

A biomimetic platelet based on assembling peptides initiates artificial coagulation for tumor therapy.
Abstract
Platelets play a critical role in the regulation of coagulation, one of the essential processes in life, attracting great attention. However, mimicking platelets for in vivo artificial coagulation is still a great challenge due to the complexity of the process. Here, we design platelet-like nanoparticles (pNPs) based on self-assembled peptides that initiate coagulation and form clots in blood vessels. The pNPs first bind specifically to a membrane glycoprotein (i.e., CD105) overexpressed on angiogenetic endothelial cells in the tumor site and simultaneously transform into activated platelet-like nanofibers (apNFs) through ligand-receptor interactions. Next, the apNFs expose more binding sites and recruit and activate additional pNPs, forming artificial clots in both phantom and animal models. The pNPs are proven to be safe in mice without systemic coagulation. The self-assembling peptides mimic platelets and achieve artificial coagulation in vivo, thus providing a promising therapeutic strategy for tumors.
INTRODUCTION
Platelets are associated not only with hemostasis and thrombosis but also with other primary pathophysiological activities. The diverse functions, mechanisms, and emerging therapeutic potency of platelets have been extensively studied (1). Recently, mimicking artificial platelets have been developed using peptide-decorated nanoparticles (2–4), surface coated with fibrinogen microspheres and microgel (5, 6), glycoprotein-conjugated liposomes (7), and polymer nanofibers (8) to mimic the platelet’s adhesion or aggregation mechanism for homeostatic application. The platelet membrane–coated biomimetic drug delivery systems have been demonstrated with improved delivery performance (9, 10). The design of artificial platelets is basically based on the features of platelet membrane including glycoprotein, combining fibrinogen, but there is less investigation on the morphology transformation of platelets to mimic function of platelets for initiating the coagulation process because of the complexity of the platelet-initiated coagulation process. The intrinsic coagulation process is initiated on the collagen underlying the damaged endothelium. Circulating platelets bind to the collagen receptor and are activated; afterward, they change the morphology from disk to stellate shape. In the meantime, the new receptor becomes exposed on the surfaces of the stellate platelet for further platelet binding, activation, and aggregation (11–14). This process leads to a self-amplifying assembly for fast construction of clots with fibrins and red blood cells (RBCs), etc. (Fig. 1A). Inspired by nature, here we design and prepare platelet-like nanoparticles (pNPs) on the basis of self-assembling peptides. From a material science point of view, the pNPs may initiate the coagulation process, involving a three-step cascaded procedure: (i) specifically targeting to the membrane receptor, (ii) ligand-receptor interaction–induced transformation, and (iii) self-amplifying self-assembly. First, pNPs target and bind to endothelial cells (ECs) in vessels and transform into activated platelet-like nanofibers (apNFs), which attach to ECs and provide new binding sites. Second, pNPs further bind and assemble with as-formed apNFs. The self-assembly process is amplified because previously formed apNFs provide more binding sites for continuous pNP activation and self-assembly, leading to the formation of artificial clots. Furthermore, pNPs in nanoparticle formulation are important for the in vivo coagulation process, which may be transported by systemic administration with high-efficiency, overcoming biological barriers and retaining in a specific region (15–17).
Fig. 1. Schematic illustration of pNPs forming artificial clots in situ by mimicking the natural coagulation process.
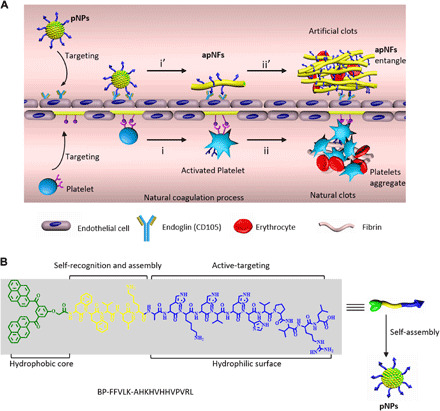
(A) Up: (i’) The biomimetic coagulation process: Intravenously injected pNP raw materials specifically target the CD105 (endoglin) receptor on human umbilical vein endothelial cells (HUVECs) and transform into apNFs by ligand-receptor interaction, similar to platelet activation and transformation. The formed apNFs provide new FFVLK binding sites. (ii′) pNPs further bind and assemble with as-formed apNFs, showing self-amplifying self-assembly and leading to artificial coagulation to construct clots, trapping the RBCs. Down: The natural coagulation process: (i) platelets target to and activate on collagen underlying the damaged endothelium by ligand-acceptor interactions with the exposure of other receptors; (ii) The platelets bind to and assemble on activated platelets to form platelet aggregates by a self-amplifying assembly, leading to the formation of clots with RBCs. (B) Molecular structures of the designed pNP monomers with hydrophobic bis-pyrene building blocks, FFVLK self-recognition and assembly, and AHKHVHHVPVRL targeting and schematic of the self-assembly process into pNPs.
CD105 (endoglin), a transmembrane glycoprotein that is specifically expressed on proliferating ECs, is not readily detectable in normal quiescent endothelium or normal organs and is an ideal target for tumor angiogenesis (i.e., new blood vessel formation) in molecular imaging and cancer therapy (18, 19). In this study, the CD105 receptor was used as an attractive vascular endothelial target for specifically binding and activating pNPs to initiate the artificial coagulation process.
Blood clots may block the blood supply and damage the corresponding tissue and, thus, are generally harmful to humans. However, clots in tumor blood may cut off the nutrition supply of tumor cells to arrest tumor growth (20). In this study, an artificial coagulation process was initiated in the vessels of a tumor-bearing mouse model. As a result, artificial clots were precisely constructed in the blood vessels of the tumor and blocked nutrition supply, exhibiting many advantages for tumor therapy (21). Artificial clots in angiogenesis tumor models based on pNPs (2.4 mg/kg) inhibited the tumor growth at a 53% inhibition rate, which is better than that of clinical angiogenesis-targeting molecular drugs, such as sunitinib (22), which can inhibit tumor growth by 32% at a dose of 8.75 mg/kg, and sorafenib (23), which can inhibit tumor growth by 54% at a dose of 15 mg/kg.
This study is the first example, to the best of our knowledge, to mimic the morphology transformation of platelets, initiating the coagulation process by peptide-based nanomaterials in blood vessels for tumor therapy. The coagulation-like process is expected to be designable and achievable to precisely construct biomedical materials in vivo for improved tumor therapy.
RESULTS AND DISCUSSION
Design and preparation of pNPs
The pNPs are self-assembled from the peptide molecule (BP-FFVLK-AHKHVHHVPVRL) with three functional modules: (i) Hydrophobic bis-pyrenes (BP) with aggregation-induced emission (AIE) effect (24, 25) can induce the formation of fluorescent pNPs in phosphate-buffered saline (PBS) to achieve systemic transportation and direct observation of biodistribution in vivo; (ii) the FFVLK sequence derived from amyloid β peptide can recognize each other and form fibrous structures for assembly and transformation (26); and (iii) the AHKHVHHVPVRL sequence can specifically target to the endoglin (CD105) receptor (27) of activated ECs to locate and induce the transformation of pNPs (Fig. 1B). The three modules are conjugated through peptide bonds to form peptide molecules, self-assembling into pNPs with the function of platelets to initiate the coagulation process.
