

Abstract
Secondary lymphedema often develops after cancer surgery, and over 250 million patients suffer from this complication. A major symptom of secondary lymphedema is swelling with fibrosis, which lowers the patient's quality of life, even if cancer does not recur. Nonetheless, the pathophysiology of secondary lymphedema remains unclear, with therapeutic approaches limited to physical or surgical therapy. There is no effective pharmacological therapy for secondary lymphedema. Notably, the lack of animal models that accurately mimic human secondary lymphedema has hindered pathophysiological investigations of the disease. Here, we developed a novel rat hindlimb model of secondary lymphedema and showed that our rat model mimics human secondary lymphedema from early to late stages in terms of cell proliferation, lymphatic fluid accumulation, and skin fibrosis. Using our animal model, we investigated the disease progression and found that transforming growth factor‐beta 1 (TGFB1) was produced by macrophages in the acute phase and by fibroblasts in the chronic phase of the disease. TGFB1 promoted the transition of fibroblasts into myofibroblasts and accelerated collagen synthesis, resulting in fibrosis, which further indicates that myofibroblasts and TGFB1/Smad signaling play key roles in fibrotic diseases. Furthermore, the presence of myofibroblasts in skin samples from lymphedema patients after cancer surgery emphasizes the role of these cells in promoting fibrosis. Suppression of myofibroblast‐dependent TGFB1 production may therefore represent an effective pharmacological treatment for inhibiting skin fibrosis in human secondary lymphedema after cancer surgery.
Keywords: fibrosis, lymphedema, myofibroblasts, pathology, transforming growth factor‐beta
Secondary lymphedema develops and causes skin fibrosis after cancer surgery. We developed a novel rat hindlimb model that accurately mimics human secondary lymphedema, and investigated disease progression. We found that myofibroblasts and transforming growth factor‐beta 1/Smad signaling play key roles in the skin fibrosis of secondary lymphedema.
Abbreviations
- ACTA2
actin alpha 2
- COL1A1
collagen type I alpha 1
- COL3A1
collagen type III alpha 1
- CTGF
connective tissue growth factor
- CTR
control
- FGF2
fibroblast growth factor 2
- HE
healthy
- HSP47
heat shock protein 47
- ICG
indocyanine green
- IHC
immunohistochemical staining
- IL1B
interleukin 1 beta
- LE
lymphedema
- LV
lymphatic vessels
- LYVE‐1
lymphatic vessel endothelial hyaluronan receptor‐1
- P4HB
prolyl 4‐hydroxylase subunit beta
- p‐Smad2/3
phosphorylation of Smad2/3
- Qdots
quantum dots
- QOL
quality of life
- qRT‐PCR
quantitative real‐time polymerase chain reaction
- TGFB1
transforming growth factor‐beta 1
- TGFR
transforming growth factor receptor
- TIMP
tissue inhibitor of metalloproteinase
- VEGFC
vascular endothelial growth factor C
1. INTRODUCTION
After cancer surgery with lymph node dissection, secondary lymphedema occurs due to damage to the lymphatic system. 1 , 2 The frequency of postsurgical lymphedema following lymph node dissection has been reported to be c. 30% in breast cancer surgery and 25%‐30% in gynecologic cancer surgery, 3 and over 250 million patients suffer from this complication. 4 The pathological condition of lymphedema consists of an excessive interstitial accumulation of lymphatic fluid in the extremities, 5 causes cellulitis and swelling followed by fibrosis, which significantly lowers the patient's QOL, even if cancer does not recur. 6 , 7 Although clinical trials on pharmacological treatments are ongoing, 8 , 9 no pharmacological treatment has been established, because the pathophysiology is not well understood. Therefore, well described experimental models that mimic the human secondary lymphedema are urgently needed. 10 , 11 , 12
Since the initial reports describing canine models, 13 , 14 several lymphedema models have been developed: canine, 10 , 12 , 15 rodent hindlimb, 16 , 17 , 18 , 19 , 20 rodent upper limb, 10 , 21 rabbit ear, 22 , 23 rodent tail, 24 and in large animals: sheep, 25 pigs, 26 , 27 and monkeys. 28 However, these previous models were impractical due to high costs, difficulties in handling, high mortality rate, and differences from human lymphedema. In this study, we generated a novel secondary lymphedema model in the rat hindlimb, described the pathophysiology of secondary lymphedema, and compared it with humans. Specifically, we focused on skin fibrosis and transforming growth factor‐beta 1 (TGFB1), a key regulator that accelerates fibrosis, 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 while inhibiting lymphangiogenesis. 37 , 38
2. MATERIALS AND METHODS
2.1. Experimental design
Experiments were conducted to evaluate the pathophysiology of secondary lymphedema. First, we developed a novel lymphedema model in the rat hindlimb and compared the rat model with the clinical stages of human lymphedema established according to the classification of the International Society of Lymphology (ISL). 39 Second, we studied the pathophysiology of fibrosis in secondary lymphedema using the rat model and determined the role of myofibroblasts in lymphedema pathophysiology using primary cultured skin fibroblasts derived from the rat model. Third, we investigated the pathophysiology of fibrosis in secondary lymphedema in humans using skin samples from lymphedema patients. All the patients had cancer‐related secondary lymphedema.
2.2. Study approval
All animal procedures were approved by the Hamamatsu University School of Medicine Ethics Committee of Animal Research (approval number H25‐069). All procedures involving humans were approved by that of Clinical Research (approval number R14‐033) and conformed to the provisions of the Declaration of Helsinki. Before the skin biopsy, each patient provided written informed consent for the use of skin samples in related research.
The details of the creation of the rat model 16 , 17 , 18 , 19 , 20 , 22 , 40 , 41 (Figure S1), evaluation methods (Figure S2), 20 , 21 , 42 , 43 , 44 , 45 , 46 , 47 , 48 , 49 , 50 , 51 , 52 , 53 , 54 , 55 , 56 , 57 , 58 , 59 , 60 , 61 diagnosis of human secondary lymphedema, 62 , 63 and statistical analysis are given in Data S1.
3. RESULTS
Data obtained in this study have been deposited in Figshare.
3.1. Changes in limb volumes and lymphatic fluid in the rat lymphedema model
In the lymphedema (LE) group, limb swelling and pooling of Evans blue solution were observed macroscopically until day 28, which was less prominent after day 84 (Figure 1A). No significant difference was observed in body weight between the control (Ctr) and LE groups (P = .896; Figure S3). However, the limb volume dramatically increased (days 3‐7), then decreased (days 7‐4), in the LE group. A significant difference was observed in limb volume between the Ctr and LE groups on days 3 and 168 (P < .01), and days 28 and 112 (P < .05) (Figure 1B).
FIGURE 1.
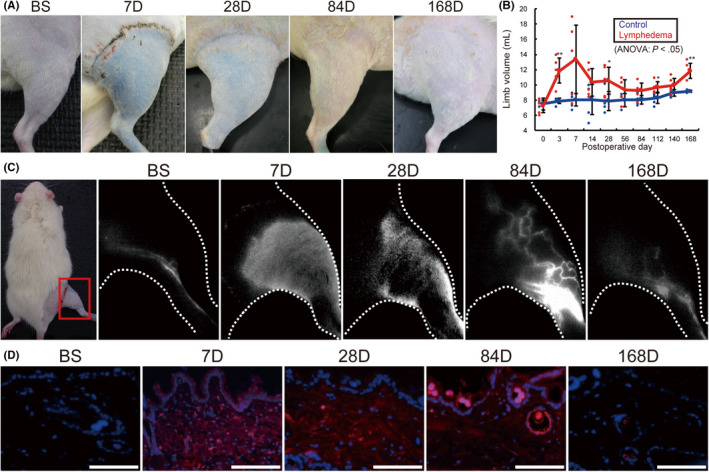
Time‐dependent changes in the rat lymphedema model. A, Macroscopic image. B, Volume changes in rat treated right hindlimbs. C, Indocyanine green near‐infrared fluorescence lymphography. D, Near‐infrared fluorescence microscopy with Qdots (blue: DAPI, red: Qdots) of lymphedema models over time. BS, before surgery; D, days after surgery. n = 5, for control and lymphedema groups. *P < .05, **P < .01 (B). Scale bars, 100 μm (D)
Indocyanine green (ICG) fluorescence lymphography showed regular collecting LV in the before surgery (BS) group. The LE group showed marked lymphatic fluid accumulation in the legs between days 7‐28. Collateral LV was observed on day 84, which gradually disappeared by day 168. Lymphatic fluid accumulation was observed throughout 168 d (Figure 1C). Fluorescence microscopy using carboxyl quantum dots (Qdots) showed no lymphatic fluid accumulation in the skin of BS rats, whereas the LE group showed diffuse lymphatic fluid accumulation (days 7‐84), and scattered accumulation (day 168) in the skin (Figure 1D).
3.2. Changes in skin fibrosis in the rat lymphedema model
Different staining procedures including Azan (Figure 2A,B), Masson trichrome (Figure S4A,B), Picrosirius red (Figure S4C,D), and immunohistochemical staining (IHC) for type I (Figure S4E,F) and III collagen fibers (Figure S4G,H) used in the study revealed marked thickening of the subcutaneous tissues, implying edema, on day 7, and an increase in collagen fibers after day 28. Azan staining revealed a significant reduction in the average collagen area fraction of subcutaneous tissues on days 3 and 7, which was increased on days 56‐168 in the LE group compared with the Ctr group (P < .01) (Figure 2B). The other 3 staining methods also revealed a similar trend of reduction in the collagen area fraction of subcutaneous tissues in the acute phase and an increase in the chronic phase in the LE group compared with the Ctr group (Figure S4B,D,F,H).
