

Abstract
A novel epidermal growth factor receptor (EGFR)‐tyrosine kinase inhibitor, osimertinib, has marked efficacy in patients with EGFR‐mutant lung cancer. While epithelial‐mesenchymal transition (EMT) plays a role in the resistance to various targeted drugs, its involvement in EGFR‐inhibitor resistance remains largely unknown. Preclinical experiments with osimertinib‐resistant lung cancer cells showed that EMT was associated with decreased microRNA‐200c and increased ZEB1 expression. In several resistant clone cells, pretreatment with the histone deacetylase inhibitor quisinostat helped overcome the resistance by reverting EMT. Furthermore, drug screening from a library of 100 kinase inhibitors indicated that Glycogen synthase kinase‐3 (GSK‐3) inhibitors, such as LY2090314, markedly inhibited the growth and induced apoptosis of resistant cells, specifically those with a mesenchymal phenotype. These results suggest that GSK‐3 inhibition could be useful to circumvent EMT‐associated resistance to osimertinib in EGFR‐mutant lung cancer.
Keywords: epidermal growth factor receptor, epithelial‐mesenchymal transition, glycogen synthase kinase‐3, osimertinib, resistance
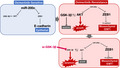
Summary scheme of the role of microRNA (miR)‐200c in the activation of the glycogen synthase kinase (GSK)‐3/AKT axis in osimertinib‐sensitive and osimertinib‐resistant cells. EMT, epithelial‐mesenchymal transition.
Abbreviations
- ddPCR
droplet digital PCR
- EGFR
epidermal growth factor receptor
- EMT
epithelial‐mesenchymal transition
- GSK
glycogen synthase kinase
- HDAC
histone deacetylase
- HER
human epidermal growth factor receptor
- miR
microRNA
- NSCLC
non‐small cell lung cancer
- TKI
tyrosine kinase inhibitor
1. INTRODUCTION
The majority of patients with NSCLC with EGFR‐activating mutations, such as exon 19 deletion and L858R point mutation, show a marked response to first‐generation reversible EGFR‐TKIs, gefitinib and erlotinib. 1 , 2 However, the acquisition of TKI resistance is almost inevitable and is commonly associated with the EGFR‐T790M gatekeeper mutation, which substitutes threonine with methionine at the amino acid position 790 of exon 20. Accordingly, the T790M mutation is detected in 50%‐60% of patients who develop clinical resistance to first‐generation EGFR‐TKIs, gefitinib or erlotinib. 3 , 4
Osimertinib is a third‐generation EGFR‐TKI that inhibits EGFR with activating mutations and/or the T790M resistance mutation. However, it does not inhibit WT EGFR or other kinases; therefore, it is called the mutant‐EGFR specific inhibitor. 5 This drug has been approved for the treatment of EGFR‐T790M‐positive NSCLC patients who acquired resistance to first‐generation or second‐generation EGFR‐TKIs. These tumors are known to acquire resistance to osimertinib through mechanisms such as the acquisition of EGFR‐C797S mutation, loss of T790M mutation, activation of a bypass pathway (such as MET and HER2), and histological transformation of cells, including small cell transformation. 6 A recent phase III clinical trial (FLAURA) reported that in patients with EGFR‐mutated NSCLC, progression‐free survival of patients treated with first‐line osimertinib was longer than that of patients treated with gefitinib or erlotinib. 7 Based on the results of the FLAURA study, first‐line treatment with osimertinib is considered as one of the standard treatments for NSCLC patients with mutated EGFR. However, notably, a fraction of the patients included in the study showed acquired resistance and an insufficient response to osimertinib treatment, similar to that observed with other EGFR‐TKIs.
Epithelial‐mesenchymal transition is an increasingly recognized driver of tumor progression and a cause of both innate and acquired resistance to various cytotoxic and targeted drugs, including EGFR‐TKIs. 8 Previous studies have reported that the prognosis of patients whose EGFR‐TKI resistance was associated with EMT was much worse than that of patients whose EGFR‐TKI resistance was due to EGFR‐T790M mutations in EGFR‐mutant NSCLC. 9 However, therapies for EMT‐associated targeted drug resistance are yet to be established. Moreover, it is not fully understood whether EMT is involved in the development of osimertinib resistance.
In this study, we investigated the underlying mechanism through which EMT was induced during the acquisition of osimertinib resistance and showed that GSK‐3 inhibition could overcome EMT‐mediated EGFR‐TKI resistance in human EGFR‐mutant lung cancer cell lines.
2. MATERIALS AND METHODS
2.1. Cell culture and reagents
The H1975 human lung adenocarcinoma cell line with the EGFR‐L858R/T790M double mutation was kindly provided by Dr Yoshitaka Sekido (Aichi Cancer Center Research Institute). All cells were maintained in RPMI‐1640 medium supplemented with 10% FBS, penicillin (100 U/mL), and streptomycin (10 μg/mL) in a humidified CO2 incubator at 37°C. Osimertinib, rociletinib, quisinostat, vorinostat, mocetinostat, belinostat, and droxinostat were obtained from Selleck Chemicals. The kinase inhibitor library, including GSK inhibitors, was also obtained from Selleck Chemicals.
