

The initial phase of the Korean Genome Project (Korea1K) releases 1094 whole genomes, along with data of 79 clinical traits.
Abstract
We present the initial phase of the Korean Genome Project (Korea1K), including 1094 whole genomes (sequenced at an average depth of 31×), along with data of 79 quantitative clinical traits. We identified 39 million single-nucleotide variants and indels of which half were singleton or doubleton and detected Korean-specific patterns based on several types of genomic variations. A genome-wide association study illustrated the power of whole-genome sequences for analyzing clinical traits, identifying nine more significant candidate alleles than previously reported from the same linkage disequilibrium blocks. Also, Korea1K, as a reference, showed better imputation accuracy for Koreans than the 1KGP panel. As proof of utility, germline variants in cancer samples could be filtered out more effectively when the Korea1K variome was used as a panel of normals compared to non-Korean variome sets. Overall, this study shows that Korea1K can be a useful genotypic and phenotypic resource for clinical and ethnogenetic studies.
INTRODUCTION
The Korean population [estimated census population size close to 85 million (M)] has been thought to be highly homogeneous with few large-scale admixture events in the past (1–4). However, little formal scrutiny has been given to these claims. Several Korean whole genomes and exomes (5, 6) have been reported since the first Korean genome data (SJK) were published in 2008 (7), including the first Korean reference genome sequence (KOREF_S) (8) and 40 unrelated individuals (KOREF_C) that formed the basis of KoVariome, the Korean genomic variation database (9). Before the current study, at least 100 whole genomes of Korean individuals were available worldwide (5, 10). However, although a global whole-genome project (the multiethnicity 1000 genome project) that aims to characterize global human genetic diversity contains over 2500 genomes, including Chinese and Japanese, it does not include Korean samples yet (11).
There has also been an effort to generate ethnicity-specific reference genome sequences, and several human variomes have been generated to expand the coverage of human genome diversity, including the UK10K (12), the Genome of the Netherlands (GoNL) project (13), and the pan-African genome (14). In 2015, the consequences of strong founder effects were demonstrated in the Icelandic population by sequencing 2636 genomes (15). In the Danish population study, 150 trios were used to de novo assemble a reference genome, and they provide detailed data on structural variations and many complex genomic regions, including the major histocompatibility complex and major regions of the Y chromosome (16). In East Asia, the 1KJPN project yielded data on 1070 Japanese genomes (17), and another recent dataset identified selection signatures in the Japanese population from 2234 Japanese whole-genome data (18). In contrast, the original KoVariome database contained only 50 Korean whole-genome sequences without clinical information at the time of publication (9), although its sample size has subsequently increased to >100 genomes. Despite these large genome sequencing projects in numerous populations, little biochemical and clinical data and limited information regarding genotype-phenotype association for the participants have been collected to characterize the population’s health and disease states.
Here, we introduce a dataset comprising 1094 Korean whole genomes of which 1007 genomes were newly generated in combination with systematically acquired clinical and biochemical measurement information from the blood and urine of the participants. This Korea1K set represents the first-phase release of the Korean Genome Project (KGP). KGP is a joint project by the Personal Genome Project at Harvard Medical School, the National Center for Standard Reference Data of Korea, Clinomics Inc., and the Korean Genomics Center of Ulsan National Institute of Science and Technology (UNIST). These genomes have been sequenced to a high sequencing depth (~31× on average) using Illumina HiSeq X10, and we used these data to characterize single-nucleotide variants (SNVs), indels, copy number variations (CNVs), transposable element (TE) insertion, and human leukocyte antigen (HLA) type in the Korean population and contrast the Korean data with similar data from other populations. The majority of the genomic data (984 samples) were from volunteers with clinical information on 79 quantitative traits that were measured at Ulsan University Hospital. To evaluate the practical utility of this large genomic dataset, we performed a genome-wide association study (GWAS) using the information of the 79 quantitative clinical traits. We also quantified the effectiveness of our dataset as a reference panel by analyzing 19 previously published Korean gastric cancer patient genomes (19).
RESULTS
SNVs and indels in Korea1K dataset
Whole-genome sequencing (WGS) data from 1007 blood or saliva samples (984 samples with clinical and biochemical information) were generated with an average sequencing depth of 31× and pooled with sequencing data from an additional 87 blood or saliva samples (without clinical information) from the KoVariome database (9). In total, 1094 complete genomes, including 916 unrelated and healthy individuals, mostly from the Ulsan metropolitan region, were compared to the human genome reference (hg38). A total of 39.2 M SNVs and 7.6 M indels were called from the dataset (table S1). We filtered out false-positive variants due to the sequencing batch effect (figs. S1 and S2) and related individuals, yielding a set of variants containing 34 M SNVs and 4.8 M indels. We divided the variants into five categories based on their allele frequency in the Korean population (singleton: allele count = 1; doubleton: allele count = 2; rare: allele count of >2 and allele frequency of ≤0.01; common: allele frequency of >0.01 but ≤0.05; and very common: allele frequency of >0.05; Fig. 1A). Highlighting the power of our large dataset, approximately half of the variants that we identified were classified as singleton or doubleton (allele count of ≤2), and unexpectedly, more than 70% of them are not reported in dbSNP (v150) (20). On the other hand, less than 20% of the variants were classified as very common (allele frequency of >0.05), with more than 94% of these variants previously reported in dbSNP (v150) (20). A total of 96.6% of the very common SNVs overlapped with KoVariome (9), compared to only 12.4% of rare SNVs (fig. S3). The number of variants that have an allele frequency of >0.01 was similar to other non-African populations (1KGP non-African and 3.5KJPN), while there was a far higher number of variants, which have an allele frequency of ≤0.01 than KoVariome because of Korea1K’s much larger sample size (fig. S4). On the basis of the final set of variants, each individual showed on average ~4.42 M variants (3.58 M very common, 0.4 M common, 0.31 M rare, 0.46 M doubleton, and 0.85 M singleton variants), of which 8928 and 918 were nonsynonymous and loss of function (LoF), respectively.
Fig. 1. Variants statistics and discovery rate of the novel variants.
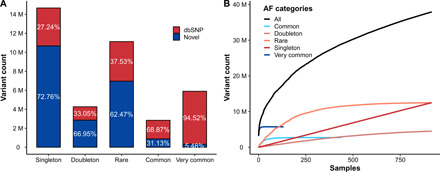
(A) Number of variants in the Korea1K dataset in all autosomal regions categorized on the basis of allele frequencies (AFs). Singleton, allele count = 1; doubleton, allele count = 2; rare, allele count of >2 and allele frequency of ≤0.01; common, allele frequency of >0.01 and allele frequency of ≤0.05; and very common, allele frequency of >0.05. (B) The number of novel variants as a function of unrelated Korean genome samples.
Next, we classified each variant into 1 of 19 different variant classes (i.e., intergenic and intronic) based on its functional impact and location in the genome (fig. S5). LoF variants (nonsense, nonstop, splicing site, and indel variants) in the Korea1K set had a higher ratio of rare, doubleton, or singleton variants than other regional classes, indicating the effect of purifying selection on these variants. In addition, the allele (site) frequency spectrum of unrelated individuals was used to estimate the fraction under selection pressure in different genomic regions (21). We confirmed that LoF variants had the highest fraction of sites under negative selection (fig. S6). We applied the same comparative analysis to the entire gene set and found that 16 genes showed high purifying selection pressure, which was even stronger than the selection for nonsynonymous variants across the genome. Four genes showed negative values suggestive of positive selection pressure (fig. S7). Regarding indels, the Korea1K set displayed more deletions (2,573,411) than insertions (2,155,644), possibly resulting from skewed variant calling (fig. S8). Indels in protein-coding regions displayed higher peaks among in-frame indels based on their length, indicating purifying selection (fig. S9) (22).
The discovery rate of newly observed variants from unrelated individual genomes is a method for quantifying genomic diversity in a given population (23). The pattern of newly observed, unshared variants of unrelated Korean genomes was investigated using the five allele frequency categories (Fig. 1B). The discovery rate of very common (allele frequency of >0.05) variants saturated after 132 samples (14.4%), while the rate of singleton and doubleton variants was still increasing after analyzing all 916 healthy unrelated samples. When we compared the count of newly observed variants in unrelated individuals against previously published KoVariome, unexpectedly, Korea1K showed a slightly higher rate of novel variant discovery than KoVariome (fig. S10; Korea1K, 101,866; KoVariome, 48,051 for 50th individual). This increase might have been caused by implementing newer versions of the variant calling pipeline and the human genome reference. As expected, this also confirms that more sequenced genomes are needed to sufficiently cover very rare variants in the Korean population.