FIGURE 2.
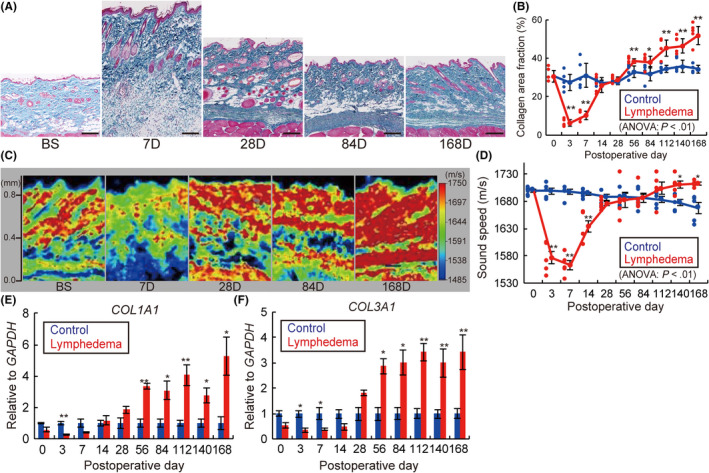
Skin fibrosis development in the rat lymphedema model. A, Azan staining of skin samples from lymphedema rat models. Scale bars, 200 μm. B, Time‐dependent changes in the collagen area fraction analyzed with Azan staining. C, Acoustic microscopy and (D) average sound speed changes in the skin samples from lymphedema rat models. Relative mRNA expression of (E) COL1A1 and (F) COL3A1 of lymphedema models over time. BS, before surgery; D, days after surgery (A, C). n = 5, for Ctr and LE groups, *P < .05, **P < .01 (B, D‐F)
Next, we evaluated the sound speed in tissues reflecting the content and quality of collagen fibers, 47 , 53 and observed an increase in the low‐sound‐speed areas in subcutaneous layers on day 7 and high‐sound‐speed areas in the dermis on days 28 to 84, and in both the dermis and subcutaneous layers on day 168 (Figure 2C). By contrast, the average sound speed of skin in the LE group significantly decreased on days 3‐14 and increased on days 140‐168 compared with the Ctr group (P < .01) (Figure 2D), indicating the onset of chronic skin fibrosis.
The level of mRNA expression of collagen type I alpha (COL1A1), and collagen type III alpha 1 (COL3A1) was significantly lower in the acute phase and higher in the chronic phase in the LE group than in the Ctr group (COL1A1, days 3, 56, 112: P < .01, days 84, 140, 168: P < .05; COL3A1, days 3, 7, 56, 84: P < .05, days 112, 140, 168: P < .01) (Figure 2E,F).
3.3. TGFB1 expression in the skin samples of the rat lymphedema model
Immunohistochemical staining showed a high expression of TGFB1 in subcutaneous tissues on days 7 and 168 in the LE group compared to the BS group (Figure 3A). Double IHC revealed TGFB1 expression on day 7 in CD68‐positive macrophages, prolyl 4‐hydroxylase subunit beta (P4HB)‐positive fibroblasts, and heat shock protein 47 (HSP47)‐positive fibroblasts only in the LE group. Conversely, on day 168, except for the CD68‐positive macrophages, TGFB1 expression was detected in P4HB‐positive and HSP47‐positive fibroblasts (Figure 3B‐D). Moreover, qRT‐PCR analysis also showed a significant increase in the levels of mRNA expression of TGFB1 on days 7, 84, 112, and 168 in the LE group (days 7, 84, 168, P < .05; day 112, P < .01; Figure 3E). In addition to detecting TGFB1 expression, we also detected the expression of TGF receptor (TGFR)1 in P4HB‐positive and HSP47‐positive fibroblasts in the BS and LE groups on days 7 and 168 (Figure S5A,B). Phosphorylation of Smad2/3 was enhanced in the subcutaneous tissues and HSP47‐positive fibroblasts on days 7 and 168 as revealed by IHC (Figure 3F) and double IHC (Figure 3G), respectively.
FIGURE 3.
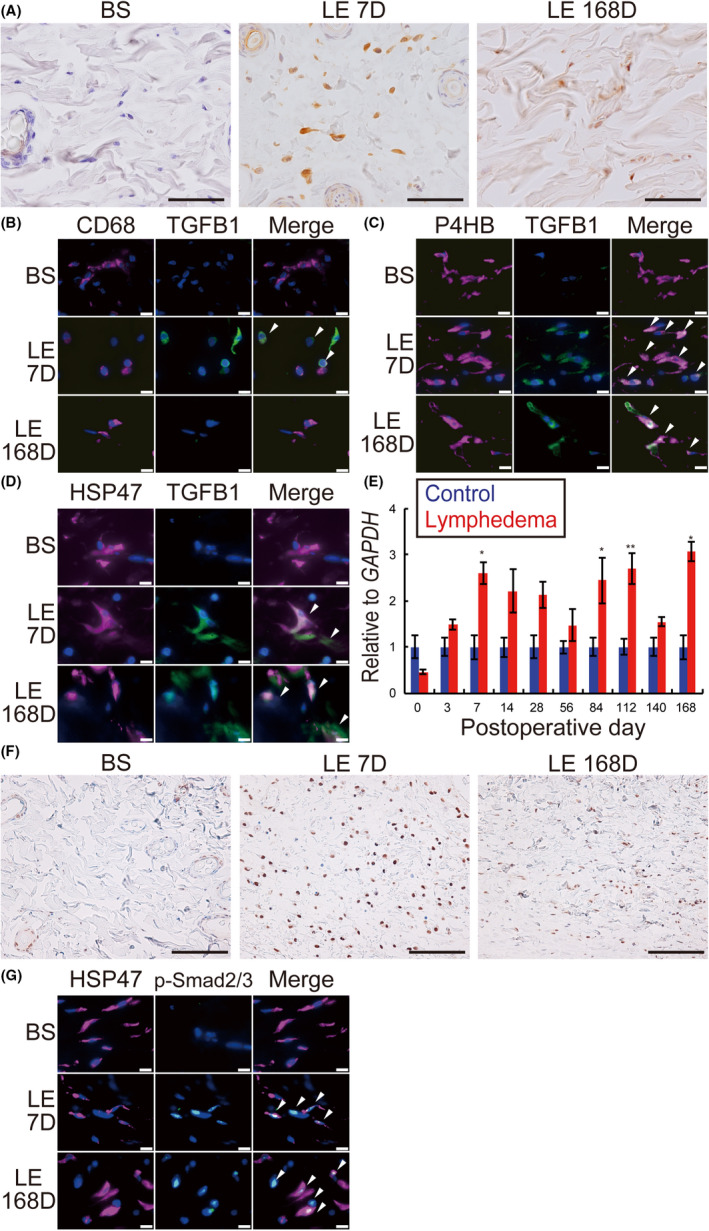
Time‐dependent changes of TGFB1 expression and Smad signaling in the rat lymphedema model. A, IHC for TGFB1. BS, before surgery; D, days after surgery; LE, lymphedema. B‐D, Double IHC of skin samples from BS, LE 7D, and LE 168D. B, Magenta: CD68; green: TGFB1; blue: DAPI. C, Magenta: P4HB; green: TGFB1; blue: DAPI. D, Magenta: HSP47; green: TGFB1; blue: DAPI. E, Relative mRNA expression levels of TGFB1 in skin samples. F, IHC for phosphorylated Smad2/3 (p‐Smad2/3) of the rat skin from lymphedema models. G, Double IHC of skin samples from BS, LE 7D, and LE 168D. Magenta: HSP47; green: p‐Smad2/3; blue: DAPI. Scale bars: (A) 50 μm, (B‐D, G) 10 μm, and (F) 100 μm. n = 5 for Ctr and LE groups, (E) *P < .05, **P < .01. White arrowheads indicate co‐localization in double IHC (B‐D, G)
qRT‐PCR analysis also showed that the mRNA expression of connective tissue growth factor (CTGF), tissue inhibitor of metalloproteinase 1 (TIMP1), and TIMP2 in the skin of the LE group increased on day 168 (Figure S6A‐C), whereas the expression of fibroblast growth factor 2 (FGF2), matrix metallopeptidase 1 (MMP1), and interleukin 1 beta (IL1B) did not (Figure S6D‐F).
3.4. Distinctive change in the number of skin fibroblasts in the rat model
Double IHC of rat skin samples showed that P4HB‐positive fibroblasts and HSP47‐positive fibroblasts in the LE group expressed actin alpha 2 (ACTA2) on both days 7 and 168, whereas those in the BS group did not (Figure 4A,B). Moreover, double IHC of the primary cultured skin fibroblasts from the Ctr group on day 84 did not express ACTA2. However, supplementing the culture medium with TGFB1 (Ctr + TGBF1) induced ACTA2 expression. In contrast, the samples from the LE group, on day 84, expressed ACTA2 without adding TGFB1 (Figure 4C). In addition, we also observed that the average cell size (Figure 4D) and mRNA expression levels of TGFB1 (Figure 4E), COL1A1 (Figure 4F), and COL3A1 (Figure 4G) in fibroblasts significantly increased in the Ctr + TGFB1 and LE groups compared to the Ctr group (cell size, Ctr vs Ctr + TGFB1, Ctr vs LE; P < .01) (TGFB1, Ctr vs LE; P < .05) (COL1A1, Ctr vs Ctr + TGFB1, Ctr vs LE; P < .01) (COL1A3, Ctr vs LE; P < .05) (Figure 4E‐G).
