

Abstract
Endotoxemia induced by lipopolysaccharide (LPS) is an extremely severe syndrome identified by global activation of inflammatory responses. Neutrophil extracellular traps (NETs) play an important role in the development of endotoxemia. Histone hypercitrullination catalyzed by peptidylarginine deiminases (PADs) is a key step of NET formation. We have previously demonstrated that simultaneous inhibition of PAD2 and PAD4 with pan PAD inhibitors can decrease NETosis and improve survival in a mouse model of LPS-induced endotoxic shock. However, the effects of PAD2 specific inhibition during NETosis and endotoxic shock are poorly understood. Therefore, in the present study, we aimed to investigate effect of the specific PAD2 or PAD4 inhibitor on LPS-induced endotoxic shock in mice. We found that PAD2 inhibition but not PAD4 inhibition improves survival. Also, the levels of proinflammatory cytokines and NETosis were significantly reduced by PAD2 inhibitor. To our knowledge this study demonstrates for the first time that PAD2 inhibition can reduce NETosis, decrease inflammatory cytokine production, and protect against endotoxin-induced lethality. Our findings provided a novel therapeutic strategy for the treatment of endotoxic shock.
Keywords: LPS-induced endotoxic shock, PAD2, Selective PAD2 inhibitor, NETosis, Acute lung injury
Introduction
Lipopolysaccharide (LPS), also known as endotoxin, is a part of the exterior cell wall of gram-negative bacteria [1]. LPS can induce excessive activation of inflammatory responses and cause endotoxemia or endotoxic shock marked by fever, dyspnea, hypotension, multiple organ dysfunction, and disseminated intravascular coagulation [2]. Endotoxemia is a life-threatening condition where the mortality rate can be as high as 30% [3]. Although the pathophysiological mechanisms underlying LPS-induced endotoxemia remain elusive, dysregulated neutrophil activation is considered to be an essential step [4].
Neutrophils are the most abundant immune cells in humans and exert indispensable anti-microbial functions [5]. After stimulation with LPS, neutrophils can extrude DNA, myeloperoxidase (MPO), neutrophil elastase (NE) and histones to form web-like structures called neutrophil extracellular traps (NETs); this process is termed NETosis [6]. NETosis is an inflammatory form of neutrophil death, which is an important anti-microbial strategy at the early stages of infections. However, excessive NETosis can lead to severe inflammatory responses and tissue damage [7]. As such, dysregulated NETosis has been implicated in the development of endotoxemia and endotoxemia-related organ dysfunction [8].
Studies have shown that histone citrullination is a vital process during NETosis [9, 10]. The citrullination of histones is catalyzed by a group of enzymes called peptidylarginine deiminases (PADs). PADs are calcium-dependent enzymes that citrullinate proteins via converting arginine residues into citrullines [11]. The PAD family consists of 5 members: PAD1, PAD2, PAD3, PAD4 and PAD6. However, only PAD2 and PAD4 have been found to be expressed in immune cells and enter the nucleus to citrullinate histones in adults [12, 13]. These findings indicate that PAD2 and PAD4 probably serve as inflammatory mediators and play important roles in the development of sepsis. In fact, our recent studies demonstrated that simultaneous inhibition of PAD2 and PAD4 with YW3–56, a pan-PAD inhibitor, rescued mice from LPS-induced lethal endotoxemia [14]. However, a study on PAD4 by Martinod et al. revealed that PAD4 deficiency does not protect mice from either CLP-induced sepsis or LPS-induced endotoxic shock [15]. The remained question is whether inhibition of PAD2 could improve animal survival in endotoxic shock.
In the present study, we first investigated whether inhibition of PAD2 with selective PAD2 inhibitor AFM32a [16], instead of PAD4 inhibitor (GSK484), could improve survival. We then determined whether AFM32a could decrease pro-inflammatory cytokine production, ameliorate acute lung injury in a mouse model of endotoxic shock, and reduce NETosis in vivo and in vitro.
Materials and Methods
Animals
Male C57BL/6J mice (8–10 weeks) were purchased form the Jackson Laboratory (Bar Harbor, ME, USA) and housed for 3 days before being used. All animal experiments were conducted in compliance with the animal protocol approved by the University of Michigan Institutional Animal Care and Use Committee.
Reagents
Lipopolysaccharide (LPS), dimethyl sulfoxide (DMSO), 4% paraformaldehyde, and 4’, 6-diamidino-2–2phenylindole (DAPI) were purchased from Sigma Aldrich (St. Louis, MO); Rabbit anti-mouse myeloperoxidase (MPO) polyclonal antibody, HRP-conjugated goat anti-rabbit IgG antibody, mouse Interleukin-1 beta (IL-1β) enzyme-linked immunosorbent assay (ELISA) kit, and mouse Tumor Necrosis Factor-alpha (TNFα) ELISA kit were purchased from Abcam (Cambridge, MA); Alexa Fluor 488-conjugated goat anti-rabbit IgG antibody, RPMI 1640 medium, fetal bovine serum (FBS) were purchased from Thermo Fisher Scientific (Waltham, MA); Neutrophil isolation kit for mouse was purchased from Miltenyi Biotech (Bergisch Gladbach, Germany); Quant-iT™ PicoGreen™ dsDNA Assay Kit was purchased from Invitrogen (San Diego, CA); Anti-citrullinated histone H3 (CitH3) rabbit polyclonal antibody (Ab5103, Abcam, Cambridge, MA); GSK484 was purchased from Cayman Chemical (Ann Arbor, MI); AFM32a was synthesized according to previously established methods [16].
LPS-induced Endotoxic Model
Mice were randomly divided into four groups:
Control: this group only received intraperitoneal (i.p.) administration of normal saline (NS) followed by DMSO (1 h later) injection i.p. as normal controls;
LPS + DMSO: mice were injected i.p. LPS (35 mg/kg, dissolved in NS), and DMSO was administered 1 h later;
LPS + AFM32a: mice were administered (i.p.) with AFM32a (20 mg/kg) dissolved in DMSO (1 μL per kg mouse body weight) 1 h after LPS insult;
LPS + GSK484: Mice were subjected to GSK484 (40 mg/kg in DMSO) treatment 1 h after LPS administration. In non-survival studies, animals were euthanized by CO2 24 h after LPS administration, and then serum and organs were harvested and stored at −80 °C. In survival observational studies, mice were monitored for 10 consecutive days and then euthanized with CO2 at the endpoint of observation or whenever they were found moribund.
Mouse Neutrophil Isolation
The neutrophils were isolated from mouse bone marrow cells (tibiae and femora) by negative magnetic cell sorting using the Neutrophil Isolation Kit (Miltenyi Biotec) according to the manufacturer’s instructions. The purity of isolated neutrophils was determined by flow cytometry (CD45+/Ly6G+ expression profile), and consistently > 95% was considered no batch to batch variation.
