

Abstract
Cerebrovascular endothelial cells (CECs) are integral components of both the blood-brain barrier (BBB) and the neurovascular unit (NVU). As the primary cell type of the BBB, CECs are responsible for the tight regulation of molecular transport between the brain parenchyma and the periphery. Additionally, CECs are essential in neurovascular coupling where they help regulate cerebral blood flow in response to regional increases in cellular demand in the NVU. CEC dysfunction occurs during both normative ageing and in cerebrovascular disease, which leads to increased BBB permeability and neurovascular uncoupling. This MiniReview compiles what is known about the molecular changes underlying CEC dysfunction, many of which are reminiscent of cells that have become senescent. In general, cellular senescence is defined as an irreversible growth arrest characterized by the acquisition of a pro-inflammatory secretory phenotype in response to DNA damage or other cellular stresses. We discuss evidence for endothelial cell senescence in ageing and cardiovascular disease, and how CEC senescence may contribute to age-related cerebrovascular dysfunction.
Keywords: Alzheimer’s disease, cellular senescence, endothelial cells, neurodegenerative disorders, neuropharmacology
1 |. INTRODUCTION
The blood-brain barrier (BBB) is the physical regulator of molecular transport between the brain parenchyma and the periphery. It is imperative that the function of the BBB is intricately controlled, as it regulates proper nutrient delivery and waste removal to maintain homeostasis. Increased permeability of the BBB is neurotoxic and has been implicated in normative cognitive ageing and in clinically diagnosed neurological deficits such as mild cognitive impairment and neurodegeneration.1–6 The neurovascular unit (NVU) is composed of a network of cerebrovascular endothelial cells (CECs), pericytes, smooth muscle cells, astrocytes and neurons working together to regulate cerebral blood flow (CBF). Alterations in CBF occur in response to cellular demand through a process known as “neurovascular coupling.” Global decreases in CBF as well as neurovascular uncoupling are hallmarks of both normal ageing and disease-associated neurodegeneration.7–9 Importantly, CEC dysfunction alone is sufficient to promote BBB disruption and neurovascular uncoupling.10 Thus, improved understanding of the molecular mechanisms capable of promoting CEC dysfunction will inform therapeutic approaches for age-related changes in the CNS and cerebrovascular diseases such as vascular dementias and Alzheimer’s disease.
Endothelial cell dysfunction has been heavily studied in cardiac ageing and atherosclerosis and has been reviewed extensively elsewhere.11,12 However, CEC dysfunction in brain ageing and cerebrovascular disease is much less well-characterized. This MiniReview compiles what is currently known about age-related changes to endothelial cells specifically in the brain and how those changes may first impair CEC function and ultimately, promote cerebrovascular dysfunction. Furthermore, we will discuss the potential role of cellular senescence in driving CEC dysfunction and cerebrovascular ageing.
Cellular senescence is characterized as a state of irreversible cell cycle arrest with a distinctive, pro-inflammatory phenotype, commonly referred to as the senescence-associated secretory phenotype (SASP). The SASP is thought to consist of various cytokines, chemokines and matrix metalloproteinases (MMPs) that have the potential to influence cells in their neighbouring environment.13 It has now been well-established that senescent cells play a causative role in a variety of age-related diseases, including neurodegenerative disease.14,15 Additionally, endothelial cell senescence promotes dysfunction underlying cardiovascular disease.16 Hypothetically, CEC senescence could have major implications in cerebrovascular disease, including vascular dementia by driving hypoperfusion, hypoxia, ischaemia and stroke.
