

SUMMARY
Problems arising during translation of mRNAs lead to ribosome stalling and collisions that trigger a series of quality control events. However, the global cellular response to ribosome collisions has not been explored. Here, we uncover a function for ribosome collisions in signal transduction. Using translation elongation inhibitors and general cellular stress conditions, including amino acid starvation and UV irradiation, we show that ribosome collisions activate the SAPK (Stress Activated Protein Kinase) and GCN2-mediated stress response pathways. We show that the MAPKKK ZAK functions as the sentinel for ribosome collisions and is required for immediate early activation of both SAPK (p38/JNK) and GCN2 signaling pathways. Selective ribosome profiling and biochemistry demonstrate that while ZAK generally associates with elongating ribosomes on polysomal mRNAs, it specifically auto-phosphorylates on the minimal unit of colliding ribosomes, the disome. Together, these results provide molecular insights into how perturbation of translational homeostasis regulates cell fate.
In Brief
Cellular stress, such as amino acid starvation and UV irradiation, causes widespread ribosome collisions. Colliding ribosomes serve as a platform that recruits ZAKa and triggers two inter-related but distinct signaling pathways- the ribotoxic stress response and the integrated stress response- to regulate cell fate decisions.
Graphical Abstract
INTRODUCTION
Cellular stress triggers a wide range of responses, from activation of pathways that promote cell survival to initiation of programmed cell death. Typically, cellular stress responses first allow cells to adapt and recover from the specific stress; and if the stimulatory insult cannot be resolved, they initiate death signaling pathways. The capacity of the cell to mount an appropriate response to environmental or intracellular stress is critical for determining cell fate.
Stress Activated Protein Kinases (SAPKs) are conserved from yeast to mammals and are critical determinants of cell fate in response to multiple environmental insults (Duch et al., 2012; Kyriakis and Avruch, 2012). There are two SAPK families in mammals: p38 and c-Jun N- terminal kinase (JNK). Activation of p38 induces cell cycle arrest while activation of JNK promotes apoptosis (Darling and Cook, 2014; Duch et al., 2012). In a process referred to as the ribotoxic stress response (RSR), SAPK signaling through p38/JNK is robustly triggered on short time scales (minutes) by agents that target the ribosome to induce elongation distress, including anisomycin (ANS), emetine (EME), and sarcin/ricin (Iordanov et al., 1997). In addition to ribosome-targeting agents, other studies established that UV irradiation is also a potent activator of the RSR (Iordanov et al., 1998, 2002), and it was proposed that ribosomal RNA (rRNA) damage caused by UV irradiation is critical to RSR activation (Iordanov et al., 1998). Both ANS- and UV-stimulated activation of p38/JNK depend specifically on the mitogen-activated protein kinase kinase kinase (MAPKKK) ZAK (also known as MAPKKK20 or MLK7) (Jandhyala et al., 2008; Wang et al., 2005). However, molecular understanding of how ZAK monitors ribotoxic stress in the cell and activates the RSR remains unknown.
A common response of cells to diverse stresses is the repression of global translation initiation. Two commonly used mechanisms for reducing initiation include the phosphorylation of eIF2a by stress-specific kinases and the phosphorylation of 4E-BPs by mTORC1 (Sonenberg and Hinnebusch, 2009). Four different eIF2a kinases exist in mammals, each initiating the integrated stress response (ISR) to promote cell survival in response to distinct stress conditions - GCN2 by amino acid limitation, PKR by double-stranded RNAs during viral infection, PERK by accumulation of unfolded proteins in the endoplasmic reticulum (ER), and HRI by heme deprivation. While there are data indicating that GCN2 phosphorylates eIF2α through sensing uncharged tRNAs that accumulate during amino acid starvation (Dong et al., 2000; Wek et al., 1989), other studies show that GCN2 is activated by the isolated ribosomal P-stalk (Harding et al., 2019; Inglis et al., 2019; Jiménez-Díaz et al., 2013) and by ribosome stalling independent of uncharged tRNAs (Ishimura et al., 2016). The degree of GCN2 activation correlates with the severity of ribosome stalling induced by amino acid starvation (Darnell et al., 2018) consistent with a model wherein GCN2 directly monitors the global status of elongating ribosomes.
Ribosome collisions have been shown to be the key cellular trigger for ribosome- mediated quality control in eukaryotic cells that targets problematic mRNAs (Yan and Zaher, 2019). These pathways serve to minimize the toxic effects of incomplete peptide products generated from incomplete or problematic mRNAs; outputs of these pathways include targeting both the mRNA (by No-Go Decay, NGD) and the nascent peptide (by the Ribosome- mediated Quality control Complex, RQC) for decay (Brandman and Hegde, 2016). While these pathways effectively manage occasional problems during translation, large-scale perturbations likely overwhelm the quality control machinery which is typically sub-stoichiometric to ribosomes across cell types (Kulak et al., 2014).
Here, we bridge these ideas and identify elongating, colliding ribosomes as key communicators of cellular homeostasis, alerting cells to general stress and activating pathways to respond to this stress. More specifically, we show that activation of eIF2α and SAPKs (p38/JNK), as induced by standard translation elongation inhibitors such as ANS and EME, is triggered by colliding ribosomes that have overwhelmed the quality control machinery. Activation of these two distinct stress response pathways depends on the MAPKKK ZAK. ZAK associates normally with elongating, transiently colliding, ribosomes, but under broad cellular stress, auto-phosphorylates on abundant more stable colliding ribosomes and activates downstream SAPK signaling pathways. Finally, we show that standard physiological stressors, including amino acid starvation and UV irradiation, strongly activate these same signaling pathways.
RESULTS
Ribosome collisions activate SAPKs (p38/JNK)
While the RSR is known to be triggered by elongation distress during translation, the molecular mechanisms associated with activation have been obscure (Iordanov et al., 1997). In light of recent studies implicating ribosome collisions in mRNA-specific quality control (QC), we first asked whether the RSR is activated by generally slow elongating ribosomes or instead by ribosome collisions. Ribosome collisions can be broadly induced in cells using translation elongation inhibitors such as ANS and EME, as previously reported (Juszkiewicz et al., 2018; Simms et al., 2017). Here we established conditions for each compound where high concentrations of inhibitor caused uniform stalling of ribosomes on mRNAs, while intermediate concentrations stalled only a subset of ribosomes, leading to widespread ribosome collisions between stalled and trailing ribosomes (Figure 1A). We directly monitored ribosome collisions by treating cellular lysates with a non-specific RNase (RNase A) and resolved the resulting ribosome population on a sucrose gradient. While the majority of polysomes are collapsed to monosomes in all treatments, a minor fraction of ribosomes resists RNase treatment and sediments as disomes in unstressed cells (Figure 1B, black) (Arpat et al., 2019; Han et al., 2020; Wolin and Walter, 1988). Importantly, however, we observe the strongest accumulation of disomes at the intermediate (Figure 1B, orange) but not the high concentration (Figure 1B, grey) of ANS as compared to the unstressed sample. Similar titrations performed for EME, a translation elongation inhibitor targeting a distinct step in elongation, also reveal an increase in colliding ribosomes (Figure S1A). These data serve as a biochemical read out for the relative amount of colliding ribosomes under different cellular conditions.
Figure 1. Phosphorylation of SAPKs (p38/JNK) in response to ribosome collisions.

(A) Schematic of ribosome collisions on treatment with varying concentrations of elongation inhibitor: untreated (unt), intermediate (int), or high dose. (B) Polysome profiles from RNaseA- digested lysates of HEK293 cells treated with ANS at 0 (black), 1 (orange), or 100 mg/L (grey). (C) Immunoblots for phosphorylation of p38 and JNK in HEK293 cells treated with ANS (0.001–100 mg/L, 15min) compared to untreated. (D) p38 phosphorylation in HeLa cells treated with EME (0.048–48 mg/L, 15min). (E) p38 phosphorylation induced by intermediate doses of ANS (1 mg/L, 15min) in HEK293 cells with or without pretreating with high doses of EME (48 mg/L, 5min). (F) p38 phosphorylation in HeLa cells transfected with pcDNA5 (Vec), A0 or A60 reporter with schematic depicting the sequence features of the reporter.
We next titrated the elongation inhibitors to determine whether activation of the SAPK signaling pathway (monitored through the phosphorylation of p38 or JNK) was sensitive to concentration. Strikingly, maximal levels of p38/JNK phosphorylation are observed in HEK293 cells at intermediate but not at high concentrations of ANS (Figure 1C). Importantly, high concentrations of ANS neither reduce cell viability over the course of the experiment (15–30 min) (Figure S1B) nor interfere with p38/JNK phosphorylation activated through TNFa (Figure S1C), a cytokine that activates p38/JNK through a distinct mechanism (Kant et al., 2011). We observe the same trends of optimal activation of p38/JNK phosphorylation at intermediate doses of EME (Figure 1D) and in multiple cell lines (Figures S1D-E). As additional validation that actively elongating ribosomes are required for the accumulation of collisions, we find that pretreating cells with high-dose EME to uniformly stall all ribosomes prevents p38 phosphorylation by subsequent addition of an intermediate dose of ANS (Figure 1E).
Problematic mRNA sequences such as extended poly(A) stretches are also known to cause ribosome collisions (Chandrasekaran et al., 2019; Juszkiewicz et al., 2018; Tesina et al., 2020). We asked whether overexpression of collision-inducing reporter constructs with either no stalling sequence (A0) or a long poly(A) sequence (A60) would activate SAPK signaling. The reporter includes 2A skipping sequences that allow nascent GFP to escape degradation by the RQC and to be used to report on mRNA levels (Figure 1F, top). We observe increased levels of p38 phosphorylation in A60-transfected cells compared to either vector-only or A0-transfected cells (Figure 1F, bottom); the modest increase in p38 phosphorylation in A0-transfected cells likely results from ribosome collisions induced at the 2A skipping sequences that we document through ribosome profiling (Figure S1F). Together, these multiple lines of evidence support a model in which colliding ribosomes, and not slow individual elongating ribosomes, trigger the RSR in mammalian cells.
ZAK is required for ribosome collision-induced SAPK activation
A previous study identified the MAPKKK - ZAK - as the upstream kinase connecting ribotoxic stress to SAPK activation (Wang et al., 2005). While ZAK has two major isoforms, a and P, the isoform that contributes to the activation of the RSR remains unknown. We generated a ZAK knockout (KO) cell line using CRISPR/Cas9 and then complemented the KO line with either the a or P isoform cDNA. Consistent with earlier studies, we show that ZAK is required for rapid p38 phosphorylation on treatment with an intermediate dose of ANS (Figure 2A, lanes 6–7). Cells complemented with ZAKα but not ZAKβ are competent to trigger p38 phosphorylation (Figure 2A, compare lanes 8 and 10), suggesting that important determinants of activation lie within the 468 amino acid (aa) long C-terminal tail of ZAKα, as recently shown (Vind et al., 2020). Next, we used mutational analysis to define regions of ZAKα responsible for activation. While an ATP-binding defective variant of ZAKα, K45M (Gross et al., 2002), fails to activate p38 phosphorylation upon ANS stimulation (Figure 2A, lanes 8–9), mutation of three different motifs within the C-terminal tail including a leucine-zipper (LZ) domain, a sterile alpha motif (SAM), and a cryptic RNA-binding motif had no impact on p38 activation (Figure S2A). Given that there is no detailed structural information on the ZAKα C-terminal tail, we systematically generated ZAKα C-terminal deletions. Deletion of the C-terminal 80 aa (ZAKα 1–720) slightly reduces the levels of p38 phosphorylation (Figure 2B, lanes 1–2 and 4–5) while deletion of the C-terminal 151 aa (ZAKα 1–649) completely abolishes p38 phosphorylation (Figure 2B, lanes 3 and 6). Taken together, we define a region of ZAKα as critical for activation of the RSR.
