

Allotetraploid cyprinids have a unique strategy for balancing subgenomic stabilization and diversification in rediploidization.
Abstract
A persistent enigma is the rarity of polyploidy in animals, compared to its prevalence in plants. Although animal polyploids are thought to experience deleterious genomic chaos during initial polyploidization and subsequent rediploidization processes, this hypothesis has not been tested. We provide an improved reference-quality de novo genome for allotetraploid goldfish whose origin dates to ~15 million years ago. Comprehensive analyses identify changes in subgenomic evolution from asymmetrical oscillation in goldfish and common carp to diverse stabilization and balanced gene expression during continuous rediploidization. The homoeologs are coexpressed in most pathways, and their expression dominance shifts temporally during embryogenesis. Homoeolog expression correlates negatively with alternation of DNA methylation. The results show that allotetraploid cyprinids have a unique strategy for balancing subgenomic stabilization and diversification. Rediploidization process in these fishes provides intriguing insights into genome evolution and function in allopolyploid vertebrates.
INTRODUCTION
Whole-genome duplication (WGD) or polyploidy provides genomic opportunities for evolutionary innovations and adaptation (1–4). Polyploidy is rare in animals, possibly because of barriers to sex determination, and physiological and developmental constraints, especially nuclear-cytoplasmic interactions and related factors (5, 6). Further, polyploid animals appear to be incapable of coping with genomic and developmental chaos resulting from the merging of two genomes because of changes in structural variation, regulatory imbalance, gene expression bias, and activation of transposable elements (TEs), as documented in many other allopolyploids (1, 3, 7–10). A newly formed allopolyploid line of fishes (11) experienced more severe chaotic changes than polyploid plants (7, 8, 10, 12, 13). These rapid and dynamic changes have genetic and epigenetic bases (7, 14). Biased subgenomic changes may help alleviate chaos from genome mergers (15), and subsequent coordination may help stabilize the subgenomic functions in newly synthesized allotetraploid plants (7, 12, 16) after the initial “genome shock” (17) between divergent subgenomes that coexist in the same cell nucleus. However, the long-term consequences for polyploid animals in rediploidization remain elusive.
Goldfish (Carassius auratus red var.) belongs to family Cyprinidae in the most specious order of fishes (Cypriniformes), which contains many polyploid species (18–20). The tetraploid carp was domesticated hundreds of years ago in China and Europe, and it is the economically most important fish in freshwater aquaculture (21). Goldfish (C. auratus red var.) is the most commonly kept pet globally, and it constitutes a model system for studying neurobiology and physiology. This allotetraploid (2n = 4x = 100) species was formed by the interspecific hybridization of two diploids (2n = 2x = 50); subsequent chromosome-doubling restored meiotic paring and disomic inheritance (18, 19). Goldfish has nearly twice as many chromosomes as zebrafish and most of other cyprinids (22, 23). Its numerous small chromosomes pose a great challenge to assembling and annotating both subgenomes at the chromosomal level. Because no extant diploid progenitors are available for study, the evolution of this complex polyploid genome remains poorly understood (21, 24, 25), despite the recently published draft genome (26).
Here, we report a reference-quality, chromosome-scale assembly of goldfish including identification of both subgenomes and analysis of the variation and expression changes between them. We also evaluate the subgenomic evolution of goldfish and common carp. Our results indicate that allotetraploid goldfish and common carp have diverse strategies for balancing dynamic subgenomic diversification during continuous rediploidization. The diverse and continuous evolutionary processes broaden our understanding of the evolution and function of genomes in allopolyploid vertebrates and may explain why most polyploid animals fail to survive.
RESULTS
Sequencing, assembly, and annotation
Allopolyploid genomes are much more complicated than diploid ones due to their polynomic inheritance and gradual random decay of progenitors’ genomes during rediploidization (2, 3). A high-quality genome assembly is required to discriminate changes from related species in allopolyploids. Our high-quality genome of a gynogenetic goldfish (C. auratus red var., n = 100) was assembled using data from a combination of three technologies (Fig. 1), including 325.34 gigabases (Gb; 203×) of Illumina sequence data (Illumina GAII, HiSeq 2000), 128.51 Gb (80×) of single-molecule real-time (SMRT) long reads (PacBio RS II and Sequel), and 231.50 Gb of clean BioNano mapping data (Bionano Genomics Irys). The final assembly consisted of 5477 scaffolds, with a scaffold N50 of 2.94 megabases (Mb) after gap filling (Table 1 and data S1_1–4), which resulted in a genome size of 1.64 Gb. This was similar to the size estimated by flow cytometry (1.71 Gb; Fig. 1) and slightly higher than the k-mer analysis (1.43 Gb; Fig. 1). In addition, using 307.46 Gb of data generated by high-throughput chromosome conformation capture sequencing (Hi-C seq) technology (Annoroad Gene Technology Co. Ltd., Beijing; data S1_5, 6), 1.59-Gb (96.95%) genome-level sequences were aligned and ordered into 50 scaffolds that potentially matched the chromosomes (Table 1, fig. S1A, and data S2_1). A genetic map consisting of 50 linkage groups was constructed in 79 F2 offspring using 7466 single-nucleotide polymorphism markers developed from 147.10 Gb of RNA sequencing (RNA-seq) data (27). Next, the genome scaffolds (1.07 Gb, 65.24%) were anchored on the genetic map (data S2_2). Sequence assemblies on the pseudochromosomes matched perfectly to the anchored 50 linkage groups, confirming the high-quality assembly of the allotetraploid goldfish genome (fig. S1, B and C, and data S2_3–5). BUSCO and CEGMA showed the assemblies to be complete (data S2_6). Our assembly conformed to Vertebrate Genome Project standards for reference genomes (https://vertebrategenomesproject.org/) and covered more sequences than the published goldfish genome by Chen et al. (26), which consisted of 1.24 Gb with an anchor ratio of ~66% from 1.82 Gb contigs (Table 1). Subsequently, we compared the completeness and collinearity between our assembly and the published genome using MUMmer (v3.23), based on the minimum clustering unit of 50 kilobases (kb), which is the smallest unit that we could check, and consistent with 50-kb resolution of Hi-C clustering. Within the total length of 1.06-Gb collinearities from 2380 clustering groups, we found general consistency, while some structural differences existed between the two. In our study, among 277 inversions and 1338 translocations identified, 63.95% were validated by PacBio long reads, optical map, and/or Hi-C data (Fig. 2A, fig.S1D, and data S2_7, 8). These efforts obtained a general improvement of quality scores in our genome assembly relative to the published data. For example, the optical map and Hi-C analyses obtained improvement at an inversion boundary (Fig. 2A).
Fig. 1. Workflow of genome assembly and subgenome identification.
