

Abstract
The inhibitory epidermal growth factor receptor (EGFR) antibody, cetuximab, is an approved therapy for head and neck squamous cell carcinoma (HNSCC). Despite tumor response observed in some HNSCC patients, cetuximab alone or combined with radio- or chemotherapy fails to yield long-term control or cures. We hypothesize that a flexible receptor tyrosine kinase coactivation signaling network supports HNSCC survival in the setting of EGFR blockade, and that drugs disrupting this network will provide superior tumor control when combined with EGFR inhibitors. In this work, we submitted EGFR-dependent HNSCC cell lines to RNA interference-based functional genomics screens to identify, in an unbiased fashion, essential protein kinases for growth and survival as well as synthetic lethal targets for combined inhibition with EGFR antagonists. Mechanistic target of rapamycin kinase (MTOR) and erythroblastosis oncogene B (ERBB)3 were identified as high-ranking essential kinase hits in the HNSCC cell lines. MTOR dependency was confirmed by distinct short hairpin RNAs (shRNAs) and high sensitivity of the cell lines to AZD8055, whereas ERBB3 dependency was validated by shRNA-mediated silencing. Furthermore, a synthetic lethal kinome shRNA screen with a pan-ERBB inhibitor, AZD8931, identified multiple components of the extracellular signal-regulated kinase (ERK) mitogen-activated protein kinase pathway, consistent with ERK reactivation and/or incomplete ERK pathway inhibition in response to EGFR inhibitor monotherapy. As validation, distinct mitogen-activated protein kinase kinase (MEK) inhibitors yielded synergistic growth inhibition when combined with the EGFR inhibitors, gefitinib and AZD8931. The findings identify ERBB3 and MTOR as important pharmacological vulnerabilities in HNSCC and support combining MEK and EGFR inhibitors to enhance clinical efficacy in HNSCC.
SIGNIFICANCE STATEMENT
Many cancers are driven by nonmutated receptor tyrosine kinase coactivation networks that defy full inhibition with single targeted drugs. This study identifies erythroblastosis oncogene B (ERBB)3 as an essential protein kinase in epidermal growth factor receptor–dependent head and neck squamous cell cancer (HNSCC) cell lines and a synthetic lethal interaction with the extracellular signal-regulated kinase mitogen-activated protein kinase pathway that provides a rationale for combining pan-ERBB and mitogen-activated protein kinase inhibitors as a therapeutic approach in subsets of HNSCC.
Introduction
Comprehensive identification of diverse, but often therapeutically-tractable oncogenes in cancers such as lung adenocarcinoma and melanoma, permits implementation of precision medicine, which matches oncogenic vulnerabilities with specific drugs, yielding greatly enhanced patient benefit (Politi and Herbst, 2015; Bivona and Doebele, 2016). By contrast, the mutational landscape of head and neck squamous cell cancer (HNSCC) defined by The Cancer Genome Atlas project has failed to provide abundant targets for effective precision medicine strategies. Although epidermal growth factor receptor (EGFR) is not subject to frequent somatic mutations in HNSCC, genomic amplification and high expression provided a rationale for clinical testing of the anti-EGFR antibody, cetuximab, and ultimately led to Food and Drug Administration approval in combination with radio- or chemotherapy (Bonner et al., 2006, 2010; Vermorken et al., 2007, 2008). EGFR-specific tyrosine kinase inhibitors (TKIs) have not exhibited clear efficacy, although pan-erythroblastosis oncogene B (ERBB)–active TKIs induce responses in HNSCC equal to cetuximab (Seiwert et al., 2014). Still, the therapeutic responses are invariably incomplete and eventually followed by tumor progression and treatment failure. Because receptor tyrosine kinases (RTKs) such as EGFR rarely signal in isolation, even when oncogenically mutated, a deeper understanding of the functional EGFR network in HNSCC is warranted.
Distinct from mutated RTKs that function as dominant oncogenes in lung adenocarcinoma, we and others have suggested that networks of nonmutated RTKs and downstream effector pathways serve as oncogene inputs in many cancers, including HNSCC (Xu and Huang, 2010; Singleton et al., 2013; Tan et al., 2017). Indeed, Singleton et al. (2013) defined a coactivation network comprised of EGFR, ERBB2, fibroblast growth factor receptor (FGFR) family members, and hepatocyte growth factor receptor (MET) in HNSCC cell lines and demonstrated maximal growth inhibition with a triple TKI cocktail. Considering the toxicity often associated when even two drugs are combined, therapeutic approaches involving such complex combinations of TKIs seem unlikely to move forward through clinical trials. Instead, an unbiased screen for novel and druggable vulnerabilities might identify safer and more effective combination therapies. In this regard, functional genomics techniques provide a path forward for dissecting the complexity of oncogene signaling in cancer cells and identifying and prioritizing therapeutic vulnerabilities (Singleton et al., 2015, 2017; Tsherniak et al., 2017).
With the goal of defining targetable vulnerabilities in HNSCC and developing TKI-based combinations, we performed functional genomics-based RNA interference (RNAi) screens for both essential protein kinases and kinases whose inhibition in combination with an ERBB-active TKI induces synergistic growth inhibition. We found that short hairpin RNAs (shRNAs) targeting mechanistic target of rapamycin kinase (MTOR) and ERBB3 were significantly lost over time in the cell lines screened. Complementary synthetic lethal screens performed with a pan-ERBB inhibitor, AZD8931 (Hickinson et al., 2010; Barlaam et al., 2013), identified multiple components of the extracellular signal-regulated kinase (ERK) mitogen-activated protein kinase (MAPK) pathway as a target for inducing synthetic lethality. Our findings demonstrate that nonmutated and druggable signal network components can be identified through the use of functional genomics techniques to guide the development of combinations of inhibitors that may be deployed as precision medicine options for the treatment of HNSCC.
Materials and Methods
Cell Culture
HNSCC cell lines were selected from our collection and cultured in Dulbecco’s mitogen-activated protein kinase Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA) supplemented with 5% FBS with 1% penicillin-streptomycin (Sigma-Aldrich, St. Louis, MO) at 37°C in a humidified 5% CO2 incubator. All cancer cell lines used in this study were submitted to fingerprint analysis by the University of Colorado Cancer Center DNA Sequencing and Analysis Core to confirm authenticity within 1 year of performing the studies described in this work.
Lentivirus Preparation
The Human Kinase TRC (The RNAi Consortium) shRNA library (obtained from the Functional Genomics Shared Resource within the University of Colorado Cancer Center) was packaged in 293T cells, as follows. The 293T cells were incubated overnight at 37°C in a 5% CO2 humidified incubator with Turbofect transfection reagent (ThermoFisher Scientific, Waltham, MA), 5.0 μg pΔ8.9, 5.0 μg pCMV-VSV-G, and 3.0 μg kinome library. The virus-containing media from the 293T cells was then filtered through a 0.45-μm filter after adding 1 μg/ml polybrene and either used immediately or stored at 4°C until ready for use.
Functional Genomics Screens
Essential Kinome Screen.