2 |. CEC DYSFUNCTION DRIVES CEREBROVASCULAR AGEING
BBB disruption and neurovascular uncoupling are hallmarks of cognitive decline in the elderly and people with cerebrovascular disease. Because CECs form the core of both the BBB and the NVU responsible for proper neurovascular coupling, it is probable that CEC dysfunction is a necessary component for these alterations. One way to gain a deeper understanding for how CEC dysfunction itself drives cerebrovascular dysfunction would be to better understand the cellular and molecular changes that occur in CECs in both normative ageing and disease (Figure 1).
FIGURE 1.
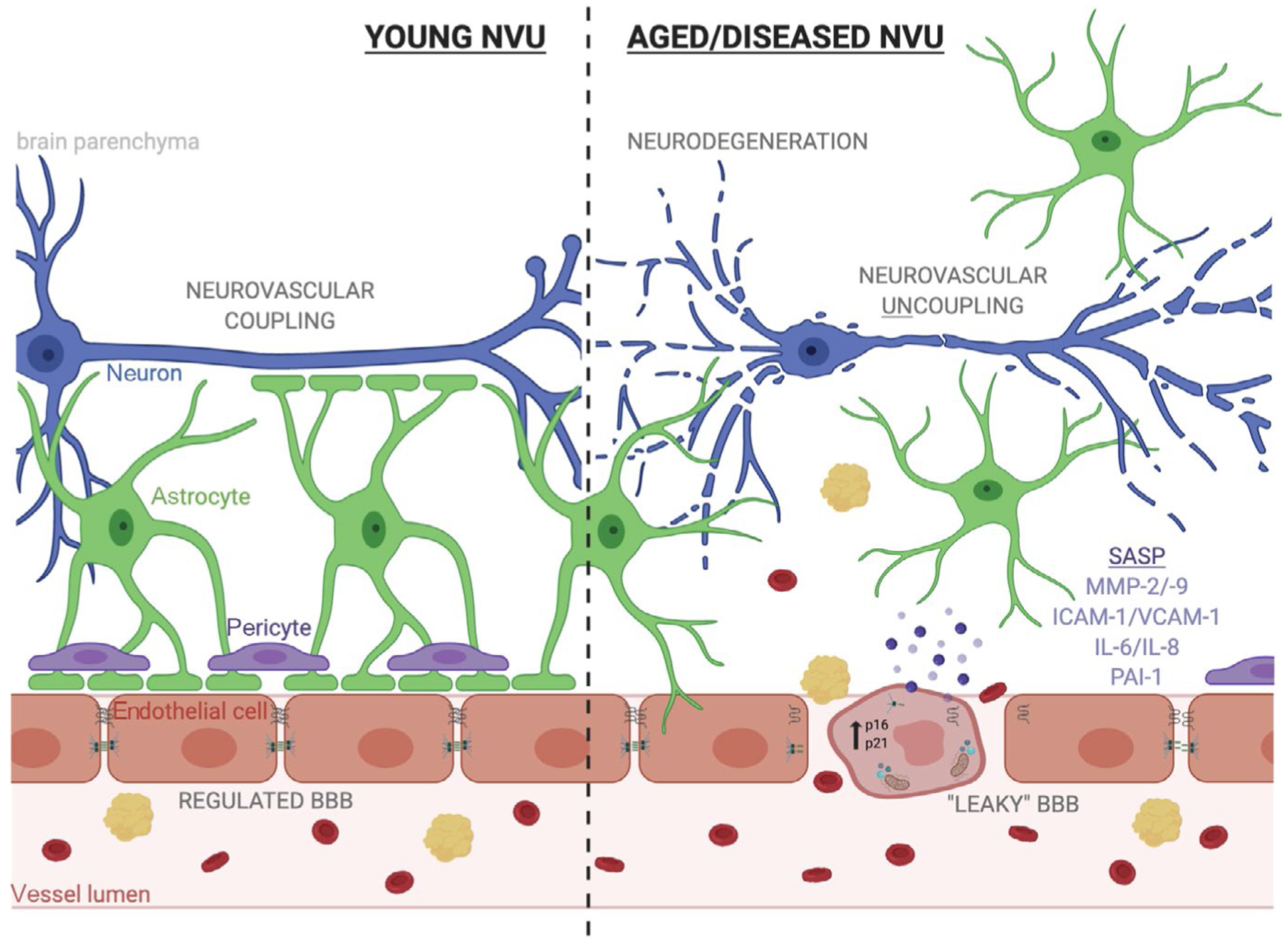
Endothelial cell senescence could drive blood-brain barrier (BBB) disruption, neurovascular uncoupling and neurodegeneration in ageing and disease. The neurovascular unit (NVU) is composed of endothelial cells, astrocytes, pericytes, and neurons which function cooperatively to regulate molecular transport from the periphery to the brain in order to maintain neuronal homeostasis and respond to changes in neuronal energy demands. Aging can induce changes in endothelial cells that disrupt the NVU’s role in the BBB and neurovascular coupling. These changes to endothelial cells are reminiscent of cellular senescence and include upregulation of cyclin-dependent kinase inhibitors, acquisition of a pro-inflammatory and degradative senescence-associated secretory phenotype (SASP), increased oxidative stress, and deregulated tight junctions. In this way, endothelial cell senescence along with age-related loss of pericytic coverage of vessels can lead to increased extravasation of red blood cells and other neurotoxic proteins. This BBB disruption causes an imbalance in the local microenvironment and can drive neuronal dysfunction. Additionally, age-related increases in astrocytic reactivity result in loss of astrocytic endfoot coverage and break the communication between neurons and the endothelium, termed “neurovascular uncoupling”, and also drives neurodegeneration
2.1 |. CEC changes underlying dysfunction
Changes to endothelial nitric oxide synthase (eNOS)/nitric oxide (NO) signalling, tight junctions, angiogenesis and neuroinflammation in CECs have the potential to drive cerebrovascular dysfunction. NO bioavailability is a crucial regulator of normal CEC function and regulates CBF in response to changes in cellular energy demands. Decreased NO bioavailability, via decreased synthesis or accumulation of reactive oxygen species (ROS), leads to ineffective CBF regulation, and hypoperfusion of the brain, and ultimately contributes to neuronal cell death and cognitive dysfunction (Figure 2).
FIGURE 2.
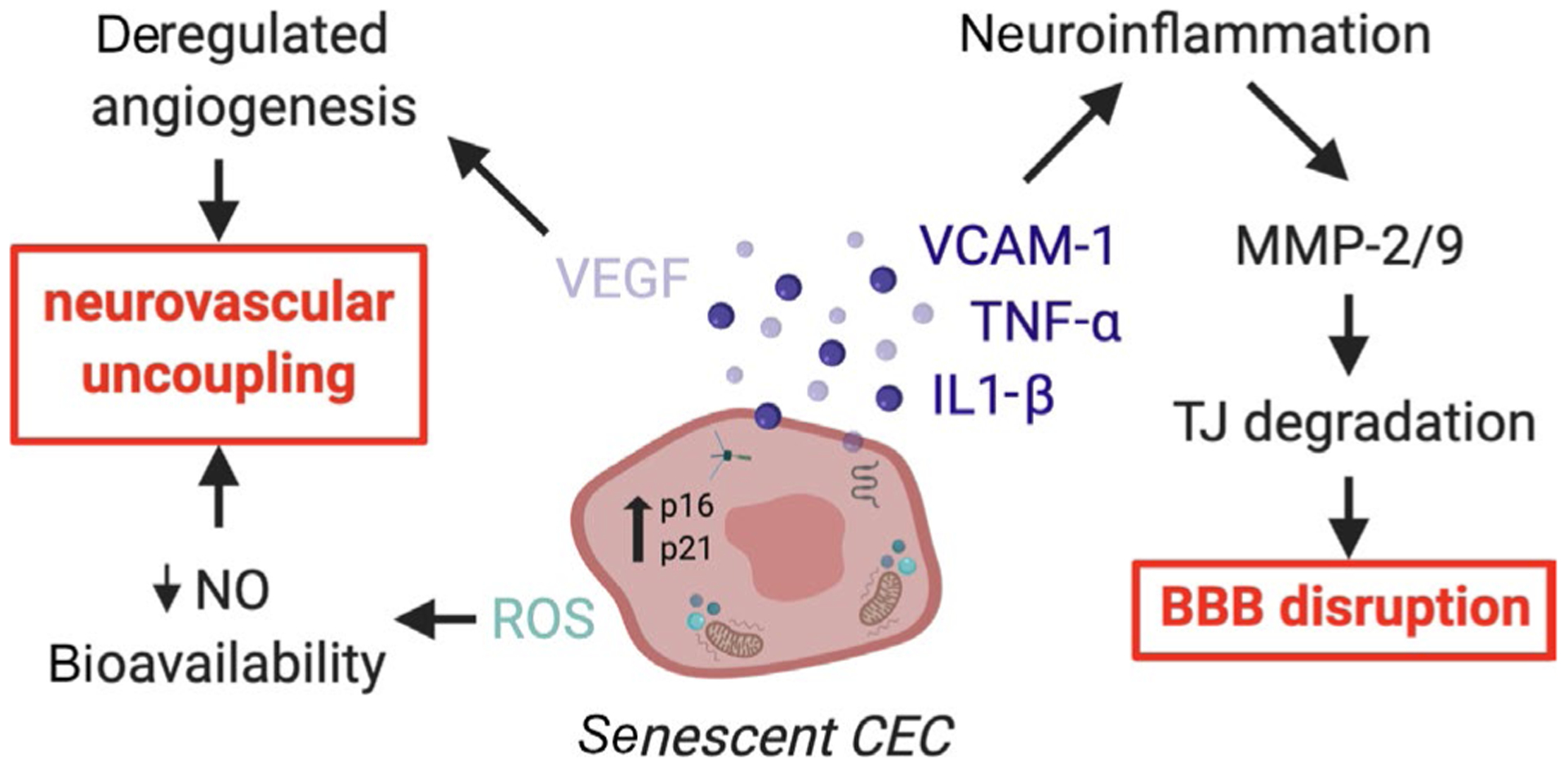
Proposed mechanisms for how senescent CECs affect the BBB and NVU. Senescent cerebrovascular endothelial cells (CECs) are hypothesized to accumulate with aging and cerebrovascular diseases like dementia by affecting neurovascular coupling and the blood-brain barrier (BBB). In general, senescent cells secrete pro-inflammatory molecules like VCAM-1, TNFα, and IL-1β, which stimulate greater neuroinflammation. Thus, one mechanism by which senescent CECs may induce BBB disruption is by stimulating chronic states of neuroinflammation and activating cytokine-inducible matrix metalloproteases (MMPs) that directly degrade tight junction (TJ) proteins. Senescent CECs may also promote neurovascular uncoupling via de-regulated VEGF/angiogenesis and/or increased reactive oxygen species (ROS)/decreased nitric oxide (NO) axes