The Korea1K set contained 266,081 nonsynonymous SNVs. Among them, 118,417 and 117,414 were categorized as protein damaging by PolyPhen (24) (possibly damaging, 46,116; probably damaging, 72,301) and SIFT (25) (deleterious, 117,414), respectively. In total, 87,671 variants were predicted as protein damaging by both programs, and their allele frequency is skewed toward rare frequencies, while benign or tolerated variants are skewed toward common frequencies, again indicating purifying selection (fig. S11).
When mitochondrial and chromosomal Y haplogroups among the Korean individuals (figs. S12 and S13) were investigated, the common types identified were D (34.19%), B (13.89%), and M (13.80%) mitochondrial and O (73.49%), C (16.9%), and N (6.58%) chromosomal Y (26, 27). The O male haplogroup is widely distributed in East Asia and Southeast Asia, while the C haplogroup is prominently distributed in East Asia and Northeast Asia (26). We also identified other fairly common mitochondrial haplogroup types (A, G, and F) in East Asia (28).
Genomic features of Koreans compared to other populations
We assessed the genetic distinctiveness of our Korea1K sample using principal components analysis (PCA) with the small size variants (SNP and indel) in our dataset and 1KGP. As previously reported, principal components PC1 and PC2 with worldwide populations showed a separate East Asian group (Fig. 2A). Although Koreans, Chinese, and Japanese are genetically very close relative to all other individuals (29), we found that those three populations clustered distinctly from each other (Fig. 2B). This pattern was replicated by ADMIXTURE analysis with K = 3 (fig. S14).
Fig. 2. Comparison with other populations.
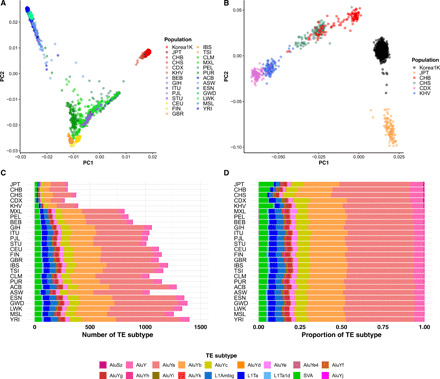
Results of PCA of Korea1K and the 1KGP set of (A) worldwide populations and (B) East Asian samples. (C) The number of TE insertions with significantly different allele frequencies between the Korea1K set and the population. (D) The proportion of differential TE insertions. Colors indicate TE subtypes. Abbreviation for populations is same population code as 1KGP (ACB, African Caribbean; ASW, African Ancestry in Southwest USA; BEB, Bengali; CDX, Dai Chinese; CEU, Utah residents with Northern and Western European ancestry; CHB, Han Chinese; CHS, Southern Han Chinese; CLM, Colombian; ESN, Esan; FIN, Finnish; GBR, British; GIH, Gujarati; GWD, Gambian Mandinka; IBS, Iberian; ITU, Telugu; JPT, Japanese; KHV, Kinh Vietnamese; LWK, Luhya; MSL, Mende; MXL, Mexican Ancestry; PEL, Peruvian; PJL, Punjabi; PUR, Puerto Rican; STU, Tamil; TSI, Toscani; and YRI, Yoruba).
To investigate functionally relevant variants, we extracted 1048 ClinVar pathogenic variants found in Korea1K. Among them, 242 variants had an allele frequency greater than 0.1 in Korea1K, which is high for pathogenic variants (fig. S15). We also found 35 drug-response variants annotated in ClinVar (fig. S16), and 11 of them displayed significantly different allele frequencies from those of the Chinese or Japanese individuals in the 1KGP set, highlighting the importance of population-specific datasets when interpreting pathogenic or drug-response variants. For example, the variant rs4961 in ADD1 had the highest frequency in the Korea1K compared to other populations and is associated with hypertension and responsiveness to furosemide and spironolactone as shown in a European study (30, 31). However, no significant association with blood pressure was found in our GWAS using the Korea1K set (see the “Genome-wide association study” section for details).
We identified CNVs, TE insertions, and HLA-1 haplotypes (see Supplementary Materials and Methods) as WGS data can identify numerous variants from complex or highly variable nongenic regions (16, 17). Korea1K contains 1441 CNV loci, and 80% of them overlapped with the CNV set from the entire 1KGP samples while not overlapping with segmental duplication regions. Four common CNVs (sample frequency of >0.05) overlapped with those in the 1KGP set and were validated by a secondary CNV caller (Supplementary Materials), which, in turn, contained five protein-coding genes (figs. S17 and S18, extended data table S1). Among the four common CNVs, Korea1K had a copy duplication of CLPS, a pancreatic colipase, which is involved in dietary lipid hydrolysis.
For TE polymorphisms, the patterns of TE insertions between the Korean and other populations were investigated by PCA (figs. S19 and S20 and table S2). PC1 and PC2 identified that four superpopulations (Africans, Asians, Americans, and Europeans) were well separated from each other, whereas subpopulations in East Asia were not. Therefore, a specific TE insertion pattern alone is insufficient to finely differentiate subpopulations in East Asia, although the genomic diversity was clearly reflected in the allele frequency distribution (figs. S21 and S22). TE insertions with significantly different allele frequencies between Koreans and 26 other populations in the 1KGP set were enumerated, and as expected, Korea1K displayed significantly fewer differential TE insertions compared to East Asian populations than non–East Asians (Fig. 2, C and D and extended data table S2). Furthermore, ALU and SINE-VNTR-ALUs (SVA) displayed a greater proportion of differential TE insertions than Long interspersed nuclear element (LINE) in JPT, CHB, and CHS, probably because of different insertion rates on the TE types.
We also compared HLA types in Korea1K with those in publicly available databases containing European, American, and Asian HLA frequencies (figs. S23 and S24). Our HLA allele frequency pattern was very similar to the HLA haplotype distribution of Korean samples from the public database. HLA types A*24:02, A*26:01, A*31:01, B*40:02, and B*52:01 displayed significantly lower allele frequencies in the Korean population relative to the Japanese population (Fisher’s exact test P = 3.61 × 10−49, 7.09 × 10−8, 1.34 × 10−12, 9.61 × 10−12, and 3.13 × 10−42, respectively), while types A*33:03 and B*44:03 had higher allele frequencies (Fisher’s exact test P = 3.10 × 10−46 and 1.00 × 10−5, respectively). Although the Japanese are genetically very close to the Korean, the HLA-type profiles of these populations are considerably different. However, we identified similarities in the Asian populations; for example, types A*33:03, A*02:06, and B*58:01 displayed relatively high allele frequencies, while types A*02:01, A*03:01, A*01:01, A*32:01, A*68:01, B*07:02, B*44:02, and B*08:01 displayed low frequencies in Asian populations (Korean, Japanese, and Chinese populations) compared to other groups.
GWAS based on clinical traits
Thanks to its extensive genomic coverage, population-scale WGS data are more effective than chip-based approaches at identifying statistically significant associations with quantitative traits and diseases (12). It is even more powerful if matched clinical or phenotypic data are available for the genomes (32). In the Korea1K set, we were able to quantify 79 quantitative clinical traits measured from 984 samples (extended data table S3) through health checks provided to the KGP participants. We analyzed associations by fitting additive genetic models with relevant covariates for 79 quantitative traits and 6,658,227 variants [5,932,215 SNVs and 726,012 indels; minor allele frequency (MAF) of >1%] from 823 unrelated individuals of the 984 samples. The analysis resulted in 467 variants that were statistically linked via GWAS to 11 quantitative traits (P < 7.5 × 10−9, the Bonferroni-corrected significance threshold). The 467 variants were clumped into 15 independent loci on eight chromosomes, and 11 of them contained previously reported variants linked to a trait. We found that 11 index variants were not present on the commonly used Illumina Omni 2.5 human SNP chip (Fig. 3, Table 1, figs. S25 to S28, and extended data tables S4 and S5). Among the 11 loci of reported variants, 9 contained variants reported in the GWAS catalog (33), but their index variants were newly identified in this study. Of the 15 independent loci, we could identify 1 previously unidentified locus in MAMASTR, which is associated with cancer marker (carbohydrate antigen 19-9 and carcinoembryonic antigen). A previously unidentified locus was also found in WDPCP, which is associated with body fat percentage. Another previously unidentified locus in SERPINA7 was found, which is associated with triiodothyronine. We also found two loci on chromosome 2 (rsID: rs28946889, trait: total bilirubin, P = 1.85 × 10−23; rs662799, neutral fat, P = 4.22 × 10−10) that have been previously identified in two Korean GWAS (34, 35). The MAFs in the previously unidentified loci were markedly lower than those previously reported when we compared the MAFs of GWAS variants in these previously reported and the unreported loci (fig. S29). This means that large-scale variomes from WGS data help identify low-frequency alleles and unreported loci via whole genome–based GWA studies.
