

Abstract
Pulmonary fibrosis occurs in a heterogeneous group of lung disorders and is characterised by an excessive deposition of extracellular matrix proteins within the pulmonary interstitium, leading to impaired gas transfer and a loss of lung function. In the past 10 years, there has been a dramatic increase in our understanding of the immune system and how it contributes to fibrogenic processes within the lung. This review will compare some of the models used to investigate the pathogenesis and treatment of pulmonary fibrosis, in particular those used to study immune cell pathogenicity in idiopathic pulmonary fibrosis, highlighting their advantages and disadvantages in dissecting human disease.
Keywords: animal models, bleomycin, fibrogenesis, inflammation, innate and adaptive immune system
This review will compare some of the models used to investigate the pathogenesis and treatment of pulmonary fibrosis, in particular those used to study immune cell pathogenicity in idiopathic pulmonary fibrosis, highlighting their advantages, disadvantages and suitability in representing the human disease.
Introduction
Idiopathic pulmonary fibrosis (IPF) is the most common of the interstitial lung diseases. It is characterised by excessive extracellular matrix (ECM) deposition in the lung with a progressive decline in lung function and a prognosis of 3–5 years after diagnosis. The disease affects men slightly more frequently than women, and the age of disease onset is from 40 to 70 years. 1 , 2 Seven distinct subtypes of interstitial lung disease have been proposed by the American Thoracic Society/European Respiratory Society. 3 , 4 Of these histopathological subtypes, usual interstitial pneumonia (UIP) is the histological pattern that characterises patients with clinical IPF.
Usual interstitial pneumonia typically demonstrates a heterogeneous appearance with areas of fibrosis within the peripheral region of the lung characterised by fibroblast proliferation, myofibroblast accumulation and excessive collagen deposition, surrounded by normal lung architecture. 5 , 6 , 7 The formation of fibroblast foci is associated with a breakdown of alveolar septal walls and enlargement of airspaces, giving the appearance of ‘honeycombing’. The damage to the alveolar structure results in decreased lung compliance and inefficient gas exchange, leading eventually to respiratory failure. UIP lungs demonstrate mild inflammation with lymphocytic infiltration in the alveolar interstitium. The aetiology of the disease is unknown but genetic studies have identified a number of susceptibility genes that play important roles in epithelial cell function and the maintenance and integrity of the epithelial barrier. 8 , 9 , 10 , 11 , 12 , 13 , 14 There is also growing evidence for the role of the innate and adaptive immune response in the initiation and/or progression of fibrotic diseases including IPF. 15 , 16 , 17 , 18
The underlying pathobiology of IPF was considered a chronic inflammatory immune response because of the damage of the airway epithelium. 2 , 19 More recently, attention has focused on intrinsic defects within the lung epithelial cells that lead to an abnormal repair process in response to damage. 6 , 20 , 21 The abnormal re‐epithelialisation and/or response to epithelial damage triggers the release of various cytokines and inflammatory mediators that results in epithelial cell hypertrophy, uncontrolled fibroblast proliferation and myofibroblast accumulation, impaired clearance of (myo)fibroblasts leading to excessive ECM deposition, tissue remodelling and angiogenesis. 22 , 23 The inflammatory response in IPF is diverse, involving innate and adaptive lymphocytes including antibody‐producing plasma B cells that are recruited to the lung tissue environment. 15 , 22 , 24 , 25 , 26 Lymphocytes accumulate within discrete foci that are distributed adjacent to areas of active fibrosis 26 , 27 , 28 but whether these cells are crucial in disease pathogenesis of IPF is unclear. IPF does not appear to behave as a single clinical disease entity but rather as a spectrum, as evidenced by differences in expression of inflammatory markers and autoantibodies. 25 , 29 , 30 , 31 , 32 , 33 Acute exacerbations of IPF have been associated with immune cell infiltration of the lung but why this occurs and how it drives the profibrotic response are unknown. 34 In this review, we examine a range of different animal models that have been used to help investigate the key immunological changes that occur in the development of lung fibrosis, and their relevance to IPF.
Animal models of pulmonary fibrosis
Some domestic animals such as cats, dogs and horses can develop spontaneous pulmonary fibrosis, which share many of the histopathological features observed with human IPF. In dogs, the West Highland white terrier is one breed that is particularly susceptible to canine IPF. 35 The disease shares many clinical features of human IPF with coarse crackling heard on thoracic auscultation, pulmonary hypertension and/or airway collapse. In dogs, ground glass opacification and traction bronchiectasis have been demonstrated with subpleural and peribronchiolar fibrosis, honeycombing and alveolar epithelial changes consistent with human UIP. 35 In cats, spontaneous IPF‐like disease is associated with interstitial fibrosis with fibroblast/myofibroblast accumulation, honeycombing and type II pneumocyte hyperplasia. 36 , 37 Horses and donkeys can develop an IPF‐like disease but unlike cats and dogs, where the disease appears to be spontaneous, the development of equine IPF is linked to equine herpesvirus 5 infection. 38
Genetic deficiency in mice can lead to the development of spontaneous pulmonary and age‐related lung disease. 39 , 40 , 41 , 42 More commonly, mouse models of pulmonary fibrosis have exposed mice to various agents including cytotoxic agents (e.g. bleomycin [BLM]), the delivery of profibrotic cytokines (e.g. transforming growth factor beta [TGF‐β] and interleukin [IL]‐13) or pharmacological agents (e.g. phorbol myristate acetate [PMA]). However, these induced mouse models do not follow the progression of human IPF as they often utilise young adult mice and elicit acute lung damage with inflammatory responses that lead to tissue fibrosis. The major concern is that each model relies on a specific known insult that will trigger an immune response that differs slightly in its cellular constitution and duration. Furthermore, these animal models are more representative of an acute lung injury, with fibrosis that in most cases eventually resolves, unlike the chronic, progressive and ultimately fatal disease observed with human IPF. Despite this, these models have provided valuable insight in the mechanisms leading to collagen regulation, fibrosis pathogenesis and possible treatments.
Modelling human lung fibrosis in a mouse model
Mouse models are widely used throughout biomedical research to study disease pathogenesis of acute and chronic diseases. Ideally, an animal model should reproduce the cardinal clinical features of the human disease/condition and its symptoms. Although animal models may not replicate all aspects of the human disease, they can nevertheless provide valuable insight into the pathobiology associated with tissue damage, cellular inflammation, immune regulation, tissue repair and fibrosis, because of the high degree of conservation in immune systems and tissue structure and function.
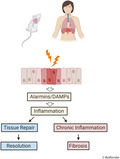
Idiopathic pulmonary fibrosis in humans is thought to arise from an environmental stimulus that causes damage to the lung epithelium. Chronic exposure to a damaging stimulus may disrupt the normal tissue repair process that results in chronic inflammation and eventually leads to collagen synthesis and tissue remodelling as summarised in Figure 1. A number of different treatments have been used to elicit pulmonary fibrosis in mice (Table 1). Some have utilised known aetiological agents including asbestos, silica and radiation, while others have used chemical agents such as BLM, monocrotaline, fluorescein isothiocyanate (FITC), oxidants and PMA. The use of these generic agents and their appropriateness as models to investigate the immune response in lung fibrosis are summarised in Table 1 and Figure 2. Studies using different animal models to investigate the immunological basis of lung fibrosis will be discussed, with the focus mainly on the BLM‐induced lung fibrosis model as it is the most widely used.
Figure 1.

Immunological outcomes of immune response to tissue damage in the lung, leading to distinct outcomes of tissue repair or pulmonary fibrosis. Innate and adaptive immune responses are central to both tissue repair and chronic inflammatory responses that lead to tissue fibrosis. Alarmins or DAMPs are released in response to epithelial damage and cell death. These host‐derived molecules are sensed by innate leukocytes (e.g. neutrophils and macrophages or innate lymphoid cells) and help in the coordinated response of adaptive immune cells including T cells and B cells. Tissue repair follows a pattern of type 1 immunity, and this switches to a type 2 dominant immune response to promote tissue repair. In contrast, under conditions of chronic tissue damage, a dominant type 2 immune response drives both innate and adaptive immune cells that can lead to the differentiation of fibroblasts to myofibroblasts, tissue remodelling and fibrosis; a characteristic feature of IPF that is responsible for the reduction in lung function.