FIGURE 4.
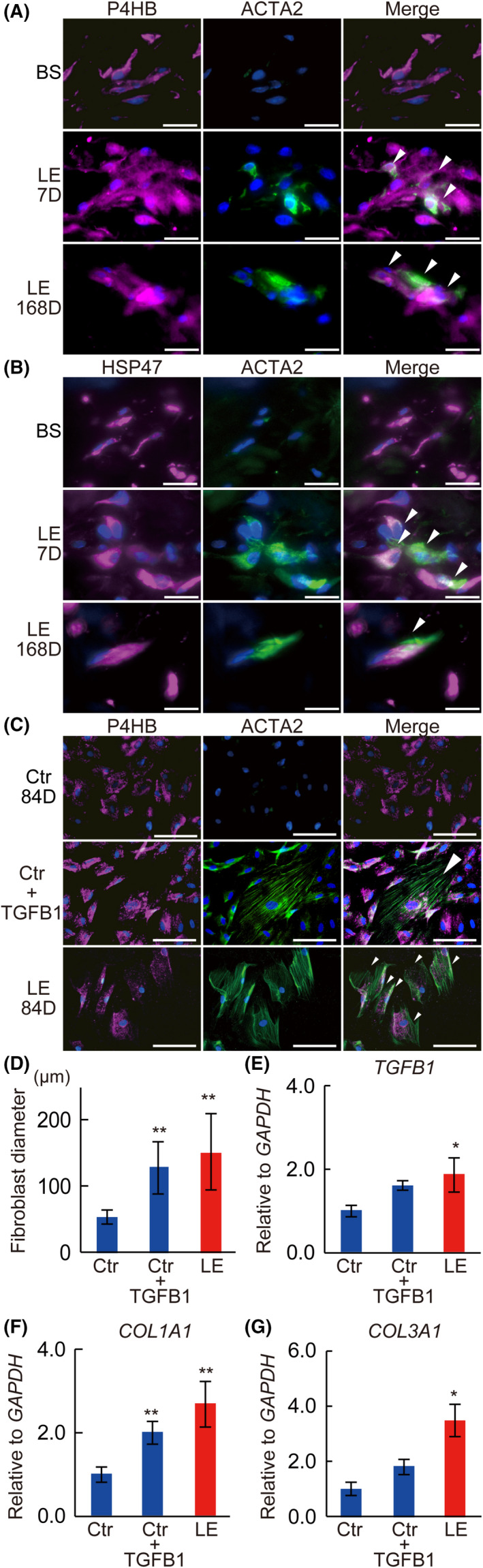
Change of skin fibroblasts in the rat model. A, B, Double IHC of rat skin samples. BS, before surgery; D, days after surgery; LE, lymphedema. A, Magenta: P4HB; green: ACTA2, blue: DAPI. B, Magenta: HSP47; green: ACTA2, blue: DAPI. C, Double IHC of primary cultured skin fibroblasts from the rat model (magenta: P4HB; green: ACTA2; blue: DAPI). D, The average diameter of primary cultured skin fibroblasts from the rat model. Relative expression of (E) TGFB1, (F) COL1A1, (G) COL3A1 mRNA of primary cultured skin fibroblasts from the rat model. (C) Ctr 84D, (F) Ctr, control on day 84; Ctr + TGFB1, Ctr fibroblasts stimulated with TGFB1; (C) LE 84D, (F) LE, lymphedema rats on day 84. n = 5 for each groups, *P < .05, **P < .01 (D‐G). Scale bars, 100 μm, and white arrowheads indicate co‐localization (A‐C)
3.5. Changes in subcutaneous lymphatic vessels and macrophages in the rat lymphedema model
Double IHC showed co‐localization of podoplanin and lymphatic vessel endothelial hyaluronan receptor‐1 (LYVE‐1) on the subcutaneous LV of BS as well as LE on days 7 and 168 (Figure S7). In contrast, IHC for podoplanin revealed a significant increase in the LV of the skin tissues in the LE group during the acute phase with a gradual decrease in the chronic phase (Figure 5A). The number of LV and the LV luminal area was significantly increased from days 14‐28, and days 3‐28, respectively, whereas they were reduced on day 168 (P < .05) and on days 140‐168 (P < .01), respectively (Figure 5B,C).
FIGURE 5.
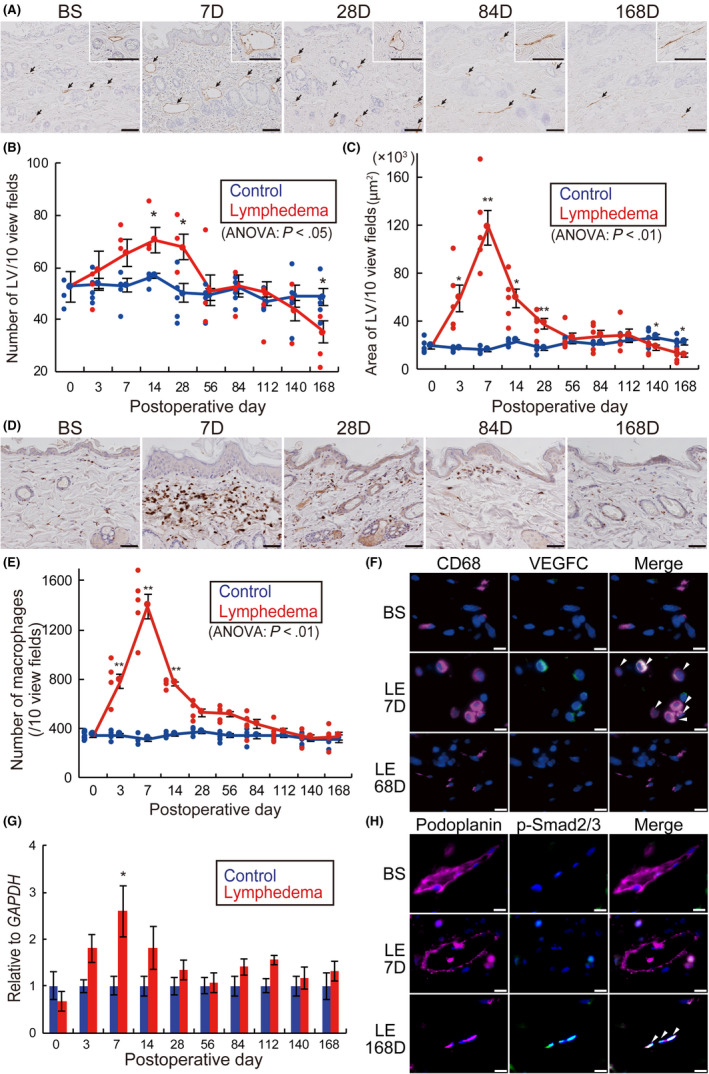
Histological time‐dependent changes of subcutaneous lymphatics in the rat lymphedema model. A, Immunohistochemistry (IHC) for podoplanin (black arrows: lymphatic vessels [LV]). B, Number of subcutaneous LV. C, Luminal area of subcutaneous LV. D, IHC for CD68. E, Number of subcutaneous macrophages of lymphedema models over time. F, Double IHC of skin samples from rats before surgery (BS), lymphedema on day 7 (LE 7D) and lymphedema on day 168 (LE 168D). Magenta: CD68; green: VEGFC; blue; DAPI. G, Relative mRNA expression levels of VEGFC in skin samples. H, Double IHC of skin samples from rats BS, LE 7D, and LE 168D. Magenta: podoplanin; green: p‐Smad2/3; blue; DAPI. n = 5, for control and lymphedema groups, *P < .05, **P < .01 (B, C, E, G). Scale bars: A, 100 μm; D, 50 μm; and F, H, 10 μm. White arrowheads indicate co‐localization in double IHC (F, H)
Marked infiltration of macrophages in the skin was observed (Figure 5D), and the number of macrophages in the LE group significantly increased between days 3‐14, as shown by IHC for CD68 (P < .01) (Figure 5E). Conversely, double IHC revealed CD68‐positive macrophages in the LE group expressing vascular endothelial growth factor (VEGFC) on day 7, which was further confirmed by the increased expression of VEGFC mRNA in the skin of the LE group on day 7 (P < .05) by qRT‐PCR analysis. In contrast, macrophages in both the BS and LE groups were negative for VEGFC on day 168 (Figure 5F,G). Moreover, phosphorylation of Smad2/3 was enhanced in podoplanin‐positive lymphatic endothelial cells on day 168, however it was not observed in the BS group and LE on day 7 (Figure 5H).
3.6. Changes in skin fibrosis in human lymphedema
All the 5 patients recruited in this study, including 4 females and 1 male, had cancer‐related secondary lymphedema. The mean age of the patients and disease duration were 74.8 ± 3.0 y and 22.4 ± 7.6 y, respectively (Table S1). Staining of the skin samples using Azan (Figure 6A,B), Masson trichrome (Figure S8A,B), Picrosirius red (Figure S8C,D) showed an increase in collagen fibers in LE legs compared with HE legs. The average collagen area fraction of subcutaneous tissues also significantly increased in LE legs compared with HE legs (Azan, P < .01, Figure 6B) (Masson trichrome, P < .01, Picrosirius red, P < .05, Figure S8B,D). Moreover, the acoustic microscopy showed increased high‐sound‐speed areas (shown in red) in the dermis and subcutaneous tissues of LE legs (Figure 6C), and the average sound speed of skin in LE legs significantly increased compared with HE legs (Figure 6D; P < .05).
FIGURE 6.
