

Abstract
Escape of cancer cells from chemotherapy is a problem in the management of cancer patients. Research on chemotherapy resistance has mainly focused on the heterogeneity of cancer cells, multiple gene mutations, and quiescence of malignant cancer cells. However, some studies have indicated that interactions between cancer cells and vascular cells promote resistance to chemotherapy. Here, we established mouse leukemia models using the cell lines THP‐1 or MEG‐1. These were derived from acute and chronic myeloid leukemias, respectively, and highly expressed DNA replication factor PSF1, a member of the GINS complex. We found that, after anti‐cancer drug administration, surviving GFP‐positive leukemia cells in the bone marrow were located adjacent to blood vessels, as previously reported in a subcutaneous solid tumor transplantation model. Treating THP‐1 and MEG‐1 cells with anti‐cancer drugs in vitro revealed that those most strongly expressing PSF1 were most chemoresistant, suggesting that PSF1 induces not only cell cycle progression but also facilitates cell survival. Indeed, when PSF1 expression was suppressed by shRNA, the growth rate was reduced and cell death was enhanced in both cell lines. Furthermore, PSF1 knockdown in leukemia cells led to a change in their location at a distance from the blood vessels in a bone marrow transplantation model. These findings potentially reflect a mechanism of escape of leukemic cells from chemotherapy and suggest that PSF1 may be a possible therapeutic target to enhance the effect of chemotherapy.
Keywords: blood vessel, cell cycle, cell death, leukemia, PSF1
Chemotherapy (AraC) in a leukemia cell transplantation model. We found that cancer cells strongly expressing PSF1 and localising near vascular areas were drug‐resistant. These findings suggest that the vascular niche might play a critical role in chemotherapy resistance. PSF1 might act as a potential therapeutic target to enhance the effect of chemotherapy and prognosis.

1. INTRODUCTION
The acquisition of chemotherapy resistance by cancer cells is a critical factor in cancer prognosis. Many studies have documented that chemotherapy resistance depends on the heterogeneity and plasticity of cancer cells, 1 , 2 , 3 , 4 and the quiescent status of malignant cancer cells. 5 , 6 For example, chemotherapy‐activated TGF‐β signaling inhibits cell cycle progression but induces cell invasion and enhances drug resistance. 7 However, more recent research has revealed that the vascular niche plays a critical role in cancer stem cell (CSC) maintenance, especially in the escape of cancer cells from chemotherapy. 8 , 9 In various solid cancers, endothelial cells promote self‐renewal of CSCs by direct cell‐cell contact or by nitric oxide production via the Notch signaling pathway. 10 , 11 Our previous study indicated that PSF1high cancer cells from solid tumors possessed stem cell characteristics and accumulated near the vascular niche. 12
PSF1 is a member of the heterotetrameric GINS (Go‐Ichi‐Ni‐San) complex, which contains PSF1, PSF2, PSF3, and SLD5. It preserves conserved sequences and regulates DNA replication in eukaryotic cells. In the process of eukaryotic DNA replication, the GINS complex and CDC45 are recruited by minichromosome maintenance (MCM) helicase 2‐7, forming the CDC45‐MCM2‐7‐GINS (C‐M‐G) complex. This then acts as a DNA helicase to unwind the DNA double strand, so that replication can start. 13 , 14 , 15 Moreover, several other studies have indicated that undifferentiated/progenitor cells, such as hematopoietic stem cells (HSCs), spermatogonial stem cells (SSCs) and neural stem cells (NSCs) strongly express PSF1. 16 , 17 , 18 We have shown that loss of PSF1 causes lethality in mice at embryonic day 6 due to lack of epiblast proliferation. 19 Additionally, PSF1+/‐ mice exhibited reduced HSC proliferation after bone marrow (BM) ablation with anti‐cancer drugs. 16 These data indicate that PSF1 is essential for acute cell proliferation, especially of undifferentiated/progenitor cells. Moreover, PSF1 not only plays a crucial role under physiological conditions, but is also highly expressed in cancer cells and is involved in cancer cell proliferation. 20 , 21 , 22 Therefore, PSF1 may represent a prognostic marker in several types of cancers. 23 , 24 However, it remains unknown whether PSF1 levels are associated with chemoresistance.
To assess chemoresistance, here we used acute and chronic myeloid human leukemia cell lines, THP‐1 and MEG‐1 cells, respectively, which highly express PSF1. Intravenous injection of these leukemia cells results in their occupation of the mouse BM and constitutes an animal model of human leukemia. Using this mouse model, we investigated whether PSF1high cells localize near blood vessels in BM and manifest chemoresistance. For subsequent in vitro analysis of chemoresistance in these leukemia cells, we conducted experiments to knock down (KD) PSF1 to determine whether it is involved in cell cycle progression and cell death.
2. MATERIALS AND METHODS
2.1. Animals
NOD.Cg‐PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice were purchased from Charles River Laboratories Japan, Inc (Yokohama, Japan). C57BL/6 mice were purchased from Japan SLC (Shizuoka, Japan). Animals were housed in environmentally controlled rooms at the animal experimentation facility at Osaka University. All experiments were performed in accordance with the guidelines of the Osaka University Committee for Animal and Recombinant DNA Experiments. Mice were handled and maintained according to Osaka University guidelines for animal experimentation.
2.2. Cell culture
Human leukemia cell lines HL60, KG‐1a, THP‐1, HEL92.1 (AML), NALM‐6 (B‐ALL), MOLT‐4, SKW3 (T‐ALL) and MEG‐1 (CML) were purchased from the RIKEN cell bank (Tsukuba, Japan). Normal human peripheral blood mononuclear cells (PBMCs) were purchased from Precision Bioservices (Frederick, MD, USA). Human leukemia cell lines were cultured in RPMI‐1640 (Sigma) supplemented with 10% or 20% (KG‐1a) fetal bovine serum (FBS; Sigma) and 1% penicillin/streptomycin (100 U/mL; Life Technologies). PBMCs were cultured in RPMI‐1640 (Sigma) supplemented with 10% FBS (Sigma), 2 mmol/L l‐glutamine (Gibco), 50 μg/mL gentamicin (Gibco) and 25 mmol/L HEPES (Gibco). Purified LSK (Lineage– Sca‐1+ c‐Kit+) cells were cultured for a short term in RPMI‐1640 (Sigma) supplemented with 1% FBS (Sigma), 100 ng/mL mouse thrombopoietin (Thpo; R&D Systems) and 10 ng/mL mouse stem cell factor (SCF; PeproTech) at 37°C in a humidified atmosphere containing 5% CO2 in air for 48 h.
2.3. Transplantation of THP‐1‐GFP cells
NSG mice (7‐8 wk of age) were irradiated sublethally with 2.5 Gy of total body irradiation 24 h before injection of THP‐1 GFP cells. These were suspended in PBS at a final concentration of 1 × 106 cells per 100 μL of PBS per mouse for tail vein injection. Daily monitoring of mice for symptoms of disease determined the time of animal sacrifice for examination.
2.4. AraC treatment
AraC (Cytarabine) was purchased from Sigma (PHR 1787) and was dissolved in distilled water. For animal experiments, AraC was administered intraperitoneally (ip) at a dose of 60 mg/kg/d consecutively for 5 d or once every 2 d. For the in vitro experiments, cells were incubated with AraC at final concentrations of 1, 2 or 4 μmol/L in culture medium for 48 h.