NO is synthesized by one of three isoforms of nitric oxide synthase (NOS), neuronal NOS (nNOS), cytokine-inducible NOS (iNOS) and eNOS. eNOS is the primary producer of NO implicated in CBF regulation, and eNOS/NO signalling dysregulation is heavily implicated in ageing and age-related cerebrovascular disease in both humans and rodents.17,18 Reduced activity of eNOS leads to decreased NO production and can occur via transcriptional down-regulation, increased expression of its inhibitors ADMA and/or caveolin-1 (CAV-1), or uncoupling from its cofactor tetrahydrobiopterin (BH4).19 Age-associated increases in CAV-1 would be expected to decrease NO bioavailability by sequestering eNOS in its inactive form and contribute to NO-dependent dysfunction.20,21 However, some studies in rat models of cerebral ischaemia suggest that CAV-1 reduction is capable of inducing NO-dependent BBB dysfunction and neuroinflammation.22,23 Accumulation of ROS occurs with ageing and can further decrease NO bioavailability as NO reacts with O22- to create peroxynitrate, which is a reactive nitrogen species that can contribute to CEC dysfunction. ROS can also oxidize BH4 to BH2 and contribute to eNOS/BH4 decoupling to further decrease NO bioavailability.19
Changes to the tight junctions that exist between CECs cause increased paracellular transport across the BBB. Several studies in mice have implicated loss or redistribution of tight junction proteins, including occludin, claudin-5 and zonula occludens-1 (ZO-1; also known as tight junction protein 1 or Tjp1), in both normative ageing and accelerated ageing.24,25 The exact mechanisms by which tight junctional proteins are down-regulated in ageing are still unclear, but one potential mechanism may be via direct degradation by brain-associated MMPs. Increases specifically in MMP-2 and MMP-9, as well as decreases in the activity or expression of their inhibitors (tissue inhibitors of metalloproteinase or TIMPs), are linked to BBB dysfunction in both humans and rat models of ischaemic reperfusion injury.21–22,26 MMP-2 and MMP-9 activity is also increased in normally aged mice.27 Altogether, tight junction degradation via MMP-2/9 activation, which can be stimulated by various pro-inflammatory cytokines produced by activated endothelial cells, may be one mechanism by which BBB disruption occurs (Figure 2). Future studies testing this proposed mechanism of BBB disruption in brain ageing and disease contexts are imperative.
Decreased angiogenesis occurs with ageing and in cerebrovascular disease, which can lead to hypoperfusion, impaired adaptation to hypoxia, compromised recovery to tissue damage and exacerbated ischaemic tissue injury.28–31 Studies in normally aged rodents indicate that expression of certain angiogenic factors, such as vascular endothelial growth factor (VEGF) and HIF1α, decreases with age.28,31 Additionally, expression of brain-derived neurotrophic factor (BDNF) decreases with age.29 BDNF is a growth factor that is secreted by both neurons and endothelial cells and contributes to synaptic plasticity. More recently, BDNF has been implicated as a driver of endothelial cell differentiation,32 which may further impair angiogenesis in the ageing brain when down-regulated. Finally, age-related decreases in NO bioavailability can alter endothelial cell sensitivity to pro-angiogenic factors such as VEGF.33 Taken together, these results suggest that decreases in VEGF and angiogenesis may induce CEC dysfunction during normative ageing. However, the opposite may be true in injury or disease contexts, where several studies have shown that increased VEGF promotes angiogenesis and BBB disruption.34–36 A strict balance of VEGF appears to be necessary to maintain proper CEC function and brain homeostasis, and future studies should seek to define this balance in order to understand whether VEGF is a useful therapeutic target in CEC dysfunction associated with ageing or disease.