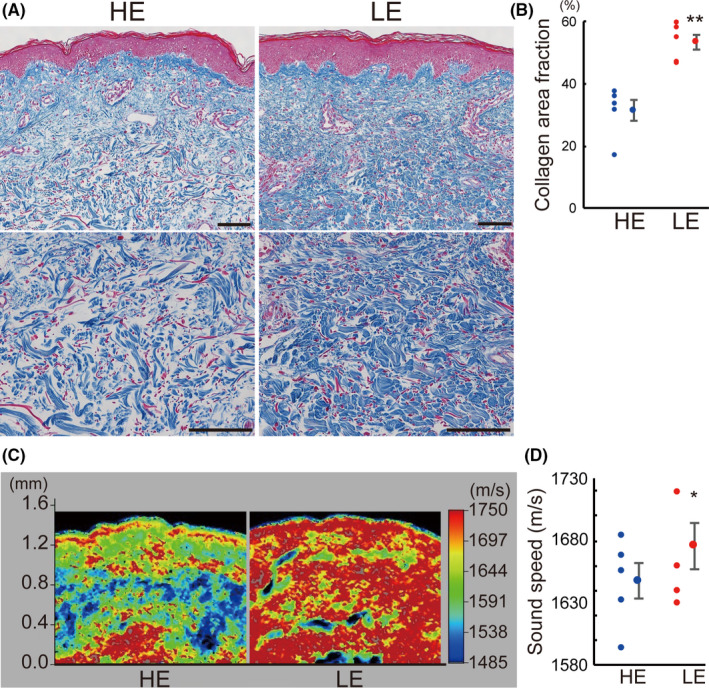
Skin fibrosis in the human lymphedema skin. A, Azan staining of skin samples from lymphedema patients. HE, healthy control leg; LE, lymphedema leg. B, Collagen area fraction analyzed with Azan staining. C, Acoustic microscopy and (D) tissue sound speeds of human skin samples from HE and LE. n = 5 for each group. *P < .05, **P < .01 (B, D). Scale bars: A, 100 μm
3.7. TGFB1 expression in the skin samples of human lymphedema patients
Immunohistochemical staining showed a high expression of TGFB1 in the subcutaneous tissues of LE legs compared with HE legs (Figure 7A). Conversely, double IHC revealed CD68‐positive macrophages express TGFB1 neither in HE nor LE legs (Figure 7B). In contrast, the HSP47‐positive fibroblasts in the LE legs expressed TGFB1, whereas those in the HE legs did not (Figure 7C). Moreover, phosphorylation of Smad2/3 was enhanced in the subcutaneous tissues in LE legs (Figure 7D) and HSP47‐positive fibroblasts (Figure 7E) compared to the HE legs.
FIGURE 7.
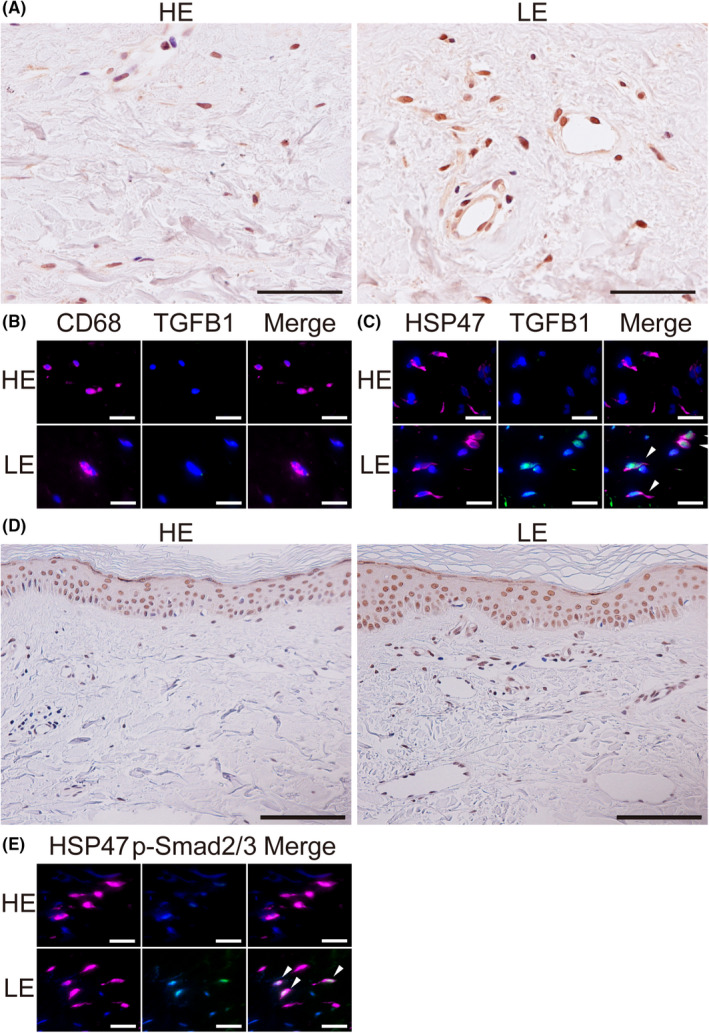
TGFB1 expression and Smad signaling in the human lymphedema skin. A, Immunohistochemistry (IHC) for TGFB1 of skin samples from lymphedema patients. HE, healthy control leg; LE, lymphedema leg. B, C, Double IHC of the skin samples from HE and LE. B, Magenta: CD68; green: TGFB1; blue: DAPI. C, Magenta: HSP47; green: TGFB1; blue: DAPI. D, IHC for p‐Smad2/3, E, Double IHC of skin samples from HE and LE. Magenta: HSP47; green: p‐Smad2/3; blue: DAPI. Scale bars: A, 50 μm; B, C, E, 20 μm; D, 100 μm. G, I, White arrowheads indicate co‐localization in double IHC
3.8. Distinctive change of skin fibroblasts in human lymphedema
The double IHC of the skin fibroblasts primarily cultured from skin samples of lymphedema patients revealed that the fibroblasts from LE legs expressed ACTA2, whereas those from HE legs did not (Figure 8A). The average cell size significantly increased in LE legs compared to HE legs (P < .05; Figure 7B). The mRNA expression of and TGFB1, COL1A1, and COL3A1 were significantly higher in fibroblasts obtained from LE legs compared to HE legs (Figure 8C‐E; P < .01).
FIGURE 8.
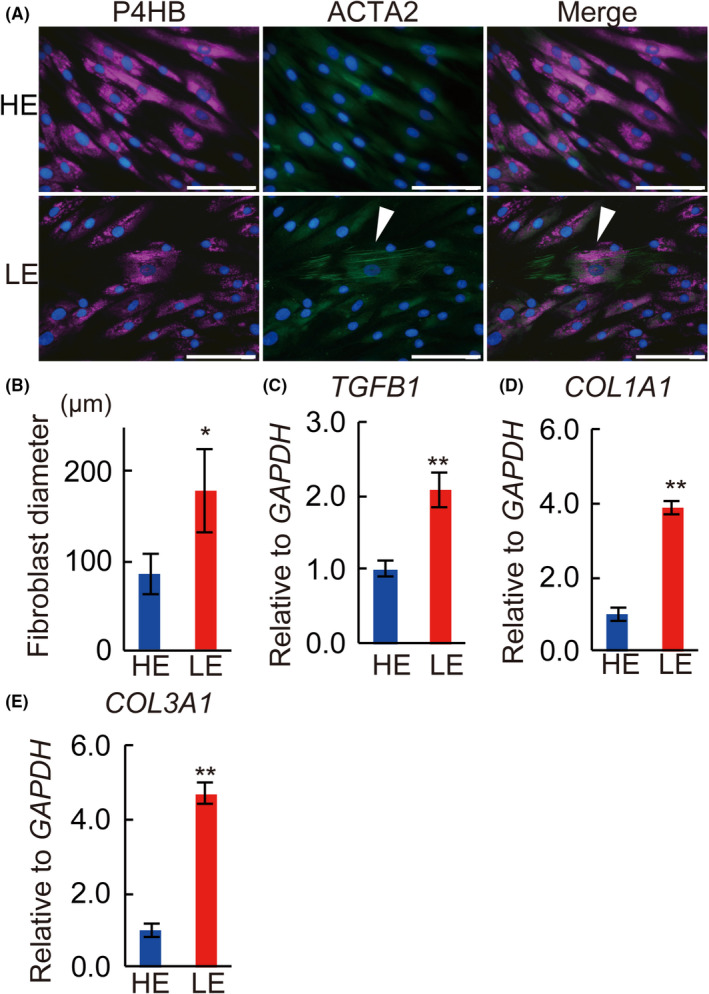
Primary cultured skin fibroblasts from lymphedema patients. A, Double immunohistochemical staining of primary cultured skin fibroblasts from lymphedema patients. HE, healthy control leg; LE, lymphedema leg. Magenta: P4HB; green: ACTA2; blue: DAPI. White arrowheads indicate co‐localization in double IHC. B, Fibroblast diameter of primary cultured skin fibroblasts from the lymphedema patients. Relative expression of (C) TGFB1, (D) COL1A1, (E) COL3A1 mRNA of primary cultured fibroblasts from lymphedema patients. n = 5 for each group, (B‐E) *P < .05, **P < .01. Scale bars: A, 100 μm
3.9. Changes in subcutaneous lymphatic vessels in human lymphedema
Immunohistochemical staining for podoplanin showed smaller and less subcutaneous LV in LE legs compared to HE legs (Figure 9A). The number and luminal areas of subcutaneous LV were lower in LE legs compared to HE legs (Figure 9B,C). Double IHC revealed an enhancement in the phosphorylation of Smad2/3 in the podoplanin‐positive lymphatic endothelial cells compared to the HE legs (Figure 9D).