2.2. Antibodies and western blot analysis
The primary Abs, including anti‐EGFR, anti‐MET, anti‐phospho‐MET, anti‐HER2, anti‐phospho‐HER2, anti‐phospho‐Akt (S473), anti‐Akt, anti‐E‐cadherin, anti‐Vimentin, anti‐ZEB1, anti‐Snail, anti‐Slug, anti‐GSK3α/β, anti‐phospho‐GSK3α/β (ser21/ser9), anti‐β‐catenin, anti‐cleaved caspase3, and anti‐β‐actin were obtained from Cell Signaling Technology. The anti‐ERK1/ERK2, anti‐p‐ERK1/ERK2 (T202/Y204), and anti‐EGFR Abs were obtained from R&D Systems. Western blot analysis was carried out as previously described. 10
2.3. Cell viability assay
Cell viability was measured using the MTT assay. Cells were plated in 96‐well plates, at a density of 2 × 103 cells per well in 100 μL RPMI‐1640 supplemented 10% FBS, and were incubated for 24 hours. Osimertinib or rociletinib were then added to each well, and incubation was continued for another 72 hours. Cell growth was measured using the MTT solution (Sigma), as previously described. 10
2.4. Immunofluorescence assay
Cells were grown on glass coverslips, and then washed with PBS and fixed with methanol chilled to −20°C. Subsequently, cells were permeabilized with 0.1% Triton X‐100 in PBS and treated with 5% BSA in PBS, followed by incubating with primary Abs (1:100) for 1 hour at 4°C. Cells were washed and further incubated with the appropriate secondary Alexa 488‐conjugated anti‐rabbit Ab (1:1000) for 1 hour. The coverslips were fixed onto a slide and the nuclei were marked with DAPI. The slides were then examined under a fluorescent microscope.
2.5. EGFR mutation analysis
Isolated DNA from cell lines was used to examine EGFR mutations within exons 18 to 21, using the peptide nucleic acid‐locked nucleic acid PCR clamp method established by Nagai and colleagues. 11 The exon 19 D761Y mutation was tested by DNA sequencing as reported by Balak and colleagues. 12
The C797S mutation was detected using digital PCR analysis. Briefly, the PCR reactions were undertaken using a Bio‐Rad QX200 ddPCR platform. Reactions were set up using ddPCR Supermix for Probes (no dUTP; Bio‐Rad) and LBx Probe EGFR C797S for the mutation analysis. The thermal cycling was undertaken on a Veriti thermal cycler (Applied Biosystems) using the following thermal cycling protocol: 10 minutes at 95°C, 40 cycles of 30 seconds at 94°C and 1 minute at 58°C, followed by 10 minutes at 98°C, with a 50% ramp rate. Results were analyzed and exported using the Quantasoft 1.6.6.0320 software.
2.6. Pretreatment with quisinostat
Cells were cultured in complete medium without inhibitor for 48 hours, followed by treatment with the HDAC inhibitor, quisinostat, in a 10‐cm dish for 24‐72 hours. Cells were then plated in 96‐well plates and treated with osimertinib, and incubated for another 72 hours for the MTT assay.
2.7. Transfection of siRNA
Small interfering RNA oligonucleotides specific to EGFR, GSK‐3α, and GSK‐3β were obtained from Santa Cruz Biotechnology. The siRNA negative control was purchased from Thermo Fisher Scientific. Transfection of siRNAs into cells was carried out using Lipofectamine RNAimax (Thermo Fisher Scientific) according to the manufacturer’s instructions.
2.8. Reporter assay and detection of miRNA
The miR‐200c‐141 promoter (1057 bp) was amplified from #4 cells and ligated into the pNL2.2 (NlucP/Hygro) vector (4832 bp) to generate the pNL2.2‐miR200c construct. The selection of positive clones was confirmed by restriction map analysis and sequencing. Cells were transfected with the pNL2.2‐miR200c construct using Lipofectamine 2000. At 24 hours posttransfection, cells were treated with drugs and incubated for 24 hours. Next, the NanoLuc luciferase activity was measured using the Nano‐Glo Luciferase Assay System (Promega). Each transfection was undertaken in triplicate in 96‐well plates. The change in miR200c expression was detected using the QuantiGene miRNA Singleplex Assay (Thermo Fisher Scientific).
3. RESULTS
3.1. Establishment of EGFR‐mutant lung cancer cells with EMT‐associated osimertinib resistance
We first sought to model acquired resistance to osimertinib by deriving polyclonal acquired resistant cell lines. The EGFR‐mutant lung cancer cell line, H1975, was initially treated with 0.03 μmol/L osimertinib. By using a stepwise dose escalation method, we generated resistant cells by administering a dose of 1 μmol/L osimertinib. In addition, we established 4 clones by limiting dilution, named H1975OR#1, OR#2, OR#3, and OR#4 (Figure 1A). Next, we examined the sensitivity of selective EGFR inhibitors to the generated cells. As shown in Figure 1B, the 4 clones were more than 100 times more resistant to osimertinib when compared with parental H1975 cells. In addition, we observed cross‐resistance to the third‐generation EGFR‐TKI, rociletinib. To elucidate the change of EGFR status in these resistant clone cells, we examined the expression and phosphorylation of EGFR through western blotting. As shown in Figure 1C, the phosphorylation of EGFR was decreased in the resistant clones, whereas the expression of EGFR was only slightly decreased. In addition, osimertinib effectively inhibited EGFR phosphorylation in both the parental H1975 cells and the 4 resistant clones. However, downstream phospho‐AKT and phospho‐ERK were inhibited in parental H1975 cells but not in the resistant clones (Figure 1C). Similarly, knockdown of EGFR by specific siRNAs inhibited the viability of parental H1975 cells but not of the 4 resistant clones (Figure 1D). The acquired EGFR C797S mutation is a known mechanism of osimertinib resistance. 6 However, we did not identify the mutation in either the parental H1975 cells or the 4 resistant clones. Notably, the resistant clones maintained the EGFR L858R and T790M mutations (Table 1). These findings indicate that the 4 clones acquire resistance through an EGFR‐independent mechanism.
FIGURE 1.