Fig. 3. Manhattan plot of the reported loci via a GWAS.
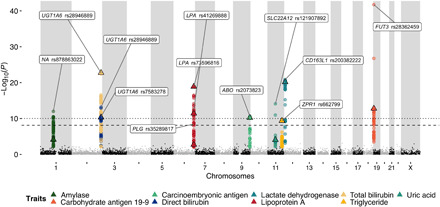
Each color indicates a different clinical trait. The most significant reported markers in the loci are denoted with triangles. The dashed line indicates the threshold for genome-wide significance (7.5 × 10−9). The dotted line indicates the threshold for study-wide significance (9.5 × 10−11).
Table 1. List of traits with index variants located in previously reported loci.
Highlighted rows indicate unreported variants with higher significance values, located in the same linkage disequilibrium block with reported variants.
| Trait | Chromosome | Position | rsID | Gene symbol | P | MAF |
| Carbohydrate antigen 19-9 |
chr19 | 5,844,781 | rs28362459 | FUT3 | 1.83 × 10−42 | 0.341 |
| Total bilirubin | chr2 | 233,762,816 | rs28946889 | UGT1A6 | 1.85 × 10−23 | 0.439 |
| Lactate dehydrogenase |
chr12 | 7,437,350 | rs200382222 | CD163L1 | 1.40 × 10−21 | 0.186 |
| Lipoprotein A | chr6 | 160,596,331 | rs73596816 | LPA | 1.31 × 10−19 | 0.038 |
| Uric acid | chr11 | 64,593,747 | rs121907892 | SLC22A12 | 7.94 × 10−15 | 0.013 |
| Direct bilirubin | chr2 | 233,762,816 | rs28946889 | UGT1A6 | 6.43 × 10−14 | 0.439 |
| Lipoprotein A | chr6 | 160,607,693 | rs41269888 | LPA | 4.30 × 10−13 | 0.454 |
| Amylase | chr1 | 103,348,267 | rs878863022 | N/A | 1.01 × 10−12 | 0.476 |
| Carcinoembryonic antigen |
chr9 | 133,257,129 | rs2073823 | ABO | 2.53 × 10−11 | 0.228 |
| Total bilirubin | chr2 | 233,708,761 | rs7583278 | UGT1A6 | 2.89 × 10−11 | 0.100 |
| Neutral fat | chr11 | 116,792,991 | rs662799 | ZPR1 | 4.22 × 10−10 | 0.315 |
| Lipoprotein A | chr6 | 160,703,093 | rs35289817 | PLG | 3.45 × 10−09 | 0.203 |
Korea1K imputation panel
Haplotype-based imputation is a cost-effective method to capture human genetic variation for clinical purposes. Crucially, the accuracy of imputation is improved when a population-specific reference panel is used (17). We first constructed a phased reference panel using the Korea1K dataset and the combination of Korea1K and 1KGP panel by using SHAPEIT2 (36). The imputation accuracy for the three reference panels [Korea1K (n = 1059), 1KGP (n = 2504), and Korea1K + 1KGP (rephased, n = 3563)] was evaluated by imputing prephased variants from the matched normal sample of the 19 Korean patients with gastric cancer. This test set was imputed with the three reference panels using Minimac3 (37). The accuracy was evaluated by comparing the squared Pearson correlation coefficient between the real genotypes and the dosage of imputed genotypes. The Korea1K panel showed better correlation with true genotypes at low allele frequencies than the 1KGP panel, and the combined Korea1K + 1KGP panel had the best accuracy overall, indicating the usefulness of Korea1K set for the imputation of Korean SNV data (Fig. 4).
Fig. 4. Imputation performance evaluation.
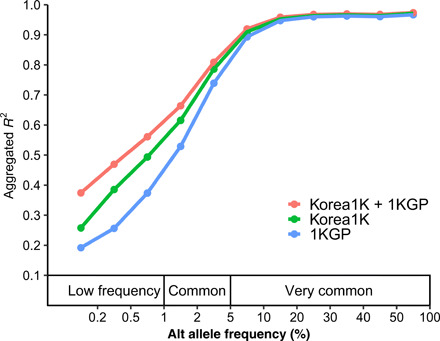
The x axis indicates alternative (Alt) allele frequency in the Korea1K set. The y axis represents the aggregated R2 values of SNVs. We used SNVs that were overlapped by imputed results across all panels.
The Korea1K dataset as a panel of normals for cancer genomics studies
Variome databases can be used as reference panels for genome-wide clinical studies (38). One of the practical examples is cancer mutation analysis. Many cancer-related studies need matched normal control samples to filter germline variants from the entire call set (39, 40). However, it is sometimes unfeasible to have matched normal sequencing data of the same patients with cancer. As an alternative to matched normal, a large-scale reference panel of variants can potentially serve as a control set for cancer genomics studies to filter out germline variants (41).
To estimate the power of our Korea1K dataset as an ethnicity-specific panel of normals, we evaluated the effectiveness of Korea1K, 3.5KJPN (17) and 1KGP (11) as a panel of normal for previously reported Korean gastric cancer datasets (19). We first identified true somatic and germline variants by comparing WGS data from cancer and matched normal tissues. Then, for the test sets, we classified the variants from the cancer tissue into tentative somatic or germline, on the basis of allele frequency cutoffs of the reference panels. When a target variant has a lower allele frequency value than the cutoff value in the reference panel, the variant was classified as a tentative somatic variant. If it has a higher allele frequency value, then it was classified as a tentative germline variant. Thereafter, we generated statistical measures of the classification performance of the datasets by comparing predicted variant categories based on multiple stepwise allele frequency cutoffs with the true variant categories (Fig. 5).
Fig. 5. Performance of the variant classification using different panels of normals.
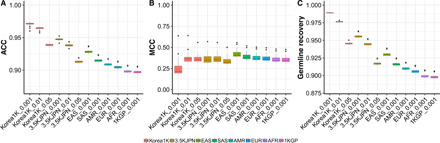
(A) Accuracy (ACC) of classification. (B) Matthews correlation coefficient (MCC) values. (C) Germline recovery rate. The x axis indicates the used reference panel and allele frequency cutoff concatenated by the underscore symbol. EAS, SAS, AMR, EUR, and AFR indicate East Asian, South Asian, American, European, and African populations in 1KGP, respectively.
Although the 3.5KJPN set contained the largest number of variants, the Korea1K dataset had the highest accuracy of prediction of germline and somatic variants (Fig. 5A; Korea1K with an allele frequency cutoff of 0.01: 96.42%; 3.5KJPN with an allele frequency cutoff of 0.01: 93.83% on average). Furthermore, the Korea1K set had similar Matthews correlation coefficient values to 3.5KJPN, indicating a similar classification performance (Fig. 5B; Korea1K with an allele frequency cutoff of 0.01: 0.38; 3.5KJPN with an allele frequency cutoff of 0.01: 0.37 on average). Germline variants were predicted with the highest accuracy using the Korea1K set (Fig. 5C), while the recovery rate for somatic variants was low (fig. S30). Since we used samples from Korean individuals with cancer, an overall increase in similarity was noted with the Korea1K dataset. This leads to the speculation that a greater number of true somatic variants were filtered out using the Korea1K set than with other datasets; nonetheless, the density of variants of cancer-related genes from the Cancer Gene Census (CGC) database was the highest in the Korea1K-filtered set (fig. S31). Moreover, when the germline filtering criterion was set to an allele frequency of 1.5%, the Korea1K set had the highest density of CGC genes. Since we used a lift-over reference panel for 3.5KJPN and 1KGP, we also applied the same approach as with the Korea1K variants on only lift-over possible regions and confirmed that there were no qualitative changes to the results (figs. S32 to S34). These results highlight the possible benefits of using an ethnicity-specific variome database for cancer genome analyses of the same or a very closely related ethnic group(s).