Target cell lines (UMSCC8, UMSCC25, HN6, HN12, Cal27, JHU011) were plated at 5 × 106 cells per 15-cm plate, two plates per cell line, in 20 ml growth medium. The following day, cells were pretreated with 1 µg/ml polybrene for 30 minutes. The media containing the packaged lentiviral kinome shRNA library supplemented with 50 shRNAs (5 per gene) targeting 10 murine genes (β-actin, Actb; aldolase A fructose-bisphosphate, Aldoa; Rho GDP dissociation inhibitor α, Arhgdia; copine I, Cpne1; glyceraldehyde-3-phosphate dehydrogenase, Gapdh; heterogeneous nuclear ribonucleoprotein D, Hnrnpd; lactate dehydrogenase A, Ldha; non-POU domain–containing octamer-binding protein, Nono; phosphoglycerate kinase 1, Pgk1; ribosomal protein L11, Rpl11) was added to each plate of target cells at a multiplicity of infection of ∼0.2, incubated for 16 hours, and then removed and replaced with fresh growth medium (lacking puromycin). Forty-eight hours postinfection, each cell line was trypsinized and plated at a density of at least 2 × 106 cells per 15-cm plate in quadruplicate. Additionally, at the same time point, four replicates of 4 × 106 cells each from each cell line were pelleted, and the genomic DNA (gDNA) was isolated using the Quick g-DNA MiniPrep kit (Zymo Research, Irvine, CA). DNA was eluted with 100 μl elution buffer and stored at −20°C. On day 14 postinfection, 4 × 106 cells per replicate from each cell line were pelleted, and the gDNA was isolated.
Synthetic Lethal Screen.
Target HNSCC cell lines (UMSCC8, UMSCC25, HN12, Cal27, JHU011) were plated at 3 × 106 cells per 15-cm plate in 20 ml growth media with two plates per cell line. Cells were maintained at cell densities that are at least 10 times the number of shRNAs in the library. Twenty-four hours later, replicate plates of target cells were pretreated with 1 μg/ml polybrene for 30 minutes, and medium containing the packaged human kinome shRNA library supplemented with shRNAs targeting 10 murine genes was added to each plate of target cells at a multiplicity of infection of ∼0.2. After a 24-hour incubation, fresh growth medium was added, and 72 hours after viral transduction, target cells expressing shRNAs were selected with 1 μg/ml puromycin for 5 days. Stable puromycin-resistant cells were plated in triplicate and treated with DMSO or AZD8931 for 72 hours. Subsequently, the DMSO/drug-containing medium was removed, and the cells were incubated in fresh medium containing puromycin without DMSO/AZD8931 for 72 hours. The rationale for this media change was to enrich for shRNA “hits” that induced cell death or strong growth arrest even with removal of the TKI. gDNA from 4 × 106 cells/replicate was isolated using the kit Quick-gDNA MiniPrep kit and eluted in 100 μl elution buffer.
Quantification of shRNAs by Illumina Sequencing.
The gDNA samples were submitted to nested polymerase chain reaction (PCR) to amplify the shRNAs and add barcodes and sequences required for Illumina sequencing, as described (Singleton et al., 2015, 2017). Samples were pooled and sequenced on an Illumina Genome Analyzer IIx. The resulting sequence data were submitted for analysis using the Bioinformatics for Next Generation Sequencing (BINGS!) pipeline, as described previously (Singleton et al., 2015, 2017). The shRNA sequencing data associated with the essential and synthetic lethal screens have been deposited in Gene Expression Omnibus (GSE67500 and GSE132788).
In Vitro Growth Assays
For experiments measuring the effect of pharmacological drugs on clonogenic growth, HNSCC cells were plated at 100–200 cells/well in 96-well tissue culture plates and treated with inhibitors at the doses indicated. When the DMSO-treated control wells became confluent (7–10 days), cell DNA content was measured with the CyQUANT Direct Cell Proliferation Assay (ThermoFisher Scientific), according to the manufacturer’s instructions, and a fluorescence plate reader set to excitation wavelength of 508 nm and an emission wavelength of 527 nm. In some experiments, the HNSCC cells were seeded at 100–200 cells/well in six-well plates. The next day, protein kinase inhibitors were added at concentrations described in the figure legends and incubated for 1 to 2 weeks. The resulting colonies were stained with crystal violet and photographed, and total colony area was quantified with MetaMorph imaging software. To measure the effect of RNAi-mediated silencing on HNSCC growth, cells (20,000/well) in six-well plates were transduced, as described above, with a control shRNA targeting GFP or shRNAs targeting MTOR or ERBB3. The cells were submitted to selection in puromycin (1 μg/ml) for 5–10 days, stained with crystal violet, and quantified by measuring the crystal violet absorbance of the fixed cells at 590 nM in a plate reader.
Immunoblot Analyses
Phospho-ERK, phospho-EGFR Y1068, phospho-EGFR Y1148, phospho-AKT S473, EGFR, ERBB2, ERBB3, and MTOR were measured by immunoblotting using antibodies obtained from Cell Signaling Technology (Danvers, MA). Aliquots of cell extracts prepared in lysis buffer [0.5% Triton X-100, 50 mM β-glycerophosphate (pH 7.2), 0.1 mM Na3VO4, 2 mM MgCl2, 1 mM EGTA, 1 mM DTT, 0.3 M NaCl, 2 μg/ml leupeptin, and 4 μg/ml aprotinin] were submitted SDS-PAGE. After electrophoretic transfer to nitrocellulose, filters were blocked in 3% bovine serum albumin (Cohn Fraction V; Gold Biotechnology, St. Louis, MO) in Tris-buffered saline with 0.1% Tween 20. The filters were then incubated overnight at 4°C with antibodies, washed three times in Tris-buffered saline with 0.1% Tween 20, and incubated for 1 hour at room temperature with horseradish peroxidase goat anti-rabbit antibodies. The filters were developed using Luminata Classico substrate (Millipore, Billerica, MA), according to the manufacturer’s instructions. When blotting phosphorylated proteins, the filters were stripped and probed the corresponding total protein EGFR, AKT (Cell Signaling Technology), ERK1/2, or Na/K-ATPase α-subunit (Santa Cruz Biotechnology, Santa Cruz, CA) as a loading control.
Chemicals
AZD8931, gefitinib, selumetinib, trametinib, AZD8055, and MK2206 were purchased from MedChemExpress (Monmouth Junction, NJ). Stocks of the drugs were prepared in DMSO at concentrations such that the final concentration of DMSO was 0.1% v/v.
Statistics Analysis
Prism 8.2 (GraphPad Software, San Diego, CA) was used to perform statistical analyses. Data are presented as the mean and S.D. A paired Student’s t test (two-tail) was used to determine statistical significance, unless otherwise noted. The P values are denoted by *(P ≤ 0.05), **(P ≤ 0.01), ***(P ≤ 0.001), and ****(P ≤ 0.0001) and were corrected for multiple comparisons (Dunnett). To derive combination index values, the CalcuSyn program (Biosoft, Cambridge, UK) was used.
Results
We previously submitted lung cancer and HNSCC cell lines with defined FGFR1 growth dependencies to functional genomics-based interrogation of protein kinase pathways that collaborate with FGFR1 to drive growth and survival (Singleton et al., 2015). In this study, a panel of human HNSCC cell lines was assembled that selectively included those with high dependency on EGFR pathway signaling as defined by sensitivity to gefitinib and the pan-ERBB inhibitor, AZD8931 (Hickinson et al., 2010; Barlaam et al., 2013). As shown in Supplemental Fig. 1, the IC50 values for growth inhibition mediated by gefitinib ranged between 16 and 23 nM. AZD8931 was a more potent inhibitor on each cell line, with IC50 values ranging between 2 and 6 nM. This increased potency most likely reflects the ability of AZD8931 to inhibit both EGFR and ERBB2 in which both RTKs are active and function within a coactivation network in HNSCC cells (Singleton et al., 2013). By contrast, two distinct HNSCC cell lines, 584-A2 and CCL30, previously defined for FGFR1 dependency (Singleton et al., 2015), were insensitive to either gefitinib or AZD8931 (Supplemental Fig. 1, A–C). The sensitivity of the HNSCC cell lines to EGFR-targeting TKIs is not strictly related to the protein expression levels of EGFR or ERBB2, although the FGFR1-dependent lines express little or no ERBB3 (Supplemental Fig. 1D). In subsequent experiments, the EGFR inhibitor–sensitive HNSCC cell lines were submitted to functional genomics screens with a kinome-targeting shRNA library using experimental formats designed to identify protein kinases essential for growth and survival, as well as specific protein kinase pathways that exhibit synthetic lethal phenotypes in the context of AZD8931.
