

Abstract
The pathogenesis of lung cancer associated with idiopathic pulmonary fibrosis (IPF) has remained largely uncharacterized. To provide insight into this condition, we undertook genomic profiling of IPF‐associated lung cancer as well as of adjacent fibrosing lung tissue in surgical specimens. Isolated DNA and RNA from 17 IPF‐associated non‐small cell lung cancer and 15 paired fibrosing lung tissue specimens were analyzed by next‐generation sequencing with a panel that targets 161 cancer‐related genes. Somatic genetic alterations were frequently identified in TP53 (n = 6, 35.3%) and PIK3CA (n = 5, 29.4%) genes in tumor samples as well as in EGFR (n = 7, 46.7%), PIK3CA (n = 5, 33.3%), ERBB3 (n = 4, 26.7%), and KDR (n = 4, 26.7%) in IPF samples. Genes related to the RAS‐RAF signaling pathway were also frequently altered in tumor (n = 7, 41.2%) and IPF (n = 3, 20.0%) samples. The number of somatic alterations identified in IPF samples was almost as large as that detected in paired tumor samples (81 vs 90, respectively). However, only 6 of the 81 somatic alterations detected in IPF samples overlapped with those in paired tumor samples. The accumulation of somatic mutations was thus apparent in IPF tissue of patients with IPF‐associated lung cancer, and the RAS‐RAF pathway was implicated in lung tumorigenesis. The finding that somatic alterations were not frequently shared between tumor and corresponding IPF tissue indicates that IPF‐associated lung cancer does not develop through the stepwise accumulation of somatic alterations in IPF.
Keywords: cancer‐related gene, idiopathic pulmonary fibrosis, lung cancer, next‐generation sequencing, somatic alteration
The genetic basis of idiopathic pulmonary fibrosis‐associated lung cancer remains largely unknown. We show that somatic alterations were frequently identified in fibrosing lung tissue as much as that in tumor tissue, and that genes related to the RAS‐RAF signaling pathway were frequently altered in both specimens. These findings can provide a basis for the development of targeted drugs for such tumors.
1. INTRODUCTION
Idiopathic pulmonary fibrosis (IPF) is a fatal lung disease characterized by the destruction of alveolar architecture associated with the proliferation and differentiation of myofibroblasts. 1 Idiopathic pulmonary fibrosis has been implicated as an independent risk factor for the development of lung cancer, with a cumulative incidence rate of lung cancer ranging from 9.8% to 38% in IPF patients and lung cancer being a major contributing factor to the death of such patients. 2 , 3 , 4 , 5 , 6 , 7 , 8 The prognosis for individuals with IPF‐associated lung cancer is worse than that for lung cancer patients without IPF, mainly as a result of complications such as exacerbation of preexisting interstitial lung disease during cancer treatment. 9
Although several somatic genetic alterations targetable by specific drugs have been identified in non‐small cell lung cancer (NSCLC), with such targeted agents having a pronounced therapeutic efficacy, 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 the pathogenesis and genetic basis of IPF‐associated lung cancer have remained largely unknown, and optimal treatment for this condition is still lacking. The frequency of KRAS or TP53 mutations has been found to be higher in lung tissue from patients with IPF and lung cancer than in lung tissue from those with IPF but without lung cancer, 19 suggesting that somatic alterations occur more frequently in patients with IPF complicated by lung cancer than in those with IPF alone. It has remained unknown, however, whether somatic alterations are shared by IPF‐associated lung cancer and the fibrosing lung tissue and whether IPF‐associated lung cancer develops through a stepwise accumulation of somatic alterations in fibrosing lung tissue.
To investigate the etiology of IPF‐associated lung cancer, we evaluated the genomic profile of lung tumors and adjacent fibrosing lung tissue from patients with IPF‐associated lung cancer with the use of targeted next‐generation sequencing (NGS).