NETosis Induction in vitro
Isolated mouse neutrophils were induced to undergo NETosis as follows. In brief, mouse neutrophils were re-suspended in RPMI 1640 medium with 5% FBS and then seeded in 4-well chamber slides or 24-well plates at concentration of 106/mL. After incubation for 30 min at 37 °C, the cells were challenged by LPS (10 μg/ml) with or without 2 μM AFM32a followed by another 1 h or 3 h incubation.
Immunofluorescent (IF) Staining of NETs in vitro
IF staining of NETs was performed as previously described [17]. In short, after a 3 h incubation in 4-well chamber slides, medium containing LPS and AFM32a was added. The remaining cells were then washed with PBS and fixed with 4% paraformaldehyde for 20 min, followed by 1 h reaction at room temperature with rabbit anti-mouse MPO polyclonal antibody at 5 μg/mL. Thereafter, unbound antibody was removed, and Alexa Fluor 488-conjugated goat anti-rabbit IgG antibody was used as secondary antibody (1 :2000) for 1 h at room temperature. For staining of neutrophil DNA, the slides were mounted with solution of DAPI after being washed with PBS. NET formation was observed under an Olympus DP70 microscope with Olympus BH2-RFL-T3 burner (Olympus, Tokyo, Japan) at the magnification of 400x. DAPI filter set and FITC filter set were applied to visualize DNA and MPO, respectively.
Quantification of NETs
NETs in supernatant of LPS-treated mouse neutrophils or blood were quantified using a Quant-iT™ PicoGreen™ dsDNA Assay Kit following manufacturer’s instructions. Briefly, the supernatant of neutrophil cell culture or mouse sera were prepared and mixed with Tris-EDTATE buffer at the ratio of 1:3. The mixtures were then added into different cuvettes (1.0 mL per cuvette). Working solution of the PicoGreen reagent were then added into the cuvettes (1.0 mL each), followed by 5-minute incubation at room temperature. Fluorescence were then read at 490 nm and interpolated using standard curve.
Determination of IL-1β and TNF-α in vivo
To determine the levels of IL-1β and TNF-α in serum and lung, a mouse IL-1β enzyme-linked immunosorbent assay (ELISA) kit and a mouse TNF- α ELISA kit were used following the manufacturer’s instructions.
Acute Lung Injury Assessment
Mouse lung sections were stained with hematoxylin and eosin (H&E) to observe the histological alterations. Acute lung injury (ALI) scores [18] were graded by a licensed pathologist who was blinded to the experiments.
Western Blot
Serum proteins were loaded and separated by SDS-PAGE. The samples were then transferred to nitrocellulose membranes. After blocking at room temperature for 20 minutes, the membranes were incubated with anti-CitH3 rabbit polyclonal antibody overnight at 4 °C. Then the membranes were washed and incubated with HRP conjugated goat anti-rabbit IgG antibody for 1 h at room temperature. The blots were imaged using chemiluminescent substrate.
Sample Size Calculation and Statistical Analysis
Power analysis is used to determine the minimum numbers of mice to be used. To achieve two-sided α=0.05, a power of 0.8, and expected difference between experimental and control groups of 0.50, a minimum of 4 mice/group was required. Analysis was performed using GraphPad Prism 7 (GraphPad Software Inc., La Jolla, CA, USA). Kaplan-Meier curves and log-rank test was performed to analyze the survival curve. To make comparisons between three or more groups, one-way analysis of variance (ANOVA) with Bonferroni’s multiple comparison test was performed. All data are presented as Mean ± SEM in figures. All the experiments were conducted three independent times with 4 replicates each time. P value less than 0.05 was considered statistically significant.
Results
PAD2 inhibition improved survival in lethal endotoxic shock
Mice were treated with DMSO or AFM32a 1 h after LPS insult. Mice in the sham group received DMSO as a vehicle control. Survival was monitored for 240 hours. No mortality was observed in sham group. Six out of eight animals survived in the LPS + AFM32a group, while LPS + DMSO mice and LPS + GSK484 mice had 100% mortality rate (Figure 1). The results indicate that a PAD2-selective inhibitor improved survival. Since treatment with specific PAD4 inhibitor GSK484 had no survival benefits, we did not study it furthermore.
Figure 1. PAD2 inhibition improved survival in lethal endotoxic shock.
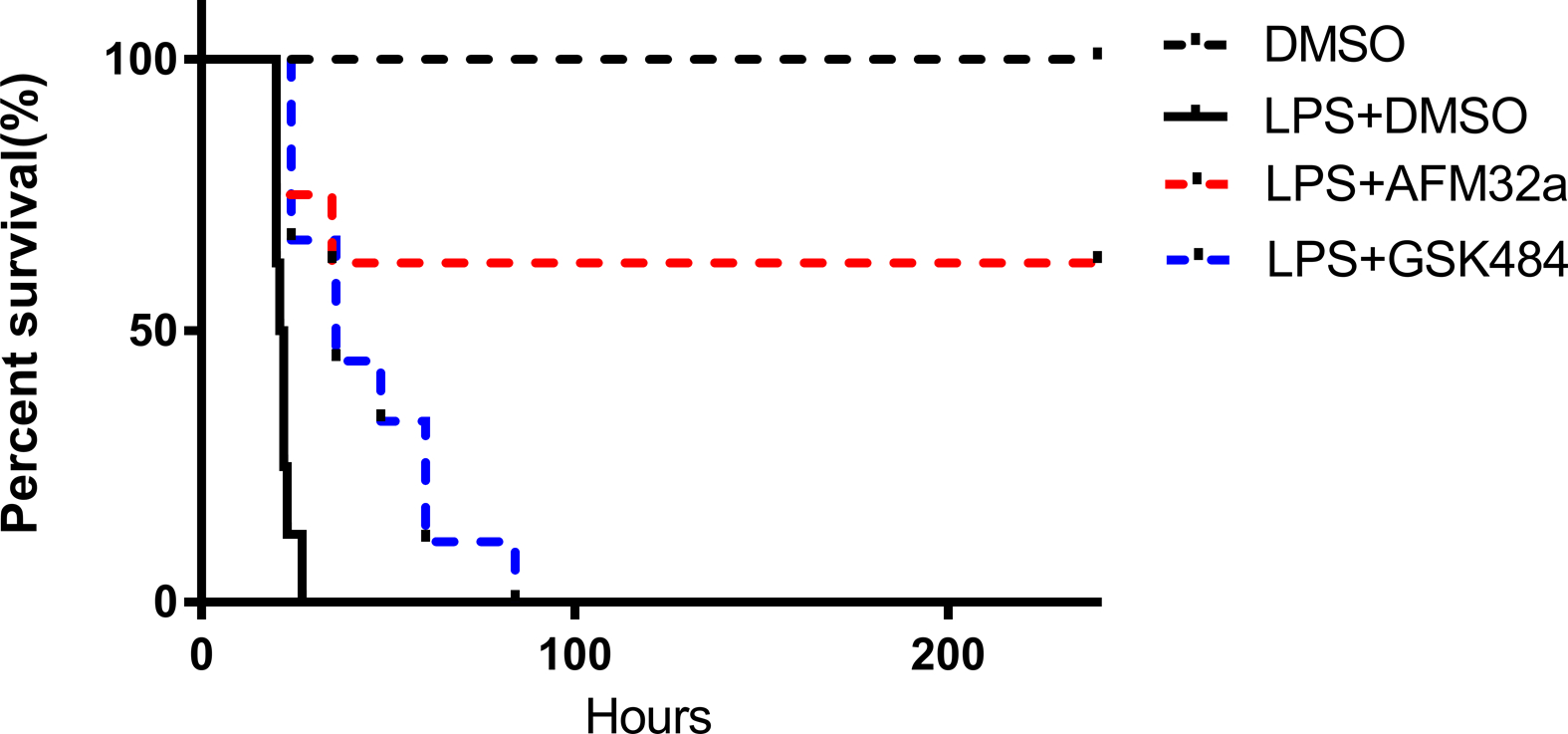
C57BL/6J mice were randomly divided into 4 groups: (1) DMSO (control, n = 8); (2) LPS (35 mg/kg) + DMSO (n = 8); (3) LPS (35 mg/kg) + AFM32a (20 mg/kg) (n= 8), and (4) LPS (35 mg/kg) + GSK484 (40 mg/kg). Survival was monitored for 10 consecutive days. AFM32a remarkably improved survival compared to LPS + DMSO (P = 0.0052) and LPS + GSK484 (P =0.0466). DMSO: dimethyl sulfoxide; LPS: lipopolysaccharide.
