

SUMMARY
The response to DNA damage is critical for cellular homeostasis, tumor suppression, immunity and gametogenesis. In order to provide an unbiased and global view of the DNA damage response in human cells, we undertook 31 CRISPR/Cas9 screens against 27 genotoxic agents in the retinal pigment epithelium-1 (RPE1) cell line. These screens identified 890 genes whose loss causes either sensitivity or resistance to DNA damaging agents. Mining this dataset, we discovered that ERCC6L2 (which is mutated in a bone-marrow failure syndrome) codes for a canonical non-homologous end-joining pathway factor, that the RNA polymerase II component ELOF1 modulates the response to transcription-blocking agents, and that the cytotoxicity of the G-quadruplex ligand pyridostatin involves trapping topoisomerase II on DNA. This map of the DNA damage response provides a rich resource to study this fundamental cellular system and has implications for the development and use of genotoxic agents in cancer therapy.
Graphical Abstract
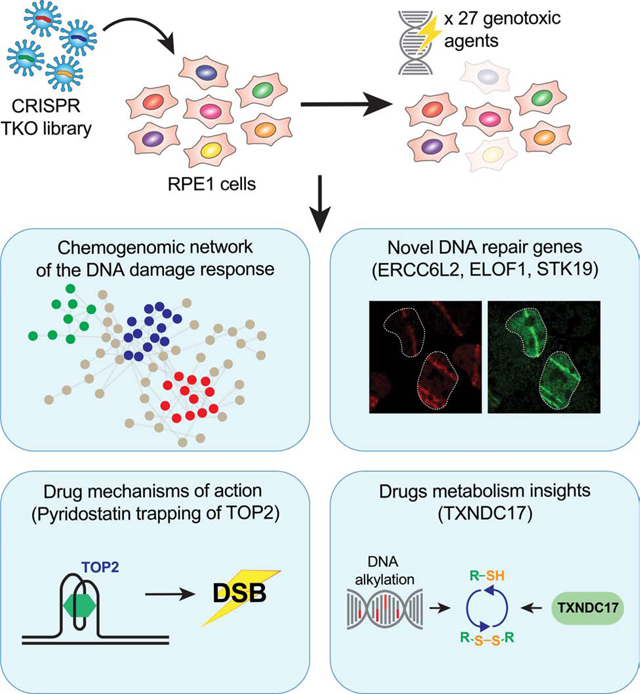
In brief
A set of CRISPR screens in cells treated with different genotoxic agents comprehensively illuminates the cellular response to DNA damage, identifying new factors in several repair pathways and pinpointing drug mechanisms
INTRODUCTION
The preservation of genetic information is a fundamental cellular process that represents a formidable challenge given that, in each cell and on a daily basis, the DNA polymer is the subject of a wide variety of chemical and physical alterations. These alterations, known collectively as DNA damage, have the potential to cause loss of genetic information, block transcription, halt DNA replication, impair chromosome segregation, generate mutations or produce chromosome rearrangements (Ciccia and Elledge, 2010; Jackson and Bartek, 2009; Lindahl and Barnes, 2000). These deleterious outcomes underlie many pathological conditions including cancer and neurodegenerative diseases and are a likely cause of cellular and organismal aging (Hoeijmakers, 2009).
To counteract the deleterious outcomes of DNA damage, a complex and interconnected network of processes detect, signal and repair DNA lesions, which is often referred to as the DNA damage response (Jackson and Bartek, 2009; Zhou and Elledge, 2000). At the centre of this genome quality-control system are the various DNA repair pathways. These include base excision repair (BER), which deals with damaged bases and uracil; nucleotide excision repair (NER), which tackles helix-distorting lesions such as UV-induced pyrimidine dimers; mismatch repair (MMR), which removes misincorporated deoxyribonucleotides; single-strand break repair (SSBR), which re-seals single-strand breaks (SSBs) and the three DNA double-strand break (DSB) repair systems: non-homologous end-joining (NHEJ), homologous recombination (HR) and microhomology-mediated end-joining (MMEJ) (Caldecott, 2008; Gupta and Heinen, 2019; Hustedt and Durocher, 2016; Lindahl and Barnes, 2000; Marteijn et al., 2014; Sfeir and Symington, 2015). Some DNA lesions require the concerted action of multiple DNA repair pathways. One example is the repair of DNA interstrand crosslinks (ICLs), which involves the dual incision of the ICL directed by a subset of the Fanconi Anemia (FA) group of proteins followed by HR, the bypass of the DNA lesion by translesion DNA polymerases and final removal of the remaining adduct by the NER machinery (Kottemann and Smogorzewska, 2013).
DNA damage detection also elicits the activation of signal transduction cascades initiated by the ATM, ATR and DNA-PK protein kinases (Blackford and Jackson, 2017). DNA damage signaling coordinates DNA repair, modulates cell cycle progression, controls DNA replication initiation and regulates a wide range of biological processes (Blackford and Jackson, 2017; Jackson and Bartek, 2009). Furthermore, since the response to DNA damage occurs on chromatin, DNA repair takes advantage of, and is modulated by, chromatin remodeling and histone modification pathways (Papamichos-Chronakis and Peterson, 2013). The presence of DNA damage can also be communicated outside the cells harboring the lesion. For example, DNA damage can lead to the activation of pattern recognition receptors, which can signal the presence of damaged cells to the immune systems for their elimination (Dhanwani et al., 2018). Therefore, the response to DNA damage is far-reaching and modulates multiple aspects of cell biology.
Despite the fact that genome maintenance mechanisms have been extensively studied, important DNA repair factors are still being uncovered. Recent examples include shieldin, which promotes NHEJ and antagonizes end-resection (Setiaputra and Durocher, 2019), and HMCES, a conserved suicide enzyme that protects abasic sites in ssDNA (Mohni et al., 2019). We also know much less about how DNA damage intersects with cellular processes that are not directly involved in the modulation of DNA repair. Therefore, there is scope in applying hypothesis-free and unbiased methods to uncover new biological features of genome maintenance control in human cells.
The emergence of CRISPR-based genetic screens has enabled genome-scale analyses of gene-gene and gene-drug interactions in human cells (Hart et al., 2015; Shalem et al., 2014; Shalem et al., 2015; Wang et al., 2015). It is therefore possible to chart the human response to genotoxic stress in an unbiased manner. Recent work illustrated the power of such screens in identifying new vulnerabilities to PARP and ATR inhibitors as well as to agents like temozolomide, a DNA alkylator (Hustedt et al., 2019a; MacLeod et al., 2019; Wang et al., 2018; Zimmermann et al., 2018). However, as these screens were carried out in isolation, they do not provide a global view of the human DNA damage response nor are they amenable to gene function inference by methods such as genetic interaction similarity profiling (Costanzo et al., 2019). We therefore aimed to map a genetic network of the response to DNA lesions and genotoxic agents, by undertaking 28 genome-scale CRISPR screens in an immortalized human diploid cell line (RPE-1 hTERT) that assessed the response to a wide variety of genotoxic agents that include multiple cancer therapeutics such as ionizing radiation, camptothecin, cisplatin and doxorubicin. The result is a dataset that uncovers new DNA repair factors, offers new insights into the mechanism-of-action of genotoxic drugs, and brings to light additional connections between genotoxic stress and extranuclear processes.
RESULTS
We undertook a set of 28 CRISPR/Cas9 dropout screens in an hTERT-immortalized RPE-1 cell line clone that expresses Flag-tagged Cas9 and in which the gene encoding p53 is knocked out (Noordermeer et al., 2018). To this set, we added three other screens in the same cell line that have been previously presented (Hustedt et al., 2019a; Hustedt et al., 2019b; Noordermeer et al., 2018). In total, the 31 screens covered 27 genotoxic agents and the main features of the screens are summarized in Table S1.
The screens were initially carried out using the TKOv2 sgRNA library (Noordermeer et al., 2018) but during the course of the study, we migrated to employing TKOv3, an all-in-one library with superior performance (Hart et al., 2017). The screens are schematized in Figure 1A and were carried out essentially as described previously (Hustedt et al., 2019b; Zimmermann et al., 2018). Briefly, cells infected with the lentiviral sgRNA library were divided into control and treated groups. The control group was left untreated and the treated group was grown in the presence of a sublethal dose of drug for approximately 10 population doublings (Table S1). Exceptions to this scheme are the “HU-acute” screen where hydroxyurea (HU) was added for 16 h every 3 d, as well as the screens probing the response to ultraviolet (UV) or ionizing radiation (IR) where cells were irradiated every 3 d at an LD20 dose (Table S1). Gene-level depletion scores were computed using DrugZ, which is optimized for chemogenomic CRISPR screens (Colic et al., 2019).
Figure 1. A chemogenomic view of the response to DNA damage.
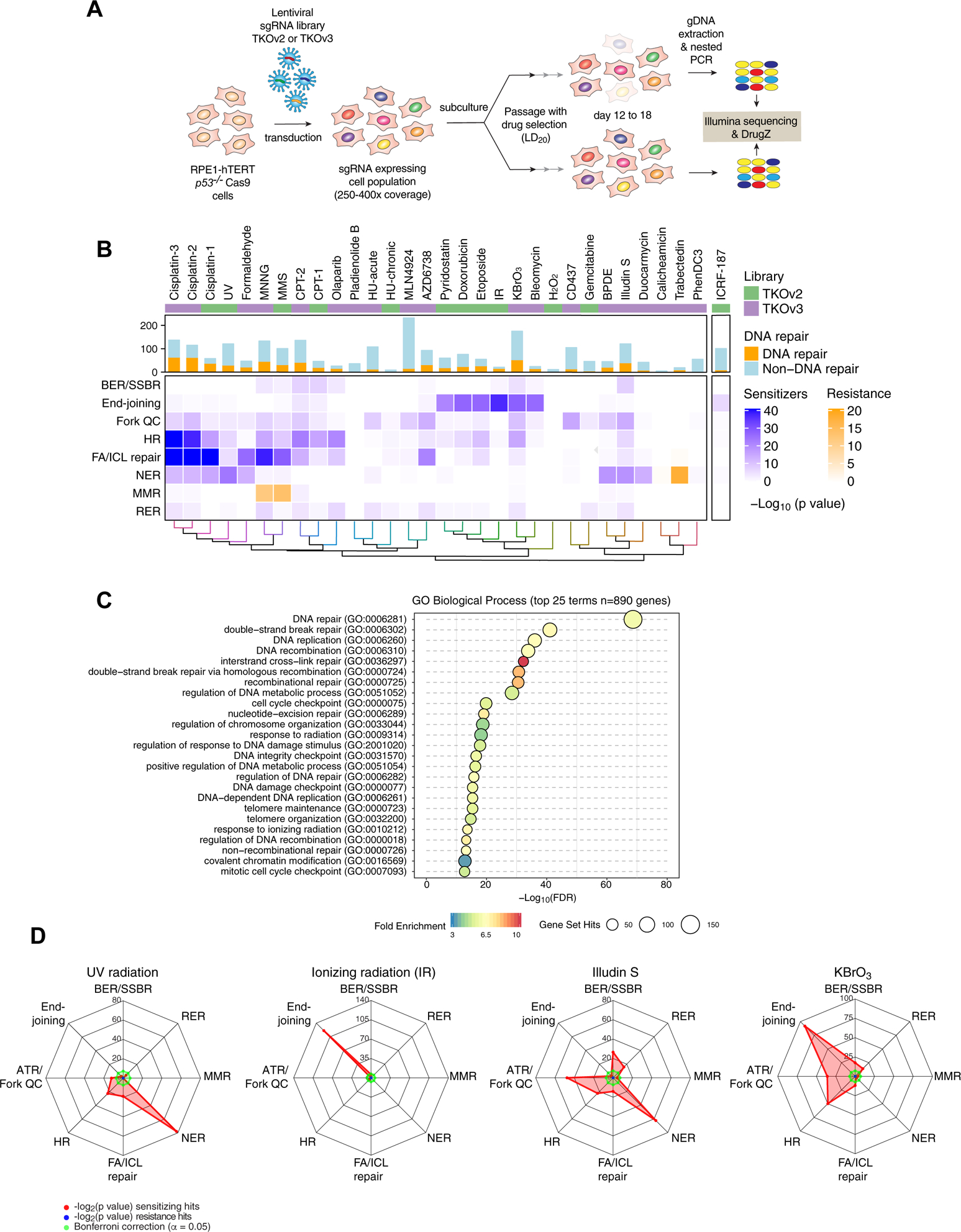
(A) Schematic of the dropout screens.
(B) Heat map representation of the 31 CRISPR screens undertaken in RPE1 hTERT p53−/− Cas9 cells. The histogram indicates the number of hits in each screen and the number of DNA repair factors identified. The lower heat map panel shows the log10-transformed p values for enrichment of different DNA repair pathways (rows) calculated using a one-sided Fisher’s exact test. The color scale indicates fold-enrichment for resistance (orange) and sensitization (blue). Pathways are defined in the text except for ribonucleotide excision repair (RER).
(C) Top 25 enriched GO terms, biological process, identified using g:Profiler (>10-fold enrichment; p < 0.05, with Benjamini-Hochberg FDR correction) among all 890 hits.
(D) Radar plots of the indicated genotoxic CRISPR screens depicting sensitization (red) or resistance (blue) for different DNA repair pathways. Values indicate the log2-transformed p values of the Fisher’s exact test score. Bonferroni thresholds are in green.
See also Figure S1.
The 27 genotoxic agents were chosen to cover most types of DNA damage (Table S1). The screens for cisplatin and camptothecin (CPT) were carried out with both TKOv2 and TKOv3, which allowed us to ascertain the reproducibility of the screens across libraries and experimenters (Figures 1B, S1 and Table S1). The gene-level normalized Z-scores (NormZ) are presented in Table S2, the readcounts are deposited in Mendeley Data (http://dx.doi.org/10.17632/gfcn2wmrpf.1), and we built a shiny app where results can be searched (https://durocher.shinyapps.io/GenotoxicScreens/). Negative NormZ values represent genes whose mutation leads to their depletion from the cell population after genotoxin exposure, whereas positive NormZ scores represent genes whose mutation leads to a selective growth advantage in the presence of the drug. The screens passed a series of quality-control tests described in the STAR methods.
Global view of the screens
We identified hits in the screens as follows: for genes whose mutation caused sensitization to the genotoxin, we selected NormZ values less than −3 with false discovery rates (FDR) lower than 15%. For genes whose mutation caused resistance to the genotoxic insult, we selected a NormZ value greater than 6. We employed this asymmetric set of thresholds since the screens were optimized to identify mutations that sensitize cells to genotoxins. These filters identified 864 genes whose loss led to sensitization and 58 genes whose mutation led to resistance, in at least one screen, for a total of 890 unique genes.
Functional term enrichment analyses using g:Profiler (Raudvere et al., 2019) showed that the 890 genes are highly enriched in DNA repair-associated terms in all categories assessed (Table S3). The DNA repair-associated terms were varied and covered most DNA repair processes ranging from DNA double-strand break repair to mismatch repair (Figures 1C, S1A and Table S3). Highly enriched terms not directly associated with DNA repair included DNA replication (GO:0006260), regulation of chromosome organization (GO:0033044) and covalent chromatin modification (GO:0016569), pointing to processes that are known to influence the response to DNA damage. While the hits from the screens were highly enriched for DNA repair-associated terms, DNA repair terms only covered 398 of the 890 genes identified, suggesting that the screens identified genes not previously associated with the response to DNA damage or that may be associated with pathways indirectly influencing the response to genotoxic agents.
To more specifically help us derive insights into the DNA repair mechanisms involved in the response to the agents screened, we curated a set of 197 DNA damage repair and signaling factors grouped according to their known function (Table S4). This list did not include genes that were not represented in our sgRNA library or that were only weakly linked to DNA repair. We next determined whether these groups were enriched in each of the screens using a Fischer exact test with Bonferroni correction and represented the data as heatmaps (Figure 1B) or radar plots (Figures 1D and S1B). For example, NER was important for survival against UV radiation, as expected, with additional contributions of FA/ICL and HR pathway genes (Figure 1B,D). Similarly, genes promoting survival against IR were enriched in NHEJ factor-coding genes, as expected (Figure 1B,D). Some less characterized agents, like illudin S, showed that multiple pathways, such as HR, DNA replication fork quality control (QC) and NER are involved in mediating cellular resistance to this agent (Jaspers et al., 2002) (Figure 1B,D). A few surprises arose from this analysis. One example was the DNA repair sensitization profile of KBrO3, an oxidizing agent and suspected carcinogen used as a food additive (IARC, 1999). KBrO3 is widely thought to oxidize DNA to produce 8-oxoguanine but whether this lesion is the root cause of bromate genotoxicity is unknown (Ballmaier and Epe, 2006). We found that KBrO3 cytotoxicity was greatly enhanced by the loss of genes acting in NHEJ, MMEJ and HR pathways, in addition to the expected sensitization by loss of BER/SSBR factors (Figure 1B,D). These observations suggest that DSBs are a relevant genotoxic lesion of KBrO3.