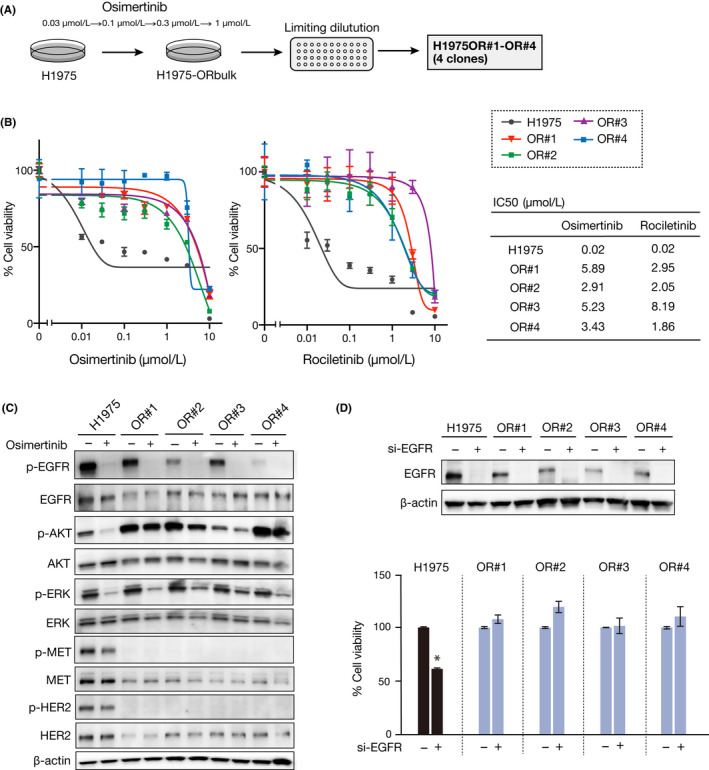
Osimertinib‐resistant H1975 clones were generated in vitro. A, H1975 parental cells and the resistant clones (OR#1, OR#2, OR#3, and OR#4) were established through stepwise dose escalation. B, MTT assay was used to assess the growth inhibition of H1975 cells and the resistant clones treated with osimertinib or rociletinib for 72 h (n = 3). Results are expressed as the mean ± SEM. IC50 values were calculated and demonstrated. C, H1975 parental cells and resistant clones were treated with 1 µmol/L osimertinib for 2 h. The changes in phosphorylation (p‐) of indicated proteins were analyzed by western blotting. D, Control and epidermal growth factor receptor (EGFR) siRNAs were transfected into parental and resistant cells, and EGFR knockdown was confirmed by western blot. Cell viability was analyzed after 72 h using the MTT assay (n = 3). *P < .001
TABLE 1.
Analysis of mutations in EGFR gene
| EGFR | H1975 | OR#1 | OR#2 | OR#3 | OR#4 |
|---|---|---|---|---|---|
| L858R | + | + | + | + | + |
| T790M | + | + | + | + | + |
| C797S | − | − | − | − | − |
To clarify the resistance mechanisms, we first examined whether alternative receptor tyrosine kinases are activated in the resistant cells. However, receptor tyrosine kinases, including MET and HER2, which have been reported to be involved in the mechanism of osimertinib resistance, 13 were not activated in the resistant cells (Figure 1C). In contrast, the resistant clones showed a typical mesenchymal phenotype, such as a changing cell shape (Figure 2A), as well as decreased epithelial marker (E‐cadherin) and increased mesenchymal marker (Vimentin and ZEB1) expression (Figure 2B,C). These findings indicate that the 4 resistant clones acquired osimertinib resistance through a mechanism associated with EMT.
FIGURE 2.
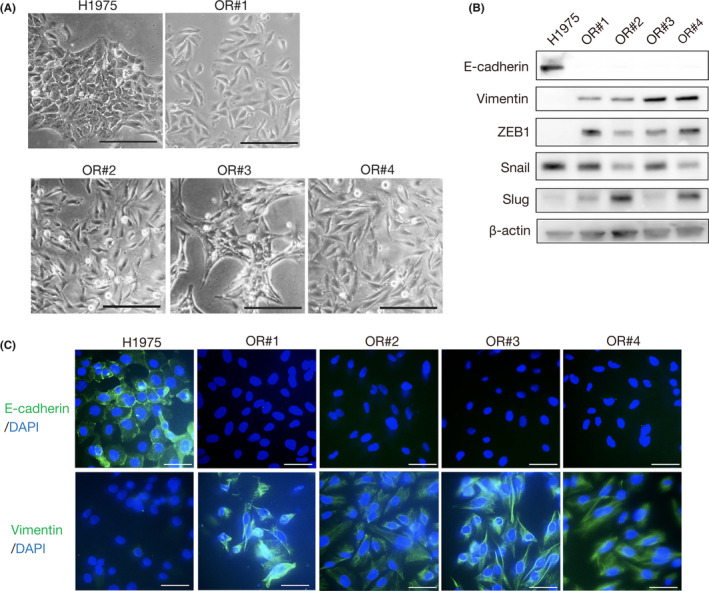
Epithelial‐mesenchymal transition (EMT) is induced in osimertinib‐resistant H1975 clones. A, H1975 parental cells and the resistant clones were evaluated to determine any morphologic changes consistent with EMT, using a light microscope. Scale bar = 200 μm. B, Expression of EMT markers was analyzed using western blotting with the indicated Abs. C, Immunofluorescence assay of EMT markers. Scale bar = 50 μm
3.2. Downregulation of miR‐200c resulted in induction of EMT in EGFR‐mutant NSCLC cells
Next, we sought to elucidate the mechanism through which the 4 resistant clones developed a mesenchymal phenotype. Recently, we reported that lung cancer cells with an EML4‐ALK rearrangement acquired crizotinib resistance through EMT, which was mediated by decreased expression of miR‐200c. 14 Thus, we first examined the expression of miRNA between the parental and OR#3 cells using the GeneChip miRNA 4.0 array. Interestingly, of all the detected miRNAs, the decrease in expression was greatest for miR‐200c in OR#3 (fold change, 215) when compared with its expression in the parental cells (Figure 3A). We then quantified the expression of miR‐200c in the osimertinib‐resistant clone cells. As shown in Figure 3B, we found that miR‐200c expression was remarkably decreased in all the resistant cells, when compared with the parental cells. MicroRNA‐200c is known to play an important role in the regulation of EMT through downregulation of ZEB1 expression. 15 In addition, we previously reported that overexpression of miR‐200c caused mesenchymal‐epithelial transition by suppression of ZEB1 in crizotinib‐resistant EML4‐ALK lung cancer cells. 14 In accordance with these findings, we observed that overexpression of miR‐200c through transfection led to decreased ZEB1 expression and increased E‐cadherin expression in all resistant cells except OR#1 (Figure 3C). Furthermore, suppression of ZEB1 by specific siRNAs led to increased E‐cadherin expression in all resistant cells except in OR#1 cells, whereas suppression of Slug did not change the expression of E‐cadherin (Figure 3D). The suppression of ZEB1 did not decrease the expression of Vimentin (Figure S1A). Microscopy examination revealed that the suppression of ZEB1 did not change cell shape (Figure S1B). These findings suggest that the OR#2, OR#3, and OR#4 clones acquired a mesenchymal phenotype through EMT, which was mediated by decreased expression of miR‐200c and increased expression of ZEB1. The findings also suggested that osimertinib sensitivity could be associated with the expression of E‐cadherin.