The peptide monomer of pNPs was prepared according to standard solid-phase peptide synthesis techniques using Fluorenylmethoxycarbonyl (Fmoc)–coupling chemistry, and the molecular structures were confirmed by matrix-assisted laser desorption/ionization–time-of-flight mass spectrometry (MALDI-TOF-MS) (fig. S1). The pNPs were obtained by rapidly precipitating the peptide monomer (28) and dispersing it well in water, assisted by 0.5% dimethyl sulfoxide (DMSO) (DMSO/H2O = 0.5/99.5, v/v) with a final concentration of 20 μM. The transmission electron microscopy (TEM) and dynamic light scattering (DLS) measurements revealed pNPs with a diameter of 23.1 ± 5.8 nm (Fig. 2A). The hydrophobic BP modules probably induced fast aggregation to form the pNPs. BP modules with AIE effect provided insight into the particulation process of pNPs by optical spectroscopy (fig. S2A). In addition, BP showed a maximum fluorescence emission at 520 nm in aggregation states (24) (Fig. 2, B and C), enabling the observation of pNPs in vitro and in vivo. pNPs were stable in water with 0.5% DMSO (fig. S2, B to D). To study the importance of the FFVLK module, which led to its transformation for attachment on ECs and further FFVLK recognition–induced assembly for forming fibrous networks, the control peptide BP-AHKHVHHVPVRL (BA) and corresponding BA NPs were designed and prepared (fig. S3).
Fig. 2. Characterization of pNPs and biomimetic coagulation self-assembly in solution.
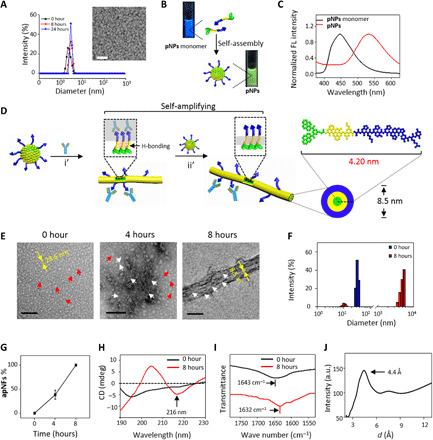
(A) DLS spectra and TEM image of pNPs (20 μM, H2O:DMSO = 99.5:0.5, v/v) indicating that pNPs are stable nanoparticles. Scale bar, 100 nm. (B) Photographs of pNP monomers in DMSO and pNPs in H2O with 0.5% DMSO under ultraviolet illumination and (C) corresponding fluorescence (FL) spectra (λex = 350 nm) showing pNPs with green fluorescence. (D) Schematic process of the structural transformation of pNPs into apNFs induced by CD105 and further self-amplifying self-assembly into fiber bundles by hydrogen bonds: i’, ligand-receptor interaction; ii’, hydrogen-bonding interaction. The diameter of self-assembled apNFs is 8.5 nm, corresponding to the length of two pNP molecules (4.2 nm). (E) TEM images of the structural transformation of pNPs by CD105 (0 to 4 hours) and further hydrogen bonds induced self-assembly (4 to 8 hours). Scale bars, 200 nm. (F) DLS measurement of pNPs cultured with CD105 for 0 and 8 hours showing size variation. (G) Statistical analysis of the ratio of apNFs from (E). The data are presented as means ± SD (n = 5). The circular dichroism (CD) (H) and Fourier transform infrared (I) spectra of pNPs cultured with CD105 for 0 and 8 hours, suggesting the secondary structure was from random coil (pNPs) to β sheet (apNFs). (J) X-ray diffraction diagram of apNFs confirming the crystalline hydrogen bonds between two adjacent β strands.
Biomimetic construction of apNFs in solution
To study the transformation and self-assembly of pNPs upon CD105 to mimic the intrinsic coagulation process, pNPs (20 μM) were incubated with CD105 (0.25 μg/ml; 2.78 × 10−3 μM) for 8 hours in water. The resulting samples exhibited time-dependent morphological changes. pNPs (30.6 ± 7.1 nm) first transformed into apNFs (8.5 ± 1.7 nm, 4 hours) and lastly formed bundles of fibrous networks (8 hours) (Fig. 2, D and E). In the first stage, the pNPs were triggered by CD105 (molar ratio of CD105 to pNP monomer, 1:7200) and transformed into apNFs, exposing the binding sites for FFVLK recognition and self-assembly, similar to platelet transformation on ECs through ligand-receptor interactions, providing new binding sites (11). In the second stage, the pNPs further assembled with the as-formed apNFs through FFVLK modules to form bundles of fibrous networks, similar to platelet aggregation. The DLS data revealed a size variation from 31.6 nm to multidisperse results when pNPs were incubated with CD105, further suggesting that pNPs transformed into apNFs (Fig. 2F). The minority of apNFs at 4 hours and the majority of apNFs bundles at 8 hours indicated that the process was self-amplifying (29) due to the ability of the as-formed apNFs to provide increasingly more FFVLK sites for further assembly (Fig. 2G). Molecular dynamic simulations were used to explore the structural characteristics in water and in CD105 solution, respectively (figs. S4 to S7 and table S1). The results revealed the peptides aggregate into a ball in water and arrange orderly when bound with CD105, supporting experimental results that peptides formed nanoparticles in water and formed nanofibers when incubated with CD105. In addition, the calculation also validated the mechanism insight of self-amplifying self-assembly biomimic process. The BA NPs with the addition of CD105 remained particulate in structure with random coils confirmed by circular dichroism (CD) and Fourier transform infrared spectroscopy (FTIR) (1643 cm−1), suggesting that FFVLK was important for in situ transformation and self-assembly (fig. S3). To study the specificity of pNPs binding to CD105, bovine serum albumin (BSA) was used to replace CD105 to study morphology and size. The results revealed that pNPs (29.5 ± 5.3 nm) remained particulate in morphology, with almost no variation in diameter (fig. S8), indicating the specificity of pNPs for CD105.
CD experiments were performed to check the secondary structure of pNPs upon CD105 incubation. As shown in Fig. 2H, the fresh pNPs had a random coil structure with a negative signal at 195 nm, most likely because the strong hydrophobic interactions of BP modules induced the fast aggregation of peptides. Incubation of pNPs with CD105 for 8 hours induced the characteristic signals of a hydrogen-bonded β sheet structure (50.5%) for apNFs, with a negative CD signal at 216 nm (30, 31). The parallel β sheet structure of apNFs was confirmed by the peak at 1632 cm−1 in the FTIR spectra (32). In contrast, pNPs exhibited a broad absorption peak at 1643 cm−1, corresponding to a random coil (Fig. 2I). The β sheet hydrogen bonds in apNFs were further confirmed by x-ray diffraction (XRD), which revealed a strong Bragg reflection at 4.4 Å (Fig. 2J), indicating crystalline hydrogen bonds between two adjacent β strands. The distance of hydrogen bonds in the apNFs was less than 4.7 Å (33, 34), which may be due to the strong π-π interaction of BP units, drawing the two adjacent β strands close. On the basis of these data and the diameter of one single apNF (8.5 nm), we proposed cylinder molecular packing structures for apNFs (Fig. 2D).