PAD2 inhibition reduced systemic proinflammatory cytokines
To determine the effect of PAD2 inhibition on systemic inflammatory responses, the levels of IL-1β and TNF-α in serum were measured after treatment. The concentration of serum IL-1β in the LPS + DMSO group (2715 ± 521.9 pg/mL) was approximately 5 times higher than that in LPS + AFM32a group (487.5 ± 210.5 pg/mL, P < 0.0001) (Figure 2A). AFM32a also decreased serum levels of TNF-α dramatically compared to LPS + DMSO group (173.5 ± 4.7 vs 353 ± 18.7 pg/mL, P < 0.001, respectively) (Figure 2B). These findings indicate that selective inhibition of PAD2 suppresses systemic inflammatory responses.
Figure 2. PAD2 inhibition decreased proinflammatory cytokines in serum.
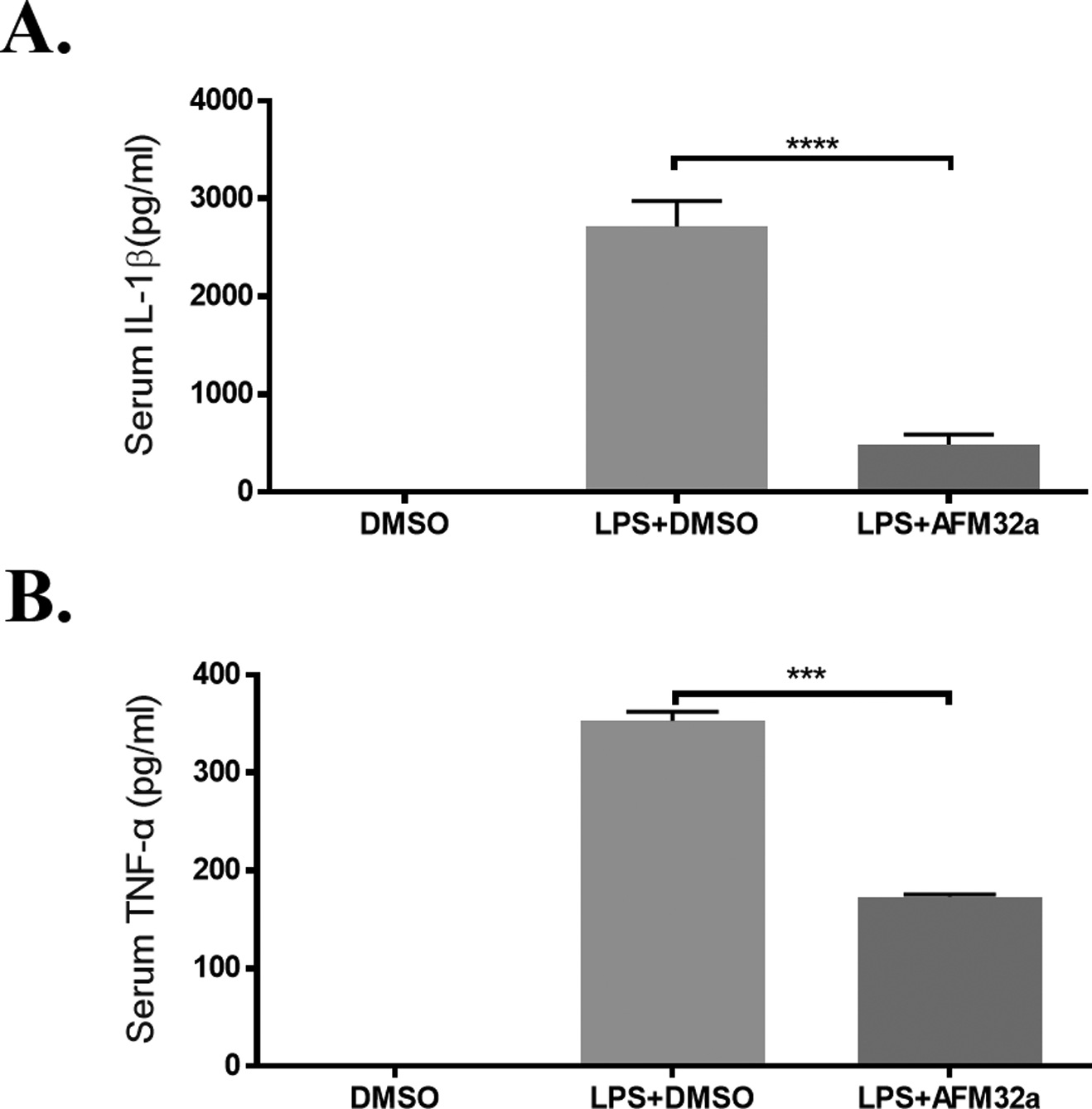
Mouse were randomly divided into 3 groups (n =3): Group DMSO, Group LPS + DMSO, and Group LPS + AFM32a. (A) ELISA results show AFM32a greatly reduced IL-1β in serum 24 h after LPS insult. (B) ELISA results show TNF-α in serum 24 h after LPS insult were reduced by AFM32a. Data were shown as a representative of three independent experiments. All data in figures were presented as mean ± SEM. IL: interleukin; TNF: tumor necrosis factor; DMSO: dimethyl sulfoxide; LPS: lipopolysaccharide; ns: no significance; ** P <0.01; *** P <0.001; **** P <0.0001.
