

Abstract
S-Nitrosothiols (RSNOs) serve as air-stable reservoirs for nitric oxide in biology. While copper enzymes promote NO release from RSNOs by serving as Lewis acids for intramolecular electron-transfer, redox-innocent Lewis acids separate these two functions to reveal the effect of coordination on structure and reactivity. The synthetic Lewis acid B(C6F5)3 coordinates to the RSNO oxygen atom, leading to profound changes in the RSNO electronic structure and reactivity. Although RSNOs possess relatively negative reduction potentials, B(C6F5)3 coordination increases their reduction potential by over 1 V into the physiologically accessible + 0.1 V vs. NHE. Outer-sphere chemical reduction gives the Lewis acid stabilized hyponitrite dianion trans-[LA-O-N=N-O-LA]2−[LA = B(C6F5)3], which releases N2O upon acidification. Mechanistic and computational studies support initial reduction to the [RSNO-B(C6F5)3] radical anion, which is susceptible to N–N coupling prior to loss of RSSR.
Keywords: bioinorganic chemistry, lewis acids, nitric oxide, S-nitrosothiols
Nitric oxide (NO) is a key endogenous gasotransmitter that plays crucial signaling roles in vasorelaxation, neurotrans-mission, and cytoprotection.[1] Nonetheless, NO has a short lifetime under biological conditions since it reacts with the omnipresent O2 and O2−.[2] S-nitrosothiols (RSNOs) serve as air-stable NO reservoirs that are capable of facile NO release due to the modest RS–NO bond strength (ca. 30 kcalmol−1).[3] Circulating at near micromolar levels, RSNOs such as S-nitrosoglutathione serve as messengers themselves and participate in protein control through S-nitrosation of proteins, an important post-translational modification connected to health and disease[4] For instance, S-nitrosation of hemoglobin at β-Cys93 is required for proper blood oxygenation.[5]
Redox-active copper enzymes such as CuZn-SOD facilitate the release of NO from RSNOs.[6] Model studies revealed that Lewis acid coordination of RSNO to a copper(I) ion to form CuI-RSNO adducts precedes intramolecular electron transfer that releases NO. For instance, addition of the synthetic S-nitrosothiol Ph3CSNO to the tris(pyrazolyl)-borate complex MesTpCu results in MesTpCuI(k1-N(O)SCPh3), which reversibly loses NO with concomitant oxidation of the copper center to MesTpCuII-SCPh3.[7] Given that the reduction potential of Ph3CSNO (−1.25 V vs. NHE)[7] is dramatically more negative than the corresponding [CuII]/[CuI] redox couple for a copper complex that releases NO with formation of the corresponding copper(II) thiolate [CuII]-SR [iPr2TpCu-(NCMe); E1/2 = 270 mV vs. NHE],[8] coordination of the RSNO to the Lewis acid is clearly required to facilitate electron transfer.[7]
RSNOs exist in a variety of protein environments, and are exposed to a wide range of chemical stimuli including Lewis acids.[9] These stimuli can have a dramatic effect on the -SNO group properties owing to the unusual nature of the RSNO electronic structure, whose description requires a combination of the three resonance structures S, D, and I (Figure 1A). Two of these structures, D and I, are antagonistic, that is, have opposite bonding patterns and formal charges on atoms.[10] This antagonistic nature of RSNOs results in the paradoxical properties of the S–N bond, which exhibits restricted rotation suggestive of a partial double-bond character while being unusually long and weak,[11] as well as the dual reactivity pattern of RSNO reactions.[11,12]
Figure 1.
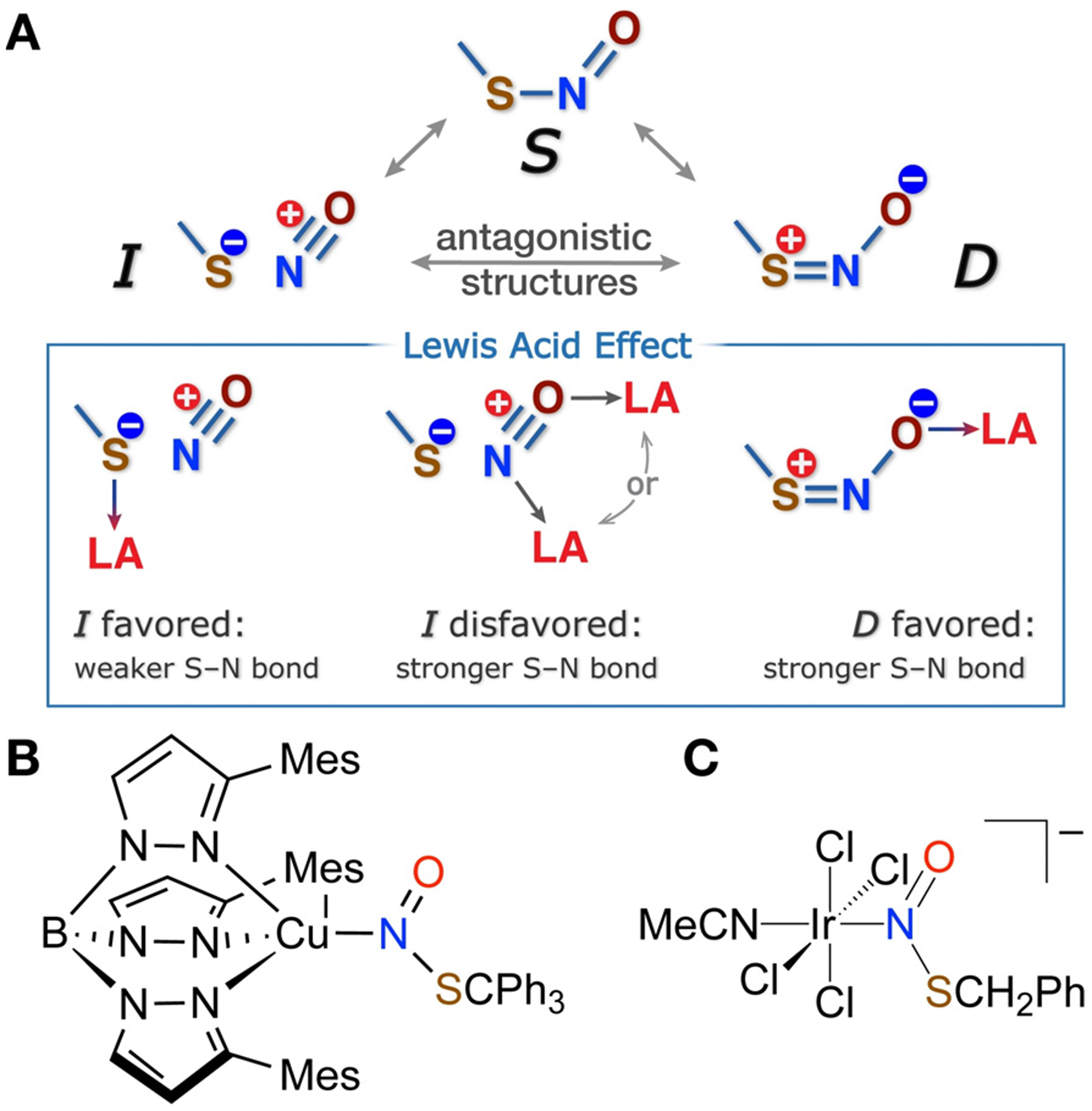
A) Resonance representation of RSNO electronic structure and its modulation by Lewis acid coordination. B) Known N-bound Lewis acid adducts of RSNOs.