Biomimetic construction of apNFs on HUVECs
To validate the biomimetic in situ transformation and self-assembly at the cellular level due to the CD105 receptors, human umbilical vein endothelial cells (HUVECs) that overexpressed CD105 were chosen as the experimental cell line, and MCF-7 cells were chosen as the negative cell line (fig. S9, A and B). The pNPs showed good biocompatibility with the concentration below 50 μM (fig. S9, C and D). Therefore, pNPs (20 μM) were incubated with HUVECs for 4 hours and observed by confocal laser scanning microscopy (CLSM) and scanning electron microscopy (SEM). As shown in Fig. 3A, the green fluorescence signals from BP aggregates were on the cell surface of the HUVECs and gradually increased with culture time (Fig. 3B and fig. S10A). This result suggested that pNPs bound to the CD105 receptors and induced the in situ transformation into apNFs on the surfaces of HUVECs. This process was similar to the way platelets bind to ECs and transform into stellate shape. Furthermore, similar to platelet aggregation, pNPs attached on the as-formed apNFs on cell surfaces through hydrogen-bonded recognition and assembly based on FFVLK. The previously formed apNFs provided increasingly more FFVLK binding sites for further assembly. The abovementioned biomimetic assembly process was self-amplifying, which was confirmed by the careful analysis of fluorescence intensity on cell surfaces that gradually increased in each time interval (Fig. 3D). The pNP-treated MCF-7 cells showed strong intracellular fluorescence (fig. S10B). In addition, HUVECs and MCF-7 cells incubated with BA NPs showed gradual increasing intracellular fluorescence due to internalization (Fig. 3, A and C and fig. S10B). These results indicated that both the targeting sequence (AHKHVHHVPVRL) and the assembling sequence (FFVLK) are important for the self-assembly processes of pNPs on HUVECs.
Fig. 3. Construction of apNFs on HUVECs.
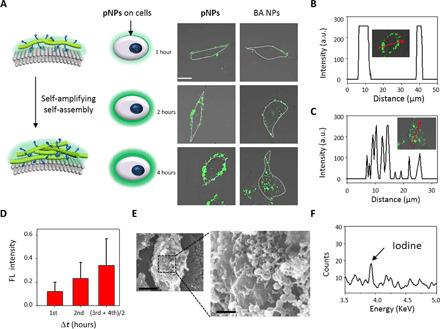
(A) Schematic and CLSM images of pNPs cultured with HUVECs for 1, 2, and 4 hours. The increasing green fluorescence signals on the cell surface indicates that pNPs transformed into apNFs induced by CD105 on the surface of HUVECs compared with internalized BA NPs. The concentration was 20 μM. Scale bar, 20 μm. (B) and (C) are the normalized intensity profiles for (A) regions of interest inserted in the picture across the red arrow in the image. (D) Quantified analysis of (A) showing the increasing fluorescence intensity at every even time interval on the cell surfaces, indicating the self-amplifying process. The data are presented as means ± SD (n = 5). (E) SEM images of HUVECs treated with pNPs for 8 hours showing a nanofibrous network on the cell surface (scale bars, 1 μm). (F) EDS-detected iodine on the surface of HUVECs 8 hours after treatment with pNP-labeled iodine. a.u., arbitrary units.
The morphology of pNP- and BA NP–treated HUVECs was studied by SEM. As shown in Fig. 3E, there was a fibrous network surrounding the cells, which potentially validated the transformation of pNPs to apNFs on the cell surfaces, in accordance with the observation in solution. Moreover, pNPs labeled with iodine were used to further differentiate apNFs from cell surfaces (the biological background) by energy-dispersive spectrometry (EDS) measurement. Elemental iodine was detected with an elemental composition of 0.27% on fibrous structures on the cell surface (Fig. 3F and fig. S10D) but not in PBS-treated HUVECs (fig. S10, C and D). However, the control BA NP–cultured cell surface exhibited some irregular protrusions, which had a morphology similar to that of untreated cells (fig. S10E).
pNPs initiate coagulation-like process and construct artificial clots in a phantom model
To test the coagulation capability, the pNPs (10 mg/ml, 3.76 mM) were incubated with CD105 (1.0 μg/ml) for 8 hours, followed by the addition of RBCs (5.0 μl). The resulting samples were placed dropwise on silicon slides to obtain the artificial clots (Fig. 4A) (35). The SEM images show fibrous networks and trapped RBCs, similar to natural clots, which were obtained using blood from BALB/c nude mice. In nature, the fibrin of natural clots would be degraded by enzymes, such as urokinase and streptokinase (36, 37). In contrast, artificial coagulation based on apNFs with an artificial peptide sequence was expected to be resistant to fibrinolytic enzymes. The as-prepared apNF-based artificial clots were not degraded, and there was almost no change before and after being treated with urokinase (0.5 g/ml; 5 μl) for 10 min. The natural clots were collapsed and dissolved, and only the RBCs were left (Fig. 4A). However, the artificial apNFs may degrade in 7 days eventually (fig. S11).
Fig. 4. Preparation and characterization of artificial clots on the plate surface in the microvasculature model.
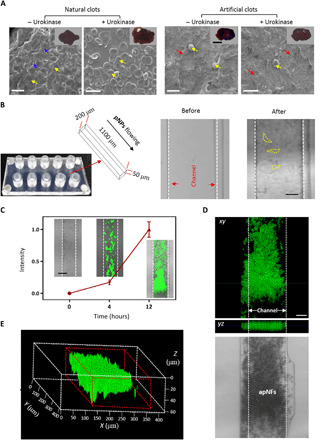
(A) SEM images of natural clots containing fibrins and RBCs, dissolving natural clots containing RBCs incubated with urokinase, artificial clots containing apNFs and RBCs, and artificial clots containing apNFs and RBCs incubated with urokinase (from left to right). apNFs (20 μM, H2O/DMSO = 99.5/0.5, v/v). The blue arrows denote fibrin, the yellow arrows denote RBC, and the red arrows denote apNFs. Scale bars, 10 μm. Inset: Photograph of clots corresponding to the SEM image. Scale bar, 0.25 cm. (B) Macroscopic view and microfluidic channel diagram of the polydimethylsiloxane microfluidic device as a microvasculature model for mimicking the platelet coagulation process (left). The brightfield images showing microfluidic channels seeded by HUVECs before or after 2 weeks to mimic the tumor vessel environment (right). Scale bar, 50 μm. (C) Quantitative fluorescence analysis by measuring the fluorescence signals of apNFs in the microfluidic channel after flowing through the microfluidic channels for 0 to 12 hours. Inset: Confocal images of the channels at different time points. A nonlinear increase in fluorescence intensity with time reveals the self-amplifying assembly of pNPs in the artificial coagulation process. The concentration of pNPs was 100 μM. Scale bar, 100 μm. (D) The CLSM image of the channels covered by HUVECs treated by flowing pNPs for 12 hours indicates the formation of apNF artificial clots with green fluorescence in the microfluidic channel (scale bar, 50 μm) and (E) three-dimensional (3D) reconstruction of segment of artificial clots based on apNFs.