DISCUSSION
This study presents a comprehensive WGS analysis of 1094 Koreans (Korea1K), which is a mixture of existing KoVariome (9) and newly added 1007 genomes with clinical information. On the basis of our analysis, Korean population is genetically homogeneous compared to other East Asians, and this is probably due to geopolitical isolation in the past thousands of years. However, we speculate that, although Koreans are fairly homogeneous, more than 1000 samples are necessary to map the Korean human genome diversity judging from the assessment of the discovery rate of newly observed variants (allele frequency of >0.05 variants saturated after 132 samples, while the rate of singleton and doubleton variants kept increasing even after analyzing all the 916 healthy unrelated samples). Despite a large amount of genomic data, coupled with clinical information, our CNV and TE analyses did not identify anything unusual or unique. This could be because short-read DNA-sequencing methods have an inherent difficulty in detecting structural variations that cannot be easily resolved bioinformatically, and we must perform long-read sequencing using the same samples in the future to map novel associations between these complex variants and phenotypically accessible traits. Furthermore, we note that our samples are mostly from the Ulsan metropolitan area and cannot reflect the whole Korean peninsula, although Ulsan has a population size of 1 M and the residents are from all across the peninsula due to rapid industrialization. Together, the sample size of 1094 from mostly Ulsan is still far from sufficient to represent the Korean population or to map latent genomic structural variations.
Our investigation of using Korea1K as a panel of normals for cancer genomics studies can be a small stepping stone for an efficient germline prefiltering process for cancer genome analyses in the future. However, it is still questionable how much actual benefit such ethnicity-specific variome-based filtering can bring to cancer genome analyses in real clinical settings, especially for rare or individual-specific variant analysis. Nevertheless, the large-scale Korean variome database constructed herein is potentially applicable in studies on various cancers and other diseases of Koreans and can indirectly help reduce the cost of certain genetic analyses. This kind of personal whole-genome dataset combined with common health check–derived clinical information is possibly a good exemplary path for an ethnicity-relevant reference panel for future personalized medical applications for Koreans.
METHODS
Sample collection and sequencing
Informed consent was obtained from all individuals for their participation in the Korean Ulsan genome project, which comprises two subprojects. All clinical information was examined by the Ulsan University Hospital. In total, 696 samples were curated in the Ulsan University Hospital Biobank, from which samples were received thereafter. Further, 311 samples were collected by us. We downloaded data from 87 Korean samples from KoVariome (9), which collected volunteers from all across the Korean peninsula. Sample collection and sequencing was approved by the Institutional Review Board (IRB) of the Ulsan National Institute of Science and Technology (UNISTIRB-15-19-A and UNISTIRB-16-13-C). Genomic DNA was isolated from human blood samples, using the DNeasy Blood & Tissue kit (Qiagen, Germany) in accordance with the manufacturer’s protocol. Genomic DNA from saliva samples was isolated using the GeneAll Exgene trademark clinic SV mini kit. Extracted DNA was quantified using the Quant-iT BR assay kit (Invitrogen). High–molecular weight genomic DNA was sheared using a Covaris S2 ultra sonicator system to obtain fragments of appropriate sizes. Libraries with short 350–base pair (bp) inserts for paired-end reads were prepared using the TruSeq Nano DNA sample prep kit in accordance with the manufacturer’s protocol for Illumina-based sequencing. The products were quantified using the Bioanalyzer 2100 (Agilent, Santa Clara, CA, USA), and raw data were generated using an Illumina HiSeq X10 platform (Illumina). Clusters were generated using paired-end 2 × 150-bp cycle sequencing reads via resequencing. Further image analysis and base calling were carried out using the Illumina real-time analysis program (https://sapac.illumina.com/informatics/sequencing-data-analysis.html) with default parameters following the manufacturer’s instructions. The quality of the base in the read was checked by FastQC (ver. 0.11.5; www.bioinformatics.babraham.ac.uk/projects/fastqc/) (table S3).
Variant calling
Adapter contamination was trimmed using Cutadapt (ver. 1.9.1) (42) with a forward adapter (′GATCGGAAGAGCACACGTCTGAACTCCAGTCAC′) and reverse adapter (′GATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT′) and with a minimum read length of 50 bp after trimming. Thereafter, trimmed reads were mapped to the hg38 reference, using BWA-MEM (ver. 0.7.16a) with the “-M” option (43). Mapped BAM files were sorted by coordination, using Picard (ver. 2. 14. 0) with the Sortsam module. Duplicate reads were marked using Picard (ver. 2. 14. 0) with the MarkDuplicates module. The mapping quality was recalibrated using the BaseRecalibrator tool in the Genome Analysis Tool Kit (GATK) (ver. 3.7) (44). gVCF files were generated by HaplotypeCaller in GATK (44) with “-stand_call_conf 30-ERC GVCF” option. SNVs and indels were jointly genotyped from the gVCF files by GenotypeGVCFs in GATK (44). The called variants were annotated using Variants Effect Predictor (VEP) ver. 92 (45), and the fraction was estimated under negative selection, using the script of Moon and Akey (https://github.com/moon-s/fraction-under-selection) (21). For estimation of a fraction under selection pressure for each protein-coding gene, we selected genes that have more than 250 alternative allele count sums, since the small number of allele count may not produce proper site frequency spectrums. The following annotated variants were assigned to LoF mutations: “Frame_Shift_Del”, “Frame_Shift_Ins”, “In_Frame_Del”, “In_Frame_Ins”, “Nonsense_Mutation”, “Nonstop_Mutation”, or “Splice_Site”. The detailed methods for calling CNV, TE insertion, and HLA typing were noted to Supplementary Materials and Methods.
Batch effect removal
Each sample was labeled in accordance with its sequencing library preparation protocol, sequencing company, and the date of sending blood samples or libraries to the company. Twelve technical batches were identified. The batch effect was assessed via PCA, using EIGENSOFT (ver. 6.1.4) (46), using variants and samples in accordance with the following criteria:
For variants:
-
1)
Biallelic SNVs with a MAF of ≥5%.
-
2)
P values of the Hardy-Weinberg Equilibrium (HWE) test >0.05.
-
3)
Genotype missing rate of <0.01.
Thereafter, filtered variants were pruned on the basis of linkage disequilibrium (LD), using PLINK (47) (ver. 1.9b) with “--indep-pairwise 200 4 0.1”,, leaving 101,326 SNVs. For individual selection, closely related individuals identified on the basis of an identity by descent (IBD), estimated in PLINK (47), were filtered. All pairs with an IBD value of >0.125 were extracted (corresponding to third-degree relatives) and clustered to a family group. Until no pairs of relatives remained, each family group was then reduced as follows:
-
1)
The sample with the highest number of pairs in the family group was eliminated.
-
2)
The sample with the highest missing calls among LD-pruned SNVs was eliminated if there are several samples with the same number of pairs in the family group.
To identify variants exhibiting the batch effect, we used logistic regression models for all variants as follows:
-
1)
The variant was eliminated if it was a batch-specific variant compared to all other batches.
-
2)
Each batch was paired with another, resulting in all possible combinations. The variant was eliminated if it was significant in any of the combinations.
In total, 6,348,049 variant positions were significantly associated with the technical batch (P ≤ 0.01) and eliminated from the original set. We used the quality by depth (QD) value in a joint VCF file for plotting variants’ quality distribution.
PCA and ADMIXTURE with the 1KGP genome data
The interpopulation genomic structure was evaluated by projecting the first two PCs determined via PCA of SNVs from Korea1K samples and 1KGP without closely related individuals. We selected and merged variants and from the Korea1K and 1KGP sets in accordance with the following criteria:
-
1)
Biallelic SNVs with a MAF of ≥5%.
-
2)
Biallelic SNVs with an HWE P >10−6.
-
3)
Biallelic SNVs with a missing genotype rate of <0.01.
Extracted variants were LD pruned using “--indep 50 5 2” in PLINK (47), yielding 153,633 sites. PCA was carried out using the EIGENSOFT program (46). ADMIXTURE (48) analysis was performed from K = 2 to K = 14 based on the same variants set as PCA. We plotted an ADMIXTRUE plot for K = 3, which showed the smallest cross-validation error rate across the Ks.
Mitochondrial and chromosome Y haplogroup analysis
Mitochondrial haplogroups were identified via Haplogrep (ver. 2.1.13) (49, 50), and the Yfitter tool (ver. 0.2) (51) was used to identify Y chromosome haplogroups. We prepared the input files for the Yfitter program by converting hg38 coordination to hg19 coordination, using CrossMap (52) (ver. 0.2.7).
Genome-wide association study
For the GWAS, 823 individuals, 6,658,227 variants, and 79 traits were selected in accordance with the following criteria:
For individuals:
-
1)
Individuals whose clinical traits were examined.