Interestingly, neuroinflammation can be driven simply due to age-related CEC changes. Recent work evidenced that the expression of vascular cell surface adhesion molecule 1 (VCAM-1) is increased in CECs of aged mice.37 Up-regulation of VCAM-1 in CECs promotes leucocyte recruitment and tethering to brain endothelium, and is hypothesized to ultimately drive glial cell activation, reduced neural precursor cell activity and cognitive decline37 (Figure 2). Additionally, soluble VCAM-1 can be shed from endothelial cells and indicates activated endothelium during neuroinflammation. This soluble form of VCAM-1 is up-regulated in human and mouse plasma with ageing37,38 and negatively correlates with cognition in elderly individuals,39 further supporting the notion that CEC activation may drive neuroinflammation and contribute to age-related cognitive decline. VCAM-1 and its association with activated CECs may also provide an interesting mechanistic link between dementia and diabetes, which are common co-morbidities.40 Similar to aged individuals, patients with diabetes have increased levels of soluble VCAM-1.41,42 Evidence suggests that high glucose levels alone are sufficient to induce VCAM-1 expression in human arterial endothelial cells (HAECs).43 Interestingly, BBB permeability induced memory loss has been seen in mouse models of type I and type II diabetes.44 Thus, one hypothetical mechanism by which diabetes could increase the risk of dementia is via hyperglycaemia-induced CEC activation which consequently drives neuroinflammation, BBB permeability and cognitive decline in a VCAM-1–dependent manner. Further studies are needed to test this hypothesis and identify which cell type (ie CECs or other peripheral endothelial cells) is the source of increased soluble VCAM-1 in patients with diabetes.
2.2 |. CEC dysfunction induces blood-brain barrier permeability and neurovascular uncoupling
Endothelial cells play a unique role in the brain compared with other tissues, where they maintain a strict barrier between peripheral circulation and the brain parenchyma through highly regulated tight junctional complexes that link the cells together and form the primary BBB. Alterations to the proteins that make up CEC tight junctions can give rise to increased paracellular transport across the BBB and contribute to the “leaky” BBB associated with ageing and cerebrovascular disease. This permeability is relevant, as it is a reliable predictor of cognitive decline in the elderly as evidenced by increased Ktrans values measured by dynamic contrast-enhanced MRI.4 Changes to tight junctions between CECs can occur due to intrinsic alterations, including down-regulation of proteins that make up tight junctions, such as claudins and occludins, or up-regulation of proteins known to rearrange tight junctions, such as phosphorylated myosin light chain.45,46 Additionally, extrinsic alterations, such as increased MMPs or decreased TIMPs in the extracellular microenvironment, can alter CEC tight junctions. CEC changes contributing to BBB defects are observed during natural ageing and in various neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, Huntington’s Disease, multiple sclerosis, frontotemporal lobular degeneration and α-synucleinopathy.4,6,47 One study in mice showed that BBB defects may actually drive neuropathology because correction of BBB leakiness reduced neuroinflammation and paralysis.48
In addition to driving BBB defects, CEC dysfunction can lead to deregulated neurovascular coupling. Neurovascular coupling is the process by which cells of the NVU co-ordinate to regionally increase CBF in response to high neuronal activity. Neurovascular uncoupling occurs when the CBF response is muted or non-existent, which occurs with normal ageing in both humans and rodents.49–52 This phenomenon has been shown to be related to disruptions in eNOS/NO signalling. eNOS knockout mice exhibit a decreased CBF response after whisker stimulation, a common experimental measure of neurovascular uncoupling, which is rescued with overexpression of eNOS.53 In this example, it is likely that decreased eNOS activity leads to decreased NO bio-availability and consequently, decreased NO-dependent CBF autoregulation. In contrast, others have reported an increased CBF response to whisker stimulation in eNOS knockout mice.54 Clearly, further characterization of the role of dysregulated eNOS in age-dependent neurovascular uncoupling is required.
In addition to CBF autoregulation, CECs also provide a neuroprotective role in response to injury by up-regulating expression of certain growth factors such as BDNF and VEGF to promote neural stem cell differentiation.32 BDNF levels are reduced in post-mortem samples from patients with Alzheimer’s disease and may contribute to deficiencies in both neurogenesis and angiogenesis.55 In a rat model of vascular dementia, rescue of BDNF deficiency restored cognitive function,56,57 suggesting that CEC down-regulation of BDNF may contribute to cognitive decline. Furthermore, BDNF expression is eNOS-dependent and therefore may be dysregulated during normative ageing.
3 |. ENDOTHELIAL CELL SENESCENCE AS A MECHANISM UNDERLYING ENDOTHELIAL DYSFUNCTION
Endothelial cell dysfunction occurs with age and contributes to age-related disease, but the mechanisms behind this impaired function are not well-characterized. Endothelial cell senescence is one mechanism by which endothelial cells may become dysfunctional with age and in certain age-related pathologies. Cellular senescence is characterized as a state of irreversible cell cycle arrest with a distinctive, pro-inflammatory senescence-associated secretory phenotype (SASP) and increased senescence-associated β-galactosidase (SA-β-gal) activity. Senescent cell burden increases with age and in sites of pathology.58–61 A handful of studies have provided evidence of cell cycle arrest and secretion of SASP members specifically in endothelial cells during normal ageing and some age-related pathologies such as atherosclerosis.62,63 Endothelial cell senescence in the brain has yet to be well-defined, but may explain much of the CEC dysfunction in age-related cerebrovascular diseases such as dementia.
3.1 |. Endothelial cell senescence in normal ageing
Stable cell cycle arrest is a distinguishing hallmark of cellular senescence and is marked by up-regulation of certain cell cycle regulators, namely p16, p21 and p53. Increased p16, p21 and p53 expression in endothelial cells of brachial arteries in healthy older individuals (aged 50–70 years), compared with younger individuals (aged 18–30 years is correlated with decreased brachial artery dilation in response to shear stress.64 Studies in mice have also linked ageing-associated increases in p16 and p21 expression to decreased endothelium-dependent vasodilation in response to acetylcholine and NO, two endogenous vasodilators.62 Long-term clearance of senescent cells reduced vasomotor dysfunction in the aortas of normally aged and atherosclerotic mice,65 linking cellular senescence causally to endothelium dysfunction, although a direct demonstration of endothelial cell–specific senescence was lacking.
Other clues into endothelial cell senescence come from studies implicating increases in endothelium-derived members of the SASP in ageing. The SASP is another major hallmark of cellular senescence. With ageing, endothelial cells shift to a more pro-inflammatory phenotype, as evidenced by up-regulation of the NF-κB pathway in endothelial cells collected from aged individuals.17 Increased NF-κB activity has also been linked to increased TNF-α, IL-1β, IL-6 and IL-17 expression in aged rat coronary arteries66 and aged mouse aortas.67 Additionally, human umbilical vein endothelial cells (HUVECs) cultured to replicative senescence have increased NF-κB signalling, ROS production and expression of a number of SASP members including E-selectin, ICAM-1, PAI-1, IGFBP-5, IL-6 and IL-8.68 Altogether, evidence is emerging to support the capacity of endothelial cells to become senescent, and consequently dysfunctional, with age. However, members of the SASP are not specific to cellular senescence and instead may be up-regulated in aged endothelial cells due to senescence-and ageing-independent factors such as oxidative stress, inflammation/activation, dyslipidaemia/atherogenesis and/or low shear stress. Thus, more comprehensive studies are needed to truly define endothelial cell senescence as a mechanism for age-related endothelium dysfunction.
