

Abstract
22q11.2 deletion syndrome (22q11.2DS) is considered one of the most frequently observed chromosomal abnormalities in association with congenital heart disease (CHD), which can also include some combination of other features. Thus, the aim of this work was to verify the profile of dysmorphic features and heart defects found in patients referred to a reference center in Southern Brazil with clinical findings suggestive of 22q11.2DS. In the overall sample group, only patients with dysmorphic facial features (skull, eyes, ear, and nose) associated with CHD (obstructive pulmonary valve ring, truncus arteriosus, and bicuspid aortic valve associated with atrial septal defect and/or right aortic arch) had a 22q11.2 deletion. These findings proved to be reliable clinical criteria for referral to perform fluorescent in situ hybridization investigation for 22q11.2 deletion.
Keywords: heart defects, congenital, 22q11 deletion syndrome, facial dysmorphism
Introduction
22q11.2 deletion syndrome (22q11.2DS or DiGeorge syndrome) (OMIM 188400) is one of the most common disorders caused by a copy number variant (CNV) and nonhomologous meiotic recombination errors. 22q11.2DS is characterized by a microdeletion in region 11.2 of the long arm of chromosome 22, with approximately 90 genes being deleted. 1 2
The prevalence and incidence in live births are still being studied due to the high phenotypic variation, which makes the immediate identification of affected individuals difficult. However, the literature reports incidences between 1 in 2,000 and 7,000 live births. 3 Currently, 22q11.2DS is considered one of the most frequently observed chromosomal abnormalities in association with congenital heart disease (CHD), only after Down syndrome. 4
CHD is a condition characterized by structural and functional abnormalities of the heart and great vessels, which originate during cardiac embryogenesis. 5 It has an incidence of 19 to 75 cases per thousand live births. CHD has a complex and multifactorial etiology, with approximately 80% of CHDs being a combination of genetic and environmental factors. 6 Aneuploidies and microdeletions of chromosomal regions are associated with the development of CHD, 7 but how this chromosomal imbalance alters cardiogenesis is unclear and probably much more complex than a gene dosage effect. The majority of CHD occurs as isolated malformations while 25 to 30% are associated with extracardiac abnormalities and are often found in association with known genetic syndromes. 8
Cardiac malformation caused by genetic or chromosomal abnormalities, such as Down and velocardiofacial syndrome, is found approximately 20% of cases”. 9 In the general population, 22q11.2 deletion is one of the most common detectable causes of several clinical conditions. A large number of individuals with CHD have 22q11.2 deletion, such as individuals with interruption of the aortic arch type B (52%), truncus arteriosus (34%), tetralogy of Fallot (ToF) (16%), and ventricular septal defects (5–10%). 10
G-band karyotype is considered the gold standard for chromosomal diseases analysis such as aneuploidy, trisomy, or monosomies, including large structural rearrangements (> 5–10 Mb). However, visualization of small submicroscopic alterations, such as microdeletions, is unable to be performed by this technique. 11 Fluorescent in situ hybridization (FISH) was the first methodology applied in cytogenetic microdeletion research, 12 and is currently one of the main techniques for clinical diagnosis as well as for microdeletion and microduplication analysis. 13 Through hybridization probing using doubly labeled fluorescent probes, the identification of aneuploidies and chromosomal rearrangements can be obtained in 48 hours. 14
Although genetic diseases are individually rare, their frequency as a group is an important field in medical care. Early diagnosis is extremely important and directly impacts on infant morbidity and mortality when associated with congenital malformations, especially in the neonatal period. Identification of chromosomal abnormalities is important in prenatal follow-up and in early diagnosis, contributing to family planning through adequate genetic counseling.
In Brazil, the unified health system (UHS) offers every Brazilian citizen full, universal, and free access to health services. Considered one of the largest and best public health systems in the world, the UHS benefits approximately 180 million Brazilians and provides medical assistance from simple outpatient procedures to highly complex care. However, molecular genetic tests are not routinely provided by this system, neglecting many individuals who have some genetic disease. Thus, research works, such as this one, are essential to help in the diagnosis of patients who need molecular investigation.
Despite the phenotypic variability of 22q11.2DS, CHD has become the main finding for clinical suspicion along with the presence or absence of facial and vertebral dysmorphic findings. 15 16 17 18 Thus, the aim of this work was to verify the profile of dysmorphic features and heart defects found in patients referred to a reference center in Southern Brazil with clinical findings suggestive of 22q11.2DS.
