

Abstract
Cholangiocarcinoma (CCA) is a highly malignant epithelial tumor of the biliary tree with poor prognosis. In the current study, we present novel evidence that the histone-lysine methyltransferase G9a is upregulated in human CCA and that G9a enhances CCA cell growth and invasiveness through regulation of the Hippo pathway kinase LATS2 and YAP signaling pathway. Kaplan–Meier survival analysis revealed that high G9a expression is associated with poor prognosis of CCA patients. In experimental systems, depletion of G9a by siRNA/shRNA or inhibition of G9a by specific pharmacological inhibitors (UNC0642 and UNC0631) significantly inhibited human CCA cell growth in vitro and in SCID mice. Increased G9a expression was also observed in mouse CCA induced by hydrodynamic tail vein injection of NICD and myr-Akt. Administration of the G9a inhibitor UNC0642 to NICD/Akt-injected mice reduced the growth of CCA, in vivo. These findings suggest that G9a inhibition may represent an effective therapeutic strategy for the treatment of CCA. Mechanistically, our data show that G9a-derived dimethylated H3K9 (H3K9me2) silenced the expression of the Hippo pathway kinase LATS2 and this effect led to subsequent activation of oncogenic YAP. Consequently, G9a depletion or inhibition reduced the level of H3K9me2 and restored the expression of LATS2 leading to YAP inhibition. Our findings provide novel evidence for an important role of G9a in cholangiocarcinogenesis through regulation of LATS2-YAP signaling and suggest that this pathway may represent a potential therapeutic target for CCA treatment.
Keywords: G9a, histone methyltransferase, cholangiocarcinoma, LATS2, YAP
INTRODUCTION
Cholangiocarcinoma (CCA) is a highly malignant epithelial neoplasm with cholangiocyte differentiation(1–4). The incidence and mortality of CCA are increasing worldwide and currently there is no effective chemoprevention or treatment. The prognosis of CCA is poor due to the inability of early diagnosis, the aggressive nature of tumor growth, and the lack of effective target therapy. Currently, surgical resection is curative for only a minority of patients with early stage of disease, whereas most CCA patients present in an unresectable state and respond poorly to conventional anti-cancer therapeutic agents. Therefore, there is an urgent need to better understand the molecular mechanisms of cholangiocarcinogenesis in order to develop more effective target therapy.
The molecular pathogenesis underlying biliary epithelial neoplasia involves genetic and epigenetic changes leading to alterations of oncogenic and tumor suppressive pathways. Recent studies with high throughput next-generation sequencing analyses have aided the identification of genetic abnormalities in CCA(5, 6). Additional evidences have shown that epigenetic alterations are also implicated in the pathogenesis of human CCA(7, 8), although the precise effect and mechanism of epigenetic modifications in biliary carcinogenesis are far from clarified.
Histone modification is an important epigenetic mechanism implicated in carcinogenesis. It is characterized by covalent post-translational modification of histone proteins through methylation, phosphorylation, acetylation, ubiquitylation or sumoylation. Studies have demonstrated that histone modifications are involved in diverse cellular processes including transcriptional regulation, chromosome packaging, and DNA damage repair. Among different forms of histone modifications, methylation of histone has been shown to play an important role in silencing the expression of tumor suppressor genes(9, 10). In particular, the methylation of histone is mediated by histone methyltransferases (HMTs), which include G9a, also known as EHMT2 (euchromatic histone-lysine N-methyltransferase 2).
G9a catalyzes histone H3 lysine 9 (H3K9) dimethylation, a modification associated with transcriptional gene silencing(9, 11). Similar to other HMTs, G9a is involved in the regulation of multiple cellular processes, including cell proliferation, differentiation, metastasis, autophagy and resistance to anti-cancer agents(12–14). Indeed, recent studies have shown that G9a expression is elevated in several human cancers which exhibit oncogenic actions(12, 14). However, it remains unknown whether G9a is implicated in cholangiocarcinogenesis.
The current study provides the first evidence that G9a is upregulated in human CCA and that G9a enhances CCA cell growth and invasiveness through regulation of the Hippo pathway kinase LATS2 and YAP signaling pathway. Our findings provide encouraging evidence that inhibition of G9a may represent an effective therapeutic strategy for the treatment of CCA.
MATERIALS AND METHODS
Human cholangiocarcinoma and non-cancerous tissues from the GEO datasets (GSE76297) and TCGA (The Cancer Genome Atlas) database were analyzed for G9a expression and the data were correlated with the patient survival.
Human primary biliary epithelial cells (BECs), noncancerous bile duct epithelial cell line (H69), and five human CCA cell lines (CCLP1, SG231, HuH28, TFK1 and EGI-1) were used in this study. The human CCA cells consist of three intrahepatic CCA cell lines (CCLP1, SG231, HuH28) and two extrahepatic CCA cell lines (TFK1 and EGI-1). CCLP1, SG231, TFK1 and H69 cells were cultured according to our previous described methods(15–18). HuH28 and EGI-1 cells were cultured in RPMI 1640 medium containing 10% fetal bovine serum. BECs cells were cultured in EpiCM medium according to the manufacture’s instruction. Lipofectamine™ 2000 reagent was used in this study for cell transfection. pSMP-G9a shRNA vector was used for establishment of the cells with G9a stable depletion. qRT-PCR and Western blotting analyses were performed to determine the expression of specific mRNAs and proteins. ChIP-PCR was performed to analyze the enrichment of G9a and H3K9me2 in the promoter region of LATS2 gene. Dual-Luciferase reporter assay system was used to evaluate YAP luciferase activity.
The athymic nude NOD CB17-Prkdc/SCID mice and wild-type FVB/NJ mice were obtained from Jackson Laboratory (Bar Harbor, ME). CCA xenograft was developed by subcutaneous inoculation of human CCA cells (CCLP1) with or without G9a depletion into the SCID mice. CCA tumor induction was developed by SB transposase-mediated integration of oncogenes (NICD/Akt) to the FVB/NJ mice via hydrodynamic tail vein (HDTV) injection. For G9a inhibitor treatment in vivo, UNC0642 was dissolved in 20% Captisol and was administrated via intraperitoneal injection to CCA-bearing mice. For all animal studies, the procedures were carried out in strict accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. The handling of the mice and all experimental procedures were approved for this study by the Institutional Animal Care and Use Committee of Tulane University (Protocol #: 4159).
Statistical analysis was performed using SPSS 13.0, GraphPad Prism7.0 and SigmaPlot statistical software. P values less than 0.05 was considered statistically significant.
Detailed methodology is described in Supplementary Materials and Methods.