2. MATERIALS AND METHODS
2.1. Patients
Seventeen patients who underwent surgical resection for NSCLC associated with pathologically diagnosed IPF at Kyushu University Hospital between January 2012 and December 2017 and for whom tumor tissue specimens were available were enrolled in the study. The study was approved by the institutional review board of Kyushu University Hospital (approval number 30‐62) and was carried out in accordance with the Declaration of Helsinki and the Ethical Guidelines for Medical Research Involving Human Subjects in Japan. We obtained the following information from clinical records of the patients: age, sex, tumor histology, clinical stage, smoking status, and the high‐resolution computed tomography (HRCT) imaging pattern of IPF.
2.2. Next‐generation sequencing analysis
DNA and RNA were isolated from formaldehyde‐fixed, paraffin‐embedded specimens of lung tumor tissue and those of tumor‐adjacent fibrosing lung tissue with the use of an AllPrep DNA/RNA FFPE Kit (Qiagen). Isolated DNA (10 ng) was subjected to multiplex PCR‐based amplification with the use of an Ion AmpliSeq Library Kit Plus (Thermo Fisher Scientific). Isolated RNA (10 ng) was subjected to reverse transcription with the use of SuperScript IV VILO Master Mix (Thermo Fisher Scientific) followed by library generation with the use of an Ion AmpliSeq Library Kit Plus (Thermo Fisher Scientific). The Oncomine Comprehensive Panel version 3 (Thermo Fisher Scientific) was used to target clinically relevant single nucleotide variants, insertions/deletions, copy number variations, and gene fusions affecting 161 unique cancer‐related genes (Table S1). The library generated from RNA was used for the detection of gene fusions. The amplification products were ligated to Ion Xpress Barcode Adapters (Thermo Fisher Scientific) and purified with the use of Agencourt AMPure XP beads (Beckman Coulter). The purified libraries were pooled and then sequenced with an Ion S5 XL instrument, Ion 540 Kit‐Chef, and Ion 540 Chip Kit (Thermo Fisher Scientific). DNA sequencing data were accessed through the Torrent Suite version 5.2.2 program (Thermo Fisher Scientific). Reads were aligned with the hg19 human reference genome, and potential mutations and copy number alterations were called with the use of Ion Reporter software version 5.2. Raw variant calls were filtered with a quality score of less than 100 and were manually checked with the use of the Integrative Genomics Viewer (Broad Institute). Somatic alterations reported in the Catalogue of Somatic Mutations in Cancer database were collected. Germline mutations were excluded with the use of the Human Genetic Variation database (http://www.genome.med.kyoto‐u.ac.jp/SnpDB) and Exome Aggregation Consortium database (http://exac.broadinstitute.org).
3. RESULTS
3.1. Patient characteristics
Tumor samples were obtained from 17 patients who underwent surgical resection of NSCLC associated with pathologically diagnosed IPF. The clinical characteristics of the patients are shown in Table 1. The HRCT of the lung showed the usual interstitial pneumonia (UIP) pattern in 16 (94%) patients and a probable UIP pattern in 1 (6%) patient. The median age was 73 years (range, 62‐78 years). Fourteen (82%) patients were male. Tumor histology was adenocarcinoma in 9 (53%) patients and squamous cell carcinoma in 8 (47%) patients. All patients had a smoking history, with 14 (82%) being former and 3 (18%) current smokers.
TABLE 1.
Characteristics of patients with idiopathic pulmonary fibrosis (n = 17)
| Characteristic | No. of patients (%) |
|---|---|
| Median age (range), y | 73 (62‐78) |
| Sex | |
| Male | 14 (82) |
| Female | 3 (18) |
| Clinical stage | |
| I | 8 (47) |
| II | 6 (35) |
| III | 2 (12) |
| IV | 1 (6) |
| Histology | |
| Adenocarcinoma | 9 (53) |
| Squamous cell carcinoma | 8 (47) |
| Smoking status | |
| Never | 0 (0) |
| Previous | 14 (82) |
| Current | 3 (18) |
| HRCT pattern | |
| UIP | 16 (94) |
| Probable UIP | 1 (6) |
3.2. Somatic alterations in NSCLC
Analysis of tumor samples from the 17 patients with IPF‐associated lung cancer uncovered somatic alterations in a total of 97 genes (including repeat counts of those affected in more than 1 patient), with the median number of affected genes per patient being 4 (range, 1‐24) (Table S2). Such alterations were frequently detected in TP53 (n = 6, 35.3%) and PIK3CA (n = 5, 29.4%) genes (Figure 1). Although 1 or more somatic alterations were identified in BRAF, RET, EGFR, ALK, ROS1, and NTRK1 (Table S2), these changes did not include those—such as the Val600Glu (V600E) mutation of BRAF, activating mutations of EGFR, and fusions of RET, ALK, or ROS1—that confer sensitivity to molecularly targeted drugs (Table S3). Non‐V600E mutations of BRAF and mutations of ARAF, KRAS, NRAS, or HRAS were detected in 4, 1, 2, 1, and 2 patients, respectively, with alterations that affect the RAS‐RAF signaling pathway being identified in 7 (41.2%) of 17 tumor samples. No gene fusions were identified in tumor samples.
