt B, Symeonidis A, Cermak J, Fenaux P, Kelaidi C, Pfeilstöcker M, Nösslinger T, Sekeres M, et al. (2012). Survival, prognostic factors and rates of leukemic transformation in 381 untreated patients with MDS and del(5q): a multicenter study. Leukemia 26, 1286–1292. [DOI] [PubMed] [Google Scholar]
- Huang CC, Weng JH, Wei TY, Wu PY, Hsu PH, Chen YH, Wang SC, Qin D, Hung CC, Chen ST, et al. (2012). Intermolecular binding between TIFA-FHA and TIFA-pT mediates tumor necrosis factor alpha stimulation and NF-κB activation. Mol. Cell. Biol 32, 2664–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jädersten M, Saft L, Smith A, Kulasekararaj A, Pomplun S, Göhring G, Hedlund A, Hast R, Schlegelberger B, Porwit A, et al. (2011). TP53 mutations in low-risk myelodysplastic syndromes with del(5q) predict disease progression. J. Clin. Oncol 29, 1971–1979. [DOI] [PubMed] [Google Scholar]
- Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, Doerks T, Julien P, Roth A, Simonovic M, et al. (2009). STRING 8–a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res 37, D412–D416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keerthivasan G, Mei Y, Zhao B, Zhang L, Harris CE, Gao J, Basiorka AA, Schipma MJ, McElherne J, Yang J, et al. (2014). Aberrant overexpression of CD14 on granulocytes sensitizes the innate immune response in mDia1 heterozygous del(5q) MDS. Blood 124, 780–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, et al. (2006). Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 442, 818–822. [DOI] [PubMed] [Google Scholar]
- Liu Y, Elf SE, Miyata Y, Sashida G, Liu Y, Huang G, Di Giandomenico S, Lee JM, Deblasio A, Menendez S, et al. (2009). p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell 4, 37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Krysiak K, Shirai CL, Kim S, Shao J, Ndonwi M, and Walter MJ (2017). Knockdown of HSPA9 induces TP53-dependent apoptosis in human hematopoietic progenitor cells. PLoS ONE 12, e0170470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long L, Thelen JP, Furgason M, Haj-Yahya M, Brik A, Cheng D, Peng J, and Yao T (2014). The U4/U6 recycling factor SART3 has histone chaperone activity and associates with USP15 to regulate H2B deubiquitination. J. Biol. Chem 289, 8916–8930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallo M, Cervera J, Schanz J, Such E, García-Manero G, Luño E, Steidl C, Espinet B, Vallesp T, Germing U, et al. (2011). Impact of adjunct cytogenetic abnormalities for prognostic stratification in patients with myelodysplastic syndrome and deletion 5q. Leukemia 25, 110–120. [DOI] [PubMed] [Google Scholar]
- Martin ME, Milne TA, Bloyer S, Galoian K, Shen W, Gibbs D, Brock HW, Slany R, and Hess JL (2003). Dimerization of MLL fusion proteins immortalizes hematopoietic cells. Cancer Cell 4, 197–207. [DOI] [PubMed] [Google Scholar]
- Matsumura T, Semba K, Azuma S, Ikawa S, Gohda J, Akiyama T, and Inoue J (2004). TIFAB inhibits TIFA, TRAF-interacting protein with a forkhead-associated domain. Biochem. Biophys. Res. Commun 317, 230–234. [DOI] [PubMed] [Google Scholar]
- Matsumura T, Kawamura-Tsuzuku J, Yamamoto T, Semba K, and Inoue J (2009). TRAF-interacting protein with a forkhead-associated domain B (TI-FAB) is a negative regulator of the TRAF6-induced cellular functions. J. Biochem 146, 375–381. [DOI] [PubMed] [Google Scholar]
- McGouran JF, Gaertner SR, Altun M, Kramer HB, and Kessler BM (2013). Deubiquitinating enzyme specificity for ubiquitin chain topology profiled by di-ubiquitin activity probes. Chem. Biol 20, 1447–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Men W, Li W, Zhao J, and Li Y (2018). TIFA promotes cell survival and migration in lung adenocarcinoma. Cell. Physiol. Biochem 47, 2097–2108. [DOI] [PubMed] [Google Scholar]