The antagonistic nature of RSNOs also suggests that Lewis acid coordination at N, S, or O atoms would radically alter the RSNO reactivity by favoring one antagonistic structure at the expense of another (Figure 1A), an effect that may underline the enzymatic control of the RSNO reactions in biological environments.[10,12b,13] Due to the sensitive nature of RSNOs, experimental support for Lewis acid modulation of RSNO properties has lagged behind computational predictions.[11,14] Seminal studies by Doctorovich and co-workers revealed that N-coordination of RSNO to an IrIII center in [IrCl4(NCMe)(k1-N(O)SR)]−(R = CH2Ph) leads to shortening of the S–N bond,[15] which was also seen in MesTpCuI(k1-N(O)SCPh3).[7] While the S–N distance in Ph3SNO is 1.792(5) Å,[16] [IrCl4(NCMe)(k1-N-(O)SCH2Ph)]−1 and MesTpCuI(k1-N(O)SCPh3) possess S–N distances of 1.737(8) and 1.755(4) Å, each consistent with reduced I character and increased D character for the k1-N-bound RSNO.
We experimentally find that O-coordination of a Lewis acid, however, has an even more profound effect on RSNO reactivity due to the interaction with the oxygen atom, which bears a formal negative charge in resonance structure D (Figure 1A). Addition of the oxophilic Lewis acid B(C6F5)3 to synthetic S-nitrosothiols AdSNO (1) and MesCH2SNO (2) in pentane at RT leads to rapid color change from green to yellow for 1 or pink to orange for 2. Crystallization from pentane provides Lewis acid adducts AdSNO-B(C6F5)3 (3) and MesCH2SNO-B(C6F5)3 (4) in 80% and 71% yield, respectively (Figure 2). X-ray diffraction analysis confirms short S–N bonds in these adducts of 1.6252(17) and 1.608-(5) Å in the syn and anti conformers of 3 and 4, respectively.[17] The observed dramatic contraction of the S–N bond is in agreement with DFT calculations at the ωB97XD-PCM-(CH2Cl2)/ma-def2-TZVPP level (Figures S42–S44). At the same time, the experimental N–O bond lengths of 1.278(2) and 1.274(5) Å in 3 and 4 are longer than typical 1.18–1.19 Å N–O distances in free RSNOs.[16,18] These observations are consistent with increased contribution of the resonance component D in O-bound RSNO-B(C6F5)3 complexes: indeed, calculations[19] suggest that the D weighting increases from circa 30% in free RSNOs to circa 55% in the B(C6F5)3 adducts 3 and 4.
Figure 2.
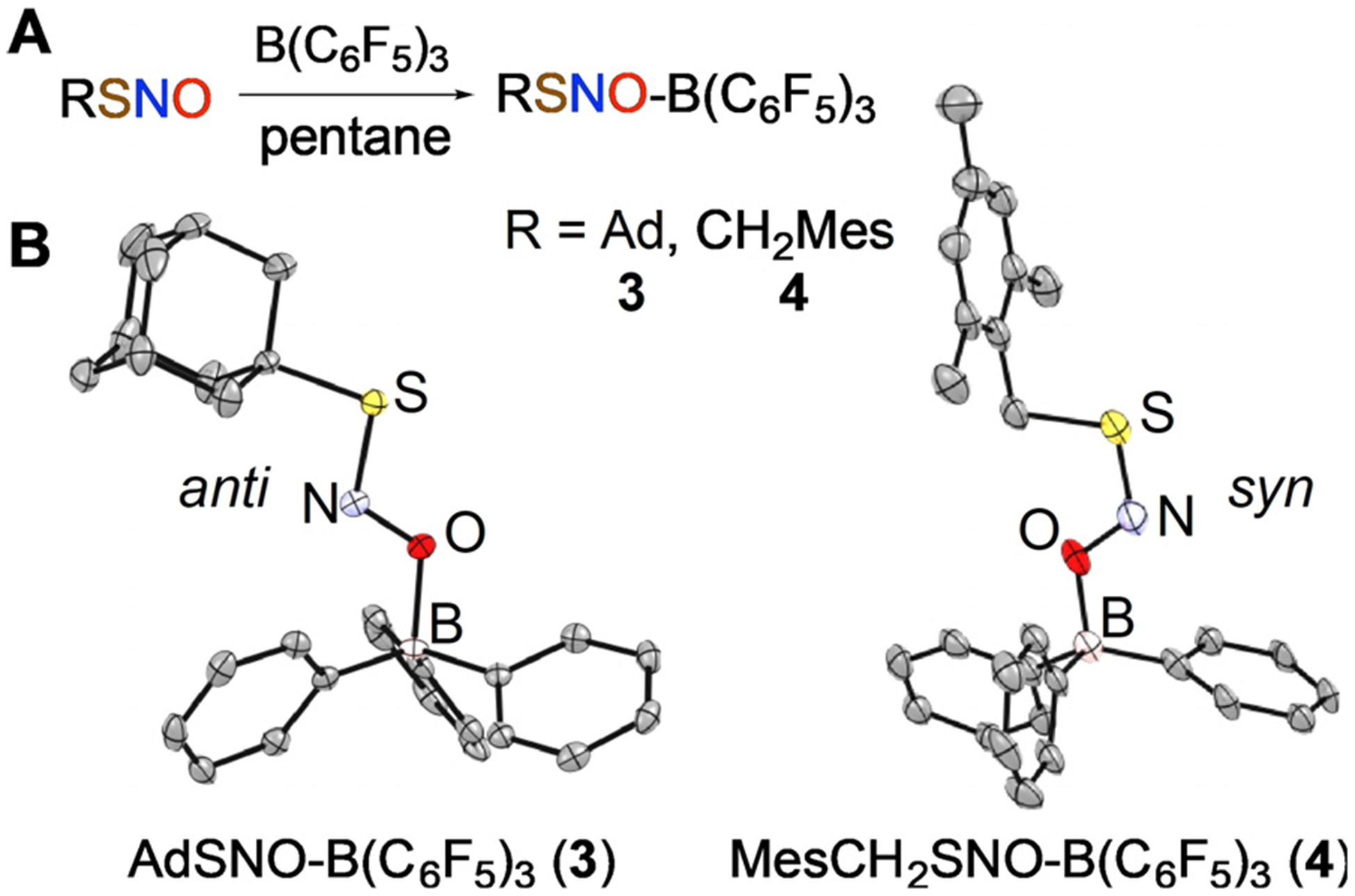
Synthesis (A) and X-ray structures (B) of AdSNO-B(C6F5)3 (3) and MesCH2SNO-B(C6F5)3 (4); H and F atoms are not shown.