Pyridostatin cytotoxicity involves TOP2 trapping
Another unsuspected insight into genotoxic drug mechanism-of-action concerns pyridostatin, a compound that binds and stabilizes G-quadruplex (G4) sequences (Muller et al., 2010; Rodriguez et al., 2012). We profiled pyridostatin along with another G-quadruplex binder, Phen-DC3 (De Cian et al., 2007). Each compound had a distinct profile, with pyridostatin displaying sensitization following loss of NHEJ (Figure 1B and Figure S1B). Furthermore, pyridostatin not only clustered with other clastogenic agents but also clustered closely to etoposide and doxorubicin, two agents that cause DSBs via the poisoning of topoisomerase II (TOP2) (Delgado et al., 2018) (Figure 1B). In fact, TDP2, NBS1 (NBN) and ZATT (ZNF451) were among the top 3 hits in the pyridostatin screen and all three proteins are involved in the removal of trapped TOP2 (Aparicio et al., 2016; Schellenberg et al., 2017) (Figure 2A). These observations hinted that the mechanism of pyridostatin cytotoxicity involves the poisoning of TOP2.
Figure 2. Pyridostatin cytotoxicity involves TOP2 trapping.
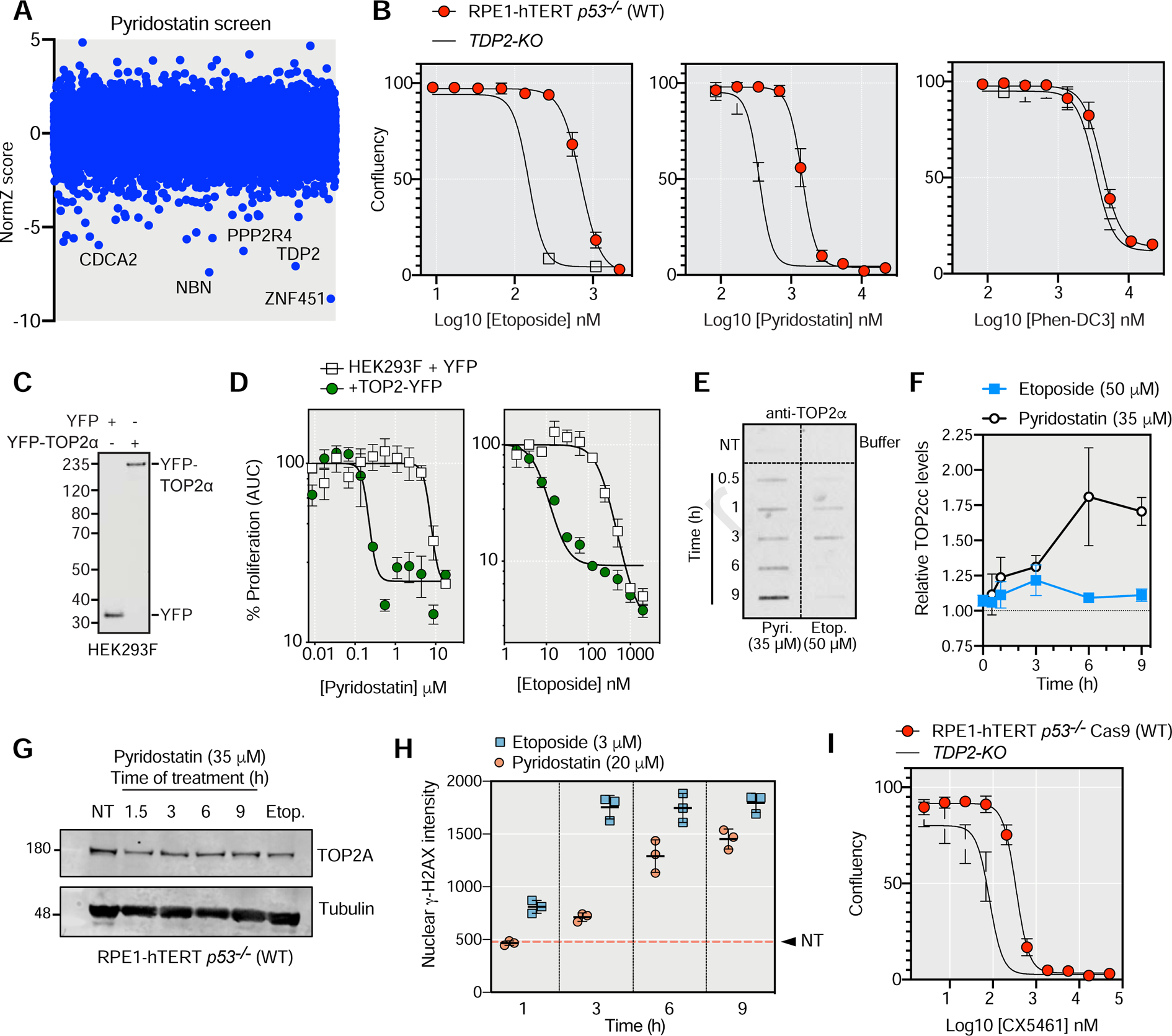
(A) CRISPR dropout screen results for RPE1 cells exposed to pyridostatin.
(B) Drug-response assays with the indicated compounds in RPE1 and isogenic TDP2-KO cells using confluency as a readout 6 d post-treatment. Data presented as mean ± SD.; N=3.
(C) Immunoblotting of YFP in HEK293F cells expressing TOP2-YFP or a control vector.
(D) Drug-response assays with pyridostatin and etoposide in HEK293F cells expressing YFP and those expressing TOP2-YFP. Data presented as mean ± SEM; N=3.
(E) RADAR assay for the detection of TOP2cc in RPE1 cells treated with etoposide (Etop.) and pyridostatin (Pyri.). The condition in upper left box represents samples that were untreated (NT). The upper right box represents buffer alone.
(F) Quantitation of the RADAR assay normalized to untreated cells. Data presented as mean ± SD; N=3.
(G) Immunoblotting for TOP2A in RPE1 cells treated with pyridostatin as indicated. As a control cells were treated with 50 μM of etoposide (Etop.) for 1.5 h. Tubulin was used as a loading control. NT, no treatment.
(H) Average γ-H2AX nuclear intensity in RPE1 cells treated with etoposide or pyridostatin determined by image segmentation. The dashed line represents the value for untreated (NT) cells. Data presented as mean ± SD; N=3.
(I) Drug-response assays with CX5461 in RPE1 and isogenic TDP2−/− cells using confluency as a readout 6 d post-treatment. Data presented as mean ± SD.; N=3.
See also Figure S2.
To test this possibility, we confirmed that loss of TDP2 hypersensitizes cells to pyridostatin but not to Phen-DC3 (Figure 2B). The extent of sensitization by TDP2 loss was similar between pyridostatin and etoposide, a bona fide TOP2 poison (Figure 2B). Furthermore, overexpression of TOP2α fused to YFP in HEK293F cells led to sensitization to both etoposide and pyridostatin (Figures 2CD) whereas depletion of TOP2β (TOP2B) by sgRNA promoted resistance to etoposide and pyridostatin to a similar extent (Figure S2B). These results are consistent with the cytotoxicity of pyridostatin involving TOP2. Assessment of TOP2 cleavage complexes (TOP2cc) using the RADAR assay (Kiianitsa and Maizels, 2014) showed that pyridostatin is a potent inducer of TOP2cc in both human (RPE1) and mouse (N2A) cells (Figures 2E,F,S2C ). We noted that the kinetics of TOP2cc formation was different between etoposide and pyridostatin, with the etoposide-induced TOP2cc being barely above the detection threshold characterized by a peak of TOP2cc 3 h post treatment (Figure 2E–G). In contrast, pyridostatin caused TOP2cc to accumulate over the duration of the experiment (9 h), reaching levels of TOP2cc that were higher than those caused by etoposide (Figure 2E,F). This increase in TOP2cc was not the consequence of higher TOP2 levels after pyridostatin treatment (Figure 2G). However, pyridostatin was less potent at evoking γ-H2AX than etoposide, with pyridostatin-induced γ-H2AX formation being delayed and blunted compared to the response caused by etoposide, which was used at lower concentrations (Figure 2H and Figure S2A). These data suggest that pyridostatin-induced TOP2cc may be converted less efficiently into DSBs than those induced by etoposide, pointing to different mechanisms of TOP2 poisoning. Since γ-H2AX formation in pyridostatin-treated cells tends to occur in regions with the propensity to form G-quadruplex structures (Rodriguez et al., 2012), one possibility is that pyridostatin promotes TOP2 trapping following G-quadruplex stabilization.
As a means to see if these results could extend to other G-quadruplex ligands, we investigated the effect CX5461, recently shown to be a G4 ligand (Xu et al., 2017). Like pyridostatin, CX5461 shows activity against HR-deficient cells and tumors (Xu et al., 2017; Zimmer et al., 2016), suggesting a common mechanism of action. We found that TDP2 loss hypersenstized cells to CX5461 (Figure 2I), consistent with the possibility that the cytotoxicity of CX5461 involves the trapping of TOP2.
Gene-level view of the screens
To visualize the activity of genes across screens, we generated fingerprint plots that give a snapshot of gene activity in cells undergoing different treatments (Figure 3A). For example, we observed that the transcription-coupled (TC)-NER factor ERCC8 (also known as CSA), promotes cellular resistance to UV, illudin S and cisplatin, as expected (Figure 3A). Some hits had exquisitely restricted profiles. For example, the dual primase and DNA polymerase PRIMPOL only scored as promoting cellular resistance to BPDE whereas ATAD5, also known as ELG1, promotes cellular resistance exclusively to the alkylating agents MMS and MNNG (Figure 3A). ATAD5 forms an alternative RFC complex that may be involved in removing DNA-bound PCNA (Kanellis et al., 2003; Kang et al., 2019). Intriguingly, DSCC1 and CHTF8, which are components of another alternative RFC complex, promote resistance to a wide variety of genotoxins and represent the group of genes that scored as hits in the highest number of screens (Figure 3A and Figure S3AB).
Figure 3. The DNA damage response network.
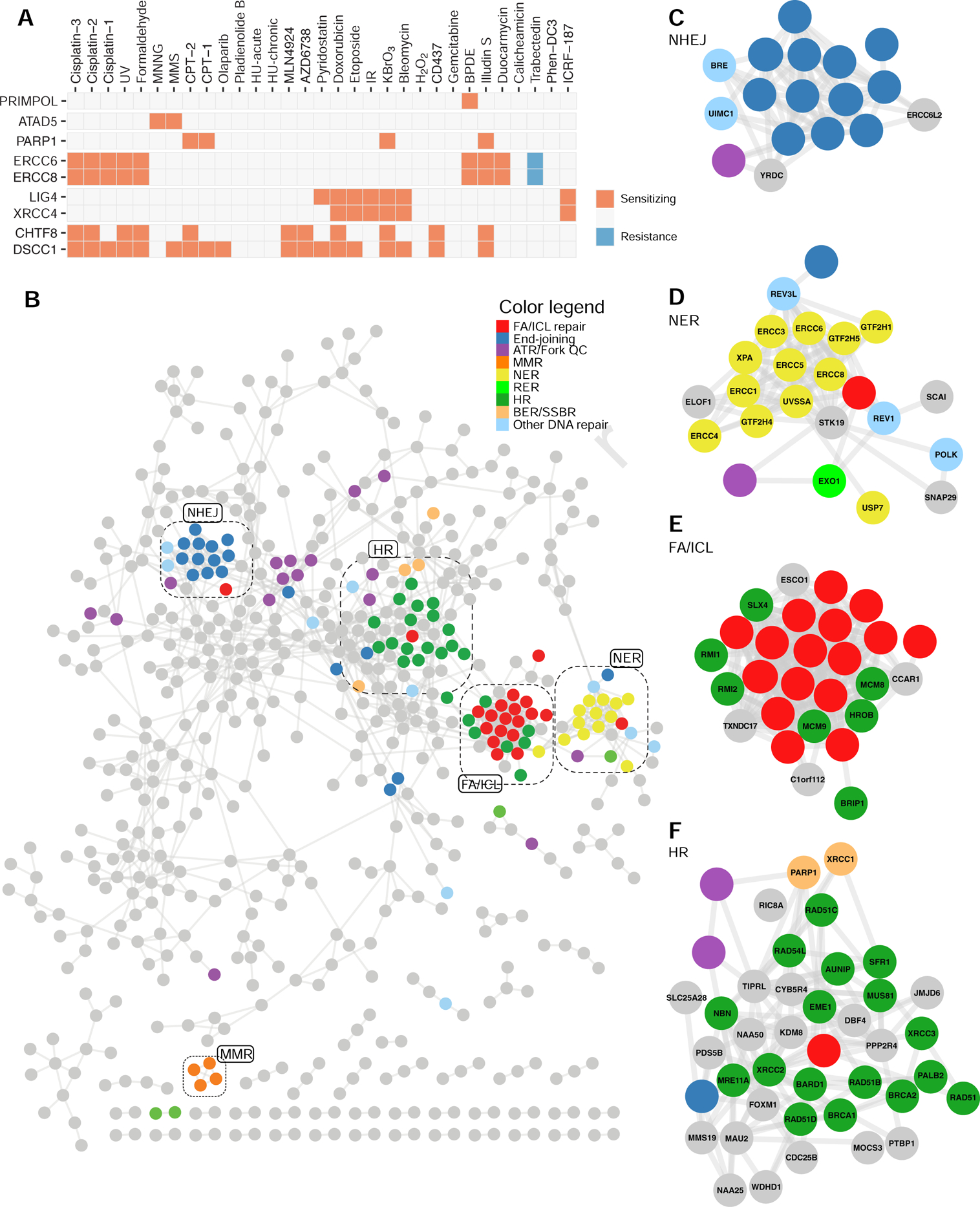
(A) Fingerprint plot of highlighted genes across the 31 screens (columns). The boxes are labeled according to whether mutations in these genes lead to sensitization (orange) or resistance (blue).
(B) Network of genes displaying at least one connection with another gene at a PCC value >0.7. Nodes are colored according to a DNA repair pathway curation (Table S4). Highlighted are the clusters enriched in DNA repair genes. A network with gene names is in Figure S3C.
(C-F) Details of the clusters enriched in NHEJ (C), NER (D), FA/ICL (E) or HR (F) pathway coding genes.
See also Figures S3 and S4.
The similarity in profiles of genes having related function (e.g. XRCC4 and LIG4 or ERCC6 and ERCC8) prompted us to undertake similarity profiling based on their Pearson correlation coefficient (PCC). This allowed us to build a network where genes are nodes and their edges are constrained by their PCC values (Figure 3B and Table S5; see Figure S3C for a network with gene names included). As expected, protein-protein interaction pairs derived from CORUM (Giurgiu et al., 2019) involving at least one of the 890 hits had higher PCC values than a randomized set of protein pairs (Figure S4A), further suggesting that the genotoxin interaction network groups together proteins of similar function.
To visualize structure within this network, we employed a PCC threshold of 0.7 in Figure 3B. The resulting 596-gene network is composed of a highly connected core subnetwork with clearly defined submodules, many of which are highly enriched in DNA repair pathways such as NHEJ, NER and ICL repair that are connected directly or indirectly to a more centrally located region enriched in HR factors (Figure 3C–F). The DNA repair-associated genes composed only 49% of the total network (291/596), suggesting that uncharacterized DNA repair factors are included in this network, and that other pathways are represented. As one example, Hippo pathway components such as NF2, FRYL, AMOTL2, LATS2 and TAOK1 clustered together in the network and their loss sensitized cells to multiple DNA damaging agents (Figure S4BC). This observation is consistent with the Hippo pathway acting as a modulator of chemosensitivity to cancer therapeutics (Nguyen and Yi, 2019) and it suggests that additional Hippo signaling modulators could be connected to these genes. Candidates for such proteins include KIRREL and FAM49B (Figure S4BC). Therefore, we expect that exploration of this network will reveal insights about a wide range of pathways that impinge on survival to genotoxic stress including, but not limited to, DNA repair.
The bone marrow failure syndrome gene ERCC6L2 encodes an NHEJ factor
We next mined the DNA damage network to uncover uncharacterized or understudied DNA repair factors. One clear example resided in a subcluster enriched in NHEJ factor-coding genes (Figure 3C). This subcluster contained core NHEJ factors (XRCC4, LIG4, NHEJ1), 53BP1-pathway modulators (53BP1, RIF1 and SHLD1–3) as well as the NHEJ regulator MRI/CYREN (Arnoult et al., 2017; Hung et al., 2018; Setiaputra and Durocher, 2019). This subcluster also contained ERCC6L2, a SWI/SNF-like ATPase (Figure 4A). ERCC6L2 was of particular interest since its mutation causes an inherited bone marrow failure (iBMF) syndrome, myelodysplastic syndrome and leukemia (Douglas et al., 2019; Jarviaho et al., 2018; Shabanova et al., 2018; Tummala et al., 2014; Zhang et al., 2016). ERCC6L2 deficiency is also associated with additional clinical phenotypes such as microcephaly, ataxia and other developmental delays that are often associated with NHEJ deficiency (Nijnik et al., 2007; Shabanova et al., 2018; Woodbine et al., 2014). While ERCC6L2 has previously been associated with genome instability, recent results point to ERCC6L2 participating in the resolution of R-loops (Tummala et al., 2018). We hypothesized that ERCC6L2 is a hitherto unrecognized NHEJ factor, a hypothesis supported by the observation that ERCC6L2 was also found to interact with MRI/CYREN (Hung et al., 2018; Rolland et al., 2014).