- Padmanabhan A, Candelaria N, Wong KK, Nikolai BC, Lonard DM, O’Malley BW, and Richards JS (2018). USP15-dependent lysosomal pathway controls p53-R175H turnover in ovarian cancer cells. Nat. Commun 9, 1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathert P, Roth M, Neumann T, Muerdter F, Roe JS, Muhar M, Deswal S, Cerny-Reiterer S, Peter B, Jude J, et al. (2015). Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 525, 543–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhyasen GW, Bolanos L, Fang J, Jerez A, Wunderlich M, Rigolino C, Mathews L, Ferrer M, Southall N, Guha R, et al. (2013). Targeting IRAK1 as a therapeutic approach for myelodysplastic syndrome. Cancer Cell 24, 90–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribezzo F, Snoeren IAM, Ziegler S, Stoelben J, Olofsen PA, Henic A, Ferreira MV, Chen S, Stalmann USA, Buesche G, et al. (2019). Rps14, Csnk1a1 and miRNA145/miRNA146a deficiency cooperate in the clinical phenotype and activation of the innate immune system in the 5q-syndrome. Leukemia 33, 1759–1772. [DOI] [PubMed] [Google Scholar]
- Saft L, Karimi M, Ghaderi M, Matolcsy A, Mufti GJ, Kulasekararaj A, Göhring G, Giagounidis A, Selleslag D, Muus P, et al. (2014). p53 protein expression independently predicts outcome in patients with lower-risk myelodysplastic syndromes with del(5q). Haematologica 99, 1041–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallman DA, Wei S, and List A (2014). PP2A: the achilles heal in MDS with 5q deletion. Front. Oncol 4, 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider RK, Schenone M, Ferreira MV, Kramann R, Joyce CE, Hartigan C, Beier F, Brümmendorf TH, Germing U, Platzbecker U, et al. (2016). Rps14 haploinsufficiency causes a block in erythroid differentiation mediated by S100A8 and S100A9. Nat. Med 22, 288–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh TR, Ali AM, Busygina V, Raynard S, Fan Q, Du CH, Andreassen PR, Sung P, and Meetei AR (2008). BLAP18/RMI2, a novel OB-fold-containing protein, is an essential component of the Bloom helicase-double Holliday junction dissolvasome. Genes Dev 22, 2856–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starczynowski DT, Kuchenbauer F, Argiropoulos B, Sung S, Morin R, Muranyi A, Hirst M, Hogge D, Marra M, Wells RA, et al. (2010). Identification of miR-145 and miR-146a as mediators of the 5q-syndrome phenotype. Nat. Med 16, 49–58. [DOI] [PubMed] [Google Scholar]
- Starczynowski DT, Morin R, McPherson A, Lam J, Chari R, Wegrzyn J, Kuchenbauer F, Hirst M, Tohyama K, Humphries RK, et al. (2011). Genome-wide identification of human microRNAs located in leukemia-associated genomic alterations. Blood 117, 595–607. [DOI] [PubMed] [Google Scholar]
- Steensma DP (2015). Myelodysplastic syndromes: diagnosis and treatment. Mayo Clin. Proc 90, 969–983. [DOI] [PubMed] [Google Scholar]
- Steensma DP (2018). Myelodysplastic syndromes current treatment algorithm 2018. Blood Cancer J 8, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoddart A, Qian Z, Fernald AA, Bergerson RJ, Wang J, Karrison T, Anastasi J, Bartom ET, Sarver AL, McNerney ME, et al. (2016). Retroviral insertional mutagenesis identifies the del(5q) genes, CXXC5, TIFAB and ETF1, as well as the Wnt pathway, as potential targets in del(5q) myeloid neoplasms. Haematologica 101, e232–e236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taganov KD, Boldin MP, Chang KJ, and Baltimore D (2006). NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 103, 12481–12486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takatsuna H, Kato H, Gohda J, Akiyama T, Moriya A, Okamoto Y, Yamagata Y, Otsuka M, Umezawa K, Semba K, and Inoue J (2003). Identification of TIFA as an adapter protein that links tumor necrosis factor receptor-associated factor 6 (TRAF6) to interleukin-1 (IL-1) receptor-associated kinase-1 (IRAK-1) in IL-1 receptor signaling. J. Biol. Chem 278, 12144–12150. [DOI] [PubMed] [Google Scholar]
- Tefferi A, and Vardiman JW (2009). Myelodysplastic syndromes. N. Engl. J. Med 361, 1872–1885. [DOI] [PubMed] [Google Scholar]