Figure 2. Ribosome collisions induce ZAtα phosphorylation.

(A) Schematic depicting two isoforms of ZAK (top). p38 phosphorylation (uppermost blot) in WT MCF10A, ZAK KO, or ZAK KO complemented with ZAKα, ZAKa-K45M, or ZAKβ under ANS treatment (1 mg/L, 15min) (bottom); control blots for total p38 and expression of ZAKα and ZAKβ. * denotes non-specific bands. (B) Schematic depicting C-terminal deletions of ZAKα (top). p38 phosphorylation in MCF10A cells expressing only FALG-tagged full-length ZAKa, a(1–720), or a(1–649). (C-D) Immunoblots of Phos-tag gels for ZAKα phosphorylation in HeLa-ZAKα-FLAG cells treated with ANS at 0, 1, or 100 mg/L (15min) (C) or with EME at 0, 0.24 or 48 mg/L (15min) (D), using FLAG antibody. Fraction of ZAKα shifted through phosphorylation is indicated. (E) ZAKα phosphorylation in MCF10A cells expressing only ZAKα or ZAKα-K45M. (F) Immunoblots for ZAKα and p38 phosphorylation in HeLa cells expressing endogenous ZAKα and FLAG-tagged ZAKα (WT or K45M mutant). Bottom most panel shows relative levels of endogenous and exogenous ZAKα after phosphatase treatment.
Given that ZAKα is a MAPKKK, a class of molecule that typically functions through autophosphorylation events (Rey et al., 2016), we asked whether ribosome collisions promote ZAKα autophosphorylation. We first probed the phosphorylation status of ZAKα as a function of elongation inhibitor concentration using Phos-tag gels that retard the migration of phosphorylated proteins (Kinoshita et al., 2009). For both endogenous ZAKα and overexpressed (OE) ZAKα-FLAG (Figure S2B), ZAKα phosphorylation is dramatically increased at intermediate but not high doses of ANS (Figures 2C and S2C); the phosphorylation status of shifted ZAKα was validated by its sensitivity to phosphatase treatment (Figure S2D). We observe similar, albeit less robust, phosphorylation of ZAKα on treatment with intermediate doses of EME (Figure 2D). Observed differences in the amplitude and pattern of ZAKα phosphorylation with these different triggers (ANS vs. EME) may impact downstream output from the signaling pathways.
We next asked whether ZAKα auto-phosphorylates as a mechanism for activation. First, ZAKα phosphorylation was completely abolished in cells that only express the ATP-binding defective variant K45M (Figure 2E), as previously shown (Rey et al., 2016); thus the kinase activity of ZAKα is essential for its own phosphorylation. To address whether ZAKα functions in cis or trans, we supplied WT or K45M epitope-tagged ZAKα to cells carrying endogenous ZAKα and followed the phosphorylation status of the exogenous ZAKα variant. These data show that exogenous ZAKα is only phosphorylated when its own kinase domain is intact (Figure 2F); importantly, the exogenous ZAKα is expressed at lower levels than the endogenous ZAKα (exogenous/endogenous= ~0.35) thus increasing the likelihood of trans activation were it to happen. These data provide compelling evidence that ZAKα functions in cis to activate the RSR.
ZAKα binds to colliding ribosomes and is activated by autophosphorylation
We next asked whether ZAKα directly associates with elongating ribosomes. Polysome profiling followed by western blot analysis reveals that substantial amounts of both ZAKα and ZAKα-K45M associate with monosomes and polysomes, while ZAKβ does not (Figure 3A). And, while the C-terminal 80 aa deletion construct (ZAKα 1–720) shows somewhat reduced affinity to polysomes, the C-terminal 151 aa deletion construct (ZAKα 1–649) fails to co-migrate with polysomes and predominantly associates with free mRNPs (Figure 3B). The correlation between polysome association of ZAKα and p38 activation argues for direct binding interactions with the ribosome being critical to pathway activation.
Figure 3. ZAtα associates with colliding ribosomes.

(A) Representative polysome profile (top) from MCF10A ZAK KO cells complemented with ZAKα, ZAKα-K45M, or ZAKβ. Fractions were analyzed by immunoblotting. (B) Similar to (A), with MCF10A ZAK KO cells complemented by FLAG-tagged ZAKα, α(1–720), or α(1–649). (C) Polysome profiles from HeLa cells treated with an intermediate (1 mg/L, orange) or high (100 mg/L, gray) dose of ANS compared to untreated sample (black). (D) Polysome profiles from HeLa cells with indicated treatment after RNase digestion. Fractions were immunoblotted with indicated antibodies. (E) Polysome profiles from ZAKα-FLAG HeLa cells with indicated treatment following RNase digestion. Fractions were analyzed by Phos-tag gels and immunoblotted using antibodies against FLAG (ZAKa) or RPL4. Fraction of ZAKα phosphorylation in monosome and disome fractions is indicated. (F) Average disome occupancies aligned at stop codons across the transcriptome from Mock IP (black) or ZAKα IP (magenta). Average normalized density (solid line) with standard deviation (shaded area) across two replicates. (G) p38 phosphorylation in WT or ZNF598 KO HEK293 cells treated with ANS (0.1–100 mg/L).
We next asked whether the association of ZAKα with ribosomes changes upon ANS stimulation and found that ZAKα associates with monosomes and polysomes independent of stimulation at the intermediate or high dose of ANS (Figure 3C). If our model that ZAKα senses ribosome collisions and activates the RSR is correct, then we would predict that ZAKα preferentially associates with colliding ribosomes relative to monosomes. When polysomes were collapsed by RNase treatment (Figure 3D, top panel), ZAKα preferentially co-sediments with disomes (Figure 3D, compare fractions 5–7), independent of ANS dose. We suspect that the deep sedimentation of ZAKα (Figure 3C) reflects interactions of ZAKα either with single elongating ribosomes or with the ubiquitous transient ribosome collisions occurring in cells (Figure 1B). We used Phos-tag gels to monitor ZAKα phosphorylation and found strong enrichment of phosphorylated ZAKα co-sedimenting with RNase-resistant disomes (Figure 3E, untreated, intermediate, and high ANS, fractions 6), though note that the majority (~70%) of the monosome-associated ZAKα species is unphosphorylated in untreated and high-dose ANS conditions (Figure 3E, untreated and high ANS, fractions 4). We also observed free and monosome-associated highly phosphorylated ZAKα in cells stimulated with intermediate-dose ANS (Figure 3E, intermediate ANS, fractions 1– 4), consistent with a recent report that phosphorylation of ZAKα reduces affinity for ribosomes (Vind et al., 2020). Given that SAPKs do not associate with polysomes (Kim et al., 2019), we propose that ZAKα dissociates from colliding disomes when phosphorylated to activate downstream signaling.
To further define ZAKα-ribosome association, we performed selective ribosome profiling (Oh et al., 2011) in ZAKα-FLAG OE cells (Figure S3A). Consistent with polysome profiles (Figures 3D and E), we observed enrichment of disome footprints (average length ~65 nts) in ZAKα IP samples (Figure S3B). Recent reports of disome profiling indicate that ribosome collisions occur preferentially at stop codons and at certain peptide motifs, even in unstressed cells (Han et al., 2020). We see strong enrichment of ZAKα at stop codons throughout the transcriptome including, for example, the TXNRD1 and RPS13 mRNAs (Figures S3C-D). And, we observe an overall ~3-fold enrichment of ZAKα-bound disome footprints at stop codons transcriptomewide (Figure 3F). In addition, ZAKα preferentially associates with colliding disomes at known problematic peptide motifs, e.g. P-P and P-D dipeptides, known to induce ribosome stalling and collisions (Han et al., 2020) (Figures S3E and S3F).
Ribosome collisions trigger mRNA-specific quality control events downstream of recognition of a composite ribosome interface by the E3 ubiquitin ligase ZNF598 (Juszkiewicz et al., 2018). Using cell lines lacking ZNF598, we find that p38 activation remains sensitive to ANS stimulation and is more potent in general (Figure 3G). This increase in p38 activation might result from overall increases in the levels of colliding ribosomes in ZNF598 KO cells (Garzia et al., 2017; Juszkiewicz et al., 2018).
Ribosome collisions trigger GCN2-mediated eIF2α phosphorylation
Cellular stresses typically down-regulate translation initiation to allow cellular recovery through activation of specific gene expression programs. Recent findings by Ishimura et al. show that ribosome stalling induces eIF2α phosphorylation by GCN2 in an uncharged-tRNA independent manner in mice harboring mutations in an Arg-tRNAAGA isodecoder and the putative ribosome rescue factor GTPBP2 (Ishimura et al., 2014). Reanalysis of ribosome profiling data from these mice reveals stacked (i.e. colliding) ribosomes upstream of pausing sites at AGA codons (Figure S4A), suggesting a potential role for collisions as the trigger of GCN2 in these earlier experiments. We asked whether ribosome collisions might trigger eIF2α phosphorylation. Similar to the patterns observed for p38/JNK phosphorylation induced by ribosome collisions (Figure 1C), we see maximal levels of eIF2α phosphorylation at intermediate doses of ANS (Figure 4A); importantly, mTOR phosphorylation was unaffected by these same treatments. Pretreatment of cells with GCN2 inhibitor, but not PERK inhibitor, abolishes the eIF2α phosphorylation induced by the intermediate dose of ANS (Figure 4B, compare lanes 1–4); GCN2 knockdown by shRNA similarly shows reduced levels of eIF2α phosphorylation upon stimulation (Figure S4B). Moreover, eIF2α phosphorylation does not occur through feedback regulation mediated through p38 since activation is insensitive to p38 inhibitor (Figure 4B, compare lanes 2 and 5). We observe similar trends for maximal eIF2α phosphorylation at intermediate levels of ANS in yeast (Figure S4C) where only one eIF2α kinase - GCN2 - exists. As the other eIF2α kinases respond to cellular stresses not directly connected to protein synthesis, a role for GCN2 in ribosome collision-mediated ISR activation makes considerable sense.
Figure 4. Ribosome collisions activate GCN2-mediated eIF2α phosphorylation.

(A) Immunoblots for phosphorylation of eIF2α and mTOR in MCF10A cells treated with ANS (0.1–100 mg/L, 0.5 h). Total eIF2α and β-actin as loading controls. (B) eIF2α phosphorylation induced by intermediate doses of ANS (0.5 mg/L, 0.5 h) in MCF10A cells pretreated with GCN2 inhibitor (A-92), PERK inhibitor (GSK 2606414), or p38 inhibitor (BIRB 796). (C) eIF2α phosphorylation induced by ANS in WT or ZAK KO MCF10A cells. (D) eIF2α phosphorylation in WT, ZAK KO, or ZAK KO MCF10A complemented with ZAKα, ZAKα -K45M, or ZAKβ under ANS treatment (0.5 mg/L, 0.5 h). (E) Polysome profiles from DSP-crosslinked WT and ZAK KO MCF10A cells. Fractions were analyzed by immunoblotting with indicated antibodies.