Figure 4. ERCC6L2 promotes canonical NHEJ.
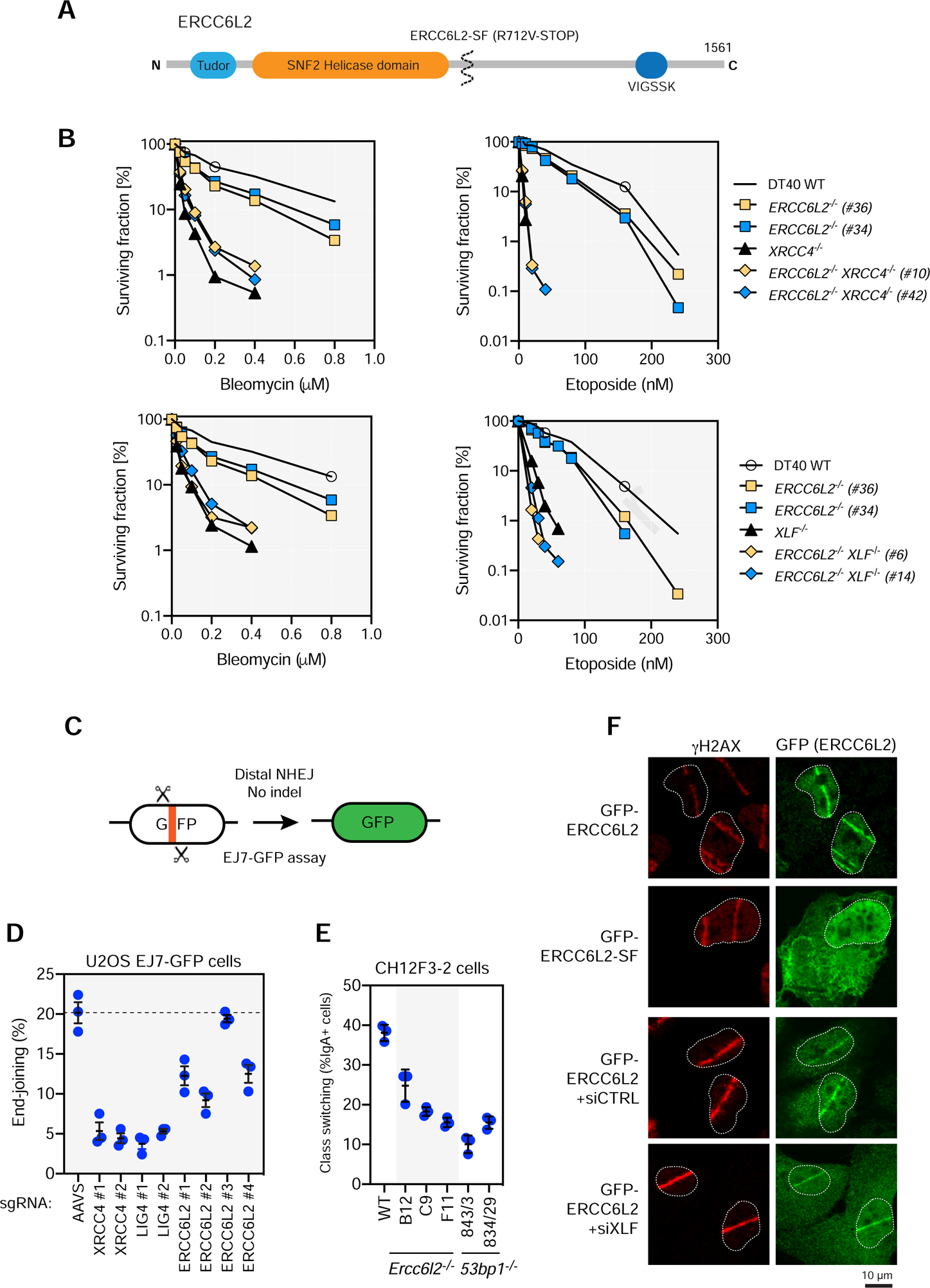
(A) Schematic overview of ERCC6L2. ERCC6L2 also possess a short isoform of 712 residues produced by alternative splicing. Most disease-associated ERCC6L2 alleles produce proteins that are truncated prior to residue 712.
(B) Cell proliferation assays of DT40 cells of the indicated genotypes treated with either etoposide or bleomycin for 3 d. Note that the bleomycin proliferation assays were done as part of the same experiment but were separated for clarity. Data presented as the mean of a technical triplicate. An independent experiment is shown in Figure S5C.
(C) Schematic of the EJ7-GFP NHEJ assay.
(D) End-joining frequency of U2OS-EJ7 cells following depletion of the indicated genes by gene editing. Dashed line represents the mean of the end-joining frequency of the AAVS1-targeted condition. Data presented as mean ± SD; N=3. TIDE analysis is shown in Table S6.
(E) Class switch recombination levels (% IgA+ cells) in CH12F3–2 cells of the indicated genotypes. Data presented as mean ± SD; N=3.
(F) U2OS cells transfected with a plasmid encoding either full-length GFP-ERCC6L2 or the short isoform (SF) were subjected to laser microirradiation and then processed for immunofluorescence with GFP and γH2AX antibodies 30 min post-irradiation. The mean percentage of cells (± SD; N=3) with γH2AX- and GFP+ stripes are indicated.
See also Figure S5.
To assess the role of ERCC6L2 in DNA repair, we first generated independent knockout clones in the chicken DT40 cell line (Figure S5A,B). The ERCC6L2 knockouts (ERCCL62−/−) were constructed in wild-type, XLF−/− and XRCC4−/− cell backgrounds to assess a possible genetic relationship between ERCC6L2 and core NHEJ factors (Xing et al., 2015). As expected from the screens, loss of ERCC6L2 led to sensitivity to etoposide and bleomycin although the extent of sensitization was milder than that caused by null mutations in XLF or XRCC4 (Figure 4B). The ERCC6L2−/− XRCC4−/− double-mutant lines showed similar sensitivity to bleomycin or etoposide as seen in XRCC4−/− cells, consistent with ERCC6L2 acting in the same genetic pathway as XRCC4 (Figure 4B and Figure S5C). The situation was similar with XLF deficiency, where the ERCC6L2−/− XLF−/− double-mutant cells showed a similar sensitivity profile as XLF−/− cells in response to bleomycin, but the double-mutant cells were slightly more sensitive to etoposide than XLF−/− cells (Figure 4B and S5C). These data indicate that ERCC6L2 acts in the canonical XRCC4-dependent NHEJ pathway but can provide support to NHEJ independently of XLF under certain conditions such as TOP2-driven DSBs.
We next employed the recently described EJ7-GFP reporter (Figure 4C) to assess the role of ERCC6L2 in NHEJ in the human U2OS cell line. The EJ7-GFP is unique among end-joining assays for being highly dependent on canonical NHEJ factors (Bhargava et al., 2018). We observed that the loss of ERCC6L2 led to a marked reduction in end-joining, albeit not as much as that caused by XRCC4 or LIG4 depletion (Figure 4D and Table S6). These results are consistent with a role for ERCC6L2 in promoting canonical NHEJ. Depletion of ERCC6L2, like that of XRCC4, does not result in PARP inhibitor resistance in RPE1-hTERT BRCA1−/− cells indicating that ERCC6L2 is unlikely act in the 53BP1-shieldin pathway (Figure S5D and Table S6) (Setiaputra and Durocher, 2019).
To assess the role of ERCC6L2 in physiological NHEJ, we examined the impact of its loss for immunoglobulin class switching (Methot and Di Noia, 2017) in mouse CH12F3–2 cells (Nakamura et al., 1996). We generated three independent Ercc6l2−/− clones and monitored switching from IgMto IgA by flow cytometry. All three Ercc6l2 knockout clones showed reduced numbers of IgA+ cells following induction of switching, with the magnitude of the defect approaching that of 53bp1−/− cells, which are highly impaired in class switching (Figure 4E). The defect was not due to reduced expression of germline transcripts (glt) at the Ig locus, or AID, which are both necessary for DSB induction, hinting at a direct role for ERCC6L2 in the repair of DNA breaks (Figure S5EF).
Lastly, we monitored recruitment of ERCC6L2 to DNA damage sites using laser microirradiation. In agreement with Zhang et al. (2016), we found that full-length ERCC6L2 fused to GFP readily localized to sites of DNA damage, in a manner that is independent of the core NHEJ factor XLF, suggesting that the localization of ERCC6L2 to damage sites is independent of the end-joining machinery (Figure 4F). However, ERCC6L-SF, which terminates the ERRCC6L2 reading fame C-terminal of the known alleles associated with iBMF (Shabanova et al., 2018) did not localize to DNA damage sites (Figures 4F and S5G). Collectively, these data suggest that ERCC6L2 is a bona fide NHEJ factor and indicates that the root cause of the bone marrow failure in individuals with mutated ERCC6L2 might be a failure to appropriately repair DSBs.
ELOF1 and STK19 modulate TC-NER
The NER cluster in the PCC network contains primarily TC-NER factors, such as ERCC8/CSB, UVSSA and various TFIIH components (ERCC3, GTF2H1, GTF2H4 and GTF2H5). Consistent with this cluster reflecting primarily TC-NER, the global-genome (GG) NER factor XPC was not connected to this group of genes even though it was a strong hit in the UV screen (Figures 3D and Table S2). Two genes within this cluster were not previously associated with TC-NER. The first is ELOF1, a CSB and RNA polymerase II-associated protein that is conserved across eukaryotes and present in archaea (Boeing et al., 2016; Daniels et al., 2009). The ELOF1 homolog in budding yeast, Elf1, collaborates with Spt5/6 (DSIF in human) to promote transcriptional elongation past the chromatin barrier (Ehara et al., 2019; Ehara et al., 2017; Prather et al., 2005). In the screens, loss of ELOF1 results in sensitivity to UV, illudin S and cisplatin, as well as resistance to trabectedin (Figure 5A). Interestingly, TC-NER factors are necessary for the cytotoxic effects of trabectedin (Takebayashi et al., 2001). This drug response profile of ELOF1 matched that of other TC-NER factors although the depletion scores for ELOF1 were generally lower than canonical TC-NER genes (Table S2).
Figure 5. Characterization of ELOF1, STK19 and TXNDC17.
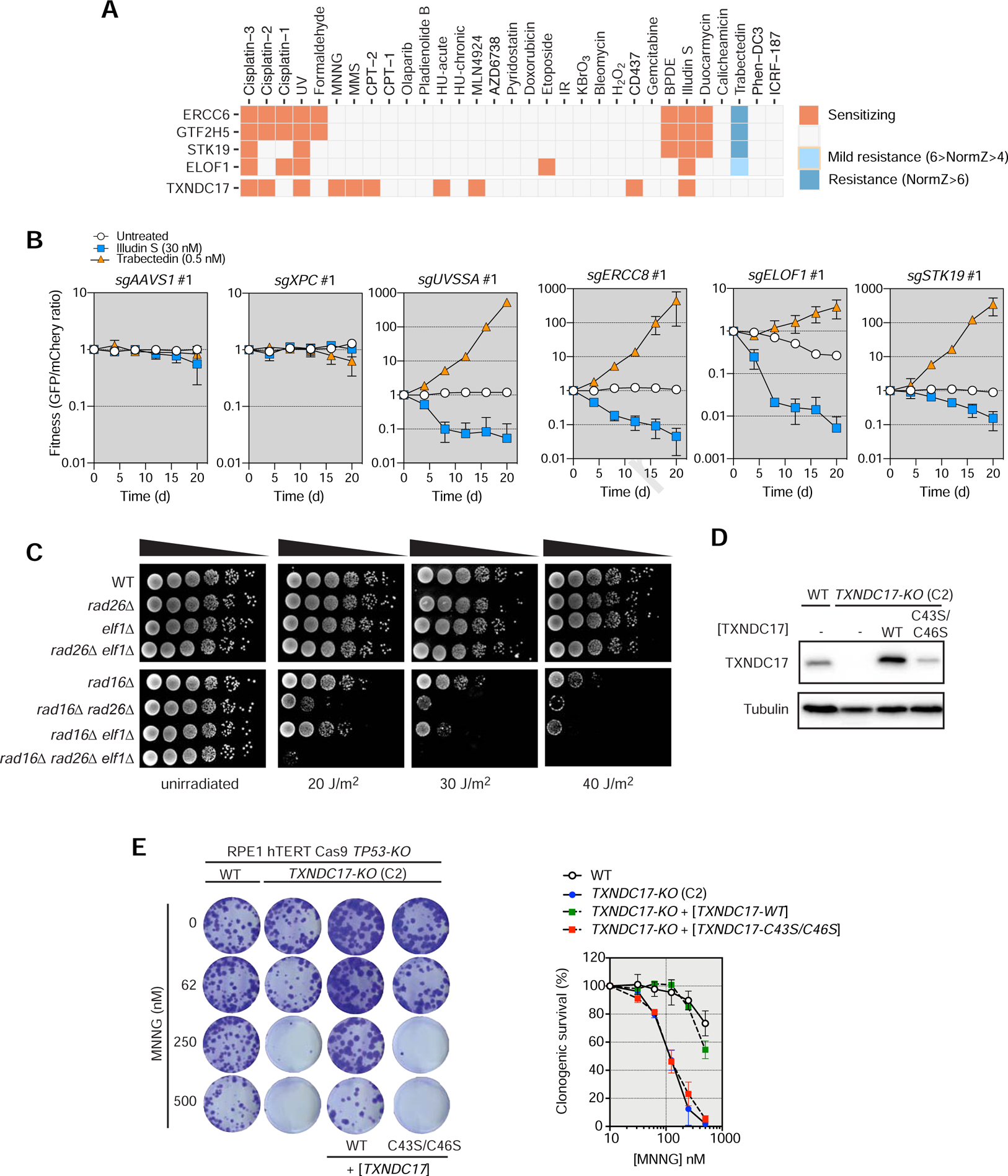
(A) Fingerprint plots of ERCC6, GTF2H5, STK19, ELOF1 and TXNDC17.
(B) Competitive growth assays with or without illudin S (30 nM) or trabectedin (0.5 nM) treatment in RPE1 cells transduced with virus expressing the indicated sgRNAs. Data represent mean fraction of GFP+ cells ± SD normalized to day 0 (N= 3, independent transductions). TIDE analysis is shown in Table S6.
(C) Cultures of S. cerevisiae strains with the indicated genotype were serially diluted and spotted onto YPD plates that were irradiated with the indicated UV dose, or left untreated. Plates were incubated at 30°C for 3 d before imaging. WT, wild type.
(D) Immunoblotting for TXNDC17 in wild type (WT) or TXNDC17-KO cells transduced with the indicated TXNDC17-encoding viruses. Tubulin was used as a loading control.
(E) Clonogenic survival of RPE1 cells of the indicated genotypes in response to MNNG. Representative images are shown (left) and quantified (right). Data represent mean ± SD (N = 3).
See also Figure S6.
To validate these results, we undertook competitive growth assays using illudin S and trabectedin. As positive controls, we used sgRNAs targeting UVSSA and ERCC8 (CSA) and, as negative controls, we used guides targeting AAVS1 and XPC. As expected, loss of UVSSA and CSA led to sensitization to illudin S and resistance to trabectedin (Figures 5B, S6A and Table S6). Independent sgRNAs targeting ELOF1 similarly led to sensitization to illudin S and resistance to trabectedin in RPE1 cells, although the magnitude of the phenotypes caused by them was generally milder than those caused by UVSSA or CSA depletion (Figures 5B, S6A and Table S6).
We next assessed whether yeast ELOF1 (Elf1) also mediates survival against UV-induced DNA lesions in fungi. We combined elf1Δ with a rad16Δ mutation that disrupts GG-NER (Prakash and Prakash, 2000). In yeast, loss of TC-NER factors only causes mild UV sensitivity on their own but their loss enhances the sensitivity of GG-NER mutants such as rad16Δ We also prepared rad26Δ rad16Δ and rad26Δ elf1Δ double mutants as well as an elf1Δ rad26Δ rad16Δ triple mutant. Rad26 is the ortholog of CSB and is involved in TC-NER. We observed that while the elf1Δ and rad26Δ mutants had no discernable sensitivity to UV, combining these mutations with rad16Δ led to an increase in UV sensitivity (Figure 5C), a phenotype that could be reversed by re-introduction of ELF1 on a plasmid (Figure S6B). Interestingly, the elf1Δ rad26Δ rad16Δ triple mutant was slightly more sensitive to UV than the rad26Δ rad16Δ mutant indicating that ELF1 promotes UV resistance at least partly independently of RAD26 (Figure 5C). Given the location of Elf1 within the elongating RNA polymerase II complex, we speculate that ELOF1 may represent a previously unassigned and evolutionarily conserved modulator of transcriptional pausing in response to RNA polymerase-blocking lesions.