2.5. Immunohistochemistry
Preparation of BM sections: fresh femoral or tibial bone from NSG mice (THP‐1 transplanted recipients) was embedded in super cryoembedding medium (Section‐lab) and frozen in liquid nitrogen. Next, 10‐μm cryostat sections were cut using the Kawamoto film method. 25 Procedures for tissue fixation and staining of sections with antibodies have been described previously. 26 Primary antibodies used were rabbit anti‐GFP (MBL, Nagoya, Japan) and rat anti‐endomucin (eBioscience). Secondary antibodies were anti‐rabbit IgG Alexa Fluor 488 (Invitrogen) and anti‐rat IgG Alexa Fluor 546 (Invitrogen). Cell nuclei were visualized with DAPI (Invitrogen). Sections were examined by upright microscopy (DM5500B; Leica) or confocal microscopy (TCS/SP5; Leica). In all analyses, an isotype‐matched Ig was used as a negative control to confirm that the positive signals were not due to nonspecific background staining. Images were processed using Photoshop CS6 software (Adobe Systems). Analysis of the distance between GFP+ leukemia cells and blood vessels was performed using Volocity software (PerkinElmer).
2.6. Flow cytometry and cell sorting
Single cell suspensions from BM were prepared as described previously. 27 Red blood cells (RBC) were removed from BM cells using RBC lysis buffer (Sigma), and cells were stained with anti‐CD16/32 antibody (93, BioLegend) for blocking. Monoclonal antibodies (mAbs) used for this assay were the anti‐lineage cocktail (BD Biosciences), Sca‐1 (E13‐161.7; BioLegend), c‐Kit (2B8; BioLegend), Flk2/CD135 (A2F10; BioLegend), CD150 (TC15‐12F12.2; BioLegend) and CD48 (HM48‐1; BioLegend). All mAbs were purified and conjugated with either PerCP‐Cy5.5, PE‐Cy7, APC‐Cy7, APC, PE, and FITC. Flow cytometry was performed on a FACSCalibur instruments (BD Biosciences) and a FACS Aria instrument (BD Biosciences) was used for cell sorting. Data analysis was performed using FlowJo v10 software (FlowJo, LLC). Dead cells were excluded by propidium iodide (PI; Sigma) staining or by analyses using the two‐dimensional profile forward scatter vs side scatter.
2.7. Quantitative real‐time PCR
Total RNA was extracted from cultured cells or sorted cells using QIAshredder and RNeasy‐plus mini kits (Qiagen). To generate cDNA, the PrimeScript RT reagent kit (TaKaRa) was used. Quantitative real‐time PCR analysis was performed using TB Green Premix Ex Taq Ⅱ (TaKaRa) and the LightCycler 96 System (Roche Diagnostics GmbH). The level of target gene expression in each sample was normalized to that of glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH). We used the following primer sets for human genes: 5′‐ACGAGGATGGACTCAGACAAG‐3′ (forward) and 5′‐TGCAGCGTCGATTTCTTAACA‐3′ (reverse) for PSF1, 5′‐CATCCCGAAGGCAGACGAAA‐3′ (forward) and 5′‐GCGCTTGTGTGAGGAAAGTC‐3′ (reverse) for PSF2, 5′‐GGAAGCGGAGAAGCTCAAGT‐3′ (forward) and 5′‐CTTGGAACCCTGTGGGACC‐3′ (reverse) for PSF3, 5′‐AGTTGGCCTTTGCCAGAGAGT‐3′ (forward) and 5′‐GAACTGCCCGAAAGAGGTCC‐3′ (reverse) for SLD5 and, finally, 5′‐GTCTCCTCTGACTTCAACAGCG‐3′ (forward) and 5′‐ACCACCCTGTTGCTGTAGCCAA‐3′ (reverse) for GAPDH.
2.8. Western blotting
Methods for western blotting have been described previously. 28 Briefly, lysates from whole cells were resolved in SDS‐PAGE. Proteins separated electrophoretically using 12.5% SDS‐PAGE gels were transferred to polyvinylidene difluoride membranes (GE Healthcare) using a wet blotting procedure and incubated with rat anti‐PSF1 (Genestem), or mouse anti‐GAPDH (Millipore). Proteins were detected using horseradish‐peroxidase‐conjugated goat anti‐rat IgG, or goat anti‐mouse IgG (Jackson Laboratories) secondary antibodies and ECL reagents (GE Healthcare). The blots were scanned with an imaging densitometer Amersham Imager 680 system (GE Healthcare).
2.9. Lentiviral shRNA transduction
Lentiviral vectors expressing mouse (E. coli glycerol stock) or human (lentiviral particles) PSF1 shRNAs and control scrambled RNA were purchased from Sigma (Table S2). For the human lentivirus‐mediated KD of PSF1, vectors were transfected into THP‐1 or MEG‐1 cells using 8 μL hexadimethrine bromide (Sigma), according to the manufacturer's instructions. In brief, cells (1 × 105) were seeded and starved for c. 8 h, and shRNA was then added at a concentration of 10 MOI together with 8 μL hexadimethrine bromide. After overnight incubation at 37°C, medium was changed the next day. To establish stable shRNA, the antibiotic puromycin (Sigma) was added as a selection marker. Transduced cells were analyzed by western blotting and real‐time PCR to confirm KD efficiency. For the mouse lentivirus‐mediated KD, the PSF1 shRNA lentiviral vector was purified from E. coli using a plasmid midi kit (Qiagen), and was packaged into Lenti‐X 293T cells (TaKaRa) using the Lentiviral High Titer Packaging Mix system (TaKaRa) according to the manufacturer's instructions. Infectious lentiviruses were harvested at 48 and 72 h post‐transfection, and the medium was centrifuged at 2300 g for 10 min at room temperature to pellet cell debris. The medium was then filtered through 0.22‐μm‐pore cellulose acetate filters. Viral particle preparations were aliquoted into cryogenic vials and stored at −80°C until use. Lentivirus concentrations were analyzed using a Lenti‐X p24 Rapid Titer Kit (TaKaRa). The transduced cells were analyzed by real‐time PCR to confirm KD efficiency.
2.10. Cell proliferation assay
Cells were divided into controls and shRNA groups. Each group had 3 replicates, each with 2.5 × 104 cells seeded into 6‐well plates and incubated at 37°C. At each time point, cells were suspended in 0.4% trypan blue (Sigma) and counted by hemocytometer (Waken).
2.11. Cell death assay
Cell death was analyzed by staining cells with 7‐aminoactinomycin D (7‐AAD; BD Biosciences) and annexin V (Invitrogen). In brief, after washing with ice‐cold PBS and centrifugation, cell pellets were resuspended in 100 μL 1× binding buffer (BD Biosciences), and stained with 5 μL 7‐AAD and 5 μL annexin V for 20 min at room temperature in the dark. Finally, 400 μL of 1× binding buffer was added and the percentage of apoptotic cells was analyzed using a FACSCalibur (BD Biosciences).
2.12. Cell cycle analysis
Cells were resuspended and fixed in cold 70% ethanol/PBS at −20°C overnight using the PI method. Fixed cells were washed twice with PBS, resuspended in 500 μL PI staining solution (50 μg/mL PI [Sigma], 0.05% Triton X‐100 [Nacalai tesque] and 0.1 mg/mL RNase A [Cell Signaling Technology]) for 20 min at room temperature in the dark. Cell cycle analysis was performed using a FACSCalibur (BD Biosciences).