Recent interactome surveys revealed that ZAKα co-localizes with GCN1, GCN20, and other factors implicated in ribosome-mediated quality control such as ZNF598 and ASCC3 (Figure S5). We asked whether ZAKα is critical for GCN2-mediated eIF2α phosphorylation induced by ribosome collisions. Indeed, under intermediate doses of ANS, we find that ZAK KO cells show marked reductions in eIF2α phosphorylation as compared to WT cells (Figure 4C). Complementation analyses show that reduction in eIF2α phosphorylation was restored by ZAKα and ZAKα-K45M, but not ZAKβ (Figure 4D). Given that both ZAKα and ZAKα-K45M associate with ribosomes (Figure 3A), we hypothesize that the role of ZAKα in this case may be structural or binding-related rather than catalytic. We provide physical evidence to support this hypothesis using polysome profiling to show that GCN1, a GCN2 activator, co-sediments with RNase-resistant disomes; moreover, GCNl-disome association was diminished in ZAK KO cells (Figure 4E, lanes 6 and 7 in ZAK KO compared to WT). While we do not capture GCN2 or GCN20 binding to colliding ribosomes, their association may be transient and/or compromised by RNase treatment. Our results suggest that colliding ribosomes function as a common platform for GCN2- and ZAKα-mediated signaling events.
CRISPR screens validate ribosome collision-induced activation of stress response pathways
We next performed cellular growth-based CRISPR/Cas9 screens (Shalem et al., 2014) under two different intermediate doses of ANS to systematically explore how mammalian cells survive stresses that induce global ribosome collisions (Figure S6A). We infected K562 cells with sgRNAs targeting each known gene (19,114 genes) and selected for growth under different ANS regimes; after selection, we deep-sequenced the remaining sgRNAs to identify genes that promote cell survival or cell death.
While overall trends observed in these two screens were similar, some different genes emerged as most critical in these two regimes (Figures S6B and S6C). Genes implicated in reducing cell survival in the presence of the higher dose of ANS include ZAK, BAX, BAK1, and ~54 other genes enriched with high statistical significance (Figure S6B and Table S1). Gene ontology analysis shows strong enrichment for genes implicated in apoptosis, RNA polymerase III transcription, and tRNA metabolism (Figure S6D). Identification of ZAK and apoptosis-related genes is readily reconciled with earlier observations that activation of ZAKα leads to JNK signaling that promotes apoptosis through the BAX/BAK-dependent pathway (Lei and Davis, 2003). We further assessed cell death under ANS treatment and found that ZAK KO completely abolished apoptosis; this loss in responsiveness to ANS is rescued by complementation with ZAKα but not ZAKα-K45M (Figure S6E).
In the screen performed at the lower concentration of ANS, genes implicated in apoptosis are again enriched (pink dots) in selection for increased survival (Figure S6C). However, we identified 14 genes that are depleted (red dots) in the screen, indicating that they normally increase survival upon ANS treatment, including p38α (MAPK14) and GCN20 (ABCF3) (Figure S6C and Table S2). p38α is known to negatively regulate JNK to promote cell survival (Miura et al., 2018) thus likely explaining the de-enrichment of p38α sgRNA. GCN20 belongs to the regulatory complex GCN1/20 for the eIF2α kinase GCN2 and is required for eIF2α phosphorylation in yeast and worms (Hirose and Horvitz, 2014; Vazquez de Aldana et al., 1995). In light of our data indicating that colliding ribosomes activate GCN2 in a ZAK-dependent manner, deletion of this pathway for eIF2α phosphorylation would effectively increase ribosome collisions, leading to increased p38/JNK activation and cell death. Together, these CRISPR screen results provide strong support for our data indicating that ribosome collisions trigger multiple distinct stress response pathways that determine cell fate.
Amino acid starvation leads to ribosome collisions that trigger the RSR
We next evaluated the implications of our mechanistic insights into ribosome collision- induced stress activation for more general cellular stresses. Recent studies showed that amino acid starvation could induce codon-specific ribosome stalling and collisions (Darnell et al., 2018). We found that when glutamine levels in the media were reduced, the levels of p38 phosphorylation increased accordingly (Figure 5A, lanes 2–4). We noted, however, that this activation was modest in comparison to that seen with intermediate doses of ANS (Figure 5A, lane 1). Since amino acid starvation induces eIF2α phosphorylation through GCN2, and a consequent block on translation initiation, we reasoned that this response would decrease ribosome collisions. When we instead pretreat starved cells with ISRIB, a compound known to block the ISR, thus allowing ongoing efficient translation initiation (Sidrauski et al., 2013), we see an increase in RNase-resistant disomes (Figure S7A) and a concomitant increase in p38 phosphorylation (Figure 5A, lanes 5–7).
Figure 5. Glutamine starvation induces ribosome collision-mediated ZAKα activation.

(A) p38 phosphorylation in HeLa cells grown in rich or glutamine-depleted media pretreated with vehicle (DMSO) or ISRIB compared to stimulation by intermediate-dose ANS (1 mg/L, 15min). (B) p38 phosphorylation in WT or ZAK KO MCF10A cells in media with or without glutamine. (C) Immunoblots of a Phos-tag gel for ZAKα phosphorylation in ZAKα-FLAG HeLa cells grown in media containing 2, 0.02 or 0 mM glutamine pretreated with DMSO or ISRIB. (D) eIF2α phosphorylation induced by glutamine starvation in WT or ZAK KO MCF10A cells pretreated with DMSO or ISRIB.
We next asked whether ZAKα and its phosphorylation are necessary for p38 activation in response to amino acid starvation. First, phosphorylation of p38 under glutamine starvation strongly depends on ZAK (Figure 5B). Second, ZAKα phosphorylation increases slightly when cells were grown in media simply depleted for glutamine (Figure 5C, lanes 2–4), and is further increased when starved cells are pretreated with ISRIB (Figure 5C, lanes 5–7). These results point to a role for ZAKα in sensing cellular amino acid levels through ribosome collisions to activate SAPK signaling.
While uncharged tRNA can function as a direct activator of GCN2 (Berlanga et al., 2006), we wondered whether ZAKα and ribosome collisions might play an additional role under amino acid starvation. We observed marked increases in eIF2α phosphorylation under glutamine starvation, and a further increase in the presence of ISRIB (Figure 5D, lanes 1–3). While the levels of eIF2α phosphorylation seen under simple glutamine starvation in ZAK KO cells remain similar to those observed in WT cells (Figure 5D, lanes 2 and 5), however, we observed strong dependence of eIF2α phosphorylation on ZAK in starved cells pretreated with ISRIB (Figure 5D, lanes 3 and 6) (Figure S7A). We propose that ZAKα is involved specifically in the ribosome- mediated pathway for GCN2 activation that depends on ribosome collisions, but not in GCN2- mediated eIF2α phosphorylation by free uncharged-tRNAs. These observations are consistent with the existence of two pathways for GCN2-mediated eIF2α phosphorylation. One pathway depends simply on free uncharged-tRNAs (Berlanga et al., 2006; Wek et al., 1989) and is unaffected by ZAK activity. A second pathway depends on ZAK-enhanced ribosome collision-mediated interactions with GCN complex (Inglis et al., 2019; Ishimura et al., 2016; Marton et al., 1997; Ramirez et al., 1991).
UV irradiation damages mRNA and triggers ribosome collisions and the RSR
UV irradiation induces the RSR and eIF2α phosphorylation on short time scales (minutes) (Deng et al., 2002; Iordanov et al., 1998). For the RSR, it was proposed that damage of rRNAs by UV irradiation causes translation elongation distress (Iordanov et al., 1998); while these studies provided strong evidence that rRNA damage occurs, it seems unlikely that a majority of this damage would be consequential to ribosome function.
DNA damage response pathways are triggered by UV irradiation that induces formation of thymine dimers or 6–4 photoproducts in DNA (Ciccia and Elledge, 2010). Given that UV irradiation induces pyrimidine dimers not only in DNA but also in RNA (Jackle and Kalthoff, 1978), we wondered whether global mRNA damage induced by UV irradiation leads to transcriptome-wide ribosome collisions and consequent ZAKα-dependent p38/JNK and eIF2α phosphorylation.
To test these ideas, we first used polysome profiling to monitor ribosome collisions upon UV irradiation and observed increased RNase-resistant disomes in UV-irradiated cells (Figure 6A). To gain higher resolution into ribosome dynamics in vivo, we performed ribosome profiling experiments with untreated and UV-irradiated cells. Our previous studies have defined two distinct sizes of ribosome-protected footprints (RPFs) in mammalian cells: long footprints (~31 nt) representing ribosomes with an occupied A site (predominantly in a pre-translocation state) and short footprints (~22 nt) representing those with an open A site (awaiting tRNA selection) (Wu et al., 2019) (Figure S7B). In UV-irradiated cells, we observe a marked increase in short RPFs and a concomitant reduction in long RPFs compared to untreated cells (Figure 6B). These data show that UV-irradiation globally increases the number of ribosomes trapped in a state waiting to decode the next aminoacyl-tRNA.
Figure 6. UV irradiation induces ribosome collision-mediated ZAKα activation.

(A) Polysome profiles from RNase-digested lysates of HeLa cells treated with UV compared to untreated. (B) Size distribution of ribosome footprints (RPFs) from UV-irradiated HeLa cells compared to untreated cells. (C) Scatter plot of codon-specific ribosome occupancies for short RPFs comparing UV-irradiated to untreated cells. Orange dots indicate codons containing two adjacent pyrimidines. (D) Metacodon analysis aligning at all CUU codons comparing UV- irradiated to untreated cells. Arrows indicate colliding ribosomes upstream of the paused ribosomes at CUU codons. (E) Phosphorylation of p38, JNK, and eIF2a in WT or ZAK KO MCF10A cells at indicated time points after UV irradiation. (F) Phosphorylation of endogenous ZAKa, p38, JNK, and eIF2α in HeLa cells transfected with untreated or UV-irradiated EGFP mRNA.
These same ribosome profiling libraries allowed us to define the sites at which ribosomes are disproportionately stalled in UV-irradiated cells. We computed ribosome occupancies of the short RPFs at all 61 sense codons and asked which codons are enriched in UV-irradiated samples. First, we observed a striking loss in the correlation of codon occupancies for the short footprints in UV-irradiated cells compared to untreated cells (Figures 6C and S7C), indicating that UV irradiation drastically perturbs the decoding step of translation elongation. Strikingly, codons containing two adjacent pyrimidines were disproportionately occupied by ribosomes in the UV-treated samples (Figure 6C); we note that these calculations will underestimate pausing that occurs on pyrimidine dimers that are shared, for example, by two adjacent codons (e.g. GGU-UAC). When we aligned ribosomes at CUU codons (Figure 6D), we observed accumulation of ribosomes at those codons but also ~30 and ~60 nt upstream of CUU codons as anticipated by the phasing of ribosome collisions.
We next explored the dependence of the RSR and eIF2α phosphorylation on ZAK; indeed, both p38/JNK and eIF2α phosphorylation are activated by UV irradiation in a ZAK- dependent manner (Figure 6E). We also see strong activation of ZAKα under these same conditions (Figure S7D), and the same dependence on the kinase activity of ZAKα for activation of the SAPKs (Figure S7E). In addition, when we trap elongating ribosomes by pretreatment with high concentrations of ANS or EME (Figures 1C and D), p38 phosphorylation induced by UV irradiation is abrogated (Figure S7F). These data provide compelling evidence that UV irradiation induces mRNA damage leading to ribosome collisions and thus activates general stress responses through ZAK.