Materials and Methods
This was a prospective study with convenience sampling developed at the cytogenetics laboratory/Universidade Federal de Ciências da Saúde de Porto Alegre (UFCSPA) from January 2017 to December 2019. The sample consisted of patients cared for by the UHS presenting clinical findings suggestive of 22q11.2DS. The patients were followed up at the Hospital da Criança Santo Antônio, Hospital Escola of Universidadede Pelotas, Hospital Universitário de Santa Maria and Instituto de Cardiologia do Rio Grande do Sul. After genetic evaluation, a blood sample was collected in a heparin tube and then sent to the cytogenetics laboratory/UFCSPA for cell culture and analysis by FISH.
Genetic evaluation consisted of a review of clinical data and medical history such as echocardiography, followed by a dysmorphological physical examination. Then, a peripheral blood sample was collected from all participants in a heparin tube for cell culture, which was based on Moorhead et al 19 protocol. Molecular cytogenetic testing by FISH was performed using the commercial Vysis LSI DiGeorge/TUPLE1 (Abbott Molecular) region dual-color probe, which identifies deletions of band 22q11.2.This probe mixture contains the OrangeSpectrum TUPLE 1 (HIRA) probe (30 regions without TUPLE 1, D22S553, D22S609, and D22S942 coding) and the GreenSpectrum LSI arylsulfatase A (ARSA) control probe that maps the telomeric end of 22q (22q13.3). The protocol suggested by the manufacturer was used to perform this methodology on fixed cell suspension samples. The analysis was performed under an Axio Imager Z2 Zeiss fluorescence microscope (Göttingen, Germany). In each case, 10 metaphase and 200 complete interphase nuclei were analyzed for respective hybridization signals of the probes. The pictures were captured using a high-resolution digital camera coupled to the fluorescence microscope and processed using Isis Fluorescence Imaging System software. The described analysis, with the count of fluorescent signals per metaphase and per interphase nucleus, were performed in the laboratory standard form.
Research ethics committee of all participant hospitals and UFCSPA approved the present study. Only patients whose parents consented to participate were included in the study.
Results
Blood samples from 47 individuals aged 3 days to 14 years with clinical findings suggestive of 22q11.2DS were referred to the cytogenetics laboratory. The final sample consisted of 46 individuals (23 females and 23 males). One patient was excluded due the absence of cellular growth.
In total, three group patients with profiles suspicious for 22q11.2DS were referred for analysis: individuals with only dysmorphic features ( n = 4), individuals with isolated heart disease ( n = 7), and individuals with both dysmorphia and CHD ( n = 35). Of all individuals analyzed, 56.52% (26/46) had karyotype results: 88.46% (23/26) with normal chromosomal constitution (46.XX/46.XY) and 11.53% (3/26) with altered chromosomal constitution. In addition, 46.47% (20/46) had no karyotype result. Alterations identified in the three altered karyotypes were 46,XX,del(16) (q12.1q22); 47,XX, + mar and 46,XY,t(6;8) (q23.3;q22.1). FISH was performed in 46 patients with clinical findings of 22q11DS, including those with altered karyotype results. In the overall sample, 15.21% (7/46) showed a 22q11.2 deletion, consistent with the clinical diagnosis of 22q11.2DS. Among the three suspect groups' profiles, only patients with dysmorphic features associated with CHD had the deletion. ( Table 1 ).
Table 1. Results of cytogenetic analysis according to the profiles of suspected individuals.
| Groups | Normal karyotype ( n = 23) | FISH results | Abnormal karyotype ( n = 3) | FISH results | Without karyotype ( n = 20) | FISH results |
|---|---|---|---|---|---|---|
| Only dysmorphisms ( n = 4) |
3 | del22 (0) | 0 | del22 (0) | 1 | del22 (0) |
| Isolated heart disease ( n = 7) | 1 | del22 (0) | 0 | del22 (0) | 6 | del22 (0) |
| CHD + dysmorphisms ( n = 35) | 19 | del22 (4) | 3 | del22 (0) | 13 | del22 (3) |
Abbreviations: CHD, congenital heart disease; FISH, fluorescent in situ hybridization.
Dysmorphia and Cardiac Alterations Analysis of Patients with 22q11.2 Deletion
The main alterations found in patients with 22q11.2 deletion were dysmorphic facial features (100%); skeletal abnormalities (57.14%); breast abnormalities (28.57%); skin abnormalities (14.28%); neurological alterations (14.28%); endocrine alterations (42.85%), and CHD (100%) ( Table 2 ).