Identification of Essential Protein Kinases in EGFR-Dependent HNSCC Cell Lines.
The human HNSCC cell lines were submitted to an essential kinome shRNA screen. Cal27, HN6, HN12, JHU011, UMSCC8, and UMSCC25 were transduced with a human kinome-targeting shRNA library that included murine-specific shRNAs targeting 10 mouse genes as negative controls (see Materials and Methods). gDNA was isolated from replicate plates (n = 4) after 48 hours and 14 days of culture postlentiviral transduction. The shRNA sequences were amplified by PCR, barcoded, and quantified by Illumina sequencing, as described in Materials and Methods. Specific shRNAs that exhibited statistically significant loss from the total shRNA pool after 14 days of culture were identified by pairwise statistical comparisons and the bioinformatics pipeline, BiNGS! (Kim and Tan, 2012). Data from the multiple shRNAs that targeted each gene were merged by weighted Z-transformation, and the kinases were subsequently ranked using the associated P value [P(wZ)]. Although the kinome library includes shRNAs targeting all kinases phosphorylating proteins, lipids, nucleosides, and sugars, we intentionally extracted and in this work present data pertaining to the protein kinases (n = 511) targeted by the shRNA library after BINGS! analysis. Figure 1A and Supplemental Table 1 show the average ranks of 511 protein kinases targeted by the human shRNA kinome library as well as the 10 control murine genes among the six HNSCC cell lines. The murine-specific shRNAs targeting mouse genes (indicated by green symbols) were ranked in the lower half and serve as an estimate of nonspecific shRNA activity in the essential screen (Fig. 1A).
Fig. 1.

Essential protein kinases identified by the functional shRNA screen. (A) The average ranks of 521 protein kinases from the essential shRNA screen performed in UMSCC25, UMSCC8, JHU011, HN6, HN12, and Cal27 cells are graphed along with the 10 murine control genes indicated with green symbols. The ranks for the protein kinases and murine control genes in the individual cell lines are presented in Supplemental Table 1. EGFR, ERBB2, MET, and FGFR1 were not identified as highly essential as single protein kinases and are shown with yellow symbols. Based on the published literature (see text in Results), the top-ranked essential kinases were assembled into interaction networks centered on MTOR (B) and ERBB3 (C) in which the average ranks of the indicated protein kinases among the six HNSCC cell lines are denoted by the colors shown in the color scale bar.
MTOR was the top-ranked protein kinase in two of the six HNSCC cell lines and, when averaged among the six cell lines, the highest ranked essential protein kinase (Fig. 1A; Supplemental Table 1). To independently validate MTOR as an essential protein kinase, two shRNAs distinct from those in the kinome library were used to silence MTOR in UMSCC8, UMSCC25, and Cal27 cells. The findings in Fig. 2 demonstrate marked reduction in MTOR protein following lentiviral transduction of the two MTOR shRNAs relative to a GFP targeting control shRNA as well as statistically significant inhibition of clonogenic growth of the three HNSCC cell lines. As a pharmacological validation, the HNSCC cell lines were treated with AZD8055, a direct MTOR kinase inhibitor (Chresta et al., 2010; Pike et al., 2013; Singleton et al., 2015), in a clonogenic growth assay format. The sensitivity of the cell line panel to AZD8055 is presented in Supplemental Fig. 2 with uniformly high potency of the drug across this panel of HNSCC cell lines (IC50 values ranging from 3 to 7 nM). Clonogenic growth was also strongly inhibited by the target of rapamycin complex (TORC) 1 selective drug, rapamycin (Supplemental Fig. 2). In addition to MTOR, the essential screen identified the AMP-dependent protein kinase α subunit, protein kinase AMP-activated catalytic subunit alpha 1, checkpoint kinase 1, and DNA-activated protein kinase, catalytic subunit as the 4th, 6th, and 26th ranked essential protein kinases, respectively (Fig. 1B; Supplemental Table 1). All three proteins have been identified as direct binding partners in the MTOR interaction network (Cheng et al., 2004; Caron et al., 2010; Srivas et al., 2016; Shen et al., 2017). DNA-activated protein kinase, catalytic subunit, and CDK1, the 17th ranked essential protein kinase, represent defined checkpoint kinase 1 interactors (Kim et al., 1999; Goudelock et al., 2003; Hein et al., 2015). Thus, the findings support MTOR and a network of interacting protein kinases as a druggable protein kinase dependency in multiple EGFR-dependent HNSCC cell lines.
Fig. 2.
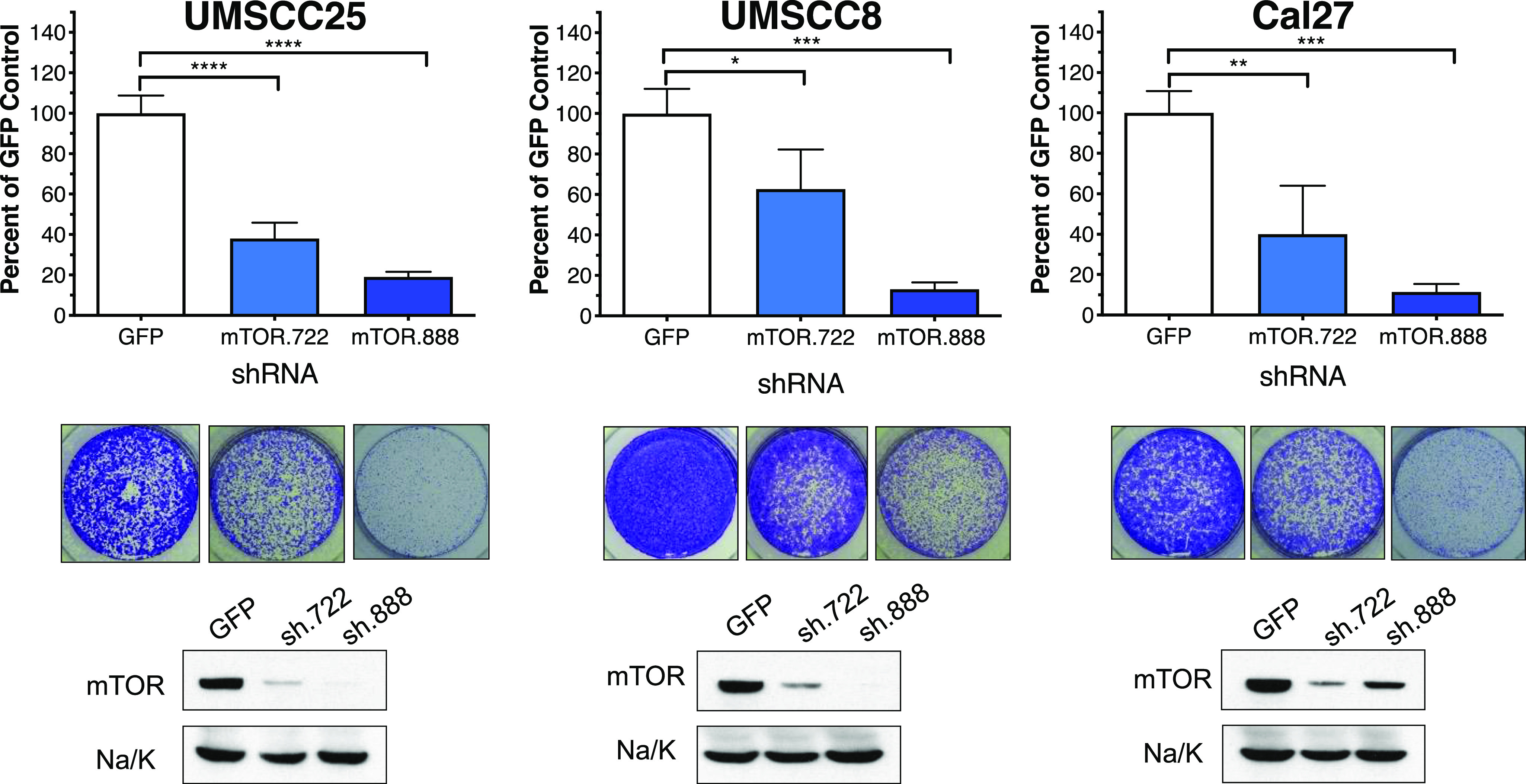
Validation of MTOR as an essential protein kinase in HNSCC cell lines. UMSCC25, UMSCC8, and Cal27 cells in six-well plates were transduced with a control shRNA targeting GFP and two MTOR shRNAs (TRCN0000363722 and TRCN0000332888) not present in the kinome shRNA library. The cells were submitted to selection in puromycin (1 μg/ml) for 5 to 10 days and stained with crystal violet. Representative images of stained wells are shown and were quantified by measuring the crystal violet absorbance of the fixed cells at 590 nM. The data are the means and S.D. of duplicate wells from two independent experiments normalized to the GFP shRNA controls. Cell extracts were prepared from replicate wells and submitted to immunoblot analysis for MTOR. The filters were stripped and reprobed for the α-subunit of NaK-ATPase (Na/K) as a loading control. The data demonstrate efficient inhibition of MTOR protein levels with the MTOR shRNAs relative to the GFP control shRNA.