RESULTS
G9a is upregulated in human CCA tissues and cells.
We first evaluated the expression of G9a in human CCA tissues by using two publically available databases, GEO datasets (GSE76297) and TCGA database. The microarray dataset GSE76297 included 90 CCA samples which were derived from intrahepatic CCA patients(19). The samples in the TCGA database were from 36 CCA patients (30 intrahepatic, 4 perihilar and 2 distal) (see summary in Supplementary Table 1). Analysis of the GEO database showed that the expression of G9a is significantly increased in CCA tissues when compared to the matched non-tumorous tissues (n = 90) (p < 0.01, Fig. 1A). Similarly, analysis of the TCGA RNA-Seq database also showed increased G9a expression in CCA tissues (n = 36) when compared to the non-cancerous tissues (n = 9) (p < 0.01, Fig. 1B). Kaplan–Meier survival analysis of the TCGA data showed that high G9a expression is associated with poorer disease-free survival (p < 0.05, Fig. 1C).
Figure 1. Upregulation of G9a histone methyltransferase in human CCA.

(A) Analysis of GEO dataset. Publically available clinical cancer gene expression data (GSE76297) were downloaded from GEO datasets to assess G9a expression in paired CCA and non-CCA samples. 90 samples were included in each group. (B) Analysis of TCGA CCA cohort. 36 CCA samples were compared with 9 non-CCA samples. Data were presented as FPKM (fragments per kilo base of transcript sequences per aligned million reads). (C) Kaplan–Meier survival curves for patients with CCA based on G9a expression from TCGA database. (D) (left) Graphic presentation of G9a staining intensity in human cholangiocarcinoma tissue (n = 28, p < 0.01). (right) Representative immunohistochemistry for G9a in human CCA tissue. Scale bar 100 μm. (E, F) Real-Time Quantitative PCR and Western blotting were used to determine the expression of G9a mRNA and protein, respectively, in CCA cell lines (CCLP1, SG231, HuH28, TFK1 and EGI-1), normal human primary biliary epithelial cells (BECs) and non-neoplastic bile duct epithelial cell line (H69). (G) Analysis of G9a localization in BECs and CCA cells by immunofluorescence staining (scale bar 20 μm). ** p < 0.01.
We next performed immunohistochemical staining for G9a in formalin-fixed, paraffin-embedded tissue specimens surgically resected from CCA patients. A total of 28 cases of human CCA samples were used for G9a immunohistochemistry. Analysis of the G9a stained tissue sections showed that the expression of G9a is significantly increased in human CCA tissues when compared to the non-neoplastic bile duct epithelia (p < 0.01, Fig. 1D). High expression of G9a (IHC score ≥ 2) was observed in 23 of 28 cases of human CCA (82%). We also examined the mRNA and protein levels of G9a in human CCA cell lines and non-cancerous human biliary epithelial cells. We observed that the G9a mRNA and protein levels in CCA cells were significantly higher than in human primary biliary epithelial cells (BECs) and the noncancerous human biliary epithelial cell line, H69 (Fig. 1E & 1F). Accordingly, the level of dimethylated H3K9 (H3K9me2), the main target for methylation by G9a, was also higher in CCA cells when compared to non-neoplastic biliary epithelial cells (Fig. 1F). Under Immunofluorescence, we observed that the expression of G9a is predominantly localized in the nuclei (Fig. 1G).
Stable depletion of G9a inhibits human CCA cell growth in vitro and in SCID mice.
To assess the functional impact of G9a in CCA cells, we established CCA cell lines (SG231 and CCLP1) with stable knockdown of G9a by shRNA. Satisfactory depletion of G9a protein in CCA cells transfected with G9a shRNA was confirmed by Western blotting (Fig. 2A). Given that G9a is a histone methyltransferase which mediates the dimethylation of H3K9, we examined the effect of G9a knockdown on H3K9 methylation. We observed that depletion of G9a by shRNA led to significant reduction of H3K9me2 (Fig. 2A). Our data indicate that G9a knockdown only slightly decreased the level of monomethylated H3K9 (H3K9me1) and did not affect the level of trimethylated H3K9 (H3K9me3) (Supplementary Fig. S1). These findings are consistent with the established role of G9a predominantly for the dimethylation of H3K9 in other cell types(20, 21).
Figure 2. shRNA depletion of G9a inhibits CCA in vitro and in SCID mice.
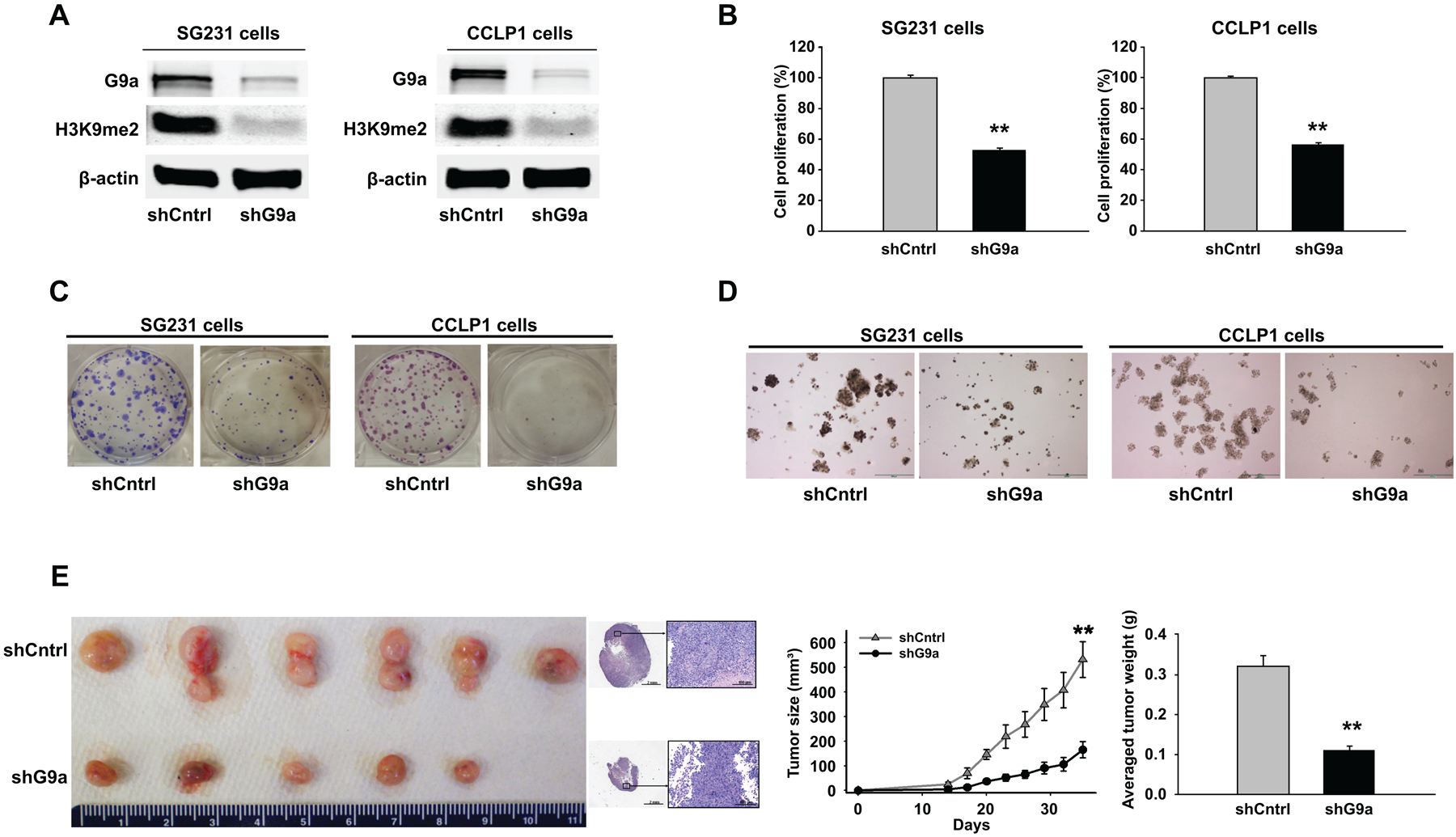
(A) SG231 and CCLP1 cells were stably transfected with the G9a shRNA vector (pSMP-G9a shRNA) or the control shRNA vector (shCntrl). The levels of G9a and H3K9me2 were analyzed by Western blotting. (B) Depletion of G9a by shRNA inhibited cell proliferation, as measured by WST-1 assay. (C) Anchorage-dependent colony formation assay in SG231 and CCLP1 cells with G9a depletion. (D) Anchorage-independent colony formation efficiency in SG231 and CCLP1 cells with G9a depletion. Scale bar 200 μm. (E) Depletion of G9a inhibits CCA progression in SCID mice. CCLP1 cells stably transfected with the G9a shRNA or control vector were injected into SCID mice subcutaneously. Tumor size was monitored every three days; tumor weight was recorded upon harvesting. (left) Representative images of xenograft tumors recovered from 6 mice in each group (no tumor was formed in 1 of 6 mice inoculated with G9a depleted cells); histological sections of the tumors (H&E staining, scale bar 100 μm). (right) growth curve and average weight of CCA tumors with or without G9a knockdown in SCID mice. ** p < 0.01.