Table 1.
Animal models of pulmonary fibrosis
| Inducing agent | Lung condition | References |
|---|---|---|
| Adenoviral‐TGF‐β | IPF | 106, 107, 108, 109, 110 |
| Asbestos | Asbestosis | 111, 112, 113 |
| Bleomycin | ARDS, IPF | 69, 73, 87, 114, 115, 116, 117 |
| Canine IPF (spontaneous) | IPF | 35, 118 |
| Feline IPF (spontaneous) | IPF | 37, 118, 119 |
| Equine IPF (spontaneous) | IPF | 120 |
| Haptens | Autoimmune‐mediated IPF | 50, 51, 121 |
| Irradiation | Radiation‐related pneumonitis | 90, 122 |
| Monocrotaline | ARDS, IPF | 43, 44, 45, 46 |
| Nanoparticles | ILD, COPD | 123, 124, 125 |
| Oxidants | ARDS, IPF | 55, 56, 57, 59 |
| Phorbol myristate acetate (PMA) | ARDS, IPF | 64 |
| Reovirus | BOOP | 126 |
| Silica | Silicosis | 127, 128, 129 |
| Welding fumes/metals | Pneumoconiosis | 130 |
ARDS, acute respiratory distress syndrome; BOOP, bronchiolitis obliterans organising pneumonia; COPD, chronic obstructive pulmonary disease; ILD, interstitial lung disease; IPF, idiopathic pulmonary fibrosis.
Figure 2.

Experimental animal models of pulmonary fibrosis. A number of different treatments have been used to elicit pulmonary fibrosis in animals. There are advantages and disadvantages of the different treatments, and although none of them elicit a pathology identical to human IPF, each model recapitulates some of the key features of IPF and provides a good model to study collagen regulation in a disease setting.
Monocrotaline
Monocrotaline (MCT) is a pyrrolizidine alkaloid that is metabolically activated by the liver and is both pneumotoxic and hepatotoxic. 43 Subcutaneous administration of a single dose of MCT induces pulmonary hypertension and respiratory distress syndrome that resembles the human conditions both morphologically and functionally. 44 Animals are usually euthanised 3–6 weeks after injection when the pathologies are fully manifested. The lungs show a severe inflammatory reaction with haemorrhage and oedema. Parenchymal changes include a reduced number of alveoli, thickened alveolar septa and accumulation of inflammatory cells, predominantly macrophages. Small arteries and arterioles show a reduced lumen, thickening of the wall with hyperproliferation of the media, inflammatory cells and increased collagen in the adventitia. 43 , 44 , 45 , 46 Mice treated with multiple subcutaneous injections of MCT for up to 18 weeks and then euthanised after 28 weeks from the start of treatment 46 showed severe interstitial pneumonia and pulmonary fibrosis, with a remarkable increase in collagen deposition in alveolar septa after 8 weeks of treatment. However, the doses used in this study were highly toxic with many mice dying within the study period. In a rat model of MCT‐induced pulmonary arterial hypertension (PAH), MCT treatment induced perivascular infiltration of macrophages, mast cells and T cells which was reduced by inhibiting CXCL12, which is a key homeostatic chemokine that controls leukocyte migration into a range of tissues including the lung. 47 Cuttica and colleagues investigated the role of T‐cell‐mediated immune pathology in MCT‐induced PAH in mice. They treated Rag1‐/‐ mice, which are devoid of mature B and T cells, and found that they were protected from vascular injury. They also provided evidence for a role of CD4+ T cells in promoting pulmonary vascular remodelling in the same model. 48
Fluorescein isothiocyanate
Fluorescein isothiocyanate (FITC) is a skin‐sensitising hapten capable of inducing specific immune responses 49 and produces fibrosis in the lungs of mice and rats following a single intratracheal instillation. 50 An advantage of this system is that the distribution of FITC in the lung can be directly visualised. However, this agent is hard to administer as the molecule is relatively insoluble and requires sonication to give a better dispersion in the lungs with a more uniform and reproducible injury. 51
After instillation of FITC, the animals develop a pattern of injury consistent with acute lung injury. This includes haemorrhage, alveolar wall oedema, eosinophilic alveolar exudate, a marked infiltrate of mononuclear cells and neutrophils principally around the bronchioles, and bronchial epithelial cell hyperplasia. 50 , 51 There is patchy focal destruction of the normal lung architecture with focal interstitial fibrosis by 21 days, which persists for at least five months with a predominantly mononuclear cell infiltrate. 50 Both the infiltrate and the scarring were confined to peribronchial areas of FITC deposition. Christensen et al. 51 examined the role of lymphocytes in FITC‐induced lung fibrosis in the different mouse strains: C57BL/6 and BALB/c. There appeared to be no difference in fibrosis onset between the two strains, suggesting that there were no background specific genes that altered pathology. It was also initially speculated that FITC‐induced pulmonary fibrosis would be driven by specific immunity to the fluorescein hapten. 50 Six months following FITC treatment, CD3+ T cells and B220+ B cells were co‐localised within lymphoid clusters in the lungs, adjacent to areas of active tissue fibrosis, 52 similar to the pattern observed in human IPF lung. 27 , 28 However, FITC‐treated T‐cell‐depleted SCID and Rag1 ‐/‐ mice still developed lung fibrosis suggesting that the adaptive immune system is not involved in fibrogenesis in these animals. 51 Interestingly, FITC treatment in the absence of the chemokine receptor CCR2, which is responsible for monocyte infiltration, reduced fibrosis, suggesting that while the adaptive immune response may not be essential, the activation of the innate immune responses may play a critical role in driving FITC‐induced lung fibrosis. 53 , 54
Ozone (O3)/nitrogen dioxide (NO2)
Pulmonary fibrosis is a common result of long‐term exposure to O3 and NO2, with more pronounced fibrosis when the gases are mixed. 55 Rats exposed to a mixture of O3 and NO2 for 6 h/day demonstrated a triphasic pattern of lesion development. Lesions developed over the first 3 weeks of exposure then partially resolved during the middle three weeks before progressing in severity in the final three weeks, with a significant increase in collagen deposition. 55 , 56 This finding was confirmed when half the concentration of O3 and NO2 was used but with continuous exposure. 57 Histological changes were mild compared with the higher oxidant concentration, despite a twofold increase in the cumulative dose. This showed that pulmonary fibrosis is more dependent on the concentration of oxidants than the cumulative dose. Both models showed some of the notable pathologic features of end‐stage IPF in humans, including severely distorted alveolar structure, increased deposition of collagen, collections of mast cells, obliteration of air spaces in fibrotic regions and focal areas of enlarged abnormal air spaces. However, the distribution of fibrosis was primarily in the centriacinar regions of the lung whereas UIP demonstrates a more peripheral fibrosis. 2 , 58 Furthermore, this response was only seen in the presence of a continuing insult, whereas in IPF, progression occurs in the apparent absence of the inciting agent.