ERBB3 was the top-ranked protein kinase in three of the six HNSCC cell lines and, when averaged among the six cell lines, the second ranked essential protein kinase overall (Fig. 1A; Supplemental Table 1). Despite the high sensitivity of the cell lines to gefitinib and AZD8931 (Supplemental Fig. 1), neither EGFR nor ERBB2 was identified as highly essential protein kinases in any of the six HNSCC cell lines, and their ranks were not statistically different from the majority of the murine control genes (Fig. 1A; Supplemental Table 1). Likewise, neither MET nor FGFR1 was found to be highly essential, despite our previous study demonstrating the functioning of EGFR, ERBB2, MET, and FGFR1 within a coactivation network involved in HNSCC growth and survival (Singleton et al., 2013). In fact, the failure of the shRNA screen to identify any of these four RTKs as essential protein kinases is consistent with ERBB3 serving as a central hub such that ERBB3 silencing simultaneously ablates growth and survival signaling mediated by all the members of the coactivation network. In addition to ERBB3 interactions with EGFR (Li et al., 2013, 2015), ERBB2 (Graus-Porta et al., 1997; Scaltriti et al., 2009), MET (Engelman et al., 2007; Liu et al., 2011), and FGFRs (Jones et al., 2006), there is ample evidence for functional lateral interactions between EGFR and ERBB2 (Li et al., 2013), MET and EGFR (Li et al., 2013, 2015), and FGFRs and ERBB2 (Kunii et al., 2008; Wang et al., 2015). These lateral interactions of EGFR and ERBB2 with other RTKs within the coactivation network likely account for the single agent sensitivity of the HNSCC cell lines to gefitinib and AZD8931. To independently validate ERBB3 as an essential protein kinase, two shRNAs were used to silence ERBB3 in UMSCC8, UMSCC25, and HN6 cells. Lentiviral transduction of UMSCC8 cells with the ERBB3 shRNAs, TRCN199364 and TRCN40108, reduced ERBB3 mRNA levels to 27.5% and 30.5%, respectively, of the levels measured in cells transduced with the GFP-targeting shRNA (Fig. 3A). ERBB3 silencing reduced clonogenic growth of UMSCC8, UMSCC25, and HN6 cells relative to a GFP-targeted control shRNA (Fig. 3, B–D). Thus, the findings validate ERBB3 as an essential protein kinase in EGFR/ERBB-dependent HNSCC cell lines driving survival and cell proliferation.
Fig. 3.

Validation of ERBB3 as an essential protein kinase in HNSCC cells. UMSCC8 (A, B), UMSCC25 (C), and HN6 cells (D) were transduced with lentiviruses encoding a control GFP shRNA or two ERBB3-targeted shRNAs (TRCN199364 and TRCN40108). After ∼10 days of culture under puromycin selection, the cells were submitted to RNA purification and reverse-transcription PCR (A) or the wells were fixed and stained with crystal violet and quantified by measuring the absorbance of the fixed cells at 590 nm (B–D). The data are the means and S.D. of duplicate wells from two independent experiments normalized to their GFP shRNA controls.
The ERK MAPK Pathway Is a Synthetic Lethal Vulnerability with ERBB-Specific TKIs in HNSCC.
To identify novel vulnerabilities that emerge in the setting of treatment with the pan-ERBB inhibitor, AZD8931, five EGFR TKI-sensitive HNSCC cell lines were transduced with the kinome shRNA library supplemented with murine control shRNAs and submitted to a synthetic lethal loss-of-function screen. The cell lines were selected for puromycin resistance to stably express the lentivirus shRNA library and then treated in triplicate for 72 hours with DMSO or AZD8931 at concentrations that inhibited growth by approximately 70% (Supplemental Fig. 1B). The treatment was followed by a 72-hour recovery period in drug-free media. gDNA was isolated from each replicate, and the shRNAs were amplified by PCR, barcoded, and analyzed by BINGS!SL (Kim and Tan, 2012; Singleton et al., 2015, 2017), as described in Materials and Methods.
The average ranks (n = 5) of protein kinases from the synthetic lethal screen are shown in Fig. 4A and Supplemental Table 2 and reveal that shRNAs targeting multiple components of the ERK MAPK pathway were reduced or eliminated in HNSCC cells treated with AZD8931. RPS6KA4 (MSK2), MAPK1 (ERK2), and RPS6KA5 (MSK1) exhibited average ranks of 48, 65, and 76, respectively, among the five HNSCC cell lines (Fig. 4; Supplemental Table 2). Although neither of the two MAP2K activators of MAPK1 was significantly ranked hits (MAP2K1, average rank = 310; MAP2K2, average rank = 239), multiple MAP3Ks exhibited synthetic lethality with AZD8931, including MAP3K2 [mitogen-activated protein kinase kinase (MEK) kinase 2, average rank = 62], MAP3K3 (MEK kinase 3, average rank = 71), MAP3K6 (ASK2, average rank = 78), MAP3K9 (Mixed-lineage protein kinase 1 (MLK1), average rank = 126), MAP3K11 (MLK3, average rank = 137), and BRAF (average rank = 132). Also, the MAPK pathway scaffold protein-encoding gene, KSR1, exhibited an average rank of 131.
Fig. 4.

Identification of protein kinases that exhibit synthetic lethality in combination with AZD8931. (A) Average ranks of 521 protein kinases from the synthetic lethal shRNA screen performed in UMSCC25, UMSCC8, JHU011, HN12, and Cal27 cells are graphed along with the 10 murine control genes indicated with green symbols. The ranks for the protein kinases and murine control genes in the individual cell lines are presented in Supplemental Table 2. (B) ERK MAPK pathway components from the synthetic lethal screen are assembled into a putative pathway, in which the strength of the hit is indicated by the color bar.