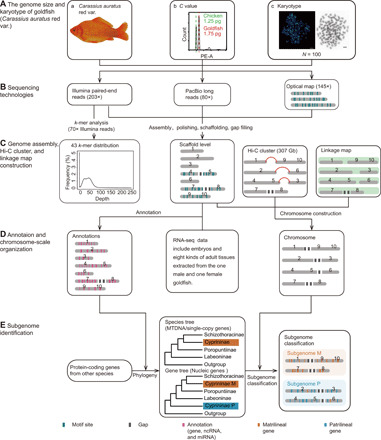
(A) Genome size and karyotype of goldfish. (a) Image of a gynogenetic goldfish (C. auratus var.). Photo credit: Shaojun Liu, Hunan Normal University, China. (b) Diagram of C value. The X axis presents the fluorescence index, and the Y axis presents the frequency of cells. Sample/calibration ratio equals the peak X value of the calibration sample divided by X value at the peak of the target sample. The first sharp peak with green dashed line displays the X axis and cell frequency of chicken blood, and the second one with red dashed line represents the X axis and cell frequencies of goldfish. C value of sample is sample/calibration ratio × calibration sample’s C value. (c) Goldfish have 100 chromosomes and 100 signals after the chromosomes are stained with DNA probe (probe A) [9468-bp fragment of 36 copies of a repetitive 263-bp fragment; adopted from Liu et al. (11)]. (B) Sequencing technologies for primary assembly. (C) Genome assembly, Hi-C cluster, and genetic map construction. Genome size assessment by k-mer analysis is performed by 40× Illumina paired-end reads after the primary assembly. Next, scaffolds are clustered into 50 pseudochromosomes by using Hi-C data obtained by chromosomes; the genetic map was constructed by using the data of Kuang et al. (27) (D) Annotation and chromosome-scale organization. Annotation of scaffolds was performed using a combination of ab initio prediction, transcript evidence gathered from RNA-seq of embryos and eight kinds of adult tissues (gonads, brain, liver, spleen, kidney, eye, epithelium, and fin), and homologous genes information from five fish genomes, by using EVidence Modeler (EVM). Final set of 50 pseudochromosomes was generated after pairwise validation among Hi-C clustering results, genetic map, and collinearity analyses. (E) Subgenome identification. After extracting the homologous genes of goldfish and other species, the species tree is constructed by using single-copy genes from 10 genomes. Gene trees were constructed by defining homologous gene clusters using whole-genome sequences/transcripts from 10 cyprinid species of Cyprininae (C. auratus, Cyprinus carpio), Labeoninae (Labeo rohita), Poropuntiinae (Poropuntius huangchuchieni), Schizothoracinae (Schizothorax oconnori, Schizothorax waltoni, Schizothorax macropogon, and Schizothorax kozlovi), Danio rerio, and Ctenopharyngodon idellus. After comparing the species tree and nucleic gene trees, the matrilineal (clustered with Schizothorax) and patrilineal markers from the gene trees were labeled back to 25 pairs of pseudochromosomes. The origin of pseudo-chromosomes was identified by most of the supported markers.
Table 1. The statistics of assembly from three versions of goldfish in this paper, goldfish by Chen et al. (26), and common carp by Xu et al. (29).
NGS, next-generation sequencing; LG, linkage group.
| V. 1 | V. 2 | V. 3 | Goldfish Chen et al. 2019 |
Hebao red carp Xu et al. 2019 |
|
| NGS | PacBio + optical map |
PacBio + optical map + Hi-C |
PacBio |
Whole-genome shotgun |
|
| Total contig size (Gb) | 1.22 | 1.54 | 1.49 | 1.85 | 1.41 |
| Contig no. | 636,670 | 6,144 | 4,433 | 8,463 | 355,804 |
| Longest contig (bp) | 74,023 | 7,661,673 | 7,650,526 | – | 207,110 |
| Contig N50 (Mb) | 0.004 | 1.11 | 1.16 | 0.82 | 0.02 |
| Contig L50 | – | 375 | 353 | 513 | 19,142 |
| Total scaffold size (Gb) | 1.46 | 1.64 | 1.59 | 1.82 | 1.46 |
| Scaffold/chr no. | 358,782 | 5,477 | 50 | 6,216 | 262,449 |
| Longest scaffold (kb) | 5,762 | 11,070 | 65,905 | – | 6,571 |
| Scaffold N50 (Mb) | 0.46 | 2.94 | 34.79 | – | 0.92 |
| Scaffold L50 | – | 160 | 18 | – | 393 |
| Scaffold N90 (kb) | 1.24 | 96.06 | 24,821.24 | – | 22.24 |
| Scaffold L90 | – | 1,452 | 40 | – | 3,290 |
| Anchor ratio on chr/LG | – | – | 97%/65% | 66% | 82% |
| Gap length on chr/LG (Mb) | – | – | 90 | 0.22 | 43 |
Fig. 2. Syntenic blocks, phylogeny, subgenome identification, and allotetraploidy.

(A) Diagrams displaying the rearrangement identified in the syntenic blocks between our assembly and the goldfish genome published by Chen et al. (26). The syntenic block between two assemblies was identified with an inversion event (a), in which the rearranged boundary on our genome was continuously covered by the long reads of the optical map (b), and supported by the strong clustering signal from the heat map (c) constructed by high-depth sequencing data from chromosome conformation capture technology. (B) Allotetraploid goldfish and common carp genome synteny. Blocks represent the genomic overview of assembled chromosomes of subgenome M and subgenome P from goldfish and subgenome B and subgenome A from common carp (Hebao red carp) (29). Colored lines indicate the orthologous sites of gene blocks and their colinear relationships between subgenomes M and B and subgenomes P and A, respectively. Numbers M01 to M25, P01 to P25, B01 to B25, and A01 to A25 indicate the homologous chromosomes of M and P subgenomes from goldfish, and B and A subgenomes from common carp, respectively, numbering in order according to the collinearity relationships to zebrafish genome (fig. S3). The numbers beside M and P chromosomes indicate the supporting rate from homoeologous gene makers with clear origin of parental ancestor determined by the gene trees. (C) Phylogenetic relationships and timing of WGD/polyploidization events in Cyprininae, with nodes based on protein-coding genes of goldfish, common carp, golden-line barbel, grass carp, and zebrafish. Dated divergence time of grass carp and the ancestor of Cyprininae was 20.9 Ma ago, and the putative matrilineal and patrilineal progenitors were 15.1 Ma ago (T1), after the WGD event (T2). Divergence of the polyploid Cyprininae radiation was dated at 13.8 Ma ago (T3), and the divergence between goldfish and common carp was dated at 10.0 Ma ago (T4). Orange and blue branches indicate the putative M and P progenitors, respectively. Photo credit: Goldfish and common carp by Shaojun Liu, Hunan Normal University, China; golden-line barbel and grass carp from FishBase; zebrafish from Wikimedia. (D) Phylogenetic tree based on protein-coding genes from single-copy orthologs, rooted with human and chicken. Alignments were performed by MUSCLE, and the maximum likelihood tree was reconstructed by PhyML.
The annotation predicted 43,144 genes with an average length of 17,025 base pairs (bp) and 9.78 exons per gene (data S1_7 and data S2_9). Of the genes, 39,205 (90.87%) were functionally annotated (data S2_9). Accuracy and completeness of the annotation were validated further through 97.78% coverage of annotated genes by RNA-seq data. The annotation included 6788 transfer RNA (tRNA), 1380 ribosomal RNA (rRNA), 1324 small nuclear RNA (snRNA), and 3385 microRNA genes (data S2_10). Repeat annotation indicated an overall repeat content of 39.49% (data S2_11), which was less than the 52.2% of zebrafish (28) and comparable to ~40% of African clawed frog (9). The most abundant TEs in the goldfish genome were type II, which represented 21.19% of the whole-genome sequence. Other annotated TEs included 174 superfamilies.
Characterization of subgenomes
To identify the goldfish’s subgenomes from two progenitors, we used phylogenetic information of the Cyprininae (18, 19). Several species in the genera Carassius (including goldfish) and Cyprinus were reported to have undergone the same allopolyploidization approximately 10 to 12 million years (Ma) ago (19). The matrilineal copy of several nuclear genes was grouped with genus Schizothorax (18, 19), and they diverged from the patrilineal ones 17 to 19 Ma ago (18, 19). By comparing phylogenies between the mitogenomes and homoeologous gene pairs of whole-genome sequences/transcripts from 10 cyprinid species, analyses confirmed that Schizothorax was monophyletic; it shared a matrilineal ancestor with all allotetraploid species of Cyprininae (Fig. 1E and data S1_8). The analysis identified 1274 homoeologous gene pairs of goldfish. Mapping these homoeologous gene pairs onto the 50 pseudochromosomes identified matrilineal (M) and patrilineal (P) subgenomes, with an average support rate of 95.34% (Fig. 2B and table S1). To clarify the relationship between each subgenome pair of goldfish and common carp (Hebao red carp) with subgenomes of A and B of Xu et al. (29), we first constructed the collinearity relationship between the two species. Analyses using MCScanX found that 9266 orthologous gene pairs had unambiguous one-to-one relationships between subgenome M (from goldfish) and subgenome B (from common carp), and 6991 orthologous gene pairs had clear one-to-one relationships between subgenome P (from goldfish) and subgenome A (from common carp; Fig. 2B). The results showed a consistency of 74.58% between subgenomes M and B and 66.97% between the subgenomes P and A. This suggests that subgenomes M and B are from the matrilineal genome of the Cyprininae ancestor, while subgenomes P and A are patrilineal ones (Fig. 2B and data S2_12, 13).