2.13. Colony‐forming unit (CFU) assay
Purified LSK cells were plated in triplicate in 35 mm Petri dishes (FALCON) containing 1 mL MethoCult GF M3434 medium (StemCell Technologies). After 10 d incubation at 37°C in 5% CO2 in air, CFU‐GM, G, M, GEMM, BFU‐E were scored under an inverted microscope (DMi8; Leica).
2.14. Statistical analysis
All data are presented as means ± standard deviation (SD). Statistical analysis was performed using Statcel version 2 software (OMS). Data were analyzed by ANOVA, followed by Tukey‐Kramer multiple comparison testing. When only 2 groups were compared, a two‐sided Student t test was used. A P‐value <.05 was considered to be statistically significant.
3. RESULTS
3.1. PSF1 is highly expressed in human leukemia cell lines
To investigate PSF1 expression levels, western blotting and real‐time PCR assays were used to quantify protein and mRNA in 8 human leukemia cell lines, namely, the B‐cell acute lymphocyte leukemia (B‐ALL) line NALM‐6, T‐cell acute lymphocyte leukemia (T‐ALL) MOLT‐4, SKW3, acute myeloid leukemia (AML) HL60, THP‐1, HEL92.1, KG‐1a and the chronic myeloid leukemia (CML)‐derived line MEG‐1. Similar to our previous report showing strong expression of PSF1 in solid cancer cells, 12 , 23 , 24 both PSF1 protein and mRNA levels were significantly higher in every leukemia cell line than in normal human PBMCs (Figure 1A,B). This suggested that PSF1 could be a potential target to control leukemia. To elucidate the function of PSF1 in leukemic cells, we selected the AML cell line THP‐1 and the CML cell line MEG‐1 for the following experiments.
Figure 1.
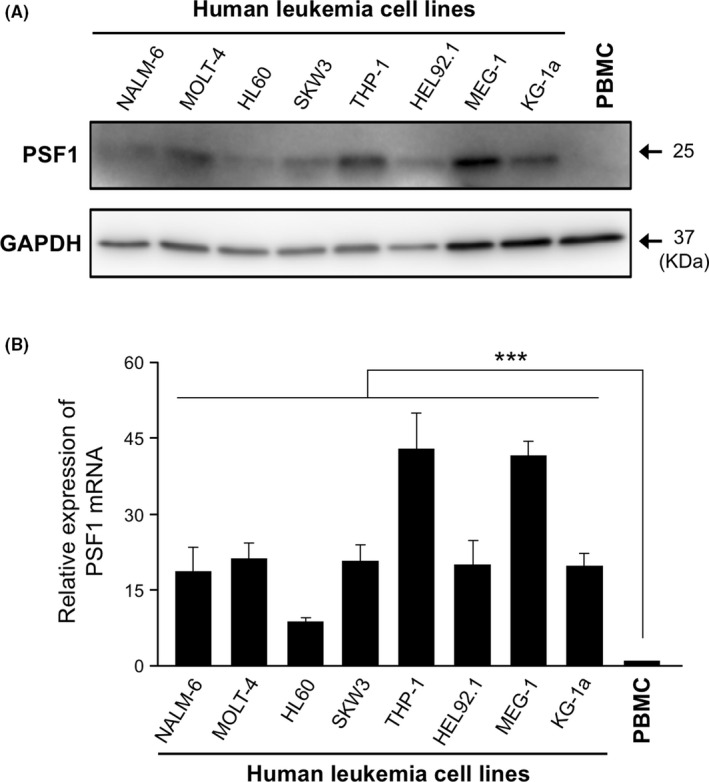
Expression of PSF1 protein and mRNA in human leukemia cell lines. PSF1 protein and mRNA expression levels were analyzed by western blotting (A) and quantitative RT‐PCR (B). NALM‐6 (B‐ALL), MOLT‐4, SKW3 (T‐ALL), HL60, THP‐1, HEL92.1, KG‐1a (acute myeloid leukemia) and MEG‐1 (chronic myeloid leukemia) were investigated. Peripheral blood mononuclear cells (PBMCs) were used as the normal human blood control. GAPDH was used as an internal control (A). The relative amounts of PSF1 mRNA were normalized to PBMCs (B) (n = 3). Error bars represent means ± SD; ***P < .001
3.2. PSF1high leukemic cells are resistant to anti‐cancer drugs
In our previous work on solid tumors (non‐small cell lung cancer and colorectal cancer), we found that PSF1high cells corresponded to a CSC population and accumulated near the blood vessels, 12 suggesting that the vascular niche supports their survival and proliferation. The vascular niche might provide an environment for cancer cell recovery and escape from chemotherapy. To test this hypothesis in leukemia, we established a mouse leukemia model using human THP‐1 GFP leukemia cells (Figure 2A). At 3 wk after THP‐1 GFP cell injection, mice were treated with the anti‐cancer drug AraC, and the distribution of surviving cancer cells and their expression of PSF1 in the BM was investigated. AraC is a pyrimidine analog that inhibits DNA replication, and is used clinically to treat different forms of leukemias, including AML and CML. The results showed that, as expected, the number of GFP+ THP‐1 cells had significantly decreased on treatment, but some survived (Figure 2B,C). Real‐time PCR results indicated that PSF1 mRNA expression was significantly increased in these surviving GFP+ THP‐1 cells (Figure 2D). Testing BM before and after treatment with AraC revealed that the remaining leukemia cells were localized near endothelial cells (Figure 2E,F), suggesting that the vascular niche was playing a similar role in leukemia as seen in solid cancers.
Figure 2.