If our model that ribosome collisions induced by damaged mRNA activates ZAKα and GCN2 under UV irradiation is correct, then DNA and rRNA damage should both be dispensable for activation of the SAPKs and GCN2-mediated eIF2α phosphorylation. To test this, we exposed in vitro-transcribed EGFP mRNA (5’-capped and 3’-polyadenylated) to UV irradiation (or not) and then transfected these mRNAs directly into cells and monitored activation of ZAKα and the downstream pathways. We observed potent activation of ZAKα and both downstream branches of signaling (Figure 6F) only in the cells transfected with the UV-irradiated sample. EGFP protein output from UV-irradiated mRNA decreased drastically, likely reflecting reduced translation on damaged mRNA. These results show that mRNA damage induced by UV irradiation is sufficient to activate ZAKα and the downstream signaling pathways, independent of any contribution from nuclear DNA-activated pathways or ribosome damage per se.
DISCUSSION
A major challenge for cells on encountering stress is to mount a measured response that balances life-or-death cell fate decisions through activation of survival or apoptotic gene expression programs. Here we show that the extremely abundant elongating ribosomes in the cytoplasm function as key sensors of general cellular stress. That the ribosome could function as such a broad sensor is appealing given recent tomographic reconstructions of yeast cells revealing extremely abundant ribosomes (20% of the cell volume) (Delarue et al., 2018). More specifically, our work reveals a mechanism by which ribosome collisions function as a key molecular trigger for the SAPKs (p38/JNK) and GCN2 (Figure 7A), leading to activation of the RSR and the ISR, respectively. Both pathways depend critically on the MAPKKK, ZAKα that binds preferentially to colliding ribosomes relative to monosomes. In addition to collisions induced by translation elongation inhibitors, such as ANS and EME, we showed that environmental perturbations — amino acid starvation and UV irradiation - also potently trigger ribosome collisions and thus the SAPK and GCN2 signaling pathways.
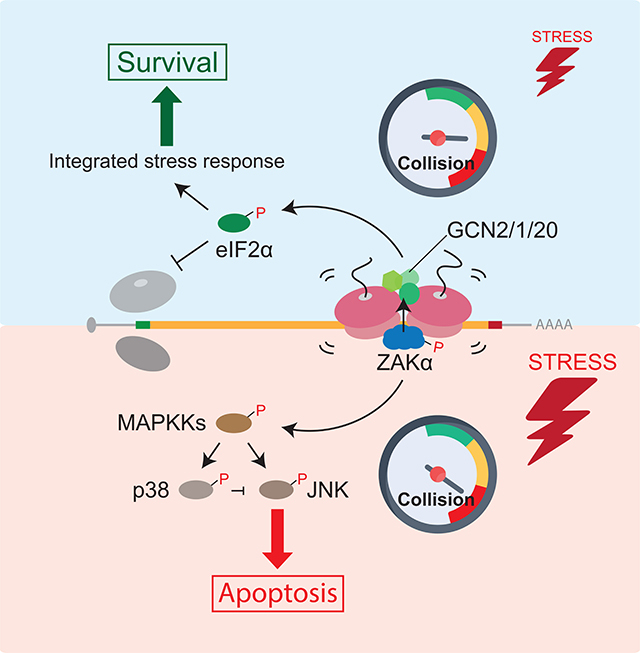
Figure 7. Molecular gauge for ribosome collisions determines cell fate.

(A) Model for ribosome collision-mediated stress response signaling through ZAKα to activate GCN2 and SAPKs (p38/JNK), leading to translation initiation block and apoptosis, respectively. Short-lived and long-lived colliding disomes are in gray and magenta, respectively. (B) Proposed model of cellar gauge for ribosome collisions that mounts measured cellular responses under stress. In unstressed cells, basal levels of ribosome collisions are handled by RQC and NGD machineries (left). Intermediate levels of collisions induced by cellular stresses trigger ZAKα- dependent GCN2-mediated translation initiation block to reduce further collisions, promoting cell survival (middle), whereas “dangerous” levels of collisions initiate ZAKα-dependent apoptosis (right).
We suggest that basal levels of ribosome collisions are processed as mRNA-specific problems by dedicated QC pathways (Yan and Zaher, 2019). However, when the number of colliding ribosomes exceeds the typically modest capacity of these QC components, the cell responds by activating more general stress responses that promote survival or trigger death. Collisions are measured on a cellular “gauge” that determines the measured response for the problem. In such a view, the ribosome through its inherent translation activity alerts the cell to the need for activation of broad cellular stress responses. The MAPKKK ZAKα effectively functions as a reader of the gauge, the sentinel, monitoring the intensity and/or duration of ribosome collisions (Figure 7B). We propose that the initial cellular response to widespread ribosome collisions would be to block global translation initiation through GCN2 to reduce ribosome loading on mRNAs and hence ribosome collisions. GCN2-mediated eIF2α phosphorylation triggers the ISR through which the expression of specific genes takes place to promote cellular survival (Pakos-Zebrucka et al., 2016). When the intensity and/or duration of ribosome collisions exceeds the cellular capacity to resolve collisions, or to recover from stress through the ISR, ZAKα activates the SAPK-mediated apoptosis pathway to mitigate damage and target the cell for death. For example, while the RSR induced by experimental elongation inhibitors such as ANS may not be resolved by activation of GCN2 and the ISR, more physiological conditions that trigger ribosome collisions, such as amino acid starvation or even UV irradiation, may be resolved by the ISR, thus preventing SAPK-mediated apoptosis.
Our finding that ribosome collisions trigger GCN2-mediated eIF2α phosphorylation is broadly consistent with numerous previous observations. First, GCN2/1/20 associates with polysomes independent of amino acid starvation (Marton et al., 1997; Ramirez et al., 1991). GCN2 was recently proposed to be activated by stalled ribosomes in cells (Ishimura et al., 2016) and the ribosomal P-stalk alone was shown to activate GCN2 in a reconstituted system in vitro (Harding et al., 2019; Inglis et al., 2019; Jiménez-Díaz et al., 2013). We add a critical layer wherein ZAKα facilitates activation of GCN2 potentially through interactions between GCN1, a GCN2 activator, and colliding ribosomes (Figures 4C and 4E). While ZAKα is critical for both SAPK and GCN2 activation, an important distinction is that the kinase activity of ZAKα is only critical for SAPK activation (Figure 2A), and not for GCN2 activation (Figure 4D). Future structural analysis of ZAKα on colliding ribosomes will further an understanding of how ZAKα influences GCN2-mediated eIF2α phosphorylation independent of its kinase activity.
Much previous work characterizing UV-induced cellular damage has focused on DNA lesions (Ciccia and Elledge, 2010) and cellular responses over relatively long-time frames. We make the striking observation that UV irradiation induces ribosome collisions through mRNA damage, leading to ZAKα-dependent activation of both GCN2 and SAPK signaling on short time scales. Using ribosome profiling, we show that short RPFs that correspond to ribosomes waiting to decode (with an empty A site) are markedly enriched under UV irradiation (Figure 6B), suggesting that ribosomes are trapped during the decoding step of elongation. These results make sense in the context of the directionality of ribosome movement on mRNAs (5’ to 3’): the first encounter of a ribosome with damaged mRNA will occur in the A site during decoding. Our data further indicate that UV irradiation results in disproportionate ribosome stalling at codons with two adjacent pyrimidines (Figure 6C) and ribosome collisions accumulate behind these codons (Figure 6D). Finally, a direct mRNA transfection experiment establishes that mRNA damage from UV irradiation is sufficient to promote the ZAK-dependent responses that we characterized (Figure 6F). More generally, these data provide a clear example of how other physiological stress such as oxidative and alkylation stresses (Yan and Zaher, 2019; Yan et al., 2019) might be sensed through the action of the ribosome, triggering rapid cellular responses that dictate cell fate decisions.
During revision of this study, Vind et al. reported that ZAKα associates with stalled ribosomes to initiate the RSR and they defined key domains and phosphorylation sites critical to ribosome binding and activation (Vind et al., 2020). These authors argued, however, that the RSR is not activated by colliding ribosomes, but instead by slowed elongating single ribosomes. Importantly, their experiments may have been complicated by use of the reversible binding inhibitor cycloheximide (Hussmann et al., 2015) that is likely to promote collisions at all doses and by not titrating ANS to the point of fully blocking elongation (our experiments used more than 3 times the amount of ANS at the high end of the titration). Interestingly, their reported activation of the RSR by the early elongation inhibitor lactimidomycin may reflect collisions between stalled initiating ribosomes and scanning 40S subunits. Further studies will be needed to resolve these different observations.
Our results are strongly consistent with a model where ribosome collisions are the key activator of both mRNA-specific QC events and of the RSR. Our model is parsimonious and easily rationalizes how so many diverse stresses (ANS, EME, aa-starvation, and UV damage) would result in the same general outcomes of SAPK and GCN2 signaling activation. We anticipate that different cell types, with different inherent demands on protein synthesis, will exhibit different sensitivities to perturbations of translational homeostasis. Defining these differences will likely reveal interesting connections between ribosome biology and cell fate, with ZAKα playing a central role.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Please direct any requests for further information or reagents to the lead contact, Rachel Green (ragreen@jhmi.edu).
Materials Availability
Reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
Raw sequencing data were deposited in the GEO database under the accession number GSE141459.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and culture conditions
Flp-In T-REx 293 and Flp-In T-REx HeLa cells were grown in Dulbecco’s modified eagle medium (DMEM) in 10% FBS, 2 mM glutamine. In the text and figures, Flp-In T-REx 293 and Flp-In T-REx HeLa cells are labeled as HEK293 and HeLa, respectively. MCF10A cells were grown in DMEM/F12 Ham’s Mixture supplemented with 5% Equine Serum, 20 μg/L EGF, 0.5 g/L hydrocortisone, 100 μg/L cholera toxin, and 10 mg/L insulin and 2mM glutamine. K562 cells were grown in Rosswell Park Memorial Institute 1640 (RPMI) media (Thermo Fisher Scientific) supplemented with 10% FBS, 1mM pyruvate and 2mM L-glutamine.
Generation of cell lines
Flp-In T-REx HeLa cells are a gift from Andrew Holland (Johns Hopkins University School of Medicine) (Gassmann et al., 2010). To generate HeLa cell line inducibly expressing ZAKα-FLAG WT or K45M variant, ZAKα-FLAG-pcDNA5 (WT or K45M variant) was co-transfected with pOG44 (Thermo Fisher Scientific, V600520) into Flp-In T-REx HeLa cells and selected in media supplemented with 100 mg/L hygromycin according to manufacturer’s instructions. Expression of ZAKα-FLAG (WT) was induced for 24 hr with the addition of doxycycline at 2 mg/L, except for Figure 2F wherein ZAKα-FLAG (WT or K45M) was induced for 5–6 hr with the addition of doxycycline at 2 μg/L.
MCF10A reporter cell lines were generated by lentiviral infection. Lentivirus was generated with lipofectamine transfection of third-generation viral packaging plasmids and lentiviral reporter constructs into HEK293FT cells. Cells were incubated in viral supernatant with polybrene (10 μg/ml) (EMD Millipore) for 48 hours before selection. MCF10A cell lines were transduced with H2B-iRFP nuclear marker (no selection marker), ERK-KTR-mCerulean3 (hygromycin B), JNK-KTR- mRuby2 (blasticidin), and p38-KTR-mClover (no selection marker) and selected with hygromycin B (100 μg/ml) (Invivogen) and blasticidin (3 μg/ml) (Invivogen) before clonal expansion. The ZAK-KO cell line was created by ligation of guide RNA targeting exon 1 (target sequence: ATGGATATCACAGGACAAGG) of the human ZAK gene into a CRISPR V2 neomycin-resistant vector (provided by A. Holland, Johns Hopkins University School of Medicine) and transduced into MCF10A cells stably expressing H2B-iRFP, ERK-KTR-mCerulean3, JNK-KTR-mRuby2, and p38-KTR-mClover. Cells were selected for neomycin resistance (0.5 g/L) and clonally expanded. To complement the ZAK KO cell line, ZAKα, ZAKα-K45M, ZAKβ, ZAKα-LZ mutant, ZAKα-ΔSAM, ZAKα-RKK mutant, ZAKα-FLAG, ZAKα-FLAG (1–720), or ZAKα-FLAG (1–649) in pLenti PGK Puro DEST vector (w529–2) (Addgene) was transduced into ZAK KO cells and selected for puromycin resistance (2 μg/ml). Cells were validated for expression and activity using live-cell imaging and western blotting.