A variety of mechanisms may contribute to endothelial cell senescence during normative ageing, including oxidative stress, decreased sirt1 expression and increased NF-κB signalling. Vasodilation defects in the vasculature of aged mice can be improved after antioxidant treatment, implicating the importance of oxidative stress in endothelial cell vascular dysfunction.62 Sirt1, a histone deacetylase that can delay cellular senescence through activation of several pro-survival pathways,69 is decreased in senescent HUVECs and human atherosclerotic plaques that display other features of senescence.70,71 Concordantly, Sirt1 overexpression in HUVECs and human coronary aortic endothelial cells (HCAECs) protects against endothelial cell senescence.71,72 Sirt1 overexpression in aged mice reduces the expression of Pai-1, an endothelium-derived regulator of thrombosis and important SASP factor that appears necessary for p53-mediated cellular senescence,73 in aortic endothelial cells, and rescues acetylcholine-induced aortic relaxation.70 Similarly, pharmacological activation of Sirt1 in aged mice ameliorates vascular endothelial cell dysfunction, normalizes aortic superoxide production and decreases NF-κB–related inflammation.74 Future studies should further explore the mechanisms by which normative ageing induces endothelial cell senescence and whether such mechanisms can be targeted therapeutically to reverse endothelium dysfunction.
3.2 |. Endothelial cell senescence in age-associated disorders
Relatively few studies have directly implicated endothelial cell senescence in age-related disease. Patients with ischaemic heart disease have increased markers of senescence in their coronary arteries, including increased SA-β-gal activity and expression of certain SASP factors such as TNF-α, IL-8 and IL-6.63,75 In a mouse model of atherosclerosis, plaque burden correlated with the expression of several senescence markers such as p21 and Pai-1.75 Increased MEF2A expression and decreased angiopoietin-2 expression are protective against senescence phenotypes in patients with cardiovascular disease.75,76
Additionally, endothelial cell senescence has been implicated in heart failure. Senescence-accelerated mice display features of endothelial cell senescence during heart failure induced by long-term high-fat and high-salt diet.77 The expression of p53 in cardiac endothelial cells increases after cardiac injury in mice, and depletion of p53 in endothelial cells after cardiac injury leads to improved cardiac function, increased angiogenesis and decreased fibrosis.78,79 Increased expression of p53 is thought to drive up-regulation of ICAM-1 and VCAM-1, thus promoting immune cell recruitment and infiltration after cardiac injury and ultimately resulting in cardiac dysfunction.80,81 While p53 is associated with cellular senescence, it is more widely recognized as a classic tumour suppressor gene with important roles in inducing cell cycle arrest, DNA repair and programmed cell death. Therefore, more studies are needed to adequately characterize endothelial cell senescence, beyond just p53 expression, in heart failure.
3.3 |. Endothelial cell senescence in the ageing brain
Limited evidence for CEC senescence exists, but it could explain many of the endothelial dysfunction phenotypes seen in ageing and cerebrovascular disease, including both leaky BBB and neurovascular uncoupling. HUVECs cultured to replicative senescence down-regulate tight junctional proteins and have compromised barrier function in culture,82 potentially implicating endothelial cell senescence in BBB dysfunction. Two studies evidenced increased leakiness of the BBB in a mouse model of accelerated senescence.83,84 Another study in the BubR1 hypomorphic mouse model of premature ageing58,60 found increased BBB leakiness in conjunction with markers of senescence in CECs.24
Clearly, additional investigations into the possible contribution of cellular senescence to endothelial cell dysfunction, both within and outside the CNS, are necessary. One can postulate on many potential mechanisms by which these changes could influence age-related cerebrovascular disease (Figure 2). For example, several established SASP factors are unregulated in CECs during ageing and cerebrovascular disease. Pro-inflammatory SASP factors such as IL-6 and IL-1β could induce a leaky BBB, which would be conducive to peripheral immune cell infiltration and inflammation. This could then drive ROS and eNOS/NO signalling defects and further contribute to CEC dysfunction, BBB disruption and neurovascular uncoupling. VEGF is an important SASP factor whose dysregulation is known to disrupt eNOS/NO signalling, which contributes to CEC dysfunction in age-related disease. Other proteases and protease-regulators such as MMPs and TIMPs that are often altered in senescent cells could exert detrimental effects on tight junctional integrity between CECs and ultimately induce blood-brain barrier leakiness. ICAM-1 is also up-regulated in senescent cells and is known to promote neuroinflammation.
Given how senescent CECs may drive cognitive decline in a myriad of ways, they are potentially relevant therapeutic targets in ageing and cerebrovascular disease. Pharmacological agents, termed “senolytics,” have been developed to eliminate senescent cells in a targeted manner. Application of these first-generation pharmacological agents in mouse models of ageing and cerebrovascular disease in well-defined studies would help causally link CEC senescence to cognitive decline. However, there are obvious limitations in such an approach because such agents lack cell- and tissue-type specificity. In theory, there would not be targetable differences in senescent CECs versus senescent endothelial cells in other tissues, and thus, such studies would need to be well-designed to truly tease apart the effects of CEC senescence versus peripheral senescence in an ageing context. Additionally, this review focused on how CEC senescence may drive cognitive decline via increased BBB disruption and neurovascular uncoupling in chronic ageing contexts, but it is even less clear what role these disturbances may have in cognitive decline associated with acute injury, such as stroke or traumatic brain injury. Looking into how BBB disruption and neurovascular uncoupling occur in such acute contexts may better inform future studies aimed at understanding the role of CEC senescence in more gradual cognitive decline.