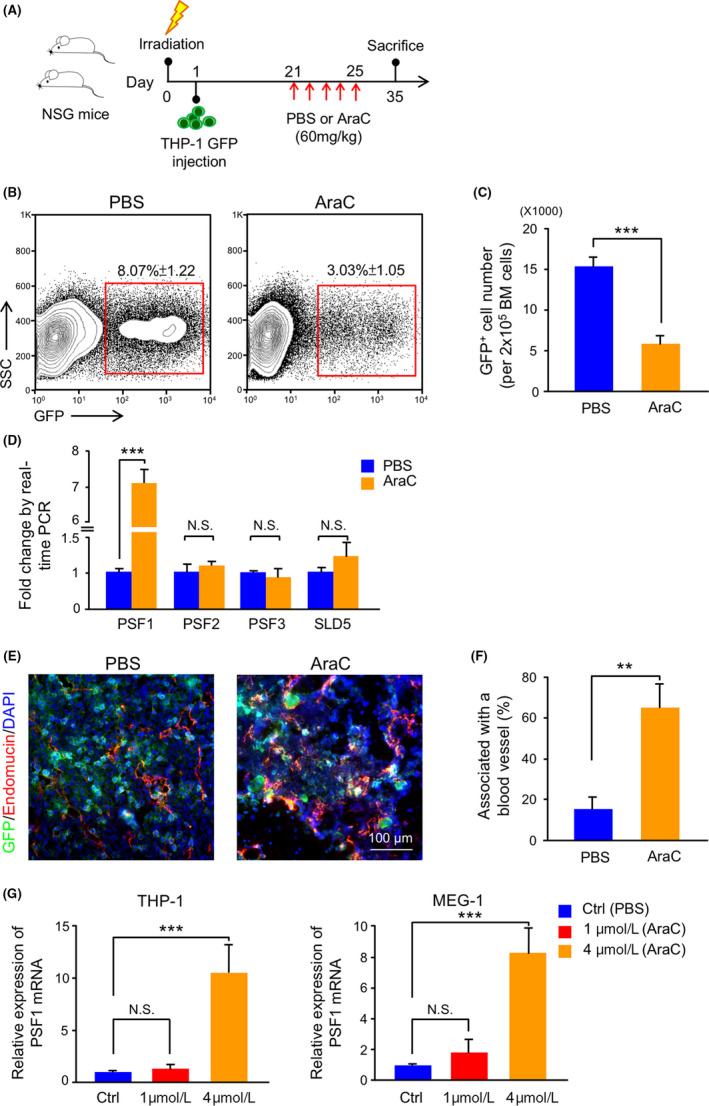
Drug resistance of leukemia cells in a mouse leukemia model using human leukemia cells. A, Experimental scheme for THP‐1 GFP cells transplantation and AraC treatment model. For details, see Section 2. B, Flow cytometric analysis of GFP+ cells in bone marrow (BM) from PBS‐treated or AraC‐treated mice. The percentage of GFP+ cells within total cells in the BM is shown in the red box (n = 6 per group). Error bars represent means ± SD. C, Quantitative evaluation of the number of GFP+ cells observed in (B) (n = 6 per group). Error bars represent means ± SD; ***P < .001. D, Quantitative real‐time PCR analysis of GINS member mRNA expression in GFP+ cells in BM from PBS‐treated or AraC‐treated mice (n = 6). Error bars represent means ± SD; ***P < .001, NS, not significant. E, Immunofluorescence staining of GFP (green), endomucin (red), and DAPI (blue) in BM sections from PBS‐treated or AraC‐treated mice. Scale bar, 100 μm. F, Percentage of GFP+ cells adjacent to blood vessels in BM sections from PBS‐treated or AraC‐treated mice. Error bars represent means ± SD; **P < .01 (5 random fields of 6 independent BM sections). G, Quantitative real‐time PCR analysis of PSF1 mRNA expression in THP‐1 and MEG‐1 cells after stimulation with PBS (Ctrl) or AraC (1 and 4 μmol/L) (n = 3). Error bars represent means ± SD; ***P < .001, NS, not significant
PSF1high leukemia cells in the BM seemed to be chemoresistant. To test this directly, we treated them with AraC in vitro and correlated PSF1 expression with chemoresistance. Further to explore the effect of different drug concentrations on PSF1 expression, we used AraC at the low concentration of 1 μmol/L and compared this dose with the half‐maximal inhibitory concentration (IC50) of 4 μmol/L. We found that cells surviving under AraC treatment had higher PSF1 expression at a level depending on the dose of drug used (Figure 2G).
Taken together, these data suggest that the vascular niche supports leukemia cells strongly expressing PSF1 and facilitates their proliferation and survival in the presence of chemotherapeutic drugs. To test this, we next analyzed whether PSF1 was indeed involved in proliferation and survival of leukemic cells.
3.3. Knockdown of PSF1 reduces growth of AML and CML cells
PSF1 plays an important role in DNA replication initiation and, therefore, complete knockout of PSF1 causes inhibition of cell proliferation. 16 , 19 Here, we partially knocked down PSF1 (PSF1‐KD) in leukemia cells to evaluate its functions. THP‐1 PSF1‐KD or MEG‐1 PSF1‐KD cells were generated by lentiviral transfection of short hairpin RNA (shRNA) followed by selection of a stable pool after 48 h. Efficiency of protein and mRNA KD was confirmed by western blotting and real‐time PCR analysis. Both PSF1 protein and mRNA levels were decreased significantly in KD cells (Figure 3A‐D). We observed that decreased amounts of PSF1 correlated with slower cell growth than that seen in the control leukemia cells (Figure 3E,F).
Figure 3.
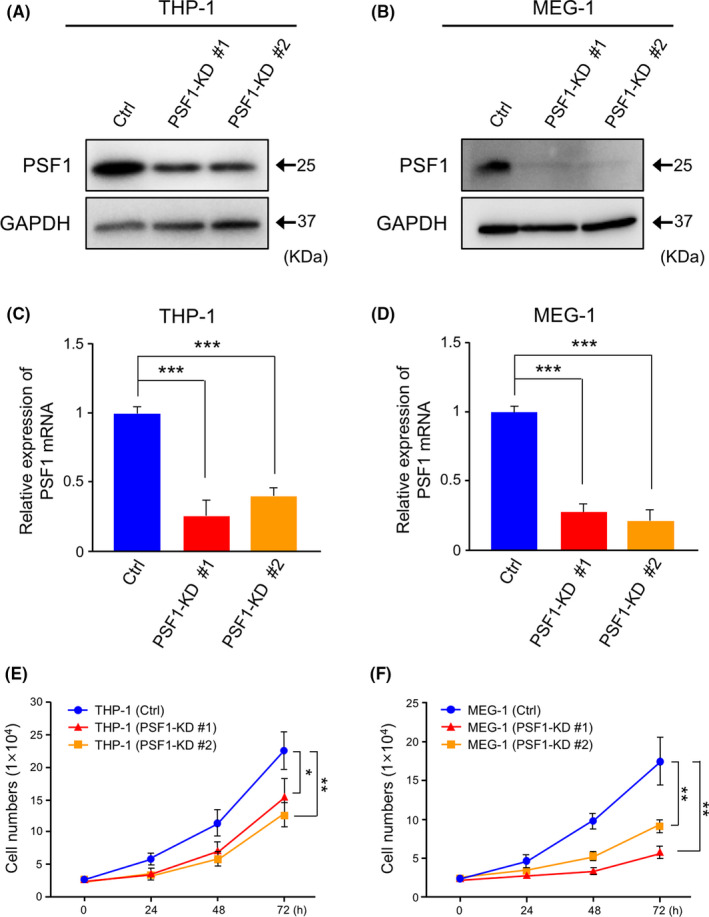
Knockdown (KD) of PSF1 inhibits the proliferation of acute myeloid leukemia (AML) and chronic myeloid leukemia (CML) cells. A, B, Western blots showing PSF1 expression in THP‐1 (A) and MEG‐1 (B) cells. Protein level of PSF1 was evaluated in control (Ctrl) and PSF1‐KD cells. GAPDH was used as the internal control. C, D, Quantitative RT‐PCR analysis of PSF1 mRNA expression in THP‐1 (C) and MEG‐1 (D) cells. mRNA levels of PSF1 were evaluated in Ctrl and PSF1‐KD cells (n = 3). Error bars represent means ± SD; ***P < .001. E, F, Effect of KD of PSF1 on growth of THP‐1 (E) and MEG‐1 (F) cells. Cells were counted at several time points as indicated (n = 3). Error bars represent means ± SD; *P < .05, **P < .01
3.4. PSF1‐KD causes cell cycle arrest at the quiescent G0/G1 phase in AML and CML cells
Further to investigate the function of PSF1, we performed cell cycle analysis. First, we examined the cell cycle status of THP‐1 and MEG‐1 by PI staining (Figure 4A,B). We found that the fraction of the population in the quiescent G0/G1 state was increased after PSF1‐KD in both cell lines. Next, we starved the cells for 48 h and then stimulated with serum for 6, 12 or 24 h. The number of cells in the G0/G1 state was significantly greater after 24 h in PSF1‐KD cells from both cell lines (Figure 4C,D), however the cell cycle rate recovered to the rate before serum starvation after 24 h of serum stimulation in both parental leukemia cells and PSF1‐KD cells from both cell lines. These findings indicated that PSF1 is involved in cell cycle progression of leukemia cells but not involved in cell cycle recovery.