Table 2. Clinical findings observed in patients with 22q11 deletion x patients without 22q11 deletion.
| Individuals diagnosed with 22q11.2 deletion | Without 22q11.2 deletion | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| P12 | P13 | P15 | P22 | P23 | P29 | P31 | n | n | |
| Age | 2 mo | 5 d | 3 mo | 16 d | 14 y | 5 d | 6 mo | ||
| Face dysmorphia | |||||||||
| Skull | |||||||||
| Large anterior fontanel | + | + | 2 | 7 | |||||
| Small anterior fontanel | + | 1 | 0 | ||||||
| Nose | |||||||||
| Nasal bridge, wide | + | + | + | 3 | 11 | ||||
| Nasal bridge, depressed | + | 1 | 2 | ||||||
| Tubular nose | + | 1 | 1 | ||||||
| Nares, anteverted | + | 1 | 1 | ||||||
| Eyes | |||||||||
| Palpebral fissure | + | + | 2 | 4 | |||||
| Epicanthal fold | + | 1 | 2 | ||||||
| Ears | |||||||||
| Low ear implantation | + | 1 | 8 | ||||||
| Microtia | + | 1 | 1 | ||||||
| Overfolded superior helix | + | + | + | + | 4 | 9 | |||
| Skeletal abnormalities | |||||||||
| Ectopic calcification in soft tissue | + | 1 | 0 | ||||||
| Sacral anomalies | + | 1 | 12 | ||||||
| Short neck | + | + | + | 3 | 6 | ||||
| Clinodactyly of the fifth fingers | + | 1 | 10 | ||||||
| Hypoplastic phalanges | + | 1 | 1 | ||||||
| Hypoplastic nails | + | 1 | 2 | ||||||
| Breast abnormalities | |||||||||
| Teletelia | + | + | 2 | 3 | |||||
| Inverted nipple | + | + | 2 | 1 | |||||
| Skin abnormalities | |||||||||
| Café au lait spots | + | 1 | 0 | ||||||
| Neurological alterations | |||||||||
| Delayed psychomotor development | + | 1 | 3 | ||||||
| Endocrine alterations | |||||||||
| Hypocalcemia | + | + | + | 3 | 1 | ||||
| Pseudohypoparathyroidism | + | 1 | 0 | ||||||
| CHD | + | + | + | + | + | + | + | 7 | 35 |
Abbreviations: CHD, congenital heart disease; d, day; mo, month; n, number of patients; P, patient; y, year.
Considering the types of alterations, some dysmorphisms and cardiac defects were identified exclusively in patients with 22q11 deletion, such as small anterior fontanel (1/7), ectopic calcifications in soft tissue (1/7), café au lait spots(1/7), pseudohypoparahyroidism (PHP) (1/7), obstructive pulmonary valve ring (1/7), truncus arteriosus (1/7), and bicuspid aortic valve (2/7) ( Tables 2 and 3 ).
Table 3. Heart defects observed in patients with 22q11 deletion x patients without 22q11 deletion.
| Individuals diagnosed with DS22q11 | Without DS22q11 diagnosis | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| CHD | P12 | P13 | P15 | P22 | P23 | P29 | P31 | n | n |
| Pulmonary valve agenesis | + | 1 | 1 | ||||||
| Right aortic arch | + | + | + | + | 4 | 5 | |||
| Pulmonary atresia | + | + | 2 | 3 | |||||
| Atrial septal defects | + | + | + | + | + | 5 | 27 | ||
| Interventricular communication | + | + | + | 3 | 19 | ||||
| Double outlet right ventricle | + | 1 | 5 | ||||||
| Interruption of the aortic arch type B | + | 1 | 1 | ||||||
| Obstructive pulmonary valve ring | + | 1 | 0 | ||||||
| Patent ductus arteriosus | + | 1 | 14 | ||||||
| Tetra logia of Fallot | + | + | 1 | 12 | |||||
| Truncus arteriosus | + | 1 | 0 | ||||||
| Bicuspid aortic valve | + | + | 2 | 0 | |||||
Abbreviations: CHD, congenital heart disease; n, number of patients; P, patient.
Unexpected Results
The three altered karyotypes identified by FISH did not show a 22q11 deletion. However, idic (22q11.2) chromosome was identified in the individual with karyotype 47,XX, + mar. In addition, one of the patients without a karyotype result showed a 22q13 (LSI ARSA × 1) deletion by FISH. The FISH analyses of both patients are presented in Fig. 1 .
Fig. 1.

FISH analysis with/without contrast: red signal, probe TUPLE1/HIRA specific to the 22q11.2 region and green signal, control probe arylsulfatase A specific to the 22q13 region. The arrows indicates the signs of hybridization. ( A ) FISH analysis in a patient with del 22q13. The normal chromosome 22 shows both red and green (marker) hybridization signal, while the deleted one shows only the green signal (marker). ( B ) FISH analysis in a patient with 47,XX, +mar. The normal chromosome 22 shows both red and green (marker) hybridization signal. Here, we can identify that an extra signal from the red probe TUPLE1/HIRA hybridized in the marker identified in the karyotype (+mar), concluding that this marker is idic (22)(q11.2). FISH, fluorescent in situ hybridization.