A second gene embedded in the NER cluster was STK19 (Figure 3D). STK19 is attractive since it was previously reported to promote transcriptional recovery following UV irradiation and is a known CSB-interacting protein (Boeing et al., 2016). In particular, STK19 scored as the top illudin S-sensitizing hit and as the top trabectedin-resistance hit (Table S2). We confirmed these observations with two independent sgRNAs in competitive growth assays with both compounds (Figures 5B, S6A and Table S6). These results strongly suggest that STK19 can be assigned to the TC-NER pathway.
TXNDC17 links redox control to genotoxin resistance
The FA/ICL cluster contains most FANC genes along with other genes coding for factors known to participate in ICL repair such as newly characterized HROB (Hustedt et al., 2019b) (Figure 3E). Two previously uncharacterized genes in this cluster were CCAR1 and TXNDC17. CCAR1 is the paralog of DBC1 and has been previously associated with DNA damage-induced apoptosis (Sekhar et al., 2019). TXNDC17, however, has no previous link to DNA damage and TXNDC17-targeting sgRNAs sensitized cells to a variety of DNA damaging agents, primarily but not limited to, MMS, MNNG and cisplatin (Figure 5A and Figure S6C). TXNDC17 is a thioredoxin-domain containing protein and is one of at least 27 such proteins encoded in the human genome (Benham, 2012). Among those, TXNDC17 was remarkable in that it was the sole member whose loss led to strong sensitivity to more than one genotoxic agent (Figure S6C).
To validate these results, we generated independent clonal knockouts of TXNDC17 (Figure S6D) and confirmed that TXNDC17-KO cells are hypersensitive to MNNG, MMS and cisplatin but not KBrO3, an oxidizing agent, suggesting selectivity towards alkylating compounds (Figure S6E). The resistance to MNNG required its catalytic cysteines, suggesting that the oxidoreductase activity of TXNDC17 is involved in resistance to these genotoxins (Figure 5DE). Co-depletion of TXNDC17 and FANCD2 resulted in higher sensitivity to MNNG than loss of either protein alone, and FANCD2 ubiquitylation following MMS treatment occurred normally TXNDC17-KO cells suggesting that TXNDC17 does not act in the FA pathway (Figure S6FG).
TXNDC17, also known as TRP14, likely controls the metabolism of thiols, including that of L-cystine, the precursor of glutathione (GSH), by promoting its reduced state (Pader et al., 2014). Since GSH can inactivate alkylating drugs by direct conjugation (Colvin et al., 1993) reduced levels of GSH in TXNDC17-KO cells could explain sensitivity to alkylating agents. We quantitated reduced (GSH) or oxidized (GSSG) glutathione levels and found that TXNDC17 loss resulted in an increase rather than decrease in GSH without altering the GSH:GSSG ratio (Figure S6H). This suggests that TXNDC17 modulates sensitivity to alkylating agents by modulating the redox state of thiols other than GSH. In support of this possibility, addition of the reducing agent N-acetylcysteine (NAC) to TXNDC17-KO cells completely reversed their sensitivity to MNNG (Figure S6I).
CYB5R4 as a candidate modulator of protein phosphatases
CYB5R4 encodes another oxidoreductase that promotes the cellular resistance to multiple agents, including CPT for which it was the top hit in the CPT-2 screen (Figure 6A and Table S2). Mice lacking CYB5R4 develop diabetes following treatment with strepotozocin, a DNA alkylator (Xie et al., 2004), which made CYB5R4 worth exploring further. We validated that CYB5R4 loss led to CPT sensitivity in RPE1 cells (Figure 6BC and Figure S7AB). Furthermore, CYB5R4-KO cells were as sensitive as TDP1 knockout cells (TDP1-KO) to both CPT and LMP-400, the latter being a TOP1 poison of the indenoisoquinoline class (Figures 6C) (Marzi et al., 2019; Pommier and Cushman, 2009). This indicates that CYB5R4 promotes viability in the face of TOP1 trapping rather than affecting CPT directly. Interestingly, in both the DNA damage network and in a co-dependency network derived from DepMap (Tsherniak et al., 2017) (Figure 6D), CYB5R4 is connected to Ser/Thr phosphatases, PPP2R4 and TIPRL, the latter two encoding proteins that regulate the assembly and disassembly of PP2A complexes, respectively (Guo et al., 2014; Wu et al., 2017). sgRNAs against PPP2R4 caused sensitization to CPT (Figures 6B, S7A and Table S6) and thus we speculate that CYB5R4 regulates the response to DNA damage via the modulation of PP2A-family phosphatases.
Figure 6. CYB5R4 promotes resistance to TOP1 poisons.
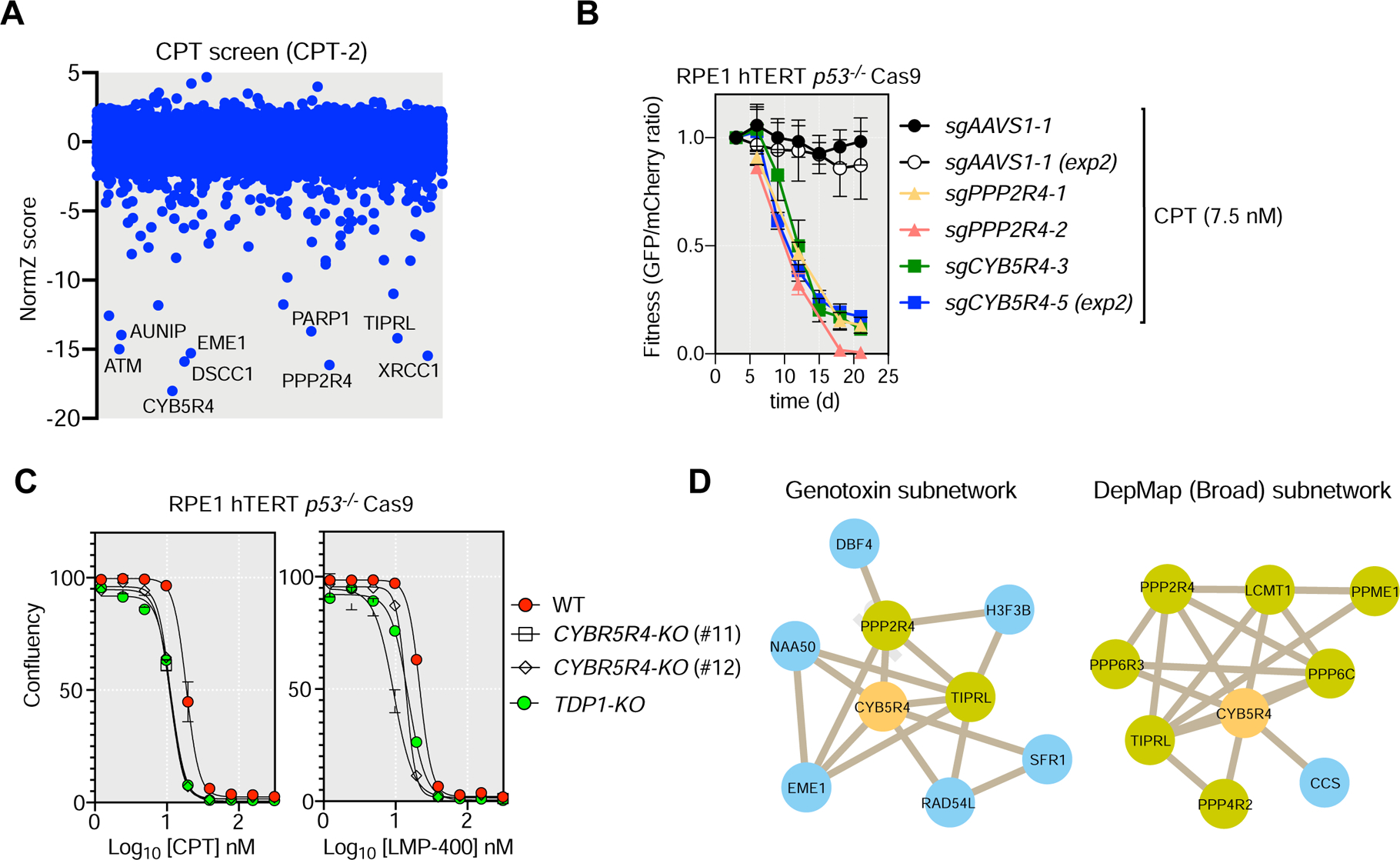
(A) CRISPR dropout screen results for RPE1 cells exposed to camptothecin (screen CPT-2).
(B) Competitive growth assays with camptothecin (CPT; 7.5 nM) treatment in RPE1 cells transduced with virus expressing the indicated sgRNAs. Data represent mean fraction of GFP-positive cells ± SD normalized to day 0 (N= 3, independent transductions). TIDE analysis is shown in Table S6. See Figure S7AB.
(C) Drug-response assays with camptothecin (CPT) and LMP-400 in RPE1 cells and isogenic TDP1-KO and CYB5R4-KO clones using confluency as a readout 6 d post-treatment. Data presented as mean ± SD.; N=3, independent experiments.
(D) Subnetworks derived from the PCC genotoxin network derived in this study or from the DepMap data.
DISCUSSION
In this work, we present a characterization of the genetic architecture of the response to DNA damaging agents in a model human cell line. Illustrating the power of this dataset to provide new insights into the organization of the DNA damage response, we uncovered previously uncharacterized DNA repair factors, revealed unsuspected drug mechanisms-of-action, and have evidence of new drug metabolism pathways. In addition to the genes highlighted in this manuscript, we discuss in the associated Mendeley Dataset additional genes of interest (TMEM2, ESD, USP37, PHF12 and BTAF1-DRAP1; file “additional genes of interest”; http://10.17632/gfcn2wmrpf.1).
With respect to DNA repair factors, the assignment of ERCC6L2 to the canonical NHEJ pathway is noteworthy and surprising given that end-joining reactions can be reconstituted in vitro with a set of purified components that does not include ERCC6L2 (Zhao et al., 2019). It remains unclear what role an enzyme of the SWI/SNF family may have during the end-joining reaction. Given that members of this enzyme family function as dsDNA translocases, one simple model may be that ERCC6L2 removes roadblocks that impair the completion of the end-joining reaction. Our results also suggest that DSBs of unknown origin underlie the bone marrow failure in individuals with ERCC6L2 mutations. Therefore, finding the endogenous sources of DSBs in the hematopoietic stem cell compartment, similarly to what was done recently for Fanconi Anemia (Pontel et al., 2015) may provide important clues as to the pathogenesis of this syndrome and may reveal paths of intervention that could benefit those patients.
This work also helps solidify a number of previous observations relating to homologous recombination. Indeed, we note a number of genes embedded within this group for which evidence of a role in HR was either circumstantial, indirect or supported by very few studies. Examples are AUNIP, KDM8/JMJD5 and JMJD6 (Figure S3C) (Amendola et al., 2017; Huo et al., 2019; Lou et al., 2017). In particular, recent work on KDM8 and JMJD6 suggest that instead of acting as lysine demethylases they are rather acting as lysyl and arginyl hydroxylases, respectively (Webby et al., 2009; Wilkins et al., 2018). It will be fascinating to assess whether this translational modification regulates this key genome maintenance pathway.
We note that our work only provides a partial view of the architecture of the human DNA damage response. Indeed, there are inherent technical limitations of dropout chemogenomic CRISPR screens (Dede et al., 2020). One such limitation is that we used an engineered RPE1 cell line in which p53 was inactivated. While this option allowed us to profile the genotoxin sensitivity of many genes whose inactivation trigger a p53-dependent cell cycle arrest, it also precluded us from probing the links between p53 and genotoxic stress responses. Another limitation of these dropout screens is that they lack the power to probe the function of many cell-essential genes. For example, the sgRNAs targeting the gene encoding the essential recombinase RAD51 are depleted too rapidly in our cell lines (with and without drugs) to produce statistically reliable results. Furthermore, our screens only used a single cell line model, RPE1-hTERT, and previous work on ATR and PARP inhibitors, among others, have revealed cell line-specific responses to these drugs that reflect their biology and tissues of origin (Hustedt et al., 2019a; Zimmermann et al., 2018). Finally, we have not explored the full range of genotoxic agents and this dataset will benefit from additional screens probing the response to compounds such as antimetabolites or environmental carcinogens.
Lastly, the work presented here has implications for the development of therapeutics and genetic toxicology. For example, the characterization of compounds using chemogenomic CRISPR screens or via the use of isogenic cell line panels derived from this work may enable the rapid identification of desired (or undesired) genotoxic effects. Furthermore, the dataset presented can be mined to elaborate patient-selection hypotheses. For example, deletions and mutations in the MCPH1 gene occur frequently in bladder and colorectal cancer (Figure S7C). Loss of MCPH1, a protein with dual function at centrosomes and as a poorly characterized reader of H2AX phosphorylation (Liu et al., 2016), sensitizes cells to etoposide, a cancer chemotherapeutic (Table S2). These results suggest that tumors with MCPH1 loss may be vulnerable to etoposide treatment, a drug already used in the clinic. Finally, our data indicates that a subset of G-quadruplex ligands have the unsuspected property of trapping TOP2 in a manner that produces signaling-competent DSBs at a slow rate. Since this mechanism of action is clearly distinct from canonical TOP2 poisons like etoposide, it is conceivable that the TOP2-trapping property of these agents could be exploited therapeutically.
STAR METHODS
LEAD CONTACT
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Daniel Durocher (durocher@lunenfeld.ca).
MATERIALS AVAILABILITY
DNA constructs and other research reagents generated by the authors will be distributed upon request to other research investigators under a Material Transfer Agreement.
DATA AND CODE AVAILABILITY
All the raw data generated is available on Mendeley (http://dx.doi.org/10.17632/gfcn2wmrpf.1). The code used for this paper is based on free accessible software described in the relevant STAR method section and uploaded on the Mendeley database.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and yeast strains
Generation of RPE1-hTERT Cas9 TP53-KO (RPE1, female human [Homo sapiens] retinal pigmented epithelium) has been previously described (Zimmermann et al., 2018). Cas9-expressing cell lines were maintained in the presence of 2 μg/mL blasticidin. RPE1 TDP2−/− cells were generated by transfecting with the sgRNA targeting TDP2 (see Table S6) using RNAiMAX (Invitrogen) according to the manufacturer’s protocol. 24 h after transfection, cells were split and seeded in a 60 mm plate. After an additional 72 h, single cells were sorted into 96-well plates on a BD FACSAria Cell Sorter instrument and grown until colonies formed. TDP2−/− clones were selected on the basis of successful gene editing determined by PCR amplification and TIDE analysis (Brinkman et al., 2014). sgRNAs used for transfection were ordered from Integrated DNA Technologies. crRNAs containing the target-specific sequence for guiding Cas9 protein to a genomic location were annealed with tracrRNA to form a functional sgRNA duplex. DT40 (female chicken [Gallus gallus] lymphoblast) knockout constructs for ERCC6L2, XRCC4 and XLF were generated as previously described using a MultiSite Gateway three-fragment vector construction kit. 5’ and 3’ arms were cloned into the pDONR P4-P1R and pDONR P2R-P3 vector, respectively. The knockout constructs were generated by attL×attR recombination of the pDONR-5′ arm, pDONR-3′ arm, resistant gene cassette-containing pDONR-211 and the pDEST R4-R3 destination vector. TXNDC17 gene knockouts were generated by electroporation of LentiGuide vectors (see Table S6) using an Amaxa II Nucleofector (Lonza, Basel). 24 h after transfection, cells were selected for 48 h with 15 μg/mL puromycin, followed by single clone isolation. Gene mutations were further confirmed by PCR amplification and TIDE analysis. Stable integration of pHIV-TXNDC17 in RPE1-hTERT TP53−/− Cas9 (RPE1) cells was achieved by lentiviral transduction. Transduced RPE1 cells were selected by culturing in the presence of 400 μg/ml nourseothricin (Jena Bioscience, cat # AB-102L). Anti-TRP14 antibody (1:1,000, Abcam #ab121725) was used to validate correct knockout and complementation of TXNDC17. CH12F3–2 B cell (male mouse [Mus musculus] lymphoma) mutant lines were edited by transient transfection with pX330 plasmid constructs expressing sgRNAs against Trp53bp1 and Ercc6l2 and were selected on the basis of successful gene editing determined by PCR amplification and TIDE analysis (Brinkman et al., 2014) (see Table S6). HEK293F cells (Invitrogen, female human [Homo sapiens] embryonic kidney) were cultured in DMEM with 10% (v/v) fetal bovine serum (Gibco), 50 U/μL penicillin, 50 μg/mL streptomycin, and 0.5 mmol/L sodium pyruvate at 37°C in 5% CO2 atmosphere. Cas9-expressing HT1080 was generated by transducing HT1080 (ATCC, male human [Homo sapiens] connective tissue) with Lenti-Cas9-2A-Blast (Addgene Cat# 73310), selecting with 7.5 μg/ml Blasticidin, and isolating individual clones. U2OS (human [Homo sapiens] bone) EJ7 cells originate from the laboratory of Jeremy Stark and were a kind gift. RPE1-hTERT-derived cell lines, 293T and derived cell lines were grown in Dulbecco’s Modified Eagle Medium (DMEM; Gibco/Thermo Fisher #11965092) supplemented with 10% fetal bovine serum (FBS; Wisent), 1x GlutaMAX, 1× non-essential amino acids (both Gibco/Thermo Fisher), 100 U/mL penicillin and 100 μg/mL streptomycin (Pen/Strep; Wisent). U2OS-derived cell lines were grown in McCoy’s 5A (Gibco #1660–082) + 10% FBS (Gibco) + 100 U/mL penicillin and 100 μg/mL streptomycin (Pen/Strep; Wisent) + 1x L-Glutamine (final 2 mM, Wako Chemical #073–05391). DT40 cells were grown in RPMI-1640 medium + 10% FBS (Gibco) + 2% Chicken serum (Gibco) + 100 U/mL penicillin and 100 μg/mL streptomycin (Pen/Strep; Wisent) + 10 mM HEPES and grown at 39°C. All other cell lines were grown at 37°C and atmospheric O2. All cell lines were routinely authenticated by STR and tested negative for mycoplasma.