We tested growth inhibition in the HNSCC cell lines with combinations of EGFR TKIs and pharmacological MEK inhibitors to validate a synthetic lethal interaction of the ERK MAPK pathway with AZD8931. UMSCC25 cells were treated with combinations of AZD8931 and the MEK inhibitor, selumetinib, as described in Materials and Methods using CyQUANT reagent to measure cellular DNA content. The resulting data (Fig. 5A) were analyzed with the CalcuSyn program based on the methods of Chou and Talalay (1984) and demonstrated evidence for broad synergy across multiple combinations of AZD8931 and selumetinib (Fig. 5B). Synergistic growth inhibition was observed in all five HNSCC cell lines in response to AZD8931–selumetinib and gefitinib–selumetinib combinations (Supplemental Fig. 3) as well as when the two TKIs were combined with the distinct MEK inhibitor, trametinib (Supplemental Fig. 4).
Fig. 5.

Synergistic growth inhibition of UMSCC25 cells with combined AZD8931 and MEK inhibitor, selumetinib. (A) UMSCC25 cells were seeded in 96-well plates at 100 cells/well and incubated in triplicate with the indicated combinations of AZD8931 and selumetinib for 7 to 10 days. Cell number was estimated by measuring DNA content with CyQUANT reagent, and the mean fluorescence values are plotted. (B) The resulting data in (A) were submitted to the CalcuSyn program (Biosoft) to calculate combination indices (CI) for inferring synergy as shown by the color scale. The data shown are representative of another independent experiment.
The ERK pathway is regulated downstream of ERBB family members and is predicted to be strongly inhibited by AZD8931. Thus, the finding that protein kinase components of the ERK MAPK pathway emerged as synthetic lethal hits in the context of AZD8931 treatment is unanticipated. To explore ERBB family regulation of the ERK pathway in HNSCC cells, UMSCC25 cells (Fig. 6) and JHU011 cells (Supplemental Fig. 5) were treated with a range of AZD8931 concentrations (0–100 nM) with or without trametinib at a dose (3 nM) that did not fully ablate phosphorylated (p)-ERK levels alone for various times (1, 6, 24, and 72 hours), and cell extracts were submitted to immunoblot analysis for phospho-EGFR (Y1068/Y1148) and phospho-ERK (T202/Y204) levels. In UMSCC25 cells, AZD8931 potently reduced but did not eliminate p-ERK levels after a 1-hour treatment (Fig. 6), whereas JHU011 cells showed complete inhibition of p-ERK at 1 hour with AZD8931 alone (Supplemental Fig. 5). Levels of p-ERK subsequently rebounded in both cell lines by 24 and 72 hours of AZD8931 treatment despite maintenance of p-EGFR inhibition similar to the 1-hour timepoint (Fig. 6; Supplemental Fig. 5). Moreover, combined treatment with AZD8931 and trametinib provided a nearly complete inhibition of p-ERK at all time points and prevented the rebound in p-ERK levels observed at 72 hours. These data are consistent with published reports of ERK pathway reactivation following TKI-mediated blockade of oncogenic RTKs in cancer cells (Ercan et al., 2012; Bruner et al., 2017; Zhou et al., 2017). The findings are also consistent with the contribution of proximal signaling distinct from EGFR and ERBB family members to ERK MAPK pathway activity such that silencing of specific protein kinases by the shRNA library and MEK inhibitors provides greater blockade of this pathway and thereby deeper growth inhibition.
Fig. 6.
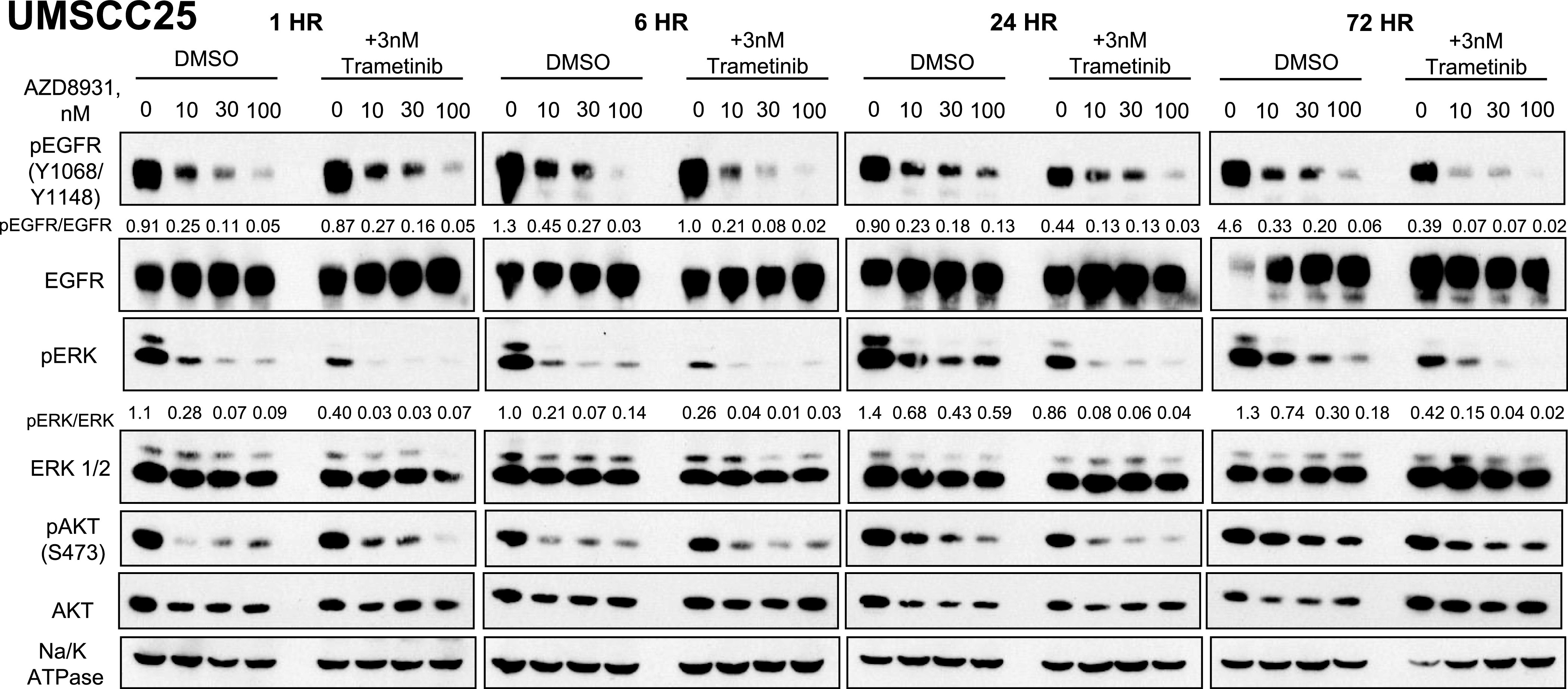
Effect of combined AZD8931 and trametinib treatment on phospho-ERK and phospho-EGFR levels in UMSCC25 cells. UMSCC25 cells were treated with AZD8931 (0–100 nM) in the presence or absence of 3 nM trametinib, and cell extracts were prepared after 1, 6, 24, and 72 hours of treatment. The extracts were submitted to SDS-PAGE and immunoblotted for phospho-EGFR (mixture of anti-Y1068 and Y1148) as well as phospho-ERK and phospho-Ser473-AKT. The filters were stripped and reprobed for total EGFR, ERK, AKT, and Na/K-ATPase α-subunit levels as loading controls. The relative levels of p-EGFR/EGFR and p-ERK/ERK following densitometry of the autoradiographic films are shown, and the data are representative of another independent experiment.