Discussion
22q11.2DS is known for the heterogeneity of clinical findings and the difficulty of establishing a reliable analysis based only on phenotype. In Brazil, the diagnosis is still a clinical one due to the difficulty of performing genetic tests, especially in UHS, which if performed could help in better follow-up and treatment of these individuals. The main medical centers, when less experienced, are not exempt from underdiagnosing patients with 22q11.2DS. 20 The overall prevalence of clinical alterations detected may differ according to the age of the patient and the specialist's experience. Currently, 180 clinical features and symptoms, such as physical and behavioral abnormalities, have already been described. However, typical findings are not found in all cases, 21 corroborating the clinical findings of this study.
In our study, we classified the patients with clinical suspicion of 22q11.2DS referred for molecular analysis into three clinical profiles. Of these, only patients with dysmorphic features associated with CHD had a 22q11.2 deletion (7/35). This result is compatible with literature. 22q11.2 deletion is more frequent in patients with CHD associated with other clinical findings (80–90%) than in patients with isolated conotruncal heart disease (29%). 22 The interindividual variability of the 22q11.2 deletion phenotype is characteristic, once individuals with significant clinical expression of the syndrome can be found, as well as in moderately affected individuals. The precise phenotypic evaluation of patients with deletion demonstrates that facial abnormalities—severely or slightly expressed—are detectable in all cases, 23 which corroborates with our findings. All individuals with 22q11.2 deletion besides heart defects showed facial dysmorphic features that ranged from skull, nose, eyes, and ears alterations ( Fig. 2 ). Among the facial abnormalities found, the small anterior fontanel was exclusively seen in deleted patients ( n = 1, Table 2 ).
Fig. 2.
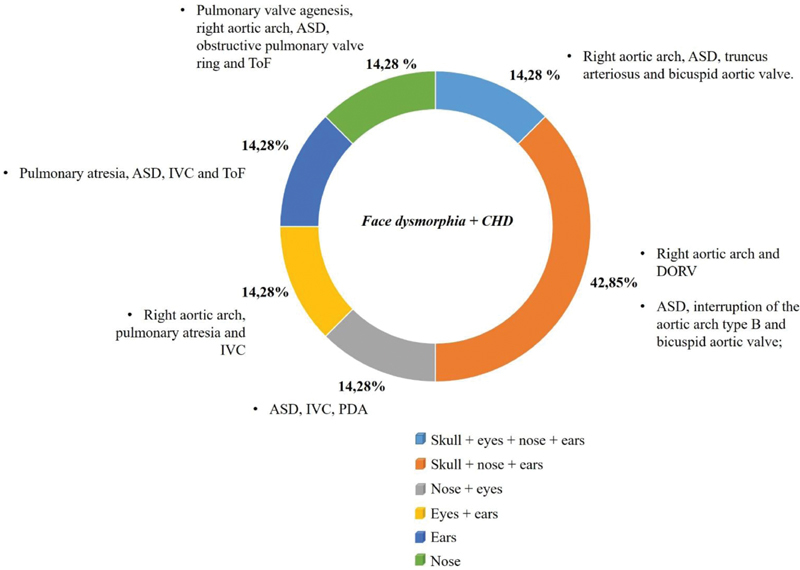
Percentage of individuals with 22q11 deletion presenting face dysmorphia associated with congenital heart disease.
In our sample, 91.3% of the patients had CHD and in 16.7%, the malformation was isolated. All individuals with 22q11.2 deletion showed extracardiac alterations and CHD (15.21%). Other studies found similar results. Fokstuen et al 15 investigated the incidence of 22q11.2 deletion in a series of patients with extracardiac alterations with CHD and isolated heart defects. All patients with the deletion presented with major or minor additional extracardiac anomalies. No deletion was detected in the group of isolated cardiac malformations. Wozniak et al 16 also analyzed children with CHD (conotruncal and nonconotruncal) who presented with at least one additional characteristic of 22q11.2DS. The deletion was detected in 14.94% of the patients examined and after CHD, an abnormal facial finding was the most evidenced clinical feature in the study (84.51%). Halder et al 17 also performed a study to determine the prevalence and capacity of clinical criteria to predict 22q11 deletion. In total, only 6.16% of individuals had the deletion and none of the cases with isolated heart defects was positive for 22q11.2DS. The most frequent alterations found in these individuals were also dysmorphic facial features. The differences in prevalence of patients with the deletion who have CHD and facial dysmorphic features can be explained by the sample size.