-
2)
Individuals having no rare diseases.
-
3)
Individuals having no kinship within the selected samples.
For variants:
-
4)
SNVs and indels having a MAF of ≥1%.
-
5)
SNVs and indels having an HWE P > 10−6.
-
6)
SNVs and indels having a missing genotype rate of <0.01.
The GWAS was performed exclusively using quantitative traits. GWA analysis was performed using linear regression under an additive genetic model. Age, age2, sex, body mass index (BMI), and the first 10 principal components were included as covariates. BMI was excluded from covariates in the GWAS for BMI itself and the degree of obesity. The genome-wide significance threshold was determined to be 7.51 × 10−9 through Bonferroni correction (0.05/6,658,227). The study-wide significance threshold was determined using the equation (0.05 / (the number of tested traits × the number of tested variants)). Variants were grouped into the loci with “--clump-p1 0.0000001 --clump-kb 1000 --clump-r2 0.1” options with PLINK (version 1.9) (47). For each locus, previously reported variants associated with the trait of interest were examined in order from the most significant variant with the National Human Genome Research Insitutue (NHGRI) GWAS catalog (33) (P ≤ 5 × 10−8, ver. 2018-12-07).
Imputation panel construction
To construct the Korea1K imputation reference panel, 1059 healthy individuals that had no rare diseases with a total of 28,692,913 autosomal biallelic variants with a missing genotype call rate of <0.1 and minor allele count of >1 (not a singleton) were selected. The variants were phased into haplotype using SHAPEIT2 (version v2.r904) (36), and the Korea1K set was used to construct a rephased imputation panel using the 1KGP reference panel. We chose an alternative allele from Korea1K if the alternative allele of Korea1K and 1KGP is discordant during the merging step. To evaluate the imputation accuracy of the reference panels, we separately processed matched normal samples from previously published 19 unrelated Korean patients with gastric cancers obtained from National Center for Biotechnology Information (NCBI; SRP014574 and SRA057772). For a test set, we extracted 1,302,490 variants that were present in Illumina Omni 2.5 chip from the 19 individuals and obtained 1,243,087 prephased SNVs using SHAPEIT2 (36). The prephased test set was imputed using the prepared reference panels by Minimac3 (ver. 2.0.1) (37). Imputation accuracies were estimated using squared Pearson correlation coefficients (R2) between the true genotypes and imputed genotype dosages.
Processing of previously reported samples from patients with cancer and classification of variants
WGS data from 19 previously reported Korean individuals with gastric cancer were obtained from NCBI (SRP014574 and SRA057772) and mapped to hg38 using BWA-MEM (ver. 0.7.15) with the “-M” option, and the SAM format was converted to the BAM format using SAMtools (ver. 1.4) (53). The BAM files were sorted using SAMtools (ver. 1.4), and duplicated reads were marked using the MarkDuplicates module in Picard tools. Base realignment and recalibration of the base quality score were carried out using GATK (ver. 3.7) (44). Variants from all samples were called using GATK HaplotypeCaller with the joint calling mode. All identified variants were annotated using the Ensembl VEP (ver. 92.1) (45). Furthermore, the variants were annotated to the cancer-related genes from the CGC database (54). If a variant was identified exclusively in a sample from an individual with cancer, then we treated these variants as somatic. Since 3.5KJPN only provides information regarding variants with hg19 coordinates, the variants were converted to the hg38 coordination, using the lift-over tool in the University of California, Santa Cruz genome browser (55). Thereafter, we merged the information regarding allele frequencies in the Korea1K and 3.5KJPN sets to the annotated variants based on genomic location and allelic information. Annotated variants were classified into tentative somatic or germline variants based on the allele frequency cutoffs in each panel (Korea1K, 3.5KJPN, and 1KGP). If a variant showed a lower allele frequency value than the cutoff value or was not presented in the reference panel, then the variant was classified as a tentative somatic variant. If not, then it was classified as a tentative germline variant. Thereafter, the classification and the true sets were compared to evaluate the performance of the datasets.
Supplementary Material
Acknowledgments
We appreciate all participants and Ulsan citizens who supported this project. We also thank M. Jung, G.-H. Kim, C.-h. Song, I.-H. Park, H. R. Cho, Y. Ham, Y. Park, B. Yoo, J. Oh, S. Shin, H.-o. Jeong, S. Hong, J. Y. Park, S. Han, S. Jho, E. Jun, and S. G. Hong, and B. Park for supporting this project. We thank the Korea Institute of Science and Technology Information (KISTI) for providing us the Korea Research Environment Open NETwork (KREONET). We thank our collaborators in NCSRD, KRISS, and C.-G. Kim. Funding: This work was supported by the U-K BRAND Research Fund (1.190007.01) of UNIST, Research Project Funded by Ulsan City Research Fund (1.190033.01) of UNIST, Research Project Funded by Ulsan City Research Fund (1.200047.01) of UNIST, and Research Project Funded by Ulsan City Research Fund (2.180016.01) of UNIST. This work was also supported by the Technology Innovation Program (20003641, Development and Dissemination on National Standard Reference Data) funded by the Ministry of Trade, Industry and Energy (MOTIE, Korea). This work was also supported by internal funding of Clinomics Inc. The Ulsan University Hospital Biobank provided DNA sample and clinical data from 696 participants (60SA2016001-002, 60SA2016001-003, 60SA2016001-005, 60SA2017002-001, and 60SA2017002-004). Author contributions: S.J., Y.B., and J.B. wrote the manuscript. S.J., Y.B., Y.C., Y.J., S.K., Jaeyoung Jang, and Jinho Jang conducted the data analysis. Y.K. and C.K. performed wet-laboratory experiments. J.S., N.K., and N.-H.P. collected the samples and clinical information. S.J., Y.S.C., Y.P., B.-C.K., E.-S.S., B.C.K., G.C., S.L., and J.B. designed the study. S.J., Y.B., Y.C., Y.J., S.K., A.B., Y.J.K., S.G P., J.K., H.-M.K., D.B., A.M., J.S.E., S.L., and J.B. revised the manuscript. G.C., S.L., and J.B. jointly supervised the study. Competing interests: Y.B., C.K., Y.S.C., H.-M.K., Y.P., B.-C.K., and D.B. are employees and B.C.K. and J.B. are the CEOs of Clinomics Inc. H.-M.K., B.C.K., J.B., and Y.S.C. have an equity interest in the company. S.J., Y.B., Y.C., S.K., Jinho Jang, C.K., Y.K., J.S., N.K., Y.J., S.G.P., Y.S.C., Y.P., B.-C.K., B.C.K., S.L., and J.B. are inventors on a patent application related to this work filed by UNIST (no. 10-2019-0116344, 20 September 2019). S.J., Y.B., Y.C., Y.S.C., B.-C.K., B.C.K., S.L., and J.B. are inventors on a patent application related to this work filed by UNIST (no. 10-2020-0037631, 27 March 2020). The authors declare no other competing interests. Data and materials availability: All the different types of data will be publicly and freely available for scientific research. Allele frequency information of SNVs, indels, CNVs, and TE insertion can be found at http://1000genomes.kr. Raw sequencing data, individual genotype information, and clinical trait data will be as easily and freely available as possible upon request and after approval from the Korean Genomics Center’s review board in UNIST. Information about the KGP and other data sharing can be found at http://koreangenome.org.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/22/eaaz7835/DC1
REFERENCES AND NOTES
- 1.Siska V., Jones E. R., Jeon S., Bhak Y., Kim H. M., Cho Y. S., Kim H., Lee K., Veselovskaya E., Balueva T., Gallego-Llorente M., Hofreiter M., Bradley D. G., Eriksson A., Pinhasi R., Bhak J., Manica A., Genome-wide data from two early Neolithic East Asian individuals dating to 7700 years ago. Sci. Adv. 3, e1601877 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.HUGO Pan-Asian SNP Consortium, Abdulla M. A., Ahmed I., Assawamakin A., Bhak J., Brahmachari S. K., Calacal G. C., Chaurasia A., Chen C. H., Chen J., Chen Y. T., Chu J., Cutiongco-de la Paz E. M., De Ungria M. C., Delfin F. C., Edo J., Fuchareon S., Ghang H., Gojobori T., Han J., Ho S. F., Hoh B. P., Huang W., Inoko H., Jha P., Jinam T. A., Jin L., Jung J., Kangwanpong D., Kampuansai J., Kennedy G. C., Khurana P., Kim H. L., Kim K., Kim S., Kim W. Y., Kimm K., Kimura R., Koike T., Kulawonganunchai S., Kumar V., Lai P. S., Lee J. Y., Lee S., Liu E. T., Majumder P. P., Mandapati K. K., Marzuki S., Mitchell W., Mukerji M., Naritomi K., Ngamphiw C., Niikawa N., Nishida N., Oh B., Oh S., Ohashi J., Oka A., Ong R., Padilla C. D., Palittapongarnpim P., Perdigon H. B., Phipps M. E., Png E., Sakaki Y., Salvador J. M., Sandraling Y., Scaria V., Seielstad M., Sidek M. R., Sinha A., Srikummool M., Sudoyo H., Sugano S., Suryadi H., Suzuki Y., Tabbada K. A., Tan A., Tokunaga K., Tongsima S., Villamor L. P., Wang E., Wang Y., Wang H., Wu J. Y., Xiao H., Xu S., Yang J. O., Shugart Y. Y., Yoo H. S., Yuan W., Zhao G., Zilfalil B. A.; Indian Genome Variation Consortium , Mapping human genetic diversity in Asia. Science 326, 1541–1545 (2009). [DOI] [PubMed] [Google Scholar]
- 3.R. O. K. M. F. Affairs, Total Number of Overseas Koreans (2017).