The ancient WGD within Cyprininae was identified via phylogenetic analyses of genome-wide markers, which integrated subgenomes M and P. Analyses compared both subgenomes of goldfish, common carp, and golden-line barbel (Sinocyclocheilus grahami) to zebrafish and grass carp. A MCMCTree analysis in the Phylogenetic Analysis by Maximum Likelihood (PAML) package (v4.8) (30) with four calibration points suggested that the progenitor-like genomes diverged approximately 15.09 Ma ago (T1; Fig. 2C). Following allopolyploidization (T2), S. grahami originated about ~11 Ma ago (T3), followed by goldfish and common carp at ~9 Ma ago (T4). These dates were more recent than those (13.75 and 9.95 Ma ago, respectively) estimated by using 568 single-copy genes (Fig. 2, C and D, and fig. S1E). The new estimate (13.75 to 15.09 Ma) of timing for the Cyprininae WGD (T2; Fig. 2C) was earlier than those (10 to 12 Ma ago) based on gene markers (19). The expansion and contraction of gene families were estimated to infer the evolutionary history after the Cyprininae-specific WGD event relative to the teleost-specific WGD event in zebrafish and grass carp (fig. S1, F and G, and data S2_14). Within 4453 expanded gene families, there were 313 transcription factors, indicating a significant abundance (P < 0.01, Fisher’s exact test). Most of them were known to be involved in embryonic development, especially organogenesis during differentiation of germinal layers (fig. S1H).
Evolution of subgenomes in Cyprininae
Evolution of subgenomes has been reported in allopolyploid angiosperms (8, 10) and vertebrates (9). They typically showed conservation of one progenitor-like genome and diversification of the other by large structural variation in collinearities compared to the ancestral genome. In contrast, comparison of allotetraploid goldfish annotated genes, repeats, and noncoding RNAs (ncRNAs) showed approximately equal representations between the two subgenomes. Approximately 1.5 Gb of chromosome-scale sequences were partitioned into subgenomes M and P, which was consistent with the number of genes in M (20,913, 52.07%) and P (19,248, 47.83%). The proportions of repeat sequences and ncRNAs were also similar between the two subgenomes (data S3_1). Thus, the two subgenomes had similar gene densities and distributions in most chromosomes, except for significantly higher densities in three M (chr1, chr20, and chr43) and one P (chr27) chromosomes (P < 0.05, two-tailed paired t test; data S3_2). The two subgenomes also contributed similar proportions of all TE families with annotation against public database (fig. S2, A and B, and data S3_3). Further, the comparison between two subgenomes of common carp in genomic contents showed nearly equal representations between subgenomes B and A (29).
To further investigate the evolution of two subgenomes after allopolyploidization in carp-like fishes, we compared subgenomes M and P of goldfish and subgenomes B and A of common carp with zebrafish to define the changes in synteny and genomic divergence. To integrate the collinearities between goldfish and common carp, we aligned 43,144 high-confidence gene models to 50 goldfish and the 25 zebrafish chromosomes. The results indicated that 12,450 genes of subgenome M and 11,042 genes of subgenome P were located on syntenic blocks (P = 1.09 × 10−5), and 7568 orthologous gene pairs had a clear two-to-one relationships to zebrafish (Fig. 3A and fig. S3). Collinearities between homologous goldfish M and common carp B chromosomes identified 15.12% inversions and 10.30% translocations, and the ones between goldfish P and common carp A chromosomes showed 22.29% inversions and 10.74% translocations, which indicated more rearrangement in the patrilineal subgenomes (data S2_12). We also validated the boundaries of all rearranged regions identified by both collinearities against goldfish from Chen et al. (26) and common carp; 69.01% of regions on subgenome M and 67.64% on subgenome P were assembled continuously in our genome by sequencing data (data S3_4). With respect to GC (guanine-cytosine) content and repeat densities, we used a sliding window of 50 kb for comparative analysis. We found that the patrilineal subgenomes P and A had greater GC content than matrilineal subgenomes M and B (Fig. 3B, a, and data S3_5), indicating subgenome-specific variation in these two species. However, the distributions of repeat densities yielded an opposite pattern; more repeats occurred in subgenome P than M in the goldfish, and in the common carp, more repeats were in B than in A. These differences may have owed to species divergence (Fig. 3B, b, and data S3_5). The distributions of GC content and repeat density might contribute to different kinds of divergence (data S3_5). In addition, we obtained 28 and 16 specific blocks with significantly biased repeat densities between subgenomes in goldfish and common carp, respectively, and labeled those potential sources of divergence between the two species on each chromosome (P < 0.05, Wilcoxon rank sum test; Fig. 3A, fig. S3, and data S3_6).
Fig. 3. Syntenic analysis and symmetrical evolution of the allopolyploid goldfish genome.
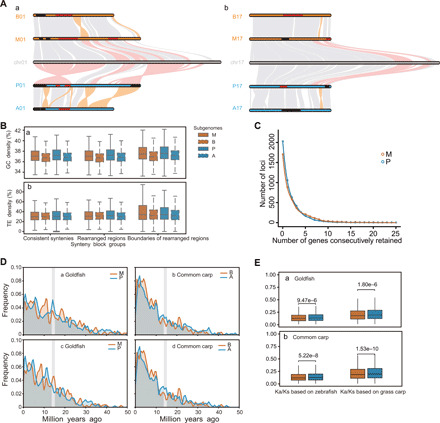
(A) Syntenic blocks between goldfish and common carp and syntenic blocks between goldfish and zebrafish with color-labeled rearrangements in two examples. The other 23 groups of syntenic analysis are shown in fig. S3. All 50 pseudochromosomes of goldfish show clear two-to-one orthologous relationships to the 25 chromosomes of zebrafish. Orange bars and numbers mark the chromosomes from matrilineal (M and B) subgenomes, while blue denotes the patrilineal (P and A) ones. Gray bars and numbers mark the chromosomes of zebrafish. Light gray lines indicate the syntenic blocks, light orange lines indicate the rearrangements between goldfish and common carp, and the pink lines indicate the rearrangements between goldfish and zebrafish. The red and black bars on each chromosome indicate the biased repeat densities, in which the red ones indicate the syntenies with significantly higher repeat densities relative to the black ones (P < 0.05, Wilcoxon rank sum test). (B) Boxplots of distributions of (a) GC content and (b) repeat density in consistent syntenies, rearranged regions, and the boundaries of rearranged regions. Orange and blue boxes mark the values from matrilineal (M and B) and patrilineal (P and A) subgenomes, respectively. Boxes with black lines indicate the distributions of goldfish, and the ones with dashed lines indicate the common carp. (C) Distributions of consecutive gene retentions in subgenomes M and P, which do not differ from each other significantly (Fisher’s exact test, P = 0.35). (D) Dating the time of pseudogene formation in subgenomes M and P of goldfish, subgenomes B and A of common carp, as described in Supplementary Methods and Analysis 6. X axis represents an estimated time of pseudogene formation; Y axis represents the frequencies of pseudogenes in every 0.5-Ma unit. Orange lines display the frequencies of pseudogenes on subgenomes M or B along time (X axis), and blue lines display the frequencies of pseudogenes on subgenomes P or A. Gray bars indicate the timing of allopolyploidization (13.8 to 15.1 Ma ago). (a) and (b) display the distributions of all pseudogenes frequencies from each subgenome along time in goldfish and common carp; (c) and (d) display pseudogenization events specific to each subgenome in goldfish and common carp. (E) Boxplot displays the distributions of Ka/Ks ratios for (a) 7568 homoeologous gene pairs between M and P subgenomes calculated against zebrafish and grass carp and (b) distributions of Ka/Ks ratios displayed in boxplot for (a) 7568 homoeologous gene pairs between subgenomes B and A calculated against zebrafish and grass carp. Central line in each boxplot indicates the median value. Top and bottom edges of the box indicate the 25th and 75th percentiles, and the dashed lines extend 1.5 times the interquartile range beyond the edges of the box.