Figure 4.
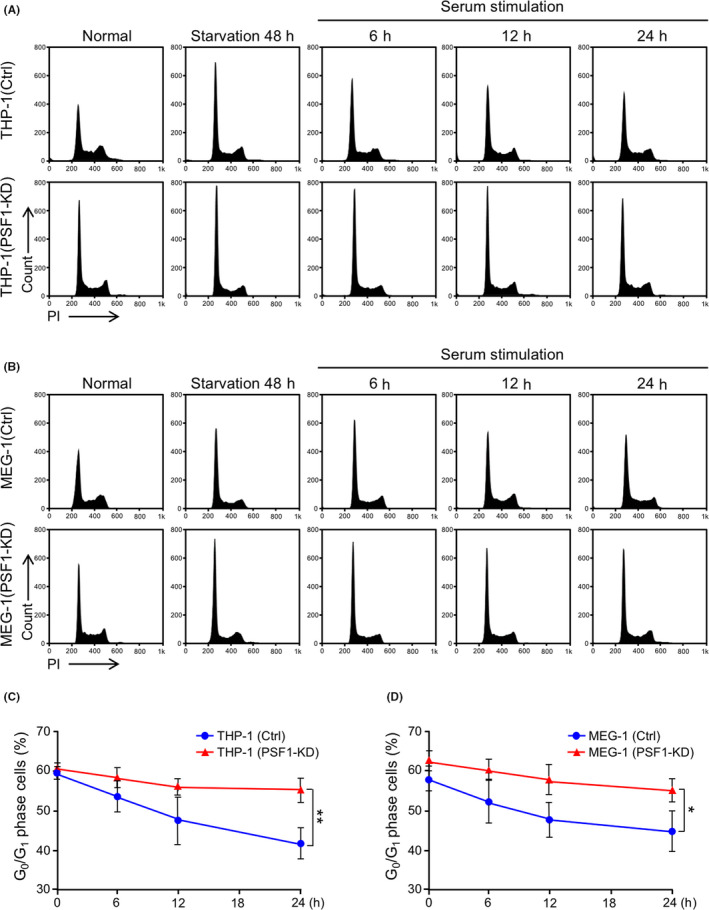
Knockdown (KD) of PSF1 causes cell cycle arrest at the G0/G1 phase in acute myeloid leukemia (AML) and chronic myeloid leukemia (CML) cells. A, B, Cell cycle analysis of PSF1 KD THP‐1 (A) and MEG‐1 (B) cells. Cells were harvested from normal cultures, after 48 h starvation, and following serum stimulation after 6, 12, 24 h. All samples were analyzed by propidium iodide (PI) staining. Representative data are shown. C, D, G0/G1 phase of cells was quantified in PSF1 KD THP‐1 (C) and MEG‐1 (D) cells (n = 3). Error bars represent means ± SD; *P < .05, **P < .01
3.5. PSF1‐KD induces death of AML and CML cells
To further confirm the influence of PSF1 on cell death, 7AAD‐annexin V analysis was performed on control and PSF1‐KD leukemic cells. The data indicated that THP‐1 PSF1‐KD and MEG‐1 PSF1‐KD cells had higher cell death rates (THP‐1 PSF1‐KD: 9.12 ± 0.71% and MEG‐1 PSF1‐KD: 8.17 ± 0.77%) compared with the control group (THP‐1:2.11 ± 0.84% and MEG‐1:1.13 ± 0.62%) (Figure 5A,B,E,F). Both apoptosis and necrosis were increased in the THP‐1 PSF1‐KD and MEG‐1 PSF1‐KD populations. These results suggested that PSF1 KD induces cell death in leukemic cells. To further determine whether PSF1‐KD leukemia cells could be useful against drug resistance, we treated THP‐1, THP‐1 PSF1‐KD, MEG‐1, and MEG‐1 PSF1‐KD cells with AraC. The results indicated that THP‐1 PSF1‐KD and MEG‐1 PSF1‐KD cells showed significantly increased cell death on treatment with AraC (THP‐1 PSF1‐KD: Figure 5C,D; MEG‐1 PSF1‐KD: Figure 5G,H). These data suggest that PSF1 inhibition could increase the therapeutic effect of chemotherapy on leukemia cells.
Figure 5.
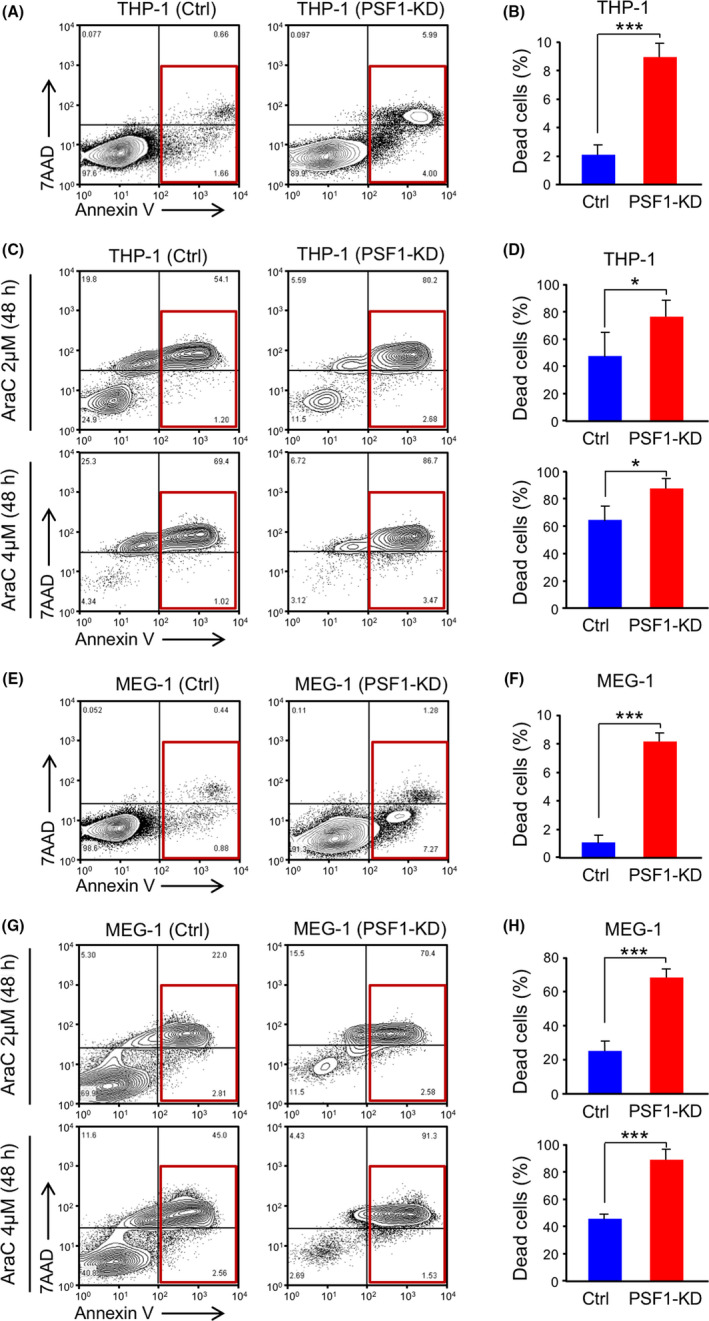
Knockdown (KD) of PSF1 induces cell death in acute myeloid leukemia (AML) and chronic myeloid leukemia (CML) cells. Cell death analysis (red box: apoptosis and necrosis) of PSF1 KD THP‐1 (A, B) and MEG‐1 (E, F) cells. Proportions of dead cells in PSF1‐KD THP‐1 (B) and MEG‐1 (F) cells were calculated and compared with the Ctrl group (n = 4). Error bars represent means ± SD; ***P < .001. Analysis of cell death in PSF1 KD THP‐1 (C, D) and MEG‐1 (G, H) cells following AraC treatment (2 and 4 μmol/L). Quantification of cell death of PSF1‐KD THP‐1 (D) and MEG‐1 (H) cells compared with the Ctrl group (n = 4). Error bars represent means ± SD; *P < .05; ***P < .001
3.6. PSF1‐KD changes the localization of leukemic cells in BM
To determine whether KD of PSF1 in leukemia cells altered their distribution around blood vessels in BM, we constructed a mouse leukemia model using THP‐1 GFP and THP‐1 GFP PSF1‐KD leukemia cells (Figure 6A). At 15 d after BM transplantation (BM‐T), we found that the number of GFP+ PSF1‐KD leukemic cells in the BM was significantly lower than in the controls (Figure 6B,C). This phenotype could have resulted from an increase in cell death of the PSF1‐KD cells. We also performed immunofluorescence staining of recipient mouse BM to determine positional relationships between leukemic cells and blood vessels. We found that the proportion of GFP+ PSF1‐KD leukemic cells that located proximate to blood vessels was significantly lower than in the controls (Figure 6D,E). We quantified the distances between leukemic cells and blood vessels in BM. The results showed that the number of GFP+ PSF1‐KD leukemic cells in close proximity to blood vessels was low (<15 μm), but abundant at a distance (>30 μm) (Figure 6F), unlike that seen with control leukemic cells. These results suggested that leukemic cells that highly expressed PSF1 maintained their interactions with blood vessels, and this may give possible protection against damage by chemotherapy.