To further test the coagulation-like process and in situ construction of artificial clots based on pNPs, a blood vessel phantom was designed and prepared using a microfluidic channel (Fig. 4B) (38). The HUVECs were cultured inside the channel for 2 weeks to cover the artificial blood vessels, which were similar to capillaries with several tens of micrometers in diameter (Fig. 4B). The flow of pNPs, the BA NPs, and the PBS solution were separately poured into the artificial blood vessels for 12 hours. The results showed that there was markedly green fluorescence in the pNP-treated channels. Moreover, nanofibrous structures with green fluorescence filled up some segments of the channels, indicating that a coagulation-like process occurred and that an artificial clot formed (Fig. 4, C to E and fig. S12, A to C). The remarkable increase in apNFs inside the channels based on quantification of time-dependent fluorescence intensity revealed that the coagulation-like process was self-amplifying (Fig. 4C). In contrast, the BA NP–treated channels showed very low fluorescence, indicating that the biomimetic process markedly increased targeting ability (fig. S12D). The results validated the coagulation-like process in the blood vessel phantom, encouraging us to investigate in vivo applications.
pNPs initiate a coagulation-like process and construct artificial clots in vivo
CD105 is overexpressed on the surface of ECs in angiogenesis. Therefore, an in vivo biomimetic coagulation process was carried out in tumor-bearing mice with abundant angiogenesis. pNPs may accumulate in tumor vessels due to active targeting of the AHKHVHHVPVRL peptide, resulting in a high concentration of pNPs for coagulation-like processes in vivo (Fig. 5A).
Fig. 5. Characterization of artificial clots in vivo.
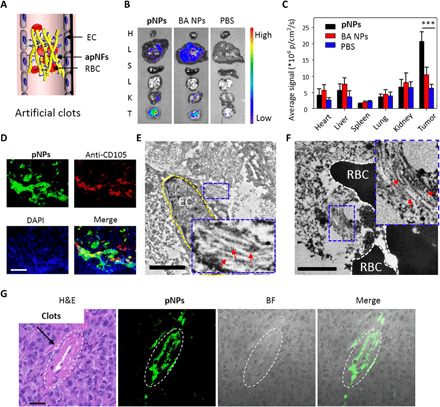
(A) Schematic representation of apNFs trapping RBCs that formed artificial clots in tumor vessels. (B) Ex vivo fluorescence images of tissues from MDA-MB-231 tumor–bearing mice treated with pNPs through intravenous injection at 72 hours after injection. The tissues from up to down are those of the heart, liver, spleen, lungs, kidney, and tumor. High fluorescence intensity is detected in the tumor tissue. (C) Corresponding quantitative analysis of (B). ***P < 0.001. (D) CLSM images of the tumor slices from MDA-MB-231 tumor–bearing mice treated with pNPs through intravenous injection after 72 hours, which were stained with anti-CD105 antibody and 4′,6-diamidino-2-phenylindole (DAPI). The pNPs (green) appear in the vessel domain (red) of the tumor, and the nuclei are denoted as blue. Scale bar, 25 μm. (E and F) Bio-TEM images of tumor slices from MDA-MB-231 tumor–bearing mice treated with pNPs through intravenous injection after 72 hours. The yellow dotted line denotes ECs, and the red arrows indicate apNFs. The black parts are the RBCs. The inserted pictures were magnified nanofibers. Scale bars, 2.0 μm. (G) Hematoxylin and eosin (H&E) staining and CLSM images of tumor slices from MDA-MB-231 tumor–bearing mice treated with pNPs through intravenous injection after 72 hours. The merged clots from H&E and the apNFs from green fluorescence reveal that apNFs form artificial clots, which may block blood. Scale bar, 50 μm. ***P < 0.001.
Nude mice bearing MDA-MB-231 human breast cancer cells as an in vivo model were treated with pNPs (200 μM, 200 μl) by intravenous injection, with BA NPs and PBS as controls. Ex vivo fluorescence imaging of the tissues (heart, liver, spleen, lung, kidney, and tumor) obtained 72 hours after pNP injection showed strong fluorescence signals in the tumors but not in the other organs (Fig. 5, B and C), indicating that the pNPs were successfully transported to the tumor sites. Furthermore, a tissue slice of the tumor was stained with red fluorescent anti-CD105, which was colocalized with green fluorescence from the pNPs, suggesting successful targeting of pNPs to the tumor vessels (Fig. 5D and fig. S13A). The bio-TEM image of the tumor slices exhibits nanofibrous structures, probably due to the apNFs, forming around the ECs and RBCs (Fig. 5, E and F, and fig. S13, B and C). The tumor tissue was further treated by homogenization and centrifugation. apNFs with a diameter of 8.5 nm were found in the supernatant and were basically identical to those in PBS, indicating the formation of apNFs in tumors (fig. S13D).
These results validated the good targeting and accumulation of pNPs in tumor vessels, resulting in coagulation-like processes based on pNPs, involving the attachment on ECs and the transformation into apNFs, followed by self-amplifying self-assembly. In addition, the coagulation-like processes increased the accumulation of pNPs in the tumor sites. As a result of these coagulation-like processes, artificial clots lastly formed in the tumor vessels, which were confirmed by hematoxylin and eosin (H&E) staining of the tumor tissues. As shown in Fig. 5G, the blood clots in the vessel lumens were merged with the fluorescence of BP, indicating that apNFs, not the BA NP and PBS groups, accumulated and formed clots (fig. S14A). Fibrin and platelets were hardly colocalized with apNFs (fig. S14, B and C), indicating that the artificial clots were mainly formed by apNFs and red cells, not fibrin and platelets. The embolization area of blood vessel for apNFs was about 45%, obviously higher than that of BA NPs and PBS (*P < 0.05, Student’s t test, n = 6) (fig. S14D). The specific CD105 in the tumor vessels enabled the precise coagulation-like process. However, BA NPs may target tumors and internalize into ECs without transformation, as demonstrated in the cellular experiments, resulting in significantly less morphology-dependent accumulation in the tumors than that of pNPs (15).
Artificial clots block tumor vessels in vivo for angiogenesis-targeting therapy
The precisely constructed artificial clots from pNPs were expected to affect oxygen supply and slow down tumor growth. The blood oxygen saturation [the hemoglobin (Hb)–bound oxygen level, sO2] in the tumors was evaluated by photoacoustic (PA) imaging (39). The MDA-MB-231–xenografted tumor-bearing mice treated with pNPs showed obvious decreases in sO2 by 33.8% in tumors 4 hours after injection compared with those before pNP injection. However, the tumor-bearing mice exhibited similar sO2 before and after PBS (−5.3%) and BA NP (7.1%) treatment (Fig. 6, A and B, and fig. S15A). Compared with that of the two control groups, the sO2 supply in the tumors of the pNP treatment group decreased significantly (***P < 0.001, Student’s t test, n = 30 slices). Therefore, the artificial clots based on pNPs effectively blocked vessels, decreasing the oxygen supply to tumors and probably inhibiting tumor growth for tumor therapy.
Fig. 6. pNP-induced tumor thrombosis inhibits the growth of MDA-MB-231 human breast cancer in vivo.