4 |. CONCLUSION
Overall, many in vitro studies support the concept that endothelial cells may become senescent, but few in vivo studies exist to connect endothelial cell senescence directly to ageing and/or age-related disease, especially in the brain. Many age-related changes in the cerebrovascular endothelial cell are reminiscent of senescence, but more direct studies need to be done to concretely establish CEC senescence as a driver of CEC dysfunction in both normal ageing and cerebrovascular disease. The use of senolytics in an in vivo model of cerebrovascular disease to clear senescent CECs and monitor effects on disease pathology could provide missing correlative link. Once linked, inducing senescence in CECs in an in vivo context and noting cerebrovascular pathology would provide strong evidence for CEC senescence as a driving force in cerebrovascular dysfunction.
Funding information
Glenn Foundation for Medical Research; Cure Alzheimer’s Fund; Ellison Medical Foundation; National Institutes of Health, Grant/Award Number: AG53229
REFERENCES
- 1.Arvanitakis Z, Capuano AW, Leurgans SE, Bennett DA, Schneider JA. Relation of cerebral vessel disease to Alzheimer’s disease dementia and cognitive function in elderly people: a cross-sectional study. Lancet Neurol. 2016;15(9):934–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Farrall AJ, Wardlaw JM. Blood–brain barrier: ageing and microvascular disease – systematic review and meta-analysis. Neurobiol Aging. 2009;30(3):337–352. [DOI] [PubMed] [Google Scholar]
- 3.Iturria-Medina Y, Sotero RC, Toussaint PJ, Mateos-Pérez JM, Evans AC. Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat Commun. 2016;7(1):11934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Montagne A, Samuel M, Matthew A, et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85(2):296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nation DA, Sweeney MD, Montagne A, et al. Blood–brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med. 2019;25(2):270–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toledo JB, Arnold SE, Raible K, et al. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s Coordinating Centre. Brain. 2013;136(9):2697–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Eulate RG, Goñi I, Galiano A, et al. Reduced cerebral blood flow in mild cognitive impairment assessed using phase-contrast MRI. J Alzheimers Dis. 2017;58(2):585–595. [DOI] [PubMed] [Google Scholar]
- 8.Leijenaar JF, Van Maurik IS, Kuijer JPA, et al. Lower cerebral blood flow in subjects with Alzheimer’s dementia, mild cognitive impairment, and subjective cognitive decline using two-dimensional phase-contrast magnetic resonance imaging. Alzheimer’s Dement (Amst). 2017;9:76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu H, Xu F, Rodrigue KM, et al. Alterations in cerebral metabolic rate and blood supply across the adult lifespan. Cereb Cortex. 2011;21(6):1426–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jia G, Aroor AR, Jia C, Sowers JR. Endothelial cell senescence in aging-related vascular dysfunction. Biochim Biophys Acta. 2019;1865(7):1802–1809. [DOI] [PubMed] [Google Scholar]
- 11.Gimbrone MA, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118(4):620–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ungvari Z, Tarantini S, Kiss T, et al. Endothelial dysfunction and angiogenesis impairment in the ageing vasculature. Nat Rev Cardiol. 2018;15(9):555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coppé J-P, Desprez P-Y, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5(1):99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baker DJ, Petersen RC. Cellular senescence in brain aging and neurodegenerative diseases: evidence and perspectives. J Clin Invest. 2018;128(4):1208–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Childs BG, Durik M, Baker DJ, Van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. 2015;21(12):1424–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Childs BG, Li H, Van Deursen JM. Senescent cells: a therapeutic target for cardiovascular disease. J Clin Invest. 2018;128(4):1217–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Donato AJ, Eskurza I, Silver AE, et al. Direct evidence of endothelial oxidative stress with aging in humans: relation to impaired endothelium-dependent dilation and upregulation of nuclear factor-kappaB. Circ Res. 2007;100(11):1659–1666. [DOI] [PubMed] [Google Scholar]
- 18.Yang Y-M, Huang A, Kaley G, Sun D. eNOS uncoupling and endothelial dysfunction in aged vessels. Am J Physiol-Heart Circu Physiol. 2009;297(5):H1829–H1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vanhoutte PM, Zhao Y, Xu A, Leung SWS. Thirty years of saying NO. Circ Res. 2016;119(2):375–396. [DOI] [PubMed] [Google Scholar]
- 20.Gratton J-P, Fontana J, O’Connor DS, Garcia-Cardena G, McCabe TJ, Sessa WC. Reconstitution of an Endothelial Nitric-oxide Synthase (eNOS), hsp90, and Caveolin-1 Complexin Vitro. J Biol Chem. 2000;275(29):22268–22272. [DOI] [PubMed] [Google Scholar]
- 21.Liu J, Jin X, Liu KJ, Liu W. Matrix metalloproteinase-2-mediated occludin degradation and caveolin-1-mediated claudin-5 redistribution contribute to blood-brain barrier damage in early ischemic stroke stage. J Neurosci. 2012;32(9):3044–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gu Y, Zheng G, Xu M, et al. Caveolin-1 regulates nitric oxide-mediated matrix metalloproteinases activity and blood-brain barrier permeability in focal cerebral ischemia and reperfusion injury. J Neurochem. 2012;120(1):147–156. [DOI] [PubMed] [Google Scholar]
- 23.Jasmin JF, Malhotra S, Singh Dhallu M, Mercier I, Rosenbaum DM, Lisanti MP. Caveolin-1 deficiency increases cerebral ischemic injury. Circ Res. 2007;100(5):721–729. [DOI] [PubMed] [Google Scholar]
- 24.Yamazaki Y, Baker DJ, Tachibana M, et al. Vascular cell senescence contributes to blood-brain barrier breakdown. Stroke. 2016;47(4):1068–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elahy M, Jackaman C, Mamo JC, et al. Blood-brain barrier dysfunction developed during normal aging is associated with inflammation and loss of tight junctions but not with leukocyte recruitment. Immun Ageing. 2015;12:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barr TL, Latour LL, Lee K-Y, et al. Blood-brain barrier disruption in humans is independently associated with increased matrix metalloproteinase-9. Stroke. 2010;41(3):e123–e128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee P, Kim J, Williams R, et al. Effects of aging on blood brain barrier and matrix metalloproteases following controlled cortical impact in mice. Exp Neurol. 2012;234(1):50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ingraham JP, Forbes ME, Riddle DR, Sonntag WE. Aging reduces hypoxia-induced microvascular growth in the rodent hippocampus. J Gerontol A Biol Sci Med Sci. 2008;63(1):12–20. [DOI] [PubMed] [Google Scholar]