- 4.Databank, Population Total (2018).
- 5.Seo J.-S., Rhie A., Kim J., Lee S., Sohn M.-H., Kim C.-U., Hastie A., Cao H., Yun J.-Y., Kim J., Kuk J., Park G. H., Kim J., Ryu H., Kim J., Roh M., Baek J., Hunkapiller M. W., Korlach J., Shin J.-Y., Kim C., De novo assembly and phasing of a Korean human genome. Nature 538, 243–247 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Lee S., Seo J., Park J., Nam J.-Y., Choi A., Ignatius J. S., Bjornson R. D., Chae J.-H., Jang I.-J., Lee S., Park W.-Y., Baek D., Choi M., Korean variant archive (KOVA): A reference database of genetic variations in the Korean population. Sci. Rep. 7, 4287 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahn S.-M., Kim T.-H., Lee S., Kim D., Ghang H., Kim D.-S., Kim B.-C., Kim S.-Y., Kim W.-Y., Kim C., Park D., Lee Y. S., Kim S., Reja R., Jho S., Kim C. G., Cha J.-Y., Kim K.-H., Lee B., Bhak J., Kim S.-J., The first Korean genome sequence and analysis: Full genome sequencing for a socio-ethnic group. Genome Res. 19, 1622–1629 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cho Y. S., Kim H., Kim H.-M., Jho S., Jun J. H., Lee Y. J., Chae K. S., Kim C. G., Kim S., Eriksson A., Edwards J. S., Lee S., Kim B. C., Manica A., Oh T.-K., Church G. M., Bhak J., An ethnically relevant consensus Korean reference genome is a step towards personal reference genomes. Nat. Commun. 7, 13637 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim J., Weber J. A., Jho S., Jang J., Jun J. H., Cho Y. S., Kim H.-M., Kim H., Kim Y., Chung O. S., Kim C. G., Lee H. J., Kim B. C., Han K., Koh I. S., Chae K. S., Lee S., Edwards J. S., Bhak J., KoVariome: Korean national standard reference variome database of whole genomes with comprehensive SNV, indel, CNV, and SV analyses. Sci. Rep. 8, 5677 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hong D., Park S. S., Ju Y. S., Kim S., Shin J. Y., Kim S., Yu S. B., Lee W. C., Lee S., Park H., Kim J. I., Seo J. S., TIARA: A database for accurate analysis of multiple personal genomes based on cross-technology. Nucleic Acids Res. 39, D883–D888 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.1000 Genomes Project Consortium, Auton A., Brooks L. D., Durbin R. M., Garrison E. P., Kang H. M., Korbel J. O., Marchini J. L., McCarthy S., McVean G. A., Abecasis G. R., A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.UK10K Consortium, Walter K., Min J. L., Huang J., Crooks L., Memari Y., McCarthy S., Perry J. R., Xu C., Futema M., Lawson D., Iotchkova V., Schiffels S., Hendricks A. E., Danecek P., Li R., Floyd J., Wain L. V., Barroso I., Humphries S. E., Hurles M. E., Zeggini E., Barrett J. C., Plagnol V., Richards J. B., Greenwood C. M., Timpson N. J., Durbin R., Soranzo N., The UK10K project identifies rare variants in health and disease. Nature 526, 82–90 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Genome of the Netherlands Consortium , Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat. Genet. 46, 818–825 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Sherman R. M., Forman J., Antonescu V., Puiu D., Daya M., Rafaels N., Boorgula M. P., Chavan S., Vergara C., Ortega V. E., Levin A. M., Eng C., Yazdanbakhsh M., Wilson J. G., Marrugo J., Lange L. A., Williams L. K., Watson H., Ware L. B., Olopade C. O., Olopade O., Oliveira R. R., Ober C., Nicolae D. L., Meyers D. A., Mayorga A., Knight-Madden J., Hartert T., Hansel N. N., Foreman M. G., Ford J. G., Faruque M. U., Dunston G. M., Caraballo L., Burchard E. G., Bleecker E. R., Araujo M. I., Herrera-Paz E. F., Campbell M., Foster C., Taub M. A., Beaty T. H., Ruczinski I., Mathias R. A., Barnes K. C., Salzberg S. L., Assembly of a pan-genome from deep sequencing of 910 humans of African descent. Nat. Genet. 51, 30–35 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gudbjartsson D. F., Helgason H., Gudjonsson S. A., Zink F., Oddson A., Gylfason A., Besenbacher S., Magnusson G., Halldorsson B. V., Hjartarson E., Sigurdsson G. T., Stacey S. N., Frigge M. L., Holm H., Saemundsdottir J., Helgadottir H. T., Johannsdottir H., Sigfusson G., Thorgeirsson G., Sverrisson J. T., Gretarsdottir S., Walters G. B., Rafnar T., Thjodleifsson B., Bjornsson E. S., Olafsson S., Thorarinsdottir H., Steingrimsdottir T., Gudmundsdottir T. S., Theodors A., Jonasson J. G., Sigurdsson A., Bjornsdottir G., Jonsson J. J., Thorarensen O., Ludvigsson P., Gudbjartsson H., Eyjolfsson G. I., Sigurdardottir O., Olafsson I., Arnar D. O., Magnusson O. T., Kong A., Masson G., Thorsteinsdottir U., Helgason A., Sulem P., Stefansson K., Large-scale whole-genome sequencing of the Icelandic population. Nat. Genet. 47, 435–444 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Maretty L., Jensen J. M., Petersen B., Sibbesen J. A., Liu S., Villesen P., Skov L., Belling K., Theil Have C., Izarzugaza J. M. G., Grosjean M., Bork-Jensen J., Grove J., Als T. D., Huang S., Chang Y., Xu R., Ye W., Rao J., Guo X., Sun J., Cao H., Ye C., van Beusekom J., Espeseth T., Flindt E., Friborg R. M., Halager A. E., le Hellard S., Hultman C. M., Lescai F., Li S., Lund O., Løngren P., Mailund T., Matey-Hernandez M. L., Mors O., Pedersen C. N. S., Sicheritz-Pontén T., Sullivan P., Syed A., Westergaard D., Yadav R., Li N., Xu X., Hansen T., Krogh A., Bolund L., Sørensen T. I. A., Pedersen O., Gupta R., Rasmussen S., Besenbacher S., Børglum A. D., Wang J., Eiberg H., Kristiansen K., Brunak S., Schierup M. H., Sequencing and de novo assembly of 150 genomes from Denmark as a population reference. Nature 548, 87–91 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Nagasaki M., Yasuda J., Katsuoka F., Nariai N., Kojima K., Kawai Y., Yamaguchi-Kabata Y., Yokozawa J., Danjoh I., Saito S., Sato Y., Mimori T., Tsuda K., Saito R., Pan X., Nishikawa S., Ito S., Kuroki Y., Tanabe O., Fuse N., Kuriyama S., Kiyomoto H., Hozawa A., Minegishi N., Douglas Engel J., Kinoshita K., Kure S., Yaegashi N.; ToMMo Japanese Reference Panel Project, Yamamoto M., Rare variant discovery by deep whole-genome sequencing of 1,070 Japanese individuals. Nat. Commun. 6, 8018 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okada Y., Momozawa Y., Sakaue S., Kanai M., Ishigaki K., Akiyama M., Kishikawa T., Arai Y., Sasaki T., Kosaki K., Suematsu M., Matsuda K., Yamamoto K., Kubo M., Hirose N., Kamatani Y., Deep whole-genome sequencing reveals recent selection signatures linked to evolution and disease risk of Japanese. Nat. Commun. 