(A) PA sO2 mapping images of subcutaneous MDA-MB-231–xenografted tumor in a BALB/c mouse that received an intravenous injection of pNPs. The 3D picture was reconstructed by 30 sheets of 2D pictures with a scan step of 0.1 mm for the tumor. The sO2 in tumors was markedly decreased at 4 hours after injection of pNPs. (B) Statistical analysis of sO2 varies from 0 to 4 hours for the injection of pNPs. The sO2 of the BA NP– and PBS-treated groups are displayed as controls. The error bars represent the means ± SD (n = 30). ***P < 0.001. (C) Time-course optical imaging of MDA-MB-231 tumor–bearing mice that received either pNPs or BA NPs as controls. Scale bar, 2 cm. (D) Corresponding quantified results. pNP-treated group exhibiting lasting fluorescence in the tumor up to 72 hours. **P < 0.01. (E to H) Tumor therapy by in situ–formed apNFs through a noninvasive method. (E) Treatment procedure of MDA-MB-231 tumor–bearing mice by pNPs through the tail vein every other day. i.v., intravenous. (F) Time-dependent relative tumor growth curves by different treatments, showing that tumor growth was inhibited by apNFs that formed artificial clots. **P < 0.01. (G) Time-course curves of body weight during the therapy. (H) Tumors harvested 15 days after treatment with pNPs were stained with H&E. The black dotted line denotes the necrotic domain in the tumor. Scale bar, 100 μm. (I) Percentage of P-selectin–positive platelets, (J) thrombin levels, and (K) platelet numbers in the plasma of tumor-bearing mice treated with pNPs (200 μl, 200 μM) at 0.1 hour, 4 hours, 1 day, and 5 days after injection, indicating safety for the blood circulation system of mice. NS, not significant.
The artificial clots based on pNPs were precisely constructed in situ in tumor vessels, not in normal tissue, due to the specific CD105 receptor. As a result, a therapeutic strategy based on pNPs could be conveniently carried out several times according to the requirements for therapy. The pharmacokinetics parameter was evaluated by measuring the time-dependent fluorescence from BP in the blood of MDA-MB-231 tumor-bearing mice after intravenous injection of pNPs with BA NPs as control. The results indicated that pNPs had a long circulation time with T1/2 (β) extended to around 14.48 hours, indicating the high accumulation of the tumor (fig. S15, B and C). To confirm the accumulation of pNPs and give the cue for the treatment protocol based on pNPs, the persistence of artificial clots in the tumors was evaluated in vivo. The real-time in vivo fluorescence of BP in the tumor sites at 4, 12, 24, 48, and 72 hours after injection of pNPs indicated that artificial clots continuously accumulated up to 24 hours and persisted for 72 hours (Fig. 6C and fig. S15D). However, the BA NP–treated group showed much less fluorescence in the tumors, and the fluorescence almost disappeared at 72 hours. Statistical analyses indicated that the pNP-treated group exhibited significantly higher fluorescence intensity in the tumors than that in the BA NP–treated groups (**P < 0.01, Student’s t test, n = 3) (Fig. 6D), indicating that artificial coagulation based on pNPs had advantages of long-time retention in tumors over BA NPs due to the stable β sheet structured fibrous networks, and the administration frequency of pNPs was determined every other day.
To evaluate therapeutic efficacy, MDA-MB-231–xenografted nude female mice were intravenously injected with pNPs (200 μM, 200 μl) every other day, and BA NPs and PBS were used as the control groups (Fig. 6E). The tumors in mice treated with pNPs showed much slower growth than those treated with BA NPs and PBS, which was ascribed to the in situ–constructed artificial clots (**P < 0.01, Student’s t test, n = 3) (Fig. 6F). The dose of pNPs was 2.4 mg/kg (200 μl, 200 μM), six times lower than that of sorafenib (15 mg/kg), which has a 53% tumor inhibition rate (23). The tumor growth inhibition of pNPs was better than that of sunitinib with 32% at a dose of 8.75 mg/kg (22). The steady weight variation indicated that the pNPs did not induce acute systematic toxicity (Fig. 6G). The H&E staining results revealed that there was tumor necrosis in the pNP-treated group over time (Fig. 6H) and nearly no necrosis in the other groups (fig. S15E). Moreover, cell necrosis was not observed in other tissues of the pNP-treated group, which validated the safety of pNPs (fig. S15F).
We further studied whether artificial clots can induce natural coagulation and interfere with normal blood circulation. Therefore, platelet activity, plasma thrombin generation, and circulating platelet numbers were carefully examined. The results indicated that pNPs did not induce obvious changes in platelet activity, plasma thrombin generation, and circulating platelet numbers when treated for 5 days, indicating that blood was circulating in the vessel (Fig. 6, I to K).
CONCLUSION
In summary, inspired by the natural coagulation process, we successfully developed peptide-based pNPs for the construction of artificial clots in tumor vessels to block the tumor blood supply to inhibit tumor growth. The pNPs first transform into apNFs in situ by ligand-receptor interactions through targeting and binding to ECs; second, the pNPs bind additional apNFs through hydrogen-bonding interactions, leading to platelet aggregation. The assembly process is amplified because previously formed apNFs provide more binding sites for the continuous self-assembly of pNPs, leading to artificial coagulation and the construction of clots. Both targeting sequence and self-assembly (fig. S16) sequence play critical roles, enabling the spatial preciseness and high efficiency of the biomimetic coagulation process to form fibrous networks. In vitro and in vivo experiments were performed to validate this hypothesis, and the artificial clots could be intelligently and precisely constructed, resulting in a remarkable antitumor growth effect. Compared with the natural coagulation to construct clots in several minutes due to long-term evolution, the efficiency of pNP-initiated artificial coagulation needs to be further improved for clinical practicality as an efficient therapeutic method.
In our study, biomimetic pNPs were used for tumor therapy, which showed great potential for several platelet-relevant dysfunctional vasculature diseases, such as traumas, hemorrhagic diseases, and acute inflammation. Moreover, pNPs could deliver various theranostic agents, such as short interfering RNA (siRNA), chemotherapeutics or peptide drugs, and contrast agents. In summary, the pNP system could precisely deliver theranostic agents in the targeted region through convenient modular molecular modification.
MATERIALS AND METHODS
Materials
DMSO was purchased from Aldrich Chemical Co. and used without further purification. The beads and amino acids for peptide synthesis were customized from GL Biochem Ltd. (Shanghai, China). The cell counting kit-8 assay (CCK-8) (Beyotime Institute of Biotechnology, China) was used without further purification. The MDA-MB-231, MCF-7, and HUVEC cell lines were purchased from the Cell Culture Center of the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences (Beijing, China). The other solvents and reagents were used as received. The cell lines had been authenticated using short tandem repeat DNA profiling. All cells tested negative for cross-contamination of other human cells and mycoplasma contamination.
Preparation of pNPs and BA NPs
First, the BP-FFVLK-AHKHVHHVPVRL and BP-AHKHVHHVPVRL peptides (corresponding to the pNPs and BA NPs, respectively) were prepared by standard solid-phase peptide synthesis techniques using Fmoc-coupling chemistry. The peptide monomers were dissolved in DMSO at a concentration of 4.0 mM, followed by quick injection into water at a volume ratio of 0.5:99.5 for DMSO and PBS to obtain the NPs (20 μM). The BA monomers and NPs were obtained by the same method. The peptides were confirmed by MALDI-TOF-MS (Bruker Daltonics).