We then performed cell proliferation and colony formation assays in CCA cells with or without G9a knockdown. As shown in Fig. 2B, knockdown of G9a by shRNA significantly decreased the proliferation of both SG231 and CCLP1 cells (p < 0.01). Consistent with these results, BrdU incorporation assay showed that G9a shRNA significantly reduced the population of BrdU positive cells (Supplementary Fig. S2). Furthermore, our data indicated that shRNA depletion of G9a inhibited the colony formation ability of CCA cells (Fig. 2C & D).
Following the above-described in vitro studies, we assessed the effect of G9a knockdown on CCA tumorigenicity in SCID mice. For this purpose, CCLP cells with or without stable knockdown of G9a were injected subcutaneously into the flank of SCID mice and the animals were monitored for tumor growth. As shown in Fig. 2E, G9a knockdown in CCA cells led to significant reduction of both tumor size and tumor volume. An approximately 3-fold decrease in tumor size and tumor weight was observed in G9a knockdown tumors when compared to the control (p < 0.01). These findings demonstrate that G9a depletion inhibited CCA cell growth in vitro and in vivo.
Transient depletion of G9a decreases the malignant characteristics of human CCA cells in vitro.
In addition to stable depletion of G9a, we employed a parallel approach to transiently deplete G9a in human CCA cells by using G9a specific siRNA duplexes. Western blotting analysis revealed that transient transfection with G9a siRNA effectively reduced the level of G9a protein as well as the level of H3K9me2 in CCA cells (Supplementary Fig. S3A). By using WST-1 cell proliferation assay, we found that siRNA depletion of G9a significantly reduced the proliferation of CCA cells (p < 0.01, Supplementary Fig. S3B). Colonogenic analysis indicated that G9a siRNA reduced the colony formation ability of CCA cells (Supplementary Fig. S4). Accordingly, G9a siRNA significantly decreased the population of BrdU positive cells (p < 0.01, Supplementary Fig. S5). Furthermore, we observed that G9a siRNA significantly reduced CCA cell migration and invasion (Supplementary Fig. S6A & B). Together, these findings demonstrate that G9a depletion reduces the malignant characteristics of human CCA cells.
Given that TGF-β-induced epithelial-mesenchymal transition (EMT) is known to be implicated in tumor cell invasion and metastasis, we had performed further studies to determine whether G9a might influence TGF-β-induced EMT in CCA cells. While TGF-β treatment induced EMT response in TFK1 and SG231 cells (as reflected by the spindle-shaped morphology, decreased epithelial marker E-cadherin, and increased mesenchymal marker Vimentin), we observed that siRNA knockdown of G9a did not alter TGF-β-induced EMT in these cells (Supplementary Fig S7). These observations suggest that G9a is not involved in TGF-β-induced EMT in CCA cells. In this context, it is worth mentioning that TGF-β-induced EMT in hepatocellular carcinoma (HCC) cells was also not suppressed by G9a knockdown (22).
Pharmacological inhibition of G9a reduces CCA cell growth in vitro and in SCID mice.
Given that depletion of G9a by shRNA and siRNA inhibited human CCA cell growth as shown in the above sections, we further evaluated the effect of pharmacological G9a inhibitors in our system. This aspect of investigation is highly significant, given that several pharmacological inhibitors of histone methyltransferases have been recently developed as new anti-cancer therapeutic agents(23, 24). In the current study, we assessed the effects of two G9a inhibitors, UNC0642 and UNC0631, which are known to bind to the catalytic domain of G9a and inhibit its enzymatic activity(25, 26). We observed that treatment with UNC0642 and UNC0631 decreased the level of H3K9me2 in human CCA cells (Fig. 3A & B, Supplementary Fig. S8A & B). WST-1 cell proliferation assay showed that both G9a inhibitors significantly inhibited the growth of CCA cells in a dose-dependent manner (p < 0.01, Fig. 3C & D, Supplementary Fig. S8C & D). Clonogenicity assay revealed that G9a inhibitors decreased the colony formation ability of CCA cells (Fig. 3E & F, Supplementary Fig. S8E). UNC0642 treatment of CCA cells decreased Ki67 staining (Supplementary Fig. S9A) and induced caspase-3 cleavage (Supplementary Fig. S9B). As UNC0642 is suitable for in vivo studies(25), we employed a CCA xenograft model in SCID mice to assess the effect of G9a inhibition by UNC0642 on CCA tumor progression. Our data show that administration of UNC0642 to SCID mice inoculated with human CCA cells reduced tumor progression in vivo, leading to approximately 2-fold reduction in mean tumor size and tumor weight (Fig. 3G). Taken together, these findings indicate that pharmacological inhibition of G9a methyltransferase decreases CCA cell growth, in vitro and in vivo.