- 29.Murugesan N, Demarest TG, Madri JA, Pachter JS. Brain regional angiogenic potential at the neurovascular unit during normal aging. Neurobiol Aging. 2012;33(5):1004.e1–1004.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bach MH, Sadoun E, Reed MJ. Defects in activation of nitric oxide synthases occur during delayed angiogenesis in aging. Mech Ageing Dev. 2005;126(4):467–473. [DOI] [PubMed] [Google Scholar]
- 31.Benderro GF, Lamanna JC. Hypoxia-induced angiogenesis is delayed in aging mouse brain. Brain Res. 2011;1389:50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Descamps B, Saif J, Benest AV, et al. BDNF (Brain-Derived Neurotrophic Factor) promotes embryonic stem cells differentiation to endothelial cells via a molecular pathway, including MicroRNA-214, EZH2 (Enhancer of Zeste Homolog 2), and eNOS (Endothelial Nitric Oxide Synthase). Arterioscler Thromb Vasc Biol. 2018;38(9):2117–2125. [DOI] [PubMed] [Google Scholar]
- 33.Ungvari Z, Tucsek Z, Sosnowska D, et al. Aging-induced dysregulation of dicer1-dependent MicroRNA expression impairs angiogenic capacity of rat cerebromicrovascular endothelial cells. J Gerontol A Biol Sci Med Sci. 2013;68(8):877–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nag S, Manias J, Eubanks JH, Stewart DJ. Increased expression of vascular endothelial growth factor-D following brain injury. Int J Mol Sci. 2019;20(7):1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang HT, Zhang P, Gao Y, et al. Early VEGF inhibition attenuates blood-brain barrier disruption in ischemic rat brains by regulating the expression of MMPs. Mol Med Rep. 2017;15(1):57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang ZG, Zhang L, Jiang Q, et al. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J Clin Invest. 2000;106(7):829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yousef H, Czupalla CJ, Lee D, et al. Aged blood impairs hippo-campal neural precursor activity and activates microglia via brain endothelial cell VCAM1. Nat Med. 2019;25(6):988–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zuliani G, Cavalieri M, Galvani M, et al. Markers of endothelial dysfunction in older subjects with late onset Alzheimer’s disease or vascular dementia. J Neurol Sci. 2008;272(1–2):164–170. [DOI] [PubMed] [Google Scholar]
- 39.Tchalla AE, Wellenius GA, Sorond FA, et al. Elevated soluble vascular cell adhesion molecule-1 is associated with cerebrovascular resistance and cognitive function. J Gerontol A Biol Sci Med Sci. 2017;72(4):560–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chatterjee S, Peters SA, Woodward M, et al. Type 2 diabetes as a risk factor for dementia in women compared with men: a pooled analysis of 2.3 million people comprising more than 100,000 cases of dementia. Diabetes Care. 2016;39(2):300–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fasching P, Veitl M, Rohac M, et al. Elevated concentrations of circulating adhesion molecules and their association with microvascular complications in insulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1996;81(12):4313–4317. [DOI] [PubMed] [Google Scholar]
- 42.Meigs JB, Hu FB, Rifai N, Manson JE. Biomarkers of endothelial dysfunction and risk of type 2 diabetes mellitus. JAMA. 2004;291(16):1978–1986. [DOI] [PubMed] [Google Scholar]
- 43.Piga R, Naito Y, Kokura S, Handa O, Yoshikawa T. Short-term high glucose exposure induces monocyte-endothelial cells adhesion and transmigration by increasing VCAM-1 and MCP-1 expression in human aortic endothelial cells. Atherosclerosis. 2007;193(2):328–334. [DOI] [PubMed] [Google Scholar]
- 44.Rom S, Zuluaga-Ramirez V, Gajghate S, et al. Hyperglycemia-driven neuroinflammation compromises BBB leading to memory loss in both diabetes mellitus (DM) Type 1 and Type 2 mouse models. Mol Neurobiol. 2019;56(3):1883–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuhlmann CR, Tamaki R, Gamerdinger M, et al. Inhibition of the myosin light chain kinase prevents hypoxia-induced blood-brain barrier disruption. J Neurochem. 2007;102(2):501–507 [DOI] [PubMed] [Google Scholar]
- 46.Haorah J, Heilman D, Knipe B, et al. Ethanol-induced activation of myosin light chain kinase leads to dysfunction of tight junctions and blood-brain barrier compromise. Alcohol Clin Exp Res. 2005;29(6):999–1009. [DOI] [PubMed] [Google Scholar]
- 47.Sweeney MD, Sagare AP, Zlokovic BV. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14(3):133–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Argaw AT, Asp L, Zhang J, et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J Clin Invest. 2012;122(7):2454–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fabiani M, Gordon BA, Maclin EL, et al. Neurovascular coupling in normal aging: a combined optical, ERP and fMRI study. NeuroImage. 2014;85:592–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lourenco CF, Ledo A, Caetano M, Barbosa RM, Laranjinha J. Age-Dependent impairment of neurovascular and neurometabolic coupling in the hippocampus. Front Physiol. 2018;9:913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Balbi M, Ghosh M, Longden TA, et al. Dysfunction of mouse cerebral arteries during early aging. J Cereb Blood Flow Metab. 2015;35(9):1445–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Park L, Anrather J, Girouard H, Zhou P, Iadecola C. Nox2-derived reactive oxygen species mediate neurovascular dysregulation in the aging mouse brain. J Cereb Blood Flow Metab. 2007;27(12):1908–1918. [DOI] [PubMed] [Google Scholar]
- 53.Toth P, Tarantini S, Davila A, et al. Purinergic glio-endothelial coupling during neuronal activity: role of P2Y1 receptors and eNOS in functional hyperemia in the mouse somatosensory cortex. Am J Physiol-Heart Circ Physiol. 2015;309(11):H1837–H1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hariharan A, Jing Y, Collie ND, Zhang H, Liu P. Altered neurovascular coupling and brain arginine metabolism in endothelial nitric oxide synthase deficient mice. Nitric Oxide. 2019;87:60–72. [DOI] [PubMed] [Google Scholar]