FIGURE 9.
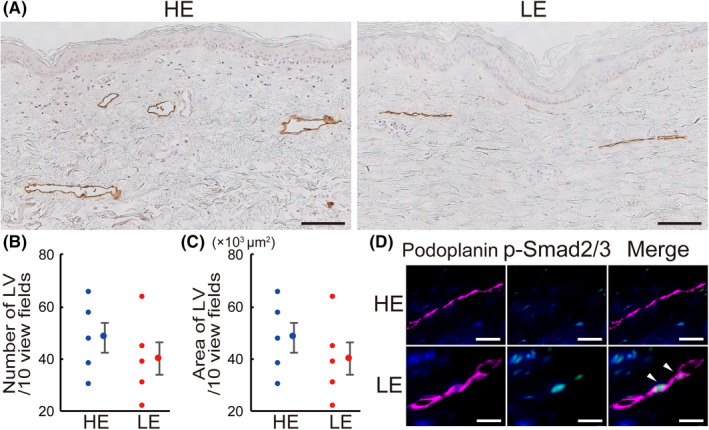
Histological changes of subcutaneous lymphatics in human lymphedema skin. A, IHC for podoplanin of human skin from healthy control leg (HE) and lymphedema leg (LE). Number (B) and luminal area (C) of subcutaneous lymphatic vessels (LV) in HE and LE. D, Double IHC of skin samples from HE and LE. Magenta: podoplanin; green: p‐Smad2/3; blue: DAPI. White arrowheads indicate co‐localization. Scale bars: A, 100 μm; D, 20 μm. B, C, n = 5 for HE and LE
3.10. Comparison of lymphedema staging between the rat model and humans
Oil Red O staining revealed a significant decrease (days 3‐7) and increase (days 84‐168) in the number of subcutaneous adipocytes in the LE group (Figure S9A,B; P < .01). Based on the classification of the ISL, 39 the secondary lymphedema in humans typically progresses from clinical stages I‐III. Based on the fluid accumulation and histological findings (proliferating cells, fibrosis, and fat deposits), the symptoms observed in our rat model were comparable with the staging of human secondary lymphedema (Table 1). The symptoms displayed in our model between days 3‐7 matched the symptoms observed in humans during clinical stage I, as represented by cell proliferation and fluid accumulation. Symptoms displayed between days 14‐56 matched the symptoms observed during clinical stage II, represented by fluid accumulation and fibrosis. Symptoms displayed between days 84‐168 matched the symptoms observed during clinical stage III, represented by fluid accumulation, fibrosis, and fat deposits.
TABLE 1.
Comparison between the symptoms observed in the rat model and the stages of human secondary lymphedema
| Stage | 0 | I | II | III | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Day | 0 | 3 | 7 | 14 | 28 | 56 | 84 | 112 | 140 | 168 |
| Proliferating cells a | – | + | + | ± | ± | – | – | – | – | – |
| Fluid accumulation | – | + | + | + | + | + | + | + | + | + |
| Fibrosis | – | – | – | – | + | + | + | + | + | + |
| Fat deposits | – | – | – | – | – | + | + | + | + | + |
+, observed; –, not observed; ±, occasionally observed.
Proliferation of inflammatory cells, mainly macrophages in our rat model.
4. DISCUSSION
In lymphedema patients, disruption of lymphatic transport induces edema and skin changes. 64 , 65 , 66 The ideal lymphedema model should satisfy the following conditions: disruption of lymphatic transport, edema, skin change from the acute to the chronic phase, been created in small animals (easier handling), human comparability, and not influenced by infection or radiation. Previous animal models have not satisfied these conditions. 10 , 11 , 67 , 68 , 69 In this study, based on detailed anatomy, 41 all lymph nodes were removed from the rat limbs following a procedure similar to lymphadenectomy in cancer surgery in human. It has been shown that the incidence of secondary lymphedema decreases in patients who have undergone sentinel node navigation surgery compared with those with lymph node dissections. 70 , 71 In addition, limited lymph node resection did not create a satisfactory rat model in preliminary studies, necessitating the removal of all lymph nodes in the tumor area. In previous models, the skin margins were sutured to the muscles, and/or postoperative radiation was performed to prevent the development of collateral LV beyond the incision (Figure S1J). 16 , 17 , 18 , 20 These procedures pose a high risk of infection and radiation disorder, resulting in a high mortality rate. In addition, inverted skin suturing prevents both the development of collateral LV beyond the incision and skin infection, thus enabling the evaluation of pathophysiology with better survival without the influence of skin infection.
To our knowledge, this is the first model corresponding to the clinical stages of human secondary lymphedema. Pathologically, we observed the infiltration of macrophages, an initial increase and then a gradual decrease in the number and lumen of LV, skin fibrosis, and fat deposition at different stages of disease progression. We compared our model with the typical staging of secondary lymphedema in humans (Table 1) using the ISL classification system, 39 the consensus, and the most reliable staging system followed by most clinicians. However, ISL staging of lymphedema has a few limitations. For example, skin lesions with several stages could be present in one leg and may have altered the lymphatic territories and, in stage I or II, various proliferating cells could be observed, but their identification and role was not defined.
Pathological evaluation in previous models was often confined to either the acute or the chronic phase and was not comparable with humans. Results from previous rodent hindlimb models often reported only in the acute phase, 72 , 73 whereas those from the large animal models were confined to the chronic phase. 13 , 14 In the present study, the developed model was evaluated over a long period (168 d) that, considering the lifespan of rats, was equivalent to several years of human life, thus enabling a consistent histological and pathological evaluation of secondary lymphedema from the acute to chronic phases.
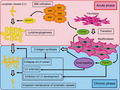
TGFB1 is secreted as a latent, high‐molecular‐weight complex (over 200 kDa) containing the mature, bioactive TGFB1 (25 kDa) and a prodomain called the latency‐associated peptide. TGFB1 also binds to the latent TGF‐binding protein in the extracellular matrix. On activation of this protein complex by proteolytic cleavage, TGFB turns highly bioactive. Bioactive TGFb1 binds to TGFB2, which then recruits and activates TGFR1. Smad2/3, the so‐called receptor‐activated Smad, binds to activated TGFR1 and is phosphorylated. Phosphorylated Smad2/3 (p‐Smad2/3) aggregates to form a heteromultimer with the common mediator Smad, and accumulates in the nucleus to regulate transcriptional responses. 33 , 74 TGFB1 stimulates the transcription of collagen proteins for the extracellular matrix in fibroblasts via Smad signaling, 30 , 31 , 33 and the transition of fibroblasts to myofibroblasts. 35 , 75 , 76 Several studies have shown the central role played by TGFB1 in the development of tissue fibrosis in the liver, lung, kidney, skin, and myocardium. 29 , 30 , 31 , 32 , 33 , 34 , 36 The schematic overview of the pathophysiology of secondary lymphedema is illustrated in Figure 10. Here, our rat model showed the development of skin fibrosis with activation of the TGFB1/Smad signaling cascade and, for the first time, we showed its involvement in the development of human secondary lymphedema. Our study revealed that, in the acute to subacute phase (days 7‐14) of secondary lymphedema, TGFB1 signaling derived from the infiltrated macrophages increased, leading to the initiation of fibroblast‐myofibroblast transition. Consequently, it enhanced the production of TGFB1 from myofibroblasts, accelerating myofibroblast–activated collagen synthesis via Smad signaling, in the chronic phase (days 56‐168). Phosphorylation of Smad2/3 is mainly enhanced by TGFB1 or activin binding to TGFR2. 77 We could not detect an increase in activin level in the skin, neither in the rat model nor in human lymphedema patients (data not shown), and speculated that phosphorylation of Smad2/3 enhanced by TGFB1 might be mainly responsible for skin fibrosis in secondary lymphedema patients. Some studies have demonstrated increased TGFB1 expression in lymphedema models 78 , 79 and, here, we confirmed this hypothesis by demonstrating the presence of myofibroblasts that released TGFB1 in vivo and in vitro. Our experimental model is the first to be significantly relevant to humans with regard to skin fibrosis.
FIGURE 10.
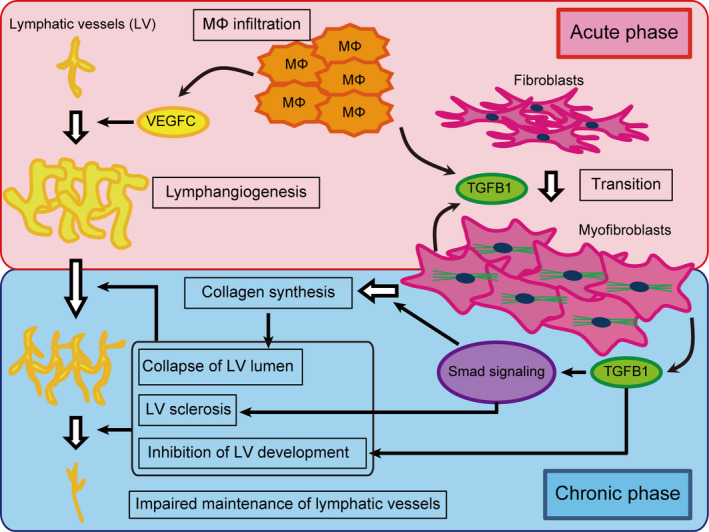
Schematic overview of the pathophysiology of secondary lymphedema. Infiltrated macrophages (MΦ) produce VEGFC and TGFB1 in the acute phase. VEGFC stimulates lymphangiogenesis, while TGFB1 promotes the transition of fibroblasts into myofibroblasts. In the chronic phase, MΦ infiltration is not prominent. Nonetheless, myofibroblasts produce TGFB1, maintaining the transition of fibroblasts into myofibroblasts. TGFB1 via Smad signaling also accelerates collagen fiber synthesis by myofibroblasts in an autocrine fashion and lymphatic vessel (LV) sclerosis by lymphatic endothelial cells. The collapse of the LV lumen resulting from the accumulation of collagen fibers and inhibition of LV development by TGFB1 leads to the impaired maintenance of lymphatic vessels
Moreover, an imbalance between collagen synthesis and degradation promotes fibrosis progression, as the complex interactions between pro‐fibrogenic and anti‐fibrogenic cytokines regulate the extracellular matrix. 31 , 32 , 80 , 81 TGFB1, CTGF, FGF2, TIMP1, and TIMP2 are considered pro‐fibrogenic, whereas MMP1 and IL1B are considered anti‐fibrogenic. 32 , 80 , 81 Our findings indicated an increase in the collagen fiber synthesis; however degradation was not affected during the pathophysiology of secondary lymphedema.