Figure 3. G9a inhibitors inhibit CCA cell growth in vitro and in SCID mice.

(A, B) The effect of the G9a inhibitors, UNC0642 (A) or UNC0631 (B), on the level of H3K9me2 in SG231 and CCLP1 cells. The cells were treated with indicated doses of G9a inhibitors for 36 h. (C, D) The effect of UNC0642 (C) and UNC0631 (D) on the growth of SG231 and CCLP1 cells. The cells were treated with indicated inhibitors for 72 h and cell proliferation was determined by WST-1 assay. NT: non-treatment. (E) The effect of UNC0642 and UNC0631 on anchorage-dependent colony formation. (F) The effect of UNC0642 and UNC0631 on anchorage-independent colony formation. Scale bar 200 μm. (G) UNC0642 inhibits the growth of CCA xenograft in SCID mice. The mice were treated with 20% Captisol (Vehicle) or UNC0642 (5 mg/kg) via daily intraperitoneal injection for 3 weeks (the treatment began 1 week after CCLP1 cell inoculation). (left) Representative images of xenograft tumors recovered from 6 mice in each group (no tumor nodule was formed in 1 of 6 UNC0642-treated mice); histological sections of the tumors (H&E staining, scale bar 100 μm). (right) growth curve and average weight of CCA tumors with or without UNC0642 treatment in SCID mice. ** p < 0.01.
To examine the potential effect of G9a inhibition in non-cancerous biliary epithelial cells, we treated BECs and H69 cells with different doses of UNC0642 and UNC0631; our data showed that G9a inhibitors had minimal effect on the growth of BECs and H69 cells (Supplementary Fig. S10A & B). Consistent with these findings, we observed that knockdown of G9a by siRNA did not significantly alter the growth of BECs and H69 cells (Supplementary Fig. S11A & B).
G9a silences the expression of the tumor suppressor LATS2 in human CCA cells.
G9a is known to confer transcriptional repressive function and silence the expression of a number of tumor suppressor genes(12, 27). To identify G9a downstream target in CCA cells, we performed qRT-PCR analyses to determine the expression levels of key tumor suppressor genes in G9a depleted CCA cells. Our data showed that, among the 31 tumor suppressor genes screened by qRT-PCR, LAST2 (Large Tumor Suppressor 2) is the most upregulated tumor suppressor in CCA cells following G9a knockdown (Fig. 4A).
Figure 4. Identification of LATS2 as a G9a downstream target in CCLP1 cells.

(A) Screening of G9a-regulated tumor suppressor gene by qRT-PCR. Total RNA was extracted from control cells and G9a siRNA treated cells. 31 tumor suppressor genes were screened by qRT-PCR. (B) ChIP-qPCR was used to analyze the enrichment of G9a and H3K9me2 in the promoter region of LATS2 gene. Five primer pairs (P1-P5) targeting the promoter region of LATS2 were used for the assay. Rabbit IgG was used as negative control. (C, D) ChIP-qPCR analyses to detect H3K9me2 association with LATS2 gene promoter (by using LATS2 promoter primer pair 1). Depletion of G9a by shRNA (C) or inhibition of G9a by UNC0642 (D) attenuates H3K9me2 association with the LATS2 gene promoter. ** p < 0.01.
LATS2 is a key kinase of the Hippo pathway and has been reported to serve as an important tumor suppressor gene in multiple cancer types(28, 29). However, very little is known about the functional role and regulatory mechanism of LATS2 in human CCA. To document the role of G9a-derived H3K9me2 for regulation of LATS2 in CCA cells, we carried out chromatin immunoprecipitation (ChIP) assay by using anti-G9a and anti-H3K9me2 antibodies. Our data showed that G9a and H3K9me2 were enriched in the promoter region of the LATS2 gene (p < 0.01, Fig. 4B) and these enrichments were significantly reduced by shRNA knockdown of G9a or by treatment with the G9a inhibitor UNC0642 (p < 0.01, Fig. 4C & D). qRT-PCR analysis showed that LATS2 mRNA expression was increased in CCA cells with stable or transient depletion of G9a (Fig. 5A & Supplementary Fig. S12A). Accordingly, the level of LATS2 protein was also significantly increased in CCA cells with G9a depletion (Fig. 5B & Supplementary Fig. S12B). These findings suggest an important role of G9a-derived H3K9me2 for regulation of LATS2 in CCA cells.
Figure 5. Depletion of G9a by shRNA upregulates LATS2 expression and reduces nuclear YAP in CCA cells.

(A) The level of LATS2 mRNA was analyzed by qRT-PCR in SG231 and CCLP1 cells with or without G9a depletion. (B) Western blotting for LATS2 and p-YAPSer127 in SG231 and CCLP1 cells with or without G9a depletion. The relative densitometry of LATS2 and p-YAPSer127 is shown in the bar graphs. (C) Western blot analysis to detect YAP nuclear accumulation. Nuclear proteins from SG231 and CCLP1 cells were utilized for the analysis. Immunoblotting for histone 3 and α-tubulin was used as control. (D) The effect of G9a on YAP activity. SG231 and CCLP1 cells with or without shRNA depletion of G9a were transfected with YAP luciferase reporter vector for 48h (with pRL-TK Renilla luciferase vector as internal control). The cell lysates were then collected to measure the luciferase reporter activity with a luminometer. (E) The mRNA levels of YAP target genes were analyzed by qRT-PCR in SG231 and CCLP1 cells with or without G9a depletion. * p < 0.05, ** p < 0.01.
We next utilized siRNA to knockdown LATS2 expression in CCA cells with G9a stable depletion and observed that LATS2 knockdown increased CCA cell growth under this condition (Supplementary Fig. S13).