- 55.Tanila H The role of BDNF in Alzheimer’s disease. Neurobiol Dis. 2017;97:114–118. [DOI] [PubMed] [Google Scholar]
- 56.Zhang N, Xing M, Wang Y, Tao H, Cheng Y. Repetitive transcranial magnetic stimulation enhances spatial learning and synaptic plasticity via the VEGF and BDNF-NMDAR pathways in a rat model of vascular dementia. Neuroscience. 2015;311:284–291. [DOI] [PubMed] [Google Scholar]
- 57.Chang J, Yao X, Zou H, et al. BDNF/PI3K/Akt and Nogo-A/RhoA/ROCK signaling pathways contribute to neurorestorative effect of Houshiheisan against cerebral ischemia injury in rats. J Ethnopharmacol. 2016;194:1032–1042. [DOI] [PubMed] [Google Scholar]
- 58.Baker DJ, Jeganathan KB, Cameron JD, et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet. 2004;36(7):744–749. [DOI] [PubMed] [Google Scholar]
- 59.Baker DJ, Wijshake T, Tchkonia T, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479(7372):232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baker DJ, Perez-Terzic C, Jin F, et al. Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat Cell Biol. 2008;10(7):825–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Baker DJ, Childs BG, Durik M, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016;530(7589):184–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bhayadia R, Schmidt BM, Melk A, Homme M. Senescence-induced oxidative stress causes endothelial dysfunction. J Gerontol A Biol Sci Med Sci. 2016;71(2):161–169. [DOI] [PubMed] [Google Scholar]
- 63.Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I. Endothelial cell senescence in human atherosclerosis. Circulation 2002;105(13):1541–1544. [DOI] [PubMed] [Google Scholar]
- 64.Rossman MJ, Kaplon RE, Hill SD, et al. Endothelial cell senescence with aging in healthy humans: prevention by habitual exercise and relation to vascular endothelial function. Am J Physiol Heart Circ Physiol. 2017;313(5):H890–H895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Roos CM, Zhang B, Palmer AK, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. 2016;15(5):973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Csiszar A, Ungvari Z, Koller A, Edwards JG, Kaley G. Aging-induced proinflammatory shift in cytokine expression profile in coronary arteries. Faseb J. 2003;17(9):1183–1185. [DOI] [PubMed] [Google Scholar]
- 67.Lesniewski LA, Durrant JR, Connell ML, et al. Aerobic exercise reverses arterial inflammation with aging in mice. Am J Physiol Heart Circ Physiol. 2011;301(3):H1025–H1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Khan SY, Awad EM, Oszwald A, et al. Premature senescence of endothelial cells upon chronic exposure to TNFα can be prevented by N-acetyl cysteine and plumericin. Sci Rep. 2017;7(1):39501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zu Y, Liu L, Lee MY, et al. SIRT1 promotes proliferation and prevents senescence through targeting LKB1 in primary porcine aortic endothelial cells. Circ Res. 2010;106(8):1384–1393. [DOI] [PubMed] [Google Scholar]
- 70.Wan YZ, Gao P, Zhou S, et al. SIRT1-mediated epigenetic downregulation of plasminogen activator inhibitor-1 prevents vascular endothelial replicative senescence. Aging Cell. 2014;13(5):890–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lin YJ, Zhen YZ, Wei J, Liu B, Yu ZY, Hu G. Effects of Rhein lysinate on H2O2-induced cellular senescence of human umbilical vascular endothelial cells. Acta Pharmacol Sin. 2011;32(10):1246–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim MY, Kang ES, Ham SA, et al. The PPARδ-mediated inhibition of angiotensin II-induced premature senescence in human endothelial cells is SIRT1-dependent. Biochem Pharmacol. 2012;84(12):1627–1634. [DOI] [PubMed] [Google Scholar]
- 73.Kortlever RM, Higgins PJ, Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol. 2006;8(8):877–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gano LB, Donato AJ, Pasha HM, Hearon CM Jr, Sindler AL, Seals DR. The SIRT1 activator SRT1720 reverses vascular endothelial dysfunction, excessive superoxide production, and inflammation with aging in mice. Am J Physiol Heart Circ Physiol. 2014;307(12):H1754–H1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Caland L, Labbe P, Mamarbachi M, et al. Knockdown of angiopoietin-like 2 induces clearance of vascular endothelial senescent cells by apoptosis, promotes endothelial repair and slows atherogenesis in mice. Aging. 2019;11(11):3832–3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu B, Wang L, Jiang W, et al. Myocyte enhancer factor 2A delays vascular endothelial cell senescence by activating the PI3K/p-Akt/SIRT1 pathway. Aging. 2019;11(11):3768–3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gevaert AB, Shakeri H, Leloup AJ, et al. Endothelial senescence contributes to heart failure with preserved ejection fraction in an aging mouse model. Circ Heart Fail. 2017;10(6):e003806. [DOI] [PubMed] [Google Scholar]
- 78.Yokoyama M, Shimizu I, Nagasawa A, et al. p53 plays a crucial role in endothelial dysfunction associated with hyperglycemia and ischemia. J Mol Cell Cardiol. 2019;129:105–117. [DOI] [PubMed] [Google Scholar]
- 79.Gogiraju R, Xu X, Bochenek ML, et al. Endothelial p53 deletion improves angiogenesis and prevents cardiac fibrosis and heart failure induced by pressure overload in mice. J Am Heart Assoc. 2015;4(2):e001770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhu C, Yu Y, Montani JP, Ming XF, Yang Z. Arginase-I enhances vascular endothelial inflammation and senescence through eNOS-uncoupling. BMC Res Notes. 2017;10(1):82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yoshida Y, Shimizu I, Katsuumi G, et al. p53-Induced inflammation exacerbates cardiac dysfunction during pressure overload. J Mol Cell Cardiol. 2015;85:183–198. [DOI] [PubMed] [Google Scholar]
- 82.Krouwer VJ, Hekking LH, Langelaar-Makkinje M, Regan-Klapisz E, Post JA. Endothelial cell senescence is associated with disrupted cell-cell junctions and increased monolayer permeability. Vasc Cell. 2012;4(1):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pelegri C, Canudas AM, del Valle J, et al. Increased permeability of blood-brain barrier on the hippocampus of a murine model of senescence. Mech Ageing Dev. 2007;128(9):522–528. [DOI] [PubMed] [Google Scholar]
- 84.Del Valle J, Duran-Vilaregut J, Manich G, et al. Time-course of blood-brain barrier disruption in senescence-accelerated mouse prone 8 (SAMP8) mice. Int J Dev Neurosci. 2009;27(1):47–52. [DOI] [PubMed] [Google Scholar]