Skin fibrosis might lead to compression of the subcutaneous tissues and the collapse of the LV lumen in the chronic phase of lymphedema. Here, we showed stenosis of subcutaneous capillary LV and TGFB1/Smad signaling in lymphatic endothelial cells. It has been shown that, in the late stage of human lymphedema, LV becomes sclerotic, causing lymphatic vessel sclerosis, 82 with decreased lymphatic pumping function. 83 Capillary lymphatic vessel sclerosis might be induced via the TGFB1/Smad signaling cascade in lymphatic endothelial cells and accumulation of myofibroblasts and collagen fibers in subcutaneous tissues, which might impair the absorption of lymphatic fluid in subcutaneous tissues, inducing lymphedema.
TGFB1 is a multifunctional cytokine and exerts a dual function in cancer progression. 33 , 74 In LV formation, TGFB1 also has a dual role. It has been reported that the suppression of TGFB1 signaling enhances lymphangiogenesis, 37 whereas its overexpression inhibits the development of lymphatic collaterals, 38 , 40 suggesting that TGFB1 is an anti‐lymphangiogenic cytokine. Moreover, TGFB1 signaling is important for sprouting and proliferation of lymphatic endothelial cells. 84 In the chronic phase of our model and human lymphedema samples, an increase in TGFB1/Smad signaling, TGFB1‐positive myofibroblasts, and a decrease in the number of LV were observed. Based on these findings, we believe that TGFB1/Smad signaling inhibits the development of subcutaneous LV in the chronic phase of secondary lymphedema.
A previous study has shown that expression of both pro‐lymphangiogenic and anti‐lymphangiogenic cytokines increases in lymphedema. 67 To verify, we focused on VEGFC as a pro‐lymphangiogenic cytokine, 21 and observed infiltration of the VEGFC‐positive macrophages into the subcutaneous tissue in the acute phase, suggesting lymphangiogenesis. Infiltration of macrophages and VEGFC expression were most prominent on day 7, and the numbers of subcutaneous LV were greatest between days 14‐28, and tended to decrease in the subacute phase, indicating that acute phase lymphangiogenesis might not be sustainable. The formation of mature LV takes 6 d, 85 and our results matched those of previously reported findings. Moreover, in the chronic phase, the number and luminal area of LV decreased, which could be due to the collapse of the LV lumen, LV sclerosis, and inhibition of LV development by TGFB1/Smad signaling.
In our study, LV density decreased in the rat model in the chronic phase, as shown in an earlier study. 21 In contrast, some studies have also reported an increase. 39 , 86 , 87 In the chronic phase of human lymphedema, the number and luminal area of capillary LV decreases, and collecting LV become stenotic. 21 , 83 , 88 This discrepancy could be due to the difference in defining the chronic phase. Here, we hypothesize that a minimum of 56 d should pass before lymphedema is considered to have entered the chronic phase to compare our model to human lymphedema.
The involvement of T cells in the pathophysiology of lymphedema has been reported. 89 , 90 However, with IHC, we did not observe prominent infiltration of CD4+ inflammatory cells in our model and in human lymphedema skin samples (data not shown), suggesting that T cells might not be the major contributors in our model and in chronic human lymphedema. Moreover, the absence of T cells in our model could be because we induced pure lymph stasis without infection, suggesting that T cells might be involved in the pathophysiology of lymphedema accompanied by infection. Skin infection might occur at any time during the long course of human lymphedema, and often worsens the condition of skin and the patient's QOL. 6 , 7 As such, skin infections might complicate the pathophysiology of human lymphedema compared to the lymphedema induced in animal models.
Recently, the therapeutic effect of the inhibition of tissue fibrosis by TGFB1 suppression was reported in several fibrotic diseases: renal, 91 pulmonary, 92 and cardiac fibrosis. 93 TGFB1 suppression might inhibit the development of skin fibrosis and slow down the progression of secondary lymphedema. At this time, complex physical and surgical therapies (lymphovenous anastomosis, vascularized lymph node transfer, and liposuction) are recommended and performed on lymphedema patients. 39 The treatment target of these therapies is limb volume reduction or improvement in lymphatic flow, but not skin fibrosis. TGFB1 suppression might be the first and only treatment method for lymphedema, as it might target skin fibrosis, in addition to inhibiting capillary LV sclerosis and improving lymphatic flow in subcutaneous tissues.
The current study has some limitations that warrant discussion. First, the number of human samples was small and obtained from stage II, but not stage I or III patients due to the difficulty in obtaining informed consent for skin biopsies from lymphedema patients. Therefore, we could not indicate a significant difference in the number and luminal area of LV in human samples. Second, although we showed changes in the expression of VEGFC and TGFB1 over time, and discussed the pathophysiology with reference to the previous reports, it is difficult to prove their direct role in the pathogenesis of the disease, as this would require the use of suppression and/or overexpression models for these cytokines. Third, the obtained human skin samples were too small. Because lymphedema is a high‐risk factor for skin infection, performing a large skin biopsy in lymphedema patients might come with ethical problems. Using small skin samples, it was impossible to perform biochemical studies regarding the direct binding of TGFB1 to TGFR2 until the accumulation of p‐Smad in the nucleus. Fourth, human lymphedema is affected by several factors such as cellulitis, treatment, and daily activities, 2 , 6 and follows a complex process that is not possible to reproduce in an animal model. However, similar limitations, from which this model was not exempt, are associated with all animal models.
In summary, we developed a novel animal model of secondary lymphedema that mimicked the disease in humans. We found a potential role for TGFB1/Smad signaling and myofibroblasts in skin fibrosis that was associated with secondary lymphedema. Suppressing TGFB1 might inhibit the development of skin fibrosis, facilitating the development of lymphatic collaterals, and represents a rational pathway in the treatment of lymphedema.
DISCLOSURE
The authors have no conflict of interest.
Supporting information
Fig S1‐S9
Table S1
Data S1
ACKNOWLEDGMENTS
We thank Dr. Hiroo Suami (Macquarie University, Australia) for his helpful comments regarding lymphatic networks in the rat hindlimb.41 This work was supported by JSPS KAKENHI (grant number 26293310 awarded to NU and grant number 15K21052 awarded to MSa).