Most genes (>88%) were retained in both subgenomes, and the distribution of consecutive (more than two contiguous genes) retentions of syntenic blocks was not biased between them (P > 0.05; Fig. 3C and fig. S2C). The tracing of the gene loss by deletion showed 1737/2727/1677 M/P/shared losses in goldfish and indicated that subgenome P experienced more small-scale deletions of genes (11.53% of 23,652) than subgenome M (7.14% of 24,327; P < 0.01). In common carp, 1009/1409/1574 B/A/shared losses indicated more small-scale deletions in subgenome A (6.98% of 20,172) than B (4.81% of 20,977). Subgenome P tended to lose more genes related to pathways of amino acid metabolism, oxidative phosphorylation, base repair, and homologous recombination (fig. S2C and data S4_1) than subgenome M. Genes lost across all subgenomes occurred in no more than two consecutive genes in all syntenic blocks of zebrafish (data S3_7). Analyses identified fewer pseudogenes in subgenome M (2.90%, 705/24,327) than P (4.33%, 1023/23,652; P < 0.01) and also fewer pseudogenes in B (2.33%, 486/20,815) than A (3.86%, 754/19,509).
According to the dating of pseudogene formation, pseudogenes accumulated continuously in both goldfish and common carp (Fig. 3D). In goldfish, pseudogenes formed asymmetrically and continuously after allotetraploidization, while in common carp, both subgenomes experienced the accelerated accumulation of pseudogenes after allotetraploidization (Fig. 3D, a and b). To test whether the functional loss continued after the divergence between the two species, we grouped the pseudogenization events into the one shared by two species, either MB or PA, and the other occurred specifically in each subgenome. Subgenomes M and B shared 79 pseudogenes, while P and A shared 156. These shared pseudogenes in the patrilineal subgenomes of goldfish and common carp were involved in pathways of base excision repair and homologous recombination (data S4_2). We found 626/867/407/598 pseudogenes specific to M/P/B/A subgenomes, respectively, which suggested that more gene loss events occurred independently in each subgenome than the ones they shared. The dating of pseudogenes specific to each subgenome showed the same distributions after the divergence of both species: continuous accumulations in goldfish (Fig. 3D, c) and accelerated accumulation of pseudogenes in common carp (Fig. 3D, d). Notably, the pseudogenes specific to subgenome P were also predicted to be involved in DNA repair and homologous recombination (data S4_3). Together, these analyses supported a bias in gene loss between goldfish and common carp.
Nonsynonymous mutation (Ka) values, synonymous mutations (Ks) values, and the ratios of these values (Ka/Ks) between the two subgenomes were compared against the reference genomes of zebrafish and grass carp to identify alterations in evolutionary rates. In goldfish, all homoeologs in subgenome M (zebrafish median Ka/Ks = 0.12; grass carp = 0.18) had a significantly lower Ka/Ks ratio than those in subgenome P (median Ka/Ks = 0.13 and 0.19; P < 0.01 for both, Wilcoxon rank sum test; Fig. 3E), while single-copy genes and all genes showed no significant bias between the two subgenomes (P > 0.05; data S3_8, 9). In common carp, both homoeologous and all genes in subgenome B had significantly lower Ka/Ks ratio than in subgenome A (P < 0.01), while single-copy genes showed no significant bias between the two subgenomes (P > 0.05; data S3_8, 9). Ka and Ks values and Ka/Ks ratios in syntenic blocks indicated that no significant correlation existed with structural changes (fig. S2, D to F). The distributions of Ka/Ks ratios between each paired M and P, or paired B and A, chromosomes also showed no difference (fig. S2G); only three syntenic blocks showed a significant biased Ka/Ks ratio between subgenomes M and P, while only seven syntenic blocks were biased significantly between subgenomes B and A (P < 0.05, Wilcoxon rank sum test). These results indicated species-specific gene fates. Signatures for positive selection occurred in 128 homoeologous genes in goldfish, symmetrically including 0.31% (65/20,913) of genes in M and 0.33% (63/19,248) in P. Statistical comparisons of both overall and pairwise homoeologous chromosomes detected significant differences between matrilineal and patrilineal subgenomes, as well as species-specific changes.
Expression changes of homoeologs
More species-specific alterations occurred between parental genomes than asymmetrical changes. This might had led to the diversities of expression in homoeologous gene pairs. To test for this, we compared transcriptome changes between the subgenomes in six adult tissues and 15 developmental stages using the homoeologous genes that were confirmed with high correlations between biological duplicates and among developmental stages (fig. S4, data S1_9, and data S3_10, 11). In goldfish, expressions of homoeologous gene pairs did not show a bias between the homoeologs among all six adult tissues or at eight developmental stages. This pattern held except in seven specific stages around the reprogramming of embryogenesis (31, 32), where M subgenomic homoeologs were expressed 4.8% higher than the P ones (Fig. 4, A and B). In common carp, expression of homoeologous gene pairs showed B-biased expression in five stages around the reprogramming of embryogenesis, one stage in pharyngula period, and two stages in hatching period (Fig. 4C).
Fig. 4. Spatiotemporal expression patterns of homoeologous gene pairs and DNA methylation levels.
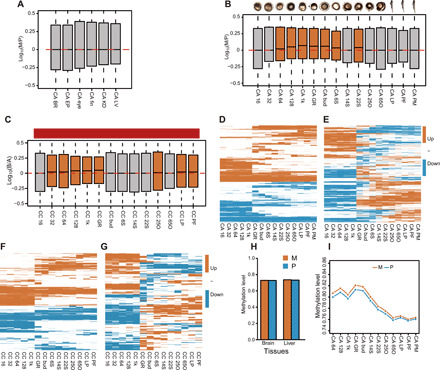
(A) Boxplot of log10(TPMM/TPMP) for homoeologous gene pairs showing medians in six adult tissues of goldfish. Red dashed line shows the equal ratio of log10(1). All adult tissues show no bias of expression between genes stemming from the subgenome M or P. (B) Boxplot of log10 [(TPMM + 0.1)/(TPMP + 0.1)] for homoeologous gene pairs showing medians in 15 developmental stages of goldfish [16 cell, 32 cell, 64 cell, 128 cell, 1000 cell; 8, 12, 16, 18, 24, 30, 46, 64, 71, and 84 hours post-fertilization (hpf)]. Red dash shows the equal ratio of log10(1). Time points from 64-cell to 22-somite stages biased expression of M homoeologs, which average 4.8% more than P genes within homoeologous gene pairs; early embryos (16- and 32-cell stages), pharyngula, and hatching period embryos show no bias of expression. (C) Boxplot of log10 [(TPMB + 0.1)/(TPMA + 0.1)] for homoeologous gene pairs from common carp shows medians in zygotically controlled developmental time points. Red dashed line shows equal ratios of log10(1). Time points from 32-cell to germ ring, 25% otic vesicle closure (OVC), long pec, and pec fin stages indicate biased B-homoeolog expression; other stages show no expression bias. (D) Expression patterns of 9090 homoeologous gene pairs from goldfish where the trend displays expression of either biased toward M or P homoeologs (EBM or EBP; 6223 genes in total, 68.46%) when gene pairs are coexpressed in at least one development stage. (E) Expression trend of 9090 homoeologous gene pairs from goldfish displaying an expression shift between two homoeologs (ES; 1644 genes in total, 18.09%) where one copy is expressed higher than the other at earlier time points, then the other copy surpasses it in later development stages. (F) Expression patterns of 4241 homoeologous gene pairs from common carp where the pattern displays either biased B- or A-homoeolog expression (2811 gene in total, 66.28%) when homoeologous gene pairs coexpressed in at least one development stage. (G) Expression patterns of 4241 homoeologous gene pairs that indicate expressional shift between two homoeologous gene pairs (414 gene in total, 9.76%); one homoeologous gene copy expressed higher than the other at an earlier time point, then the other copy surpasses it later in development. Patterns include two groups: genes (185, 4.36%) with ES before germ ring stage and genes (229, 5.40%) with ES post-germ ring stage, which accounts for most genes. Among the ES genes, 32 (0.75%) have more than two time shifts. (H) Comparison of DNA methylation levels between the two subgenomes in brain and liver tissues. (I) Comparison of DNA methylation levels between the two subgenomes in embryos of 12 developmental stages.