Figure 6.
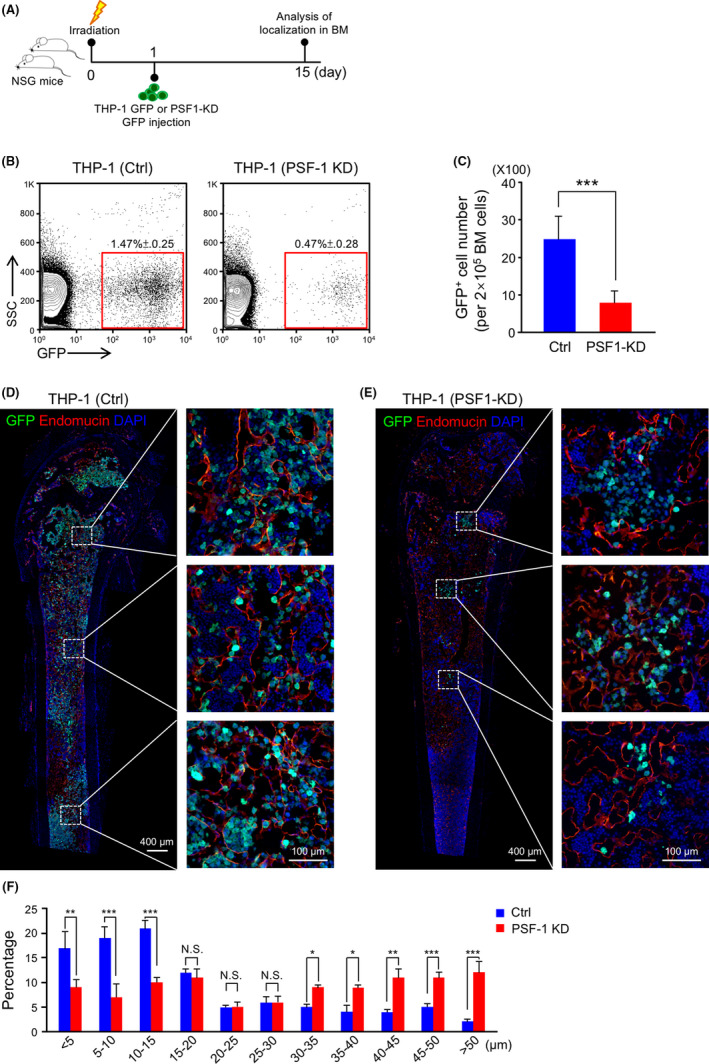
Knockdown (KD) of PSF1 changes the distance between leukemia cells and blood vessels in bone marrow (BM). A, Experimental scheme for THP‐1 GFP (Ctrl) or THP‐1 GFP (PSF1‐KD) cells used in the BM‐T model. B, Flow cytometric analysis of GFP+ cells in BM from Ctrl or PSF1‐KD transplanted mice. The percentage of GFP+ cells within all cells in the BM is shown in the red box (n = 4 per group). Error bars represent means ± SD. C, Quantitative evaluation of the number of GFP+ cells observed in (B) (n = 4 per group). Error bars represent means ± SD; ***P < .001. D, E, Immunofluorescence staining of GFP (green) and endomucin (red) in BM sections from Ctrl (D) or PSF1‐KD (E) recipient mice. Endomucin was selected as an endothelial cell marker in BM. DAPI (blue) was used to detect nuclei and dashed boxes indicate areas shown at higher magnification. Scale bars, 400 μm and 100 μm (inset). F, Distances between Ctrl or PSF1‐KD GFP+ cells and blood vessels in the metaphysis and diaphysis area of recipient mouse BM. Error bars represent means ± SD; *P < .05, **P < .01, ***P < .001, NS, not significant (5 random fields of 6 independent BM sections)
4. DISCUSSION
One of the biggest barriers in cancer chemotherapy is drug resistance, which is associated with cancer cell plasticity and heterogeneity. 1 , 2 Regarding the issue of heterogeneity, malignant cancer cells such as CSCs may sustain their proliferative capacity even on exposure to anti‐cancer drugs. 29 , 30 , 31 With increasing understanding of the cancer microenvironment, the notion that cancer cells can be maintained in the vascular niche has gradually gained traction. In the present study, we treated mice with an anti‐cancer drug in a leukemia transplantation model, and found that cancer cells that strongly expressed PSF1, a DNA initiation regulating factor, localized to vascular areas and were drug resistant. These findings suggested that the vascular niche might play a critical role in chemotherapy resistance. Moreover, PSF1 might act as a potential therapeutic target to enhance the effect of chemotherapy and prognosis.
Our results indicated that PSF1‐KD causes cell cycle arrest in leukemic cells, but that the cytotoxic effect of AraC was enhanced. We hypothesize that there are 2 possible reasons to account for this phenomenon. The first is that AraC might mediate its action by other mechanisms, eg AraC‐induced cell death is associated with the accumulation of reactive oxygen species in AML cells. 32 , 33 The other reason is that cancer cells tend to undergo cell division despite a paucity of DNA replication factors, because cell cycle checkpoint systems are disrupted. Therefore, once the cell cycle is arrested, leukemic cells nonetheless soon start a new cell cycle, and thus death may be induced because of the lack of a full set of DNA replication factors.