Dramatic structural alterations of RSNOs induced by O-coordination of a Lewis acid result in distinct spectroscopic changes. Consistent with trends revealed through X-ray structures, IR spectra report higher energy S–N stretches at 853 and 863 cm−1 for 3 and 4 relative to 645 and 630 cm−1 for the corresponding free RSNOs 1 and 2. Conversely, the N–O stretching frequencies decrease to 1257 and 1282 cm−1 for 3 and 4 relative to 1486 and 1484 cm−1 in 1 and 2. Analysis of 15N isotopomers of 1–4 confirms these assignments (Figures S5, S9, S16 and S23 in the Supporting Information). As the oxygen atom lone pair becomes involved in dative bonding with B(C6F5)3, the n→π* adsorption characteristic to the UV/Vis spectra of RSNOs shifts to higher energies. For instance, low intensity bands at 560 (5m−1cm−1) and 600 nm (13m−1cm−1) for AdSNO (1) shift to 510 nm (9m−1cm−1) in AdSNO-B(C6F5)3 (3; Figures S4 and S15).
15N NMR spectra of RSNOs typically reveal two signals for anti and syn conformers that require low-temperature acquisition due to a relatively low barrier for S–N bond rotation (10–14 kcalmol−1).[16] For instance, the low temperature 15N NMR spectrum of AdS15NO in CD2Cl2 at −60 °C exhibits major and minor signals at δ 845.4 and 786.6 ppm (relative to NH3), which are attributed to anti and syn conformers. Low-temperature 15N NMR spectra of crystallized adducts 3 and 4 dissolved in CD2Cl2 each exhibit three upfield-shifted resonances. The major resonance for O-anti-AdSNO-B(C6F5)3 (3) appears at δ 741 ppm, with two minor signals at 681 and 553 ppm. DFT calculations suggest that among the possible forms of AdSNO-B(C6F5)3 the O-syn and N-syn species are closest in energy to the most stable O-anti form (enthalpies higher by 1.3 and 3.5 kcalmol−1, Figures S43, S44), with predicted 15N shifts at 755 and 605 ppm (compared to O-anti at 838 ppm). This allows us to tentatively assign the minor signals experimentally observed at d 681 and 553 ppm to the O-syn and N-syn forms of 3, respectively.
Besides significant alteration of the RSNO geometry and spectral properties, Lewis acid coordination has a dramatic effect on the reduction potentials of RSNOs. Cyclic voltammograms (CVs) of free RSNOs 1 and 2 in CH2Cl2 each reveal two irreversible reduction features at rather negative potentials (Figures S24 and S28). The first corresponds to one-electron reduction of the free RSNO to the [RSNO] radical-anion appearing at −1.13 and −1.01 V vs. NHE for 1 and 2, respectively. The second peak in each at −1.32 V corresponds to the reduction of free NO released from reduced RSNO. Remarkably, AdSNO-B(C6F5)3 (3) exhibits a quasi-reversible wave centered at + 0.08 V vs. NHE, which is nearly 1 V higher than free AdSNO (1) that is also present under CV conditions (Figure 3). MesCH2SNO-B(C6F5)3 (4) also undergoes reduction at a potential ≈ 1 V higher than free MesCH2SNO (2), but is largely irreversible under these conditions signaled by a reduction wave centered at + 0.1 V vs. NHE (Figures S26 and S29).
Figure 3.