FIGURE 1.
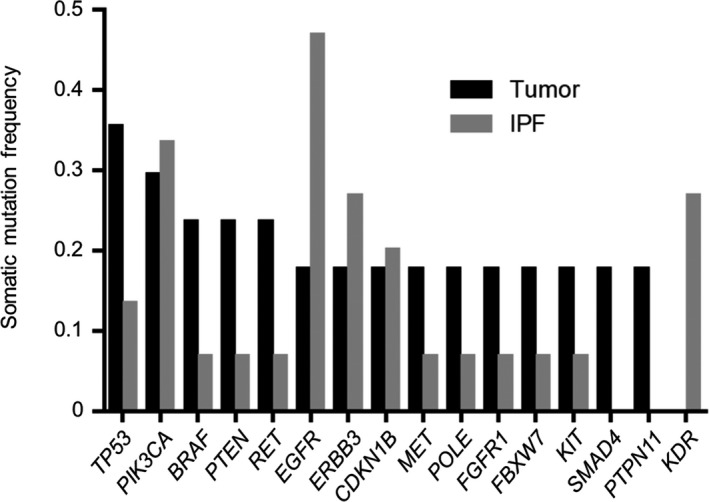
Frequently detected genes with somatic alterations in tumor or idiopathic pulmonary fibrosis (IPF) tissue specimens of patients with IPF‐associated lung cancer. Those genes with a somatic alteration frequency of 0.15 or higher, as detected by next‐generation sequencing, are shown
3.3. Somatic alterations in fibrosing lung tissue
Fibrosing lung tissue located adjacent to tumor tissue and resected at the time of surgery was available for 15 of the 17 patients enrolled in the study. All such tissue samples were pathologically confirmed as positive for fibrosis with the UIP pattern and to contain no tumor cells. Next‐generation sequencing analysis of these 15 IPF samples detected somatic alterations in 69 genes (including repeat counts of those affected in more than 1 patient), with the median number of genes in which somatic alterations were identified per sample being 2 (range, 0‐16) (Table S2). Such alterations frequently occurred in EGFR (n = 7, 46.7%), PIK3CA (n = 5, 33.3%), ERBB3 (n = 4, 26.7%), and KDR (n = 4, 26.7%) (Figure 1). No somatic alterations—such as the V600E mutation of BRAF or activating mutations of EGFR—that confer sensitivity to molecularly targeted drugs for NSCLC and no gene fusions were identified in the IPF samples (Table S3). Somatic alterations that affect the RAS‐RAF pathway were identified in 3 (20.0%) of the 15 IPF samples (a non‐V600E mutation of BRAF as well as mutations in KRAS and ARAF in 1 patient each).
3.4. Overlap of somatic alterations in paired samples
In the case of the 15 patients for whom paired tumor and fibrosing lung tissue samples were available, 90 and 81 somatic alterations were detected in 81 and 69 genes in tumor and IPF samples, respectively, with a total of 19 genes overlapping in 8 of the sample pairs (Figures 2 and 3). Among these 19 genes, only 6 somatic alterations (CDKN1B amplification in 2; Val1578del of NOTCH1, Ala232Val of ERBB3, Arg465Cys of FBXW7, and Arg295Trp of AXL in 1 each) overlapped in 4 paired samples. Most somatic alterations, such as those affecting TP53, PIK3CA, and RET, were thus not shared by corresponding tumor and IPF tissue (Tables S2 and S3).