It has to be noted that AML treatment has not changed markedly over the past 3 decades. Standard induction chemotherapy relies on the combination of the nucleoside analog AraC with an anthracycline such as daunorubicin or idarubicin, sometimes in association with other drugs, such as etoposide. AraC has been utilized not only for induction therapy for AML but also consolidation therapy, because it has more durable effects on leukemia cells. 34 , 35 , 36
4.1. The relationship between PSF1 and cancer stem cells
Many studies have indicated that CSCs are the main culprits in chemotherapy resistance. One explanation for this is that due to the ATP‐binding cassette (ABC) transporters expressed on the surface of CSCs, 37 anti‐cancer drugs are excluded to protect the cells from injury. However, another possibility is the existence of a specific niche allowing CSCs to escape chemotherapy by entering a dormant state in such a niche. 38 , 39 Our previous studies have shown that PSF1 is highly expressed in undifferentiated cells such as HSCs, SSCs, and NSCs. 16 , 17 , 18 Moreover, PSF1‐deficiency in mice leads to lethally at embryonic day 6.5. 19 These findings suggested that PSF1 plays an important role in the regulation of stem cell proliferation. Furthermore, some previous studies revealed that PSF1 is expressed abnormally in several types of cancers, such as lung, breast, and prostate cancer. 20 , 21 , 23 Patients harboring PSF1high cancer cells had a poorer prognosis. 23 , 24 In a mouse model, PSF1high cancer cells showed higher invasive and metastatic ability compared with PSF1low cells, and their characteristic gene expression patterns were similar to those of embryonic stem cells. 12 Hence, we concluded that PSF1high cells were more similar to CSCs. To test this hypothesis, we analyzed the stemness genes in surviving leukemia cells after treatment with AraC. We found that Oct 4 and Nanog, which are characteristic stemness gene, were strongly expressed at the mRNA level in such cells (Supporting Information Figure S1 and Table S1). These results indicated that leukemia cells that highly expressed PSF1 might have stem cell characteristics.
4.2. The relationship between PSF1 and the vascular niche
The HSC niche was the first defined microenvironment maintaining stem cells in a quiescent or undifferentiated state. 40 Following the rapid development of CSC surface marker research, CSCs can be isolated from different types of cancers, enabling interactions between the tumor microenvironment and CSCs to be investigated. In our previous work on lung and colorectal cancers, we found that cells that highly expressed PSF1 also localized near the tumor blood vessels. 12 Moreover, we also observed that, after AraC treatment, surviving cancer cells localized near the vascular region and these cells had a higher proliferation rate than other cancer cells at some distance from the blood vessels (data not shown). On this basis, we suggested that, after chemotherapy, surviving cancer cells near the vascular region need to interact with vascular endothelial cells for self‐maintenance and proliferation.
4.3. Potential therapeutic exploitation of PSF1
We conducted a survey of GINS1 (PSF1) expression using PrognoScan (http://dna00.bio.kyutech.ac.jp/PrognoScan‐cgi/PrognoScan.cgi). The results indicated that multiple myeloma patients who had higher PSF1 expression had a poor prognosis relative to other hematological malignancies. While more extensive investigations are required, this finding suggests that PSF1 may be a target for leukemia treatment.
In this work, we have shown that PSF1 is highly expressed in several types of leukemias, suggesting that it may be a pan‐leukemia marker, and might be a potential therapeutic target to enhance the effect of chemotherapy. The most successful targeted therapies are mediated by chemical entities that target or preferentially target a protein or enzyme that carries a mutation or other genetic alteration that is specific to cancer cells and not found in normal host tissue. Based on this theory, we evaluated damage to normal HSCs after PSF1 targeted therapy in a mouse model. First, we analyzed PSF1 expression in different hematopoietic cell populations from wild‐type mouse BM and found that quiescent LT‐HSCs and Lin+ mature blood cells expressed lower levels of PSF1. In contrast, PSF1 expression was higher in progenitors (multipotent progenitors (MPPs) and LSK) to assist cell differentiation and proliferation (Figure S2A). Therefore, damage might occur mainly in the progenitors rather than in LT‐HSCs if PSF1 was to be utilized as a therapeutic target. We also evaluated the effect of PSF1‐KD on hematopoietic function of LSK HSCs. The results revealed that PSF1‐KD increased cell death and reduced the colony‐forming ability of LSK cells (Figure S2B‐D). Therefore, we need to take this effect into account when PSF1 is utilized as a target for therapy in leukemia. Finally, in our previous research on breast cancers, we identified a PSF1 peptide that bound human leukocyte antigen (HLA‐A02) and could be recognized by antigen‐specific cytotoxic T lymphocytes, which could then attack PSF1high cancer cells. 41 Further analysis is warranted to determine whether targeting of PSF1 may offer any benefit in terms of decreasing chemoresistance and eliminating CSCs, including leukemia CSCs.
DISCLOSURE STATEMENT
The authors have no conflicting financial interest.
Supporting information
Figure S1
Figure S2
Table S1
Table S2
ACKNOWLEDGMENTS
We thank N. Fujimoto for the preparation of plasmid DNA and Y. Mori for administrative assistance. This work was partly supported by the Japan Agency for Medical Research and Development (AMED) under Grant number (19cm0106508h0004, 19gm5010002h0003) and the Japan Society for the Promotion of Science (JSPS) Grants‐in‐Aid for Scientific Research (A) (16H02470) and an IPBS Grant‐in‐Aid for Interdisciplinary Research Fiscal Year 2018.
Hsieh H‐Y, Jia W, Jin Z‐C, Kidoya H, Takakura N. High expression of PSF1 promotes drug resistance and cell cycle transit in leukemia cells. Cancer Sci. 2020;111:2400–2412. 10.1111/cas.14452
These authors contributed equally to this work.
Contributor Information
Weizhen Jia, Email: jiawz@biken.osaka-u.ac.jp.
Nobuyuki Takakura, Email: ntakaku@biken.osaka-u.ac.jp.