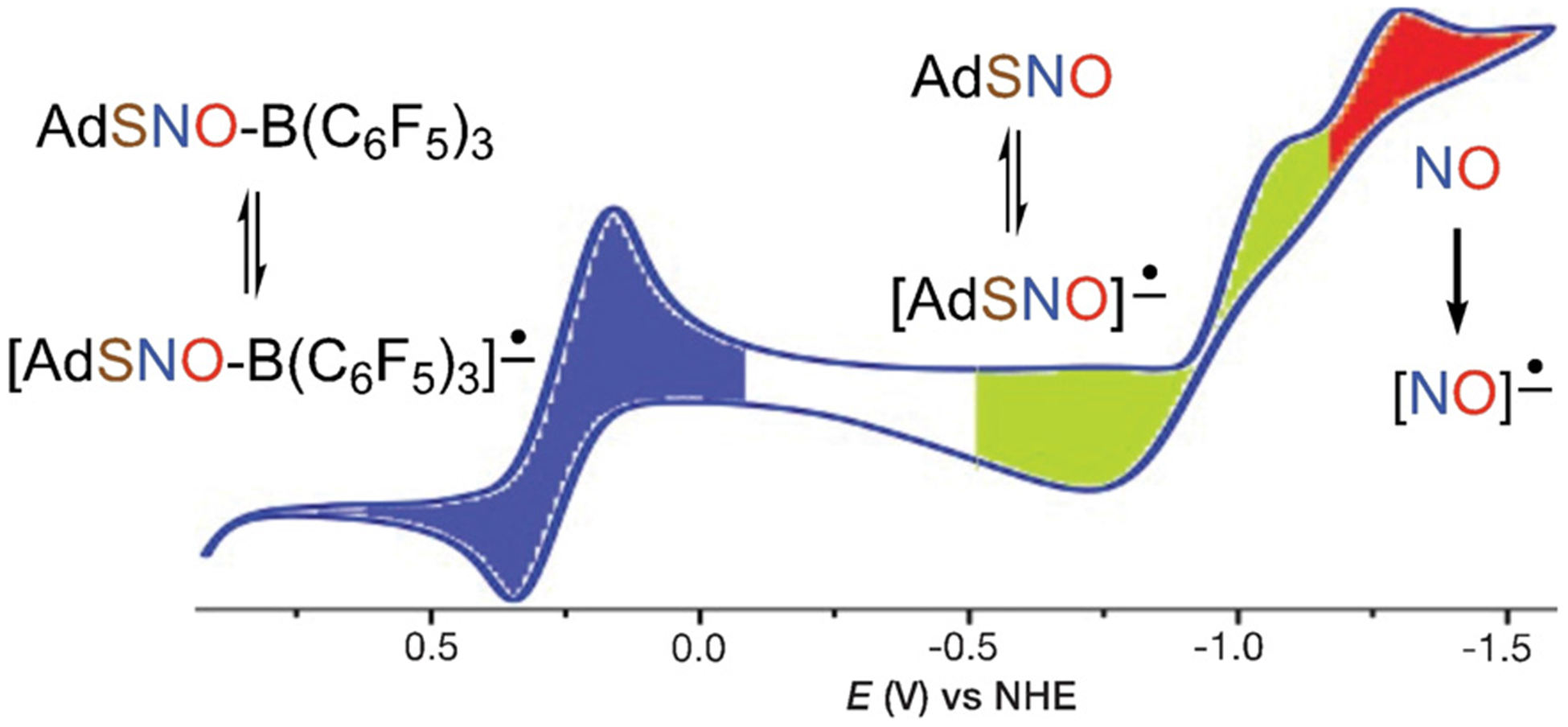
Cyclic voltammogram of AdSNO-B(C6F5)3 (3; 7 mm) in CH2Cl2 at 25°C with [nBu4N][BPh4] (0.1m).
To provide greater insight into the species generated under CV conditions, we carried out chemical reduction with outer-sphere reductants. Addition of Cp2*Co (Ered = −1.24 V vs. NHE in CH2Cl2)[20] to free RSNOs 1 and 2 results in NO generation in 82% and 71% yield, respectively (Figure 4A). This experimental finding is consistent with DFT calculations that suggest the [RSNO]•− structure as a weakly bound complex of RS− and NO• (S–N distance > 2.6 Å) with more than 95% of the spin density on the NO moiety (Figure S45).
Figure 4.
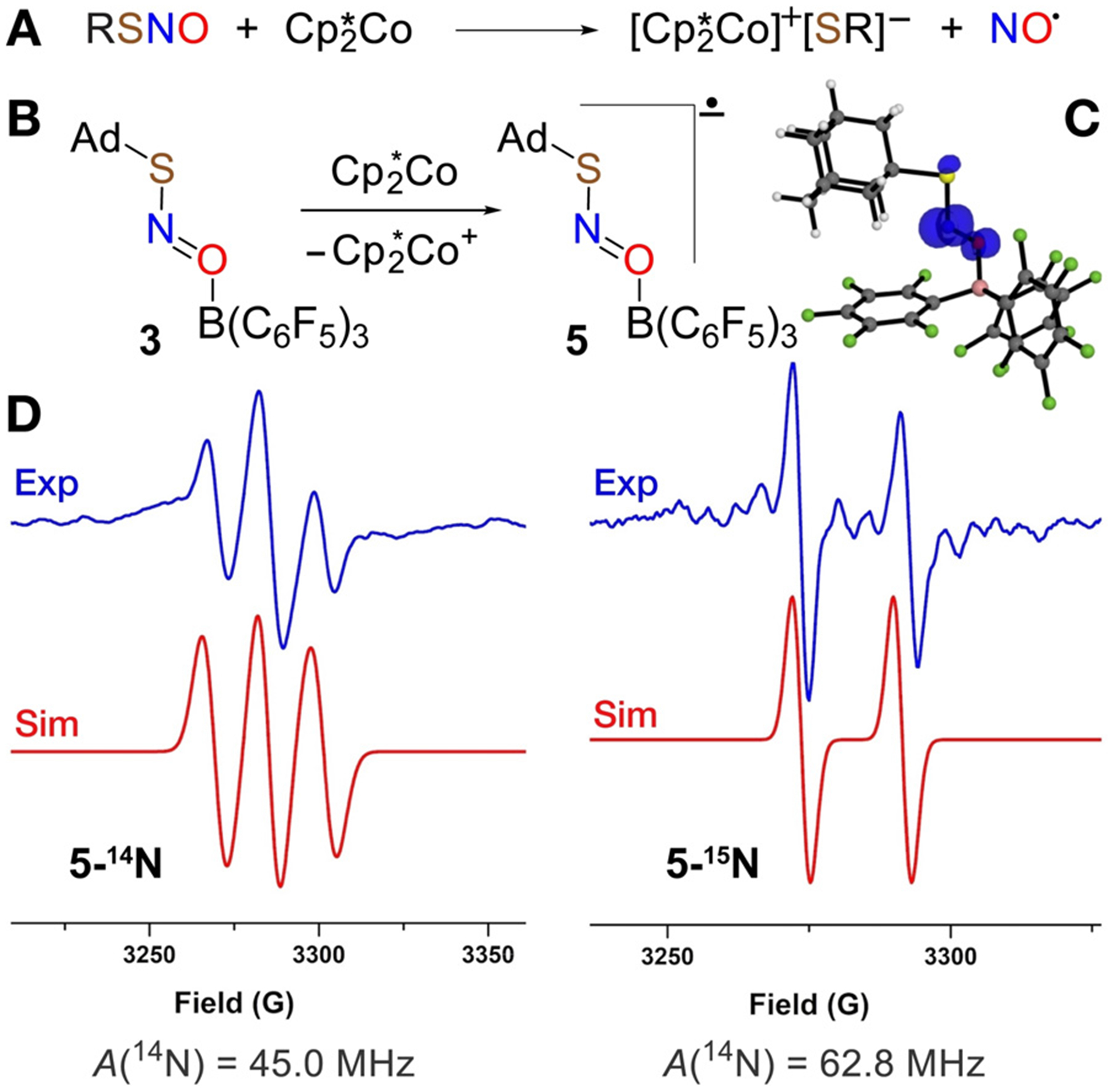
A) Outer-sphere reduction of RSNO (R=Ad, MesCH2) by Cp2*Co. B) Chemical reduction of AdSNO-B(C6F5)3 by Cp2*Co. C) Spin density plot of [AdSNO-B(C6F5)3]•–* (5). D) X-band EPR spectra of radical anions 5-14N and 5-15N recorded in a mixture of fluorobenzene/pentane at 25°C.
The quasi-reversible reduction of AdSNO-B(C6F5)3 (3) observed by cyclic voltammetry suggests that the radical anion [AdSNO-B(C6F5)3]•− (5) may have a long enough lifetime to enable characterization by EPR. Indeed, mixing a fluorobenzene solution of 3 with a pentane solution of Cp2*Co generates an intermediate with a three-line EPR signal at g = 1.999 (Figure 4D). This resonance is consistent with the N-centered radical anion 5 predicted by DFT to exhibit extensive spin density on N (0.7 e−, Figure 4C and Figure S45B in the Supporting Information). The three-line pattern indicates strong coupling to the 14N nucleus (I = 1; A(14N) = 45.0 MHz), which was confirmed by isotopic substitution with 15N that gives a two line pattern (I=½; A(15N) = 62.5 MHz; Figure 4D). Attempts to observe [MesCH2SNO-B(C6F5)3]•− by reduction of 4 with Cp2*Co under similar conditions did not allow EPR observation of the corresponding radical anion, which is consistent with its shorter lifetime as indicated by the irreversible reduction peak in the CV of 4.