CD spectra
The CD spectra of pNPs (20 μM) (cultured with or without protein for 0 and 8 hours) were collected at room temperature using a CD spectrometer (JASCO-1500, Tokyo, Japan) with a cell path length of 1 mm. The measurements were implemented between 190 and 230 nm with a resolution of 1.0 nm and a scanning speed of 300 nm/min. For each measurement, three spectra were collected and averaged.
FTIR measurements and XRD measurements
The pNPs and BA NPs with a concentration of 20 μM in water with 0.5% DMSO were freshly prepared and further cocultured with and without CD105 (0.25 μg/ml) for 0 and 8 hours, separately. The solution was lyophilized to obtain the powders for FTIR and XRD measurements. The FTIR spectrum was recorded on a spectrometer (Spectrum One, PerkinElmer Instruments Co. Ltd.) using KBr pellets. The XRD spectrum was recorded on a Xeuss SAXS/WAXS system (Xenocs Asia Pacific Pte. Ltd.).
TEM confirmed the morphology
pNPs with a concentration of 20 μM (10 μl) induced by CD105 (0.25 μg/ml) at 0, 4, and 8 hours were placed dropwise onto a copper mesh for 15 min; subsequently, most of the liquid was removed through a filter paper. Ten microliters of uranyl acetate solution was used to stain the samples for 15 min, followed by drying the spare liquid with the filter. Last, the copper mesh was washed with 10 μl of deionized water, which was blotted after staining and drying at room temperature. The pNPs without being added CD105, the BA NPs with/without CD105, and the pNPs with BSA incubation as controls were prepared with the same method. All of the samples were observed by TEM (Tecnai G2 20 S-TWIN) at an accelerating voltage of 200 kV.
Cytotoxicity assay for HUVECs and MCF-7 cells
The HUVECs and MCF-7 cell lines were purchased from the Cell Culture Center of the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences (Beijing, China). The cytotoxicity of the pNPs and BA NPs was evaluated by the CCK-8 assay. Cells were seeded at a density of 5.0 × 103 cells per well in 96-well plates in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin in a humidified atmosphere with 5% CO2 and then cultured at 37°C overnight. Ten microliters of pNPs or BA NPs was dispersed in DMEM with a series of different concentrations (10, 20, 40, and 80 μM) and cultured with the cells for 24 hours. Subsequently, 10 μl of the CCK-8 solutions was added to each well and cultured for 4 hours. The ultraviolet-visible absorption of the sample wells (Asample), Ablank, and control wells (Acontrol) was measured by a microplate reader at a test wavelength of 450 nm and a reference wavelength of 690 nm. Cell viability (%) was equal to (Asample − Ablank)/(Acontrol − Ablank) × 100%. All experiments were performed in triplicate.
CLSM observation
HUVECs cultured with and without pNPs and BA NPs (20 μM) were imaged using a Zeiss LSM 710 CLSM (Jena, Germany). The HUVECs were seeded in complete DMEM in a humidified atmosphere with 5% CO2 and then cultured at 37°C overnight. Then, 1 ml of serum-free fresh medium containing pNPs (20 μM) was used for replacing the medium, and the cells were cultured for 1, 2, and 4 hours and washed with PBS three times before being imaged using a Zeiss LSM 710 CLSM with a 63× objective lens. The MCF-7 cells and HUVECs cultured with pNPs and BA NPs were imaged in the same way.
Ex vivo fluorescence imaging, H&E staining, immunofluorescence, and bio-TEM
Animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of the National Center for Nanoscience and Technology, China. The pNPs (200 μl, 200 μM) were injected into tumor-bearing mice via the tail vein, and the organs (heart, liver, spleen, lung, and kidney) and tumors were collected after 72 hours for ex vivo fluorescence imaging. The BA NPs and PBS were used as the control groups.
The tumor, heart, liver, spleen, lung, and kidney tissues were fixed in paraformaldehyde (4% in PBS buffer) overnight for H&E staining and immunostaining. All tissues and tumors were further embedded in paraffin and sectioned. The H&E-stained slices were measured using an optical microscope (Nikon ECLIPSE Ci, Nikon) with an image system (Nikon DS-U3, Nikon) to observe the obstruction of blood vessels from the tumor sections and to monitor possible histological changes from normal tissues.
For immunostaining, the paraffin-embedded slices were deparaffinized and subjected to antigen retrieval with EDTA buffer (pH 8.0) through microwave analysis. The sections were immunostained as follows: primary antibodies (anti-CD105 antibody, EPR10145-12, Abcam, China) and dye-labeled secondary antibodies [Cy3-conjugated goat anti-rabbit IgG (immunoglobulin G) (H + L), Servicebio, Wuhan, China] were applied, followed by DAPI (4′,6-diamidino-2-phenylindole) counterstaining. The slices were used for immunofluorescence measurement through an optical microscope (Nikon ECLIPSE Ti-SR, Nikon) with an image analysis system (Nikon DS-U3, Nikon).
For bio-TEM, the tumor tissues were fixed overnight at 4°C in PBS buffer with 2.5% glutaraldehyde. After washing with PBS buffer (100 mM) three times, the tumor tissues were fixed at room temperature with 1% osmium–containing PBS buffer for 2 hours, washed three times with PBS buffer, and dehydrated with a graded series of acetone (50, 70, 80, 90, 95, and 100%) for 15 min each step. After infiltration with a graded series of mixtures (acetone/EPON 812 resin: 2/1, 1/1, and 1/2) at room temperature for 1 hour, pure resin was added and incubated overnight at 4°C. Last, gelatine capsules were used to cover the tissues and were incubated with pure EPON 812 resin at 37°, 45°, and 60°C for 24 hours. The tissues were cut into ultrathin sections by a diamond knife and picked up with formvar-coated copper grids (300 mesh). All of the sections were counter-stained with osmic acid (1%) for 1 hour and uranyl acetate (4%) for 20 min. A HITACHI HT7700 electron microscope (HITACHI, Tokyo, Japan) was used to observe tissues.
SEM for imaging the coassembly of apNFs and RBCs
SEM was used to examine the morphology of apNFs and RBCs. Fresh blood was collected from healthy nude mice through the tail vein. Forty microliters of blood was centrifuged with a centrifugal force of 2000g for 3 min to obtain the underlayer RBCs. For natural clots, blood (10 μl) was placed dropwise on a silicon wafer for 10 min to form clotting. Urokinase (0.5 g/ml, 5 μl) was further added dropwise onto the clots, and the latter were dissolved within 5 min. For artificial clots, apNFs (10 mg/ml, 5 μl) and RBCs (5 μl) were incubated on silicon wafers to form clots, which were further fixed with 4% paraformaldehyde in PBS buffer overnight and dehydrated with a graded series of alcohol (10, 30, 50, 70, and 90%) for 10 min for each step. Then, the samples were coated with gold for 120 s and observed by SEM (NOVA NanoSEM 430 + EDS).