To generate GCN2 knockdown MCF10A cell lines, scramble shRNA, GCN2-shRNA1 (GAATGGTTGGCACTGCTCTCTATGTAAGC) or GCN2-shRNA2 (AGTCATCTCCTGGCTGTTGAACCACGATC) was cloned into pGFP-C-shLenti (OriGene), transduced, and selected under puromycin following manufacturer’s instructions.
To generate cell line cBZ056 for CRISPR screening, a K562 line containing a tandem tagBFP- tdTomato reporter (irrelevant to the current study) was lentivirally transduced with lentiCas9- Blast (addgene #52962) and after a 24-hour recovery, selected with 20 mg/L blasticidin for 4 days, at which point untransduced cells showed no viability. Cas9 integration was confirmed by western blotting and by sgRNA targeting of test genes.
METHOD DETAILS
Polysome profiling
Cells were grown to ~70% confluency and then treated with indicated antibiotic for 15 min. Following treatment, cells were rinsed once with PBS and lysed in lysis buffer (20 mM Tris-Cl [pH 8], 150 mM KCl, 15 mM MgCl2, 1% Triton X100, 1mM DTT, phosphatase inhibitor cocktail [Cell Signaling, 5870], EDTA-free Protease inhibitor cocktail [Roche Diagnostics, 5056489001]). Lysates containing 200 ĝ of total RNA were run through 10–50% sucrose gradients using Beckmann Coulter SW41 Ti rotor at 40,000 rpm for 4°C for 3 hr. For RNase-digested profiles, lysates containing 100 μg of total RNA were treated with RNase A (Thermo Fisher Scientific, EN0531) at 4 mg/L for 15 min at RT and quenched by adding 200 U of SUPERaseIn (Thermo Fisher Scientific, AM2694). Digested lysates were run through 10–35% sucrose gradients (20 mM Tris-Cl [pH 8], 150 mM NaCl, 5 mM MgCl2) using Beckmann Coulter SW41 Ti rotors at 40,000 rpm for 4°C for 2 hr. For Figure 4F, cells were rinsed once with PBS and treated with 0.5 mM DSP (Thermo Fisher Scientific, 22585) in PBS for 10 min at RT. After crosslinking, cells were lysed in HEPES lysis buffer (20 mM HEPES [pH 7.5], 150 mM KCl, 15 mM MgCl2, 1% Triton X100, phosphatase inhibitor cocktail, EDTA-free Protease inhibitor cocktail). Digested lysates were run through 10–35% sucrose gradients (20 mM HEPES [pH 7.5], 150 mM NaCl, 5 mM MgCl2). Gradients were fractionated using a Biocomp piston gradient fractionator. The absorbance at 254nm was recorded. Protein was TCA-precipitated from the fractions and methanol- precipitated to remove residual sucrose. Pellets were resuspended in Laemmli loading buffer containing DTT and boiled at 99°C for 5 min.
Immunoblot analysis
Culture medium was refreshed ~ 2 hr before treatment. For antibiotic or cytokine treatments, cells were incubated with antibiotics and/or cytokine at indicated concentrations for 15 or 30 min before harvest. Anisomycin (Sigma-Aldrich, A9789) and emetine (Cayman Chemical, 21048) were dissolved in ethanol and H2O, respectively. TNFα (Sigma, PHP051A) was reconstituted in H2O. For phosphatase treatment, lysates were incubated with lambda phosphatase (NEB, P0753S) following the manufacturer’s instructions. For glutamine starvation, cells were pretreated with DMSO or 1.1 μM ISRIB (dissolved in DMSO) for 30 min, then washed twice with DMEM media without L-glutamine (Thermo Fisher Scientific, 11960–044), and cultured in the same media with DMSO or 1.1 μM ISRIB for 1 hr before harvest. For A60 reporter experiments (Fig. 1E), ~ 400,000 HeLa cells were transfected with 10 μg of pcDNA5, A0, or A60 reporters using Lipofectamine 3000 ~24 hr prior to harvest. For inhibitor experiments, cells were pretreated with eIF2α kinase inhibitors (GSK2606414 at 200 nM or A-92 at 1 μM) or p38 inhibitor (BIRB 796 at 5 μM) for 30 min following anisomycin treatment for 30 min. For UV irradiation, cells were irradiated in a Stratagene UV Stratalinker 2400 at 1,200 J/m2 (UV-C) in culture media with the lid open. Following irradiation, cells were recovered at 37°C for indicated time. For EGFP mRNA transfection experiments, 5 μg of EGFP mRNA (TriLink Biotech, L-7601–100) was reconstituted in nuclease-free water and irradiated with UVC at 3,600 J/m2 twice and transfected into ~ 2×105 HeLa cells using TransIT-mRNA transfection kit (Mirus). Non-irradiated EGFP mRNA was transfected as an untreated control. Transfected cells were harvested 5 hr after transfection.
In general, cells were rinsed with PBS once and lysed in lysis buffer. Lysates were kept on ice during preparation and clarified by centrifugation at 15,000 rpm for 5 min. After clarification, supernatants were boiled in Laemmli loading buffer containing DTT at 99°C for 5 min. Equal amount of proteins were resolved by 4–12% Criterion XT Bis-Tris protein gels (Bio-Rad, 3450124) and transferred to PVDF membranes using a Trans-Blot Turbo transfer system (Bio-Rad). Membranes were blocked with 5% nonfat dry milk (Bio-Rad, 1706404XTU) in PBST for 1 hr at RT with gentle rocking. Blots were washed in PBST and then incubated with indicated primary antibodies overnight at 4°C with gentle rocking. Blots were washed with PBST, 3–4 times over 45–60 min at RT with gentle rocking, then incubated with secondary antibodies (if needed) diluted in PBST for 1 hr with gentle rocking. Membranes were washed again with PBST, 3–4 times over 45–60 min at RT, developed using SuperSignal West Pico Plus ECL substrate (Thermo Fisher Scientific, 34580) and SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific, 34095), and imaged on a G:Box Chemi XRQ system. Shown are representative results from at least two independent experiments.
Phos-tag gels
For Phos-tag gel immunoblotting, cells were lysed in lysis buffer, resolved by 7% SDS-PAGE containing 10.7 μM Phos-tag (Wako, AAL-107) and 21.3 μM MnCl2, and transferred to PVDF membranes following manufacturer’s instructions. Membranes were blocked with 5% nonfat dry milk in for 1 hr at RT with gentle rocking. Blots were washed in PBST and then incubated with indicated antibodies overnight at 4°C or for 1 hr at RT with gentle rocking. Blots were washed with PBST, 3–4 times over 45–60 min at RT with gentle rocking, then developed using SuperSignal West Pico Plus ECL substrate (Thermo Fisher Scientific, 34580), and imaged on a G:Box Chemi XRQ system.
Antibodies
The following antibodies were used in this study. Antibodies for phospho-p38 (Thr180/Tyr182, 9211), p38 (9212), phospho-JNK (Thr183/Tyr185, 4668), β-actin (51255), phospho-mTOR (Ser2448, 2971) and eIF2α (9722S) were from Cell Signaling Technology. Antibodies for ZAKα (A301–933A) and RPS2 (A303–794A) were purchased from Bethyl Laboratories Inc. Phospho- eIF2α (Ser51, ab32157) from Abcam. Antibodies for FLAG (A8592) and ZAKP (HPA017205) were from Sigma-Aldrich. RPL4 antibody (11302–1-AP) was from Proteintech. GFP antibody (632381) was from Takara. Mouse anti-rabbit IgG-HRP (sc-2357) was from Santa Cruz Biotechnology. Goat anti-mouse IgG2a-HRP (115–035-2006) was from Jackson ImmunoResearch Laboratories Inc.
Cell lysis assays
HeLa cells were plated (10,000 cells per well) onto 96-well plates prior to treatment with anisomycin for 30 min. Cell lysis was measure using CellTox Green Cytotoxicity Assay kit (Promega, G8741) following manufacturer’s instructions. 9% Triton X100 was used as a positive control for cell lysis.
Cell viability assays
To measure cell death, MCF10A cells were plated (27,000 cells per well) onto glass-bottom 96 well imaging plates (Thermo Fisher Scientific) coated with 10 mg/L fibronectin (Sigma-Aldrich) and left to adhere and expand for 24 hr. The next day, cells were washed once with PBS and switched to imaging media (phenol-red free DMEM F12 (Thermo Fisher), 5% Horse Serum, and 1% penicillin-streptomycin (Gibco). 24 hours later, the cells were incubated with 10 μM of CellEvent™ Caspase-3/7 Green Detection Reagent (Thermo Fisher Scientific, C10423) and imaged on a Nikon Eclipse Ti-E inverted fluorescence microscope with an Andor Neo 5.5 or Hamamatsu sCMOS camera. Light-emitting diode (LED) excitation light source (SPECTRA X) was used at 500/20 nm (green fluorescent dye) and 640/30 nm (iRFP670). Cells were imaged for 24 hours after the addition of anisomycin or media and changes in green fluorescence were measured.
Selective ribosome profiling
HeLa cells expressing ZAKα-FLAG from eight 15-cm plates (~ 2.5 × 106/plate) were transfected with A60-reporter (10 μg/plate) two days prior to harvest using Lipofectamin 3000 according to manufacturer’s instructions. ZAKα-FLAG was induced by the addition of doxycycline at 2 mg/L 24 hr prior to cell lysis. Cells were washed with PBS and collected in 700 μL of footprint lysis buffer (20 mM Tris-Cl [pH 8], 150 mM KCl, 15 mM MgCl2, 1% Triton X100, 1mM DTT, 0.1 g/L cycloheximide with vigorous scrapping. Lysates were digested with TURBO DNase (2 U per mL) for 15 min on ice and clarified by centrifugation at 15,000 rpm for 5 min. Lysates containing 150 μg of total RNA were digested with 187.5 U of MNase at 25 °C for 1 hr, quenched by adding EGTA (pH 8) to 1.16 mM, and run through 10–30% sucrose gradients (20 mM Tris-Cl [pH 8], 150 mM NaCl, 5 mM MgCl2) using Beckmann Coulter SW41 Ti rotors at 40,000 rpm for 4°C for 2.5 hr. Gradients were fractionated using a Biocomp piston gradient fractionator. Fractions were pooled and incubated without (mock) or with ZAKa antibody (Bethyl Laboratories Inc., A301–994A) at 62.5 mg/L and 500 U SUPERaseIn overnight at 4 °C with gentle nutation. 200 μL of sheep anti-rabbit IgG Dynabeads M-280 (Thermo Fisher Scientific, 11203D) were added to each condition and incubated at 4 °C for 6 hr. Magnetic beads were washed 3 times with footprint lysis buffer without DTT. Immunoprecipitates were eluted in 250 μL of elution buffer (100 mM glycine [pH 2.8], 50U SUPERaseIn). RNA was extracted by hot acid phenol and size-selected from 15% denaturing PAGE gels, cutting between 24–90 nt for monosome and disome footprints. Library construction was as carried out as described previously (Wu et al., 2019). Libraries were sequenced on a HiSeq2500 machine at facilities at the Johns Hopkins Institute of Genetic Medicine.