FIGURE 3.
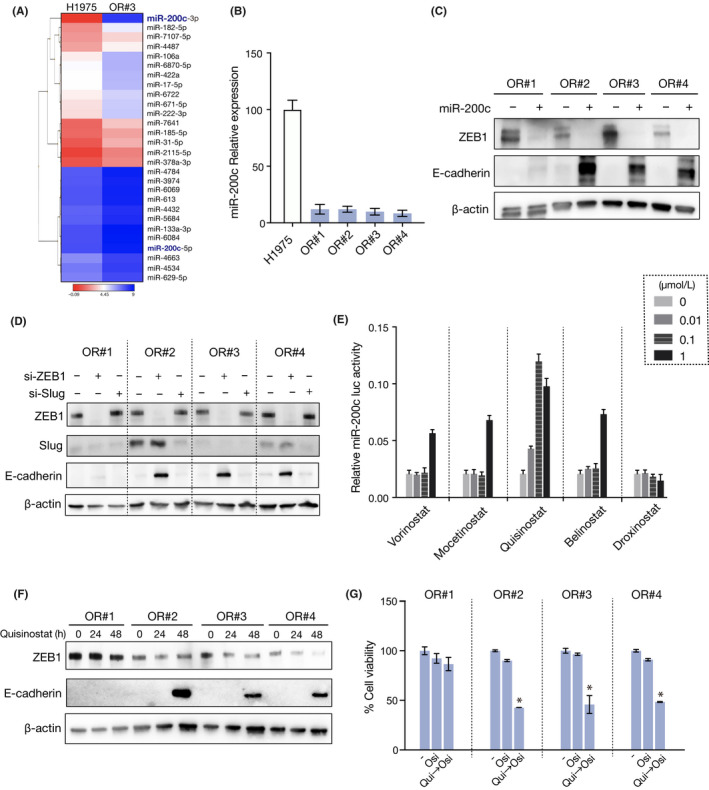
Quisinostat reversed epithelial‐mesenchymal transition by upregulating microRNA (miR)‐200c expression through inhibition of ZEB1. A, Heatmap representation of miRNA array data showing the expression levels of miRNAs in decreasing order of fold change more than 2 between parental H1975 and OR#3 cells (n = 2). B, Relative expression of miR‐200c was measured in H1975 parental cells and resistant clones (n = 3). Error bars represent SEM. *P < .0001. C, H1975‐resistant clone cells were treated with a miR‐200c mimic. Change in ZEB1 and E‐cadherin expression was analyzed by western blotting. D, ZEB1 or Slug siRNA was introduced into H1975‐resistant clone cells, and cell lysates were analyzed by western blotting with the indicated Abs 48 h later. E, Change of miR‐200c promoter activity in OR#3 cells treated with 5 HDAC inhibitors (n = 3). Error bars represent SEM. *P < .01. F, Change in ZEB1 and E‐cadherin expression in resistant clones treated with 0.03 µmol/L quisinostat was analyzed by western blotting. G, Resistant clones were pretreated with 0.03 µmol/L quisinostat for 48 h, followed by treatment with osimertinib for 72 h. Cell viability was measured using MTT assay (n = 3). Error bars represent SEM. *P < .0001
3.3. Quisinostat reversed EMT and restored sensitivity to osimertinib
We then hypothesized that the chemical restoration of miR‐200c expression could reverse EMT and resensitize the tumor cells to osimertinib. In the EML4‐ALK lung cancer cells, we have previously screened drugs using a 200 kinase inhibitor library and found that HDAC inhibitors 16 , 17 showed the highest potential to increase miR‐200c promoter activity. 14 Thus, we first transfected OR#3 cells with a NanoLuc expression vector 14 and undertook a reporter assay on 5 HDAC inhibitors. Consistent with the results obtained in EML4‐ALK lung cancer cells, quisinostat, which has a high potency toward class I HDACs, increased miR‐200c promotor activity with the highest potency among the tested HDAC inhibitors (Figure 3E). Furthermore, we confirmed that treatment with quisinostat decreased ZEB1 expression and increased E‐cadherin expression in all resistant cells, with the exception of OR#1 (Figure 3F). To further investigate whether quisinostat could reverse osimertinib resistance, we pretreated the clone cells with quisinostat for 48 hours and then treated them with osimertinib for another 3 days. Importantly, pretreatment with quisinostat restored the sensitivity to osimertinib in the three clones to the level observed in the parental H1975 cells. However, it did not change the sensitivity of OR#1 (Figure 3G). We also confirmed that in the resistant bulk cells, such as a changing cell shape (Figure S2A), EMT markers (Figure S2B) and expression of miR‐200c (Figure S2C). Furthermore, both overexpression of miR‐200c and quisinostat treatment also led to decreased ZEB1 expression and increased E‐cadherin (Figure S2D,E), and that quisinostat treatment restored sensitivity to osimertinib (Figure S2F). These results indicate that EGFR‐mutant lung cancer cells could acquire osimertinib resistance through EMT and that quisinostat can resensitize EGFR‐mutant lung cancer cells with a mesenchymal phenotype to osimertinib by reverting EMT.