PAD2 inhibition alleviates endotoxic shock-induced acute lung injury
Due to the unique blood supply, the lung is one of the most vulnerable organs during endotoxemia [19]. Therefore, we further investigated therapeutic effects of AFM32a on LPS-induced pulmonary injury. The levels of IL-1β and TNF-α in lung tissues were determined. Similar to the results in serum, AFM32a significantly reduced IL-1β (333 ± 125.1 vs 559.6 ± 32.2 pg/mg, P = 0.0104, respectively) and TNF-α (25.1± 1.2 vs 40.9 ± 6.6 pg/mg, P = 0.0020, respectively) compared to LPS + DMSO group (Figure 3A & 3B). Additionally, AFM32a notably alleviated acute lung injury according to morphological analyses. In the LPS + DMSO group, increased inflammatory cell infiltration, thickening of alveolar wall, and erythrocyte leakage were observed which was significantly reversed by AFM32a treatment (Figure 3C). These findings indicate that PAD2 inhibition alleviates endotoxic shock-induced lung inflammation and restores pulmonary function.
Figure 3. PAD2 inhibition alleviated endotoxic shock-induced Lung Inflammation.
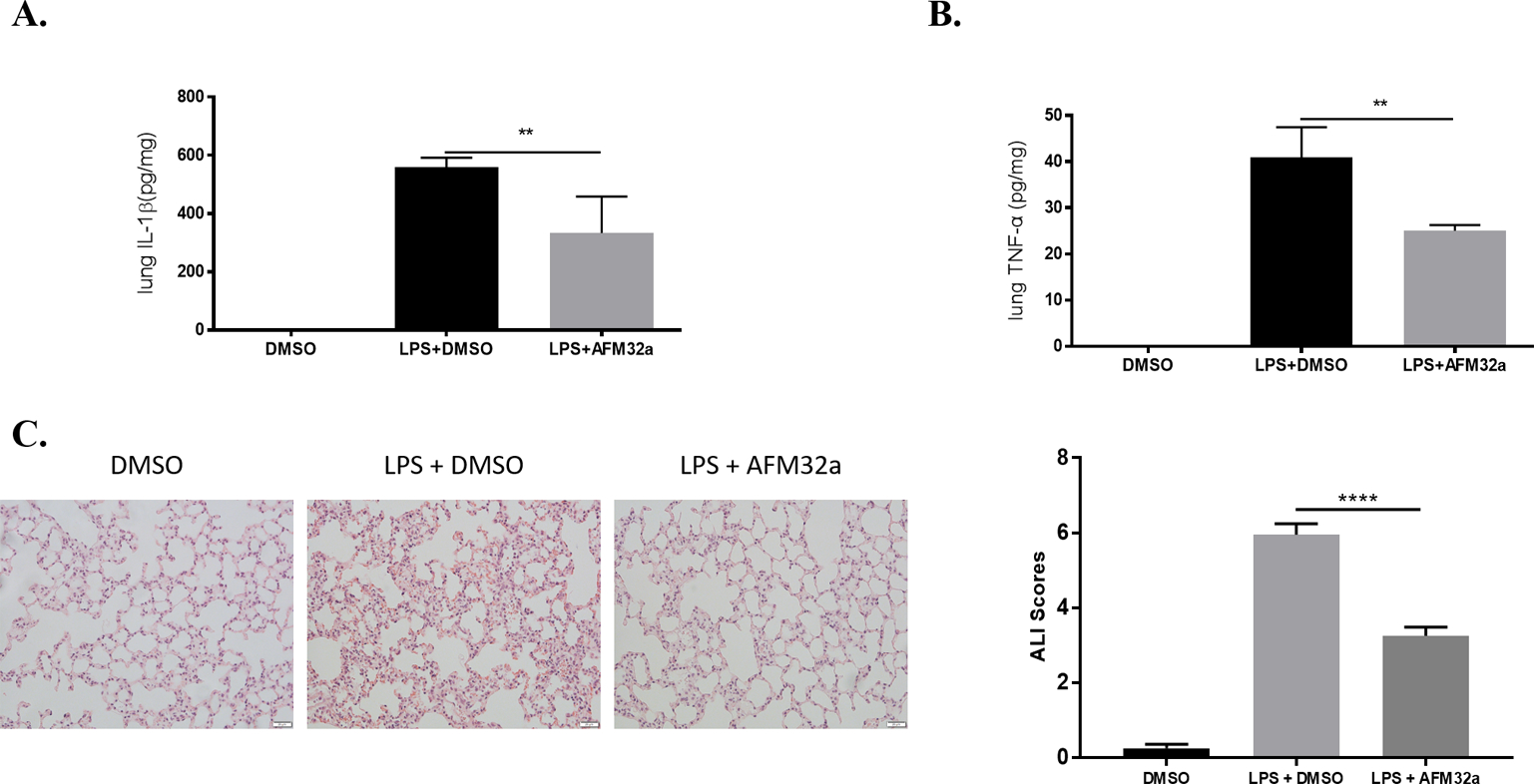
Lung tissues were harvested 24 h after LPS insult. (A & B) ELISA results showed that proinflammatory cytokines (IL-1β and TNF-α) in lung 24 h after LPS insult were significantly reduced by AFM32a. All data in figures were presented as mean ± SEM. (C) H & E staining of lung sections and ALI scores showed that AFM32a alleviated lung injury caused by LPS-induced endotoxic shock. Data were shown as a representative of three independent experiments. Scale bar: 20 μm. DMSO: dimethyl sulfoxide; LPS: lipopolysaccharide; ALI: acute lung injury; IL: interleukin; TNF: tumor necrosis factor. ns: no significance; * P <0.05; ** P <0.01; *** P <0.001; **** P <0.0001.
PAD2 inhibition decreases NET formation in vitro
Next, we investigated the effect of PAD2 inhibition on NET formation, which may mechanistically explain how PAD2 inhibition suppresses inflammation. Neutrophils isolated from mice were challenged with LPS (100 ng/mL), and then treated with AFM32a or DMSO (2 μM). Supernatants were collected to measure extracellular DNA levels, an indicator of NETosis [20]. We found that LPS stimulation for 3 h significantly increases NETosis compared to the sham group [(4695 ± 1979) vs (2081 ± 132.1) arbitrary units (au), P =0.0007, respectively] and that AFM32a treatment leads to a remarkable decrease in extracellular DNA levels compared to the LPS + DMSO group [(2831 ± 275.5) vs (4695 ± 1979) au, P =0.0115, respectively] (Figure 4A). We also observed increased extracellular DNA in the control group. This increase could be attributed to the neutrophils undergoing other forms of death such as apoptosis and necrosis. To directly demonstrate that NETosis was decreased by PAD2 inhibition, we co-localized DNA and MPO (a component of NETs) to visualize NETs using immunofluorescence. As shown in Figure 4B, LPS-induced NET formation was obviously diminished by AFM32a treatment, whereas there was limited NETosis in sham group. These results suggest that PAD2 inhibition prevents LPS-stimulated neutrophils from releasing NETs.