A single exposure to O3 can induce desquamation of lung epithelial cells with concomitant secretion of chemokines CXCL1, CCL2/MIP1 and IL‐6, which promote macrophage and neutrophil recruitment to the site of damaged lung tissue. 59 Antibody depletion of neutrophils reduced inflammation and parenchymal injury, which was associated with increased levels of amphiregulin, a cytokine secreted by type 2 ILCs and CD4+ Tregs to promote tissue repair. 60 , 61 Ozone exposure elicited increased expression of type 2 cytokines IL‐4, IL‐5, IL‐13, eotaxin and monocyte chemoattractant protein (MCP)‐2 (also known as CCL8) mRNA in wild‐type C57BL/6 and Rag2 ‐/‐ mice. 62 In contrast, ozone‐exposed ILC‐deficient Rag2 ‐/‐/Il2rg ‐/‐ mice had no nasal lesions or overexpression of Th2‐ or ILC2‐related transcripts. 62 In a similar manner, Michaudel et al. used this model to examine the role of IL‐33 cytokine signalling in a model of O3‐induced airway hyper‐responsiveness. The authors determined that O3 exposure led to an immediate increase in IL‐33 which was followed by increased numbers of neutrophils and alveolar macrophages within 6–18 h post‐O3 treatment, while lymphocyte and eosinophil numbers were increased by 18–24 h post‐treatment. Genetic deletion of IL‐33 (Il‐33 ‐/‐) or its receptor ST2 (also known as IL‐1RL1) (St2 ‐/‐) in mice that were exposed to a single dose of O3 displayed exacerbated epithelial injury and neutrophilic inflammation. Enhanced neutrophilic inflammation mediated via O3 treatment was also observed following anti‐IL‐33R blockade. 63 Conversely, the administration of IL‐33 protein to IL33 ‐/‐ mice reduced neutrophil recruitment in response to O3 exposure. These data suggest that the IL‐33 signalling pathway may play an important role in the regulation of lung homeostasis and in particular to promote the maintenance of the epithelial barrier. 63
Phorbol myristate acetate
Phorbol myristate acetate (PMA) is a potent activator of leukocytes including neutrophils, macrophages, lymphocytes and platelets. A single intravenous dose of PMA in rabbits induced an acute haemorrhagic pneumonitis followed by a phase of interstitial inflammation involving neutrophils and macrophages. 64 Progressive interstitial fibrosis did not occur, and the lungs were virtually normal after 14 days. The development of histologically determined fibrosis required the continued administration of PMA (5 of every 7 days) in this model. Although the amount of fibrosis increased with the number of PMA injections, the amount of interstitial inflammation decreased, suggesting that the chronic phase of the lung reaction is not simply the consequence of acute lung injury, which is more consistent with human IPF than the other models described.
Radiation‐induced pulmonary fibrosis
Thoracic exposure to radiation in humans can induce strong inflammatory responses that lead to alveolitis or even fibrosing alveolitis. Radiation‐induced pulmonary fibrosis is characterised by tissue damage, epithelial and fibroblast cell proliferation and remodelling of the lung interstitium. To mimic the pathologic changes that occur with radiation exposure in humans, mice were exposed to a single 18 Gy dose of radiation. In this model, lung fibrosis developed within 6 months of treatment. Fox and colleagues exposed C57BL/6, A/J and C3HeJ strains of mice to a single full thoracic exposure of 18 Gy of radiation and observed that C57BL/6 mice were most susceptible to developing lung fibrosis compared to the other strains. 65 However, compared to the other lung fibrosis models discussed, the radiation model is time consuming and requires a high degree of monitoring as exposure to irradiation increases the susceptibility to lymphopenia and thus predisposes the animals to infection.
Schistosoma mansoni eggs
Chronic infection with the blood fluke Schistosoma mansoni causes a severe pathology including liver fibrosis and splenomegaly. The fibrotic response is caused by an immune response to the parasite eggs rather than the parasite itself. The parasite egg antigens induce a delayed type hypersensitivity response characterised by a CD4+ Th2 immune response with production of type 2 cytokines IL‐4, IL‐5 and IL‐13, a rise in IgE antibody levels, activation of alternative macrophages and the recruitment of eosinophils. 66 Following intraperitoneal sensitisation and intravenous challenge, S. mansoni eggs are transported to the lung via the pulmonary arteries where they become trapped within the lung parenchyma with subsequent formation of granulomas composed of lymphocytes, eosinophils and alternatively activated macrophages. These granulomas are associated with inflammation in the broncho‐alveolar spaces, expansion of the draining lymph nodes and CD4+ T‐cell activation which in turn can lead to pulmonary fibrosis. 66 More recently, activation and recruitment of ILC2 cells have been shown to contribute to the pulmonary fibrosis in response to S. mansoni egg sensitisation through secretion of IL‐25. 66 Hams and colleagues showed that antibody‐mediated depletion of ILC2 cells in lymphocyte‐deficient Rag1 ‐/‐ mice could reduce the size of S. mansoni egg‐induced granulomas and the degree of pulmonary fibrosis. 66 This result indicates that pulmonary fibrosis in this disease model does not rely on CD4+ T cells but emphasises an important role for the innate immune response to orchestrate the lung fibrotic response. To relate these findings in mice back to human IPF, the authors identified increased pulmonary expression of IL‐25 and recruitment of ILC2 cells to the lung of IPF patients. 66
Bleomycin (BLM)
The best characterised and most widely used agent to induce pulmonary fibrosis in mice and rats is BLM, an anti‐cancer drug that induces DNA damage within target cells. 67 , 68 BLM is usually administered in saline or PBS as a single dose via intratracheal, intranasal, intraperitoneal, oropharyngeal or intravenous routes; the concentration administered depending on the route of administration and the species and strain of animal used.
Following a single BLM challenge, the animals often experience weight loss within the first few days, which is associated with the acute lung injury. After 5–7 days, weights begin to increase and animals eat and behave normally. The lung weight is usually maximal at about 7 days after BLM treatment. 69 The histologic pattern of lung injury is similar for all species but does vary slightly depending on the route of administration. 70 , 71 , 72 The initial injury is predominantly focused around bronchioles, with areas of microvascular leakage and early hyperplastic changes in type II pneumocytes in regions of inflammatory cell influx. By day 7 following BLM treatment, the injury is more widespread and involves the distal lung parenchyma with multiple inflammatory foci and oedema present within alveolar septa. By day 14, the lungs show a more mature regional interstitial fibrosis with an increase in macrophages and focal lymphocytosis and lymphoid expansion. Focal alveolar re‐epithelialisation is present with extensive collagen deposition and remodelling of the alveolar unit, and there is often a low‐grade focal ‘honeycombing’ reaction present, consistent with emphysematous changes. By days 21–30, there is evidence of focal condensation of ECM. Macrophage and lymphocyte margination is marked, particularly within the periphery of the fibrotic areas, and regional re‐epithelialisation of the alveolar septa is pronounced. By 120 days, there is bronchiolar and peribronchiolar fibrosis, together with inflammation and emphysematous changes, with large areas of normal lung. These changes are consistent across studies and are thought to reflect similar changes observed with IPF. 70 , 72 Intravenous or intraperitoneal administration of BLM gives a similar pattern of lung fibrosis although the initial site of injury is the endothelium of capillaries and larger vessels and the perivascular lung structures of the subpleural parenchyma. 73 Aran and colleagues have used single‐cell RNA sequencing (scRNAseq) to characterise the heterogeneity of macrophages following BLM‐induced fibrosis in mice. They identified a putative profibrogenic macrophage population that displayed a signature gene profile (CX3CR1 +, CCR2 +, MHCII +), intermediate between a monocyte‐derived macrophage and alveolar macrophage, that produced high levels of platelet‐derived growth factor AA that directs fibroblast proliferation. 74 Depletion of CXCR3+ macrophages in mice from day 8 following BLM administration revealed a significant reduction in SiglecF+ macrophages and fibroblasts in the lung and decreased collagen synthesis. These findings suggest an important role for CX3CR1+ macrophages in lung fibrosis. Furthermore, they were able to demonstrate the existence of a similar population of transitional macrophages within lung samples from human IPF patients. 74 Reyfman and colleagues used RNAseq to analyse lung biopsy samples from IPF patients, compared to healthy lung tissue obtained from transplant donors. They compared scRNAseq data sets obtained from immune cells, epithelial cells and fibroblasts. They identified a similar novel profibrogenic macrophage population in IPF patients and were able to establish a single‐cell atlas of pulmonary fibrosis. 22 This novel and unique resource revealed for the first time a significant level of heterogeneity of alveolar macrophages and lung epithelial cells within IPF patients.
A continuous or repetitive delivery method of BLM appears to produce more fibrosis in the lung, and a fibrotic phenotype more closely resembling IPF than the single BLM delivery method. 73 , 74 , 75 , 76 , 77 Heterogeneous areas of inflammation and fibrosis, with persistent deposition of collagen and collapse of alveolar structures, likely leading to reduced lung function 5–6 weeks after BLM exposure, support the progressive nature of the pulmonary lesion in this model, thereby more closely resembling the human UIP pattern. 69 , 73 , 78 Aged mice have also been used in different models of fibrosis, including BLM‐induced lung fibrosis, to try and more closely resemble human IPF. 79 , 80 , 81 However, there is limited information on the effect of aging on the immune response within these models.