REFERENCES
- 1. Marusyk A, Almendro V, Polyak K. Intra‐tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer. 2012;12:323‐334. [DOI] [PubMed] [Google Scholar]
- 2. Yates LR, Gerstung M, Knappskog S, et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med. 2015;21:751‐759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Roerink SF, Sasaki N, Lee‐Six H, et al. Intra‐tumour diversification in colorectal cancer at the single‐cell level. Nature. 2018;556:457‐462. [DOI] [PubMed] [Google Scholar]
- 4. McKenzie MD, Ghisi M, Oxley EP, et al. Interconversion between tumorigenic and differentiated states in acute myeloid leukemia. Cell Stem Cell. 2019;25:258‐272. [DOI] [PubMed] [Google Scholar]
- 5. Ebinger S, Özdemir EZ, Ziegenhain C, et al. Characterization of rare, dormant, and therapy‐resistant cells in acute lymphoblastic leukemia. Cancer Cell. 2016;30:849‐862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Puig I, Tenbaum SP, Chicote I, et al. TET2 controls chemoresistant slow‐cycling cancer cell survival and tumor recurrence. J Clin Invest. 2018;128:3887‐3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Oshimori N, Oristian D, Fuchs E. TGF‐β promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell. 2015;160:963‐976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pietras A, Katz AM, Ekström EJ, et al. Osteopontin‐CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell. 2014;14:357‐369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ghajar CM, Peinado H, Mori H, et al. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol. 2013;15:807‐817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang R, Li Y, Tsung A, et al. iNOS promotes CD24+CD133+ liver cancer stem cell phenotype through a TACE/ADAM17‐dependent Notch signaling pathway. Proc Natl Acad Sci USA. 2018;115:E10127‐E10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Charles N, Ozawa T, Squatrito M, et al. Perivascular nitric oxide activates notch signaling and promotes stem‐like character in PDGF‐induced glioma cells. Cell Stem Cell. 2010;6:141‐152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nagahama Y, Ueno M, Miyamoto S, et al. PSF1, a DNA replication factor expressed widely in stem and progenitor cells, drives tumorigenic and metastatic properties. Cancer Res. 2010;70:1215‐1224. [DOI] [PubMed] [Google Scholar]
- 13. Kamada K. The GINS complex: structure and function. Subcell Biochem. 2012;62:135‐156. [DOI] [PubMed] [Google Scholar]
- 14. Onesti S, MacNeill SA. Structure and evolutionary origins of the CMG complex. Chromosoma. 2013;122:47‐53. [DOI] [PubMed] [Google Scholar]
- 15. Yuan Z, Bai L, Sun J, et al. Structure of the eukaryotic replicative CMG helicase suggests a pumpjack motion for translocation. Nat Struct Mol Biol. 2016;23:217‐224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ueno M, Itoh M, Sugihara K, et al. Both alleles of PSF1 are required for maintenance of pool size of immature hematopoietic cells and acute bone marrow regeneration. Blood. 2009;113:555‐562. [DOI] [PubMed] [Google Scholar]
- 17. Han Y, Ueno M, Nagahama Y, et al. Identification and characterization of stem cell‐specific transcription of PSF1 in spermatogenesis. Biochem Biophys Res Commun. 2009;380:609‐613. [DOI] [PubMed] [Google Scholar]
- 18. Jia W, Hsieh HY, Kidoya H, et al. Embryonic expression of GINS members in the development of the mammalian nervous system. Neurochem Int. 2019;129:104465. [DOI] [PubMed] [Google Scholar]
- 19. Ueno M, Itoh M, Kong L, et al. PSF1 is essential for early embryogenesis in mice. Mol Cell Biol. 2005;25:10528‐10532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang J, Wu Q, Wang Z, et al. Knockdown of PSF1 expression inhibits cell proliferation in lung cancer cells in vitro. Tumour Biol. 2015;36:2163‐2168. [DOI] [PubMed] [Google Scholar]
- 21. Nakahara I, Miyamoto M, Shibata T, et al. Up‐regulation of PSF1 promotes the growth of breast cancer cells. Genes Cells. 2010;15:1015‐1024. [DOI] [PubMed] [Google Scholar]
- 22. Kimura T, Cui D, Kawano H, et al. Induced expression of GINS complex is an essential step for reactivation of quiescent stem‐like tumor cells within the peri‐necrotic niche in human glioblastoma. J Cancer Res Clin Oncol. 2019;145:363‐371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tahara H, Naito H, Kise K, et al. Evaluation of PSF1 as a prognostic biomarker for prostate cancer. Prostate Cancer Prostatic Dis. 2015;18:56‐62. [DOI] [PubMed] [Google Scholar]
- 24. Kanzaki R, Naito H, Kise K, et al. PSF1 (Partner of SLD Five 1) is a prognostic biomarker in patients with non‐small cell lung cancer treated with surgery following preoperative chemotherapy or chemoradiotherapy. Ann Surg Oncol. 2016;23:4093‐4100. [DOI] [PubMed] [Google Scholar]
- 25. Kawamoto T, Kawamoto K. Preparation of thin frozen sections from nonfixed and undecalcified hard tissues using Kawamot's film method (2012). Methods Mol Biol. 2014;1130:149‐164. [DOI] [PubMed] [Google Scholar]
- 26. Kidoya H, Naito H, Muramatsu F, et al. APJ regulates parallel alignment of arteries and veins in the skin. Dev Cell. 2015;33:247‐259. [DOI] [PubMed] [Google Scholar]
- 27. Takakura N, Watanabe T, Suenobu S, et al. A role for hematopoietic stem cells in promoting angiogenesis. Cell. 2000;102:199‐209. [DOI] [PubMed] [Google Scholar]
- 28. Jia W, Kidoya H, Yamakawa D, et al. Galectin‐3 accelerates M2 macrophage infiltration and angiogenesis in tumors. Am J Pathol. 2013;182:1821‐1831. [DOI] [PubMed] [Google Scholar]
- 29. Eun K, Ham SW, Kim H. Cancer stem cell heterogeneity: origin and new perspectives on CSC targeting. BMB Rep. 2017;50:117‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. 2017;23:1124‐1134. [DOI] [PubMed] [Google Scholar]
- 31. Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14:611‐629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bossis G, Sarry JE, Kifagi C, et al. The ROS/SUMO axis contributes to the response of acute myeloid leukemia cells to chemotherapeutic drugs. Cell Rep. 2014;7:1815‐1823. [DOI] [PubMed] [Google Scholar]
- 33. Hosseini M, Rezvani HR, Aroua N, et al. Targeting myeloperoxidase disrupts mitochondrial redox balance and overcomes cytarabine resistance in human acute myeloid leukemia. Cancer Res. 2019;79:5191‐5203. [DOI] [PubMed] [Google Scholar]
- 34. Preisler H, Davis RB, Kirshner J, et al. Comparison of three remission induction regimens and two postinduction strategies for the treatment of acute nonlymphocytic leukemia: a cancer and leukemia group B study. Blood. 1987;69:1441‐1449. [PubMed] [Google Scholar]
- 35. Mayer RJ, Davis RB, Schiffer CA, et al. Intensive postremission chemotherapy in adults with acute myeloid leukemia. Cancer and Leukemia Group B. N Engl J Med. 1994;331:896‐903. [DOI] [PubMed] [Google Scholar]
- 36. Byrd JC, Dodge RK, Carroll A, et al. Patients with t(8;21)(q22;q22) and acute myeloid leukemia have superior failure‐free and overall survival when repetitive cycles of high‐dose cytarabine are administered. J Clin Oncol. 1999;17:3767‐3775. [DOI] [PubMed] [Google Scholar]
- 37. Begicevic RR, Falasca M. ABC transporters in cancer stem cells: beyond chemoresistance. Int J Mol Sci. 2017;18:E2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Goddard ET, Bozic I, Riddell SR, et al. Dormant tumour cells, their niches and the influence of immunity. Nat Cell Biol. 2018;20:1240‐1249. [DOI] [PubMed] [Google Scholar]
- 39. Senft D, Jeremias I. Tumor cell dormancy‐triggered by the niche. Dev Cell. 2019;49:311‐312. [DOI] [PubMed] [Google Scholar]
- 40. Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505:327‐334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yoshida M, Ishioka Y, Ozawa T, et al. Soluble HLA‐associated peptide from PSF1 has a cancer vaccine potency. Sci Rep. 2017;7:11137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Table S1
Table S2