Figure 4. PAD2 inhibition decreased NET formation in vitro.
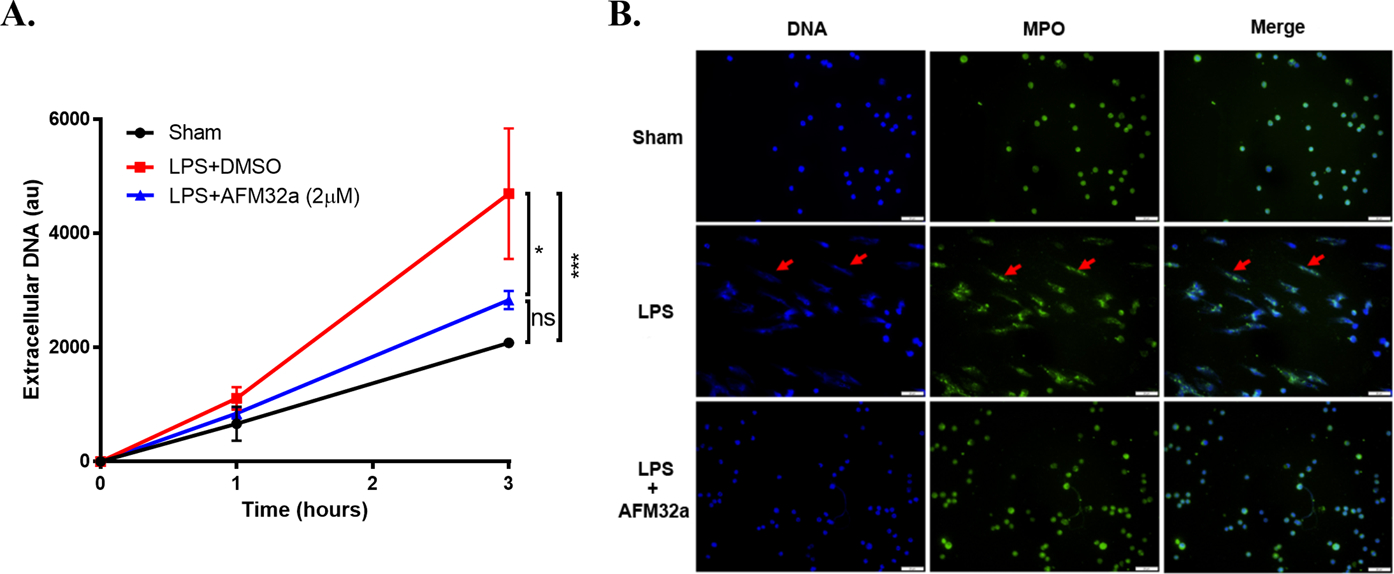
(A). The graph showed the levels of extracellular DNA in supernatant of cultured mouse neutrophils 1 h and 3 h after LPS treatment (100 ng/ml) with or without AFM32a (2 μM). Extracellular DNA were significantly reduced by AFM32a compared to Group LPS + DMSO. (B). Represent results of immunofluorescence staining showed that AFM32a significantly prevented NET formation. DNA was stained with DAPI (blue), MPO were stained with rabbit anti-mouse MPO antibody and Alexa Fluor 488-conjugated goat anti-rabbit IgG antibody (Green). NETs were recognized as scattered DNA co-localized with MPO (red arrows). Scale bar: 50 μm. DMSO: dimethyl sulfoxide; LPS: lipopolysaccharide; MPO: myeloperoxidase; au: arbitrary units; ns: no significance; * P <0.05; *** P <0.001.
PAD2 Inhibition Decreases NET Formation in vivo
Next, we evaluated the inhibitory effect of AFM32a on NETosis in vivo. Circulating double strand DNA (dsDNA) is a widely used marker of NETosis [21]. The results show that serum levels of circulating dsDNA in LPS + DMSO group are notably increased (6.483 ± 0.522 μg/mL) compared to DMSO group (0.589 ± 0.131 μg/mL) (P <0.0001). A modest but significant decrease in circulating dsDNA was observed in the LPS + AFM32a group (4.817 ± 0.679 μg/mL), relative to the LPS + DMSO group (P <0.05) (Figure 5A). We further detected the levels of serum CitH3, a marker of NETosis in vivo. Consistent with the levels of dsDNA, LPS induced CitH3 production were significantly reduced by AFM32a (Figure 5B). These results suggest that PAD2 inhibition also reduces NET formation in vivo.
Figure 5. PAD2 inhibition decreased serum levels of NET and CitH3 in vivo.