- Varney ME, Niederkorn M, Konno H, Matsumura T, Gohda J, Yoshida N, Akiyama T, Christie S, Fang J, Miller D, et al. (2015). Loss of Tifab, a del(5q) MDS gene, alters hematopoiesis through derepression of Toll-like receptor-TRAF6 signaling. J. Exp. Med 212, 1967–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varney ME, Choi K, Bolanos L, Christie S, Fang J, Grimes HL, Maciejewski JP, Inoue JI, and Starczynowski DT (2017). Epistasis between TI-FAB and miR-146a: neighboring genes in del(5q) myelodysplastic syndrome. Leukemia 31, 491–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeneuve NF, Tian W, Wu T, Sun Z, Lau A, Chapman E, Fang D, and Zhang DD (2013). USP15 negatively regulates Nrf2 through deubiquitination of Keap1. Mol. Cell 51, 68–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward SJ, Gratton HE, Indrayudha P, Michavila C, Mukhopadhyay R, Maurer SK, Caulton SG, Emsley J, and Dreveny I (2018). The structure of the deubiquitinase USP15 reveals a misaligned catalytic triad and an open ubiquitin-binding channel. J. Biol. Chem 293, 17362–17374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weh HJ, Kuse R, Seeger D, Suciu S, and Hossfeld DK (1991). Cytogenetic studies in patients with acute myeloid leukemia following a myelodysplastic syndrome. Leuk. Lymphoma 3, 423–427. [DOI] [PubMed] [Google Scholar]
- Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, Shahab A, Yong HC, Fu Y, Weng Z, et al. (2006). A global map of p53 transcription-factor binding sites in the human genome. Cell 124, 207–219. [DOI] [PubMed] [Google Scholar]
- Wei S, Chen X, McGraw K, Zhang L, Komrokji R, Clark J, Caceres G, Billingsley D, Sokol L, Lancet J, et al. (2013). Lenalidomide promotes p53 degradation by inhibiting MDM2 auto-ubiquitination in myelodysplastic syndrome with chromosome 5q deletion. Oncogene 32, 1110–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg EL, Schauer NJ, Yang J, Lamberto I, Doherty L, Bhatt S, Nonami A, Meng C, Letai A, Wright R, et al. (2017). Inhibition of USP10 induces degradation of oncogenic FLT3. Nat. Chem. Biol 13, 1207–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiu Y, Dong Q, Li Q, Li F, Borcherding N, Zhang W, Boyce B, Xue HH, and Zhao C (2018). Stabilization of NF-κB-inducing kinase suppresses MLL-AF9-induced acute myeloid leukemia. Cell Rep 22, 350–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Menendez S, Schlegelberger B, Bae N, Aplan PD, Göhring G, Deblasio TR, and Nimer SD (2012). Loss of p53 accelerates the complications of myelodysplastic syndrome in a NUP98-HOXD13-driven mouse model. Blood 120, 3089–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Wang D, Zhong H, Luo R, Shang M, Liu D, Chen H, Fang L, and Xiao S (2015). Ubiquitin-specific protease 15 negatively regulates virus-induced type I interferon signaling via catalytically-dependent and -independent mechanisms. Sci. Rep 5, 11220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao N, Stoffel A, Wang PW, Eisenbart JD, Espinosa R 3rd, Larson RA, and Le Beau MM (1997). Molecular delineation of the smallest commonly deleted region of chromosome 5 in malignant myeloid diseases to 1–1.5 Mb and preparation of a PAC-based physical map. Proc. Natl. Acad. Sci. USA 94, 6948–6953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann S, Pfannkuch L, Al-Zeer MA, Bartfeld S, Koch M, Liu J, Rechner C, Soerensen M, Sokolova O, Zamyatina A, et al. (2017). ALPK1- and TIFA-dependent innate immune response triggered by the Helicobacter pylori type IV secretion system. Cell Rep 20, 2384–2395. [DOI] [PubMed] [Google Scholar]
- Zou Q, Jin J, Hu H, Li HS, Romano S, Xiao Y, Nakaya M, Zhou X, Cheng X, Yang P, et al. (2014). USP15 stabilizes MDM2 to mediate cancer-cell survival and inhibit antitumor T cell responses. Nat. Immunol 15, 562–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The gene-set enrichment analyses and RNA-sequencing of Tifab+/+ and Tifab−/− HSPCs analyzed during this study have been deposited in NCBI’s Gene Expression Omnibus and are accessible by the following GEO Series accession numbers: GSE72936 (Varney et al., 2015) and GSE87453 (Varney et al., 2017). Mass-spectrometry data for TIFAB-interacting proteins is available in Table S1.