3.4. Glycogen synthase kinase‐3 inhibitor suppressed growth of EMT‐associated resistant cells
To reverse the resistance to osimertinib in OR#1 cells, we treated the cells with several HDAC inhibitors. However, other HDAC inhibitors had a weaker effect on E‐cadherin expression in OR#1 than that observed for OR#2, OR#3, or OR#4 (FigureS3). These results imply that in OR#1, EMT could be induced not by miR‐200c/ZEB1 but other signaling pathways. Therefore, to identify compounds other than HDAC inhibitors that could overcome osimertinib resistance in OR#1 cells, we screened drugs, using a library of 100 kinase inhibitors, by administering 1 µmol/L of each drug to OR#1 cells, followed by incubation for 72 hours. Interestingly, we found that several GSK‐3‐targeted inhibitors markedly suppressed cell viability when compared with other inhibitors (Figure 4A). Thus, we focused on the effect of GSK‐3 inhibition and treated the cells with 16 different GSK‐3‐targeted inhibitors. As shown in Figure 4B,C, LY2090314, a potent GSK‐3α/β inhibitor, showed the highest effect on the suppression of cell growth and similar effects were also confirmed in OR#3 (Figure S4). Furthermore, LY2090314 strongly decreased the cell growth of all resistant clone cells, whereas it only slightly decreased the growth of the parental cells (Figure 4D). To elucidate its synergistic effect with osimertinib, we treated cells with 0.1 μmol/L LY2090314 in combination with 1 μmol/L osimertinib for 72 hours. As shown in Figure 4E, treatment with LY2090314 alone decreased the growth of resistant cells, whereas osimertinib did not exert an effect on cell growth. These results indicate that the growth of resistant cells is dependent on the GSK‐3α/β‐associated signaling pathway. In addition, to exclude the possibility of lack of specificity of LY2090314, we treated cells with GSK‐3α/β specific siRNAs in combination with 1 μmol/L osimertinib for 72 hours. We found that transfection of GSK‐3α/β specific siRNA decreased the cell growth of all resistant cells, whereas it did not decrease the growth of the parent cells, which is consistent with the results obtained by LY2090314 treatment alone (Figure 5A). These results suggest that EGFR signaling, which is dominant in the growth of parental cells, could switch to GSK‐3‐associated signaling during the process through which cells acquire resistance to osimertinib and change to a mesenchymal phenotype.
FIGURE 4.
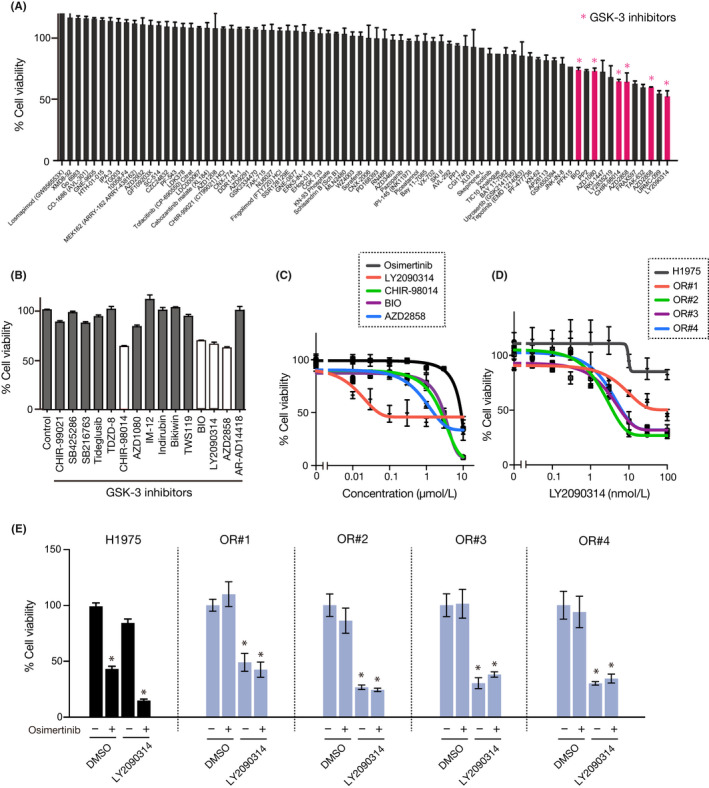
Glycogen synthase kinase (GSK)‐3 inhibitor suppressed the growth of epithelial‐mesenchymal transition‐associated resistant cells. A, Drug screening by treatment with 1 μmol/L each drug to identify compounds that suppress the growth of OR#1 cells (n = 3). Pink columns represent GSK‐3 inhibitors. B, C, OR#1 cells were treated with 1 μM GSK inhibitors for 72 h. Cell viability was measured using MTT assay (n = 3) D, H1975 parental cells and resistant clones were treated with LY2090314 for 72 h. Cell viability was measured using MTT assay (n = 3). E, H1975 parental cells and the resistant clones were treated with 0.1 μmol/L LY2090314 in combination with 1 μmol/L osimertinib (n = 3). Error bars represent SEM. *P < .001
FIGURE 5.