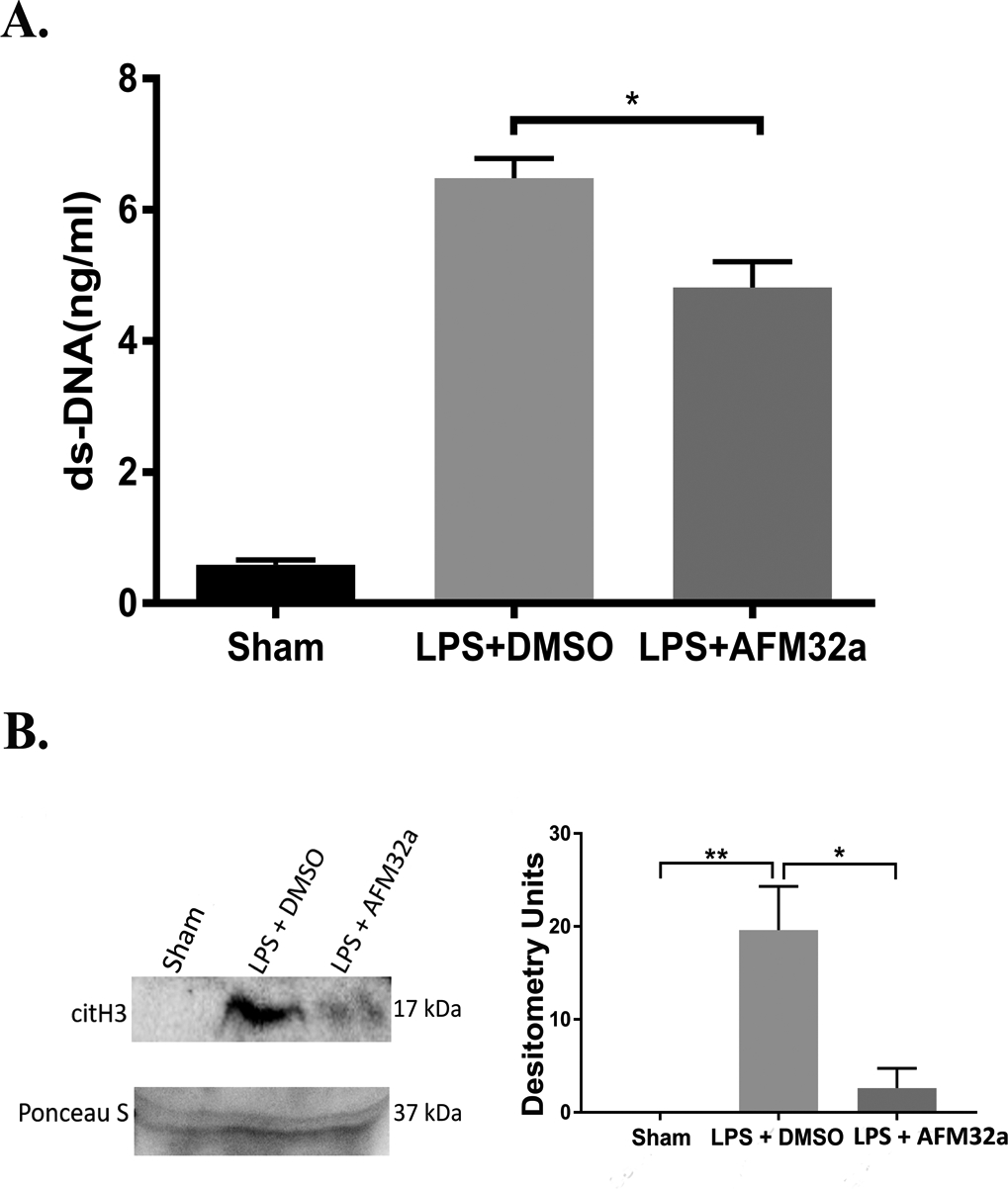
Treatment with AFM32a significantly decreased serum levels of NETs (A) and CitH3 (B) 24 h post LPS insult. DMSO: dimethyl sulfoxide; LPS: lipopolysaccharide; ds-DNA: double strand DNA. * P <0.05; ** P <0.01.
Discussion
Host responses to LPS-induced endotoxic shock are marked by systemic inflammation [22]. These responses are predominantly mediated by the activation of neutrophils. Neutrophils represent the foremost line of the innate immune system and dysregulated neutrophil activation plays an important role in the development of endotoxic shock [23, 24]. During endotoxic shock, neutrophils can over-produce proinflammatory cytokines and cast excessive NETs, which subsequently cause multiple organ dysfunction and death [25, 26]. In the present study, we sought to investigate the effects of selective PAD2 inhibition in a mouse model of LPS-induced endotoxic shock. Consistent with previous findings, LPS-induced endotoxic shock notably increased the level of circulating NETs, proinflammatory cytokines, and resulted in severe acute lung injury and high mortality [15, 27]. Notably, selective PAD2 inhibition instead of PAD4 inhibition improved survival. Moreover, we showed that AFM32a also alleviates systemic inflammation and acute lung injury, and decreases NET levels in vivo and in vitro. These data imply that PAD2 plays a key role in the onset of endotoxic shock and secondary acute lung injury.
NETs are regarded as ‘a double-edged sword’ [6, 28]. On the one hand, NETs entrap pathogens (e.g., bacteria and fungi) to immobilize and eliminate them, which serves as a vital mechanism of pathogen clearance [7]. On the other hand, DNA and citrullinated histones in NETs are implicated in causing damage to the host [20, 29–31]. Dysregulated NETosis was observed during endotoxemia, which is responsible for the progression of systemic inflammation to organ injury [32, 33]. As such, previous studies have found that decreasing the NET levels can improve survival and attenuate organ injury [30, 34, 35]. NET formation is generally associated with chromatin decondensation and histone hypercitrullination, which is catalyzed by PADs [12, 36]. Within recent years, it has been displayed that both PAD2 and PAD4 can translocate into nucleus and citrullinate histones [12, 13]. The effect of PAD4 inhibition on NETosis has been studied frequently [37–39], whereas the effect of PAD2 inhibition on NETosis has barely been explored. Here, we used AFM32a, a PAD2-selective inhibitor developed by the Thompson lab [16], and showed that this compound reduces NET formation in LPS-treated neutrophils. Previous studies showed that PAD4 plays an important role in NET formation and the knockout of pad4 gene can largely reduce the production of NETs [38, 39]. However, pad4-deficiency cannot improve survival during endotoxic shock in the absence of NETs [15], which is consistent with our findings. Besides, notable levels of extracellular DNA and histones were still detected in LPS-insulted pad4−/− animals, which were likely from necrotic cells [15]. In our study, we observed a significant decrease of circulating dsDNA after PAD2 inhibition in mice with endotoxic shock, suggesting that PAD2 inhibition not only suppresses NETosis but also is likely to enhances the clearance of dsDNA in circulation, which is possibly why PAD2 inhibition has a survival benefit.
PAD2 is the most widely expressed member among the PAD family and can citrullinate hundreds of proteins beyond histones [40, 41]. These citrullinated proteins often serve as inflammatory mediators that trigger inflammatory responses in vivo. PAD2 hyper-activation can result in dysregulation of immune responses in the host and is associated with multiple diseases including rheumatoid arthritis (RA), systemic lupus erythematosus (SLE) and even cancer [42–44]. Endotoxic shock is also caused by dysregulated inflammatory responses to LPS. Thus, inhibition of PAD2 can possibly decrease the citrullination of certain proteins and result in attenuated systemic inflammation [45]. This is another possible reason why inhibition of PAD2 can decrease levels of IL-1β and TNF-α in serum of AFM32a treated endotoxic mice than those non-treated endotoxic shock mice.
IL-1β and TNF-α, as pro-inflammatory cytokines, can mediate the innate immune response and cause systemic inflammation that contribute to the pathogenesis of many diseases [46] as well as pulmonary damage (e.g., ALI). In this study, we demonstrated that PAD2 inhibition significantly reduced the levels of IL-1β and TNF-α in lung and blood, which may explain why AFM32a can attenuate LPS-induced ALI.
In conclusion, our results show that selective PAD2 inhibition with AFM32a can improve survival and alleviate lung injury during LPS-induced endotoxic shock via attenuating systemic inflammation and decreasing levels of circulating NETs. Our findings provide the evidence that inhibition of PAD2 could be a novel therapeutic strategy for treatment of endotoxic shock.