The S. cerevisiae strains in used this study are all derivatives of S288c BY4741 (Brachmann et al., 1998) and are described in Table S7. BY4741-derived elf1::KANMX and rad26::KANMX deletion strains as well as a Y7092-derived rad16::NATMX strain were kind gifts of Charlie Boone and were used as starting strains or sources of disrupted alleles. We constructed all strains using standard genetic techniques. The original elf1::KANMX strain we received was discovered to have unusual growth properties on synthetic media, therefore we recreated the elf1 deletion strain by amplifying the elf1::KANMX genomic locus by PCR, transforming the reaction product into ELF1 wild-type yeast and screening G418-resistant colonies for normal growth rates and by diagnostic PCR for correct integration.
METHODS DETAILS
CRISPR screens
RPE1-hTERT Cas9 TP53−/− cells were transduced with the lentiviral TKOv2 or TKOv3 library (Hart et al., 2015; Hart et al., 2017) at a low MOI (~0.35) and puromycin-containing medium was added the next day to select for transductants (see Table S1 for more details). The next day cells were trypsinized and replated in the same plates while maintaining the puromycin selection. 3 d after infection, which was considered the initial time point (t0), cells were pooled together and divided in 2 technical replicates (usually labelled as “A” and “B”). Negative-selection screens were performed by subculturing cells at days 3 and 6 (t3 and t6), at which point each replicate was divided into different treatments, including one that was left untreated as a control (NT), and then subcultured every 3 days (t9, t12 and t15) until the final timepoint at t18. For all screens, drugs and treatments were applied according to previous evaluation of LD20 concentrations in uninfected RPE1 cells treated for 12 d (except for gemcitabine, which was determined over 3 d). Screens done using duocarmycin SA, trabectedin and calicheamicin resulted in higher lethality than expected due to a narrow toxicity window and thus the screens were done at LD50 concentration. Cells were subcultured and medium with and without drugs was refreshed every 3 d. UV and IR treatments were applied one day after subculturing cells (t7, t10, t13 and t16). The “HU-acute” screen was performed by adding hydroxyurea (HU) for 16 h ever 3 d. Cell pellets were frozen at t18 for gDNA isolation. Screens were performed in technical duplicates and library coverage of ≥ 375 cells per sgRNA was maintained at every step with the exception of the gemcitabine screen which was carried out with a 250-fold coverage. gDNA from cell pellets was isolated using the QIAamp Blood Maxi Kit (Qiagen, Cat# 51194) and genome-integrated sgRNA sequences were amplified by PCR using Q5 Mastermix Next Ultra II (New England Biolabs, Cat# M5044L). Samples preparations was done amplifying the sgRNA with primers forward: V3_2_F: 5’-CTGCGTGCGCCAATTCTG-3’, reverse: V3_1_R2: 5’-AGAACCGGTCCTGTGTTCTG-3’, follow by a second PCR reaction containing i5 and i7 multiplexing barcodes and final gel-purified products were sequenced on Illumina NextSeq500 systems to determine sgRNA representation in each sample.
We initiated the screening campaign with the TKOv2 library, which is an sgRNA-only library (Brown et al., 2019; Noordermeer et al., 2018). During the campaign, the TKOv3 library became available (Hart et al., 2017) and was used thereafter given its superior performance. TKOv3 is an all-in-one library, meaning it expresses both the sgRNA and Cas9. TKOv2 contains 70 697 guides targeting 17 943 genes while TKOv3 contains 71 090 guides targeting 18 056 genes, 113 genes more than TKOv2. Comparing the sgRNAs, 32 684 guides in the TKOv3 are also present in the TKOv2 (i.e. a 45.9% overlap).
Immunoblots
Immunoblots for TOP2A was done using mouse monoclonal anti-human TOP2α (1:1000, 5% milk, TBST, Santa Cruz, Cat# 365916) incubated over night at 4°C. To confirm successful knock-out of CYB5R4 and TXNDC17, mouse monoclonal anti-CYB5R4 (1:500, 5% milk, TBST, Santa Cruz, Cat# sc-100529) and rabbit polyclonal anti-TRP14 (1:1000, 5% milk, TBS, TXNDC17) (Abcam, Cat# ab121725) were used, respectively. AID protein was detected by immunoblotting with mouse mAb L7E7 (Cell Signaling #4975) and normalised to β-actin (Sigma #A2066) levels. As loading control, immunoblots were performed with mouse monoclonal anti-α-Tubulin (1:1000, 5% milk, TBST, Millipore, Cat# CP06). Immunoblot to evaluate FANCD2 ubiquitylation was done using rabbit monoclonal anti-FANCD2 (1:5000, 5% milk, TBST, Novus Biologicals #NB100–182). To evaluate TOP2-YFP, immunoblot was performed using rabbit polyclonal anti-YFP (1:2500, 5% mil, TBST, Santa Cruz # sc-32897).
EJ7 assay
U2OS-EJ7 cells were infected with viruses produced in HEK293T using second generation vectors psPAX2 (Addgene #12259) as packaging plasmid, pMD2.G (Addgene #12259) as envelope and pLentiCRISPRv2-mCherry (Addgene #99154) as transfer vectors. All sgRNAs cloned into pLentiCRISPRv2-mCherry are reported in Table S6. After 6 d in culture, cells were plated at 70–80% confluence in 6 well plates and transfected with 1μg of each plasmid containing 7a and 7b sgRNAs (Addgene #113620 and #113624) using Lipofectamine 2000 (ThermoFisher, #11668030). 48 h after transfection cells were resuspended, washed in PBS and analysed using a BD LSR Fortessa.
Immunofluorescence
Cells were seeded (~10,000 cells/well) in 96-well plates and cultured for 24 h. 20 μmol EdU (5-ethynyl-2-deoxyuridine, Life Technologies) was added 30 min prior washing with PBS and fixation with 4% paraformaldehyde (PFA) in PBS for 10 min. Cells were rinsed with PBS and permeabilized using 0.3% TritonX-100/ PBS for 30 min. Cells were washed with PBS and incubated in blocking buffer (0.2% fish skin gelatin, 0.5% BSA/PBS) for 30 min. Fresh blocking buffer containing mouse anti-γH2AX (Millipore #JBW301, 1:5,000) was added for 2 h. Cells were rinsed three times with PBS and blocking buffer with AlexaFluor 488-coupled goat anti-mouse antibody (Life Technologies, 1:1000) and 0.8 μg/mL DAPI (4,6-diamidino-2-phenylindole, Sigma) was added for 1h. After rinsing with PBS, immunocomplexes were fixed again using 4% PFA/PBS for 5 min. Cells were rinsed with PBS and incubated with EdU staining buffer (150 mM Tris/HCl pH8.8, 1mM CuSO4, 100 mM ascorbic acid and 10 μM AlexaFluor azide 647 (Life Technologies)) for 30 min. After rinsing with PBS, images were acquired on an In Cell Analyzer 6000 automated microscope (GE Life Sciences) with a 60X objective. Image analysis was performed using Columbus (PerkinElmer). Cell cycle profiling and analysis was evaluated based on EdU and DAPI staining. For GFP-ERCC6L2 laser stripe analysis, 450 000 U2OS cells were seeded on glass coverslips in a 6 well plate and either reverse transfect with siCTRL or siXLF. The day after cells were transfected with 3 μg of pcDNA5-FRT-TO-EGFP-ERCC6L2 or pcDNA5-FRT-TO-EGFPERCC6L2-SF vector. 48 h post transfection, cells were micro-irradiated, pre-extracted 10 min on ice with NuEx buffer (20 mM HEPES, pH 7.4, 20 mM NaCl, 5 mM MgCl2, 0.5% NP-40, 1 mM DTT and protease inhibitors) followed by 10 min 2% PFA fixation 30 minutes after micro-irradiation. Antibody staining and blocking were performed in PBS + 0.1% Tween-20 + 5% BSA using GFP (1:3000) and γH2AX (1:1000) antibodies. siRNA efficiency was analysed by qPCR using the primers indicated in the STAR table (see also supplementary data on Mendeley).
RADAR assay for detection of TOP2 cleavage complexes
The assay was performed as previously described (Anand et al., 2018). RPE1-hTERT Cas9 TP53−/− cells were seeded in 6 well plates (1×106 cells each). 24 h later cells were lysed directly in the plates using 1 mL of buffer containing 5 M guanidinium isothiocyanate, 10 mM Tris-Cl (pH 6.5), 20 mM EDTA, 4% TritonX-100/PBS, 1% Sarkosyl (Sigma-Aldrich #L9150), 1% DTT. Genomic DNA was precipitated by incubating the lysate at −20°C for 5 min with 0.5 volumes of 100% ethanol, shaking vigorously, and centrifuging at 14,000 rpm for 15 min at 4 °C. The DNA pellet was washed 2 times in 75% EtOH by vortexing and centrifuging again at 14,000 rpm for 15 min at 4 °C. The final DNA pellet was resuspended in 100 μL of in freshly prepared 8 mM NaOH solution. The samples were incubated at 65°C for 5 min, passed through a 26G needle 10 times and then quantified using a Nanodrop (ThermoFisher). For each sample, 7 μg of DNA was resuspended in 200 μL of 25 mM NaPO4 (pH 6.5) buffer and loaded in a slot blot apparatus (Bio-Dot® SF Microfiltration Apparatus, Bio-Rad). Membranes were blocked for 1 h in 5% milk/TBS Tween-20 solution and incubated for 1 h with anti-TOP2β (1:5,000, mouse anti-human, Clone 40/Topo IIβ, RUO – 611492, BD Biosciences-US) or anti-TOP2α (1:10,000, mouse anti-human, Topo IIα antibody (F-12), sc-365916, Santa Cruz) diluted in 5% milk/TBS-Tween-20.
Class switch recombination assays
To induce switching in CH12F3–2 murine B cell lymphoma cell or their derivatives, 2×105 cells were cultured in CH12 medium supplemented with a mixture of IL4 (10 ng/mL, R&D Systems #404- ML-050, Minneapolis, MN, USA), TGFβ (1 ng/mL, R&D Systems #7666-MB-005) and anti-CD40 antibody (1 μg/mL, #16-0401-86, eBioscience, Thermo Fisher) for 48 h. Cells were then stained with anti-IgA-PE (IgA Monoclonal Antibody (mA-6E1), PE, eBioscience from Thermo Fisher Scientific, catalog # 12–4204-82, RRID AB_465917) and the fluorescence signal was acquired on an LSR II or Fortessa X-20 flow cytometer (BD Biosciences).
Quantitative PCR was performed with qPCRBIO SyGreen Blue mix (PCR Biosystems) and CFX384 Real-Time PCR Detection System (Bio-Rad) according to manufacturers’ instructions. The Iμ sterile transcript was detected with primers 5’-GAACATGCTGGTTGGTGGTT-3’ and 5’-TCACACAGAGCATGTGGACT-3’; the Iα transcript was detected with primers 5’-GGGACAAGAGTCTGCGAGAA-3’ and 5’-TCAGGCAGCCGATTATCACT-3’, and normalised to HPRT (primers 5’-CCCAGCGTCGTGATTAGC-3’ and 5’-GGAATAAACACTTTTTCCAAAT-3’). All samples were collected 48 h after stimulation with anti-CD40, IL4, and TGFβ as described above.
Clonogenic survival assays
RPE1 and RPE TXNDC17-KO cells were seeded in 6-well plates (250 cells per well) and treated with indicated concentrations of MMS, cisplatin, MNNG, or KBrO3. After 14 days, colonies were stained with crystal violet solution (0.4% (w/v) crystal violet, 20% methanol) and manually counted. Relative survival was calculated for the drug treatments by setting the number of colonies in non-treated controls at 100%.
Plasmids and viral vectors
DNA corresponding to sgRNAs was cloned into LentiGuide-Puro (Addgene: 52963), LentiCRISPRv2 (Addgene: 52961), or a modified form of LentiGuide-Puro in which Cas9 was replaced by NLS-tagged GFP or mCherry using AgeI and BamHI (designated as LentiGuide-NLS– GFP or –mCherry respectively), as described (Noordermeer et al., 2018). Lentiviral particles were produced in 293T cells by co-transfection of the targeting vector with vectors expressing VSV-G, RRE and REV using calcium-phosphate, PEI (Sigma-Aldrich) or Lipofectamine 2000 (ThermoFisher). Viral transductions were performed in the presence of 8 μg/mL polybrene (Sigma-Aldrich) at an MOI <1, unless stated otherwise. Transduced RPE1 cells were selected by culturing in the presence of 20 μg/mL puromycin for 3 d. The TXNDC17 coding sequence was obtained from the ORFeome collection (http://horfdb.dfci.harvard.edu/), archived in the Lunenfeld-Tanenbaum Research Institute’s OpenFreezer (Olhovsky et al., 2011). The complete TXNDC17 coding sequence was PCR-amplified and cloned into the pHIV-NAT-T2A-hCD52 vector using NotI and EcoRI restriction enzymes. To generate a catalytic dead TXNDC17 mutant, the active-site Cys residues were replaced by Ser (C43/46S) using PCR-mediated site-directed mutagenesis. ERCC6L2 expressing vectors were cloned using the following sequences: pcDNA5-FRT-TO-EGFP-ERCC6L2 from uniprot: PRO_0000326086 and pcDNA5-FRT-TO-EGFP-ERCC6L2-SF from ENST00000665077.1.
Cell proliferation assays
RPE1 cells were seeded in 96-well plates (2,000 cells per well) and treated with sequential serial dilutions of genotoxic agents as indicated. After 4 days of treatment, the cell confluency was measured using an IncuCyte Live-Cell Analysis imager (Essen/Sartorius). Confluence growth inhibition was calculated as the relative confluency compared to untreated cells.
YFP-TOP2α overexpression and pyridostatin sensitivity
To generate cells that overexpress YFP or YFP-TOP2α, HEK293F were transfected with a plasmid encoding YFP-TOP2α or its parental YFP vector (Schellenberg et al., 2018), followed by growth under selection with 20 μg/mL blasticidin for 3 weeks. Cells with high levels of YFP expression were selected by FACS. A single YFP-positive cell was propagated to generate the cell line that overexpresses YFP–TOP2α. 96-well plates were seeded with 2000 cells per well of the indicated cell line in 200 μL DMEM media. After growth for 24 h, the indicated concentration of DNA damaging agent was added. For proliferation (AUC) assays, cells were grown in an IncuCyte Zoom live cell imager (Essen/Sartorius) with images recorded every 2 h for 4 d after addition of drug. The confluence percentage for each image was calculated using the IncuCyte Zoom software and summed over the course of the experiment for each well. Viability measurements were normalized to that of untreated cells and fit to a 4-parameter dose-response curve to calculate IC50 values.
Yeast spot assay
ELF1 rescue plasmids were created by ligating PCR products from wild-type genomic DNA template into the BamHI/XhoI sites of either pRS416 (Sikorski R. S. and Heiter P., 1989) to insert the ELF1 locus (including the open reading frame with 133 and 109 bp of up- and downstream flanking genomic DNA, respectively), or into a derivative of pRS416-ADH (Mumberg et al., 1995) where the ELF1 open reading frame was placed under control of the ADH1 promoter. To measure UV sensitivity, cells were grown at 30°C overnight in 2% glucose-containing non-selective rich (XY) media or synthetic complete media lacking uracil to maintain plasmid selection. The next day serial ten-fold dilutions of cells were spotted on XY plates and exposed to the indicated dose of UV treatment. Plates were imaged after 3 days of incubation at 30°C in the dark.