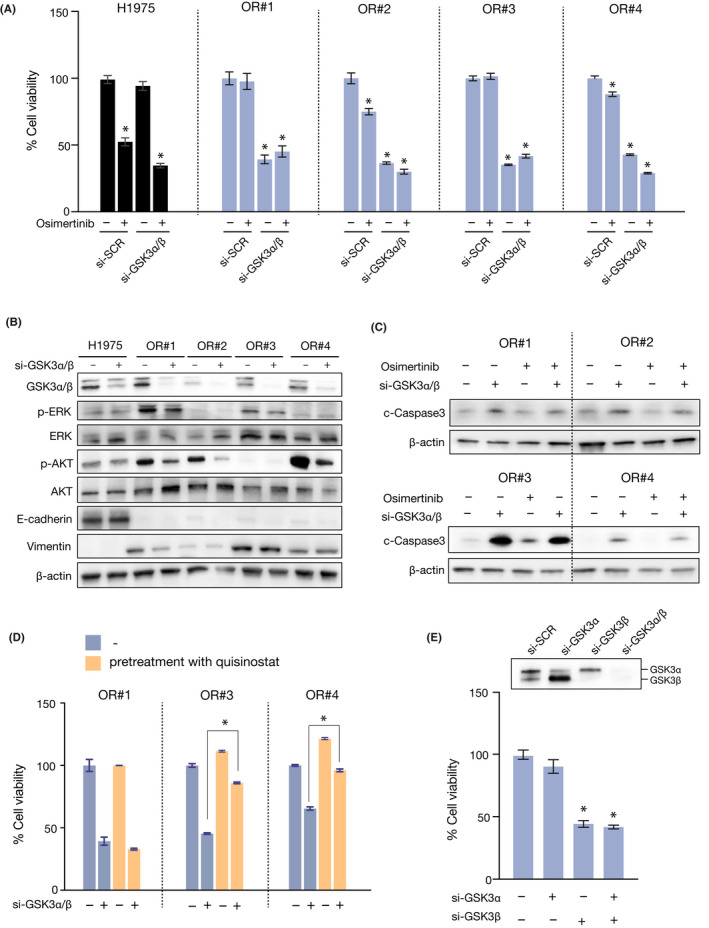
Glycogen synthase kinase (GSK)‐3 inhibition induced apoptosis of epithelial‐mesenchymal transition‐associated resistant cells. A, Control and GSK‐3 siRNAs were transfected into H1975 parental cells and resistant clone cells. At 24 h posttransfection, cells were treated with 1 μmol/L osimertinib and cell viability was measured after 72 h using MTT assay (n = 3). Error bars represent SEM. *P < .0001. B, Control and GSK‐3 siRNAs were transfected into H1975 parental and resistant clone cells. Cell lysates were analyzed using western blotting with the indicated Abs at 48 h posttransfection. C, Control and GSK‐3 siRNAs were transfected into resistant clone cells. At 24 h post‐transfection, cells were treated with 1 μmol/L osimertinib and cell lysates were analyzed using western blotting with the indicated Abs after 72 h (n = 4). D, OR#1, OR#3, and OR#4 cells were pretreated with 0.03 μmol/L quisinostat for 48 h, followed by GSK‐3 siRNA for 72 h. Cell viability was measured using MTT assay (n = 3). Error bars represent SEM. *P < .0001. E, Control, GSK‐3α, and GSK‐3β siRNAs were transfected into OR#3 cells and cell viability was measured after 72 h using MTT assay (n = 3). Error bars represent SEM. *P < .0001
3.5. Inhibition of GSK‐3β decreases AKT signaling and induces apoptosis in mesenchymal phenotype cells
To further elucidate the mechanisms through which the suppression of GSK‐3α/β reduced the growth of resistant cells, we examined cellular growth signaling using western blot analysis. As shown in Figure 5B, treatment with a GSK‐3α/β specific siRNA for 72 hours decreased the phosphorylation of AKT in all the resistant clone cells, whereas the expression of EMT markers, such as E‐cadherin and Vimentin, was not changed. In addition, we found that treatment with GSK‐3α/β specific siRNA for 72 hours increased cleaved caspase 3, indicating that apoptosis was induced in all resistant clone cells (Figure 5C). These data suggest that GSK‐3 could suppress apoptosis signaling in resistant cells. To further clarify whether the effect of GSK‐3 knockdown is specific to a mesenchymal phenotype, we first reverted the resistant cells from a mesenchymal to an epithelial phenotype by treatment with quisinostat for 48 hours. Next, the cells were treated with a GSK‐3α/β specific siRNA for 72 hours. Interestingly, pretreatment with quisinostat significantly diminished the effects of GSK‐3 inhibition, suggesting that GSK‐3 contributes to cell viability after EMT occurrence (Figure 5D). These data suggest that GSK‐3 inhibition induces apoptosis and suppresses the growth of cells with EMT‐associated acquired resistance.
4. DISCUSSION
In the present study, we found that tumor cells with a mesenchymal phenotype were cross‐resistant to the new generation of EGFR‐TKIs, such as osimertinib and rociletinib. Importantly, inhibition of GSK‐3β circumvented EMT‐associated osimertinib resistance by suppressing AKT signaling and inducing apoptosis in osimertinib‐resistant cells.
We have previously reported a novel strategy to overcome EMT‐associated resistance in EML4‐ALK lung cancer cells, namely by reverting from a mesenchymal phenotype to an epithelial phenotype using an HDAC inhibitor, through upregulation of miR‐200c. 14 In the present study, we showed that miR‐200c was also downregulated in osimertinib‐resistant EGFR‐mutant lung cancer cells and that the HDAC inhibitor quisinostat was effective in restoring the sensitivity to osimertinib in 3 resistant clone cells. Quisinostat is a second‐generation HDAC inhibitor that specifically inhibits class1 HDACs, including HDAC2 (IC50 = 0.33 μmol/L), compared with other HDAC inhibitors. 18 Histone deacetylase 2 has been reported to play a critical role in suppression of the miR‐200 family. 19 These reports suggest the reason why quisinostat effectively enhanced the expression of miR‐200c.
In contrast, quisinostat treatment did not induce a transition from a mesenchymal to epithelial phenotype in 1 of the clone cells. Thus, this resistant clone was not sensitized to osimertinib following treatment with the HDAC inhibitor. This is likely due to the fact that EMT is not controlled by the miR‐200c/ZEB1 axis in OR#1 cells. Hence, we sought to develop another strategy to overcome EMT‐associated osimertinib resistance by targeting mesenchymal‐specific survival signaling without reverting to an epithelial phenotype.
Interestingly, our drug screening results indicated that GSK‐3 inhibition suppressed the growth of EMT‐associated‐resistant clone cells but not of the parental cells. GSK‐3β reportedly plays important roles in the regulation of EMT. In bladder cancer cell lines, PI3K/Akt has been reported to target GSK‐3β to control ZEB1 transcription. 20 In NSCLC and pancreatic ductal adenocarcinoma, the Akt/GSK‐3β/Snail pathway has been reported to be involved in the EMT process. 21 Moreover, in prostate cancer, miR‐493‐5p inhibition induces EMT through expression of EGFR, followed by Akt/GSK‐3β/Snail signaling. 22 However, in the current study, we have shown that the expression of EMT markers and ZEB1, which is an EMT‐associated transcriptional factor, remained unchanged in the mesenchymal‐resistant clone cells after GSK‐3β inhibition. These results indicate that the effects of GSK‐3β inhibition are not a consequence of reversing EMT, as was the case with the HDAC inhibitors.