Funding
This work was funded by grants from Kickstart N022142 to Dr. Yongqing Li, UMHS-PUHSC Joint Institute U050150 and the National Institutes of Health Grant 5 R01 GM084127 to Dr. Hasan B. Alam and by National Institutes of Health Grant R35 GM118112 to Paul R. Thompson.
Footnotes
Ethics Statement
The protocol for the animal experiments was approved by the University of Michigan Institutional Animal Care and Use Committee (PRO00008861). All experiments complied with animal welfare and research regulations.
References
- 1.Kulp A, and Kuehn MJ. 2010. Biological functions and biogenesis of secreted bacterial outer membrane vesicles. Annu Rev Microbiol 64:163–184. doi: 10.1146/annurev.micro.091208.073413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Opal SM 2010. Endotoxins and other sepsis triggers. Contrib Nephrol 167:14–24. doi: 10.1159/000315915. [DOI] [PubMed] [Google Scholar]
- 3.Ramnath RD, Ng SW, Guglielmotti A, and Bhatia M. 2008. Role of MCP-1 in endotoxemia and sepsis. Int Immunopharmacol 8 (6):810–818. doi: 10.1016/j.intimp.2008.01.033. [DOI] [PubMed] [Google Scholar]
- 4.Grigoleit JS, Oberbeck JR, Lichte P, Kobbe P, Wolf OT, Montag T, del Rey A, Gizewski ER, Engler H, and Schedlowski M. 2010. Lipopolysaccharide-induced experimental immune activation does not impair memory functions in humans. Neurobiol Learn Mem 94 (4):561–567. doi: 10.1016/j.nlm.2010.09.011. [DOI] [PubMed] [Google Scholar]
- 5.Hirz T, and Dumontet C. 2016. Neutrophil Isolation and Analysis to Determine their Role in Lymphoma Cell Sensitivity to Therapeutic Agents. J Vis Exp (109):e53846. doi: 10.3791/53846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yipp BG, and Kubes P. 2013. NETosis: how vital is it? Blood 122 (16):2784–2794. doi: 10.1182/blood-2013-04-457671. [DOI] [PubMed] [Google Scholar]
- 7.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, and Zychlinsky A. 2004. Neutrophil extracellular traps kill bacteria. Science 303 (5663):1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 8.Camicia G, Pozner R, and de Larranaga G. 2014. Neutrophil extracellular traps in sepsis. Shock 42 (4):286–294. doi: 10.1097/shk.0000000000000221. [DOI] [PubMed] [Google Scholar]
- 9.Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, Kahn CR, and Wagner DD. 2015. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med 21 (7):815–819. doi: 10.1038/nm.3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li RHL, Ng G, and Tablin F. 2017. Lipopolysaccharide-induced neutrophil extracellular trap formation in canine neutrophils is dependent on histone H3 citrullination by peptidylarginine deiminase. Vet Immunol Immunopathol 193–194:29–37. doi: 10.1016/j.vetimm.2017.10.002. [DOI] [PubMed] [Google Scholar]
- 11.Wang S, and Wang Y. 2013. Peptidylarginine deiminases in citrullination, gene regulation, health and pathogenesis. Biochim Biophys Acta 1829 (10):1126–1135. doi: 10.1016/j.bbagrm.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, Hayama R et al. 2009. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol 184 (2):205–213. doi: 10.1083/jcb.200806072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang X, Bolt M, Guertin MJ, Chen W, Zhang S, Cherrington BD, Slade DJ et al. 2012. Peptidylarginine deiminase 2-catalyzed histone H3 arginine 26 citrullination facilitates estrogen receptor alpha target gene activation. Proc Natl Acad Sci U S A 109 (33):13331–13336. doi: 10.1073/pnas.1203280109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liang Y, Pan B, Alam HB, Deng Q, Wang Y, Chen E, Liu B et al. 2018. Inhibition of peptidylarginine deiminase alleviates LPS-induced pulmonary dysfunction and improves survival in a mouse model of lethal endotoxemia. Eur J Pharmacol 833:432–440. doi: 10.1016/j.ejphar.2018.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martinod K, Fuchs TA, Zitomersky NL, Wong SL, Demers M, Gallant M, Wang Y, and Wagner DD. 2015. PAD4-deficiency does not affect bacteremia in polymicrobial sepsis and ameliorates endotoxemic shock. Blood 125 (12):1948–1956. doi: 10.1182/blood-2014-07-587709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muth A, Subramanian V, Beaumont E, Nagar M, Kerry P, McEwan P, Srinath H, Clancy K, Parelkar S, and Thompson PR. 2017. Development of a Selective Inhibitor of Protein Arginine Deiminase 2. J Med Chem 60 (7):3198–3211. doi: 10.1021/acs.jmedchem.7b00274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park SY, Shrestha S, Youn YJ, Kim JK, Kim SY, Kim HJ, Park SH et al. 2017. Autophagy Primes Neutrophils for Neutrophil Extracellular Trap Formation during Sepsis. Am J Respir Crit Care Med 196 (5):577–589. doi: 10.1164/rccm.201603-0596OC. [DOI] [PubMed] [Google Scholar]
- 18.Deng Q, Zhao T, Pan B, Dennahy IS, Duan X, Williams AM, Liu B et al. 2018. Protective Effect of Tubastatin A in CLP-Induced Lethal Sepsis. Inflammation 41 (6):2101–2109. doi: 10.1007/s10753-018-0853-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gotts JE, and Matthay MA. 2016. Sepsis: pathophysiology and clinical management. Bmj 353:i1585. doi: 10.1136/bmj.i1585. [DOI] [PubMed] [Google Scholar]
- 20.Pan B, Alam HB, Chong W, Mobley J, Liu B, Deng Q, Liang Y et al. 2017. CitH3: a reliable blood biomarker for diagnosis and treatment of endotoxic shock. Sci Rep 7 (1):8972. doi: 10.1038/s41598-017-09337-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gloude NJ, Khandelwal P, Luebbering N, Lounder DT, Jodele S, Alder MN, Lane A et al. 2017. Circulating dsDNA, endothelial injury, and complement activation in thrombotic microangiopathy and GVHD. Blood 130 (10):1259–1266. doi: 10.1182/blood-2017-05-782870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van den Boogaard M, Ramakers BP, van Alfen N, van der Werf SP, Fick WF, Hoedemaekers CW, Verbeek MM, Schoonhoven L, van der Hoeven JG, and Pickkers P. 2010. Endotoxemia-induced inflammation and the effect on the human brain. Crit Care 14 (3):R81. doi: 10.1186/cc9001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Okeke EB, Mou Z, Onyilagha N, Jia P, Gounni AS, and Uzonna JE. 2017. Deficiency of Phosphatidylinositol 3-Kinase delta Signaling Leads to Diminished Numbers of Regulatory T Cells and Increased Neutrophil Activity Resulting in Mortality Due to Endotoxic Shock. J Immunol 199 (3):1086–1095. doi: 10.4049/jimmunol.1600954. [DOI] [PubMed] [Google Scholar]