FIGURE 2.
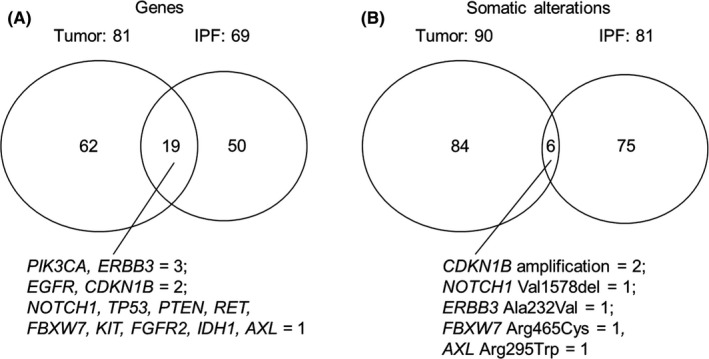
Overlap in genes with somatic alterations (A) or in somatic alterations themselves (B) between tumor and idiopathic pulmonary fibrosis (IPF) tissue in paired samples from patients with IPF‐associated lung cancer
FIGURE 3.
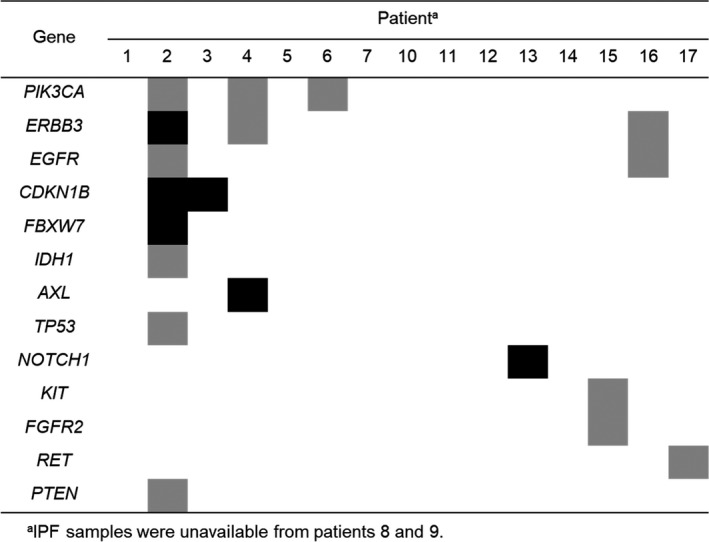
Overlapping genetic alterations in tumor and idiopathic pulmonary fibrosis (IPF) samples from the 15 patients for whom paired samples were available. The presence of any or the same genetic alterations of each gene in both tumor and IPF samples is indicated by gray or black shading, respectively
4. DISCUSSION
With the use of NGS with a panel that targets 161 cancer‐related genes, we have here investigated somatic alterations in both tumor and adjacent fibrosing lung tissue from patients with IPF‐associated lung cancer. Somatic alterations were identified in 97 genes in 17 tumor samples. Although somatic alterations—such as activating mutations of EGFR, rearrangement of ALK, or the V600E mutation of BRAF—that confer sensitivity to molecularly targeted drugs for NSCLC were not identified, non‐V600E mutations of BRAF were frequently detected in IPF‐associated tumor samples (4 of 17, 23.5%), consistent with previous results. 20 , 21 All of these 4 mutations of BRAF were located in the kinase domain (Thr599Ile in 2; Gly464Arg and Gly466Glu in 1 each). The Thr599Ile and Gly464Arg mutants of BRAF were found to have increased kinase activity compared with the WT protein. 22 , 23 Although the kinase activity of the Gly466Glu mutant of BRAF is impaired, this mutant increases signaling by the downstream kinase ERK as a result of an increased affinity for the GTP‐bound form of RAS compared with WT BRAF. 24 This type of BRAF mutant has been found to coexist with RAS mutants. 24 Consistent with this previous observation, we identified a tumor with both the Gly466Glu mutation of BRAF and a mutation in HRAS. Somatic alterations that affect the RAS‐RAF pathway were identified in 7 (41.2%) of the 17 tumor samples in the present study, suggesting that this pathway plays an important role in tumorigenesis in patients with IPF.