In vitro coagulation-like process of pNPs in the phantom
The HUVECs (100 μl, 500,000 cells/ml) in DMEM were infused separately into the channels of the microfluidic device, and the medium was replaced by fresh medium every other 12 hours. After 2 weeks, the microchannels coated with cells were circularly infused in an oven (37°C, 5% CO2) by pNPs in serum-free medium with a concentration of 100 μM for 0, 4, and 12 hours; BA NPs and PBS were used as controls. The flow rate was 0.2 μl/min.
sO2 of tumors treated with NPs
Oxyhemoglobin (HbO2) and Hb in tumor blood were detected by a real-time multispectral optoacoustic tomography scanner (iThera Medical inVision 128, Germany). The MDA-MB-231 tumor-bearing mice were placed in a glass box and further anesthetized with isoflurane. The data were collected with wavelengths of 715 (Hb, blue) and 850 nm (HbO2, red) in the near-infrared window before and 4 hours after intravenous injection (pNPs and BA NPs, 200 μl, 200 μM; PBS, 200 μl).
Starving tumor therapy in vivo
Mice bearing MDA-MB-231 cells were randomly divided into three groups. The three groups were injected with pNPs (200 μl, 200 μM), BA NPs (200 μl, 200 μM), and PBS (200 μl, 200 μM) every other day. The tumor volume and body weight of the mice were measured every other day. After treatment for 10 days, the mice were euthanized, and the tumors were collected.
Animal fluorescence imaging
The MDA-MB-231–bearing BALB/c nude mice were intravenously injected with pNPs (200 μl, 200 μM), BA NPs (200 μl, 200 μM), and PBS (200 μl). The in vivo images were acquired at 4, 12, 24, 48, and 72 hours after intravenous injection. The excitation wavelength was λex = 500 nm, and the emission wavelength was λem = 520 nm.
Evaluation of safety
The MD-MBA-231–xenografted tumor-bearing mice were treated with pNPs (200 μl, 200 μM) by intravenous injection. Mouse whole blood was collected with 3.8% sodium citrate at different time points (0, 1, 4, 24, and 120 hours) after injection. Platelet-rich plasma (PRP) was obtained through incubation of blood and with an equal volume of 2% paraformaldehyde in PBS buffer for 30 min, followed by centrifugation (1000 rpm, 10 min). The amount of P-selectin was measured through flow cytometry after incubating PRP with FITC (fluorescein isothiocyanate)–conjugated P-selectin–specific monoclonal antibodies (Abcam, ab33279) for 30 min. The thrombin levels were measured using enzyme-linked immunosorbent assay kits (Abcam, ab157527). The platelet numbers were measured by flow cytometry.
Statistical analysis
All data are reported as the means ± SD. The in vitro experiments were performed in three independent experiments with at least three technical replicates. The in vivo experiments were performed with three to four mice for each group. Statistical analysis of the samples was performed using unpaired two-tailed Student’s t test, and a P value of <0.05 was considered significant.
Supplementary Material
Acknowledgments
Funding: This work was supported by the National Natural Science Foundation of China (51890891, 51890894, 51725302, 21807020, 51573031, and 51573032), Science Fund for Creative Research Groups of the National Natural Science Foundation of China (11621505), CAS Interdisciplinary Innovation Team, and Jilin Province Key Laboratory of Organic Functional Molecular Design and Synthesis (no. 130028911). Author contributions: P.-P.Y., K.Z., J.-P.Z., L.W., and H.W. conceived the project and wrote the manuscript. P.-P.Y., K.Z., J.-P.Z., L.W., and H.W. planned and designed the experiments. P.-P.Y. and K.Z. performed most of the experiments. X.J.G. and X.G. performed the quantum calculation. Z.-M.C. and D.-B.C. assisted with the chemical synthesis, and P.-P.H., Y.F., and Y.L. assisted with the cell culture studies. Y.Y., D.-Y.H., and Y.L. assisted with the animal studies and performed the experiments. L.S. and X.-Z.Z. helped to supervise the revision. Competing interests: The authors filed patents pertaining to the results presented in the paper. The authors declare the following competing financial interest(s): L.W., H.W., P.-P.Y., and K.Z. are the coinventors of a pending patent on the biomimetic coagulation strategy [National Center for Nanoscience and Technology, China, H.W., L.W., and P.-P.Y., not submitted due to the COVID-19 epidemic (SARS-CoV-2 virus) in China]. The remaining authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/22/eaaz4107/DC1
REFERENCES AND NOTES
- 1.van der Meijden P. E. J., Heemskerk J. W. M., Platelet biology and functions: New concepts and clinical perspectives. Nat. Rev. Cardiol. 16, 166–179 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Onwukwe C., Maisha N., Holland M., Varley M., Groynom R., Hickman D., Uppal N., Shoffstall A., Ustin J., Lavik E., Engineering intravenously administered nanoparticles to reduce infusion reaction and stop bleeding in a large animal model of trauma. Bioconjug. Chem. 29, 2436–2447 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bertram J. P., Williams C. A., Robinson R., Segal S. S., Flynn N. T., Lavik E. B., Intravenous hemostat: Nanotechnology to halt bleeding. Sci. Transl. Med. 1, 11ra22 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anselmo A. C., Modery-Pawlowski C. L., Menegatti S., Kumar S., Vogus D. R., Tian L. L., Chen M., Squires T. M., Sen Gupta A., Mitragotri S., Platelet-like nanoparticles: Mimicking shape, flexibility, and surface biology of platelets to target vascular injuries. ACS Nano 8, 11243–11253 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hickman D. A., Pawlowski C. L., Shevitz A., Luc N. F., Kim A., Girish A., Marks J., Ganjoo S., Huang S., Niedoba E., Sekhon U. D. S., Sun M., Dyer M., Neal M. D., Kashyap V. S., Sen Gupta A., Intravenous synthetic platelet (SynthoPlate) nanoconstructs reduce bleeding and improve 'golden hour' survival in a porcine model of traumatic arterial hemorrhage. Sci. Rep. 8, 3118 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown A. C., Stabenfeldt S. E., Ahn B., Hannan R. T., Dhada K. S., Herman E. S., Stefanelli V., Guzzetta N., Alexeev A., Lam W. A., Lyon L. A., Barker T. H., Ultrasoft microgels displaying emergent platelet-like behaviours. Nat. Mater. 13, 1108–1114 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Modery-Pawlowski C. L., Tian L. L., Ravikumar M., Wong T. L., Gupta A. S., In vitro and in vivo hemostatic capabilities of a functionally integrated platelet-mimetic liposomal nanoconstruct. Biomaterials 34, 3031–3041 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Chan L. W., Wang X., Wei H., Pozzo L. D., White N. J., Pun S. H., A synthetic fibrin cross-linking polymer for modulating clot properties and inducing hemostasis. Sci. Translat. Med. 7, 277ra29 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu C.-M. J., Fang R. H., Wang K. C., Luk B. T., Thamphiwatana S., Dehaini D., Nguyen P., Angsantikul P., Wen C. H., Kroll A. V., Carpenter C., Ramesh M., Qu V., Patel S. H., Zhu J., Shi W., Hofman F. M., Chen T. C., Gao W., Zhang K., Chien S., Zhang L., Nanoparticle biointerfacing by platelet membrane cloaking. Nature 526, 118–121 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farokhzad O. C., Platelet mimicry. Nature 526, 47–48 (2015). [DOI] [PubMed] [Google Scholar]