Although DFT calculations predict elongated S–N and N–O bonds in [AdSNO-B(C6F5)3]•− (5; 1.71 Å and 1.33 Å; Figure S45) vs. AdSNO-B(C6F5)3 (3; 1.61 Å and 1.25 Å), 1-electron reduction of AdSNO-B(C6F5)3 does not seem to destabilize the S–N bond enough to encourage loss of NO• as occurs in the reduction of free RSNOs (Figure 4A). Instead, reduction of Lewis acid adducts 3 and 4 ultimately delivers the Lewis acid stabilized hyponitrite dianion trans-[(C6F5)3BO-N=N-OB(C6F5)3]]2− (7; Figure 5). Addition of Cp2*Co to 3 or 4 gives dianion 7 charge-stabilized by 2 equiv Cp2*Co+ in 73% and 65% yield, respectively, along with the corresponding disulfides AdS-SAd and MesCH2S-SCH2Mes in 80% and 74% yield, respectively. Due to the mild reduction potentials of 3 and 4, a range of metallocene reductants provide [M]2[(C6F5)3BO-N=N-OB(C6F5)3] (7-[M]2; M = Cp2*Co+, Cp2Fe+). The X-ray crystal structure of [Cp2*Co+]2[(C6F5)3BO-N=N-OB(C6F5)3]2− (7-[Cp2*Co+]2) reveals N–N coupling to give a trans disposition of the [O-N=N-O]2− hyponitrite core located at a center of inversion with N–N’ and O–N distances of 1.257(9) and 1.381(6) Å, respectively. These distances are similar to those found in other structures of the trans-hyponitrite anion.[21] 15N NMR analysis of 7-[Cp2*Co+]2 shows signals at δ 429.9 (major) and 384.9 (minor). We assign these as the trans and cis isomers of 7 based on DFT-predicted 15N chemical shifts at δ 425 and 380 ppm, respectively. DFT also indicates a small difference of 1.6 kcalmol−1 in free energies for these isomers that favors the trans form (Figure S46).
Figure 5.