‘Humanised’ immunodeficient mice have proven a valuable addition to study the role of human cells/tissues in disease pathobiology associated with lung fibrosis. These studies have been restricted to the transfer of human IPF versus control lung fibroblasts to investigate the profibrotic potential of these mesenchymal cells. Two recipient mouse strains have been used that included the C.B.‐17 SCID/beige 82 , 83 and the NOD‐scid‐IL2Rγc–/– [NSG] mouse strain. 84 Pierce et al. first demonstrated that human IPF fibroblasts but not control fibroblasts could drive pulmonary fibrosis in recipient mice that elicited a disease pathology similar to that observed in human IPF patients. Furthermore, they targeted human CCR7 or CCL21 proteins using specific antibodies to neutralise the respective protein and showed that the immunotherapy could attenuate the progression of lung fibrosis in mice. 83 Jones et al. 82 investigated the role of the MAP3K19 enzyme that has been shown to be upregulated in IPF patients. They transferred cultured human IPF fibroblasts into C.B‐17SCID/bg mice and showed that targeting the MAP3K19 enzyme with either siRNA or a small molecule inhibitor could attenuate lung fibrosis in recipient mice. 82 Geng et al. 84 also used the humanised mouse model to identify two distinct fibroblast populations from IPF lung based on the expression of the check point molecule programmed death ligand‐1 (PDL‐1, also called CD274). PD‐L1+ fibroblasts demonstrated greater motility and invasive properties compared to PD‐L1‐ fibroblasts. NSG mice that received PDL1+ cells developed pulmonary fibrosis to a greater extent than NSG mice that received PDL1‐ fibroblasts. Furthermore, they showed that antibody‐mediated targeting of PD‐L1 or CRISPR‐mediated depletion of PD‐L1 both reduced the level of pulmonary fibrosis in NSG recipient mice. 84 Habiel and colleagues have suggested a role for CD28null T cells in the pathogenesis of lung fibrosis by transferring T cells from an IPF patient to a NSG mouse. The mice experienced unresolved lung remodelling 63–65 days after injection. The authors suggest that this is due to injury to type II alveolar epithelial cells, shown by loss in BAL surfactant protein C. 85
It is also necessary to consider the selection of the appropriate mouse strain, as there is strong evidence that the genetic background can influence the degree of lung fibrosis following BLM treatment. 86 C57BL/6J mice are the most commonly used strain for BLM treatment, because of the reproducibly high levels of inducible lung collagen deposition, that are maintained for at least 12 weeks. It has been reported that other inbred strains such as DBA/2 show more prolonged fibrosis in multiple exposure studies. 87 In contrast, inbred stains such as A/J, C3Hf/KAM or C3H/HeJ are protected from BLM‐induced fibrosis. 88 , 89 , 90 This disparity in the fibrotic response between mouse strains prompted scientists to examine whether there may be BLM susceptibility genes for lung fibrosis. Using a series of genetic crosses, a region on chromosome 17 named the bleomycin‐induced pulmonary fibrosis 1 (BLMpf1) locus was identified. 90 , 91 The locus was further refined down to a 0.71 Mb region by using subcongenic mice that contained 17 C3H/HeJ alleles in the BLMpf1interval on a C57BL/6J background. 92
Identifying genes that promote susceptibility to BLM‐induced pulmonary fibrosis
The BLMpf1 interval contains 40 known proteins and 17 of the genes contain single nucleotide polymorphisms (SNPs) that affect the encoded protein. Some of the key proteins within the interval, which showed linkage to fibrotic disease in 23 inbred mouse strains, were Butyrophilin‐like (BTNL)6, activating transcription factor (ATF)6, Notch4, Tenascin XB and complement components 4a (C4a) and C4b. 90 Although the protective alleles led to an increase in macrophage numbers in BAL fluid in response to BLM treatment compared to the wild‐type mice, subcongenic mice showed a significantly reduced number of lymphocytes in broncho‐alveolar lavage (BAL) compared to wild‐type C57BL/6J mice. Likewise, examination of gene expression in lung epithelial cells in response to BLM treatment in vivo showed a significant increase in expression of two butyrophilin‐like molecules BTLN4, BTLN6 and the complement protein C4b. 92
Butyrophilin‐like proteins
The butyrophilins (BTN) and BTNL genes are part of the immunoglobulin superfamily. The BTNL family comprises six mouse and five human genes. 93 They are structurally related to the costimulatory proteins CD80 (CD80.CD86, ICOS) and the inhibitory molecule PD‐L1, and they are evolutionarily related to major histocompatibility complex (MHC) molecules. The BTN family is composed of two mouse and six human genes. 93 Although the function of BTN/BTNL genes remains poorly characterised, recent studies have indicated a role in immune regulation of TCRγδ+ cell subsets within both mouse TCR Vγ7+ and human TCRVγ4 cells within intestinal epithelium. 93 Human BTN3A1 directs blood TCRγδ cells to low molecular weight microbial and endogenous metabolites, but it is not known if this requires recognition of TCR‐BTN3A1 binding. In addition, Di Marco Barros and colleagues recently demonstrated that intestinal epithelial cells can express BTNL molecules and these are used to activate TCRγδ cells via their TCR, and this can shape the local TCRγδ compartment in the gut. 93 The BTNL1, 2, 4 and 6 genes were identified within the BLMpf1 interval. Of those, BTNL1 and 4 were shown to be expressed on lung epithelium of C57BL/6J mice but it was only the BTNL6 gene which was identified in a genomewide association study (GWAS) to significantly associate with fibrotic lung disease in C57BL/6J mice. 90 Given the emerging role of BTNL proteins and TCRγδ cells in human and mouse mucosal tissue homeostasis, 93 , 94 , 95 , 96 it will be interesting to determine how these molecules might function in the human lung as well as in diseases such as IPF. The restricted expression of BTNL molecules may be used to sustain cognate intra‐epithelial lymphocytes in the gut, and by extension, they may help to orient lymphocytes to their correct anatomical location and to the health status of the tissue. Therefore, BTNL molecules may have a broader function outside of the gut to enable resting epithelial cells and innate T cells to interact, and this could facilitate tissue surveillance by TCRγδ cells at mucosal surfaces which play a critical role in immune stress surveillance. 93
There is an emergence of subsets of other innate‐like T‐cell populations in the peripheral immune system, including invariant NK T cells (iNKT), mucosal‐associated invariant T (MAIT) cells and germline‐encoded mycolyl lipid‐reactive (GEM) T cells that express TCRαβ receptors with a characteristically restricted TCRV gene usage. 97 , 98 These innate T‐cell populations do not recognise peptide antigens but instead recognise a range of microbial products from lipids, vitamins or glycolipids in the context of non‐classical MHC proteins. 99 These cells are believed to play an important role together with TCRγδ cells in mucosal immune surveillance. TCRγδ+ cells have been linked to an immunoregulatory role at mucosal surfaces for some time. McMenamin and colleagues first described an immunoregulatory role for CD8+ TCRγδ+ cells in the murine lung in response to the aerosolised protein antigen ovalbumin that were important to control IgE responses and allergic sensitisation. 100 As the understanding of the innate and adaptive immune response has grown, we are on the verge of unravelling one of the key roles that innate immune cells play in the control of pulmonary fibrosis. This could be explored not from just a cellular level, but with the power of genomics, it will be possible to elucidate the response of different cell subsets during pulmonary fibrosis at a single‐cell resolution. The increasing number of scRNAseq studies in IPF is allowing us to identify specific immune cell populations that can be manipulated in animal models to determine their clinical relevance.