When comparing our results with other studies, it can be seen that screening for 22q11.2 microdeletion in cases of cardiac malformations associated with extracardiac manifestations, in the form of facial dysmorphism, should be considered. It is also known that it is difficult to carry out an efficient dysmorphological assessment in newborns or in infants in the intensive care unit (ICU). These characteristics are often subtle or less evident, making CHD one of the main clinical suspicions for 22q11DS in these individuals. In these situations, it is important to observe if the patient does not have any other alterations compatible with the syndrome since isolated heart disease has not been demonstrated to be a good parameter for molecular analysis referral.
Conotruncal heart defects are often found in these individuals. ToF is the most described alteration, 24 but interruption of the aortic arch type B, truncus arteriosus, and ventricular septal defects are also frequently observed. 10 We analyzed the types of heart defects present in patients with 22q11.2 deletion and verified if there were any exclusive alteration in these individuals. In total, 12 heart defects were observed ( Table 3 ). Obstructive pulmonary valve ring, truncus arteriosus, and bicuspid aortic valve were present only in patients with the deletion.
Study of a specific “cardiac phenotype” in patients with 22q11.2 deletion shows that a particular cardiac anatomy can be identified. 18 In fact, patients with conotruncal heart defect often have additional heart defects that may lead to the suspicion of the presence of the 22q11 deletion. 23 In our sample, the most frequent alterations in this individuals were atrial septal defect (ASD) (5/7), followed by right aortic arch (4/7). However, these findings were also observed in the nondeleted individuals associated with other heart defects. Coincidentally, the three exclusive alterations in the deleted individuals were associated with an ASD and right aortic arch. Aortic arch alterations are observed in 85% of 22q11.2DS cases, and when there is an association with other heart defects, it is one of the main reasons to be referred for a 22q11DS investigation. 7 Association between interruption of the aortic arch type B, persistent truncus arteriosus, ToF, and ventricular septal defect with ASD have also been described in patients with 22q11.2DS. 25
Other clinical features were also seen exclusively in the deleted patients, such as ectopic calcification in soft tissue ( n = 1), café au lait spots ( n = 1), and PHP ( n = 1). Ectopic calcification was a consequence of PHP in only one patient of this study, a rare case of association of PHP with 22q11.2 deletion. 26
FISH is the most widely used genetic diagnostic procedure for 22q11.2 deletion analysis. 27 28 29 However, commercially available FISH probes cannot detect atypical deletions. 30 In total, seven deleted individuals (15.21%) were confirmed, but it is important to take into account that although the rest of the individuals have clinical signs of the syndrome and have no detectable deletion, other causes may be responsible for the phenotype. Mutations in genes associated with heart disease, such as TBX1 (located in the deleted region of chromosome 22), 31 GATA4 (8p23.1), 32 and DGS2 (10p13–14), 33 are other possible causes.
Others diagnostic methods can also be used to identify 22q11.2 deletion and determine microdeletion size and chromosomal breakpoints, such as high-resolution comparative genomic hybridization, multiplex ligation dependent probe amplification (MLPA), short tandem repeats, as well as quantitative polymerase chain reaction (qPCR). 34 Kuo et al 35 described a case of a fetus with CHD diagnosed by ultrasonography (singleton fetus with heart defects including overriding aorta, small pulmonary artery, and ventricular septal defect) associated with extracardiac defects, and referred for molecular analysis for 22q11.2 deletion. MLPA, a-CGH, and FISH techniques confirmed the deletion. Conventional cytogenetic analysis revealed a normal male karyotype. Prenatal identification of CHD is beginning to play an important role, since early diagnosis is crucial for the treatment and follow-up of these individuals, improving the life quality and reducing premature deaths. 36
In our study, 22q11.2 deletion was not detected by karyotype analysis in any case, reinforcing the idea that this technique is ineffective for micro deletion research. However, karyotype examination is useful to detect other chromosomal alterations that may or may not be related to clinical findings that resemble 22q11.2DS. The three individuals with altered karyotype were referred for 22q11.2 deletion molecular cytogenetic analysis, as they had clinical findings compatible with 22q11.2DS. However, no deletion was detected by FISH.
Patients with altered karyotypes (46,XX,del(16) (q12.1q22), 46,XY,t(6;8) (q23.3;q22.1), 47,XX, + mar) had heart defects and dysmorphic features that were also observed in our deleted patients (interventricular communication, ASD, patent ductus arteriosus, right aortic arch, double outlet right ventricle, sacral anomalies, large anterior fontanel, wide nose, and low ear implantation). We have not found any description in the literature of 16q12.1q22 deletion cases, as well as t(6;8) (q23.3;q22.1). Interstitial deletions near the 16q region (16q12.2q21 37 and 16q12.2-q13 38 ) have already been reported in cases of skeletal abnormalities and cardiac malformations, respectively.