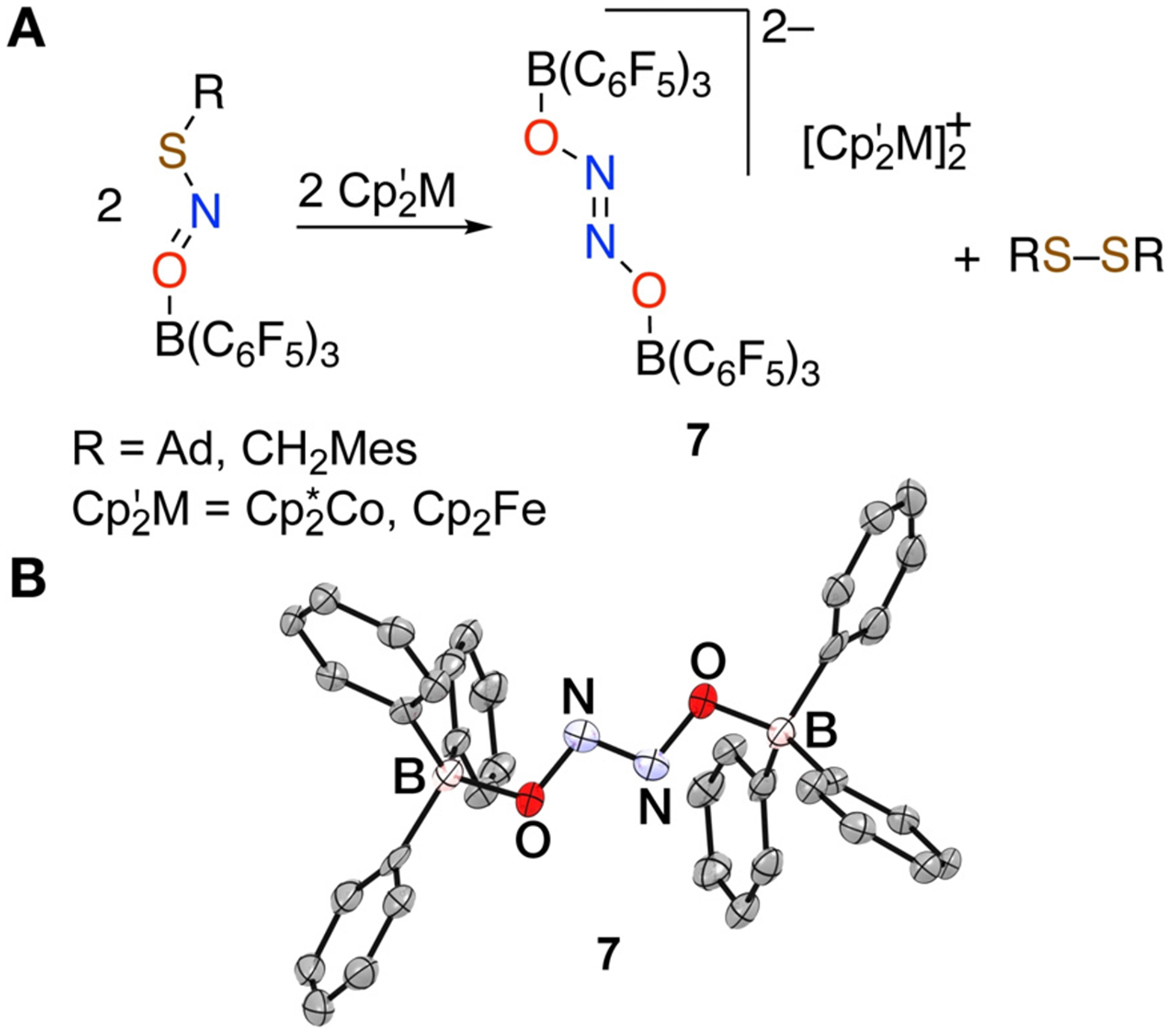
A) Reduction of RSNO-B(C6F5)3 along with B) the X-ray structure of the trans-hyponitrite dianion [(C6F5)3B-ON=NO-B(C6F5)3]2− (7); H and F atoms are not shown.
DFT calculations suggest that the radical anions [RSNOB(C6F5)3]•– [R = Ad (5), MesCH2 (6)] formed by 1-electron reduction of 3 and 4 undergo thermodynamically favorable dimerization via N–N bond formation to give dianions 8 and 9 (“Δ~GR° = −6.5 and −6.7 kcalmol−1, respectively). These dianions then lose the corresponding disulfide RS-SR (Δ~GR° = −39.0 and −41.3 kcalmol−1, respectively) to give the doubly borane-capped hyponitrite dianion 7 (Figure 6 and Figures S47,S48). Full reaction pathway calculations for a small model system (Figure S49) support the proposed mechanism with reasonably low barriers (< 17 kcalmol−1) for the dimer formation and the subsequent RS− elimination, and a barrierless RS− attack of to give disulfide RS-SR and Lewis acid capped hyponitrite.
Figure 6.
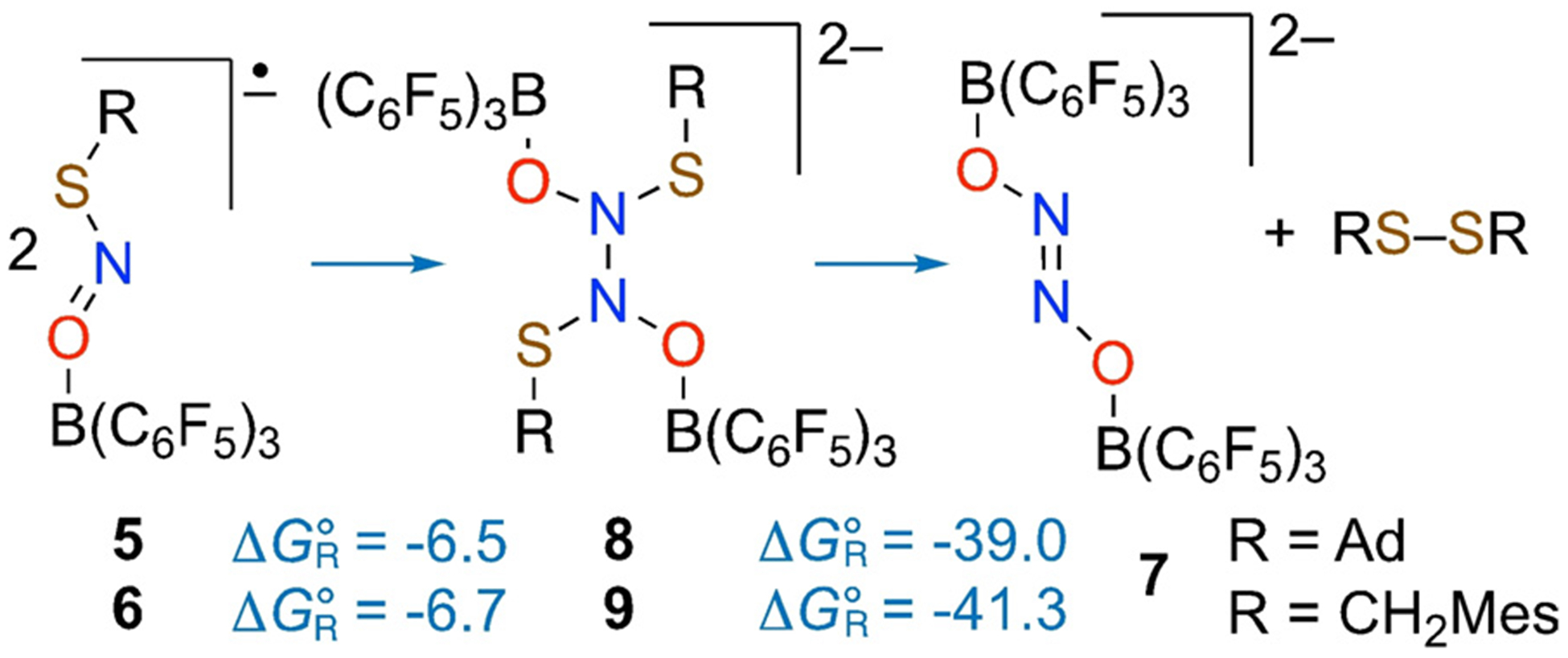
Stepwise conversion of [RSNO-B(C6F5)3] radical anions to dianion 7 and disulfide RS-SR. Reaction free energies (in kcalmol−1 at 298 K) calculated at the wB97XD-PCM(CH2Cl2)/red-ma-def2-SV(P) level of theory.