Genome editing using clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 technology has provided scientists with the ability to evaluate the role of candidate genes in disease specific processes in vivo. CRISPR/Cas9 editing can be used to either delete a specific target gene or alternatively, to introduce specific point mutations of a clinically relevant candidate gene. The benefit of this approach is that it could give rise to novel animal models that could be more ‘clinically relevant’ as lung fibrosis may occur spontaneously without the need of intervening agents. This approach has been applied to the study of tissue fibrosis more broadly in the heart and cystic fibrosis in the lung. Galectin 3 has been associated with pulmonary hypertension and fibrosis, and Barman and colleagues have used CRISPR‐mediated deletion of Gal3 to examine its role in pulmonary hypertension in rats. 101 McCarron et al. 102 used CRISPR/Cas9‐mediated editing in rats to generate two separate strains with either a deletion of the CFTR gene or to generate a CFTR disease‐specific allele (Phe508 del) to evaluate the role of both mutations in causing cystic fibrosis in vivo. A number of gene variants have been identified in IPF patients that were identified through GWAS. Some variants are more specifically related to the epithelial function, 9 , 11 , 13 , 18 while a number of variants have been associated with the TERT/PARN pathway that regulates telomere length in cells. 8 , 103 Disease‐specific alleles of the Muc5b 9 , 104 gene have been evaluated using CRSPR‐mediated gene editing in human airway epithelial cells in vitro. 105 The next step would be to determine whether gene editing could be used to validate whether one of these alleles could drive spontaneous pulmonary fibrosis in mice, and thus provide a clinically relevant animal model of IPF where pre‐clinical therapeutic interventions could be evaluated.
Conclusions
Developing appropriate models to reflect human chronic disease is a challenge, particularly in diseases such as IPF where the aetiology of the disease is unknown, and the disease has likely developed over a long period of time. Many different animal models are currently used to study pulmonary fibrosis, and each model has its particular strengths and weaknesses. For this reason, care must be taken when extrapolating data from any single animal model to the human disease.
Use of animal models has led to a major growth in our understanding of the innate immune system and how it coordinates adaptive immune responses. The development of single‐cell technologies to examine gene expression and epigenetic changes, combined with the ability to analyse cells with multicolour flow cytometry and cell sorting capacity, is helping to identify different cell subpopulations within different normal and diseased tissues that have not been previously possible. It is hoped that these types of studies in animal models will inspire further research to better understand the pathogenesis of IPF.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
Tylah Miles: Visualization; Writing‐original draft; Writing‐review & editing. Gerard Hoyne: Conceptualization; Supervision; Visualization; Writing‐original draft; Writing‐review & editing. Darryl Knight: Writing‐review & editing. Mark Fear: Supervision; Writing‐review & editing. Steven Mutsaers: Conceptualization; Visualization; Writing‐original draft; Writing‐review & editing. Cecilia Prele: Conceptualization; Supervision; Visualization; Writing‐original draft; Writing‐review & editing.
Acknowledgments
CMP, DK and MF are supported by National Health and Medical Research Council of Australia Project Grant #1127337. Ms Tylah Miles is supported by a Research Training Program Scholarship from UWA and the Lung Foundation Australia Bill van Nierop PhD Scholarship. MF is supported by the Stan Perron Centre of Excellence for Childhood Burns and the Perth Children’s Hospital Foundation.
REFERENCES
- 1. Jo HE, Glaspole I, Grainge C et al Baseline characteristics of idiopathic pulmonary fibrosis: analysis from the Australian Idiopathic Pulmonary Fibrosis Registry. Eur Respir J 2017; 49: 1601592. [DOI] [PubMed] [Google Scholar]
- 2. Katzenstein AL, Myers JL. Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am J Respir Crit Care Med 1998; 157 (4 Pt 1): 1301–1315. [DOI] [PubMed] [Google Scholar]
- 3. Jo HE, Prasad JD, Troy LK et al Diagnosis and management of idiopathic pulmonary fibrosis: Thoracic Society of Australia and New Zealand and Lung Foundation Australia position statements summary. Med J Australia 2018; 208: 82–88. [DOI] [PubMed] [Google Scholar]
- 4. Raghu G, Remy‐Jardin M, Myers JL et al Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med 2018; 198: e44–e68. [DOI] [PubMed] [Google Scholar]
- 5. King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet 2011; 378: 1949–1961. [DOI] [PubMed] [Google Scholar]
- 6. Plantier L, Crestani B, Wert SE et al Ectopic respiratory epithelial cell differentiation in bronchiolised distal airspaces in idiopathic pulmonary fibrosis. Thorax 2011; 66: 651–657. [DOI] [PubMed] [Google Scholar]
- 7. Vyalov SL, Gabbiani G, Kapanci Y. Rat alveolar myofibroblasts acquire alpha‐smooth muscle actin expression during bleomycin‐induced pulmonary fibrosis. Am J Pathol 1993; 143: 1754–1765. [PMC free article] [PubMed] [Google Scholar]
- 8. Armanios MY, Chen JJ, Cogan JD et al Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med 2007; 356: 1317–1326. [DOI] [PubMed] [Google Scholar]
- 9. Fingerlin TE, Murphy E, Zhang W et al Genome‐wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet 2013; 45: 613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lawson WE, Grant SW, Ambrosini V et al Genetic mutations in surfactant protein C are a rare cause of sporadic cases of IPF. Thorax 2004; 59: 977–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thomas AQ, Lane K, Phillips J 3rd et al Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med 2002; 165: 1322–1328. [DOI] [PubMed] [Google Scholar]
- 12. Tsakiri KD, Cronkhite JT, Kuan PJ et al Adult‐onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci USA 2007; 104: 7552–7557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. van Moorsel CH, van Oosterhout MF, Barlo NP et al Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a dutch cohort. Am J Respir Crit Care Med 2010; 182: 1419–1425. [DOI] [PubMed] [Google Scholar]
- 14. Wang Y, Kuan PJ, Xing C et al Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am J Hum Genet 2009; 84: 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Celada LJ, Kropski JA, Herazo‐Maya JD et al PD‐1 up‐regulation on CD4+ T cells promotes pulmonary fibrosis through STAT3‐mediated IL‐17A and TGF‐β1 production. Sci Transl Med 2018; 10: 1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fingerlin TE, Zhang W, Yang IV et al Genome‐wide imputation study identifies novel HLA locus for pulmonary fibrosis and potential role for auto‐immunity in fibrotic idiopathic interstitial pneumonia. BMC Genet 2016; 17: 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Noth I, Zhang Y, Ma SF et al Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome‐wide association study. Lancet Respir Med 2013; 1: 309–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Oldham JM, Ma SF, Martinez FJ et al TOLLIP, MUC5B, and the response to N‐Acetylcysteine among Individuals with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2015; 192: 1475–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Scadding JG, Hinson KF. Diffuse fibrosing alveolitis (diffuse interstitial fibrosis of the lungs). Correlation of histology at biopsy with prognosis. Thorax 1967; 22: 291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chilosi M, Poletti V, Zamò A et al Aberrant Wnt/β‐Catenin pathway activation in idiopathic pulmonary fibrosis. Am J Pathol 2003; 162: 1495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Königshoff M, Balsara N, Pfaff EM et al Functional Wnt signaling is increased in idiopathic pulmonary fibrosis. PLoS One 2008; 3: e2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reyfman PA, Walter JM, Joshi N et al Single‐cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am J Respir Crit Care Med 2019; 199: 1517–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu Y, Mizuno T, Sridharan A et al Single‐cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight 2016; 1: e90558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hou Z, Ye Q, Qiu M, Hao Y, Han J, Zeng H. Increased activated regulatory T cells proportion correlate with the severity of idiopathic pulmonary fibrosis. Respir Res 2017; 18: 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Moore BB, Fry C, Zhou Y et al Inflammatory leukocyte phenotypes correlate with disease progression in idiopathic pulmonary fibrosis. Front Med 2014; 1: 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schiller HB, Mayr CH, Leuschner G et al Deep proteome profiling reveals common prevalence of MZB1‐positive plasma B cells in human lung and skin fibrosis. Am J Respir Crit Care Med 2017; 196: 1298–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Marchal‐Sommé J, Uzunhan Y, Marchand‐Adam S et al Cutting edge: nonproliferating mature immune cells form a novel type of organized lymphoid structure in idiopathic pulmonary fibrosis. J Immunol 2006; 176: 5735–5739. [DOI] [PubMed] [Google Scholar]
- 28. Wallace WA, Howie SE, Krajewski AS, Lamb D. The immunological architecture of B‐lymphocyte aggregates in cryptogenic fibrosing alveolitis. J Pathol 1996; 178: 323–329. [DOI] [PubMed] [Google Scholar]
- 29. Collard HR, Cool CD, Leslie KO, Curran‐Everett D, Groshong S, Brown KK. Organizing pneumonia and lymphoplasmacytic inflammation predict treatment response in idiopathic pulmonary fibrosis. Histopathology 2007; 50: 258–265. [DOI] [PubMed] [Google Scholar]
- 30. DePianto DJ, Chandriani S, Abbas AR et al Heterogeneous gene expression signatures correspond to distinct lung pathologies and biomarkers of disease severity in idiopathic pulmonary fibrosis. Thorax 2015; 70: 48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kahloon RA, Xue J, Bhargava A et al Patients with idiopathic pulmonary fibrosis with antibodies to heat shock protein 70 have poor prognoses. Am J Respir Crit Care Med 2013; 187: 768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nicholson AG, Fulford LG, Colby TV, du Bois RM, Hansell DM, Wells AU. The relationship between individual histologic features and disease progression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2002; 166: 173–177. [DOI] [PubMed] [Google Scholar]
- 33. Taillé C, Grootenboer‐Mignot S, Boursier C et al Identification of periplakin as a new target for autoreactivity in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011; 183: 759–766. [DOI] [PubMed] [Google Scholar]
- 34. Balestro E, Calabrese F, Turato G et al Immune inflammation and disease progression in idiopathic pulmonary fibrosis. PLoS One 2016; 11: e0154516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Clercx C, Fastres A, Roels E. Idiopathic pulmonary fibrosis in West Highland white terriers: An update. Vet J 2018; 242: 53–58. [DOI] [PubMed] [Google Scholar]
- 36. Roman J, Brown KK, Olson A, Corcoran BM, Williams KJ. An official American thoracic society workshop report: comparative pathobiology of fibrosing lung disorders in humans and domestic animals. Ann Am Thorac Soc 2013; 10: S224–S229. [DOI] [PubMed] [Google Scholar]
- 37. Secrest SA, Bailey MQ, Williams KJ, Smarick SD. Imaging diagnosis–Feline idiopathic pulmonary fibrosis. Vet Radiol Ultrasound 2008; 49: 47–50. [DOI] [PubMed] [Google Scholar]
- 38. Tashiro J, Rubio GA, Limper AH et al Exploring animal models that resemble idiopathic pulmonary fibrosis. Front Med 2017; 4: 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chung KP, Hsu CL, Fan LC et al Mitofusins regulate lipid metabolism to mediate the development of lung fibrosis. Nat Commun 2019; 10: 3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Laucho‐Contreras ME, Polverino F, Rojas‐Quintero J, Wang X, Owen CA. Club cell protein 16 (Cc16) deficiency increases inflamm‐aging in the lungs of mice. Physiol Rep 2018; 6: e13797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nureki SI, Tomer Y, Venosa A et al Expression of mutant Sftpc in murine alveolar epithelia drives spontaneous lung fibrosis. J Clin Invest 2018; 128: 4008–4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Van Dyken SJ, Liang HE, Naikawadi RP et al Spontaneous chitin accumulation in airways and age‐related fibrotic lung disease. Cell 2017; 169: 497–509.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Baybutt RC, Molteni A. Dietary β‐‐carotene protects lung and liver parenchyma of rats treated with monocrotaline. Toxicology 1999; 137: 69–80. [DOI] [PubMed] [Google Scholar]
- 44. Czer GT, Marsh J, Konopka R, Moser KM. Low‐dose PGI2 prevents monocrotaline‐induced thromboxane production and lung injury. J Appl Physiol 1986; 60: 464–471. [DOI] [PubMed] [Google Scholar]
- 45. Baybutt RC, Rosales C, Brady H, Molteni A. Dietary fish oil protects against lung and liver inflammation and fibrosis in monocrotaline treated rats. Toxicology 2002; 175: 1–13. [DOI] [PubMed] [Google Scholar]
- 46. Hayashi S, Mitsumori K, Imaida K et al Establishment of an animal model for pulmonary fibrosis in mice using monocrotaline. Toxicol Pathol 1995; 23: 63–71. [DOI] [PubMed] [Google Scholar]
- 47. Savai R, Pullamsetti SS, Kolbe J et al Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 2012; 186: 897–908. [DOI] [PubMed] [Google Scholar]
- 48. Cuttica MJ, Langenickel T, Noguchi A, Machado RF, Gladwin MT, Boehm M. Perivascular T‐cell infiltration leads to sustained pulmonary artery remodeling after endothelial cell damage. Am J Respir Cell Mol Biol 2011; 45: 62–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sikic BI, Young DM, Mimnaugh EG, Gram TE. Quantification of bleomycin pulmonary toxicity in mice by changes in lung hydroxyproline content and morphometric histopathology. Cancer Res 1978; 38: 787–792. [PubMed] [Google Scholar]
- 50. Roberts SN, Howie SE, Wallace WA et al A novel model for human interstitial lung disease: hapten‐driven lung fibrosis in rodents. J Pathol 1995; 176: 309–318. [DOI] [PubMed] [Google Scholar]
- 51. Christensen PJ, Goodman RE, Pastoriza L, Moore B, Toews GB. Induction of lung fibrosis in the mouse by intratracheal instillation of fluorescein isothiocyanate is not T‐cell‐dependent. Am J Pathol 1999; 155: 1773–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fisher CE, Ahmad SA, Fitch PM, Lamb JR, Howie SE. FITC‐induced murine pulmonary inflammation: CC10 up‐regulation and concurrent Shh expression. Cell Biol Int 2005; 29: 868–876. [DOI] [PubMed] [Google Scholar]
- 53. Moore BB, Murray L, Das A, Wilke CA, Herrygers AB, Toews GB. The role of CCL12 in the recruitment of fibrocytes and lung fibrosis. Am J Respir Cell Mol Biol 2006; 35: 175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Moore BB, Paine R 3rd, Christensen PJ et al Protection from pulmonary fibrosis in the absence of CCR2 signaling. J Immunol 2001; 167: 4368–4377. [DOI] [PubMed] [Google Scholar]
- 55. Last JA, Gelzleichter TR, Pinkerton KE, Walker RM, Witschi H. A new model of progressive pulmonary fibrosis in rats. Am Rev Respir Dis 1993; 148: 487–494. [DOI] [PubMed] [Google Scholar]
- 56. Farman CA, Watkins K, van Hoozen B, Last JA, Witschi H, Pinkerton KE. Centriacinar remodeling and sustained procollagen gene expression after exposure to ozone and nitrogen dioxide. Am J Respir Cell Mol Biol 1999; 20: 303–311. [DOI] [PubMed] [Google Scholar]
- 57. Ishii Y, Hirano K, Morishima Y et al Early molecular and cellular events of oxidant‐induced pulmonary fibrosis in rats. Toxicol Appl Pharmacol 2000; 167: 173–181. [DOI] [PubMed] [Google Scholar]
- 58. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias . This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med 2002; 165: 277–304. [DOI] [PubMed] [Google Scholar]