Notably, GSK‐3β plays a role in the activation of the Wnt/β‐catenin pathway, which has been previously reported to confer gefitinib resistance in lung cancer cell lines. 23 , 24 In this study, we investigated whether Wnt/β‐catenin signaling could play a role in acquiring resistance to osimertinib. However, β‐catenin, which is localized in the nucleus (Figure S5A), was not increased in the resistant cells, whereas the phosphorylation levels of GSK‐3α/β were higher in the resistant cells (Figure S5B). These results exclude the possibility that the Wnt/β‐catenin signaling pathway contributes to osimertinib resistance.
Recent studies have reported that miR‐200c can inhibit EMT by suppressing phospho‐AKT in endometrial carcinoma cells. 25 Furthermore, in osteosarcoma, AKT2 has been validated as the novel target of miR‐200c. 26
MicroRNA‐200c was strongly downregulated in osimertinib‐resistant cells, indicating that the downregulation of miR‐200c might induce AKT phosphorylation. In addition, GSK‐3 has been identified as the first AKT substrate and AKT phosphorylates GSK‐3β(Ser9). 27 These results imply that downregulated miR‐200c activates the GSK‐3/AKT axis in osimertinib‐resistant cells (Figure 6). This needs to be further tested.
FIGURE 6.
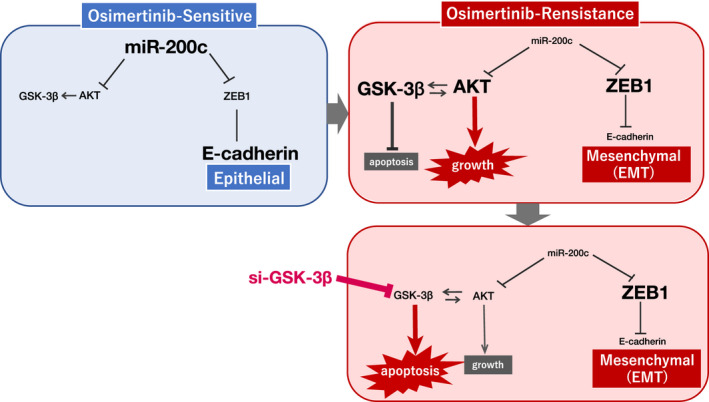
Summary scheme of the role of microRNA (miR)‐200c in the activation of the glycogen synthase kinase (GSK)‐3/AKT axis in osimertinib‐sensitive and osimertinib‐resistant cells. EMT, epithelial‐mesenchymal transition
Glycogen synthase kinase‐3 has been reported to encode different isoforms by distinct genes, namely, GSK‐3α (51 kDa) and GSK‐3β (47 kDa); however, the functions of GSK‐3 isoforms are unknown. 28 Therefore, we treated cells with either GSK‐3α or GSK‐3β specific siRNAs and examined their effect on the growth of resistant cells. We found that the knockdown of GSK‐3β, but not GSK‐3α, significantly suppressed cell growth, indicating that GSK‐3β is associated with cell growth and survival of resistant cells (Figure 5E). LY209031 inhibits not only GSK‐3β but also GSK‐3α, therefore, development of GSK‐3β specific inhibitor could become an effective strategy.
To develop this GSK‐3 inhibitor therapy for clinical use, we sought to establish a tumor xenograft mouse model with the resistant clone cells, using SCID mice. However, the cells were unable to grow and did not lead to the development of tumors by either s.c. or orthotopic injection. Therefore, in future studies we need to identify other lung cancer cells that acquire EMT‐associated osimertinib resistance.
In summary, we showed that EGFR‐mutant lung cancer cells acquired EMT‐associated osimertinib resistance, which was accompanied by downregulation of miR‐200c and upregulation of ZEB1. Moreover, GSK‐3 inhibitors, such as LY2090314, were shown to markedly inhibit the growth and induce apoptosis of resistant cells with a mesenchymal phenotype, suggesting that GSK‐3 inhibition could be useful to circumvent EMT‐associated resistance to osimertinib in EGFR‐mutant lung cancer.
DISCLOSURE
Seiji Yano obtained speaker’s fees from AstraZeneca, Chugai Pharma, Boehringer‐Ingelheim Japan, Novartis, and Pfizer, and research grants from Chugai Pharma, Boehringer‐Ingelheim Japan, and Novartis. The other authors have nothing to disclose.
Supporting information
Fig S1‐S5
ACKNOWLEDGEMENT
This work was supported by grants JSPS KAKENHI (grant numbers 18K07261 [to KF] and 19H03665 [to Seiji Yano]).