In goldfish, the number of differentially expressed homoeologs differed among developmental stages (fig. S5), and their expression levels exhibited spatial and temporal variation throughout development. Expression of 9090 homoeologous gene pairs showed three patterns. First, most gene pairs (68.46%, 6223/9090) displayed an expression bias toward either M (39.69%, 3608) or P (28.77%, 2615) homoeologs (Fig. 4D and Table 2). Second, expression shifted between two homoeologs (18.09%, 1644/9090) during different developmental stages (Fig. 4E and Table 2). Among them, 11.88% (1080) displayed a shift after the reprogramming of embryogenesis, while 184 genes shifted more than once through various developmental stages. Third, 13.45% of homoeologs (1223/9090) were expressed equally throughout all developmental stages. Approximately 39.69% homoeologous pairs displayed biased expression toward the M homoeologs throughout all development stages with a slightly higher expression levels in stages around germ ring, while 18.09% homoeologous gene pairs were equally expressed. These trends were consistent with those in the common carp using the same analysis with 4241 homoeologous pairs (Fig. 4, F and G, and Table 2). The expression-bias shift of both M/B and P/A genes occurred most frequently in the germ ring of the gastrula stage, which is crucial for germ-layer development in both goldfish (395 shifts) and common carp (117 shifts; data S3_12).
Table 2. The numbers and ratios of homoeologous gene pairs with each expression patterns in common carp.
EBM, expression bias toward matrilineal (M and B) homoeologs; EBP, expression bias toward patrilineal (P and A) homoeologs; ES, expression shift between two homoeologs.
| Goldfish | Common carp | |||||||
| EBM | EBP | EBM | EBP | |||||
| Trend 1 | N | % | N | % | N | % | N | % |
| 3608 | 39.69 | 2615 | 28.77 | 1522 | 35.89 | 1289 | 30.39 | |
| Trend 2 | N | % | N | % | ||||
| ES total no. | 1644 | 18.09 | 414 | 9.76 | ||||
| Group 1: ES before germ ring stage |
564 | 6.20 | 185 | 4.36 | ||||
| Group 2: ES after germ ring stage |
1080 | 11.88 | 229 | 5.40 | ||||
In goldfish, Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses found overrepresentation of the M- and P-preferentially expressed genes of goldfish in 91 pathways (data S4_4, 5) and of expression-bias shifted genes in 61 pathways (data S4_6). Most of these preferentially expressed homoeologs shared enrichment of 13 pathways, while some M- and P-preferred genes were overrepresented in DNA replication and repair and in spliceosome and phototransduction, respectively. The results indicated that homoeologous genes from both subgenomes contributed similarly to biological pathways (fig. S6, A to D).
DNA methylation and correlation to expression
DNA methylation could have occurred during polyploidization, and the methylation-associated genes could have been inherited as epialleles (33). In goldfish, the subgenomes had indistinguishable levels of DNA methylation (difference less than 20%) in both gene body and promotor regions (fig. S7) in brain and liver tissues and among 12 developmental stages (Fig. 4, H and I, and data S1_10). A high level of DNA methylation in early stages of embryos was inherited from the sperm rather than eggs and decreased over time during development (fig. S7, A and B). Expression levels of homoeologous gene pairs correlated negatively with DNA methylation patterns, especially in the proximal promotor regions (figs. S7, C and D, and S8). Thus, DNA methylation may have played a role in the regulation of homoeologous gene expression. Analyses of co-DNA methylation by weighted gene coexpression network analyses (WGCNA) (34) identified 182 homoeologs (12.5%, 182/1454), with the DNA methylation–level shifts between M and P homoeologs corresponding to their expression shifts but with no significant difference in either promoter or gene body regions among embryos of 12 time points (P > 0.05, Fisher’s exact test; fig. S7, E and F, and data S3_13).
DISCUSSION AND CONCLUSION
Polyploid vertebrates such as goldfish and common carp might have experienced chaotic changes in their early stages of polyploidization (15 Ma ago), as reported in newly formed allopolyploid fishes (11) and in other newly synthesized polyploid plants (7, 12, 13). Our analyses of goldfish and common carp indicate similarities and differences in structural change. Significant bias (P < 0.05, Wilcoxon rank sum test) occurs in Ka/Ks ratios in homoeologous gene pairs between subgenomes M and P in the goldfish (M < P), and yet no significant bias occurs in Ka/Ks in single-copy genes. This suggests that the goldfish genome was prone to retain functionally constrained genes after its WGD (35). Specifically, the goldfish genome usually retains only one copy of homologous DNA repair–related genes, which is consistent with the pattern in plants (36). Further, goldfish subgenome P continues to lose these genes via pseudogenization, yet this does not occur in common carp subgenome A, suggesting variable strategies in different species. This provides functional flexibility for both subgenomes during evolution and adaptation. Likewise, biases in post-polyploidization gene loss have been studied in plants (37, 38), and more work is necessary to elucidate this both in plants and animals. Our analyses reveal that short fragment loss only involved one or two consecutive genes of subgenomes M and P; this differs from flowering plants and African clawed frog, which have lost more and longer fragments (9, 10, 38). Considering gene expression, a few surviving animals could have evolved a balanced strategy to maintain genome stability. This would limit structural changes and genomic diversification by reprogramming homoeologous gene expression during embryonic development, which is critical for survival in both natural and controlled environments. Cyprinines can serve as models for investigating the evolution of vertebrate polyploidization, and they may explain why polyploidization events are far less common in animals than in plants. The dosage balance hypothesis is an attractive explanation for the patterns of post-polyploidization gene retention and loss (39), and future functional work is necessary to completely paint the picture.
MATERIALS AND METHODS
Sequencing and assembly
Three gynogenetic goldfish from the same inbred line were collected to extract genomic DNA (data S1). DNA from one fish was used in whole-genome sequencing by Illumina and SMRT (Pacific Biosciences) sequencing platforms. We used wtdbg (v1.1.006) (40) to assemble the long reads and polished the resulting contigs with short reads. Another goldfish was sampled for optical mapping (BioNano Genomics Irys) and Hi-C library construction, which produced chromosome-level scaffolds. For the RNA-seq, eight adult tissues were sampled from one male and one female goldfish. Two groups of mature eggs and embryos were taken from 15 developmental stages of goldfish [16 cell, 32 cell, 64 cell, 128 cell, 1000 cell, 8 hours post-fertilization (hpf), 12 hpf, 16 hpf, 18 hpf, 24 hpf, 30 hpf, 46 hpf, 64 hpf, 71 hpf, and 84 hpf] and 14 developmental stages of common carp for RNA-seq. Further, brain and liver tissues and embryos from 12 developmental stages of goldfish were sampled for whole-genome bisulfite sequencing (WGBS). All experiments were approved by Animal Care Committee of Hunan Normal University (2014278) and followed guidelines of the Administration of Affairs Concerning Experimental Animals of China.
Chromosome-scale organization
On the basis of the scaffolds linked by the Irys optical map, 50 pseudochromosomes were clustered with the Hi-C data. Next, a genetic map of goldfish based on genotyping was constructed by adopting the pooling-sequenced transcriptomic data of Kuang et al. (27), which were based on an inbred line of two parents and 79 F2 individuals. Subsequently, the two-to-one colinear relationships between goldfish and zebrafish were identified by using MCScanX (41). Last, 25 homologous chromosome pairs were generated after pairwise validation among Hi-C clustering results, genetic map, and collinearity analyses.