- 59. Sokolowska M, Quesniaux VFJ, Akdis CA, Chung KF, Ryffel B, Togbe D. Acute respiratory barrier disruption by ozone exposure in mice. Front Immunol 2019; 10: 2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Arpaia N, Green JA, Moltedo B et al A distinct function of regulatory T cells in tissue protection. Cell 2015; 162: 1078–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Monticelli LA, Sonnenberg GF, Abt MC et al Innate lymphoid cells promote lung‐tissue homeostasis after infection with influenza virus. Nat Immunol 2011; 12: 1045–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kumagai K, Lewandowski R, Jackson‐Humbles DN et al Ozone‐induced nasal type 2 immunity in mice is dependent on innate lymphoid cells. Am J Respir Cell Mol Biol 2016; 54: 782–791. [DOI] [PubMed] [Google Scholar]
- 63. Michaudel C, Mackowiak C, Maillet I et al Ozone exposure induces respiratory barrier biphasic injury and inflammation controlled by IL‐33. J Allergy Clin Immunol 2018; 142: 942–958. [DOI] [PubMed] [Google Scholar]
- 64. Taylor RG, McCall CE, Thrall RS, Woodruff RD, O'Flaherty JT. Histopathologic features of phorbol myristate acetate‐induced lung injury. Lab Invest 1985; 52: 61–70. [PubMed] [Google Scholar]
- 65. Fox J, Bergeron ME, Haston CK. Genetic deficiency in complement component 4b does not alter radiation‐induced lung disease in mice. Radiat Res 2013; 179: 146–150. [DOI] [PubMed] [Google Scholar]
- 66. Hams E, Armstrong ME, Barlow JL et al IL‐25 and type 2 innate lymphoid cells induce pulmonary fibrosis. Proc Natl Acad Sci USA 2014; 111: 367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Allawzi A, Elajaili H, Redente EF, Nozik‐Grayck E. Oxidative toxicology of bleomycin: role of the extracellular redox environment. Curr Opin Toxicol 2019; 13: 68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hay J, Shahzeidi S, Laurent G. Mechanisms of bleomycin‐induced lung damage. Arch Toxicol 1991; 65: 81–94. [DOI] [PubMed] [Google Scholar]
- 69. Mutsaers SE, Foster ML, Chambers RC, Laurent GJ, McAnulty RJ. Increased endothelin‐1 and its localization during the development of bleomycin‐induced pulmonary fibrosis in rats. Am J Respir Cell Mol Biol 1998; 18: 611–619. [DOI] [PubMed] [Google Scholar]
- 70. Gross TJ, Hunninghake GW. Idiopathic pulmonary fibrosis. N Engl J Med 2001; 345: 517–525. [DOI] [PubMed] [Google Scholar]
- 71. Lederer DJ, Martinez FJ. Idiopathic pulmonary fibrosis. N Engl J Med 2018; 378: 1811–1823. [DOI] [PubMed] [Google Scholar]
- 72. Nuovo GJ, Hagood JS, Magro CM et al The distribution of immunomodulatory cells in the lungs of patients with idiopathic pulmonary fibrosis. Modern Pathol 2012; 25: 416–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Harrison JH Jr, Lazo JS. High dose continuous infusion of bleomycin in mice: a new model for drug‐induced pulmonary fibrosis. J Pharmacol Exp Ther 1987; 243: 1185–1194. [PubMed] [Google Scholar]
- 74. Aran D, Looney AP, Liu L et al Reference‐based analysis of lung single‐cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol 2019; 20: 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Aono Y, Ledford JG, Mukherjee S et al Surfactant protein‐D regulates effector cell function and fibrotic lung remodeling in response to bleomycin injury. Am J Respir Crit Care Med 2012; 185: 525–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Degryse AL, Tanjore H, Xu XC et al Repetitive intratracheal bleomycin models several features of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 2010; 299: L442–L552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lee R, Reese C, Bonner M et al Bleomycin delivery by osmotic minipump: similarity to human scleroderma interstitial lung disease. Am J Physiol Lung Cell Mol Physiol 2014; 306: L736–L748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gabazza EC, Taguchi O, Adachi Y. Bleomycin‐induced lung fibrosis: the authors should have used another method to induce pulmonary lesions resembling human idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2002; 165: 845–846. [DOI] [PubMed] [Google Scholar]
- 79. Hecker L, Logsdon NJ, Kurundkar D et al Reversal of persistent fibrosis in aging by targeting Nox4‐Nrf2 redox imbalance. Sci Transl Med 2014; 6: 231ra47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lok SS, Haider Y, Howell D, Stewart JP, Hasleton PS, Egan JJ. Murine gammaherpes virus as a cofactor in the development of pulmonary fibrosis in bleomycin resistant mice. Eur Respir J 2002; 20: 1228–1232. [DOI] [PubMed] [Google Scholar]
- 81. Stout‐Delgado HW, Cho SJ, Chu SG et al Age‐dependent susceptibility to pulmonary fibrosis is associated with NLRP3 inflammasome activation. Am J Respir Cell Mol Biol 2016; 55: 252–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Jones IC, Espindola MS, Narayanan R et al Targeting MAP3K19 prevents human lung myofibroblast activation both in vitro and in a humanized SCID model of idiopathic pulmonary fibrosis. Sci Rep 2019; 9: 19796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Pierce EM, Carpenter K, Jakubzick C et al Therapeutic targeting of CC ligand 21 or CC chemokine receptor 7 abrogates pulmonary fibrosis induced by the adoptive transfer of human pulmonary fibroblasts to immunodeficient mice. Am J Pathol 2007; 170: 1152–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Geng Y, Liu X, Liang J et al PD‐L1 on invasive fibroblasts drives fibrosis in a humanized model of idiopathic pulmonary fibrosis. JCI Insight 2019; 4: e12532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Habiel DM, Espindola MS, Kitson C et al Characterization of CD28(null) T cells in idiopathic pulmonary fibrosis. Mucosal Immunol 2019; 12: 212–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kolb M, Bonniaud P, Galt T et al Differences in the fibrogenic response after transfer of active transforming growth factor‐β1 gene to lungs of "fibrosis‐prone" and "fibrosis‐resistant" mouse strains. Am J Respir Cell Mol Biol 2002; 27: 141–150. [DOI] [PubMed] [Google Scholar]
- 87. Chung MP, Monick MM, Hamzeh NY, Butler NS, Powers LS, Hunninghake GW. Role of repeated lung injury and genetic background in bleomycin‐induced fibrosis. Am J Respir Cell Mol Biol 2003; 29 (3 Pt 1): 375–380. [DOI] [PubMed] [Google Scholar]
- 88. Haston CK, Tomko TG, Godin N, Kerckhoff L, Hallett MT. Murine candidate bleomycin induced pulmonary fibrosis susceptibility genes identified by gene expression and sequence analysis of linkage regions. J Med Genet 2005; 42: 464–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lemay AM, Haston CK. Bleomycin‐induced pulmonary fibrosis susceptibility genes in AcB/BcA recombinant congenic mice. Physiol Genomics 2005; 23: 54–61. [DOI] [PubMed] [Google Scholar]
- 90. Paun A, Lemay AM, Haston CK. Gene expression profiling distinguishes radiation‐induced fibrosing alveolitis from alveolitis in mice. Radiat Res 2010; 173: 512–521. [DOI] [PubMed] [Google Scholar]
- 91. Haston CK, Wang M, Dejournett RE et al Bleomycin hydrolase and a genetic locus within the MHC affect risk for pulmonary fibrosis in mice. Hum Mol Genet 2002; 11: 1855–1863. [DOI] [PubMed] [Google Scholar]
- 92. Bergeron ME, Stefanov A, Haston CK. Fine mapping of the major bleomycin‐induced pulmonary fibrosis susceptibility locus in mice. Mamm Genome 2018; 29: 670–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Di Marco Barros R, Roberts NA, Dart RJ et al Epithelia use butyrophilin‐like molecules to shape organ‐specific γδ T cells compartments. Cell 2016; 167: 203–218.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Melandri D, Zlatareva I, Chaleil RAG et al The γδTCR combines innate immunity with adaptive immunity by utilizing spatially distinct regions for agonist selection and antigen responsiveness. Nat Immunol 2018; 19: 1352–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Willcox CR, Vantourout P, Salim M et al Butyrophilin‐like 3 directly binds a human Vγ4+ T cell receptor using a modality distinct from clonally‐restricted antigen. Immunity 2019; 51: 813–825.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wu Y, Kyle‐Cezar F, Woolf RT et al An innate‐like Vδ1+ γδ T cell compartment in the human breast is associated with remission in triple‐negative breast cancer. Sci Transl Med 2019;11:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Borger JG, Lau M, Hibbs ML. The influence of innate lymphoid cells and unconventional T cells in chronic inflammatory lung disease. Front Immunol 2019; 10: 1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Hinks TSC, Marchi E, Jabeen M et al Activation and in vivo evolution of the MAIT cell transcriptome in mice and humans reveals tissue repair functionality. Cell Rep 2019; 28: 3249–3262.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lantz O, Legoux F. MAIT cells: an historical and evolutionary perspective. Immunol Cell Biol 2018; 96: 564–572. [DOI] [PubMed] [Google Scholar]