9, 1631 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoon K., Lee S., Han T. S., Moon S. Y., Yun S. M., Kong S. H., Jho S., Choe J., Yu J., Lee H. J., Park J. H., Kim H. M., Lee S. Y., Park J., Kim W. H., Bhak J., Yang H. K., Kim S. J., Comprehensive genome- and transcriptome-wide analyses of mutations associated with microsatellite instability in Korean gastric cancers. Genome Res. 23, 1109–1117 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sherry S. T., Ward M. H., Kholodov M., Baker J., Phan L., Smigielski E. M., Sirotkin K., dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moon S., Akey J. M., A flexible method for estimating the fraction of fitness influencing mutations from large sequencing data sets. Genome Res. 26, 834–843 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clark T. G., Andrew T., Cooper G. M., Margulies E. H., Mullikin J. C., Balding D. J., Functional constraint and small insertions and deletions in the ENCODE regions of the human genome. Genome Biol. 8, R180 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Telenti A., Pierce L. C. T., Biggs W. H., di Iulio J., Wong E. H. M., Fabani M. M., Kirkness E. F., Moustafa A., Shah N., Xie C., Brewerton S. C., Bulsara N., Garner C., Metzker G., Sandoval E., Perkins B. A., Och F. J., Turpaz Y., Venter J. C., Deep sequencing of 10,000 human genomes. Proc. Natl. Acad. Sci. U.S.A. 113, 11901–11906 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adzhubei I., Jordan D. M., Sunyaev S. R., Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 76, 7.20.1–7.20.41 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sim N. L., Kumar P., Hu J., Henikoff S., Schneider G., Ng P. C., SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 40, W452–W457 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jin H. J., Kwak K. D., Hammer M. F., Nakahori Y., Shinka T., Lee J. W., Jin F., Jia X., Tyler-Smith C., Kim W., Y-chromosomal DNA haplogroups and their implications for the dual origins of the Koreans. Hum. Genet. 114, 27–35 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Jin H. J., Tyler-Smith C., Kim W., The peopling of Korea revealed by analyses of mitochondrial DNA and Y-chromosomal markers. PLOS One 4, e4210 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanaka M., Cabrera V. M., González A. M., Larruga J. M., Takeyasu T., Fuku N., Guo L. J., Hirose R., Fujita Y., Kurata M., Shinoda K., Umetsu K., Yamada Y., Oshida Y., Sato Y., Hattori N., Mizuno Y., Arai Y., Hirose N., Ohta S., Ogawa O., Tanaka Y., Kawamori R., Shamoto-Nagai M., Maruyama W., Shimokata H., Suzuki R., Shimodaira H., Mitochondrial genome variation in eastern Asia and the peopling of Japan. Genome Res. 14, 1832–1850 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y., Lu D., Chung Y. J., Xu S., Genetic structure, divergence and admixture of Han Chinese, Japanese and Korean populations. Hereditas 155, 19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cusi D., Barlassina C., Azzani T., Casari G., Citterio L., Devoto M., Glorioso N., Lanzani C., Manunta P., Righetti M., Rivera R., Stella P., Troffa C., Zagato L., Bianchi G., Polymorphisms of α-adducin and salt sensitivity in patients with essential hypertension. Lancet 349, 1353–1357 (1997). [DOI] [PubMed] [Google Scholar]
- 31.Psaty B. M., Smith N. L., Heckbert S. R., Vos H. L., Lemaitre R. N., Reiner A. P., Siscovick D. S., Bis J., Lumley T., Longstreth W. T. Jr., Rosendaal F. R., Diuretic therapy, the α-Adducin gene variant, and the risk of myocardial infarction or stroke in persons with treated hypertension. JAMA 287, 1680–1689 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Bycroft C., Freeman C., Petkova D., Band G., Elliott L. T., Sharp K., Motyer A., Vukcevic D., Delaneau O., O’Connell J., Cortes A., Welsh S., Young A., Effingham M., McVean G., Leslie S., Allen N., Donnelly P., Marchini J., The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.MacArthur J., Bowler E., Cerezo M., Gil L., Hall P., Hastings E., Junkins H., McMahon A., Milano A., Morales J., Pendlington Z. M., Welter D., Burdett T., Hindorff L., Flicek P., Cunningham F., Parkinson H., The new NHGRI-EBI catalog of published genome-wide association studies (GWAS catalog). Nucleic Acids Res. 45, D896–D901 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kang T.-W., Kim H.-J., Ju H., Kim J.-H., Jeon Y.-J., Lee H.-C., Kim K.-K., Kim J.-W., Lee S., Kim J. Y., Kim S.-Y., Kim Y. S., Genome-wide association of serum bilirubin levels in Korean population. Hum. Mol. Genet. 19, 3672–3678 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim Y. J., Go M. J., Hu C., Hong C. B., Kim Y. K., Lee J. Y., Hwang J. Y., Oh J. H., Kim D. J., Kim N. H., Kim S., Hong E. J., Kim J. H., Min H., Kim Y., Zhang R., Jia W., Okada Y., Takahashi A., Kubo M., Tanaka T., Kamatani N., Matsuda K.; MAGIC consortium, Park T., Oh B., Kimm K., Kang D., Shin C., Cho N. H., Kim H. L., Han B. G., Lee J. Y., Cho Y. S., Large-scale genome-wide association studies in East Asians identify new genetic loci influencing metabolic traits. Nat. Genet. 43, 990–995 (2011). [DOI] [PubMed] [Google Scholar]
- 36.McCarthy S., Das S., Kretzschmar W., Delaneau O., Wood A. R., Teumer A., Kang H. M., Fuchsberger C., Danecek P., Sharp K., Luo Y., Sidore C., Kwong A., Timpson N., Koskinen S., Vrieze S., Scott L. J., Zhang H., Mahajan A., Veldink J., Peters U., Pato C., van Duijn C., Gillies C. E., Gandin I., Mezzavilla M., Gilly A., Cocca M., Traglia M., Angius A., Barrett J. C., Boomsma D., Branham K., Breen G., Brummett C. M., Busonero F., Campbell H., Chan A., Chen S., Chew E., Collins F. S., Corbin L. J., Smith G. D., Dedoussis G., Dorr M., Farmaki A. E., Ferrucci L., Forer L., Fraser R. M., Gabriel S., Levy S., Groop L., Harrison T., Hattersley A., Holmen O. L., Hveem K., Kretzler M., Lee J. C., McGue M., Meitinger T., Melzer D., Min J. L., Mohlke K. L., Vincent J. B., Nauck M., Nickerson D., Palotie A., Pato M., Pirastu N., McInnis M., Richards J. B., Sala C., Salomaa V., Schlessinger D., Schoenherr S., Slagboom P. E., Small K., Spector T., Stambolian D., Tuke M., Tuomilehto J., Berg L., van Rheenen W., Volker U., Wijmenga C., Toniolo D., Zeggini E., Gasparini P., Sampson M. G., Wilson J. F., Frayling T., de Bakker P. I., Swertz M. A., McCarroll S., Kooperberg C., Dekker A., Altshuler D., Willer C., Iacono W., Ripatti S., Soranzo N., Walter K., Swaroop A., Cucca F., Anderson C. A., Myers R. M., Boehnke M., McCarthy M. I., Durbin R.; Haplotype Reference Consortium , A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 48, 1279–1283 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Das S., Forer L., Schönherr S., Sidore C., Locke A. E., Kwong A., Vrieze S. I., Chew E. Y., Levy S., McGue M., Schlessinger D., Stambolian D., Loh P. R., Iacono W. G., Swaroop A., Scott L. J., Cucca F., Kronenberg F., Boehnke M., Abecasis G. R., Fuchsberger C., Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dou Y., Gold H. D., Luquette L. J., Park P. J., Detecting somatic mutations in normal cells. Trends Genet. 34, 545–557 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cibulskis K., Lawrence M. S., Carter S. L., Sivachenko A., Jaffe D., Sougnez C., Gabriel S., Meyerson M., Lander E. S., Getz G., Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alioto T. S., Buchhalter I., Derdak S., Hutter B., Eldridge M. D., Hovig E., Heisler L. E., Beck T. A., Simpson J. T., Tonon L., Sertier A. S., Patch A. M., Jäger N., Ginsbach P., Drews R., Paramasivam N., Kabbe R., Chotewutmontri S., Diessl N., Previti C., Schmidt S., Brors B., Feuerbach L., Heinold M., Gröbner S., Korshunov A., Tarpey P. S., Butler A. P., Hinton J., Jones D., Menzies A., Raine K., Shepherd R., Stebbings L., Teague J. W., Ribeca P., Giner F. C., Beltran S., Raineri E., Dabad M., Heath S. C., Gut M., Denroche R. E., Harding N. J., Yamaguchi T. N., Fujimoto A., Nakagawa H., Quesada V., Valdés-Mas R., Nakken S., Vodák D., Bower L., Lynch A. G., Anderson C. L., Waddell N., Pearson J. V., Grimmond S. M., Peto M., Spellman P., He M., Kandoth C., Lee S., Zhang J., Létourneau L., Ma S., Seth S., Torrents D., Xi L., Wheeler D. A., López-Otín C., Campo E., Campbell P. J., Boutros P. C., Puente X. S., Gerhard D. S., Pfister S. M., McPherson J. D., Hudson T. J., Schlesner M., Lichter P., Eils R., Jones D. T. W., Gut I. G., A comprehensive assessment of somatic mutation detection in cancer using whole-genome sequencing. Nat. Commun. 6, 10001 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hiltemann S., Jenster G., Trapman J., van der Spek P., Stubbs A., Discriminating somatic and germline mutations in tumor DNA samples without matching normals. Genome Res. 25, 1382–1390 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin M., Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011). [Google Scholar]