Sano M, Hirakawa S, Suzuki M, et al. Potential role of transforming growth factor‐beta 1/Smad signaling in secondary lymphedema after cancer surgery. Cancer Sci. 2020;111:2620–2634. 10.1111/cas.14457
REFERENCES
- 1. Beesley V, Janda M, Eakin E, Obermair A, Battistutta D. Lymphedema after gynecological cancer treatment: prevalence, correlates, and supportive care needs. Cancer. 2007;109:2607‐2614. [DOI] [PubMed] [Google Scholar]
- 2. Rockson SG. Lymphedema. Am J Med. 2001;110:288‐295. [DOI] [PubMed] [Google Scholar]
- 3. Shaitelman SF, Cromwell KD, Rasmussen JC, et al. Recent progress in the treatment and prevention of cancer‐related lymphedema. CA Cancer J Clin. 2015;65:55‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Földi MFE. Földi's Textbook of Lymphology: For Physicians and Lymphedema Therapists , 3rd rev edn. Munchen, Germany: Urban & Fischer; 2012. [Google Scholar]
- 5. Reed RK, Laurent TC, Taylor AE. Hyaluronan in prenodal lymph from skin: changes with lymph flow. Am J Physiol. 1990;259:H1097‐H1100. [DOI] [PubMed] [Google Scholar]
- 6. Jager G, Doller W, Roth R. Quality‐of‐life and body image impairments in patients with lymphedema. Lymphology. 2006;39:193‐200. [PubMed] [Google Scholar]
- 7. Kim SI, Lim MC, Lee JS, et al. Impact of lower limb lymphedema on quality of life in gynecologic cancer survivors after pelvic lymph node dissection. Eur J Obstet Gynecol Reprod Biol. 2015;192:31‐36. [DOI] [PubMed] [Google Scholar]
- 8. Kilarski WW. Physiological perspective on therapies of lymphatic vessels. Adv Wound Care (New Rochelle). 2018;7:189‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tian W, Rockson SG, Jiang X, et al. Leukotriene B4 antagonism ameliorates experimental lymphedema. Sci Transl Med. 2017;9:eaal3920. [DOI] [PubMed] [Google Scholar]
- 10. Frueh FS, Gousopoulos E, Rezaeian F, Menger MD, Lindenblatt N, Giovanoli P. Animal models in surgical lymphedema research–a systematic review. J Surg Res. 2016;200:208‐220. [DOI] [PubMed] [Google Scholar]
- 11. Hadamitzky C, Pabst R. Acquired lymphedema: an urgent need for adequate animal models. Cancer Res. 2008;68:343‐345. [DOI] [PubMed] [Google Scholar]
- 12. Shin WS, Szuba A, Rockson SG. Animal models for the study of lymphatic insufficiency. Lymphat Res Biol. 2003;1:159‐169. [DOI] [PubMed] [Google Scholar]
- 13. Danese CA, Georgalas‐Bertakis M, Morales LE. A model of chronic postsurgical lymphedema in dogs' limbs. Surgery. 1968;64:814‐820. [PubMed] [Google Scholar]
- 14. Olszewski W, Machowski Z, Sokolowski J, Nielubowicz J. Experimental lymphedema in dogs. J Cardiovasc Surg (Torino). 1968;9:178‐183. [PubMed] [Google Scholar]
- 15. Hadrian R, Palmes D. Animal models of secondary lymphedema: new approaches in the search for therapeutic options. Lymphat Res Biol. 2017;15:2‐16. [DOI] [PubMed] [Google Scholar]
- 16. Kanter MA, Slavin SA, Kaplan W. An experimental model for chronic lymphedema. Plast Reconstr Surg. 1990;85:573‐580. [DOI] [PubMed] [Google Scholar]
- 17. Kawahira T, Sugimoto T, Okada M, Maeda S. An experimental study of surgical treatment for lymphedema in rats: A modified Kinmonth procedure and autologous lymph node capsule‐venous anastomosis with lymph node transfer. Ann Thorac Cardiovasc Surg. 1999;5:86‐93. [PubMed] [Google Scholar]
- 18. Lee‐Donaldson L, Witte MH, Bernas M, Witte CL, Way D, Stea B. Refinement of a rodent model of peripheral lymphedema. Lymphology. 1999;32:111‐117. [PubMed] [Google Scholar]
- 19. Oashi K, Furukawa H, Oyama A, et al. A new model of acquired lymphedema in the mouse hind limb: a preliminary report. Ann Plast Surg. 2012;69:565‐568. [DOI] [PubMed] [Google Scholar]
- 20. Wang GY, Zhong SZ. A model of experimental lymphedema in rats' limbs. Microsurgery. 1985;6:204‐210. [DOI] [PubMed] [Google Scholar]
- 21. Tammela T, Saaristo A, Holopainen T, et al. Therapeutic differentiation and maturation of lymphatic vessels after lymph node dissection and transplantation. Nat Med. 2007;13:1458‐1466. [DOI] [PubMed] [Google Scholar]
- 22. Huang GK, Hsin YP. An experimental model for lymphedema in rabbit ear. Microsurgery. 1983;4:236‐242. [DOI] [PubMed] [Google Scholar]
- 23. Kubo M, Li TS, Kamota T, Ohshima M, Shirasawa B, Hamano K. Extracorporeal shock wave therapy ameliorates secondary lymphedema by promoting lymphangiogenesis. J Vasc Surg. 2010;52:429‐434. [DOI] [PubMed] [Google Scholar]
- 24. Aschen S, Zampell JC, Elhadad S, Weitman E, De Brot M, Mehrara BJ. Regulation of adipogenesis by lymphatic fluid stasis: part II. Expression of adipose differentiation genes. Plast Reconstr Surg. 2012;129:838‐847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tobbia D, Semple J, Baker A, Dumont D, Semple A, Johnston M. Lymphedema development and lymphatic function following lymph node excision in sheep. J Vasc Res. 2009;46:426‐434. [DOI] [PubMed] [Google Scholar]
- 26. Lahteenvuo M, Honkonen K, Tervala T, et al. Growth factor therapy and autologous lymph node transfer in lymphedema. Circulation. 2011;123:613‐620. [DOI] [PubMed] [Google Scholar]
- 27. Visuri MT, Honkonen KM, Hartiala P, et al. VEGF‐C and VEGF‐C156S in the pro‐lymphangiogenic growth factor therapy of lymphedema: a large animal study. Angiogenesis. 2015;18:313‐326. [DOI] [PubMed] [Google Scholar]
- 28. Wu GJ, Xu H, Zhou WH, et al. Rhesus monkey is a new model of secondary lymphedema in the upper limb. Int J Clin Exp Patho. 2014;7:5665‐5673. [PMC free article] [PubMed] [Google Scholar]
- 29. Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen SJ, Yuan W, Mori Y, Levenson A, Trojanowska M, Varga J. Stimulation of type I collagen transcription in human skin fibroblasts by TGF‐beta: involvement of Smad 3. J Invest Dermatol. 1999;112:49‐57. [DOI] [PubMed] [Google Scholar]
- 31. Ihn H. Pathogenesis of fibrosis: role of TGF‐beta and CTGF. Curr Opin Rheumatol. 2002;14:681‐685. [DOI] [PubMed] [Google Scholar]
- 32. Leask A, Abraham DJ. TGF‐beta signaling and the fibrotic response. FASEB J. 2004;18:816‐827. [DOI] [PubMed] [Google Scholar]
- 33. Miyazono K. TGF‐beta signaling by Smad proteins. Cytokine Growth Factor Rev. 2000;11:15‐22. [DOI] [PubMed] [Google Scholar]
- 34. Waltenberger J, Lundin L, Oberg K, et al. Involvement of transforming growth factor‐beta in the formation of fibrotic lesions in carcinoid heart disease. Am J Pathol. 1993;142:71‐78. [PMC free article] [PubMed] [Google Scholar]
- 35. Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199‐210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang K, Garner W, Cohen L, Rodriguez J, Phan S. Increased types I and III collagen and transforming growth factor‐beta 1 mRNA and protein in hypertrophic burn scar. J Invest Dermatol. 1995;104:750‐754. [DOI] [PubMed] [Google Scholar]
- 37. Oka M, Iwata C, Suzuki HI, et al. Inhibition of endogenous TGF‐beta signaling enhances lymphangiogenesis. Blood. 2008;111:4571‐4579. [DOI] [PubMed] [Google Scholar]
- 38. Vittet D, Merdzhanova G, Prandini MH, Feige JJ, Bailly S. TGFbeta1 inhibits lymphatic endothelial cell differentiation from mouse embryonic stem cells. J Cell Physiol. 2012;227:3593‐3602. [DOI] [PubMed] [Google Scholar]
- 39. Executive C. The diagnosis and treatment of peripheral lymphedema: 2016 Consensus Document of the International Society of Lymphology. Lymphology. 2016;49:170‐184. [PubMed] [Google Scholar]
- 40. Arnfinn E, Enok T. Lymphatic pathways from the tail in rats and mice. Cancer Res. 1960;20:613‐614. [Google Scholar]
- 41. Suami H, Chang DW, Matsumoto K, Kimata Y. Demonstrating the lymphatic system in rats with microinjection. Anat Rec (Hoboken). 2011. [DOI] [PubMed] [Google Scholar]
- 42. Banyasz T, Lozinskiy I, Payne CE, et al. Transformation of adult rat cardiac myocytes in primary culture. Exp Physiol. 2008;93:370‐382. [DOI] [PubMed] [Google Scholar]
- 43. Bustin SA. Absolute quantification of mRNA using real‐time reverse transcription polymerase chain reaction assays. J Mol Endocrinol. 2000;25:169‐193. [DOI] [PubMed] [Google Scholar]
- 44. Evans RA, Tian YC, Steadman R, Phillips AO. TGF‐beta1‐mediated fibroblast‐myofibroblast terminal differentiation‐the role of Smad proteins. Exp Cell Res. 2003;282:90‐100. [DOI] [PubMed] [Google Scholar]
- 45. Faarvang AS, Rordam Preil SA, Nielsen PS, Beck HC, Kristensen LP, Rasmussen LM. Smoking is associated with lower amounts of arterial type I collagen and decorin. Atherosclerosis. 2016;247:201‐206. [DOI] [PubMed] [Google Scholar]
- 46. Galie PA, Westfall MV, Stegemann JP. Reduced serum content and increased matrix stiffness promote the cardiac myofibroblast transition in 3D collagen matrices. Cardiovasc Pathol. 2011;20:325‐333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hagiwara Y, Saijo Y, Chimoto E, et al. Increased elasticity of capsule after immobilization in a rat knee experimental model assessed by scanning acoustic microscopy. Ups J Med Sci. 2006;111:303‐313. [DOI] [PubMed] [Google Scholar]
- 48. Kim S, Lim YT, Soltesz EG, et al. Near‐infrared fluorescent type II quantum dots for sentinel lymph node mapping. Nat Biotechnol. 2004;22:93‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kosaka N, Ogawa M, Sato N, Choyke PL, Kobayashi H. In vivo real‐time, multicolor, quantum dot lymphatic imaging. J Invest Dermatol. 2009;129:2818‐2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kuroda K, Tajima S. HSP47 is a useful marker for skin fibroblasts in formalin‐fixed, paraffin‐embedded tissue specimens. J Cutan Pathol. 2004;31:241‐246. [DOI] [PubMed] [Google Scholar]