Annotation of gene structures and functions
Protein-coding genes were annotated using a combination of ab initio prediction, transcript evidence gathered from RNA-seq of embryos and 16 adult tissues (ovary/testis, brain, liver, spleen, kidney, eye, epithelium, and fin for both female and male), and homologous genes prediction from five fish genomes (Ctenopharyngoden idellus, Danio rerio, Gasterosteus aculeatus, Tetraodon nigroviridi, and Sinocyclocheilus anshuiensis), with EVidence Modeler (v1.1.1). Functional annotations mainly included the following methods: (i) searching against known sequence data (Swiss-Prot/Gene Ontology) by BLASTP with E value at 1 × 10−5 and online comparison against the KEGG database by KEGG Automatic Annotation Server (KAAS) and (ii) InterProScan (v5.21-60.0) predicted conservative motifs and domains.
Identification of subgenome
OrthoMCL (42) was used to cluster gene families for zebrafish, grass carp, golden-line barbel, common carp, goldfish, and species of Schizothorax. PhyML (v3.1) (43) was then used to build the phylogenetic trees for each gene family. A species tree was also constructed by using single-copy genes from the above 10 genomes. In the topology of gene trees, homoeologs located in the same clade with Schizothorax were considered to be M markers, while the remaining P copies constituted the hypothetical P species. The M/P markers were labeled back to 25 pairs of pseudochromosomes. The origins of pseudochromosomes were thereby identified by most of the M/P markers.
Times of speciation and progenitors’ divergence were estimated by the divergence time using MCMCTree in the PAML package (v4.8) (30). The general time reversible (GTR) nucleotide substitution model was used with a relaxed clock analysis. The multiple calibration points based on literature and fossil records were listed in detail in Supplementary Methods and Analysis 5. We used the divergence time of putative M and P progenitors in Cyprininae, and synonymous substitution levels between putative maternal and paternal homoeologs in goldfish, common carp, and S. grahami, respectively, to estimate an absolute substitution rate. The Ks values were measured with the method as implemented by using the yn00 program in PAML (v4.8) (30).
Deletions and pseudogenes
Putative gene-loss events were traced from the syntenic blocks between zebrafish and the two subgenomes of goldfish. In the triples of consecutive genes within syntenic blocks from the zebrafish genome and two goldfish subgenomes, missing genes were considered as deletions or pseudogenes. Sequences of potential missing genes were confirmed with The BLAST-Like Alignment Tool (BLAT) alignment and mapping coverage of Illumina short reads. Deletions had little support, and pseudogenes contained various defects including premature stop codons, frameshifts, disrupted splicing, and/or partial coding deletions. More details were provided in Supplementary Methods and Analysis 6.
Gene expression
RNA-seq data of six adult tissues, mature eggs, and all embryos with two biological duplicates were mapped to reference genome using Tophat (v2.1.1) (44). Gene expressions in each sample were estimated by RSEM (v1.2.19) (45) and quantified as values of transcripts per million (TPM). Gene expressions with TPM > 0.5 were considered to be detectable. Then, we analyzed expression variation among homoeologous genes in 15 developmental stages by developing coexpression networks with WGCNA (v1.63) (34), following the workflow of Session et al. (9).
Analysis of DNA methylation
We analyzed DNA methylation level of brain and liver tissues from WGBS data using Bismark (v0.19.0) (46) with three steps. More details about the methylation differences in functional elements between two subgenomes are provided in Supplementary Methods and Analysis 8.
Supplementary Material
Acknowledgments
We thank high-performance computing teams at Beijing Institute of Genomics (BIG) and Chinese Academy of Sciences for their computing and bioinformatic support. Funding: This work was supported by the National Natural Science Foundation of China (grant nos. 91631305, U1902201, 91631302, 91331105, 31360514, 31430088, 31872551, 31760306, and 31872561), Yunnan Provincial Science and Technology Department (grant nos. 2017HA003 and 2019FY003027), the fund from China Agriculture Research System (grant no. CARS-45), Hunan Provincial Natural Science and Technology Major Project (grant no. 2017NK1031), The Second Tibetan Plateau Scientific Expedition and Research Program (STEP; grant no. 2019QZKK0501), CAS “Light of West China” Program, National Postdoctoral Program for Innovative Talents (grant no. BX201600130), and China Postdoctoral Science Foundation (grant no. 2017 M613015). Author contributions: J.L., A.M., J.R., Z.J.C., S. Liu, X.Lu, and Y.Z. conceived and designed the study. J.L., J.Cha., S. Liu, X.Lu, and Y.Z. led sequencing of the goldfish genome. S. Liu led the breeding. M.T., L.R., S. Li, Y.Wa., Q.Q., S.Wa., M.L., J.Che., H.H., and L.Y. collected blood, adult tissues, and embryos of fish. J.Cha., F.H., and J.W. extracted the total DNA and RNA. L.W. and L.Z. contributed to library construction and sequencing. Y.We. and Z.C. performed the library construction and sequencing of DNA methylation. Z.M. and J.R. led the genome assembly. X.Li., S.Wu, C.A., X.W., L.L., and C.Y. performed the genome assembly and quality control. Y.We. and Z.D. performed the annotation. Y.We. conducted the most analyses of genomic data. J.Cha., G.L., X.Li., Z.C., H.Y., and J.X. contributed to the data analyses and quality control. J.Cha., M.T., G.L., L.R., and Y.G. contributed reagents/materials/analysis tools. P.X. provided the genomic data of common carp. J.L., J.Cha., and Y.We. wrote the manuscript. J.L., J.Cha., Y.We., R.W.M., A.M., J.G., Z.J.C., S. Liu, X.Lu, and Y.Z. revised the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The genome assembly V.1 of one gynogenetic goldfish (C. auratus red var.) was deposited in the DNA Data Bank of Japan/European Molecular Biology Laboratory/GenBank (project accession no. PRJNA289059). The whole-genome sequence data of assembly V.2 reported here have been deposited in the Genome Warehouse in BIG Data Center (47), BIG, Chinese Academy of Sciences, under accession number GWHAAIA00000000 (BioProject No.: PRJCA001234; BioSample No.: SAMC056914), which is publicly accessible at http://bigd.big.ac.cn/gwh. The raw data of SMRT sequencing, BioNano Irys data, DNA read libraries by Illumina platform, datasets of the goldfish and common carp RNA-seq short reads, and the epigenetic data were also deposited in Genome Warehouse in BIG Data Center, under accession number CRA001423. All the accession numbers of downloaded data were listed in data S1. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/22/eaaz7677/DC1
REFERENCES AND NOTES
- 1.Comai L., The advantages and disadvantages of being polyploid. Nat. Rev. Genet. 6, 836–846 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Muller H. J., Why polyploidy is rare in animals than in plants. Am. Nat. 59, 346–353 (1925). [Google Scholar]
- 3.Otto S. P., Whitton J., Polyploid incidence and evolution. Annu. Rev. Genet. 34, 401–437 (2000). [DOI] [PubMed] [Google Scholar]
- 4.Van de Peer Y., Maere S., Meyer A., The evolutionary significance of ancient genome duplications. Nat. Rev. Genet. 10, 725–732 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Mable B. K., ‘Why polyploidy is rarer in animals than in plants’: Myths and mechanisms. Biol. J. Linn. Soc. 82, 453–466 (2004). [Google Scholar]