Although genomic profiling of IPF‐associated lung cancer by NGS has been undertaken in previous studies, 20 , 25 that of IPF tissue in patients with IPF‐associated tumors has not. In the present study, somatic alterations in cancer‐related genes were identified almost as frequently in IPF samples (n = 15) as in paired tumor samples (81 vs 90 alterations), suggestive of susceptibility to somatic mutation in the IPF component of individuals with IPF‐associated lung cancer and suggesting that such susceptibility might contribute to the development of lung cancer.
Among the 81 somatic alterations identified in IPF samples, 6 changes overlapped with those detected in paired tumor samples. These 6 alterations occurred in 5 genes that encode receptor tyrosine kinases (ERBB3 and AXL), a cell cycle regulator (CDKN1B), an activator of oncogene expression (NOTCH1), and a protein that contributes to the degradation of oncoproteins (FBXW7), all of which regulate cell differentiation, growth, proliferation, or survival. 26 , 27 , 28 , 29 , 30 CDKN1B amplification was apparent in 2 of the sample pairs. Although CDKN1B encodes a cyclin‐dependent kinase inhibitor (p27Kip1), which plays a role in tumor suppression, its amplification is thought to contribute to oncogenesis by promoting tumor cell migration, invasion, and metastasis. 31 , 32 CDKN1B amplification might therefore also contribute to the pathogenesis of IPF‐associated lung cancer.
Although 6 overlapping somatic alterations were apparent in 4 of the 15 paired tumor and IPF samples, the remaining sample pairs did not show overlap in somatic alterations. TP53 mutations were frequently detected in both tumor and IPF samples in the present study, consistent with previous findings. 19 , 33 , 34 Although all of the TP53 mutations were located in the DNA binding domain of the encoded protein, which recognizes a consensus sequence in the promoters of several genes related to DNA repair and apoptosis, 35 , 36 they were diverse and did not overlap between tumor and IPF tissue. Somatic alterations in most of the genes, including TP53, PIK3CA, and RET, identified in both tumor and IPF samples thus differed between these 2 types of sample. These results suggest that most cases of IPF‐associated lung cancer do not develop through the stepwise accumulation of somatic alterations in fibrosing lung tissue.
There are several limitations to the present study. First, the sample size was relatively small. Second, we analyzed only surgical samples obtained from patients with early‐stage lung cancer. There is thus a possibility that our findings might not be applicable to advanced lung cancer. However, surgical or transbronchial biopsy of lung tissue is not recommended for diagnosis of IPF when HRCT of the lung demonstrates the UIP pattern. 1 It is therefore difficult to obtain paired biopsy samples of IPF‐associated lung cancer and IPF, which limited us to the use of only surgically resected specimens containing both lung cancer and fibrosing lung tissue. Third, NGS analysis of IPF samples was carried out for IPF tissue in only 1 location for each patient. We thus cannot exclude the possibility that somatic alterations were shared between tumor samples and IPF tissue in locations other than those examined in the present study. Finally, the gene panel adopted for the present study targets only cancer‐related genes. Analysis of other genes, such as those related to the pulmonary surfactant system, in paired IPF‐associated tumor and fibrosing lung tissue might also provide insight into the etiology of IPF‐associated lung cancer. 25
In conclusion, as far as we are aware, our study is the first to investigate somatic genetic alterations in paired tumor and tumor‐adjacent fibrosing lung tissue specimens by targeted NGS. Although somatic alterations were frequently detected in several genes in both sample types, such alterations were not frequently shared within the sample pairs. Further comprehensive NGS analysis of paired tumor and IPF samples is warranted to provide further insight into the etiology of IPF‐associated lung cancer and to provide a basis for the development of targeted drugs for such tumors as well as for IPF.