Ribosome profiling
Following UV irradiation, HeLa cells were recovered at 37°C for 15 min (replicate 1) or 30 min (replicate 2). Cells were washed with PBS once and collected in 700 μL of footprint lysis buffer (see above) with vigorous scrapping. Lysates were digested with TURBO DNase (2 units per mL) for 15 min on ice and clarified by centrifugation at 15,000 rpm for 10 min. Lysates containing 10 ĝ of total RNA were digested with 750 units of RNase I (Thermo Fisher Scientific, 2295) at 25 °C for 1 hr with gentle shaking and quenched by adding 200 units of SUPERaseIn. Nuclease-treated lysates were layered on a sucrose cushion [20 mM Tris-Cl (pH8), 150mM KCl, 5 mM MgCl2, 1 mM DTT, 1 M sucrose]. Ribosomes were pelleted by centrifugation in a TLA100.3 rotor at 100,000 rpm for 1 hr at 4°C and RNA was extracted by miRNeasy mini kit (Qiagen). Library construction was as carried out as described previously (Wu et al., 2019), except using TGIRT (InGex) to perform the reverse transcription reactions.
CRISPR/Cas9 screening
Lentivirus was produced from the Brunello Human CRISPR knockout pooled library (Addgene, 73179) (Sanson et al., 2018) in by cotransfecting seven 15cm dishes of 70% confluent HEK293FT cells (in 20ml DMEM+10% FBS) with a cocktail of 28ml Opti-MEM media, 840 μL lipofectamine 3000 (Thermo Fisher Scientific, L3000015), 64 μg of the library, 119.4 μg psPAX2 (Addgene, 12259), 51.6 μg pMD2.G (Addgene, 12259) and 644 μL P3000 (Joung et al., 2017). Virus-containing media was collected, replaced, and passed through a 0.45 micron filter at 48, 72, and 96 hours. ~420ml of pooled media was concentrated to ~4ml with 100K centricon plus-70 filters and frozen. The library was transduced by spinfection into 200 million cBZ056 cells (see Generation of Cell Lines) at a 30% infection rate. 2 days after transduction, the cells were selected with 0.5 g/L puromycin for 2 days, and aliquots of 108 cells were frozen. To conduct growth-based screens, 120 million cells were seeded in a T225 flask for each condition prior to selection. All screens were performed in duplicate. The cells were treated with ethanol (vehicle) or anisomycin at 0.1 or 1 mg/L. Ethanol-treated cells were maintained in flasks by daily dilution to ~ 0.5× 106 cells per mL. After 12 days of selection, cells were harvested by centrifugation. Genomic DNA was extracted from ~150 million cells using NucleoSpin columns (MACHEREY- NAGEL, 740950) according to manufacturer’s instructions, except that RNase A was added after proteinase K inactivation, and incubated for 2 hour at RT. Guide RNA sequences were amplified from genomic DNA in 30×50 μL PCR reactions containing 25μL NEBNext Ultra II Q5 Master mix (NEB M0544), 5 μg genomic DNA, 1.25 μL F primer cocktail (2.5 μM each of oBZ475-oBZ478) and 1.25 μL of one barcoded reverse primer (oBZ479–490) per library. Primers are listed in Table S3. Thermal cycling was performed as follows: 3 minutes at 98°C, 20 cycles of (10 seconds at 98°C, 10 seconds at 68°C, 25 seconds at 72°C), 2 minutes at 72°C. PCR products were pooled, and 300 μl of pooled PCR product was cleaned up with SPRIselect beads (Beckmann Coulter, B23317) using 0.55× and 0.95× cuts to isolate the expected product size. Purified libraries were quantified with an Agilent Bioanalyzer using a high sensitivity DNA chip (Agilent 5067–4627), pooled at equimolar ratios, and sequenced with 66nt single-end reads on a HiSeq2500 sequencer (Illumina).
QUANTIFICATION AND STATISTICAL ANALYSIS
General data analysis
Statistical details can be found in figure legends.
Western blot quantification
The intensity of the protein bands/smears was quantified using Fiji (Schindelin et al., 2012).
Analysis of ribosome profiling data
Hg19 reference genome assembly from UCSC was used for alignment. A transcriptome file was generated to include canonical transcript of known genes from UCSC table browser. 3’ adapter (NNNNNNCACTCGGGCACCAAGGA) was trimmed with skewer (Jiang et al., 2014), and 4 random nucleotides included in the RT primer oBZ408 (RNNNAGATCGGAAGAGCGTCGTGTAGGGAAAGA GTGTAGATCTCGGTGGTCGC/iSP18/TTCAGACGTGTGCTCTTCCGATCTGTCCTTGGTGCCCGAGTG) were removed from the 5’ end of reads. Trimmed reads longer than 24 nt (for ZAKα selective ribosome profiling) or 15 nt (for regular ribosome profiling experiments) were aligned to human ribosomal and non-coding RNA sequence using STAR (Dobin et al., 2013) with the option ‘-- outFilterMismatchNoverLmax 0.3’. Unmapped reads were then mapped to human genome with ‘--outFilterIntronMotifs RemoveNoncanonicalUnannotated --outFilterMultimapNmax 1 -- outFilterMismatchNoverLmax 0.1’. For monosome footprints, offsets (20:[16], 21:[17], 22:[17], 23:[17]) were used to infer the A site of short RPFs whereas offsets (30:[16], 31:[16], 32:[17], 33:[17]) were used to infer the A site of long RPFs. Short RPFs were used to calculate the codon- specific ribosome occupancies (Figures 6C and S7C) while combined short and long RPFs were used for the meta-codon analysis (Figure 6D). For disome footprints (Figures 3F and S3C-F), offsets (61:[44], 62:[44], 63:[44], 64:[44], 65:[44], 66:[44], 67:[44], 68:[44], 69:[44], 70:[44], 71:[43], 72:[43], 73:[43], 74:[43], 75:[43], 76:[43], 77:[43], 78:[43], 79:[43], 80:[43]) were used to infer the A site of the lead ribosome. All other analyses were performed using software custom written in Python 2.7 and R 3.3.1.
Analysis of CRISPR/Cas9 screening
Sequencing reads were aligned to the Brunello library sequences and quantified using custom- written Python script. Effect size and false discovery rates were calculated by MAGeCK (Li et al., 2014) using ‘--adjust-method fdr’ and available in Table S1 and S2.
Supplementary Material
Figure S1. Ribosome collisions activate SAPKs (p38/JNK) (related to Figure 1) (A) Representative polysome profiles from RNaseA-digested lysates of HEK293 cells treated with emetine (EME) at 0 (black), 0.24 (blue), or 48 mg/L (grey). (B) Measurement of lysis of HeLa cells under ANS treatment at indicated concentration for 30 min. 9% Triton X-100 used as a positive control for cell lysis. (C) Phosphorylation of p38/JNK in HEK293 cells stimulated by TNFα (100 μg/L), ANS (100 mg/L), or both TNFα and ANS compared to untreated cells. (D) Phosphorylation of p38/JNK in HeLa cells treated with ANS (0.001-100 mg/L) for 15 min. (E) Phosphorylation of p38 in U2OS cells treated with ANS (0.01-100 mg/L) for 15 min. (F) T2A skipping sequence in yeast induces ribosome collisions. Ribosome stalling at the PGP motif of the T2A sequence was visualized by short RPFs (orange trace) indicating defective decoding; stacked ribosomes (i.e. collisions) were observed ~30 nt upstream of the stalling site (long RPFs in green trace). Amino acid sequences in gray with T2A sequence highlighted in black and PGP in red. Data from GEO database (GSE129128).
Figure S2. ZAKα phosphorylation under ANS stimulation (related to Figure 2) (A) Mutational analysis of ZAK. Immunoblots for p38 phosphorylation in WT MCF10A, ZAK KO, or ZAK KO cells complemented with ZAKα, ZAKβ, ZAKα-K45M, ZAKα leucine-zipper mutation (LZmut), ZAKα SAM domain truncation (ΔSAM), or ZAKα R774A K775A K776A (RKKmut) under intermediate doses of ANS (1 mg/L). Total p38 as a loading control. ZAKα and ZAKβ immunoblots indicate the relative expression levels. * denotes non-specific bands. (B) Induction of ZAKα-FLAG in ZAK OE HeLa cells by doxycycline (Dox) at 2 mg/L for 24 hr. (C) Immunoblots of a Phos-tag gel for phosphorylation of endogenous ZAKa in HeLa cells treated with ANS at 0, 1, or 100 mg/L. (D) Collapse of phosphorylated ZAKα on a Phos-tag gel by phosphatase treatment following intermediate doses of ANS stimulation (1 mg/L).
Figure S3. Enrichment of ZAKα-associated disome footprints at stop codons and known problematic peptide motifs (related to Figure 3) (A) Schematic for selective ribosome profiling of ZAKα-associated disomes. After collapsing polysomes with RNase, monosomes and RNase-resistant disomes from sucrose gradients were isolated. ZAKα-bound ribosomes from within these combined samples were immunoprecipitated, and deep-sequenced. (B) Percentage of ZAKα-associated disome footprints (average size ~ 65 nts) (relative to monosome and disome footprints combined) from Mock or ZAKα IP samples. (C and D) Enrichment of ZAKα-associated disome footprints at stop codons of several representative transcripts, TXNRD1 (C) and RPS13 (D). (E and F) Average disome occupancies centered at PD (E) and PP (F) di-peptide motifs from Mock IP (black) or ZAKα IP (magenta).
Figure S4. Activation of GCN2-mediated eIF2α phosphorylation independent of uncharged tRNAs (related to Figure 4) (A) Metacodon analysis centered at AGA codons from indicated mouse cerebella. Data from GEO database (GSE56127). (B) Phosphorylation of eIF2α in WT MCF10A cells compared to those transduced with scrambled or two distinct shRNAs against GCN2. Total eIF2α as loading control. * denotes a non-specific band. (C) Phosphorylation of eIF2α in BY4741 Saccharomyces cerevisiae strain treated with ANS at 0, 0.16, 0.8, 4, 20, or 20 mg/L for 30 min.
Figure S5. ZAK, GCN1 and GCN20 share a similar interactome (related to Figures 4 and 5) (A) Heat map of correlation coefficients for 280 common preys from ZAK, GCN1, or GCN20 Bio- ID surveys (data obtained from http://cell-map.org). (B) Zoom-in view from the bracket in (A). Black arrowheads indicate ZAK, GCN1, and GCN20 and blue arrowheads indicate RPS3 and RPS10.
Figure S6. CRISPR screens for anisomycin-sensitive genes (related to Figures 4 and 5) (A) Schematic for CRISPR screens performed under ANS selection. ANS treatment at 1 mg/L reduces cell viability ~40% while ANS treatment at 0.1 mg/L decreases cell viability ~ 20% over a 24hr time period. (B and C) Volcano plots showing effect size (log2 fold change, x axis) versus FDR (-log(fdr), y axis) with enriched genes in green and de-enriched genes in red at the higher (5% FDR, adjusted p-value < 1.64× 10−4 at 1 mg/L) (B) or the lower (5% FDR, adjusted p-value < 2.87× 10−5 at 0.1 mg/L) (C) collision-inducing dose of ANS. Pinks dots indicate genes implicated in apoptosis. (D) Over-representation analysis for genes in (B) using WebGestalt (Liao et al., 2019). (E) Cellular apoptosis measured by CellEvent caspase dye (y-axis) in indicated MCF10A cells treated with ANS at 1 mg/L for 24 hr.