Discussion
The essential protein kinase screen identified MTOR as a top-ranked hit in the HNSCC cell lines tested. This finding was validated with distinct MTOR-targeting shRNAs and by AZD8055, a direct kinase inhibitor, both of which inhibited clonogenic growth. Importantly, MTOR is a highly essential gene across the majority of cancer cell lines assessed by RNAi and CRISPR-based screens in the Dependency Map project [(Tsherniak et al., 2017) and https://depmap.org/portal/gene/MTOR?tab=overview]. Indeed, the findings show that MTOR is a highly essential protein kinase in diverse cancer cell lines and is consistent with the extensive MTOR protein interactome and the multitude of cell signal pathways that it controls (Cheng et al., 2004; Caron et al., 2010; Srivas et al., 2016; Shen et al., 2017). MTOR exists in two signaling complexes, TORC1 and TORC2, which associate with the interacting proteins, RAPTOR and RICTOR, respectively. Our shRNA screen does not discern which of these complexes may be dominant due to the absence of RAPTOR/RICTOR-targeting shRNAs in the kinome library. However, the six HNSCC cell lines tested were highly sensitive to both AZD8055 (IC50 = 3–7 nM), a direct MTOR kinase inhibitor that blocks signaling through TORC1 and TORC2 (Chresta et al., 2010; Pike et al., 2013) and rapamycin (IC50 = 2–20 nM) with selectivity for TORC1. TORC2 functions as a hydrophobic motif kinase for AKT and PKC isoforms (Jhanwar-Uniyal et al., 2017), although none of the AKT isoforms were identified as high-ranking essential protein kinase hits (Supplemental Table 1), despite their established function within the MTOR interaction network (Caron et al., 2010). Although the tested HNSCC cell lines were highly sensitive to AZD8055 (Supplemental Fig. 2), UMSCC8 and HN6 cells are not sensitive to the AKT inhibitor, MK2206, either as a single agent or in combination with gefitinib (Supplemental Fig. 6). Thus, we conclude that the essential function of MTOR unveiled in the RNAi screen can be largely attributed to TORC1 activity in these HNSCC cell lines.
MTOR was not highly ranked in the synthetic lethal screen with AZD8931 (Supplemental Table 2), although our previous study identified MTOR as synthetic lethal in the context of FGFR-specific TKIs in FGFR1-dependent lung cancer and HNSCC cell lines (Singleton et al., 2015). The distinction between MTOR functioning as a synthetic lethal interactor with FGFR1 but not with EGFR/ERBB complexes may be related to the more limited effector pathways driven by FGFR1 relative to EGFR and ERBB family members in the HNSCC cell lines tested in this study. FGFR-specific TKIs fail to reduce AKT-pS473 levels in FGFR1-dependent lung cancer and HNSCC cell lines (Singleton et al., 2015). However, this TORC2-mediated phospho-AKT site is inhibited by AZD8931 in UMSCC25 cells (Fig. 6), indicating that TORC2 is regulated downstream of EGFR/ERBB but not FGFR1 signaling. Importantly, AKT was not identified as an essential kinase (Supplemental Table 1) or as a high-ranking synthetic lethal interactor in the setting of AZD8931 treatment (Supplemental Table 2). Thus, unlike AKT inhibitors that synergize with FGFR-specific TKIs in FGFR1-dependent lung cancer and HNSCC cell lines (Singleton et al., 2015), AKT is neither an essential nor a synthetic lethal vulnerability in EGFR/ERBB-dependent HNSCC cell lines (Supplemental Fig. 6).
The present shRNA screen identified ERBB3 as the second highest–ranked essential protein kinase in EGFR/ERBB TKI-sensitive HNSCC cell lines. This finding is supported by the Dependency Map Project (https://depmap.org/portal/gene/ERBB3?tab=overview) in which HNSCC/upper aerodigestive cell lines along with esophageal cancer and HER2 amplified breast cancer cell lines exhibit significantly greater dependency for ERBB3 relative to other cancer cell lines. The HNSCC cell lines used in this study were not included in the Dependency Map Project and were selected for high sensitivity to gefitinib, an EGFR-specific TKI, as well as the pan-ERBB inhibitor, AZD8931 (Supplemental Fig. 1). Yet, neither EGFR nor ERBB2 was identified as essential kinases by the screen, supporting our previous study showing that these RTKs function within a coactivation network along with MET and FGFR family members such that significant growth inhibition is only achieved with a combination of distinct TKIs (Singleton et al., 2013). Important lateral interactions of EGFR and ERBB2 with other RTKs within the coactivation network likely account for the single agent sensitivity of the HNSCC cell lines to gefitinib and AZD8931 (Graus-Porta et al., 1997; Engelman et al., 2007; Kunii et al., 2008; Scaltriti et al., 2009; Liu et al., 2011; Li et al., 2013, 2015; Wang et al., 2015). The fact that ERBB3 exhibits an essential phenotype in HNSCC cells supports its function as an obligate partner for signaling through EGFR, ERBB2, MET, and, potentially, FGFR family members. Moreover, coactivation networks are not static, but highly dynamic in response to blockade of single RTKs with inhibitors (Tan et al., 2017). As an example, wild-type EGFR functions as a bypass signaling pathway stimulated within minutes following treatment with TKIs targeting oncogenic RTKs in lung adenocarcinoma (Vaishnavi et al., 2017). This RTK switching has also been observed in glioblastoma following inhibition of EGFR (Stommel et al., 2007; Huang et al., 2009). Thus, our results and the literature support the existence of a dynamic, multi-RTK complex as a dominant growth driver in HNSCC.
The synthetic lethal screen identified multiple components of the ERK MAPK pathway in the context of the pan-ERBB inhibitor, AZD8931. Although this result was unanticipated because the ERK MAPKs are direct downstream effectors of RTKs, we hypothesize that this finding derives from ERK reactivation through feedback regulation, as has been described in the literature (Ercan et al., 2012; Bruner et al., 2017; Zhou et al., 2017), as well as incomplete ERK pathway inhibition by AZD8931 due to residual stimulation through other proximal receptors that remain undefined. The ability of the MEK inhibitor, trametinib, to fully inhibit the residual phospho-ERK levels observed after 1 hour of AZD8931 treatment (Fig. 6; Supplemental Fig. 5) supports ERK MAPK pathway inputs distinct from EGFR/ERBB activity. With the exception of BRAF, the MAP3Ks identified as synthetic lethal hits by the screen (MAP3K2, MAP3K3, MAP3K6, MAP3K9, MAP3K11) are generally considered to be proximal activators of the p38 MAPK, JNK, and ERK5 MAPK pathways (Johnson, 2011). However, MAP3K11/MLK3 is required for EGFR activation of BRAF and ERK MAPKs (Chadee and Kyriakis, 2004). Moreover, both MAP3K9/MLK1 and MAP3K11 have been shown to function as MEK kinases independent of BRAF to stimulate ERK MAPK activity (Marusiak et al., 2014). Finally, acquisition of gain-of-function mutations in the MLK family of MAP3Ks is a candidate mechanism for acquired resistance to RAF protein kinase inhibitors (Marusiak et al., 2014).