DISCLOSURE
The authors declare that they have no conflicts of interest.
Supporting information
Table S1‐S3
ACKNOWLEDGMENTS
No funding was received for this study.
Otsubo K, Iwama E, Ijichi K, et al. Paired genetic analysis by next‐generation sequencing of lung cancer and associated idiopathic pulmonary fibrosis. Cancer Sci. 2020;111:2482–2487. 10.1111/cas.14488
Kohei Otsubo and Eiji Iwama contributed equally to this study.
REFERENCES
- 1. Raghu G, Remy‐Jardin M, Myers JL, et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198:e44‐e68. [DOI] [PubMed] [Google Scholar]
- 2. Haddad R, Massaro D. Idiopathic diffuse interstitial pulmonary fibrosis (fibrosing alveolitis), atypical epithelial proliferation and lung cancer. Am J Med. 1968;45:211‐219. [DOI] [PubMed] [Google Scholar]
- 3. Fraire AE, Greenberg SD. Carcinoma and diffuse interstitial fibrosis of lung. Cancer. 1973;31:1078‐1086. [DOI] [PubMed] [Google Scholar]
- 4. Turner‐Warwick M, Lebowitz M, Burrows B, Johnson A. Cryptogenic fibrosing alveolitis and lung cancer. Thorax. 1980;35:496‐499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kawai T, Yakumaru K, Suzuki M, Kageyama K. Diffuse interstitial pulmonary fibrosis and lung cancer. Acta Pathol Jpn. 1987;37:11‐19. [DOI] [PubMed] [Google Scholar]
- 6. Nagai A, Chiyotani A, Nakadate T, Konno K. Lung cancer in patients with idiopathic pulmonary fibrosis. Tohoku J Exp Med. 1992;167:231‐237. [DOI] [PubMed] [Google Scholar]
- 7. Tomassetti S, Gurioli C, Ryu JH, et al. The impact of lung cancer on survival of idiopathic pulmonary fibrosis. Chest. 2015;147:157‐164. [DOI] [PubMed] [Google Scholar]
- 8. Tzouvelekis A, Spagnolo P, Bonella F, et al. Patients with IPF and lung cancer: diagnosis and management. Lancet Respir Med. 2018;6:86‐88. [DOI] [PubMed] [Google Scholar]
- 9. Kudoh S, Kato H, Nishiwaki Y, et al. Interstitial lung disease in Japanese patients with lung cancer: a cohort and nested case‐control study. Am J Respir Crit Care Med. 2008;177:1348‐1357. [DOI] [PubMed] [Google Scholar]
- 10. Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non‐small‐cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129‐2139. [DOI] [PubMed] [Google Scholar]
- 11. Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin‐paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947‐957. [DOI] [PubMed] [Google Scholar]
- 12. Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK‐positive lung cancer. N Engl J Med. 2013;368:2385‐2394. [DOI] [PubMed] [Google Scholar]
- 13. Solomon BJ, Mok T, Kim DW, et al. First‐line crizotinib versus chemotherapy in ALK‐positive lung cancer. N Engl J Med. 2014;371:2167‐2177. [DOI] [PubMed] [Google Scholar]
- 14. Shaw AT, Ou SH, Bang YJ, et al. Crizotinib in ROS1‐rearranged non‐small‐cell lung cancer. N Engl J Med. 2014;371:1963‐1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Planchard D, Besse B, Groen HJM, et al. Dabrafenib plus trametinib in patients with previously treated BRAF(V600E)‐mutant metastatic non‐small cell lung cancer: an open‐label, multicentre phase 2 trial. Lancet Oncol. 2016;17:984‐993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yoh K, Seto T, Satouchi M, et al. Vandetanib in patients with previously treated RET‐rearranged advanced non‐small‐cell lung cancer (LURET): an open‐label, multicentre phase 2 trial. Lancet Respir Med. 2017;5:42‐50. [DOI] [PubMed] [Google Scholar]
- 17. Ou SH, Kwak EL, Siwak‐Tapp C, et al. Activity of crizotinib (PF02341066), a dual mesenchymal‐epithelial transition (MET) and anaplastic lymphoma kinase (ALK) inhibitor, in a non‐small cell lung cancer patient with de novo MET amplification. J Thorac Oncol. 2011;6:942‐946. [DOI] [PubMed] [Google Scholar]