- 6.Mable B. K., Polyploids and hybrids in changing environments: Winners or losers in the struggle for adaptation? Heredity 110, 95–96 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen Z. J., Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Annu. Rev. Plant Biol. 58, 377–406 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu S., Liu Y., Yang X., Tong C., Edwards D., Parkin I. A., Zhao M., Ma J., Yu J., Huang S., Wang X., Wang J., Lu K., Fang Z., Bancroft I., Yang T.-J., Hu Q., Yue Z., Li H., Yang L., Wu J., Zhou Q., Wang W., King G. J., Pires J. C., Lu C., Wu Z., Sampath P., Wang Z., Guo H., Pan S., Min J., Zhang D., Jin D., Li W., Belcram H., Tu J., Guan M., Qi C., Du D., Li J., Jiang L., Batley J., Sharpe A. G., Park B.-S., Ruperao P., Cheng F., Waminal N. E., Huang Y., Dong C., Wang L., Hu Z., Zhuang M., Huang J., Shi J., Mei D., Liu J., Lee T.-H., Jin H., Li Z., Li X., Zhang J., Xiao L., Zhou Y., Liu Z., Liu X., Qin R., Tang X., Liu W., Wang Y., Zhang Y., Lee J., Kim H. H., Denoeud F., Xu X., Liang X., Hua W., Chalhoub B., Paterson A. H., The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 5, 3930 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Session A. M., Uno Y., Kwon T., Chapman J. A., Toyoda A., Takahashi S., Fukui A., Hikosaka A., Suzuki A., Kondo M., van Heeringen S. J., Quigley I., Heinz S., Ogino H., Ochi H., Hellsten U., Lyons J. B., Simakov O., Putnam N., Stites J., Kuroki Y., Tanaka T., Michiue T., Watanabe M., Bogdanovic O., Lister R., Georgiou G., Paranjpe S. S., van Kruijsbergen I., Shu S., Carlson J., Kinoshita T., Ohta Y., Mawaribuchi S., Jenkins J., Grimwood J., Schmutz J., Mitros T., Mozaffari S. V., Suzuki Y., Haramoto Y., Yamamoto T. S., Takagi C., Heald R., Miller K., Haudenschild C., Kitzman J., Nakayama T., Izutsu Y., Robert J., Fortriede J., Burns K., Lotay V., Karimi K., Yasuoka Y., Dichmann D. S., Flajnik M. F., Houston D. W., Shendure J., DuPasquier L., Vize P. D., Zorn A. M., Ito M., Marcotte E. M., Wallingford J. B., Ito Y., Asashima M., Ueno N., Matsuda Y., Veenstra G. J., Fujiyama A., Harland R. M., Taira M., Rokhsar D. S., Genome evolution in the allotetraploid frog Xenopus laevis. Nature 538, 336–343 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang T., Hu Y., Jiang W., Fang L., Guan X., Chen J., Zhang J., Saski C. A., Scheffler B. E., Stelly D. M., Hulse-Kemp A. M., Wan Q., Liu B., Liu C., Wang S., Pan M., Wang Y., Wang D., Ye W., Chang L., Zhang W., Song Q., Kirkbride R. C., Chen X., Dennis E., Llewellyn D. J., Peterson D. G., Thaxton P., Jones D. C., Wang Q., Xu X., Zhang H., Wu H., Zhou L., Mei G., Chen S., Tian Y., Xiang D., Li X., Ding J., Zuo Q., Tao L., Liu Y., Li J., Lin Y., Hui Y., Cao Z., Cai C., Zhu X., Jiang Z., Zhou B., Guo W., Li R., Chen Z. J., Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 33, 531–537 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Liu S., Luo J., Chai J., Ren L., Zhou Y., Huang F., Liu X., Chen Y., Zhang C., Tao M., Lu B., Zhou W., Lin G., Mai C., Yuan S., Wang J., Li T., Qin Q., Feng H., Luo K., Xiao J., Zhong H., Zhao R., Duan W., Song Z., Wang Y., Wang J., Zhong L., Wang L., Ding Z., Du Z., Lu X., Gao Y., Murphy R. W., Liu Y., Meyer A., Zhang Y.-P., Genomic incompatibilities in the diploid and tetraploid offspring of the goldfish x common carp cross. Proc. Natl. Acad. Sci. U.S.A. 113, 1327–1332 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hegarty M. J., Barker G. L., Wilson I. D., Abbott R. J., Edwards K. J., Hiscock S. J., Transcriptome shock after interspecific hybridization in senecio is ameliorated by genome duplication. Curr. Biol. 16, 1652–1659 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Xu Y., Zhao Q., Mei S., Wang J., Genomic and transcriptomic alterations following hybridisation and genome doubling in trigenomic allohexaploid Brassica carinata x Brassica rapa. Plant Biol. 14, 734–744 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Z. J. Chen, H. H. Yu, in Polyploid and Hybrid Genomics, Z. J. Chen, J. A. Birchler, Eds. (John Wiley & Sons, 2013), pp. 335–354. [Google Scholar]
- 15.Wendel J. F., Lisch D., Hu G., Mason A. S., The long and short of doubling down: Polyploidy, epigenetics, and the temporal dynamics of genome fractionation. Curr. Opin. Genet. Dev. 49, 1–7 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Hegarty M. J., Hiscock S. J., Genomic clues to the evolutionary success of polyploid plants. Curr. Biol. 18, R435–R444 (2008). [DOI] [PubMed] [Google Scholar]
- 17.McClintock B., The significance of responses of the genome to challenge. Science 226, 792–801 (1984). [DOI] [PubMed] [Google Scholar]
- 18.Luo J., Gao Y., Ma W., Bi X.-y., Wang S.-y., Wang J., Wang Y.-q., Chai J., Du R., Wu S.-f., Meyer A., Zan R.-g., Xiao H., Murphy R. W., Zhang Y.-p., Tempo and mode of recurrent polyploidization in the Carassius auratus species complex (Cypriniformes, Cyprinidae). Heredity 112, 415–427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma W., Zhu Z.-H., Bi X.-Y., Murphy R. W., Wang S.-Y., Gao Y., Xiao H., Zhang Y.-P., Luo J., Allopolyploidization is not so simple: Evidence from the origin of the tribe cyprinini (Teleostei: Cypriniformes). Curr. Mol. Med. 14, 1331–1338 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Yang L., Sado T., Hirt M. V., Pasco-Viel E., Arunachalam M., Li J., Wang X., Freyhof J., Saitoh K., Simons A. M., Miya M., He S. P., Mayden R. L., Phylogeny and polyploidy: Resolving the classification of cyprinine fishes (Teleostei: Cypriniformes). Mol. Phylogenet. Evol. 85, 97–116 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Wang S.-Y., Luo J., Murphy R. W., Wu S.-F., Zhu C.-L., Gao Y., Zhang Y.-P., Origin of Chinese goldfish and sequential loss of genetic diversity accompanies new breeds. PLOS ONE 8, e59571 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.J. S. Nelson, Fishes of the World (John Wiley & Sons, ed. 3, 1994). [Google Scholar]
- 23.X. J. Yu, Chromosome of Chinese Freshwater Fish (Science Press, 1989). [Google Scholar]
- 24.Y. D. Liu, M. Bai, Goldfish (Ocean Press, 2014). [Google Scholar]
- 25.C. Y. Wang, Chinese Goldfish (Jindun Press, 1994). [Google Scholar]
- 26.Chen Z., Omori Y., Koren S., Shirokiya T., Kuroda T., Miyamoto A., Wada H., Fujiyama A., Toyoda A., Zhang S., Wolfsberg T. G., Kawakami K., Phillippy A. M.; NISC Comparative Sequencing Program, Mullikin J. C., Burgess S. M., De novo assembly of the goldfish (Carassius auratus) genome and the evolution of genes after whole-genome duplication. Sci. Adv. 5, eaav0547 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuang Y.-Y., Zheng X.-H., Li C.-Y., Li X.-M., Cao D.-C., Tong G.-X., Lv W.-H., Xu W., Zhou Y., Zhang X.-F., Sun Z.-P., Mahboob S., Al-Ghanim K. A., Li J.-T., Sun X.-W., The genetic map of goldfish (Carassius auratus) provided insights to the divergent genome evolutions in the Cyprinidae family. Sci. Rep. 6, 34849 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Howe K., Clark M. D., Torroja C. F., Torrance J., Berthelot C., Muffato M., Collins J. E., Humphray S., McLaren K., Matthews L., McLaren S., Sealy I., Caccamo M., Churcher C., Scott C., Barrett J. C., Koch R., Rauch G.-J., White S., Chow W., Kilian B., Quintais L. T., Guerra-Assunção J. A., Zhou Y., Gu Y., Yen J., Vogel J.