Taken together, our experimental findings presented in the above sections suggest that G9a silences the expression of LATS2 in CCA cells and that this regulatory mechanism is implicated in the regulation of CCA cell growth.
G9a regulates YAP activity in human CCA cells.
LATS2 is a key component of the Hippo pathway which exerts tumor-suppressor function through phosphorylation of YAP (yes-associated protein). Mechanistically, LATS2-mediated YAP phosphorylation leads to YAP cytoplasmic retention and degradation, thus preventing YAP nuclear accumulation and transcriptional activation(29–31). The importance of YAP inhibition by LATS2 is highlighted by the fact that YAP is a crucial oncogene implicated in the tumorigenesis of various human cancers(31, 32) including CCA(33, 34). On the basis of the existing knowledge, we performed further experiments to determine the status of YAP phosphorylation and nuclear accumulation in CCA cells with or without G9a depletion. Our data showed that G9a depletion led to increased YAP phosphorylation and decreased YAP nuclear accumulation (Fig. 5B & C). Consistent with these observations, immunofluorescence staining revealed that the level of nuclear YAP accumulation was reduced in G9a-depleted cells (Supplementary Fig. S14). The regulation of YAP by LAST2 is further indicated by the fact that LATS2 knockdown enhanced YAP nuclear accumulation in cells with G9a depletion (Supplementary Fig. S15).
To further assess the effect of G9a on YAP transcription activity, we transfected CCA cells with a well-established YAP luciferase reporter vector, 8xGTIIC TEAD luciferase(35), and the cell lysates were then obtained for luciferase reporter activity assay. Our data showed that the YAP transcription activity was significantly decreased in SG231 and CCLP1 cells transfected with G9a shRNA (Fig. 5D) or siRNA (Supplementary Fig. S16). Furthermore, the expression of YAP downstream genes (including AXL, CTGF, CYR61 and EDN1) in CCA cells was reduced by G9a depletion or inhibition (Fig. 5E, Supplementary Fig. S17). Collectively, these results indicate an important role of G9a for regulation of LATS2/YAP in CCA cells.
The effect of forced overexpression of G9a in non-neoplastic biliary epithelial cells.
We next sought to evaluate the effect of G9a in non-cancerous human biliary epithelial cells. As the level of G9a and H3K9me2 is low in non-neoplastic biliary epithelial cells, we performed experiments with forced overexpression of G9a in human BECs and H69 cells. Our data showed that forced overexpression of G9a in these cells led to increased level of H3K9me2 with decreased LATS2 expression, reduced YAP phosphorylation, and increased YAP nuclear accumulation (Fig. 6A & B). We observed that G9a overexpression promoted the growth and invasion of human BECs and H69 cells (Fig. 6C–E). These findings further support a potential oncogenic role of G9a in biliary epithelial cells.
Figure 6. The effects of forced overexpression of G9a in human biliary epithelial cells.

BECs and H69 cells were transfected with the G9a expression or control vector (pT3-EF1a). (A) Western blotting for H3K9me2, LATS2 and p-YAPSer127 in cells with or without G9a overexpression. The densitometry data for LATS2 and p-YAPSer127 are shown in the bar graphs. (B) Nuclear proteins from cells with or without G9a overexpression were subjected to Western blot analysis to detect the level of nuclear YAP. (C) WST-1 cell proliferation assay in cells with or without G9a overexpression. (D) Colony formation assay in cells with or without G9a overexpression (grown on ultra-low attachment plates). Scale bar 200 μm. (E) Transwell cell invasion assay in cells with or without G9a overexpression. * p < 0.05, ** p < 0.01.
The role of G9a in a separate murine model of CCA induced by NICD/AKT.
To further evaluate the role of G9a in cholangiocarcinogenesis, we examined the expression level of G9a in a mouse model of CCA induced by hydrodynamic tail vein (HDTV) injection of NICD (notch intracellular domain) and myr-Akt in conjunction with the SB transposon system as described(36). For this purpose, we utilized wild type FVB/NJ mice for HDTV injection. As shown in Fig. 7A, intrahepatic CCA lesions occupied almost the entire liver 5 weeks following tail vein injection. The CCA tumor tissues harvested from the mice receiving HDTV injection of NICD/Akt showed increased level of G9a expression when compared to the controls (Fig. 7B). Increased G9a expression in CCA cells was further confirmed by immumohistochemical staining (Fig. 7C). The above findings in mouse CCA tissues are corroborated by increased G9a in human CCA tissues as documented in the earlier section.
Figure 7. The expression and function of G9a in a separate mouse model of CCA.

FVB/NJ mice were subjected to HDTV injection of NCID, Akt and transposase plasmids, and the animals were sacrificed 5 weeks after tail vein injection. (A) Representative gross photographs of livers from mice with or without HDTV injection (upper panels). Histological images (H&E stain) from the corresponding livers are shown at the lower panels. Scale bar 200 μm. (B) Western blotting analysis for G9a protein in the liver tissues from mice with or without HDTV injection. (C) Immunohistochemical stain for G9a in CCA tissues from NCID/Akt-injected mice. Scale bar 200 μm. For Panels D-G, FVB/NJ mice receiving HDTV injection of NCID, Akt and transposase plasmids were treated with 2.5 mg/kg UNC0642 every other day via intraperitoneal injection (6 mice were included in each group). The treatment began one week after HDTV injection and continued for 3 weeks. Five weeks after HDTV injection, the animals were sacrificed and the livers were harvested. (D, E) CCA tumor burden. The gross photographs from each group are shown in Panel D; the liver/body weight ratio is shown in Panel E. * p < 0.05. (F) Immunohistochemistry for SOX9 (scale bar 500 μm). * p < 0.05. (G) Immunohistochemistry for Ki67 (scale bar 50 μm). ** p < 0.01. (H) Schematic summary of the effect and mechanism of G9a in CCA.
To further document the role of G9a in cholangiocarcinogenesis, we subjected the NICD/Akt-injected mice to G9a inhibitor treatment. Specifically, the mice receiving HDTV injection of NICD and Akt were treated with 2.5 mg/kg UNC0642 every other day via intraperitoneal injection; the treatment began one week after HDTV injection and continued for 3 weeks. Five weeks after HDTV injection, the animals were sacrificed to determine CCA tumor burden. Macroscopic examination revealed that UNC0642 treatment inhibited tumor growth in this model of cholangiocarcinogenesis (Fig. 7D). The liver/body weight ratio in UNC0642-treated mice was significantly lower than that in the control mice (treated with 20% Captisol) (Fig. 7E). Decreased CCA cell mass was confirmed by immunohistochemical staining for the biliary marker SOX9 (with quantitative analysis) (Fig. 7F). UNC0642 treatment reduced the numbers of Ki67-positive tumor cells (Fig. 7G). The CCA cells harvested from UNC0642-treated mice exhibited increased LATS2 expression with reduced YAP nuclear accumulation (Supplementary Fig. S18). These findings, along with the data from the CCA xenograft in SCID as shown in the above section, provide encouraging evidence for utilization of G9a inhibitor for CCA treatment.