Metabolite profiling by LC-MS/MS
Cells were seeded in 6-well plates with 6 technical replicates per experimental condition and cultured for 24 h. Following two quick washes of the wells with warm PBS (37°C), the plates were placed on dry ice. The metabolites were immediately extracted by adding 1 mL of extraction solution (40% acetonitrile, 40% methanol, and 20% water) containing internal standards (500μg/ml and 300μg/mL of D7-Glucose and 13C915N-Tyrosine, respectively) and then the cells were scraped and collected in 1.5 mL vials. The mixture was shaken at 1400 rpm for 1 hour at 4°C in a Thermomixer (Eppendorf, Germany). The samples were centrifuged down at 14000 rpm for 10 min at 4°C (Eppendorf, Germany), and supernatants were transferred into fresh tubes to be evaporated to dryness in a CentreVap concentrator at 40°C (Labconco, MO). The dry extract samples were stored at −80°C until LC/MS analysis. Following reconstituted in 100 μL of water, samples were injected by auto-sampler (Dionex Corporation, CA) onto an HPLC column in line with a triple-quadrupole mass spectrometer (AB Sciex 5500Qtrap, Toronto, ON, Canada). Metabolites were separated through a guard column (Inertsil ODS-3, 4 mm internal diameter × 10 mm length, 3 μM particle size) and analytical column (Inertsil ODS-3, 4.6 mm internal diameter, 150 mm length, and 3-μM particle size; GL Sciences, Japan) for both polarity modes. In positive mode analysis, the organic portion (acetonitrile) of the mobile phase (0.1% acetic acid) ramped from 5% to 90% in 16 min, held for 1 min at 90%, then returned within 1 minute to 5% acetonitrile in mobile phase for column regeneration. In negative mode, the acetonitrile composition ramped from 5 to 90% in 10 min, held for 1 min at 90%, then the gradient ramped back over 3 minutes to 5% acetonitrile in mobile phase (0.1% tributylamine, 0.03% acetic acid, 10% methanol), to regenerate the column for the next run. The total run time for each sample in both modes was 20 min. The samples were maintained at 4°C in the auto-sampler, and the injection volume was 10 μL. An automated washing procedure was developed before and after each sample to avoid sample carryover. The mass spectrometric data acquisition time for each run was 20 min, and the dwell time for each MRM channel was 10 ms. Settings for negative and positive modes with electrospray ionization (ESI) were optimized. MultiQuant software (AB Sciex, Version 2.1) was used for peak analysis of targeted metabolites, with standards run consecutively. Peaks representing targeted masses and LC retention times were confirmed manually. Peak height data was analyzed using MetaboAnalyst 4.0 software44. Common mass spectrometric parameters: GS1 and GS2 were 50 psi; CUR was 20 psi, and CAD was 3 and 7 for positive and negative modes, respectively, and source temperature (TEM) was 400°C. Signal was normalized to internal standard and cell number. Metaboanalyst was used to analyze the data (http://www.metaboanalyst.ca/MetaboAnalyst/).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data processing and gene essentiality scores
The sgRNA read count files were computed from the raw CRISPR fastq files, for each experiment, using the ‘count’ function from the MAGeCK software (Li et al., 2014). The gene scores (normZ values) were estimated from the read count files using the DrugZ (Colic et al., 2019) algorithm, testing the drug treated replicates against their respective non-treated replicates.
Data quality control
To assess data quality, for each screen, the main data diagnostic tools used were the MAGeCK (Li et al., 2014) standard generated countsummary.Rnw R script and the BAGEL precision-recall curves, calculated through BAGEL.py ‘pr’ function, using the core essential (CEGv2.txt) and nonessential (NEGv1.txt) gene lists from (https://github.com/hart-lab/bagel). From the MAGeCK count summary it was observed that most replicates had a mapped reads percentage above 78%, with only calicheamicin replicate B (67%) and trabectedin replicate B (68%) having mapped reads percentages below 70%. Gini indexes were acceptably low, with most screens scoring below 0.15; only CPT-1 replicate B had a high Gini index of 0.21 in comparison. From the precision-recall curves it was possible to verify that known essential and nonessential genes behaved as expected, even though some replicates had steeper curves than others, with all areas under the curve (AUC) ranging from 0.93 to 0.99. Additionally, to provide an overall degree of correlation between experiments and assure consistency, the following steps were conducted, in similarity to the method used by project Achilles (Dempster et al., 2019). For each CRISPR screen batch, the fold change of non-treated replicates at t18 against their respective t0 replicates was calculated. From the fold change matrix, a subset of the top 3% high variance genes was selected and the correlation across experiments was plotted at the gene level (“correlation analysis” file, http://dx.doi.org/10.17632/gfcn2wmrpf.1), including both TKOv2 and TKOv3 libraries. The experiments presented consistent correlation values ranging from 0.89 to 0.96, for batches from the same libraries, and from 0.62 to 0.71 for batches of different libraries. The exception was experiment S10 (ICRF-187 screen), where both replicates showed correlations from 0.25 to 0.31 with all other screens, thus this particular screen was excluded from certain analyses. Given the consistency of all correlation values, no further replicates were excluded. The correlation between drug treated technical replicate pairs (A against B) was also calculated at the read count level (“correlation analysis” file, http://dx.doi.org/10.17632/gfcn2wmrpf.1), presenting high correlation values (range 0.68–0.98 and a median of 0.92). The gemcitabine screen did not have its t0 (initial time point) sequenced, thus its AUC value could not be calculated, nor could it be included in analysis of fold change correlation. However, at the read count level, both the non-treated and the treated replicates presented consistent and high correlation values (above 0.84) with other screens, including pyridostatin and MMS treatments.
Network analysis
The Network Analysis can be divided in two phases: the exploratory phase and the design phase. For the exploratory phase we developed an R shiny app (https://durocher.shinyapps.io/ScreenClustering/) that would allow us to visualize several distance measures, clustering techniques and networks graphs, with dynamically changing parameters to provide insights into our research. The design phase consisted of choosing how best to illustrate a single meaningful network of the 890 gene hits. For this step, the PCC for each pair of genes was calculated using the function ‘cor’, from the stats package, setting ‘use = pairwise.complete.obs’, R version 3.5.2, from a data frame with 842 genes and 27 normZ scores, which is cut down from the initial 890 by 31 configuration, since data and hits from the ICRF-187 screen were excluded and also duplicated reagents (in this case Cisplatin-1, Cisplatin-2 and CPT-1) were excluded when calculating the network Pearson’s correlation coefficient (PCC) to avoid bias towards a specific agent. The criteria used for selecting which screen to keep for the network was simply to select the one with the highest number of hits (Cisplatin-3 and CPT-2). The interaction network was then created using Cytoscape version 3.7.1, where the 596 nodes represent genes and the 1312 edges represent all pairwise connections that had a PCC greater than 0.70, explaining why the network has fewer nodes than the initial 842 genes. These threshold values were chosen to ensure that we captured known genetic interactions while being able to observe structure in the network.
Pathway enrichment, radar plots and fingerprint plots
Radar charts were drawn in R version 3.5.2, using the fmsb package version 0.7.0, based on our curated list of DNA damage repair and signaling factors (Table S4), considering each drug treatment independently. The one-sided alternative hypothesis Fisher Exact Test p-value was calculated by testing for overexpression enrichment (FDR < 15% for sensitizing genes and FDR < 1% for resistance genes) of each single pathway individually, against the overall expected enrichment for that drug treatment in the gene library, by creating two-dimensional contingency tables and using the ‘fisher.test’ function from the stats package. The Bonferroni correction threshold, adjusted for the number of pathways tested [for 8 pathways and significance α < 0.05, the threshold was therefore −log2(0.05/8) ≅ 7.32], was also added to the graph in green. The Fisher Exact Test p-values obtained for the radar plots were combined and used to plot the heatmap (Figure 1B). Gene fingerprint plots (Figures 3A, 5A, S4C, S6C, S7D) were drawn using the ggplot2 package version 3.2.1.
Functional Enrichment Analysis
g:Profiler (Raudvere et al., 2019) g:GOSt tool for functional profiling was used to identify functional enrichment analysis of the 890 hit genes (NormZ < −3 and FDR < 15% for sensitizing genes; NormZ > 6 for resistance genes), resulting in a list (Table S3) of 425 statistically significant enriched terms (Benjamini-Hochberg FDR < 0.05). The gene hit list was treated as an unordered query, and statistical tests were conducted within a statistical domain scope of only annotated genes, selecting terms with sizes between 4 and 500 genes, considering the GO molecular function, GO cellular component, GO biological process, KEGG, Reactome and WikiPathways data sources, removing electronic GO annotations. The Ensembl ID with the most GO annotations were chosen for all 5 ambiguous genes (BABAM1, CHTF8, FOXP1, MATR3, SOD2).
DNA repair-associated terms gene list
The list of 398 genes considered as ‘DNA repair genes’ was assembled by combining all genes present in at least one of 100 manually selected terms present in the g:Profiler analysis (Table S3) mentioned under ‘Functional Enrichment Analysis’. The terms were selected by their association with DNA damage and repair such as Homologous recombination (KEGG:03440), DNA Double-Strand Break Repair (REAC:R-HSA-5693532), response to UV (GO:0009411); DNA damage response (WP:WP707) and negative regulation of cell cycle phase transition (GO:1901988). The curated list of 197 DNA repair genes was assembled by the authors and assessed by the lead author. Strength of evidence for an involvement in DNA repair or DNA damage response was the key criterion for inclusion.
Known complexes comparison to random pairs
To analyze whether known complexes exhibit higher correlation than random pairs of genes, the ‘All Complexes CORUM Protein-Protein Interaction Database’ was downloaded (release 03.09.2018 Corum 3.0, https://mips.helmholtz-muenchen.de/corum/#) and genes belonging to the same complex were grouped pairwise. Only pairs in which at least one of the genes belonged to our list of 890 hits were selected for analysis. The Pearson correlation for each of these pairs was calculated in our DrugZ dataset (Table S2) and plotted as a density function, overlaid by the density of 10.000 correlation values from random pairs of genes. To test if the difference among the two distributions was significant, the Kolmogorov-Smirnov test was calculated in R (Figure S4A), using the function ‘ks.test’, with resulting p-value < 2.2*10−16.
ADDITIONAL RESOURCES
An R shiny app was developed for easier and faster exploration of the drug treatment screen data. It has been made available in the url: https://durocher.shinyapps.io/GenotoxicScreens/.
Supplementary Material
Figure S1. Gene term and DNA repair pathway enrichments. Relates to Figure 1. (A) Top 25 enriched GO terms, for molecular function (left) and cellular component (right), identified using g:Profiler (adjusted p < 0.05, with Benjamini-Hochberg FDR correction) among the 890 gene hits. Circle size indicates the number of genes from the common gene hit list included in each GO term, x-axis position indicates −log10-transformed FDR score and colors indicate the fold enrichment compared with the whole-genome reference set.
(B) Radar plots of the genotoxic CRISPR screens performed with genotoxic agents not shown in Figure 1, depicting sensitization (red) or resistance (blue) for different DNA repair pathways (axes). Values indicate the log2-transformed p-values of the Fisher’s exact test score. Bonferroni thresholds are indicated in green.
Figure S2. Clustering of screens and control images. Relates toFigure 2. (A) Representative images depicting automated quantitation of γ-H2AX foci using the IN Cell Analyzer 6000 coupled with Columbus image data analysis system. The initial nuclear masks were generated using images acquired from the DAPI channel. Upon selection of only completely in-frame nuclei, the number of foci and γH2AX intensity was evaluated in the FITC (γ-H2AX) channel (additional details in Methods).
(B) Competitive growth assays with or without pyridostatin (1 μM) or etoposide (500 nM) treatment in RPE1 cells transduced with virus expressing the indicated sgRNAs. Data represent mean fraction of GFP-positive cells ± SD normalized to day 0 (N= 3, independent transductions). TIDE analysis is shown in Table S6.
(C) RADAR assay for the detection of TOP2cc in N2A cells treated with pyridostatin (50 μM). Cell lysates were applied with a slot blot apparatus and immunoblotted with antibodies to TOP2α and TOP2β. NT, not treated.
Figure S3. The DNA damage network with gene names. Relates toFigure 3. (A) Histogram that bins the number of genes and how many times they were identified as hits in the screens.
(B) Histogram of the 10 genes that scored as hits in the highest number of screens.
(C) The same PCC-based genetic network as in Figure 3 with gene names annotated.
Figure S4. Hippo signaling promotes resistance to DNA damaging agents. Relates toFigure 3. (A) Density plot of the correlation between random pairs (green) or known complexes (red) present among the 890 hits. Known protein-protein interactions estimated using the CORUM database.
(B) Subnetwork highlighting NF2, FRYL, AMOTL2, LATS2, TAOK1 with genes that have PCC values > 0.70. The network was generated with Cytoscape (v3.7.1) using yFiles Organic layout.
(C) Fingerprint plots of NF2, FRYL, AMOTL2, LATS2, KIRREL, FAM49B and TAOK1. The boxes are colored according to whether mutations in these genes lead to sensitization (orange) or resistance (blue) in any of the 31 CRISPR screens.
Figure S5. Additional characterization of ERCC6L2. Relates toFigure 4. (A) Schematic of the strategy used to knock out ERCC6L2 in DT40 cells. Shown are the location of primers (arrows) used to confirm targeting.
(B) Agarose gel images of the PCR-mediated amplification of the internal ERCC6L2 sequence to confirm gene-targeting of ERCC6L2, using the primers indicated in (A). XRCC4 PCR amplification was used as a positive control.
(C) Cell proliferation assays of DT40 cells of the indicated genotypes treated with either etoposide or bleomycin for 3 d. Data is presented as the mean of a technical triplicate. An independent experiment is shown in Figure 4.
(D) Competitive growth assays with or without olaparib (16 nM) treatment in RPE1 cells transduced with virus expressing the indicated sgRNAs. Data represent mean fraction of GFP-positive cells ± SD normalized to day 0 (N= 3, independent transductions). TIDE analysis is shown in Table S6.
(E) Quantitative PCR assessing the levels of Iμ sterile transcript and Iα transcript (αGLT; normalised to HPRT) in CH12F3–2 cell lines of the indicated genotypes. Anti-CD40, IL-4, and TGFβ (CIT) treatment was used to activate switching.
(F) Immunoblotting of AID in independent CH12F3–2 cells of the indicated genotypes. Samples were collected 3 d after stimulation (N = 3). β-actin was used as loading control.
(G) Validation of XLF silencing by qPCR. Values are normalized for the average of GAPDH signal (N =3, independent transfections).
Figure S6. Further characterization of ELOF1, STK19 and TXNDC17. Relates toFigures 5 and 6. (A) Competitive growth assays with or without illudin S (30 nM) or trabectedin (0.5 nM) in RPE1 cells transduced with virus expressing the indicated sgRNAs. Data represent log10-transformed mean fraction of GFP-positive cells ± SD normalized to day 0 (N= 3, independent transductions). TIDE analysis is shown in Table S6.
(B) Overnight cultures of S. cerevisiae strains with the indicated genotype and transformed with the indicated plasmids were serially diluted and spotted onto YPD plates that were irradiated with the indicated UV dose, or left untreated. Plates were incubated at 30°C for 3 d before being imaged. WT, wild-type. ADHp, ADH1 promoter.
(C) Fingerprint plots of the 25 thioredoxin-domain containing genes screened. The boxes are colored according to whether mutations in these genes leads to sensitization (orange) or resistance (blue) in any of the 31 CRISPR screens.
(D) Immunoblotting to assess successful knock-out of TXNDC17 in two independent clones of RPE1 hTERT p53−/− Cas9 cells. Tubulin was used as a loading control.
(E) Quantitation of clonogenic survival of RPE1 cells of the indicated genotypes in response to increasing doses of MNNG, MMS, cisplatin and KBrO3.
(F) Viability assay of RPE1 WT and TXNDC17-KO cells infected with sgRNA targeting FANCD2, in response to MNNG. Data represent mean cell confluency ± SD (N= 3, independent transductions).
(G) Immunoblotting of FANCD2 ubiquitylation in parental (RPE1 hTERT p53−/− Cas9) or TXNDC17-KO cells following treatment with the indicated dose of MMS. Actinin was used as loading control.
(H) Quantification of reduced (GSH; left panel) or oxidized (GSSG; middle panel) glutathione levels by liquid chromatography coupled to mass spectrometry (LC-MS) in RPE1 WT, TXNDC17-KO and TXNDC17-KO complemented with TXNDC17 or a predicted catalytic-dead mutant (C43S/C46S). Right panel, ratio of reduced to oxidized glutathione. Data represent mean of 6 technical replicates.
(I) Quantitation of clonogenic survival of RPE1 cells of the indicated genotypes with or without N-acetylcysteine (NAC) treatment. Data represent mean ± SD (N= 3, independent experiments).
Figure S7. Control experiments relating to CYB5R4. Relates toFigure 6. (A) Competitive growth assays in untreated RPE1 cells transduced with virus expressing the indicated sgRNAs. Data represent mean fraction of GFP-positive cells ± SD normalized to day 0 (N= 3, independent transductions). TIDE analysis is shown in Table S6. See Figure 6B.
(B) Immunoblotting for CYB5R4 in RPE1 wild type (WT) and CYB5R4-KO cells. Tubulin was used as a loading control.