- 24.Stiel L, Meziani F, and Helms J. 2018. Neutrophil Activation During Septic Shock. Shock 49 (4):371–384. doi: 10.1097/SHK.0000000000000980. [DOI] [PubMed] [Google Scholar]
- 25.Delabranche X, Stiel L, Severac F, Galoisy AC, Mauvieux L, Zobairi F, Lavigne T et al. 2017. Evidence of Netosis in Septic Shock-Induced Disseminated Intravascular Coagulation. Shock 47 (3):313–317. doi: 10.1097/SHK.0000000000000719. [DOI] [PubMed] [Google Scholar]
- 26.Czaikoski PG, Mota JM, Nascimento DC, Sonego F, Castanheira FV, Melo PH, Scortegagna GT et al. 2016. Neutrophil Extracellular Traps Induce Organ Damage during Experimental and Clinical Sepsis. PLoS One 11 (2):e0148142. doi: 10.1371/journal.pone.0148142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pieterse E, Rother N, Yanginlar C, Hilbrands LB, and van der Vlag J. 2016. Neutrophils Discriminate between Lipopolysaccharides of Different Bacterial Sources and Selectively Release Neutrophil Extracellular Traps. Front Immunol 7:484. doi: 10.3389/fimmu.2016.00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gupta S, and Kaplan MJ. 2016. The role of neutrophils and NETosis in autoimmune and renal diseases. Nat Rev Nephrol 12 (7):402–413. doi: 10.1038/nrneph.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Porto BN, and Stein RT. 2016. Neutrophil Extracellular Traps in Pulmonary Diseases: Too Much of a Good Thing? Front Immunol 7:311. doi: 10.3389/fimmu.2016.00311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McDonald B, Davis RP, Kim SJ, Tse M, Esmon CT, Kolaczkowska E, and Jenne CN. 2017. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 129 (10):1357–1367. doi: 10.1182/blood-2016-09-741298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen XF, Cao K, Jiang JP, Guan WX, and Du JF. 2017. Neutrophil dysregulation during sepsis: an overview and update. J Cell Mol Med 21 (9):1687–1697. doi: 10.1111/jcmm.13112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaufman T, Magosevich D, Moreno MC, Guzman MA, D’Atri LP, Carestia A, Fandino ME, Fondevila C, and Schattner M. 2017. Nucleosomes and neutrophil extracellular traps in septic and burn patients. Clin Immunol 183:254–262. doi: 10.1016/j.clim.2017.08.014. [DOI] [PubMed] [Google Scholar]
- 33.Sakurai K, Miyashita T, Okazaki M, Yamaguchi T, Ohbatake Y, Nakanuma S, Okamoto K et al. 2017. Role for Neutrophil Extracellular Traps (NETs) and Platelet Aggregation in Early Sepsis-induced Hepatic Dysfunction. In Vivo 31 (6):1051–1058. doi: 10.21873/invivo.11169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jimenez-Alcazar M, Rangaswamy C, Panda R, Bitterling J, Simsek YJ, Long AT, Bilyy R et al. 2017. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science 358 (6367):1202–1206. doi: 10.1126/science.aam8897. [DOI] [PubMed] [Google Scholar]
- 35.Biron BM, Chung CS, O’Brien XM, Chen Y, Reichner JS, and Ayala A. 2017. Cl-Amidine Prevents Histone 3 Citrullination and Neutrophil Extracellular Trap Formation, and Improves Survival in a Murine Sepsis Model. J Innate Immun 9 (1):22–32. doi: 10.1159/000448808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leshner M, Wang S, Lewis C, Zheng H, Chen XA, Santy L, and Wang Y. 2012. PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structures. Front Immunol 3:307. doi: 10.3389/fimmu.2012.00307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kolaczkowska E, Jenne CN, Surewaard BG, Thanabalasuriar A, Lee WY, Sanz MJ, Mowen K, Opdenakker G, and Kubes P. 2015. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat Commun 6:6673. doi: 10.1038/ncomms7673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, and Wang Y. 2010. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med 207 (9):1853–1862. doi: 10.1084/jem.20100239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rohrbach AS, Slade DJ, Thompson PR, and Mowen KA. 2012. Activation of PAD4 in NET formation. Front Immunol 3:360. doi: 10.3389/fimmu.2012.00360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Asaga H, Nakashima K, Senshu T, Ishigami A, and Yamada M. 2001. Immunocytochemical localization of peptidylarginine deiminase in human eosinophils and neutrophils. J Leukoc Biol 70 (1):46–51. [PubMed] [Google Scholar]
- 41.Takahara H, Tsuchida M, Kusubata M, Akutsu K, Tagami S, and Sugawara K. 1989. Peptidylarginine deiminase of the mouse. Distribution, properties, and immunocytochemical localization. J Biol Chem 264 (22):13361–13368. [PubMed] [Google Scholar]
- 42.Horibata S, Rogers KE, Sadegh D, Anguish LJ, McElwee JL, Shah P, Thompson PR, and Coonrod SA. 2017. Role of peptidylarginine deiminase 2 (PAD2) in mammary carcinoma cell migration. BMC Cancer 17 (1):378. doi: 10.1186/s12885-017-3354-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Knight JS, Subramanian V, O’Dell AA, Yalavarthi S, Zhao W, Smith CK, Hodgin JB, Thompson PR, and Kaplan MJ. 2015. Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann Rheum Dis 74 (12):2199–2206. doi: 10.1136/annrheumdis-2014-205365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muller S, and Radic M. 2015. Citrullinated Autoantigens: From Diagnostic Markers to Pathogenetic Mechanisms. Clin Rev Allergy Immunol 49 (2):232–239. doi: 10.1007/s12016-014-8459-2. [DOI] [PubMed] [Google Scholar]
- 45.Mishra N, Schwerdtner L, Sams K, Mondal S, Ahmad F, Schmidt RE, Coonrod SA, Thompson PR, Lerch MM, and Bossaller L. 2019. Cutting Edge: Protein Arginine Deiminase 2 and 4 Regulate NLRP3 Inflammasome-Dependent IL-1beta Maturation and ASC Speck Formation in Macrophages. J Immunol 203 (4):795–800. doi: 10.4049/jimmunol.1800720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ott LW, Resing KA, Sizemore AW, Heyen JW, Cocklin RR, Pedrick NM, Woods HC et al. 2007. Tumor Necrosis Factor-alpha- and interleukin-1-induced cellular responses: coupling proteomic and genomic information. J Proteome Res 6 (6):2176–2185. doi: 10.1021/pr060665l. [DOI] [PMC free article] [PubMed] [Google Scholar]