Importantly, reaction of 7-[Cp2*Co+]2 with 2 equiv CF3COOH triggers release of N2O in 42–43% yield (Figure 7). Release of N2O, the prototypical reactivity of hyponitrite upon protonation,[22] underscores how Lewis acids can redirect the chemical outcome of RSNO reduction, which typically generates NO.
Figure 7.
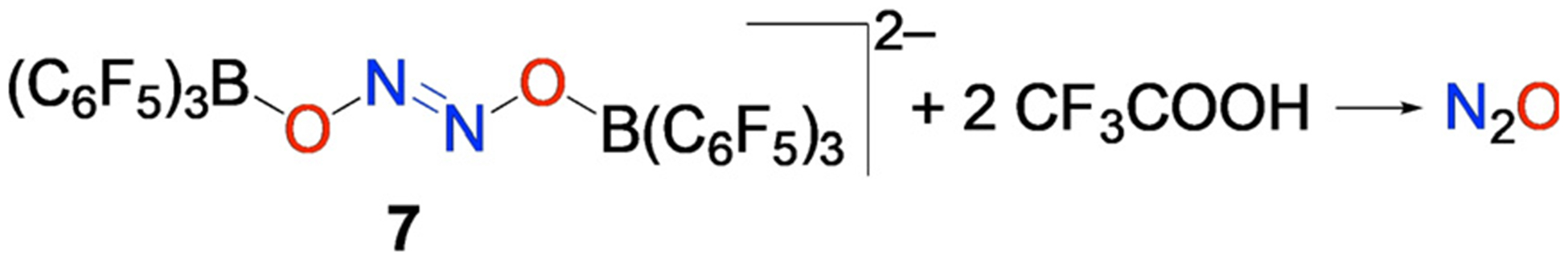
Reaction of (7-[Cp*2Co]2) with trifluoroacetic acid releases N2O.
Lewis acid coordination via the O atom of S-nitrosothiols results in significant changes in the structure and chemical properties of the RSNO by strongly promoting its RS+=N-O− resonance component. Most profoundly, this mode of Lewis acid coordination redirects reduction of RSNOs to the hyponitrite dianion [ON=NO]2− rather than NO. Moreover, Lewis acid coordination significantly raises the reduction potential of RSNOs, enabling reduction by mild reductants. This represents a general strategy to facilitate reduction of small molecules, previously illustrated in the reduction of disulfides RSSR by ferrocene that only occurs in presence of a Lewis acid such as B(C6F5)3.[23] Thus, Lewis acids could serve as a signaling motif to turn on outer-sphere RSNO reduction in the vicinity of electron-transfer proteins that function in the range of + 700 to −800 mV vs. NHE.[24] Future studies will examine the connection between Lewis acid strength and reduction potential, including Lewis acids that serve as H-bond donors that could directly promote N2O formation.
Supplementary Material
Acknowledgements
T.H.W. acknowledges the NIH (R01GM126205) and the Georgetown Environment Initiative. Q.K.T. acknowledges NSF CAREER award CHE-1255641, and Marquette University Way-Klinger Sabbatical Award. V.H. thanks Mahdi Raghibi Boroujeni and Dr. Pokhraj Ghosh for assistance with CV experiments. This work used the high-performance computing cluster at the Extreme Science and Engineering Discovery Environment (XSEDE) supported by NSF grant ACI-1053575.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under: https://doi.org/10.1002/anie.202001450.
Contributor Information
Valiallah Hosseininasab, Department of Chemistry, Georgetown University, Box 571227, Washington, DC 20057-1227 (USA).
Alison C. McQuilken, Department of Chemistry, Georgetown University, Box 571227, Washington, DC 20057-1227 (USA)
Abolghasem (Gus) Bakhoda, Department of Chemistry, Georgetown University, Box 571227, Washington, DC 20057-1227 (USA).