- 11.George J. N., Platelets. Lancet 355, 1531–1539 (2000). [DOI] [PubMed] [Google Scholar]
- 12.Clemetson K. J., Platelets and primary haemostasis. Thromb. Res. 129, 220–224 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Kisucka J., Butterfield C. E., Duda D. G., Eichenberger S. C., Saffaripour S., Ware J., Ruggeri Z. M., Jain R. K., Folkman J., Wagner D. D., Platelets and platelet adhesion support angiogenesis while preventing excessive hemorrhage. Proc. Natl. Acad. Sci. U.S.A. 103, 855–860 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Furie B., Furie B. C., Mechanisms of thrombus formation. New Engl. J. Med. 359, 938–949 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Yang P.-P., Luo Q., Qi G. B., Gao Y. J., Li B. N., Zhang J. P., Wang L., Wang H., Host materials transformable in tumor microenvironment for homing theranostics. Adv. Mater. 29, 1605869 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Zhang D., Qi G. B., Zhao Y. X., Qiao S. L., Yang C., Wang H., In situ formation of nanofibers from purpurin18-peptide conjugates and the assembly induced retention effect in tumor sites. Adv. Mater. 27, 6125–6130 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Feng Z., Zhang T., Wang H., Xu B., Supramolecular catalysis and dynamic assemblies for medicine. Chem. Soc. Rev. 46, 6470–6479 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dallas N. A., Samuel S., Xia L., Fan F., Gray M. J., Lim S. J., Ellis L. M., Endoglin (CD105): A marker of tumor vasculature and potential target for therapy. Clin. Cancer Res. 14, 1931–1937 (2008). [DOI] [PubMed] [Google Scholar]
- 19.Li D. Y., Sorensen L. K., Brooke B. S., Urness L. D., Davis E. C., Taylor D. G., Boak B. B., Wendel D. P., Defective angiogenesis in mice lacking endoglin. Science 284, 1534–1537 (1999). [DOI] [PubMed] [Google Scholar]
- 20.Ruoslahti E., Specialization of tumour vasculature. Nat. Rev. Cancer 2, 83–90 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Zhang C., Ni D., Liu Y., Yao H., Bu W., Shi J., Magnesium silicide nanoparticles as a deoxygenation agent for cancer starvation therapy. Nat. Nanotechnol. 12, 378–386 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Nowak-Sliwinska P., Weiss A., van Beijnum J. R., Wong T. J., Kilarski W. W., Szewczyk G., Verheul H. M. W., Sarna T., van den Bergh H., Griffioen A. W., Photoactivation of lysosomally sequestered sunitinib after angiostatic treatment causes vascular occlusion and enhances tumor growth inhibition. Cell Death Dis. 6, e1641 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilhelm S., Carter C., Lynch M., Lowinger T., Dumas J., Smith R. A., Schwartz B., Simantov R., Kelley S., Discovery and development of sorafenib: A multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 5, 835–844 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Wang L., Li W., Lu J., Zhao Y. X., Fan G., Zhang J. P., Wang H., Supramolecular nano-aggregates based on bis(pyrene) derivatives for lysosome-targeted cell imaging. J. Phys. Chem. C 117, 26811–26820 (2013). [Google Scholar]
- 25.Hong Y., Lam J. W. Y., Tang B. Z., Aggregation-induced emission. Chem. Soc. Rev. 40, 5361–5388 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Luo Q., Lin Y. X., Yang P. P., Wang Y., Qi G. B., Qiao Z. Y., Li B. N., Zhang K., Zhang J. P., Wang L., Wang H., A self-destructive nanosweeper that captures and clears amyloid β-peptides. Nat. Commun. 9, 1802 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fonsatti E., Jekunen A. P., Kairemo K. J., Coral S., Snellman M., Nicotra M. R., Natali P. G., Altomonte M., Maio M., Endoglin is a suitable target for efficient imaging of solid tumors: In vivo evidence in a canine mammary carcinoma model. Clin. Cancer Res. 6, 2037–2043 (2000). [PubMed] [Google Scholar]
- 28.Yang P.-P., Zhao X. X., Xu A. P., Wang L., Wang H., Reorganization of self-assembled supramolecular materials controlled by hydrogen bonding and hydrophilic-lipophilic balance. J. Mater. Chem. B 4, 2662–2668 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Simberg D., Duza T., Park J. H., Essler M., Pilch J., Zhang L., Derfus A. M., Yang M., Hoffman R. M., Bhatia S., Sailor M. J., Ruoslahti E., Biomimetic amplification of nanoparticle homing to tumors. Proc. Natl. Acad. Sci. U.S.A. 104, 932–936 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kelly S. M., Jess T. J., Price N. C., How to study proteins by circular dichroism. Biochim. Biophys. Acta 1751, 119–139 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Nowak A. P., Breedveld V., Pakstis L., Ozbas B., Pine D. J., Pochan D., Deming T. J., Rapidly recovering hydrogel scaffolds from self-assembling diblock copolypeptide amphiphiles. Nature 417, 424–428 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Miyazawa T., Blout E. R., The infrared spectra of polypeptides in various conformations: Amide I and II Bands. J. Am. Chem. Soc. 83, 712–719 (1961). [Google Scholar]
- 33.Sawaya M. R., Sambashivan S., Nelson R., Ivanova M. I., Sievers S. A., Apostol M. I., Thompson M. J., Balbirnie M., Wiltzius J. J. W., McFarlane H. T., Madsen A. Ø., Riekel C., Eisenberg D., Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature 447, 453–457 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Makin O. S., Atkins E., Sikorski P., Johansson J., Serpell L. C., Molecular basis for amyloid fibril formation and stability. Proc. Natl. Acad. Sci. U.S.A. 102, 315–320 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aleman M. M., Walton B. L., Byrnes J. R., Wolberg A. S., Fibrinogen and red blood cells in venous thrombosis. Thromb. Res. 133, S38–S40 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Camiolo S. M., Thorsen S., Astrup T., Fibrinogenolysis and fibrinolysis with tissue plasminogen activator, urokinase, streptokinase-activated human globulin, and plasmin. Proc. Soc. Exp. Biol. Med. 138, 277–280 (1971). [DOI] [PubMed] [Google Scholar]
- 37.Smith R. A. G., Dupe R. J., English P. D., Green J., Fibrinolysis with acyl-enzymes: A new approach to thrombolytic therapy. Nature 290, 505–508 (1981). [DOI] [PubMed] [Google Scholar]
- 38.Qiu Y., Tong S., Zhang L., Sakurai Y., Myers D. R., Hong L., Lam W. A., Bao G., Magnetic forces enable controlled drug delivery by disrupting endothelial cell-cell junctions. Nat. Commun. 8, 15594 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yin G., Xing D., Yang S., Dynamic monitoring of blood oxygen saturationin vivousing double-ring photoacoustic sensor. J. Appl. Phys. 106, 013109 (2009). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/22/eaaz4107/DC1