- 18. Paik PK, Drilon A, Fan PD, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015;5:842‐849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Takahashi T, Munakata M, Ohtsuka Y, et al. Expression and alteration of ras and p53 proteins in patients with lung carcinoma accompanied by idiopathic pulmonary fibrosis. Cancer. 2002;95:624‐633. [DOI] [PubMed] [Google Scholar]
- 20. Hwang JA, Kim D, Chun SM, et al. Genomic profiles of lung cancer associated with idiopathic pulmonary fibrosis. J Pathol. 2018;244:25‐35. [DOI] [PubMed] [Google Scholar]
- 21. Guyard A, Danel C, Theou‐Anton N, et al. Morphologic and molecular study of lung cancers associated with idiopathic pulmonary fibrosis and other pulmonary fibroses. Respir Res. 2017;18:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wan PT, Garnett MJ, Roe SM, et al. Mechanism of activation of the RAF‐ERK signaling pathway by oncogenic mutations of B‐RAF. Cell. 2004;116:855‐867. [DOI] [PubMed] [Google Scholar]
- 23. Houben R, Becker JC, Kappel A, et al. Constitutive activation of the Ras‐Raf signaling pathway in metastatic melanoma is associated with poor prognosis. J Carcinog. 2004;3:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yao Z, Yaeger R, Rodrik‐Outmezguine VS, et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature. 2017;548:234‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Honda T, Hiroyuki S, Msai K, et al. Deleterious pulmonary surfactant system gene mutations in lung adenocarcinomas associated with usual interstitial pneumonia. JCO Precision Oncology. 2018;2:1‐24. [DOI] [PubMed] [Google Scholar]
- 26. Soltoff SP, Carraway KL 3rd, Prigent SA, Gullick WG, Cantley LC. ErbB3 is involved in activation of phosphatidylinositol 3‐kinase by epidermal growth factor. Mol Cell Biol. 1994;14:3550‐3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ben‐Batalla I, Schultze A, Wroblewski M, et al. Axl, a prognostic and therapeutic target in acute myeloid leukemia mediates paracrine crosstalk of leukemia cells with bone marrow stroma. Blood. 2013;122:2443‐2452. [DOI] [PubMed] [Google Scholar]
- 28. Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. 2008;8:253‐267. [DOI] [PubMed] [Google Scholar]
- 29. Bray SJ. Notch signalling in context. Nat Rev Mol Cell Biol. 2016;17:722‐735. [DOI] [PubMed] [Google Scholar]
- 30. Mao JH, Kim IJ, Wu D, et al. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science. 2008;321:1499‐1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhao D, Besser AH, Wander SA, et al. Cytoplasmic p27 promotes epithelial‐mesenchymal transition and tumor metastasis via STAT3‐mediated Twist1 upregulation. Oncogene. 2015;34:5447‐5459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Minarikova P, Benesova L, Halkova T, et al. Prognostic importance of cell cycle regulators cyclin D1 (CCND1) and cyclin‐dependent kinase inhibitor 1B (CDKN1B/p27) in sporadic gastric cancers. Gastroenterol Res Pract. 2016;2016:9408190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sozzi G, Miozzo M, Donghi R, et al. Deletions of 17p and p53 mutations in preneoplastic lesions of the lung. Cancer Res. 1992;52:6079‐6082. [PubMed] [Google Scholar]
- 34. Kawasaki H, Ogura T, Yokose T, Nagai K, Nishiwaki Y, Esumi H. p53 gene alteration in atypical epithelial lesions and carcinoma in patients with idiopathic pulmonary fibrosis. Hum Pathol. 2001;32:1043‐1049. [DOI] [PubMed] [Google Scholar]
- 35. Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323‐331. [DOI] [PubMed] [Google Scholar]
- 36. Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53‐regulated genes. Nat Rev Mol Cell Biol. 2008;9:402‐412. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1‐S3