DISCUSSION
Epigenetic dysregulation is importantly implicated in cellular transformation and evolution of cancer cells. Some well-characterized cancer epigenetic changes include post-translational modifications of histone proteins which are known to regulate the loosening of the nucleosome, affecting the binding of transcription factors and RNA polymerase to their target genes(37, 38). As one of the most well studied patterns among histone modifications, histone methylation modifiers have been shown to play crucial roles in regulating the transcription of oncogenes and tumor suppressor genes in a variety of cancer types(9, 12, 39). With respect to CCA, however, the effect and mechanism of histone methylation in cholangiocarcinogenic processes remain largely undefined. The current study provides novel evidence that the histone lysine methyltransferase G9a enhances CCA cell growth and migration/invasion in vitro and in complementary mouse models of cholangiocarcinogenesis. Mechanistic studies show that G9a-derived H3K9me2 silenced the expression of the Hippo pathway kinase LATS2 which led to decrease in YAP phosphorylation and subsequent activation of YAP (Fig. 7H). The therapeutic potential of inhibiting G9a for CCA treatment was demonstrated by studies in vitro and in mouse models of CCA induction and tumor xenografts.
Increasing evidences have shown that G9a is overexpressed in a number of cancers and is attributable to tumor development and/or progression(9, 12, 14, 27, 40). The expression of G9a in CCA has been described recently(41), although the mechanism by which G9a regulates CCA growth has not been defined. In the current study, we employed complementary analyses to document the expression of G9a in CCA cells and tissues and to investigate its functional impact and mechanism of action. Our data indicate that the expression of G9a is increased in both human and mouse CCA tissues. We show that the level of G9a is increased in human CCA cells when compared to the non-neoplastic biliary epithelial cells. These observations are consistent with the increased G9a expression in human CCA tissues, as documented by analysis of human CCA samples in the TCGA and GEO databases. Notably, our analysis revealed that patients with high G9a expression exhibited poorer prognosis when compared to patient with low G9a expression. These results, along with the functional and mechanistic studies as detailed in the current paper, provide important evidence indicating an oncogenic action of G9a in cholangiocarcinogenesis.
Several recent studies suggest that G9a may be targeted for the treatment of human cancers(20, 40, 42). In the current study, we treated the CCA cells with two G9a inhibitors, UNC0631 and UNC0642, respectively. We observed that both UNC0631 and UNC0642 significantly inhibited the proliferation of CCA cells. Consistent with these effects, knockdown of G9a by siRNA or shRNA inhibited the proliferation, colony formation, migration and invasion of CCA cells. Notably, the growth of CCA tumors was significantly inhibited by treatment with the G9a inhibitor UNC0642 and by shRNA knockdown of G9a in vivo. These findings further support an oncogenic role of G9a in cholangiocarcinogenesis and provide encouraging pre-clinical evidence for targeting G9a in CCA treatment.
G9a is the primary enzyme for dimethylation of histone H3 at the Lys 9 residue(12, 20). In the current study, we observed that G9a depletion or inhibition decreased the level of H3K9me2, confirming the role of G9a for formation of H3K9me2 in human CCA cells. Consistent with the documented role of H3K9 methylation for transcriptional repression of gene expression in other cells(11, 43), our data provide novel evidence implicating G9a in suppression of the Hippo pathway kinase LATS2 in CCA cells. We observed that G9a and H3K9me2 were associated with the promoter region of LATS2 and their associations were disrupted when G9a was depleted or inhibited. Furthermore, we noted that G9a depletion enhanced the expression of both LATS2 mRNA and protein. These findings suggest that G9a-derived H3K9me2 suppresses LAST2 expression in human CCA cells.
LATS2 is a key tumor suppressor gene involved in the regulation of cell proliferation, cell death, and cell migration(29, 44). It is a crucial Hippo signaling kinase which phosphorylates and inactivates the YAP transcription coactivator by inhibiting the translocation of YAP into the nucleus(30, 45). The importance of LATS2-mediated inactivation of YAP is attested by the fact that YAP is a potent driver of cancer development and progression in multiple cancer types(31, 46). Indeed, recent evidences have shown that YAP is an important oncogene in CCA(33, 34). In our study, we have analyzed the nuclear accumulation and activity of YAP in CCA cells. Our data showed that knockdown of G9a significantly decreased the level of nuclear YAP and attenuated its transcription activity. Furthermore, we observed that knockdown of LATS2 restored YAP nuclear accumulation and reversed the growth-inhibition by G9a-depletion. Our data, along with the existing studies, suggest a potentially important role of the G9a-LATS2-YAP signaling axis in cholangiocarcinogenesis.
The function and mechanism of G9a in mammalian cells are rather complicated. Besides histone methylation and transcriptional repression, G9a is known to regulate cellular processes by methylating several non-histone substrates (including CDYL1 and MEF2, among others)(47, 48). G9a has also been reported to act as a co-activator for some nuclear receptors(49, 50). In the setting of carcinogenesis, G9a overexpression has been reported to enhance tumor progression through silencing Ep-CAM(9), suppressing RARRES3(12), or activating the serine-glycine synthesis pathway(14). In this context, while our study has identified Hippo pathway as a key mechanism for G9a function in CCA cells, further studies are warranted to investigate whether these and/or other mechanisms may also be implicated in the regulation of cholangiocarcinogenesis.
In the current study, we utilized both intra-hepatic and extra-hepatic CCA cells to assess the effect and mechanism of G9a. Our data showed that G9a signaling is implicated similarly in both intra- and extrahepatic CCA cells. High G9a mRNA and protein levels were found in both intra- and extrahepatic CCA cells; accordingly, the G9a downstream target, H3K9me2, was also elevated in both intra- and extrahepatic CCA cells. We observed that pharmacological inhibitors of G9a significantly reduced the malignant characteristics of both intra- and extrahepatic CCA cells; similarly, G9a knockdown also inhibited the growth of both intra- and extrahepatic CCA cells. Furthermore, our data showed that knockdown of G9a by siRNA restored the mRNA and protein levels of the Hippo pathway kinase LATS2 in both intra- and extrahepatic CCA cells. Together, our findings support the concept that G9a silences the expression of LATS2 in both intra- and extrahepatic CCA cells.