- 43.Li H., Durbin R., Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Poplin R., Ruano-Rubio V., DePristo M. A., Fennell T. J., Carneiro M. O., Van der Auwera G. A., Kling D. E., Gauthier L. D., Levy-Moonshine A., Roazen D., Shakir K., Thibault J., Chandran S., Whelan C., Lek M., Gabriel S., Daly M. J., Neale B., MacArthur D. G., Banks E., Scaling accurate genetic variant discovery to tens of thousands of samples.bioRxiv, 201178 (2017). [Google Scholar]
- 45.McLaren W., Gil L., Hunt S. E., Riat H. S., Ritchie G. R. S., Thormann A., Flicek P., Cunningham F., The Ensembl variant effect predictor. Genome Biol. 17, 122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Price A. L., Patterson N. J., Plenge R. M., Weinblatt M. E., Shadick N. A., Reich D., Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M. A. R., Bender D., Maller J., Sklar P., de Bakker P. I. W., Daly M. J., Sham P. C., PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alexander D. H., Novembre J., Lange K., Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van Oven M., PhyloTree build 17: Growing the human mitochondrial DNA tree. Forensic Sci. Int.-Gen. Supp. Ser. 5, e392–e394 (2015). [Google Scholar]
- 50.Weissensteiner H., Pacher D., Kloss-Brandstätter A., Forer L., Specht G., Bandelt H. J., Kronenberg F., Salas A., Schönherr S., HaploGrep 2: Mitochondrial haplogroup classification in the era of high-throughput sequencing. Nucleic Acids Res. 44, W58–W63 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jostins L., Xu Y., McCarthy S., Ayub Q., Durbin R., Barrett J., Tyler-Smith C., YFitter: Maximum likelihood assignment of Y chromosome haplogroups from low-coverage sequence data. arXiv Preprint arXiv, 1407.7988 (2014). [Google Scholar]
- 52.Zhao H., Sun Z., Wang J., Huang H., Kocher J. P., Wang L., CrossMap: A versatile tool for coordinate conversion between genome assemblies. Bioinformatics 30, 1006–1007 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.; 1000 Genome Project Data Processing Subgroup , The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tate J. G., Bamford S., Jubb H. C., Sondka Z., Beare D. M., Bindal N., Boutselakis H., Cole C. G., Creatore C., Dawson E., Fish P., Harsha B., Hathaway C., Jupe S. C., Kok C. Y., Noble K., Ponting L., Ramshaw C. C., Rye C. E., Speedy H. E., Stefancsik R., Thompson S. L., Wang S., Ward S., Campbell P. J., Forbes S. A., COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 47, D941–D947 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kent W. J., Sugnet C. W., Furey T. S., Roskin K. M., Pringle T. H., Zahler A. M., Haussler D., The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Abyzov A., Urban A. E., Snyder M., Gerstein M., CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 21, 974–984 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Csardi M. G., Package. igraph 3, 214–217 (2013). [Google Scholar]
- 58.Zerbino D. R., Achuthan P., Akanni W., Amode M. R., Barrell D., Bhai J., Billis K., Cummins C., Gall A., Girón C. G., Gil L., Gordon L., Haggerty L., Haskell E., Hourlier T., Izuogu O. G., Janacek S. H., Juettemann T., To J. K., Laird M. R., Lavidas I., Liu Z., Loveland J. E., Maurel T., McLaren W., Moore B., Mudge J., Murphy D. N., Newman V., Nuhn M., Ogeh D., Ong C. K., Parker A., Patricio M., Riat H. S., Schuilenburg H., Sheppard D., Sparrow H., Taylor K., Thormann A., Vullo A., Walts B., Zadissa A., Frankish A., Hunt S. E., Kostadima M., Langridge N., Martin F. J., Muffato M., Perry E., Ruffier M., Staines D. M., Trevanion S. J., Aken B. L., Cunningham F., Yates A., Flicek P., Ensembl 2018. Nucleic Acids Res. 46, D754–D761 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sudmant P. H., Rausch T., Gardner E. J., Handsaker R. E., Abyzov A., Huddleston J., Zhang Y., Ye K., Jun G., Fritz M. H., Konkel M. K., Malhotra A., Stütz A. M., Shi X., Casale F. P., Chen J., Hormozdiari F., Dayama G., Chen K., Malig M., Chaisson M. J. P., Walter K., Meiers S., Kashin S., Garrison E., Auton A., Lam H. Y. K., Mu X. J., Alkan C., Antaki D., Bae T., Cerveira E., Chines P., Chong Z., Clarke L., Dal E., Ding L., Emery S., Fan X., Gujral M., Kahveci F., Kidd J. M., Kong Y., Lameijer E. W., McCarthy S., Flicek P., Gibbs R. A., Marth G., Mason C. E., Menelaou A., Muzny D. M., Nelson B. J., Noor A., Parrish N. F., Pendleton M., Quitadamo A., Raeder B., Schadt E. E., Romanovitch M., Schlattl A., Sebra R., Shabalin A. A., Untergasser A., Walker J. A., Wang M., Yu F., Zhang C., Zhang J., Zheng-Bradley X., Zhou W., Zichner T., Sebat J., Batzer M. A., McCarroll S. A.; 1000 Genomes Project Consortium, Mills R. E., Gerstein M. B., Bashir A., Stegle O., Devine S. E., Lee C., Eichler E. E., Korbel J. O., An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boeva V., Popova T., Bleakley K., Chiche P., Cappo J., Schleiermacher G., Janoueix-Lerosey I., Delattre O., Barillot E., Control-FREEC: A tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics 28, 423–425 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gardner E. J., Lam V. K., Harris D. N., Chuang N. T., Scott E. C., Pittard W. S., Mills R. E.; 1000 Genomes Project Consortium, Devine S. E., The mobile element locator tool (MELT): Population-scale mobile element discovery and biology. Genome Res. 27, 1916–1929 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rishishwar L., Tellez Villa C. E., Jordan I. K., Transposable element polymorphisms recapitulate human evolution. Mob. DNA 6, 21 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.1000 Genomes Project Consortium, Abecasis G. R., Altshuler D., Auton A., Brooks L. D., Durbin R. M., Gibbs R. A., Hurles M. E., McVean G. A., A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Szolek A., Schubert B., Mohr C., Sturm M., Feldhahn M., Kohlbacher O., OptiType: Precision HLA typing from next-generation sequencing data. Bioinformatics 30, 3310–3316 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.González-Galarza F. F., Takeshita L. Y. C., Santos E. J. M., Kempson F., Maia M. H. T., da Silva A. L. S., Teles e Silva A. L., Ghattaoraya G. S., Alfirevic A., Jones A. R., Middleton D., Allele frequency net 2015 update: New features for HLA epitopes, KIR and disease and HLA adverse drug reaction associations. Nucleic Acids Res. 43, D784–D788 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/22/eaaz7835/DC1