The 47,XX, + mar patient's karyotype revealed the presence of a nondefined chromosome origin isodicentric marker. Through FISH, it was observed that TUPLE1/HIRA probe hybridized on the marker chromosome allowing identification of the idic (22q11.2). This alteration may be associated with cat eye syndrome (OMIM 115470), a rare malformation syndrome whose diagnosis is based on the presence of an extra marker chromosome derived from chromosome 22. 39
FISH also detected a 22q13 deletion in a patient with clinical characteristics similar to 22q11.2DS. This region has been associated with Phelan-McDermid syndrome (OMIM 606232) 40 and metachromatic leukodystrophy (OMIM 250100), 41 but our patient did not have clinical characteristics compatible with these two disorders.
Conclusion
More assertive clinical criteria for molecular testing are crucial for optimizing spending on testing, better diagnostics, treatment, and follow-up of patients. Furthermore, appropriate genetic counseling can be offered to families to aid them with risk of recurrence as well as prenatal care in future pregnancies.
Facial dysmorphic features (skull, eyes, ear, and nose) associated with CHD proved to be a reliable clinical criterion for referral to undergo molecular analysis. Among heart defects, we suggest that individuals with obstructive pulmonary valve ring, truncus arteriosus, and bicuspid aortic valve associated with ASD and/or right aortic arch should undergo FISH investigation for 22q11.2 deletion. Particularly in newborns, the detection of these cardiac defects is crucial for the early diagnosis of this syndrome, as dysmorphisms associated with 22q11.2DS are often more difficult to evaluate.
Acknowledgments
We thank the patients and their families for their participation in this study. We are also thankful to the partner institutions that forwarded the samples for molecular analysis. We would also like to acknowledge Desirée Deconte for her contribution to the language editing.
Funding Statement
Funding This study was supported by the Programa de Extensão Universitária do Ministério da Educação e Cultura (PROEXT), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, 001), and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, 302931/2019-8).
Conflict of Interest None declared.
Authors' Contributions
B.L.D, A.S.S, A.B.G., and P.R.G.Z performed genetic testing and wrote the manuscript. B.L.D., and P.R.G.Z supervised genetic testing. B.L.D, B.B.G., and R.F.M.R reviewed clinical data and edited the manuscript. A.S.S, A.B.G., B.B.G, C.F.L, and J.A.J reviewed the medical records. B.L.D, P.R.G.Z., and R.F.M.R designed the study, supervised genetic tests, wrote, and edited the manuscript.
References
- 1.McDonald-McGinn D M, Kirschner R, Goldmuntz E et al. The Philadelphia story: the 22q11.2 deletion: report on 250 patients. Genet Couns. 1999;10(01):11–24. [PubMed] [Google Scholar]
- 2.Shprintzen R J. Velo-cardio-facial syndrome: 30 years of study. Dev Disabil Res Rev. 2008;14(01):3–10. doi: 10.1002/ddrr.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mastroiacovo P, Rossi P, Cancrini Cet al. 2005. Available at:http://www.aieop.org/stdoc/prot/rec_del22_en_06.pdf. Accessed May 2005
- 4.Botto L D, May K, Fernhoff P Met al. A population-based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population Pediatrics 2003112(1 Pt 1):101–107. [DOI] [PubMed] [Google Scholar]
- 5.Fahed A C, Gelb B D, Seidman J G, Seidman C E. Genetics of congenital heart disease: the glass half empty. Circ Res. 2013;112(04):707–720. doi: 10.1161/CIRCRESAHA.112.300853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bruneau B G.The developmental genetics of congenital heart disease Nature 2008451(7181):943–948. [DOI] [PubMed] [Google Scholar]
- 7.Brennan P, Young I D. Congenital heart malformations: aetiology and associations. Semin Neonatol. 2001;6(01):17–25. doi: 10.1053/siny.2000.0032. [DOI] [PubMed] [Google Scholar]
- 8.Blue G M, Kirk E P, Sholler G F, Harvey R P, Winlaw D S. Congenital heart disease: current knowledge about causes and inheritance. Med J Aust. 2012;197(03):155–159. doi: 10.5694/mja12.10811. [DOI] [PubMed] [Google Scholar]