Fukuda K, Takeuchi S, Arai S, et al. Glycogen synthase kinase‐3 inhibition overcomes epithelial‐mesenchymal transition‐associated resistance to osimertinib in EGFR‐mutant lung cancer. Cancer Sci. 2020;111:2374–2384. 10.1111/cas.14454
REFERENCES
- 1. Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non‐small‐cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380‐2388. [DOI] [PubMed] [Google Scholar]
- 2. Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first‐line treatment for European patients with advanced EGFR mutation‐positive non‐small‐cell lung cancer (EURTAC): a multicentre, open‐label, randomised phase 3 trial. Lancet Oncol. 2012;13:239‐246. [DOI] [PubMed] [Google Scholar]
- 3. Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non‐small‐cell lung cancer to gefitinib. N Engl J Med. 2005;352:786‐792. [DOI] [PubMed] [Google Scholar]
- 4. Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Medicine. 2005;2:e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Finlay MR, Anderton M, Ashton S, et al. Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J Med Chem. 2014;57:8249‐8267. [DOI] [PubMed] [Google Scholar]
- 6. Minari R, Bordi P, Tiseo M. Third‐generation epidermal growth factor receptor‐tyrosine kinase inhibitors in T790M‐positive non‐small cell lung cancer: review on emerged mechanisms of resistance. Translational Lung Cancer Research. 2016;5:695‐708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Soria JC, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR‐mutated advanced non‐small‐cell lung cancer. N Engl J Med. 2018;378:113‐125. [DOI] [PubMed] [Google Scholar]
- 8. Nieto MA, Huang RY, Jackson RA, Thiery JP. Emt: 2016. Cell. 2016;166:21‐45. [DOI] [PubMed] [Google Scholar]
- 9. Uramoto H, Yamada T, Yano S, Kondo N, Hasegawa S, Tanaka F. Prognostic value of acquired resistance‐related molecules in Japanese patients with NSCLC treated with an EGFR‐TKI. Anticancer Res. 2012;32:3785‐3790. [PubMed] [Google Scholar]
- 10. Taniguchi H, Takeuchi S, Fukuda K, et al. Amphiregulin triggered epidermal growth factor receptor activation confers in vivo crizotinib‐resistance of EML4‐ALK lung cancer and circumvention by epidermal growth factor receptor inhibitors. Cancer Sci. 2017;108:53‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miyazawa H, Tanaka T, Nagai Y, et al. Peptide nucleic acid‐locked nucleic acid polymerase chain reaction clamp‐based detection test for gefitinib‐refractory T790M epidermal growth factor receptor mutation. Cancer Sci. 2008;99:595‐600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Balak MN, Gong Y, Riely GJ, et al. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor‐mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res. 2006;12:6494‐6501. [DOI] [PubMed] [Google Scholar]
- 13. Planchard D, Loriot Y, Andre F, et al. EGFR‐independent mechanisms of acquired resistance to AZD9291 in EGFR T790M‐positive NSCLC patients. Ann Oncol. 2015;26:2073‐2078. [DOI] [PubMed] [Google Scholar]
- 14. Fukuda K, Takeuchi S, Arai S, et al. Epithelial‐to‐mesenchymal transition is a mechanism of ALK inhibitor resistance in lung cancer independent of ALK mutation status. Can Res. 2019;79:1658‐1670. [DOI] [PubMed] [Google Scholar]
- 15. Gregory PA, Bert AG, Paterson EL, et al. The miR‐200 family and miR‐205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593‐601. [DOI] [PubMed] [Google Scholar]
- 16. Cai X, Zhai HX, Wang J, et al. Discovery of 7‐(4‐(3‐ethynylphenylamino)‐7‐methoxyquinazolin‐6‐yloxy)‐N‐hydroxyheptanamide (CUDc‐101) as a potent multi‐acting HDAC, EGFR, and HER2 inhibitor for the treatment of cancer. J Med Chem. 2010;53:2000‐2009. [DOI] [PubMed] [Google Scholar]
- 17. Shimizu T, LoRusso PM, Papadopoulos KP, et al. Phase I first‐in‐human study of CUDC‐101, a multitargeted inhibitor of HDACs, EGFR, and HER2 in patients with advanced solid tumors. Clin Cancer Res. 2014;20:5032‐5040. [DOI] [PubMed] [Google Scholar]
- 18. Roy SS, Gonugunta VK, Bandyopadhyay A, et al. Significance of PELP1/HDAC2/miR‐200 regulatory network in EMT and metastasis of breast cancer. Oncogene. 2014; 33:3707‐3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Arts J, King P, Marien A, et al. JNJ‐26481585, a novel "second‐generation" oral histone deacetylase inhibitor, shows broad‐spectrum preclinical antitumoral activity. Clin Cancer Res. 2009;15:6841‐6850. [DOI] [PubMed] [Google Scholar]
- 20. Wu K, Fan J, Zhang L, et al. PI3K/Akt to GSK3beta/beta‐catenin signaling cascade coordinates cell colonization for bladder cancer bone metastasis through regulating ZEB1 transcription. Cell Signal. 2012;24:2273‐2282. [DOI] [PubMed] [Google Scholar]
- 21. Meng Q, Shi S, Liang C, et al. Abrogation of glutathione peroxidase‐1 drives EMT and chemoresistance in pancreatic cancer by activating ROS‐mediated Akt/GSK3beta/Snail signaling. Oncogene. 2018;37:5843‐5857. [DOI] [PubMed] [Google Scholar]
- 22. Wang S, Wang X, Li J, et al. c‐Met, CREB1 and EGFR are involved in miR‐493‐5p inhibition of EMT via AKT/GSK‐3beta/Snail signaling in prostate cancer. Oncotarget. 2017;8:82303‐82313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Casas‐Selves M, Kim J, Zhang Z, et al. Tankyrase and the canonical Wnt pathway protect lung cancer cells from EGFR inhibition. Can Res. 2012;72(16):4154‐4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nakayama S, Sng N, Carretero J, et al. beta‐catenin contributes to lung tumor development induced by EGFR mutations. Can Res. 2014;74:5891‐5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li F, Liang A, Lv Y, Liu G, Jiang A, Liu P. MicroRNA‐200c inhibits epithelial‐mesenchymal transition by targeting the BMI‐1 gene through the phospho‐AKT pathway in endometrial carcinoma cells in vitro. Med Sci Monit. 2017;23:5139‐5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu Y, Zhu ST, Wang X, et al. MiR‐200c regulates tumor growth and chemosensitivity to cisplatin in osteosarcoma by targeting AKT2. Sci Rep. 2017;7:13598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dajani R, Fraser E, Roe SM, et al. Crystal structure of glycogen synthase kinase 3 beta: structural basis for phosphate‐primed substrate specificity and autoinhibition. Cell. 2001;105:721‐732. [DOI] [PubMed] [Google Scholar]
- 28. Tejeda‐Munoz N, Robles‐Flores M. Glycogen synthase kinase 3 in Wnt signaling pathway and cancer. IUBMB Life. 2015;67:914‐922. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S5