Jeffery A. Bertke, Department of Chemistry, Georgetown University, Box 571227, Washington, DC 20057-1227 (USA)
Qadir K. Timerghazin, Department of Chemistry, Marquette University, P.O. Box 1881, Milwaukee, WI 53201-1881 (USA)
Timothy H. Warren, Department of Chemistry, Georgetown University, Box 571227, Washington, DC 20057-1227 (USA)
References
- [1].Ignarro L, Nitric Oxide Biology and Pathobiology, 2nd ed., Elsevier, Amsterdam, 2009. [Google Scholar]
- [2].Rassaf T, Kleinbongard P, Preik M, Dejam A, Gharini P, Lauer T, Erckenbrecht J, Duschin A, Schulz R, Heusch G, Feelisch M, Kelm M, Circ. Res 2002, 91, 470–477. [DOI] [PubMed] [Google Scholar]
- [3].a) Smith BC, Marletta MA, Curr. Opin. Chem. Biol 2012, 16, 498–506; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bartberger MD, Mannion JD, Powell SC, Stamler JS, Houk KN, Toone EJ, J. Am. Chem. Soc 2001, 123, 8868–8869. [DOI] [PubMed] [Google Scholar]
- [4].a) Nakamura T, Tu S, Akhtar MW, Sunico CR, Okamoto SI, Lipton SA, Neuron 2013, 78, 596–614; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) McMahon TJ, Ahearn GS, Moya MP, Gow AJ, Huang YC, Luchsinger BP, Nudelman R, Yan Y, Krichman AD, Bashore TM, Califf RM, Singel DJ, Piantadosi CA, Tapson VF, Stamler JS, Proc. Natl. Acad. Sci. USA 2005, 102, 14801–14806; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS, Nat. Rev. Mol. Cell Biol 2005, 6, 150–166. [DOI] [PubMed] [Google Scholar]
- [5].Allen BW, Stamler JS, Piantadosi CA, Trends Mol. Med 2009, 15, 452–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Johnson MA, Macdonald TL, Mannick JB, Conaway MR, Gaston B, J. Biol. Chem 2001, 276, 39872–39878. [DOI] [PubMed] [Google Scholar]
- [7].Zhang S, Melzer MM, Nermin SS, Çelebi-Ölçüm N, Warren TH, Nat. Chem 2016, 8, 663–669. [DOI] [PubMed] [Google Scholar]
- [8].Fujisawa K, Ono T, Ishikawa Y, Amir N, Miyashita Y, Okamoto K, Lehnert N, Inorg. Chem 2006, 45, 1698–1713. [DOI] [PubMed] [Google Scholar]
- [9].a) Stomberski CT, Hess DT, Stamler JS, Antioxid. Redox Signaling 2019, 30, 1331–1351; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Seth D, Hess DT, Hausladen A, Wang L, Wang Y-J, Stamler JS, Molecular Cell 2018, 69, 451–464; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Anand P, Stamler JS, J. Mol. Med 2012, 90, 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Talipov MR, Timerghazin QK, J. Phys. Chem. B 2013, 117, 1827–1837. [DOI] [PubMed] [Google Scholar]
- [11].Timerghazin QK, Peslherbe GH, English AM, Org. Lett 2007, 9, 3049–3052. [DOI] [PubMed] [Google Scholar]
- [12].a) Ivanova LV, Cibich D, Deye G, Talipov MR, Timerghazin QK, ChemBioChem 2017, 18, 726–738; [DOI] [PubMed] [Google Scholar]; b) Timerghazin QK, Talipov MR, J. Phys. Chem. Lett 2013, 4, 1034–1038. [DOI] [PubMed] [Google Scholar]
- [13].Moran EE, Timerghazin QK, Kwong E, English AM, J. Phys. Chem. B 2011, 115, 3112–3126. [DOI] [PubMed] [Google Scholar]
- [14].a) Baciu C, Cho K-B, Gauld JW, J. Phys. Chem. B 2005, 109, 1334–1336; [DOI] [PubMed] [Google Scholar]; b) Talipov MR, Khomyakov DG, Xian M, Timerghazin QK, J. Comput. Chem 2013, 34, 1527–1530. [DOI] [PubMed] [Google Scholar]
- [15].Perissinotti LL, Estrin DA, Leitus G, Doctorovich F, J. Am. Chem. Soc 2006, 128, 2512–2513. [DOI] [PubMed] [Google Scholar]
- [16].Arulsamy N, Bohle DS, Butt JA, Irvine GJ, Jordan PA, Sagan E, J. Am. Chem. Soc 1999, 121, 7115–7123. [Google Scholar]
- [17].The X-ray structure of an FLP-bound R–N=S=O linkage isomer has been reported: Longobardi LE, Wolter V, Stephan DW, Angew. Chem. Int. Ed 2015, 54, 809–812; Angew. Chem. 2015, 127, 823 – 826. [DOI] [PubMed] [Google Scholar]
- [18].a) Goto K, Hino Y, Kawashima T, Kaminaga M, Yano E, Yamamoto G, Takagi N, Nagase S, Tetrahedron Lett. 2000, 41, 8479–8483; [Google Scholar]; b) Spivey AC, Colley J, Sprigens L, Hancock SM, Cameron DS, Chigboh KI, Veitch G, Frampton CS, Adams H, Org. Biomol. Chem 2005, 3, 1942–1952. [DOI] [PubMed] [Google Scholar]
- [19].Glendening ED, Landis CR, Weinhold F, J. Am. Chem. Soc 2019, 141, 4156–4166. [DOI] [PubMed] [Google Scholar]
- [20].Connelly NG, Geiger WE, Chem. Rev 1996, 96, 877–910. [DOI] [PubMed] [Google Scholar]
- [21].a) Chen LH, Laan J, J. Raman Spectrosc 1983, 14, 284–287; [Google Scholar]; b) Wijeratne GB, Hematian S, Siegler MA, Karlin KD, J. Am. Chem. Soc 2017, 139, 13276–13279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wright AM, Hayton TW, Inorg. Chem 2015, 54, 9330–9341. [DOI] [PubMed] [Google Scholar]
- [23].Liu LL, Cao LL, Shao Y, Stephan DW, J. Am. Chem. Soc 2017, 139, 10062–10071. [DOI] [PubMed] [Google Scholar]
- [24].Liu J, Chakraborty S, Hosseinzadeh P, Yu Y, Tian S, Petrik I, Bhagi A, Lu Y, Chem. Rev 2014, 114, 4366–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.