Figure S7. Amino acid starvation or UV irradiation induces ribosome collisions (related to Figures 5 and 6) (A) RNase-digested polysome profiles from HeLa cells starved for glutamine without or with ISRIB pre-treatment. (B) Schematic representation of the eukaryotic translation elongation cycle. Short ribosome-protected footprints (RPFs) correspond to decoding ribosomes with an empty A site (top of triangle) while long RPFs correspond to ribosomes with a full A site (bottom two corners of triangle). (C) Biological replicates for codon-specific ribosome occupancies of short RPFs comparing UV-irradiated to untreated cells (Figure 6C). Orange dots indicate codons containing two adjacent pyrimidines. (D) Phosphorylation of ZAKα, p38, JNK and eIF2α in HeLa cells after 30 min recovery from UV-irradiation compared to untreated cells. (E) Immunoblots for p38 phosphorylation in WT MCF10A, ZAK KO, or ZAK KO cells complemented with ZAKα, ZAKα-K45M, or ZAKβ after 15 min recovery from UV irradiation compared to unstressed cells. (F) Immunoblots for p38 phosphorylation after 15 min recovery from UV irradiation in HEK293 cells with or without pretreatment with high concentrations of EME (48 mg/L) or ANS (110 mg/L) for 5 min prior to UV irradiation.
Table S1. Table for CRISPR screen hits at the higher ANS dose (related to STAR Methods)
Table S2. Table for CRISPR screen hits at the lower ANS dose (related to STAR Methods)
Table S3. Oligonucleotides used for the CRISPR/Cas9 screens (related to STAR Methods)
Highlights.
Ribosome collisions trigger the ribotoxic stress response
Ribosome collisions activate the GCN2-mediated integrated stress response
The MAPKKK ZAKa functions as a sentinel for colliding ribosomes
ACKNOWLEDGEMENTS
We thank Jamie Wangen, Niladri Sinha, and Kamena Kostova for critical reading; Andrew Holland for Flp-In T-REx HeLa cells; Thomas Tuschl for ZNF598 knockout cells; Christopher Shoemaker for advice on CRISPR screens; David Mohr and the Johns Hopkins Genetic Resources Core Facility for sequencing assistance; Jeff Corden for discussions. Supported by NIH (R37GM059425 to R.G.) and HHMI (R.G. and C.C.W.). B.Z. is an HHMI fellow of the Damon Runyon Cancer Research Foundation (DRG-2250–16). S.R. is supported by an NSF CAREER award (MCB-1844994), NIGMS R35 (1R35GM133499), American Cancer Society Research Scholar Grant (133537-RSG-19–005-01-CCG), and a Jerome L. Greene Foundation Discovery Award.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Arpat AB, Liechti A, Matos M. De, Dreos R, Janich P, and Gatfield D (2019). Transcriptome-wide sites of collided ribosomes reveal principles of translational pausing. BioRxiv 710061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlanga JJ, Ventoso I, Harding HP, Deng J, Ron D, Sonenberg N, Carrasco L, and de Haro C (2006). Antiviral effect of the mammalian translation initiation factor 2a kinase GCN2 against RNA viruses. EMBO J. 25, 1730–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekaran V, Juszkiewicz S, Choi J, Puglisi JD, Brown A, Shao S, Ramakrishnan V, and Hegde RS (2019). Mechanism of ribosome stalling during translation of a poly(A) tail. Nat. Struct. Mol. Biol 26, 1132–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, and Elledge SJ (2010). The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 40, 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling NJ, and Cook SJ (2014). The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim. Biophys. Acta - Mol. Cell Res. 1843, 2150–2163. [DOI] [PubMed] [Google Scholar]
- Darnell AM, Subramaniam AR, and O’Shea EK (2018). Translational Control through Differential Ribosome Pausing during Amino Acid Limitation in Mammalian Cells. Mol. Cell 71, 229–243.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delarue M, Brittingham GP, Pfeffer S, Surovtsev IV, Pinglay S, Kennedy KJ, Schaffer M, Gutierrez JI, Sang D, Poterewicz G, et al. (2018). mTORC1 Controls Phase Separation and the Biophysical Properties of the Cytoplasm by Tuning Crowding. Cell 174, 338–349.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Harding HP, Raught B, Gingras A-C, Berlanga JJ, Scheuner D, Kaufman RJ, Ron D, and Sonenberg N (2002). Activation of GCN2 in UV-Irradiated Cells Inhibits Translation. Curr. Biol 12, 1279–1286. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Qiu H, Garcia-Barrio M, Anderson J, and Hinnebusch AG (2000). Uncharged tRNA Activates GCN2 by Displacing the Protein Kinase Moiety from a Bipartite tRNA-Binding Domain. Mol. Cell 6, 269–279. [DOI] [PubMed] [Google Scholar]
- Duch A, de Nadal E, and Posas F (2012). The p38 and Hog1 SAPKs control cell cycle progression in response to environmental stresses. FEBS Lett. 586, 2925–2931. [DOI] [PubMed] [Google Scholar]
- Garzia A, Jafarnejad SM, Meyer C, Chapat C, Gogakos T, Morozov P, Amiri M, Shapiro M, Molina H, Tuschl T, et al. (2017). The E3 ubiquitin ligase and RNA-binding protein ZNF598 orchestrates ribosome quality control of premature polyadenylated mRNAs. Nat. Commun 8, 16056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gassmann R, Holland AJ, Varma D, Wan X, Civril F, Cleveland DW, Oegema K, Salmon ED, and Desai A (2010). Removal of Spindly from microtubule-attached kinetochores controls spindle checkpoint silencing in human cells. Genes Dev. 24, 957–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross EA, Callow MG, Waldbaum L, Thomas S, and Ruggieri R (2002). MRK, a Mixed Lineage Kinase-related Molecule That Plays a Role in Y-Radiation-induced Cell Cycle Arrest. J. Biol. Chem 277, 13873–13882. [DOI] [PubMed] [Google Scholar]
- Han P, Shichino Y, Schneider-Poetsch T, Mito M, Hashimoto S, Udagawa T, Kohno K, Yoshida M, Mishima Y, Inada T, et al. (2020). Genome-wide Survey of Ribosome Collision. Cell Rep. 31, 107610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Ordonez A, Allen F, Parts L, Inglis AJ, Williams RL, and Ron D (2019). The ribosomal P-stalk couples amino acid starvation to GCN2 activation in mammalian cells. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose T, and Horvitz HR (2014). The Translational Regulators GCN-1 and ABCF-3 Act Together to Promote Apoptosis in C. elegans. PLoS Genet. 10, e1004512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussmann JA, Patchett S, Johnson A, Sawyer S, and Press WH (2015). Understanding Biases in Ribosome Profiling Experiments Reveals Signatures of Translation Dynamics in Yeast. PLoS Genet 11, e1005732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglis AJ, Masson GR, Shao S, Perisic O, McLaughlin SH, Hegde RS, and Williams RL (2019). Activation of GCN2 by the ribosomal P-stalk. Proc. Natl. Acad. Sci 116, 4946–4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iordanov MS, Pribnow D, Magun JL, Dinh TH, Pearson JA, Chen SL, and Magun BE (1997). Ribotoxic stress response: activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the alpha-sarcin/ricin loop in the 28S rRNA. Mol. Cell. Biol 17, 3373–3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iordanov MS, Pribnow D, Magun JL, Dinh T-H, Pearson JA, and Magun BE (1998). Ultraviolet Radiation Triggers the Ribotoxic Stress Response in Mammalian Cells. J. Biol. Chem 273, 15794–15803. [DOI] [PubMed] [Google Scholar]
- Iordanov MS, Choi RJ, Ryabinina OP, Dinh T-H, Bright RK, and Magun BE (2002). The UV (Ribotoxic) Stress Response of Human Keratinocytes Involves the Unexpected Uncoupling of the Ras-Extracellular Signal-Regulated Kinase Signaling Cascade from the Activated Epidermal Growth Factor Receptor. Mol. Cell. Biol 22, 5380–5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimura R, Nagy G, Dotu I, Zhou H, Yang X-L, Schimmel P, Senju S, Nishimura Y, Chuang JH, and Ackerman SL (2014). Ribosome stalling induced by mutation of a CNS- specific tRNA causes neurodegeneration. Science (80-. ). 345, 455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimura R, Nagy G, Dotu I, Chuang JH, and Ackerman SL (2016). Activation of GCN2 kinase by ribosome stalling links translation elongation with translation initiation. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäckle H, and Kalthoff K (1978). Photoreactivation of RNA in UV-irradiated insect eggs (Smittia sp., Chironomidae, Diptera) I. Photosensitized production and light-dependent disappearance of pyrimidine dimers. Photochem. Photobiol 27, 309–315. [DOI] [PubMed] [Google Scholar]
- Jandhyala DM, Ahluwalia A, Obrig T, and Thorpe CM (2008). ZAK: a MAP3Kinase that transduces Shiga toxin- and ricin-induced proinflammatory cytokine expression. Cell. Microbiol 10, 1468–1477. [DOI] [PubMed] [Google Scholar]
- Jiang H, Lei R, Ding SW, and Zhu S (2014). Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics 15, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-Díaz A, Remacha M, Ballesta JPG, and Berlanga JJ (2013). Phosphorylation of Initiation Factor eIF2 in Response to Stress Conditions Is Mediated by Acidic Ribosomal P1/P2 Proteins in Saccharomyces cerevisiae. PLoS One 8, e84219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung J, Konermann S, Gootenberg JS, Abudayyeh OO, Platt RJ, Brigham MD, Sanjana NE, and Zhang F (2017). Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc 12, 828–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juszkiewicz S, Chandrasekaran V, Lin Z, Kraatz S, Ramakrishnan V, and Hegde RS (2018). ZNF598 Is a Quality Control Sensor of Collided Ribosomes. Mol. Cell 72, 469–481.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kant S, Swat W, Zhang S, Zhang Z-Y, Neel BG, Flavell RA, and Davis RJ (2011). TNF- stimulated MAP kinase activation mediated by a Rho family GTPase signaling pathway. Genes Dev. 25, 2069–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T-S, Kim HD, Park YJ, Kong E, Yang HW, Jung Y, Kim Y, and Kim J (2019). JNK activation induced by ribotoxic stress is initiated from 80S monosomes but not polysomes. BMB Rep. 52, 502–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita E, Kinoshita-Kikuta E, and Koike T (2009). Separation and detection of large phosphoproteins using Phos-tag SDS-PAGE. Nat Protoc 4, 1513–1521. [DOI] [PubMed] [Google Scholar]
- Kulak NA, Pichler G, Paron I, Nagaraj N, and Mann M (2014). Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 11, 319–324. [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, and Avruch J (2012). Mammalian MAPK Signal Transduction Pathways Activated by Stress and Inflammation: A 10-Year Update. Physiol. Rev 92, 689–737. [DOI] [PubMed] [Google Scholar]
- Lei K, and Davis RJ (2003). JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci 100, 2432–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Wang J, Jaehnig EJ, Shi Z, and Zhang B (2019). WebGestalt 2019: gene set analysis toolkit with revamped Uls and APIs. Nucleic Acids Res. 47, W199–W205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marton MJ, Vazquez de Aldana CR, Qiu H, Chakraburtty K, and Hinnebusch AG (1997). Evidence that GCN1 and GCN20, translational regulators of GCN4, function on elongating ribosomes in activation of eIF2alpha kinase GCN2. Mol. Cell. Biol. 17, 4474–4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura H, Kondo Y, Matsuda M, and Aoki K (2018). Cell-to-Cell Heterogeneity in p38- Mediated Cross-Inhibition of JNK Causes Stochastic Cell Death. Cell Rep. 24, 2658–2668. [DOI] [PubMed] [Google Scholar]