- 9.Sun R, Liu M, Lu L, Zheng Y, Zhang P. Congenital heart disease: causes, diagnosis, symptoms, and treatments. Cell Biochem Biophys. 2015;72(03):857–860. doi: 10.1007/s12013-015-0551-6. [DOI] [PubMed] [Google Scholar]
- 10.Peyvandi S, Lupo P J, Garbarini J et al. 22q11.2 deletions in patients with conotruncal defects: data from 1,610 consecutive cases. Pediatr Cardiol. 2013;34(07):1687–1694. doi: 10.1007/s00246-013-0694-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bui T H. Prenatal cytogenetic diagnosis: gone FISHing, BAC soon! Ultrasound Obstet Gynecol. 2007;30(03):247–251. doi: 10.1002/uog.5142. [DOI] [PubMed] [Google Scholar]
- 12.Gall J G, Pardue M L. Formation and detection of RNA-DNA hybrid molecules in cytological preparations. Proc Natl Acad Sci U S A. 1969;63(02):378–383. doi: 10.1073/pnas.63.2.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.El-Ella S S, El Gendy F, Tawfik M A et al. Chromosome 22 microdeletion in children with syndromic congenital heart disease by fluorescent in situ hybridization (FISH) Egypt J Med Hum Genet. 2012;13(03):313–322. [Google Scholar]
- 14.Shaffer L G, Bejjani B A, Torchia B, Kirkpatrick S, Coppinger J, Ballif B C. The identification of microdeletion syndromes and other chromosome abnormalities: cytogenetic methods of the past, new technologies for the future. Am J Med Genet C Semin Med Genet. 2007;145C(04):335–345. doi: 10.1002/ajmg.c.30152. [DOI] [PubMed] [Google Scholar]
- 15.Fokstuen S, Arbenz U, Artan S et al. 22q11.2 deletions in a series of patients with non-selective congenital heart defects: incidence, type of defects and parental origin. Clin Genet. 1998;53(01):63–69. doi: 10.1034/j.1399-0004.1998.531530113.x. [DOI] [PubMed] [Google Scholar]
- 16.Wozniak A, Wolnik-Brzozowska D, Wisniewska M et al. Frequency of 22q11.2 microdeletion in children with congenital heart defects in western poland. BMC Pediatr. 2010;10(01):88. doi: 10.1186/1471-2431-10-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halder A, Jain M, Chaudhary I, Kabra M. Prevalence of 22q11.2 microdeletion in 146 patients with cardiac malformation in a referral hospital of North India. BMC Med Genet. 2010;11(01):101. doi: 10.1186/1471-2350-11-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marino B, Digilio M C, Toscano A et al. Anatomic patterns of conotruncal defects associated with deletion 22q11. Genet Med. 2001;3(01):45–48. doi: 10.1097/00125817-200101000-00010. [DOI] [PubMed] [Google Scholar]
- 19.Moorhead P S, Nowell P C, Mellman W J, Battips D M, Hungerford D A. Chromosome preparations of leukocytes cultured from human peripheral blood. Exp Cell Res. 1960;20(03):613–616. doi: 10.1016/0014-4827(60)90138-5. [DOI] [PubMed] [Google Scholar]
- 20.Bassett A S, McDonald-McGinn D M, Devriendt K et al. Practical guidelines for managing patients with 22q11.2 deletion syndrome. J Pediatr. 2011;159(02):332–90. doi: 10.1016/j.jpeds.2011.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simmons M A, Brueckner M. The genetics of congenital heart disease… understanding and improving long-term outcomes in congenital heart disease: a review for the general cardiologist and primary care physician. Curr Opin Pediatr. 2017;29(05):520–528. doi: 10.1097/MOP.0000000000000538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Belangero S I, Bellucco F T, Kulikowski L D, Christofolini D M, Cernach M C, Melaragno M I. 22q11.2 deletion in patients with conotruncal heart defect and del22q syndrome phenotype. Arq Bras Cardiol. 2009;92(04):307–311. doi: 10.1590/s0066-782x2009000400010. [DOI] [PubMed] [Google Scholar]
- 23.Digilio M, Marino B, Capolino R, Dallapiccola B. Clinical manifestations of deletion 22q11.2 syndrome (DiGeorge/Velo-Cardio-Facial syndrome) Images Paediatr Cardiol. 2005;7(02):23–34. [PMC free article] [PubMed] [Google Scholar]
- 24.Grassi M S, Jacob C M, Kulikowski L D et al. Congenital heart disease as a warning sign for the diagnosis of the 22q11.2 deletion. Arq Bras Cardiol. 2014;103(05):382–390. doi: 10.5935/abc.20140145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huber J, Peres V C, de Castro A L et al. Molecular screening for 22Q11.2 deletion syndrome in patients with congenital heart disease. Pediatr Cardiol. 2014;35(08):1356–1362. doi: 10.1007/s00246-014-0936-0. [DOI] [PubMed] [Google Scholar]