Among the protein kinase vulnerabilities identified in this study, several might be considered for further preclinical exploration. The potency of the MTOR inhibitor, AZD8055, and the TORC1 inhibitor, rapamycin, in the HNSCC cell lines makes MTOR an attractive target for further investigation. A search of clinicaltrials.gov reveals that a clinical-grade MTOR kinase inhibitor, AZD2014, is being tested in patients bearing solid tumors, although not apparently in HNSCC. The TORC1-specific inhibitor, rapamycin, has been tested as a neoadjuvant therapy in HNSCC patients (Shirai et al., 2015). Among the 16 patients tested, rapamycin therapy (21 days) yielded one complete response and one partial response with the remaining patients experiencing stable disease. In addition, therapeutic-grade ERBB3-inhibitory antibodies have been developed and are being tested in early-phase clinical trials in HNSCC patients [(Aurisicchio et al., 2012; Reynolds et al., 2017) and NCT02473731, NCT01966445, NCT03254927]. If, in fact, ERBB3 does function as a central hub in a RTK coactivation network, then single-agent activity is predicted in patients. Finally, the strong synergism shown with combinations of gefitinib or AZD8931 and MEK inhibitors highlights the importance of clinically testing these particular combination therapies. The availability of multiple therapeutic-grade MEK inhibitors that have already been extensively tested in patients provides ample opportunity for clinical testing of the hypothesis that MEK/ERK pathway inhibitors will greatly enhance efficacy of EGFR inhibitors in HNSCC patients through deeper blockade of ERK MAPK signaling.
Acknowledgments
The shRNAs amplified from gDNA were sequenced by the University of Colorado Cancer Center Genomics core facility.
Abbreviations
- BINGS!
Bioinformatics for Next Generation Sequencing
- EGFR
epidermal growth factor receptor
- ERBB
erythroblastosis oncogene B
- ERK
extracellular signal-regulated kinase
- FGFR
fibroblast growth factor receptor
- gDNA
genomic DNA
- HNSCC
head and neck squamous cell cancer
- MAPK
mitogen-activated protein kinase
- MEK
mitogen-activated protein kinase kinase
- MLK1
mixed-lineage protein kinase 1
- MTOR
mechanistic target of rapamycin kinase
- p
phosphorylated
- PCR
polymerase chain reaction
- RNAi
RNA interference
- RTK
receptor tyrosine kinase
- shRNA
short hairpin RNA
- TKI
tyrosine kinase inhibitor
Authorship Contributions
Participated in research design: Hinz, Kleczko, Singleton, Tan, Heasley.
Conducted experiments: Hinz, Kleczko, Singleton, Calhoun, Marek.
Contributed new reagents or analytic tools: Hinz, Kleczko, Singleton, Kim, Tan.
Performed data analysis: Hinz, Kleczko, Singleton, Marek, Kim, Tan, Heasley.
Writing or contributed to the writing of the manuscript: Hinz, Kleczko, Singleton, Tan, Heasley.
Note Added in Proof—One of the groups supported by a VA grant was accidentally not included in the Fast Forward version published September 25, 2019. The funding footnote has now been corrected.
Footnotes
The studies were supported by a VA grant (Merit Award 1BX001994-01) to LEH and the UC Cancer Center Support Grant (P30 CA046934).
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Aurisicchio L, Marra E, Roscilli G, Mancini R, Ciliberto G. (2012) The promise of anti-ErbB3 monoclonals as new cancer therapeutics. Oncotarget 3:744–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlaam B, Anderton J, Ballard P, Bradbury RH, Hennequin LF, Hickinson DM, Kettle JG, Kirk G, Klinowska T, Lambert-van der Brempt C, et al. (2013) Discovery of AZD8931, an equipotent, reversible inhibitor of signaling by EGFR, HER2, and HER3 receptors. ACS Med Chem Lett 4:742–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bivona TG, Doebele RC. (2016) A framework for understanding and targeting residual disease in oncogene-driven solid cancers. Nat Med 22:472–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, Jones CU, Sur R, Raben D, Jassem J, et al. (2006) Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med 354:567–578. [DOI] [PubMed] [Google Scholar]
- Bonner JA, Harari PM, Giralt J, Cohen RB, Jones CU, Sur RK, Raben D, Baselga J, Spencer SA, Zhu J, et al. (2010) Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol 11:21–28. [DOI] [PubMed] [Google Scholar]
- Bruner JK, Ma HS, Li L, Qin ACR, Rudek MA, Jones RJ, Levis MJ, Pratz KW, Pratilas CA, Small D. (2017) Adaptation to TKI treatment reactivates ERK signaling in tyrosine kinase-driven leukemias and other malignancies. Cancer Res 77:5554–5563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron E, Ghosh S, Matsuoka Y, Ashton-Beaucage D, Therrien M, Lemieux S, Perreault C, Roux PP, Kitano H. (2010) A comprehensive map of the mTOR signaling network. Mol Syst Biol 6:453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadee DN, Kyriakis JM. (2004) MLK3 is required for mitogen activation of B-Raf, ERK and cell proliferation. Nat Cell Biol 6:770–776. [DOI] [PubMed] [Google Scholar]
- Cheng SW, Fryer LG, Carling D, Shepherd PR. (2004) Thr2446 is a novel mammalian target of rapamycin (mTOR) phosphorylation site regulated by nutrient status. J Biol Chem 279:15719–15722. [DOI] [PubMed] [Google Scholar]
- Chou TC, Talalay P. (1984) Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22:27–55. [DOI] [PubMed] [Google Scholar]
- Chresta CM, Davies BR, Hickson I, Harding T, Cosulich S, Critchlow SE, Vincent JP, Ellston R, Jones D, Sini P, et al. (2010) AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res 70:288–298. [DOI] [PubMed] [Google Scholar]
- Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, et al. (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316:1039–1043. [DOI] [PubMed] [Google Scholar]
- Ercan D, Xu C, Yanagita M, Monast CS, Pratilas CA, Montero J, Butaney M, Shimamura T, Sholl L, Ivanova EV, et al. (2012) Reactivation of ERK signaling causes resistance to EGFR kinase inhibitors. Cancer Discov 2:934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudelock DM, Jiang K, Pereira E, Russell B, Sanchez Y. (2003) Regulatory interactions between the checkpoint kinase Chk1 and the proteins of the DNA-dependent protein kinase complex. J Biol Chem 278:29940–29947. [DOI] [PubMed] [Google Scholar]
- Graus-Porta D, Beerli RR, Daly JM, Hynes NE. (1997) ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J 16:1647–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein MY, Hubner NC, Poser I, Cox J, Nagaraj N, Toyoda Y, Gak IA, Weisswange I, Mansfeld J, Buchholz F, et al. (2015) A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 163:712–723. [DOI] [PubMed] [Google Scholar]
- Hickinson DM, Klinowska T, Speake G, Vincent J, Trigwell C, Anderton J, Beck S, Marshall G, Davenport S, Callis R, et al. (2010) AZD8931, an equipotent, reversible inhibitor of signaling by epidermal growth factor receptor, ERBB2 (HER2), and ERBB3: a unique agent for simultaneous ERBB receptor blockade in cancer. Clin Cancer Res 16:1159–1169. [DOI] [PubMed] [Google Scholar]
- Huang PH, Xu AM, White FM. (2009) Oncogenic EGFR signaling networks in glioma. Sci Signal 2:re6. [DOI] [PubMed] [Google Scholar]
- Jhanwar-Uniyal M, Amin AG, Cooper JB, Das K, Schmidt MH, Murali R. (2017) Discrete signaling mechanisms of mTORC1 and mTORC2: connected yet apart in cellular and molecular aspects. Adv Biol Regul 64:39–48. [DOI] [PubMed] [Google Scholar]