(C) Histogram plot generated from data at cBioPortal (http://www.cbioportal.org) showing the frequency of MCPH1 deletions and mutations across different types of cancer.
Supplementary Table S1. Genotoxic CRISPR screens Summary. Relates to Figure 1. This table describes the CRISPR screens performed, including details about the genotoxic agents and their mechanism of action, the concentrations used, the supplier information for the compounds, the experimenters and in which screen set they were performed.
Supplementary Table S2. CRISPR screens NormZ results. Relates to Figure 1. NormZ values calculated with DrugZ for each CRISPR screen reported in this work.
Supplementary Table S3. Gene ontology analysis of the 890 screens hits. Relates to Figure 1 Here are listed all the gene ontology terms for the 890 hits from the 31 genotoxic screens. Only terms with adjusted p-value <0.05 are reported.
Supplementary Table S4. Curated DNA repair factor list. Relates to Figure 1. This table contains the list of DNA repair factors curated by the authors used for the radar plots. Note that bona fide DNA repair factor genes not present in the sgRNA libraries were not included.
Supplementary Table S5. Correlation values for screens hits. Relates to Figure 3. PCC values calculated for the 842 hits in the 27 screens. 48 hits from the ICRF-187 screen were excluded from the analysis. The hits from the Cisplatin-1, Cisplatin-2 and CPT-1 screens were also excluded from correlation calculations.
Supplementary Table S6. sgRNA efficiency as measured by TIDE. Relates to Figure 5. This table contain the list of sgRNA used in the manuscript with relative primers used for performing TIDE. Indel formation is reported for each guide.
Supplementary Table S7. Yeast strains. Relates to Figure 5 Genotype of the Saccharomyces cerevisiae strains used in this work.
Highlights.
Resource of 31 genome-scale CRISPR screens against DNA damaging agents
Cytotoxicity of G-quadruplex ligand pyridostatin involves TOP2 trapping
The bone marrow failure syndrome gene ERCC6L2 codes for an NHEJ factor
The ELOF1 and STK19 proteins are candidate TC-NER factors
ACKNOWLEDGEMENTS
We thank JY Yuan for technical assistance as well as J. Moffat, S. Angers and T. Hart for the TKO libraries and protocols and C. Boone for yeast strains. We also thank F. Meng, S. Rottenberg and M. Luijsterburg for sharing unpublished results. AAQ, GSM and AMH are recipients of long-term EMBO fellowships (ALTF 910-2017, 795-2017 and 124-2019), SER was supported by a fellowship from AIRC and SA was a Banting post-doctoral fellow. ASB was supported by a PhD Studentship from Asociación Española Familia Ataxia Telangiectasia and an EMBO short-term fellowship. The ICRF-187 was funded by the Spanish Government (SAF2017-89619-R, European Regional Development Fund) and European Research Council (ERC-CoG-2014-647359). Work in the RSW laboratory was supported in part by the US National Institutes of Health Intramural Program, US National Institute of Environmental Health Sciences (NIEHS, 1Z01ES102765). DD is a Canada Research Chair (Tier I) and the work was supported by grants from the CIHR (FDN143343 to DD; FRN 123518, PJT-156330 to AM) and Canadian Cancer Society grant (705644 to DD) with additional support to DD from the Krembil Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
Michal Zimmermann is an employee and shareholder of Repare Therapeutics. Daniel Durocher is a founder of Repare Therapeutics and a member of its scientific advisory board.
REFERENCES
- Amendola PG, Zaghet N, Ramalho JJ, Vilstrup Johansen J, Boxem M, and Salcini AE (2017). JMJD-5/KDM8 regulates H3K36me2 and is required for late steps of homologous recombination and genome integrity. PLoS Genet 13, e1006632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand J, Sun Y, Zhao Y, Nitiss KC, and Nitiss JL (2018). Detection of Topoisomerase Covalent Complexes in Eukaryotic Cells. Methods Mol Biol 1703, 283–299. [DOI] [PubMed] [Google Scholar]
- Aparicio T, Baer R, Gottesman M, and Gautier J (2016). MRN, CtIP, and BRCA1 mediate repair of topoisomerase II-DNA adducts. J Cell Biol 212, 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnoult N, Correia A, Ma J, Merlo A, Garcia-Gomez S, Maric M, Tognetti M, Benner CW, Boulton SJ, Saghatelian A, et al. (2017). Regulation of DNA repair pathway choice in S and G2 phases by the NHEJ inhibitor CYREN. Nature 549, 548–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballmaier D, and Epe B (2006). DNA damage by bromate: mechanism and consequences. Toxicology 221, 166–171. [DOI] [PubMed] [Google Scholar]
- Benham AM (2012). The protein disulfide isomerase family: key players in health and disease. Antioxid Redox Signal 16, 781–789. [DOI] [PubMed] [Google Scholar]
- Bhargava R, Sandhu M, Muk S, Lee G, Vaidehi N, and Stark JM (2018). C-NHEJ without indels is robust and requires synergistic function of distinct XLF domains. Nat Commun 9, 2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackford AN, and Jackson SP (2017). ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol Cell 66, 801–817. [DOI] [PubMed] [Google Scholar]
- Boeing S, Williamson L, Encheva V, Gori I, Saunders RE, Instrell R, Aygun O, Rodriguez-Martinez M, Weems JC, Kelly GP, et al. (2016). Multiomic Analysis of the UV-Induced DNA Damage Response. Cell Rep 15, 1597–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, and Boeke JD (1998). Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14, 115–132. [DOI] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, and van Steensel B (2014). Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42, e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KR, Mair B, Soste M, and Moffat J (2019). CRISPR screens are feasible in TP53 wild-type cells. Mol Syst Biol 15, e8679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldecott KW (2008). Single-strand break repair and genetic disease. Nat Rev Genet 9, 619–631. [DOI] [PubMed] [Google Scholar]
- Chong J, Soufan O, Li C, Caraus I, Li S, Bourque G, Wishart DS, and Xia J (2018). MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res 46, W486–W494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, and Elledge SJ (2010). The DNA damage response: making it safe to play with knives. Molecular cell 40, 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colic M, Wang G, Zimmermann M, Mascall K, McLaughlin M, Bertolet L, Lenoir WF, Moffat J, Angers S, Durocher D, et al. (2019). Identifying chemogenetic interactions from CRISPR knockout screens with drugZ. bioRxiv, 232736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin OM, Friedman HS, Gamcsik MP, Fenselau C, and Hilton J (1993). Role of glutathione in cellular resistance to alkylating agents. Adv Enzyme Regul 33, 19–26. [DOI] [PubMed] [Google Scholar]
- Costanzo M, Kuzmin E, van Leeuwen J, Mair B, Moffat J, Boone C, and Andrews B (2019). Global Genetic Networks and the Genotype-to-Phenotype Relationship. Cell 177, 85–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels JP, Kelly S, Wickstead B, and Gull K (2009). Identification of a crenarchaeal orthologue of Elf1: implications for chromatin and transcription in Archaea. Biol Direct 4, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cian A, Delemos E, Mergny JL, Teulade-Fichou MP, and Monchaud D (2007). Highly efficient G-quadruplex recognition by bisquinolinium compounds. J Am Chem Soc 129, 1856–1857. [DOI] [PubMed] [Google Scholar]
- Dede M, Kim E, and Hart T (2020). Biases and Blind-Spots in Genome-Wide CRISPR Knockout Screens. bioRxiv, 2020.2001.2016.909606 [Google Scholar]
- Delgado JL, Hsieh CM, Chan NL, and Hiasa H (2018). Topoisomerases as anticancer targets. Biochem J 475, 373–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster JM, Rossen J, Kazachkova M, Pan J, Kugener G, Root DE, and Tsherniak A (2019). Extracting Biological Insights from the Project Achilles Genome-Scale CRISPR Screens in Cancer Cell Lines. bioRxiv, 720243. [Google Scholar]
- Dhanwani R, Takahashi M, and Sharma S (2018). Cytosolic sensing of immuno-stimulatory DNA, the enemy within. Curr Opin Immunol 50, 82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas SPM, Siipola P, Kovanen PE, Pyorala M, Kakko S, Savolainen ER, Salmenniemi U, Orte K, Kytola S, Pitkanen E, et al. (2019). ERCC6L2 defines a novel entity within inherited acute myeloid leukemia. Blood 133, 2724–2728. [DOI] [PubMed] [Google Scholar]
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, and Naldini L (1998). A third-generation lentivirus vector with a conditional packaging system. J Virol 72, 8463–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehara H, Kujirai T, Fujino Y, Shirouzu M, Kurumizaka H, and Sekine SI (2019). Structural insight into nucleosome transcription by RNA polymerase II with elongation factors. Science 363, 744–747. [DOI] [PubMed] [Google Scholar]
- Ehara H, Yokoyama T, Shigematsu H, Yokoyama S, Shirouzu M, and Sekine SI (2017). Structure of the complete elongation complex of RNA polymerase II with basal factors. Science 357, 921–924. [DOI] [PubMed] [Google Scholar]
- Gheiratmand L, Coyaud E, Gupta GD, Laurent EM, Hasegan M, Prosser SL, Goncalves J, Raught B, and Pelletier L (2019). Spatial and proteomic profiling reveals centrosome-independent features of centriolar satellites. EMBO J 38, e101109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giurgiu M, Reinhard J, Brauner B, Dunger-Kaltenbach I, Fobo G, Frishman G, Montrone C, and Ruepp A (2019). CORUM: the comprehensive resource of mammalian protein complexes-2019. Nucleic Acids Res 47, D559–D563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. [DOI] [PubMed] [Google Scholar]
- Guo F, Stanevich V, Wlodarchak N, Sengupta R, Jiang L, Satyshur KA, and Xing Y (2014). Structural basis of PP2A activation by PTPA, an ATP-dependent activation chaperone. Cell Res 24, 190–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta D, and Heinen CD (2019). The mismatch repair-dependent DNA damage response: Mechanisms and implications. DNA Repair (Amst) 78, 60–69. [DOI] [PubMed] [Google Scholar]
- Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G, Mis M, Zimmermann M, Fradet-Turcotte A, Sun S, et al. (2015). High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 163, 1515–1526. [DOI] [PubMed] [Google Scholar]
- Hart T, Tong AHY, Chan K, Van Leeuwen J, Seetharaman A, Aregger M, Chandrashekhar M, Hustedt N, Seth S, Noonan A, et al. (2017). Evaluation and Design of Genome-Wide CRISPR/SpCas9 Knockout Screens. G3 (Bethesda) 7, 2719–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeijmakers JH (2009). DNA damage, aging, and cancer. N Engl J Med 361, 1475–1485. [DOI] [PubMed] [Google Scholar]
- Hung PJ, Johnson B, Chen BR, Byrum AK, Bredemeyer AL, Yewdell WT, Johnson TE, Lee BJ, Deivasigamani S, Hindi I, et al. (2018). MRI Is a DNA Damage Response Adaptor during Classical Non-homologous End Joining. Mol Cell 71, 332–342 e338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo D, Chen H, Cheng Y, Song X, Zhang K, Li MJ, and Xuan C (2019). JMJD6 modulates DNA damage response through downregulating H4K16ac independently of its enzymatic activity. Cell Death Differ. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hustedt N, Alvarez-Quilon A, McEwan A, Yuan JY, Cho T, Koob L, Hart T, and Durocher D (2019a). A consensus set of genetic vulnerabilities to ATR inhibition. Open Biol 9, 190156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hustedt N, and Durocher D (2016). The control of DNA repair by the cell cycle. Nat Cell Biol 19, 1–9. [DOI] [PubMed] [Google Scholar]
- Hustedt N, Saito Y, Zimmermann M, Alvarez-Quilon A, Setiaputra D, Adam S, McEwan A, Yuan JY, Olivieri M, Zhao Y, et al. (2019b). Control of homologous recombination by the HROB-MCM8-MCM9 pathway. Genes Dev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IARC (1999). Potassium bromate. IARC Monogr Eval Carcinog Risks Hum 73, 481–496. [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, and Bartek J (2009). The DNA-damage response in human biology and disease. Nature 461, 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarviaho T, Halt K, Hirvikoski P, Moilanen J, Mottonen M, and Niinimaki R (2018). Bone marrow failure syndrome caused by homozygous frameshift mutation in the ERCC6L2 gene. Clin Genet 93, 392–395. [DOI] [PubMed] [Google Scholar]
- Jaspers NG, Raams A, Kelner MJ, Ng JM, Yamashita YM, Takeda S, McMorris TC, and Hoeijmakers JH (2002). Anti-tumour compounds illudin S and Irofulven induce DNA lesions ignored by global repair and exclusively processed by transcription- and replication-coupled repair pathways. DNA Repair (Amst) 1, 1027–1038. [DOI] [PubMed] [Google Scholar]
- Kanellis P, Agyei R, and Durocher D (2003). Elg1 forms an alternative PCNA-interacting RFC complex required to maintain genome stability. Curr Biol 13, 1583–1595. [DOI] [PubMed] [Google Scholar]
- Kang MS, Ryu E, Lee SW, Park J, Ha NY, Ra JS, Kim YJ, Kim J, Abdel-Rahman M, Park SH, et al. (2019). Regulation of PCNA cycling on replicating DNA by RFC and RFC-like complexes. Nat Commun 10, 2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiianitsa K, and Maizels N (2014). Ultrasensitive isolation, identification and quantification of DNA-protein adducts by ELISA-based RADAR assay. Nucleic Acids Res 42, e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottemann MC, and Smogorzewska A (2013). Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 493, 356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15, 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl T, and Barnes DE (2000). Repair of endogenous DNA damage. Cold Spring Harb Symp Quant Biol 65, 127–133. [DOI] [PubMed] [Google Scholar]
- Liu X, Zhou ZW, and Wang ZQ (2016). The DNA damage response molecule MCPH1 in brain development and beyond. Acta Biochim Biophys Sin (Shanghai) 48, 678–685. [DOI] [PubMed] [Google Scholar]
- Lou J, Chen H, Han J, He H, Huen MSY, Feng XH, Liu T, and Huang J (2017). AUNIP/C1orf135 directs DNA double-strand breaks towards the homologous recombination repair pathway. Nat Commun 8, 985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod G, Bozek DA, Rajakulendran N, Monteiro V, Ahmadi M, Steinhart Z, Kushida MM, Yu H, Coutinho FJ, Cavalli FMG, et al. (2019). Genome-Wide CRISPR-Cas9 Screens Expose Genetic Vulnerabilities and Mechanisms of Temozolomide Sensitivity in Glioblastoma Stem Cells. Cell Rep 27, 971–986 e979. [DOI] [PubMed] [Google Scholar]
- Marteijn JA, Lans H, Vermeulen W, and Hoeijmakers JH (2014). Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol 15, 465–481. [DOI] [PubMed] [Google Scholar]
- Marzi L, Szabova L, Gordon M, Weaver Ohler Z, Sharan SK, Beshiri ML, Etemadi M, Murai J, Kelly K, and Pommier Y (2019). The Indenoisoquinoline TOP1 Inhibitors Selectively Target Homologous Recombination-Deficient and Schlafen 11-Positive Cancer Cells and Synergize with Olaparib. Clin Cancer Res 25, 6206–6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methot SP, and Di Noia JM (2017). Molecular Mechanisms of Somatic Hypermutation and Class Switch Recombination. Adv Immunol 133, 37–87. [DOI] [PubMed] [Google Scholar]
- Mohni KN, Wessel SR, Zhao R, Wojciechowski AC, Luzwick JW, Layden H, Eichman BF, Thompson PS, Mehta KPM, and Cortez D (2019). HMCES Maintains Genome Integrity by Shielding Abasic Sites in Single-Strand DNA. Cell 176, 144–153 e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller S, Kumari S, Rodriguez R, and Balasubramanian S (2010). Small-molecule-mediated G-quadruplex isolation from human cells. Nat Chem 2, 1095–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumberg D, Muller R, and Funk M (1995). Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156, 119–122. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Kondo S, Sugai M, Nazarea M, Imamura S, and Honjo T (1996). High frequency class switching of an IgM+ B lymphoma clone CH12F3 to IgA+ cells. International immunology 8, 193–201. [DOI] [PubMed] [Google Scholar]
- Nguyen CDK, and Yi C (2019). YAP/TAZ Signaling and Resistance to Cancer Therapy. Trends Cancer 5, 283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijnik A, Woodbine L, Marchetti C, Dawson S, Lambe T, Liu C, Rodrigues NP, Crockford TL, Cabuy E, Vindigni A, et al. (2007). DNA repair is limiting for haematopoietic stem cells during ageing. Nature 447, 686–690. [DOI] [PubMed] [Google Scholar]
- Noordermeer SM, Adam S, Setiaputra D, Barazas M, Pettitt SJ, Ling AK, Olivieri M, Alvarez-Quilon A, Moatti N, Zimmermann M, et al. (2018). The shieldin complex mediates 53BP1-dependent DNA repair. Nature 560, 117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olhovsky M, Williton K, Dai AY, Pasculescu A, Lee JP, Goudreault M, Wells CD, Park JG, Gingras AC, Linding R, et al. (2011). OpenFreezer: a reagent information management software system. Nat Methods 8, 612–613. [DOI] [PubMed] [Google Scholar]