In summary, the current study provides novel evidence that G9a plays an important oncogenic role in cholangiocarcinogenesis through regulation of the LATS2-YAP signaling pathway. Our findings provide important pre-clinical evidence for targeting the G9a-LATS2-YAP signaling axis for the treatment of human CCA.
Supplementary Material
Grant Support:
The work in the authors’ laboratory is supported by the National Institutes of Health grants CA219541, CA102325 and CA226281.
Abbreviations:
- CCA
Cholangiocarcinoma
- HMT
histone lysine methyltransferase
- H3K9me2
dimethylated histone H3 lysine 9
- BECs
human intrahepatic biliary epithelial cells
- BrdU
Bromodeoxyuridine
- LATS2
Large Tumor Suppressor 2
- YAP
yes-associated protein
Footnotes
No conflicts of interest exist
REFERENCES
- 1.Rizvi S, Khan SA, Hallemeier CL, Kelley RK, Gores GJ. Cholangiocarcinoma - evolving concepts and therapeutic strategies. Nat Rev Clin Oncol 2018;15:95–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sirica AE, Gores GJ, Groopman JD, Selaru FM, Strazzabosco M, Wang XW, Zhu AX. Intrahepatic Cholangiocarcinoma: Continuing Challenges and Translational Advances. Hepatology 2018;69:1803–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Razumilava N, Gores GJ. Cholangiocarcinoma. Lancet 2014;383:2168–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Banales JM, Cardinale V, Carpino G, Marzioni M, Andersen JB, Invernizzi P, Lind GE, et al. Expert consensus document: Cholangiocarcinoma: current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat Rev Gastroenterol Hepatol 2016;13:261–280. [DOI] [PubMed] [Google Scholar]
- 5.Zou S, Li J, Zhou H, Frech C, Jiang X, Chu JS, Zhao X, et al. Mutational landscape of intrahepatic cholangiocarcinoma. Nat Commun 2014;5:5696. [DOI] [PubMed] [Google Scholar]
- 6.Nakamura H, Arai Y, Totoki Y, Shirota T, Elzawahry A, Kato M, Hama N, et al. Genomic spectra of biliary tract cancer. Nat Genet 2015;47:1003–1010. [DOI] [PubMed] [Google Scholar]
- 7.Kwon H, Song K, Han C, Zhang J, Lu L, Chen W, Wu T. Epigenetic Silencing of miRNA-34a in Human Cholangiocarcinoma via EZH2 and DNA Methylation: Impact on Regulation of Notch Pathway. Am J Pathol 2017;187:2288–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Rourke CJ, Munoz-Garrido P, Aguayo EL, Andersen JB. Epigenome dysregulation in cholangiocarcinoma. Biochim Biophys Acta Mol Basis Dis 2018;1864:1423–1434. [DOI] [PubMed] [Google Scholar]
- 9.Chen MW, Hua KT, Kao HJ, Chi CC, Wei LH, Johansson G, Shiah SG, et al. H3K9 Histone Methyltransferase G9a Promotes Lung Cancer Invasion and Metastasis by Silencing the Cell Adhesion Molecule Ep-CAM. Cancer Research 2010;70:7830–7840. [DOI] [PubMed] [Google Scholar]
- 10.Au SL, Wong CC, Lee JM, Fan DN, Tsang FH, Ng IO, Wong CM. Enhancer of zeste homolog 2 epigenetically silences multiple tumor suppressor microRNAs to promote liver cancer metastasis. Hepatology 2012;56:622–631. [DOI] [PubMed] [Google Scholar]
- 11.Shinkai Y, Tachibana M. H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev 2011;25:781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wei L, Chiu DK, Tsang FH, Law CT, Cheng CL, Au SL, Lee JM, et al. Histone methyltransferase G9a promotes liver cancer development by epigenetic silencing of tumor suppressor gene RARRES3. J Hepatol 2017;67:758–769. [DOI] [PubMed] [Google Scholar]
- 13.Dutta A, Le Magnen C, Mitrofanova A, Ouyang XS, Califano A, Abate-Shen C. Identification of an NKX3.1-G9a-UTY transcriptional regulatory network that controls prostate differentiation. Science 2016;352:1576–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ding J, Li T, Wang X, Zhao E, Choi JH, Yang L, Zha Y, et al. The histone H3 methyltransferase G9A epigenetically activates the serine-glycine synthesis pathway to sustain cancer cell survival and proliferation. Cell Metab 2013;18:896–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu D, Han C, Wu T. Microsomal prostaglandin E synthase-1 inhibits PTEN and promotes experimental cholangiocarcinogenesis and tumor progression. Gastroenterology 2011;140:2084–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yao L, Han C, Song K, Zhang J, Lim K, Wu T. Omega-3 Polyunsaturated Fatty Acids Upregulate 15-PGDH Expression in Cholangiocarcinoma Cells by Inhibiting miR-26a/b Expression. Cancer Research 2015;75:1388–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han C, Leng J, Demetris AJ, Wu T. Cyclooxygenase-2 promotes human cholangiocarcinoma growth: evidence for cyclooxygenase-2-independent mechanism in celecoxib-mediated induction of p21waf1/cip1 and p27kip1 and cell cycle arrest. Cancer Research 2004;64:1369–1376. [DOI] [PubMed] [Google Scholar]
- 18.Zhang JQ, Han C, Wu T. MicroRNA-26a Promotes Cholangiocarcinoma Growth by Activating beta-catenin. Gastroenterology 2012;143:246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chaisaingmongkol J, Budhu A, Dang H, Rabibhadana S, Pupacdi B, Kwon SM, Forgues M, et al. Common Molecular Subtypes Among Asian Hepatocellular Carcinoma and Cholangiocarcinoma. Cancer Cell 2017;32:57–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tu WB, Shiah YJ, Lourenco C, Mullen PJ, Dingar D, Redel C, Tamachi A, et al. MYC Interacts with the G9a Histone Methyltransferase to Drive Transcriptional Repression and Tumorigenesis. Cancer Cell 2018;34:579–595. [DOI] [PubMed] [Google Scholar]
- 21.Chen X, Skutt-Kakaria K, Davison J, Ou YL, Choi E, Malik P, Loeb K, et al. G9a/GLP-dependent histone H3K9me2 patterning during human hematopoietic stem cell lineage commitment. Genes Dev 2012;26:2499–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yokoyama M, Chiba T, Zen Y, Oshima M, Kusakabe Y, Noguchi Y, Yuki K, et al. Histone lysine methyltransferase G9a is a novel epigenetic target for the treatment of hepatocellular carcinoma. Oncotarget 2017;8:21315–21326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Italiano A, Soria JC, Toulmonde M, Michot JM, Lucchesi C, Varga A, Coindre JM, et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol 2018;19:649–659. [DOI] [PubMed] [Google Scholar]