- Johnson GL. (2011) Defining MAPK interactomes. ACS Chem Biol 6:18–20. [DOI] [PubMed] [Google Scholar]
- Jones RB, Gordus A, Krall JA, MacBeath G. (2006) A quantitative protein interaction network for the ErbB receptors using protein microarrays. Nature 439:168–174. [DOI] [PubMed] [Google Scholar]
- Kim J, Tan AC. (2012) BiNGS!SL-seq: a bioinformatics pipeline for the analysis and interpretation of deep sequencing genome-wide synthetic lethal screen. Methods Mol Biol 802:389–398. [DOI] [PubMed] [Google Scholar]
- Kim ST, Lim DS, Canman CE, Kastan MB. (1999) Substrate specificities and identification of putative substrates of ATM kinase family members. J Biol Chem 274:37538–37543. [DOI] [PubMed] [Google Scholar]
- Kunii K, Davis L, Gorenstein J, Hatch H, Yashiro M, Di Bacco A, Elbi C, Lutterbach B. (2008) FGFR2-amplified gastric cancer cell lines require FGFR2 and Erbb3 signaling for growth and survival. Cancer Res 68:2340–2348. [DOI] [PubMed] [Google Scholar]
- Li J, Bennett K, Stukalov A, Fang B, Zhang G, Yoshida T, Okamoto I, Kim JY, Song L, Bai Y, et al. (2013) Perturbation of the mutated EGFR interactome identifies vulnerabilities and resistance mechanisms. Mol Syst Biol 9:705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Puliyappadamba VT, Chakraborty S, Rehman A, Vemireddy V, Saha D, Souza RF, Hatanpaa KJ, Koduru P, Burma S, et al. (2015) EGFR wild type antagonizes EGFRvIII-mediated activation of Met in glioblastoma. Oncogene 34:129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wang Q, Yang G, Marando C, Koblish HK, Hall LM, Fridman JS, Behshad E, Wynn R, Li Y, et al. (2011) A novel kinase inhibitor, INCB28060, blocks c-MET-dependent signaling, neoplastic activities, and cross-talk with EGFR and HER-3. Clin Cancer Res 17:7127–7138. [DOI] [PubMed] [Google Scholar]
- Marusiak AA, Edwards ZC, Hugo W, Trotter EW, Girotti MR, Stephenson NL, Kong X, Gartside MG, Fawdar S, Hudson A, et al. (2014) Mixed lineage kinases activate MEK independently of RAF to mediate resistance to RAF inhibitors. Nat Commun 5:3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike KG, Malagu K, Hummersone MG, Menear KA, Duggan HM, Gomez S, Martin NM, Ruston L, Pass SL, Pass M. (2013) Optimization of potent and selective dual mTORC1 and mTORC2 inhibitors: the discovery of AZD8055 and AZD2014. Bioorg Med Chem Lett 23:1212–1216. [DOI] [PubMed] [Google Scholar]
- Politi K, Herbst RS. (2015) Lung cancer in the era of precision medicine. Clin Cancer Res 21:2213–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds KL, Bedard PL, Lee SH, Lin CC, Tabernero J, Alsina M, Cohen E, Baselga J, Blumenschein G, Jr, Graham DM, et al. (2017) A phase I open-label dose-escalation study of the anti-HER3 monoclonal antibody LJM716 in patients with advanced squamous cell carcinoma of the esophagus or head and neck and HER2-overexpressing breast or gastric cancer. BMC Cancer 17:646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaltriti M, Verma C, Guzman M, Jimenez J, Parra JL, Pedersen K, Smith DJ, Landolfi S, Ramon y Cajal S, Arribas J, et al. (2009) Lapatinib, a HER2 tyrosine kinase inhibitor, induces stabilization and accumulation of HER2 and potentiates trastuzumab-dependent cell cytotoxicity. Oncogene 28:803–814. [DOI] [PubMed] [Google Scholar]
- Seiwert TY, Fayette J, Cupissol D, Del Campo JM, Clement PM, Hitt R, Degardin M, Zhang W, Blackman A, Ehrnrooth E, et al. (2014) A randomized, phase II study of afatinib versus cetuximab in metastatic or recurrent squamous cell carcinoma of the head and neck. Ann Oncol 25:1813–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen JP, Zhao D, Sasik R, Luebeck J, Birmingham A, Bojorquez-Gomez A, Licon K, Klepper K, Pekin D, Beckett AN, et al. (2017) Combinatorial CRISPR-Cas9 screens for de novo mapping of genetic interactions. Nat Methods 14:573–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirai K, Day TA, Szabo E, Van Waes C, O’Brien PE, Matheus MG, Godwin K, Sood AJ, Vick JA, Martin D, et al. (2015) A pilot, single arm, prospective trial using neoadjuvant rapamycin prior to definitive therapy in head and neck squamous cell carcinoma. J Clin Oncol 33:6071. [Google Scholar]
- Singleton KR, Earley KT, Heasley LE. (2017) Analysis of drug resistance using kinome-wide functional screens. Methods Mol Biol 1636:163–177. [DOI] [PubMed] [Google Scholar]
- Singleton KR, Hinz TK, Kleczko EK, Marek LA, Kwak J, Harp T, Kim J, Tan AC, Heasley LE. (2015) Kinome RNAi screens reveal synergistic targeting of MTOR and FGFR1 pathways for treatment of lung cancer and HNSCC. Cancer Res 75:4398–4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton KR, Kim J, Hinz TK, Marek LA, Casás-Selves M, Hatheway C, Tan AC, DeGregori J, Heasley LE. (2013) A receptor tyrosine kinase network composed of fibroblast growth factor receptors, epidermal growth factor receptor, v-erb-b2 erythroblastic leukemia viral oncogene homolog 2, and hepatocyte growth factor receptor drives growth and survival of head and neck squamous carcinoma cell lines. Mol Pharmacol 83:882–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivas R, Shen JP, Yang CC, Sun SM, Li J, Gross AM, Jensen J, Licon K, Bojorquez-Gomez A, Klepper K, et al. (2016) A network of conserved synthetic lethal interactions for exploration of precision cancer therapy. Mol Cell 63:514–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R, Stegh AH, Bradner JE, Ligon KL, Brennan C, et al. (2007) Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 318:287–290. [DOI] [PubMed] [Google Scholar]
- Tan AC, Vyse S, Huang PH. (2017) Exploiting receptor tyrosine kinase co-activation for cancer therapy. Drug Discov Today 22:72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. (2017) Defining a cancer dependency map. Cell 170:564–576.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaishnavi A, Schubert L, Rix U, Marek LA, Le AT, Keysar SB, Glogowska MJ, Smith MA, Kako S, Sumi NJ, et al. (2017) EGFR mediates responses to small-molecule drugs targeting oncogenic fusion kinases. Cancer Res 77:3551–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, Erfan J, Zabolotnyy D, Kienzer HR, Cupissol D, et al. (2008) Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med 359:1116–1127. [DOI] [PubMed] [Google Scholar]
- Vermorken JB, Trigo J, Hitt R, Koralewski P, Diaz-Rubio E, Rolland F, Knecht R, Amellal N, Schueler A, Baselga J. (2007) Open-label, uncontrolled, multicenter phase II study to evaluate the efficacy and toxicity of cetuximab as a single agent in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck who failed to respond to platinum-based therapy. J Clin Oncol 25:2171–2177. [DOI] [PubMed] [Google Scholar]
- Wang J, Mikse O, Liao RG, Li Y, Tan L, Janne PA, Gray NS, Wong KK, Hammerman PS. (2015) Ligand-associated ERBB2/3 activation confers acquired resistance to FGFR inhibition in FGFR3-dependent cancer cells. Oncogene 34:2167–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu AM, Huang PH. (2010) Receptor tyrosine kinase coactivation networks in cancer. Cancer Res 70:3857–3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Wu Z, Wong G, Pectasides E, Nagaraja A, Stachler M, Zhang H, Chen T, Zhang H, Liu JB, et al. (2017) CDK4/6 or MAPK blockade enhances efficacy of EGFR inhibition in oesophageal squamous cell carcinoma. Nat Commun 8:13897. [DOI] [PMC free article] [PubMed] [Google Scholar]