- Oh E, Becker AH, Sandikci A, Huber D, Chaba R, Gloge F, Nichols RJ, Typas A, Gross CA, Kramer G, et al. (2011). Selective Ribosome Profiling Reveals the Cotranslational Chaperone Action of Trigger Factor In Vivo. Cell 147, 1295–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, and Gorman AM (2016). The integrated stress response. EMBO Rep. 17, 1374–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez M, Wek RC, and Hinnebusch AG (1991). Ribosome association of GCN2 protein kinase, a translational activator of the GCN4 gene of Saccharomyces cerevisiae. Mol. Cell. Biol 11, 3027–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey C, Faustin B, Mahouche I, Ruggieri R, Brulard C, Ichas F, Soubeyran I, Lartigue L, and De Giorgi F (2016). The MAP3K ZAK, a novel modulator of ERK-dependent migration, is upregulated in colorectal cancer. Oncogene 35, 3190–3200. [DOI] [PubMed] [Google Scholar]
- Sanson KR, Hanna RE, Hegde M, Donovan KF, Strand C, Sullender ME, Vaimberg EW, Goodale A, Root DE, Piccioni F, et al. (2018). Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat. Commun 9, 5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: An open-source platform for biologicalimage analysis. Nat. Methods [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, et al. (2014). Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science (80-. ). 343, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidrauski C, Acosta-Alvear D, Khoutorsky A, Vedantham P, Hearn BR, Li H, Gamache K, Gallagher CM, Ang KKH, Wilson C, et al. (2013). Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simms CL, Yan LL, and Zaher HS (2017). Ribosome Collision Is Critical for Quality Control during No-Go Decay. Mol. Cell 68, 361–373.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonenberg N, and Hinnebusch AG (2009). Regulation of Translation Initiation in Eukaryotes: Mechanisms and Biological Targets. Cell 136, 731–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesina P, Lessen LN, Buschauer R, Cheng J, Wu CC, Berninghausen O, Buskirk AR, Becker T, Beckmann R, and Green R (2020). Molecular mechanism of translational stalling by inhibitory codon combinations and poly(A) tracts. EMBO J. 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez de Aldana CR, Marton MJ, and Hinnebusch AG (1995). GCN20, a novel ATP binding cassette protein, and GCN1 reside in a complex that mediates activation of the eIF-2 alpha kinase GCN2 in amino acid-starved cells. EMBO J. 14, 3184–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vind AC, Snieckute G, Blasius M, Tiedje C, Krogh N, Bekker-Jensen DB, Andersen KL, Nordgaard C, Tollenaere MAX, Lund AH, et al. (2020). ZAKa Recognizes Stalled Ribosomes through Partially Redundant Sensor Domains. Mol. Cell 78, 700–713.e7. [DOI] [PubMed] [Google Scholar]
- Wang X, Mader MM, Toth JE, Yu X, Jin N, Campbell RM, Smallwood JK, Christe ME, Chatterjee A, Goodson T, et al. (2005). Complete Inhibition of Anisomycin and UV Radiation but Not Cytokine Induced JNK and p38 Activation by an Aryl-substituted Dihydropyrrolopyrazole Quinoline and Mixed Lineage Kinase 7 Small Interfering RNA. J. Biol. Chem 280, 19298–19305. [DOI] [PubMed] [Google Scholar]
- Wek RC, Jackson BM, and Hinnebusch AG (1989). Juxtaposition of domains homologous to protein kinases and histidyl-tRNA synthetases in GCN2 protein suggests a mechanism for coupling GCN4 expression to amino acid availability. Proc. Natl. Acad. Sci 86, 4579–4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolin SL, and Walter P (1988). Ribosome pausing and stacking during translation of a eukaryotic mRNA. EMBO J. 7, 3559–3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CC-C, Zinshteyn B, Wehner KA, and Green R (2019). High-Resolution Ribosome Profiling Defines Discrete Ribosome Elongation States and Translational Regulation during Cellular Stress. Mol. Cell 73, 959–970.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan LL, and Zaher HS (2019). How do cells cope with RNA damage and its consequences? J. Biol. Chem 294, 15158–15171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan LL, Simms CL, McLoughlin F, Vierstra RD, and Zaher HS (2019). Oxidation and alkylation stresses activate ribosome-quality control. Nat. Commun 10, 5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Ribosome collisions activate SAPKs (p38/JNK) (related to Figure 1) (A) Representative polysome profiles from RNaseA-digested lysates of HEK293 cells treated with emetine (EME) at 0 (black), 0.24 (blue), or 48 mg/L (grey). (B) Measurement of lysis of HeLa cells under ANS treatment at indicated concentration for 30 min. 9% Triton X-100 used as a positive control for cell lysis. (C) Phosphorylation of p38/JNK in HEK293 cells stimulated by TNFα (100 μg/L), ANS (100 mg/L), or both TNFα and ANS compared to untreated cells. (D) Phosphorylation of p38/JNK in HeLa cells treated with ANS (0.001-100 mg/L) for 15 min. (E) Phosphorylation of p38 in U2OS cells treated with ANS (0.01-100 mg/L) for 15 min. (F) T2A skipping sequence in yeast induces ribosome collisions. Ribosome stalling at the PGP motif of the T2A sequence was visualized by short RPFs (orange trace) indicating defective decoding; stacked ribosomes (i.e. collisions) were observed ~30 nt upstream of the stalling site (long RPFs in green trace). Amino acid sequences in gray with T2A sequence highlighted in black and PGP in red. Data from GEO database (GSE129128).
Figure S2. ZAKα phosphorylation under ANS stimulation (related to Figure 2) (A) Mutational analysis of ZAK. Immunoblots for p38 phosphorylation in WT MCF10A, ZAK KO, or ZAK KO cells complemented with ZAKα, ZAKβ, ZAKα-K45M, ZAKα leucine-zipper mutation (LZmut), ZAKα SAM domain truncation (ΔSAM), or ZAKα R774A K775A K776A (RKKmut) under intermediate doses of ANS (1 mg/L). Total p38 as a loading control. ZAKα and ZAKβ immunoblots indicate the relative expression levels. * denotes non-specific bands. (B) Induction of ZAKα-FLAG in ZAK OE HeLa cells by doxycycline (Dox) at 2 mg/L for 24 hr. (C) Immunoblots of a Phos-tag gel for phosphorylation of endogenous ZAKa in HeLa cells treated with ANS at 0, 1, or 100 mg/L. (D) Collapse of phosphorylated ZAKα on a Phos-tag gel by phosphatase treatment following intermediate doses of ANS stimulation (1 mg/L).
Figure S3. Enrichment of ZAKα-associated disome footprints at stop codons and known problematic peptide motifs (related to Figure 3) (A) Schematic for selective ribosome profiling of ZAKα-associated disomes. After collapsing polysomes with RNase, monosomes and RNase-resistant disomes from sucrose gradients were isolated. ZAKα-bound ribosomes from within these combined samples were immunoprecipitated, and deep-sequenced. (B) Percentage of ZAKα-associated disome footprints (average size ~ 65 nts) (relative to monosome and disome footprints combined) from Mock or ZAKα IP samples. (C and D) Enrichment of ZAKα-associated disome footprints at stop codons of several representative transcripts, TXNRD1 (C) and RPS13 (D). (E and F) Average disome occupancies centered at PD (E) and PP (F) di-peptide motifs from Mock IP (black) or ZAKα IP (magenta).
Figure S4. Activation of GCN2-mediated eIF2α phosphorylation independent of uncharged tRNAs (related to Figure 4) (A) Metacodon analysis centered at AGA codons from indicated mouse cerebella. Data from GEO database (GSE56127). (B) Phosphorylation of eIF2α in WT MCF10A cells compared to those transduced with scrambled or two distinct shRNAs against GCN2. Total eIF2α as loading control. * denotes a non-specific band. (C) Phosphorylation of eIF2α in BY4741 Saccharomyces cerevisiae strain treated with ANS at 0, 0.16, 0.8, 4, 20, or 20 mg/L for 30 min.
Figure S5. ZAK, GCN1 and GCN20 share a similar interactome (related to Figures 4 and 5) (A) Heat map of correlation coefficients for 280 common preys from ZAK, GCN1, or GCN20 Bio- ID surveys (data obtained from http://cell-map.org). (B) Zoom-in view from the bracket in (A). Black arrowheads indicate ZAK, GCN1, and GCN20 and blue arrowheads indicate RPS3 and RPS10.
Figure S6. CRISPR screens for anisomycin-sensitive genes (related to Figures 4 and 5) (A) Schematic for CRISPR screens performed under ANS selection. ANS treatment at 1 mg/L reduces cell viability ~40% while ANS treatment at 0.1 mg/L decreases cell viability ~ 20% over a 24hr time period. (B and C) Volcano plots showing effect size (log2 fold change, x axis) versus FDR (-log(fdr), y axis) with enriched genes in green and de-enriched genes in red at the higher (5% FDR, adjusted p-value < 1.64× 10−4 at 1 mg/L) (B) or the lower (5% FDR, adjusted p-value < 2.87× 10−5 at 0.1 mg/L) (C) collision-inducing dose of ANS. Pinks dots indicate genes implicated in apoptosis. (D) Over-representation analysis for genes in (B) using WebGestalt (Liao et al., 2019). (E) Cellular apoptosis measured by CellEvent caspase dye (y-axis) in indicated MCF10A cells treated with ANS at 1 mg/L for 24 hr.
Figure S7. Amino acid starvation or UV irradiation induces ribosome collisions (related to Figures 5 and 6) (A) RNase-digested polysome profiles from HeLa cells starved for glutamine without or with ISRIB pre-treatment. (B) Schematic representation of the eukaryotic translation elongation cycle. Short ribosome-protected footprints (RPFs) correspond to decoding ribosomes with an empty A site (top of triangle) while long RPFs correspond to ribosomes with a full A site (bottom two corners of triangle). (C) Biological replicates for codon-specific ribosome occupancies of short RPFs comparing UV-irradiated to untreated cells (Figure 6C). Orange dots indicate codons containing two adjacent pyrimidines. (D) Phosphorylation of ZAKα, p38, JNK and eIF2α in HeLa cells after 30 min recovery from UV-irradiation compared to untreated cells. (E) Immunoblots for p38 phosphorylation in WT MCF10A, ZAK KO, or ZAK KO cells complemented with ZAKα, ZAKα-K45M, or ZAKβ after 15 min recovery from UV irradiation compared to unstressed cells. (F) Immunoblots for p38 phosphorylation after 15 min recovery from UV irradiation in HEK293 cells with or without pretreatment with high concentrations of EME (48 mg/L) or ANS (110 mg/L) for 5 min prior to UV irradiation.
Table S1. Table for CRISPR screen hits at the higher ANS dose (related to STAR Methods)
Table S2. Table for CRISPR screen hits at the lower ANS dose (related to STAR Methods)
Table S3. Oligonucleotides used for the CRISPR/Cas9 screens (related to STAR Methods)
Data Availability Statement
Raw sequencing data were deposited in the GEO database under the accession number GSE141459.