- 100. McMenamin C, Pimm C, McKersey M, Holt PG. Regulation of IgE responses to inhaled antigen in mice by antigen‐specific γδ T cells. Science 1994; 265: 1869–1871. [DOI] [PubMed] [Google Scholar]
- 101. Barman SA, Li X, Haigh S et al Galectin‐3 is expressed in vascular smooth muscle cells and promotes pulmonary hypertension through changes in proliferation, apoptosis, and fibrosis. Am J Physiol Lung Cell Mol Physiol 2019; 316: L784–L797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. McCarron A, Cmielewski P, Reyne N et al Phenotypic characterization and comparison of Phe508del and cystic fibrosis transmembrane conductance regulator (CFTR) knockout rat models of cystic fibrosis generated by clustered regularly interspaced short palindromic repeats/clustered regularly interspaced short palindromic repeats‐associated protein 9 gene editing. Am J Pathol 2020; 190: 977–993. [DOI] [PubMed] [Google Scholar]
- 103. Codd V, Nelson CP, Albrecht E et al Identification of seven loci affecting mean telomere length and their association with disease. Nat Genet 2013; 45: 422–427, 7e1‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Seibold MA, Wise AL, Speer MC et al A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med 2011; 364: 1503–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Chen G, Ribeiro CMP, Sun L et al XBP1S regulates MUC5B in a promoter variant‐dependent pathway in idiopathic pulmonary fibrosis airway epithelia. Am J Respir Crit Care Med 2019; 200: 220–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Boutanquoi PM, Burgy O, Beltramo G et al TRIM33 prevents pulmonary fibrosis by impairing TGF‐β1 signaling. Eur Respir J 2020; 55: 1901346. [DOI] [PubMed] [Google Scholar]
- 107. Lam AP, Herazo‐Maya JD, Sennello JA et al Wnt coreceptor Lrp5 is a driver of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2014; 190: 185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Marudamuthu AS, Bhandary YP, Fan L et al Caveolin‐1‐derived peptide limits development of pulmonary fibrosis. Sci Transl Med 2019; 11. [DOI] [PubMed] [Google Scholar]
- 109. Sime PJ, Xing Z, Graham FL, Csaky KG, Gauldie J. Adenovector‐mediated gene transfer of active transforming growth factor‐β1 induces prolonged severe fibrosis in rat lung. J Clin Invest 1997; 100: 768–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. van der Velden JL, Alcorn JF, Chapman DG et al Airway epithelial specific deletion of Jun‐N‐terminal kinase 1 attenuates pulmonary fibrosis in two independent mouse models. PLoS One 2020; 15: e0226904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Coin PG, Osornio‐Vargas AR, Roggli VL, Brody AR. Pulmonary fibrogenesis after three consecutive inhalation exposures to chrysotile asbestos. Am J Respir Crit Care Med 1996; 154: 1511–1519. [DOI] [PubMed] [Google Scholar]
- 112. Cummins AB, Palmer C, Mossman BT, Taatjes DJ. Persistent localization of activated extracellular signal‐regulated kinases (ERK1/2) is epithelial cell‐specific in an inhalation model of asbestosis. Am J Pathol 2003; 162: 713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Warshamana GS, Pociask DA, Sime P, Schwartz DA, Brody AR. Susceptibility to asbestos‐induced and transforming growth factor‐β1‐induced fibroproliferative lung disease in two strains of mice. Am J Respir Cell Mol Biol 2002; 27: 705–713. [DOI] [PubMed] [Google Scholar]
- 114. Borzone G, Moreno R, Urrea R, Meneses M, Oyarzun M, Lisboa C. Bleomycin‐induced chronic lung damage does not resemble human idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2001; 163: 1648–1653. [DOI] [PubMed] [Google Scholar]
- 115. Gad ES, Salama AAA, El‐Shafie MF, Arafa HMM, Abdelsalam RM, Khattab M. The anti‐fibrotic and anti‐inflammatory potential of bone marrow‐derived mesenchymal stem cells and nintedanib in bleomycin‐induced lung fibrosis in rats. Inflammation 2020; 43: 123–134. [DOI] [PubMed] [Google Scholar]
- 116. O'Donoghue RJ, Knight DA, Richards CD et al Genetic partitioning of interleukin‐6 signalling in mice dissociates Stat3 from Smad3‐mediated lung fibrosis. EMBO Mol Med 2012; 4: 939–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Yara S, Kawakami K, Kudeken N et al FTS reduces bleomycin‐induced cytokine and chemokine production and inhibits pulmonary fibrosis in mice. Clin Exp Immunol 2001; 124: 77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Reinero C. Interstitial lung diseases in dogs and cats part I: the idiopathic interstitial pneumonias. Vet J 2019; 243: 48–54. [DOI] [PubMed] [Google Scholar]
- 119. Williams K, Malarkey D, Cohn L, Patrick D, Dye J, Toews G. Identification of spontaneous feline idiopathic pulmonary fibrosis: morphology and ultrastructural evidence for a type II pneumocyte defect. Chest 2004; 125: 2278–2288. [DOI] [PubMed] [Google Scholar]
- 120. Williams KJ. Gammaherpesviruses and pulmonary fibrosis: evidence from humans, horses, and rodents. Vet Pathol 2014; 51: 372–384. [DOI] [PubMed] [Google Scholar]
- 121. Zhang‐Hoover J, Sutton A, Stein‐Streilein J. CD40/CD40 ligand interactions are critical for elicitation of autoimmune‐mediated fibrosis in the lung. J Immunol 2001; 166: 3556–3563. [DOI] [PubMed] [Google Scholar]
- 122. Pauluhn J, Baumann M, Hirth‐Dietrich C, Rosenbruch M. Rat model of lung fibrosis: comparison of functional, biochemical, and histopathological changes 4 months after single irradiation of the right hemithorax. Toxicology 2001; 161: 153–163. [DOI] [PubMed] [Google Scholar]
- 123. Lai X, Zhao H, Zhang Y et al Intranasal delivery of copper oxide nanoparticles induces pulmonary toxicity and fibrosis in C57BL/6 mice. Sci Rep 2018; 8: 4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Roda E, Bottone MG, Biggiogera M, Milanesi G, Coccini T. Pulmonary and hepatic effects after low dose exposure to nanosilver: early and long‐lasting histological and ultrastructural alterations in rat. Toxicol Rep 2019; 6: 1047–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Zhou L, Li P, Zhang M et al Carbon black nanoparticles induce pulmonary fibrosis through NLRP3 inflammasome pathway modulated by miR‐96 targeted FOXO3a. Chemosphere 2020; 241: 125075. [DOI] [PubMed] [Google Scholar]
- 126. Majeski EI, Paintlia MK, Lopez AD, Harley RA, London SD, London L. Respiratory reovirus 1/L induction of intraluminal fibrosis, a model of bronchiolitis obliterans organizing pneumonia, is dependent on T lymphocytes. Am J Pathol 2003; 163: 1467–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Helal MG, Said E. Carvedilol attenuates experimentally induced silicosis in rats via modulation of P‐AKT/mTOR/TGFβ1 signaling. Int Immunopharmacol 2019; 70: 47–55. [DOI] [PubMed] [Google Scholar]
- 128. Li N, Feng F, Wu K, Zhang H, Zhang W, Wang W. Inhibitory effects of astragaloside IV on silica‐induced pulmonary fibrosis via inactivating TGF‐β1/Smad3 signaling. Biomed Pharmacother 2019; 119: 109387. [DOI] [PubMed] [Google Scholar]
- 129. Porter DW, Millecchia LL, Willard P et al Nitric oxide and reactive oxygen species production causes progressive damage in rats after cessation of silica inhalation. Toxicol Sci 2006; 90: 188–197. [DOI] [PubMed] [Google Scholar]
- 130. Yu IJ, Song KS, Chang HK et al Recovery from manual metal arc‐stainless steel welding‐fume exposure induced lung fibrosis in Sprague‐Dawley rats. Toxicol Lett 2003; 143: 247–259. [DOI] [PubMed] [Google Scholar]