- 24.Kim Y, Kim YS, Kim DE, Lee JS, Song JH, Kim HG, Cho DH, et al. BIX-01294 induces autophagy-associated cell death via EHMT2/G9a dysfunction and intracellular reactive oxygen species production. Autophagy 2013;9:2126–2139. [DOI] [PubMed] [Google Scholar]
- 25.Liu F, Barsyte-Lovejoy D, Li F, Xiong Y, Korboukh V, Huang XP, Allali-Hassani A, et al. Discovery of an in vivo chemical probe of the lysine methyltransferases G9a and GLP. J Med Chem 2013;56:8931–8942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim Y, Lee HM, Xiong Y, Sciaky N, Hulbert SW, Cao X, Everitt JI, et al. Targeting the histone methyltransferase G9a activates imprinted genes and improves survival of a mouse model of Prader-Willi syndrome. Nat Med 2017;23:213–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Casciello F, Al-Ejeh F, Kelly G, Brennan DJ, Ngiow SF, Young A, Stoll T, et al. G9a drives hypoxia-mediated gene repression for breast cancer cell survival and tumorigenesis. Proc Natl Acad Sci U S A 2017;114:7077–7082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu FX, Zhao B, Guan KL. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015;163:811–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murakami H, Mizuno T, Taniguchi T, Fujii M, Ishiguro F, Fukui T, Akatsuka S, et al. LATS2 Is a Tumor Suppressor Gene of Malignant Mesothelioma. Cancer Research 2011;71:873–883. [DOI] [PubMed] [Google Scholar]
- 30.Hong AW, Meng Z, Guan KL. The Hippo pathway in intestinal regeneration and disease. Nat Rev Gastroenterol Hepatol 2016;13:324–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016;29:783–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moroishi T, Hayashi T, Pan WW, Fujita Y, Holt MV, Qin J, Carson DA, et al. The Hippo Pathway Kinases LATS1/2 Suppress Cancer Immunity. Cell 2016;167:1525–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pei TM, Li YJ, Wang JB, Wang HL, Liang YJ, Shi HW, Sun BS, et al. YAP is a critical oncogene in human cholangiocarcinoma. Oncotarget 2015;6:17206–17220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marti P, Stein C, Blumer T, Abraham Y, Dill MT, Pikiolek M, Orsini V, et al. YAP promotes proliferation, chemoresistance, and angiogenesis in human cholangiocarcinoma through TEAD transcription factors. Hepatology 2015;62:1497–1510. [DOI] [PubMed] [Google Scholar]
- 35.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, et al. Role of YAP/TAZ in mechanotransduction. Nature 2011;474:179–183. [DOI] [PubMed] [Google Scholar]
- 36.Fan B, Malato Y, Calvisi DF, Naqvi S, Razumilava N, Ribback S, Gores GJ, et al. Cholangiocarcinomas can originate from hepatocytes in mice. Journal of Clinical Investigation 2012;122:2911–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 2009;10:295–304. [DOI] [PubMed] [Google Scholar]
- 38.Morris MR, Latif F. The epigenetic landscape of renal cancer. Nat Rev Nephrol 2017;13:47–60. [DOI] [PubMed] [Google Scholar]
- 39.Wong M, Tee AEL, Milazzo G, Bell JL, Poulos RC, Atmadibrata B, Sun YT, et al. The Histone Methyltransferase DOT1L Promotes Neuroblastoma by Regulating Gene Transcription. Cancer Research 2017;77:2522–2533. [DOI] [PubMed] [Google Scholar]
- 40.Hua KT, Wang MY, Chen MW, Wei LH, Chen CK, Ko CH, Jeng YM, et al. The H3K9 methyltransferase G9a is a marker of aggressive ovarian cancer that promotes peritoneal metastasis. Molecular Cancer 2014;13:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mayr C, Helm K, Jakab M, Ritter M, Shrestha R, Makaju R, Wagner A, et al. The histone methyltransferase G9a: a new therapeutic target in biliary tract cancer. Hum Pathol 2018;72:117–126. [DOI] [PubMed] [Google Scholar]
- 42.Zhang KQ, Wang JH, Yang L, Yuan YC, Tong TR, Wu J, Yun XW, et al. Targeting histone methyltransferase G9a inhibits growth and Wnt signaling pathway by epigenetically regulating HP1 alpha and APC2 gene expression in non-small cell lung cancer. Molecular Cancer 2018;17:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang YF, Zhang J, Su Y, Shen YY, Jiang DX, Hou YY, Geng MY, et al. G9a regulates breast cancer growth by modulating iron homeostasis through the repression of ferroxidase hephaestin. Nat Commun 2017;8:274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Furth N, Ben-Moshe NB, Pozniak Y, Porat Z, Geiger T, Domany E, Aylon Y, et al. Down-regulation of LATS kinases alters p53 to promote cell migration. Genes Dev 2015;29:2325–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yi J, Lu L, Yanger K, Wang WQ, Sohn BH, Stanger BZ, Zhang M, et al. Large tumor suppressor homologs 1 and 2 regulate mouse liver progenitor cell proliferation and maturation through antagonism of the coactivators YAP and TAZ. Hepatology 2016;64:1757–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moroishi T, Hansen CG, Guan KL. The emerging roles of YAP and TAZ in cancer. Nature Reviews Cancer 2015;15:73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rathert P, Dhayalan A, Murakami M, Zhang X, Tamas R, Jurkowska R, Komatsu Y, et al. Protein lysine methyltransferase G9a acts on non-histone targets. Nature Chemical Biology 2008;4:344–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choi J, Jang H, Kim H, Lee JH, Kim ST, Cho EJ, Youn HD. Modulation of lysine methylation in myocyte enhancer factor 2 during skeletal muscle cell differentiation. Nucleic Acids Research 2014;42:224–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang X, Peng D, Xi Y, Yuan C, Sagum CA, Klein BJ, Tanaka K, et al. G9a-mediated methylation of ERalpha links the PHF20/MOF histone acetyltransferase complex to hormonal gene expression. Nat Commun 2016;7:10810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bittencourt D, Wu DY, Jeong KW, Gerke DS, Herviou L, Ianculescu I, Chodankar R, et al. G9a functions as a molecular scaffold for assembly of transcriptional coactivators on a subset of Glucocorticoid Receptor target genes. Proc Natl Acad Sci U S A 2012;109:19673–19678. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.