- 26.Diniz B L, Glaeser A B, Deconte Det al. Pseudohypoparathyroidism with Ectopic calcification and 22q11 deletion syndrome: a rare caseJ Pediatr Genet2020. Doi: 10.1055/s-0040-1701640 [DOI] [PMC free article] [PubMed]
- 27.Rosa R F, Pilla C B, Pereira V L et al. 22q11.2 deletion syndrome in patients admitted to a cardiac pediatric intensive care unit in Brazil. Am J Med Genet A. 2008;146A(13):1655–1661. doi: 10.1002/ajmg.a.32378. [DOI] [PubMed] [Google Scholar]
- 28.Ramírez-Velazco A, Rivera H, Vásquez-Velázquez A I, Aguayo-Orozco T A, Delgadillo-Pérez S, Domínguez M G. 22q11.2 deletion detected by in situ hybridization in Mexican patients with velocardiofacial syndrome-like features . Colomb Med (Cali) 2018;49(03):219–222. doi: 10.25100/cm.v49i2.3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McDonald-McGinn D M, Tonnesen M K, Laufer-Cahana A et al. Phenotype of the 22q11.2 deletion in individuals identified through an affected relative: cast a wide FISHing net! Genet Med. 2001;3(01):23–29. doi: 10.1097/00125817-200101000-00006. [DOI] [PubMed] [Google Scholar]
- 30.Mademont-Soler I, Morales C, Soler A et al. Prenatal diagnosis of chromosomal abnormalities in fetuses with abnormal cardiac ultrasound findings: evaluation of chromosomal microarray-based analysis. Ultrasound Obstet Gynecol. 2013;41(04):375–382. doi: 10.1002/uog.12372. [DOI] [PubMed] [Google Scholar]
- 31.Jerome L A, Papaioannou V E. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet. 2001;27(03):286–291. doi: 10.1038/85845. [DOI] [PubMed] [Google Scholar]
- 32.Zhou L, Liu J, Xiang M et al. Gata4 potentiates second heart field proliferation and Hedgehog signaling for cardiac septation. Proc Natl Acad Sci U S A. 2017;114(08):E1422–E1431. doi: 10.1073/pnas.1605137114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lichtner P, König R, Hasegawa T, Van Esch H, Meitinger T, Schuffenhauer S. An HDR (hypoparathyroidism, deafness, renal dysplasia) syndrome locus maps distal to the DiGeorge syndrome region on 10p13/14. J Med Genet. 2000;37(01):33–37. doi: 10.1136/jmg.37.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fernández L, Lapunzina P, Arjona D et al. Comparative study of three diagnostic approaches (FISH, STRs and MLPA) in 30 patients with 22q11.2 deletion syndrome. Clin Genet. 2005;68(04):373–378. doi: 10.1111/j.1399-0004.2005.00493.x. [DOI] [PubMed] [Google Scholar]
- 35.Kuo Y L, Chen C P, Wang L K et al. Prenatal diagnosis and molecular cytogenetic characterization of chromosome 22q11.2 deletion syndrome associated with congenital heart defects. Taiwan J Obstet Gynecol. 2014;53(02):248–251. doi: 10.1016/j.tjog.2014.04.021. [DOI] [PubMed] [Google Scholar]
- 36.Moore J W, Binder G A, Berry R. Prenatal diagnosis of aneuploidy and deletion 22q11.2 in fetuses with ultrasound detection of cardiac defects. Am J Obstet Gynecol. 2004;191(06):2068–2073. doi: 10.1016/j.ajog.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 37.Yamamoto T, Shimojima K, Yamazaki S, Ikeno K, Tohyama J. A 16q12.2q21 deletion identified in a patient with developmental delay, epilepsy, short stature, and distinctive features. Congenit Anom (Kyoto) 2016;56(06):253–255. doi: 10.1111/cga.12172. [DOI] [PubMed] [Google Scholar]
- 38.Elder F FB, Ferguson J W, Lockhart L H. Identical twins with deletion 16q syndrome: evidence that 16q12.2-q13 is the critical band region. Hum Genet. 1984;67(02):233–236. doi: 10.1007/BF00273010. [DOI] [PubMed] [Google Scholar]
- 39.Denavit T M, Malan V, Grillon C et al. A new case of a severe clinical phenotype of the cat-eye syndrome. Genet Couns. 2004;15(04):443–448. [PubMed] [Google Scholar]
- 40.Delahaye A, Toutain A, Aboura A et al. Chromosome 22q13.3 deletion syndrome with a de novo interstitial 22q13.3 cryptic deletion disrupting SHANK3. Eur J Med Genet. 2009;52(05):328–332. doi: 10.1016/j.ejmg.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 41.Chen L, Yan H, Cao B et al. Identification of novel ARSA mutations in Chinese patients with metachromatic leukodystrophy. Int J Genomics. 2018;2018:2.361068E6. doi: 10.1155/2018/2361068. [DOI] [PMC free article] [PubMed] [Google Scholar]