- 51. Li XH, Fang ZN, Guan TM, et al. A novel collagen area fraction index to quantitatively assess bowel fibrosis in patients with Crohn's disease. BMC Gastroenterol. 2019;19:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Malmstrom E, Sennstrom M, Holmberg A, et al. The importance of fibroblasts in remodelling of the human uterine cervix during pregnancy and parturition. Mol Hum Reprod. 2007;13:333‐341. [DOI] [PubMed] [Google Scholar]
- 53. Miyasaka M, Sakai S, Kusaka A, et al. Ultrasonic tissue characterization of photodamaged skin by scanning acoustic microscopy. Tokai J Exp Clin Med. 2005;30:217‐225. [PubMed] [Google Scholar]
- 54. Ordonez NG. Immunohistochemical endothelial markers: a review. Adv Anat Pathol. 2012;19:281‐295. [DOI] [PubMed] [Google Scholar]
- 55. Rittie L, Fisher GJ. Isolation and culture of skin fibroblasts. Methods Mol Med. 2005;117:83‐98. [DOI] [PubMed] [Google Scholar]
- 56. Schmittgen TD, Livak KJ. Analyzing real‐time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101‐1108. [DOI] [PubMed] [Google Scholar]
- 57. Schoppmann SF, Birner P, Studer P, Breiteneder‐Geleff S. Lymphatic microvessel density and lymphovascular invasion assessed by anti‐podoplanin immunostaining in human breast cancer. Anticancer Res. 2001;21:2351‐2355. [PubMed] [Google Scholar]
- 58. Singh R, Artaza JN, Taylor WE, Gonzalez‐Cadavid NF, Bhasin S. Androgens stimulate myogenic differentiation and inhibit adipogenesis in C3H 10T1/2 pluripotent cells through an androgen receptor‐mediated pathway. Endocrinology. 2003;144:5081‐5088. [DOI] [PubMed] [Google Scholar]
- 59. Vermeulen PB, Gasparini G, Fox SB, et al. Quantification of angiogenesis in solid human tumours: an international consensus on the methodology and criteria of evaluation. Eur J Cancer. 1996;32A:2474‐2484. [DOI] [PubMed] [Google Scholar]
- 60. Zhou L, Yang K, Randall Wickett R, Zhang Y. Dermal fibroblasts induce cell cycle arrest and block epithelial‐mesenchymal transition to inhibit the early stage melanoma development. Cancer Med. 2016;5:1566‐1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jun H, Lee JY, Kim JH, et al. Modified mouse models of chronic secondary lymphedema: tail and hind limb models. Ann Vasc Surg. 2017;43:288‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Maegawa J, Mikami T, Yamamoto Y, Satake T, Kobayashi S. Types of lymphoscintigraphy and indications for lymphaticovenous anastomosis. Microsurgery. 2010;30:437‐442. [DOI] [PubMed] [Google Scholar]
- 63. Unno N, Inuzuka K, Suzuki M, et al. Preliminary experience with a novel fluorescence lymphography using indocyanine green in patients with secondary lymphedema. J Vasc Surg. 2007;45:1016‐1021. [DOI] [PubMed] [Google Scholar]
- 64. Daroczy J. Pathology of lymphedema. Clin Dermatol. 1995;13:433‐444. [DOI] [PubMed] [Google Scholar]
- 65. Sano M, Hirakawa S, Yamanaka Y, et al. Development of a noninvasive skin evaluation method for lower limb lymphedema. Lymphat Res Biol. 2020;18:7‐15. [DOI] [PubMed] [Google Scholar]
- 66. Szuba A, Rockson SG. Lymphedema: anatomy, physiology and pathogenesis. Vasc Med. 1997;2:321‐326. [DOI] [PubMed] [Google Scholar]
- 67. Gardenier JC, Hespe GE, Kataru RP, et al. Diphtheria toxin‐mediated ablation of lymphatic endothelial cells results in progressive lymphedema. JCI Insight. 2016;1:e84095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yang CY, Nguyen DH, Wu CW, et al. Developing a lower limb lymphedema animal model with combined lymphadenectomy and low‐dose radiation. Plast Reconstr Surg Glob Open. 2014;2:e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yarnold J, Brotons MC. Pathogenetic mechanisms in radiation fibrosis. Radiother Oncol. 2010;97:149‐161. [DOI] [PubMed] [Google Scholar]
- 70. Golshan M, Martin WJ, Dowlatshahi K. Sentinel lymph node biopsy lowers the rate of lymphedema when compared with standard axillary lymph node dissection. Am Surg. 2003;69:209‐211; discussion 212. [PubMed] [Google Scholar]
- 71. Yahata H, Kobayashi H, Sonoda K, et al. Prognostic outcome and complications of sentinel lymph node navigation surgery for early‐stage cervical cancer. Int J Clin Oncol. 2018;23:1167‐1172. [DOI] [PubMed] [Google Scholar]
- 72. Honkonen KM, Visuri MT, Tervala TV, et al. Lymph node transfer and perinodal lymphatic growth factor treatment for lymphedema. Ann Surg. 2013;257:961‐967. [DOI] [PubMed] [Google Scholar]
- 73. Tabibiazar R, Cheung L, Han J, et al. Inflammatory manifestations of experimental lymphatic insufficiency. PLoS Med. 2006;3:e254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Miyazono K. Transforming growth factor‐beta signaling in epithelial‐mesenchymal transition and progression of cancer. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85:314‐323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Serini G, Gabbiani G. Mechanisms of myofibroblast activity and phenotypic modulation. Exp Cell Res. 1999;250:273‐283. [DOI] [PubMed] [Google Scholar]
- 76. Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18:1028‐1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Shimizu A, Kato M, Nakao A, et al. Identification of receptors and Smad proteins involved in activin signalling in a human epidermal keratinocyte cell line. Genes Cells. 1998;3:125‐134. [DOI] [PubMed] [Google Scholar]
- 78. Hespe GE, Nores GG, Huang JJ, Mehrara BJ. Pathophysiology of lymphedema‐Is there a chance for medication treatment? J Surg Oncol. 2017;115:96‐98. [DOI] [PubMed] [Google Scholar]
- 79. Torrisi JS, Joseph WJ, Ghanta S, et al. Lymphaticovenous bypass decreases pathologic skin changes in upper extremity breast cancer‐related lymphedema. Lymphat Res Biol. 2015;13:46‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gomez DE, Alonso DF, Yoshiji H, Thorgeirsson UP. Tissue inhibitors of metalloproteinases: structure, regulation and biological functions. Eur J Cell Biol. 1997;74:111‐122. [PubMed] [Google Scholar]
- 81. Woessner JF Jr. Matrix metalloproteinases and their inhibitors in connective tissue remodeling. FASEB J. 1991;5:2145‐2154. [PubMed] [Google Scholar]
- 82. Hara H, Mihara M, Seki Y, Todokoro T, Iida T, Koshima I. Comparison of indocyanine green lymphographic findings with the conditions of collecting lymphatic vessels of limbs in patients with lymphedema. Plast Reconstr Surg. 2013;132:1612‐1618. [DOI] [PubMed] [Google Scholar]
- 83. Unno N, Nishiyama M, Suzuki M, et al. A novel method of measuring human lymphatic pumping using indocyanine green fluorescence lymphography. J Vasc Surg. 2010;52:946‐952. [DOI] [PubMed] [Google Scholar]
- 84. James JM, Nalbandian A, Mukouyama YS. TGFbeta signaling is required for sprouting lymphangiogenesis during lymphatic network development in the skin. Development. 2013;140:3903‐3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kim ES, Kim MS, Moon A. TGF‐beta‐induced upregulation of MMP‐2 and MMP‐9 depends on p38 MAPK, but not ERK signaling in MCF10A human breast epithelial cells. Int J Oncol. 2004;25:1375‐1382. [PubMed] [Google Scholar]
- 86. Liu Y, Fang Y, Dong P, et al. Effect of vascular endothelial growth factor C (VEGF‐C) gene transfer in rat model of secondary lymphedema. Vascul Pharmacol. 2008;48:150‐156. [DOI] [PubMed] [Google Scholar]
- 87. Rutkowski JM, Moya M, Johannes J, Goldman J, Swartz MA. Secondary lymphedema in the mouse tail: Lymphatic hyperplasia, VEGF‐C upregulation, and the protective role of MMP‐9. Microvasc Res. 2006;72:161‐171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Mihara M, Hara H, Hayashi Y, et al. Pathological steps of cancer‐related lymphedema: histological changes in the collecting lymphatic vessels after lymphadenectomy. PLoS One. 2012;7:e41126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Gousopoulos E, Proulx ST, Scholl J, Uecker M, Detmar M. Prominent lymphatic vessel hyperplasia with progressive dysfunction and distinct immune cell infiltration in lymphedema. Am J Pathol. 2016;186:2193‐2203. [DOI] [PubMed] [Google Scholar]
- 90. Zampell JC, Yan A, Elhadad S, Avraham T, Weitman E, Mehrara BJ. CD4(+) cells regulate fibrosis and lymphangiogenesis in response to lymphatic fluid stasis. PLoS One. 2012;7:e49940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Liu R, Das B, Xiao W, et al. A novel inhibitor of homeodomain interacting protein kinase 2 mitigates kidney fibrosis through inhibition of the TGF‐beta1/Smad3 Pathway. J Am Soc Nephrol. 2017;28:2133‐2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Li L, Huang W, Li K, et al. Metformin attenuates gefitinib‐induced exacerbation of pulmonary fibrosis by inhibition of TGF‐beta signaling pathway. Oncotarget. 2015;6:43605‐43619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Li B, Chen H, Yang X, Wang Y, Qin L, Chu Y. Knockdown of eIF3a ameliorates cardiac fibrosis by inhibiting the TGF‐beta1/Smad3 signaling pathway. Cell Mol Biol (Noisy‐le‐Grand). 2016;62:97‐101. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S9
Table S1
Data S1