- Pader I, Sengupta R, Cebula M, Xu J, Lundberg JO, Holmgren A, Johansson K, and Arner ES (2014). Thioredoxin-related protein of 14 kDa is an efficient L-cystine reductase and S-denitrosylase. Proc Natl Acad Sci U S A 111, 6964–6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papamichos-Chronakis M, and Peterson CL (2013). Chromatin and the genome integrity network. Nat Rev Genet 14, 62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y, and Cushman M (2009). The indenoisoquinoline noncamptothecin topoisomerase I inhibitors: update and perspectives. Mol Cancer Ther 8, 1008–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontel LB, Rosado IV, Burgos-Barragan G, Garaycoechea JI, Yu R, Arends MJ, Chandrasekaran G, Broecker V, Wei W, Liu L, et al. (2015). Endogenous Formaldehyde Is a Hematopoietic Stem Cell Genotoxin and Metabolic Carcinogen. Mol Cell 60, 177–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash S, and Prakash L (2000). Nucleotide excision repair in yeast. Mutat Res 451, 13–24. [DOI] [PubMed] [Google Scholar]
- Prather D, Krogan NJ, Emili A, Greenblatt JF, and Winston F (2005). Identification and characterization of Elf1, a conserved transcription elongation factor in Saccharomyces cerevisiae. Mol Cell Biol 25, 10122–10135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran S, Haddad D, Li C, Le MX, Ling AK, So CC, Nepal RM, Gommerman JL, Yu K, Ketela T, et al. (2016). The SAGA Deubiquitination Module Promotes DNA Repair and Class Switch Recombination through ATM and DNAPK-Mediated gammaH2AX Formation. Cell Rep 15, 1554–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, and Vilo J (2019). g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res 47, W191–W198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez R, Miller KM, Forment JV, Bradshaw CR, Nikan M, Britton S, Oelschlaegel T, Xhemalce B, Balasubramanian S, and Jackson SP (2012). Small-molecule-induced DNA damage identifies alternative DNA structures in human genes. Nat Chem Biol 8, 301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolland T, Tasan M, Charloteaux B, Pevzner SJ, Zhong Q, Sahni N, Yi S, Lemmens I, Fontanillo C, Mosca R, et al. (2014). A proteome-scale map of the human interactome network. Cell 159, 1212–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellenberg MJ, Lieberman JA, Herrero-Ruiz A, Butler LR, Williams JG, Munoz-Cabello AM, Mueller GA, London RE, Cortes-Ledesma F, and Williams RS (2017). ZATT (ZNF451)-mediated resolution of topoisomerase 2 DNA-protein cross-links. Science 357, 1412–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellenberg MJ, Petrovich RM, Malone CC, and Williams RS (2018). Selectable high-yield recombinant protein production in human cells using a GFP/YFP nanobody affinity support. Protein Sci 27, 1083–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekhar SC, Venkatesh J, Cheriyan VT, Muthu M, Levi E, Assad H, Meister P, Undyala VV, Gauld JW, and Rishi AK (2019). A H2AX(−)CARP-1 Interaction Regulates Apoptosis Signaling Following DNA Damage. Cancers (Basel) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setiaputra D, and Durocher D (2019). Shieldin - the protector of DNA ends. EMBO Rep 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfeir A, and Symington LS (2015). Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem Sci 40, 701–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabanova I, Cohen E, Cada M, Vincent A, Cohn RD, and Dror Y (2018). ERCC6L2-associated inherited bone marrow failure syndrome. Mol Genet Genomic Med 6, 463–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, et al. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, and Zhang F (2015). High-throughput functional genomics using CRISPR-Cas9. Nat Rev Genet 16, 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, and Hieter P (1989). A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takebayashi Y, Pourquier P, Zimonjic DB, Nakayama K, Emmert S, Ueda T, Urasaki Y, Kanzaki A, Akiyama SI, Popescu N, et al. (2001). Antiproliferative activity of ecteinascidin 743 is dependent upon transcription-coupled nucleotide-excision repair. Nat Med 7, 961–966. [DOI] [PubMed] [Google Scholar]
- Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. (2017). Defining a Cancer Dependency Map. Cell 170, 564–576 e516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tummala H, Dokal AD, Walne A, Ellison A, Cardoso S, Amirthasigamanipillai S, Kirwan M, Browne I, Sidhu JK, Rajeeve V, et al. (2018). Genome instability is a consequence of transcription deficiency in patients with bone marrow failure harboring biallelic ERCC6L2 variants. Proc Natl Acad Sci U S A 115, 7777–7782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tummala H, Kirwan M, Walne AJ, Hossain U, Jackson N, Pondarre C, Plagnol V, Vulliamy T, and Dokal I (2014). ERCC6L2 mutations link a distinct bone-marrow-failure syndrome to DNA repair and mitochondrial function. Am J Hum Genet 94, 246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Wang G, Feng X, Shepherd P, Zhang J, Tang M, Chen Z, Srivastava M, McLaughlin ME, Navone NM, et al. (2018). Genome-wide CRISPR screens reveal synthetic lethality of RNASEH2 deficiency and ATR inhibition. Oncogene. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, and Sabatini DM (2015). Identification and characterization of essential genes in the human genome. Science 350, 1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webby CJ, Wolf A, Gromak N, Dreger M, Kramer H, Kessler B, Nielsen ML, Schmitz C, Butler DS, Yates JR 3rd, et al. (2009). Jmjd6 catalyses lysyl-hydroxylation of U2AF65, a protein associated with RNA splicing. Science 325, 90–93. [DOI] [PubMed] [Google Scholar]
- Wilkins SE, Islam MS, Gannon JM, Markolovic S, Hopkinson RJ, Ge W, Schofield CJ, and Chowdhury R (2018). JMJD5 is a human arginyl C-3 hydroxylase. Nat Commun 9, 1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodbine L, Gennery AR, and Jeggo PA (2014). The clinical impact of deficiency in DNA non-homologous end-joining. DNA Repair (Amst) 16, 84–96. [DOI] [PubMed] [Google Scholar]
- Wu CG, Zheng A, Jiang L, Rowse M, Stanevich V, Chen H, Li Y, Satyshur KA, Johnson B, Gu TJ, et al. (2017). Methylation-regulated decommissioning of multimeric PP2A complexes. Nat Commun 8, 2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Zhu H, Larade K, Ladoux A, Seguritan A, Chu M, Ito S, Bronson RT, Leiter EH, Zhang CY, et al. (2004). Absence of a reductase, NCB5OR, causes insulin-deficient diabetes. Proc Natl Acad Sci U S A 101, 10750–10755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing M, Yang M, Huo W, Feng F, Wei L, Jiang W, Ning S, Yan Z, Li W, Wang Q, et al. (2015). Interactome analysis identifies a new paralogue of XRCC4 in non-homologous end joining DNA repair pathway. Nat Commun 6, 6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Di Antonio M, McKinney S, Mathew V, Ho B, O’Neil NJ, Santos ND, Silvester J, Wei V, Garcia J, et al. (2017). CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat Commun 8, 14432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Pondarre C, Pennarun G, Labussiere-Wallet H, Vera G, France B, Chansel M, Rouvet I, Revy P, Lopez B, et al. (2016). A nonsense mutation in the DNA repair factor Hebo causes mild bone marrow failure and microcephaly. J Exp Med 213, 1011–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Watanabe G, Morten MJ, Reid DA, Rothenberg E, and Lieber MR (2019). The essential elements for the noncovalent association of two DNA ends during NHEJ synapsis. Nat Commun 10, 3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BB, and Elledge SJ (2000). The DNA damage response: putting checkpoints in perspective. Nature 408, 433–439. [DOI] [PubMed] [Google Scholar]
- Zimmer J, Tacconi EMC, Folio C, Badie S, Porru M, Klare K, Tumiati M, Markkanen E, Halder S, Ryan A, et al. (2016). Targeting BRCA1 and BRCA2 Deficiencies with G-Quadruplex-Interacting Compounds. Mol Cell 61, 449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Murina O, Reijns MAM, Agathanggelou A, Challis R, Tarnauskaite Z, Muir M, Fluteau A, Aregger M, McEwan A, et al. (2018). CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 559, 285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Gene term and DNA repair pathway enrichments. Relates to Figure 1. (A) Top 25 enriched GO terms, for molecular function (left) and cellular component (right), identified using g:Profiler (adjusted p < 0.05, with Benjamini-Hochberg FDR correction) among the 890 gene hits. Circle size indicates the number of genes from the common gene hit list included in each GO term, x-axis position indicates −log10-transformed FDR score and colors indicate the fold enrichment compared with the whole-genome reference set.
(B) Radar plots of the genotoxic CRISPR screens performed with genotoxic agents not shown in Figure 1, depicting sensitization (red) or resistance (blue) for different DNA repair pathways (axes). Values indicate the log2-transformed p-values of the Fisher’s exact test score. Bonferroni thresholds are indicated in green.
Figure S2. Clustering of screens and control images. Relates toFigure 2. (A) Representative images depicting automated quantitation of γ-H2AX foci using the IN Cell Analyzer 6000 coupled with Columbus image data analysis system. The initial nuclear masks were generated using images acquired from the DAPI channel. Upon selection of only completely in-frame nuclei, the number of foci and γH2AX intensity was evaluated in the FITC (γ-H2AX) channel (additional details in Methods).
(B) Competitive growth assays with or without pyridostatin (1 μM) or etoposide (500 nM) treatment in RPE1 cells transduced with virus expressing the indicated sgRNAs. Data represent mean fraction of GFP-positive cells ± SD normalized to day 0 (N= 3, independent transductions). TIDE analysis is shown in Table S6.
(C) RADAR assay for the detection of TOP2cc in N2A cells treated with pyridostatin (50 μM). Cell lysates were applied with a slot blot apparatus and immunoblotted with antibodies to TOP2α and TOP2β. NT, not treated.
Figure S3. The DNA damage network with gene names. Relates toFigure 3. (A) Histogram that bins the number of genes and how many times they were identified as hits in the screens.
(B) Histogram of the 10 genes that scored as hits in the highest number of screens.
(C) The same PCC-based genetic network as in Figure 3 with gene names annotated.
Figure S4. Hippo signaling promotes resistance to DNA damaging agents. Relates toFigure 3. (A) Density plot of the correlation between random pairs (green) or known complexes (red) present among the 890 hits. Known protein-protein interactions estimated using the CORUM database.
(B) Subnetwork highlighting NF2, FRYL, AMOTL2, LATS2, TAOK1 with genes that have PCC values > 0.70. The network was generated with Cytoscape (v3.7.1) using yFiles Organic layout.
(C) Fingerprint plots of NF2, FRYL, AMOTL2, LATS2, KIRREL, FAM49B and TAOK1. The boxes are colored according to whether mutations in these genes lead to sensitization (orange) or resistance (blue) in any of the 31 CRISPR screens.
Figure S5. Additional characterization of ERCC6L2. Relates toFigure 4. (A) Schematic of the strategy used to knock out ERCC6L2 in DT40 cells. Shown are the location of primers (arrows) used to confirm targeting.
(B) Agarose gel images of the PCR-mediated amplification of the internal ERCC6L2 sequence to confirm gene-targeting of ERCC6L2, using the primers indicated in (A). XRCC4 PCR amplification was used as a positive control.
(C) Cell proliferation assays of DT40 cells of the indicated genotypes treated with either etoposide or bleomycin for 3 d. Data is presented as the mean of a technical triplicate. An independent experiment is shown in Figure 4.
(D) Competitive growth assays with or without olaparib (16 nM) treatment in RPE1 cells transduced with virus expressing the indicated sgRNAs. Data represent mean fraction of GFP-positive cells ± SD normalized to day 0 (N= 3, independent transductions). TIDE analysis is shown in Table S6.
(E) Quantitative PCR assessing the levels of Iμ sterile transcript and Iα transcript (αGLT; normalised to HPRT) in CH12F3–2 cell lines of the indicated genotypes. Anti-CD40, IL-4, and TGFβ (CIT) treatment was used to activate switching.
(F) Immunoblotting of AID in independent CH12F3–2 cells of the indicated genotypes. Samples were collected 3 d after stimulation (N = 3). β-actin was used as loading control.
(G) Validation of XLF silencing by qPCR. Values are normalized for the average of GAPDH signal (N =3, independent transfections).
Figure S6. Further characterization of ELOF1, STK19 and TXNDC17. Relates toFigures 5 and 6. (A) Competitive growth assays with or without illudin S (30 nM) or trabectedin (0.5 nM) in RPE1 cells transduced with virus expressing the indicated sgRNAs. Data represent log10-transformed mean fraction of GFP-positive cells ± SD normalized to day 0 (N= 3, independent transductions). TIDE analysis is shown in Table S6.
(B) Overnight cultures of S. cerevisiae strains with the indicated genotype and transformed with the indicated plasmids were serially diluted and spotted onto YPD plates that were irradiated with the indicated UV dose, or left untreated. Plates were incubated at 30°C for 3 d before being imaged. WT, wild-type. ADHp, ADH1 promoter.
(C) Fingerprint plots of the 25 thioredoxin-domain containing genes screened. The boxes are colored according to whether mutations in these genes leads to sensitization (orange) or resistance (blue) in any of the 31 CRISPR screens.
(D) Immunoblotting to assess successful knock-out of TXNDC17 in two independent clones of RPE1 hTERT p53−/− Cas9 cells. Tubulin was used as a loading control.
(E) Quantitation of clonogenic survival of RPE1 cells of the indicated genotypes in response to increasing doses of MNNG, MMS, cisplatin and KBrO3.
(F) Viability assay of RPE1 WT and TXNDC17-KO cells infected with sgRNA targeting FANCD2, in response to MNNG. Data represent mean cell confluency ± SD (N= 3, independent transductions).
(G) Immunoblotting of FANCD2 ubiquitylation in parental (RPE1 hTERT p53−/− Cas9) or TXNDC17-KO cells following treatment with the indicated dose of MMS. Actinin was used as loading control.
(H) Quantification of reduced (GSH; left panel) or oxidized (GSSG; middle panel) glutathione levels by liquid chromatography coupled to mass spectrometry (LC-MS) in RPE1 WT, TXNDC17-KO and TXNDC17-KO complemented with TXNDC17 or a predicted catalytic-dead mutant (C43S/C46S). Right panel, ratio of reduced to oxidized glutathione. Data represent mean of 6 technical replicates.
(I) Quantitation of clonogenic survival of RPE1 cells of the indicated genotypes with or without N-acetylcysteine (NAC) treatment. Data represent mean ± SD (N= 3, independent experiments).
Figure S7. Control experiments relating to CYB5R4. Relates toFigure 6. (A) Competitive growth assays in untreated RPE1 cells transduced with virus expressing the indicated sgRNAs. Data represent mean fraction of GFP-positive cells ± SD normalized to day 0 (N= 3, independent transductions). TIDE analysis is shown in Table S6. See Figure 6B.
(B) Immunoblotting for CYB5R4 in RPE1 wild type (WT) and CYB5R4-KO cells. Tubulin was used as a loading control.
(C) Histogram plot generated from data at cBioPortal (http://www.cbioportal.org) showing the frequency of MCPH1 deletions and mutations across different types of cancer.
Supplementary Table S1. Genotoxic CRISPR screens Summary. Relates to Figure 1. This table describes the CRISPR screens performed, including details about the genotoxic agents and their mechanism of action, the concentrations used, the supplier information for the compounds, the experimenters and in which screen set they were performed.
Supplementary Table S2. CRISPR screens NormZ results. Relates to Figure 1. NormZ values calculated with DrugZ for each CRISPR screen reported in this work.
Supplementary Table S3. Gene ontology analysis of the 890 screens hits. Relates to Figure 1 Here are listed all the gene ontology terms for the 890 hits from the 31 genotoxic screens. Only terms with adjusted p-value <0.05 are reported.
Supplementary Table S4. Curated DNA repair factor list. Relates to Figure 1. This table contains the list of DNA repair factors curated by the authors used for the radar plots. Note that bona fide DNA repair factor genes not present in the sgRNA libraries were not included.
Supplementary Table S5. Correlation values for screens hits. Relates to Figure 3. PCC values calculated for the 842 hits in the 27 screens. 48 hits from the ICRF-187 screen were excluded from the analysis. The hits from the Cisplatin-1, Cisplatin-2 and CPT-1 screens were also excluded from correlation calculations.
Supplementary Table S6. sgRNA efficiency as measured by TIDE. Relates to Figure 5. This table contain the list of sgRNA used in the manuscript with relative primers used for performing TIDE. Indel formation is reported for each guide.
Supplementary Table S7. Yeast strains. Relates to Figure 5 Genotype of the Saccharomyces cerevisiae strains used in this work.
Data Availability Statement
All the raw data generated is available on Mendeley (http://dx.doi.org/10.17632/gfcn2wmrpf.1). The code used for this paper is based on free accessible software described in the relevant STAR method section and uploaded on the Mendeley database.