-H., Eyre T., Redmond S., Banerjee R., Chi J., Fu B., Langley E., Maguire S. F., Laird G. K., Lloyd D., Kenyon E., Donaldson S., Sehra H., Almeida-King J., Loveland J., Trevanion S., Jones M., Quail M., Willey D., Hunt A., Burton J., Sims S., McLay K., Plumb B., Davis J., Clee C., Oliver K., Clark R., Riddle C., Elliot D., Threadgold G., Harden G., Ware D., Begum S., Mortimore B., Kerry G., Heath P., Phillimore B., Tracey A., Corby N., Dunn M., Johnson C., Wood J., Clark S., Pelan S., Griffiths G., Smith M., Glithero R., Howden P., Barker N., Lloyd C., Stevens C., Harley J., Holt K., Panagiotidis G., Lovell J., Beasley H., Henderson C., Gordon D., Auger K., Wright D., Collins J., Raisen C., Dyer L., Leung K., Robertson L., Ambridge K., Leongamornlert D., McGuire S., Gilderthorp R., Griffiths C., Manthravadi D., Nichol S., Barker G., Whitehead S., Kay M., Brown J., Murnane C., Gray E., Humphries M., Sycamore N., Barker D., Saunders D., Wallis J., Babbage A., Hammond S., Mashreghi-Mohammadi M., Barr L., Martin S., Wray P., Ellington A., Matthews N., Ellwood M., Woodmansey R., Clark G., Cooper J., Tromans A., Grafham D., Skuce C., Pandian R., Andrews R., Harrison E., Kimberley A., Garnett J., Fosker N., Hall R., Garner P., Kelly D., Bird C., Palmer S., Gehring I., Berger A., Dooley C. M., Ersan-Ürün Z., Eser C., Geiger H., Geisler M., Karotki L., Kirn A., Konantz J., Konantz M., Oberlander M., Rudolph-Geiger S., Teucke M., Lanz C., Raddatz G., Osoegawa K., Zhu B., Rapp A., Widaa S., Langford C., Yang F., Schuster S. C., Carter N. P., Harrow J., Ning Z., Herrero J., Searle S. M. J., Enright A., Geisler R., Plasterk R. H. A., Lee C., Westerfield M., de Jong P. J., Zon L. I., Postlethwait J. H., Nüsslein-Volhard C., Hubbard T. J. P., Crollius H. R., Rogers J., Stemple D. L., The zebrafish reference genome sequence and its relationship to the human genome. Nature 496, 498–503 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu P., Xu J., Liu G., Chen L., Zhou Z., Peng W., Jiang Y., Zhao Z., Jia Z., Sun Y., Wu Y., Chen B., Pu F., Feng J., Luo J., Chai J., Zhang H., Wang H., Dong C., Jiang W., Sun X., The allotetraploid origin and asymmetrical genome evolution of the common carp Cyprinus carpio. Nat. Commun. 10, 4625 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang Z., PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Aanes H., Winata C. L., Lin C. H., Chen J. P., Srinivasan K. G., Lee S. G. P., Lim A. Y. M., Hajan H. S., Collas P., Bourque G., Gong Z., Korzh V., Aleström P., Mathavan S., Zebrafish mRNA sequencing deciphers novelties in transcriptome dynamics during maternal to zygotic transition. Genome Res. 21, 1328–1338 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langley A. R., Smith J. C., Stemple D. L., Harvey S. A., New insights into the maternal to zygotic transition. Development 141, 3834–3841 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Song Q., Zhang T., Stelly D. M., Chen Z. J., Epigenomic and functional analyses reveal roles of epialleles in the loss of photoperiod sensitivity during domestication of allotetraploid cottons. Genome Biol. 18, 99 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langfelder P., Horvath S., WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hakes L., Pinney J. W., Lovell S. C., Oliver S. G., Robertson D. L., All duplicates are not equal: The difference between small-scale and genome duplication. Genome Biol. 8, R209 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Smet R., Adams K. L., Vandepoele K., Van Montagu M. C. E., Maere S., Van de Peer Y., Convergent gene loss following gene and genome duplications creates single-copy families in flowering plants. Proc. Natl. Acad. Sci. U.S.A. 110, 2898–2903 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garsmeur O., Schnable J. C., Almeida A., Jourda C., D'Hont A., Freeling M., Two evolutionarily distinct classes of paleopolyploidy. Mol. Biol. Evol. 31, 448–454 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Tang H., Woodhouse M. R., Cheng F., Schnable J. C., Pedersen B. S., Conant G., Wang X., Freeling M., Pires J. C., Altered patterns of fractionation and exon deletions in Brassica rapa support a two-step model of paleohexaploidy. Genetics 190, 1563–1574 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Birchler J. A., Veitia R. A., Gene balance hypothesis: Connecting issues of dosage sensitivity across biological disciplines. Proc. Natl. Acad. Sci. U.S.A. 109, 14746–14753 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ruan J., Li H., Fast and accurate long-read assembly with wtdbg2. Nat. Methods 17, 155–158 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Y., Tang H., DeBarry J. D., Tan X., Li J., Wang X., Lee T.-h., Jin H. Z., Marler B., Guo H., Kissinger J. C., Paterson A. H., MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li L., Stoeckert C. J. Jr., Roos D. S., OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guindon S., Dufayard J.-F., Lefort V., Anisimova M., Hordijk W., Gascuel O., New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010). [DOI] [PubMed] [Google Scholar]
- 44.Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S. L., TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li B., Dewey C. N., RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krueger F., Andrews S. R., Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.BIG Data Center Members , Database resources of the BIG data center in 2018. Nucleic Acids Res. 46, D14–D20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hastie A. R., Dong L., Smith A., Finklestein J., Lam E. T., Huo N., Cao H., Kwok P.-Y., Deal K. R., Dvorak J., Luo M.-C., Gu Y., Xiao M., Rapid genome mapping in nanochannel arrays for highly complete and accurate de novo sequence assembly of the complex Aegilops tauschii genome. PLOS ONE 8, e55864 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burton J. N., Liachko I., Dunham M. J., Shendure J., Species-level deconvolution of metagenome assemblies with Hi-C–based contact probability maps. G3 (Bethesda) 4, 1339–1346 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Steinkamp J. A., Flow cytometry. Rev. Sci. Instrum. 55, 1375–1400 (1984). [Google Scholar]
- 51.Luo J., Lang M., Salzburger W., Siegel N., Stölting K. N., Meyer A., A BAC library for the goldfish Carassius auratus auratus (Cyprinidae, cypriniformes). J. Exp. Zool. Part B 306B, 567–574 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Li R., Zhu H., Ruan J., Qian W., Fang X., Shi Z., Li Y., Li S., Shan G., Kristiansen K., Li S., Yang H., Wang J., Wang J., De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20, 265–272 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu D., Dong L., Yan F., Mu H., Tang B., Yang X., Zeng T., Zhou Q., Gao F., Wang Z., Hao Z., Kang H., Zheng Y., Huang H., Wei Y., Pan W., Xu Y., Zhu J., Zhao S., Wang C., Wang P., Dai L., Li M., Lan L., Wang Y., Chen H., Li Y.-X., Fu Y.-X., Shao Z., Bao Y., Zhao F., Chen L.-N., Zhang G.-Q., Zhao W., Li H., eGPS 1.0: Comprehensive software for multi-omic and evolutionary analyses. Natl. Sci. Rev. 6, 867–869 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.T. M. Cavender, in Cyprinid Fishes, Fish & Fisheries Series, I. J. Winfield, J. S. Nelson, Eds. (Springer, Dordrecht, 1991), pp. 34–54. [Google Scholar]
- 55.Parfrey L. W., Lahr D. J. G., Knoll A. H., Katz L. A., Estimating the timing of early eukaryotic diversification with multigene molecular clocks. Proc. Natl. Acad. Sci. U.S.A. 108, 13624–13629 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Broughton R. E., Betancur-R R., Li C., Arratia G., Ortí G., Multi-locus phylogenetic analysis reveals the pattern and tempo of bony fish evolution. PLOS Curr. 5, ecurrents.tol.2ca8041495ffafd0c92756e75247483e (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/22/eaaz7677/DC1